De detectie van Lynch syndroom middels tumor analyse
Uitgangsvraag
Wat is het optimale beleid voor moleculair onderzoek op populatieniveau naar Lynch syndroom en bij onverklaarde MMR-deficiëntie?
De uitgangsvraag omvat de volgende deelvragen:
- Wat is de opbrengst van standaard tumoronderzoek (IHC MMR) en aanvullend moleculair onderzoek naar Lynch syndroom op populatieniveau bij patiënten >70 jaar met darm- en baarmoederkanker?
- Is er uitbreiding nodig van standaard tumoronderzoek (IHC MMR) en aanvullend moleculair onderzoek naar Lynch syndroom bij patiënten met andere Lynch-geassocieerde tumoren (ovariumkanker, pancreascarcinoom, galweg- en galblaascarcinoom, urotheelcarcinoom, dunnedarmcarcinoom, maagcarcinoom, hersentumoren, talgkliertumoren) en prostaatcarcinoom en naar andere leeftijdsgroepen?
- Welke adviezen zijn optimaal voor onverklaard (unexplained) MMRd (UMMRd) patiënten?
Aanbeveling
Aanbeveling-1
Verricht voor de detectie van Lynch syndroom routinematig IHC van de MMR-eiwitten en/of moleculaire MSI-analyse bij de volgende tumoren gediagnostiseerd < 70 jaar:
- Adenocarcinoom van de dikke en dunne darm
- Endometriumcarcinoom
- Niet-sereus epitheliaal ovariumcarcinoom
- Urotheelcelcarcinoom van de hogere urinewegen
- Talgklier tumoren
Verricht voor de detectie van Lynch syndroom routinematig IHC van de MMR-eiwitten en/of moleculaire MSI-analyse bij de volgende tumoren gediagnostiseerd < 50 jaar:
- Adenocarcinoom van de alvleesklier of galwegen (inclusief papil van Vater) en galblaas
- Adenocarcinoom van de maag
- Urotheelcelcarcinoom van de blaas
Aanbeveling 2
Voor routinematig onderzoek naar aanwijzing voor Lynch syndroom voldoet IHC-analyse van MLH1, MSH2, PMS2 en MSH6. In laboratoria kan bij CRC en EC ook gekozen worden voor initieel IHC PMS2 en MSH6 met in 2e instantie bij afwijkende of onduidelijke kleuring ook MLH1 en MSH2 analyse.
In geval van immunohistochemisch MLH1 (en PMS2) verlies, dient bij de onderzochte tumoren <70 jaar aanvullend MLH1 promoter methylatie (PM) onderzoek in de tumor te worden gedaan.
Verwijs voor kiembaandiagnostiek <50 jaar:
- Bij afwezige kleuring van MLH1, MSH2, MSH6 en/of PMS2
Verwijs voor kiembaandiagnostiek 50-70 jaar:
- Bij afwezige kleuring van, MSH2, MSH6 en/of PMS2
- Bij afwezige MLH1 kleuring, tenzij sprake is van MLH1-PM.
Indien bij IHC-onderzoek (dat wordt ingezet om andere redenen dan Lynch syndroom detectie) >70 jaar afwijkende MMR-kleuring wordt gevonden, verwijs dan voor kiembaan diagnostiek:
- Bij afwezige kleuring van alleen (geïsoleerd) MSH6 of PMS2
In algemeen geldt dat buiten deze criteria een belaste voorgeschiedenis of familieanamnese een reden kan zijn om toch te verwijzen naar de klinische genetica, zie de flowchart en submodule Verwijscriteria voor klinisch genetisch onderzoek bij CRC en genpanelanalyse
NB: omdat het aantonen van Lynch syndroom soms gevolgen kan hebben voor chirurgische behandelkeuzes (subtotale colectomie of segmentresectie en bij vrouwen eventueel gelijktijdige preventieve operatie van uterus en/of ovaria, zie submodule Chirurgische behandeling van Lynch syndroom), moet in deze gevallen patiënt met spoed worden verwezen naar de Klinische genetica.
Gebruik voor het rapporteren van de IHC MMR uitslag door de pathologie de standaard teksten via PALGA.
Aanbeveling-3
Bij negatieve IHC MMR zonder MLH1-PM in de tumor en geen kiembaan MMR-defect dient aanvullend somatisch DNA-analyse van de MMR genen in de tumor gedaan te worden in patiënten met een leeftijd van diagnose <60 jaar of bij blijvende aanwijzing voor een mogelijk kiembaan MMR-gendefect vanwege een belaste voor- of familieanamnese zoals beschreven in de flowchart en submodule Verwijscriteria voor klinisch genetisch onderzoek bij CRC en genpanelanalyse.
In geval dat er geen kiembaan of somatische verklaring voor afwijkende IHC MMR en/of MSI-High profiel in de tumor wordt gevonden, spreekt men van onverklaard MMRd (UMMRd). Afhankelijk van leeftijd en familieanamnese kan bij sterke verdenking gemist Lynch syndroom, Lynch-syndroom advies worden gegeven, of als voldaan wordt aan FCC-criteria een FCC-advies (zie submodule Incidentie, risico’s en surveillance bij familiair colorectaal carcinoom, submodule Colorectaal kankerrisico en surveillance bij het Lynch syndroom en submodule Extracolonische kankerrisico en surveillance bij het Lynch syndroom), In andere gevallen is het advies deel te nemen aan het bevolkingsonderzoek voor darmkanker.
Overwegingen
Er is literatuuronderzoek gedaan naar de opbrengst van standaard en aanvullend moleculair (tumor)onderzoek naar Lynch syndroom op populatieniveau. De vraag werd gesteld of moleculair tumoronderzoek moet worden uitgebreid naar >70 jaar bij CRC en EC en naar andere Lynch-geassocieerde maligniteiten en prostaatkanker. Voor deze uitgangsvraag is voor darm-, baarmoedercarcinoom gebruik gemaakt van recente reviews en meta-analyses aangevuld met bij de commissie bekende relevante literatuur, voor de andere tumor typen is een analyse gedaan van studies van de afgelopen 10 jaar. Wat betreft adviezen voor onverklaard MMRd (UMMRD) cases is ook gebruikt gemaakt van recente reviews en meta-analyses aangevuld met bij de commissie bekende relevante literatuur.
Balans tussen gewenste en ongewenste effecten
Algemene tekst
Lynch syndroom is de meest voorkomende vorm van erfelijke darmkanker, waarbij ook een verhoogd risico op baarmoederkanker en in mindere mate ook op andere vormen van kanker aanwezig is. Dit syndroom erft autosomaal-dominant over. Het wordt veroorzaakt door kiembaan pathogene varianten (PV) in genen betrokken bij een DNA-herstel systeem, genaamd mismatch repair (MMR). De genen waar het om gaat zijn MLH1, MSH2, PMS2 en MSH6. Deleties van het EPCAM-gen kunnen ook leiden tot MSH2 functieverlies en daarmee Lynch syndroom. Bij het MMR-systeem zijn nog meer eiwitten betrokken, maar tot nu toe is niet duidelijk aangetoond dat mono-allelische varianten in deze genen ook leiden tot een hoger kanker risico. Wel is er voor MSH3 en MLH3 bekend dat bi-allelische mutatie dragers een verhoogd risico hebben op onder andere polyposis. Hier wordt in de submodule over genetische testen bij adenomateuze polyposis (zie submodule Genetische testen bij adenomateuze polyposis) kort ingegaan. De dragerschapsfrequentie in de bevolking van één van de vier vormen van Lynch syndroom is 1 op 274 (Win, 2017); voor de individuele genen is dit: MLH1: 1 op 1,946, MSH2: 1 op 2,841, MSH6: 1 op 758, PMS2: 1 op 714.
In een cel treedt verlies van het MMR-systeem op als een of meerdere MMR-eiwitten niet meer worden aangemaakt. Dit gebeurt als beide gen kopieën coderend voor het betreffende MMR-eiwit worden uitgeschakeld. Uitschakeling van een gen in een tumor kan worden veroorzaakt door (i) een constitutionele (kiembaan) PV, (ii) een somatische PV, (iii) somatische uitschakeling door methylatie van de promotersequentie (iv) kiembaan uitschakeling door promoter methylatie (v) of verlies van het hele allel (LOH) waar het gen op ligt. Bij iemand met een kiembaan PV (Lynch syndroom) is er alleen uitschakeling van de tweede gen kopie nodig voordat MMR-deficiëntie ontstaat -bij iemand zonder kiembaan PV moeten in beiden gen kopieën nieuwe events optreden.
Technieken om MMR-deficiëntie aan te tonen zijn moleculaire microsatelliet (MSI) analyse en immunohistochemisch onderzoek van de MMR-eiwitten (IHC MMR). Beide technieken zijn vergelijkbaar wat betreft het opsporen van Lynch syndroom. Voor routinematig screenen van tumoren voor Lynch syndroom werd in de richtlijn van 2015 immunohistochemisch onderzoek geadviseerd, omdat dit goedkoper en sneller is en daarbij makkelijker te implementeren in ieder pathologisch laboratorium. Wel wordt het traditionele MSI-onderzoek met gebruik van met name voor darm- en baarmoederkanker gevalideerde markers in toenemende mate vervangen door op NGS gebaseerde MSI-analyse. Zie ook de landelijke richtlijn darmkanker Richtlijn colorectaal carcinoom voor beschrijving MMR/MSI onderzoek bij CRC.
Sinds 2015 is het in Nederland standaard beleid om in elke darmkanker (CRC) en baarmoederkanker (EC) bij patiënten onder de 70 jaar na te gaan of er sprake is van mismatch repair deficiëntie (MMRd) middels immunohistochemisch onderzoek (IHC MMR) van de tumor. Indien er geen aankleuring is van 1 of 2 van de MMR-eiwitten, en er in geval van afwezige kleuring van MLH1 en PMS2 geen methylatie van de MLH1 promoter aanwezig is in de tumor, is er indicatie om na te gaan of sprake is van een kiembaan pathogene variant in één van de MMR genen via de klinisch geneticus. Door dit beleid is het aantal families met de diagnose Lynch syndroom toegenomen.
Inmiddels is er een andere belangrijke reden voor MMR-deficiëntie (MMRd)-onderzoek bij gekomen. Het is namelijk ook van belang de MMR-status van tumoren te weten omdat behandelkeuzes hiervan af kunnen hangen: MMRd tumoren (zowel bij sporadisch als in het kader van Lynch syndroom) kunnen gevoeliger zijn voor immuuntherapie en minder voor reguliere chemotherapie. Veel laboratoria doen daarom inmiddels ook MMRd-analyse middels IHC MMR of moleculair onderzoek bij CRC en EC-patiënten >70 jaar en andere tumoren. Een richtlijn voor informatie en informed consent bij moleculaire tumor diagnostiek en een leidraad voor verwijzing voor kiembaan diagnostiek bij het vinden van een aanwijzing voor een erfelijke aanleg voor kanker zijn ontwikkeld, zie ook: Erfelijke-aanleg-voor-kanker. Deze submodule richt zich op de rol van tumordiagnostiek bij de detectie van Lynch syndroom.
De vraag nu is wat de opbrengst van MMRd-analyse in verschillende leeftijdsgroepen en tumortypen is en of een aanpassing van het huidige richtlijn advies nodig is. Ook is de vraag in hoeverre kiembaan en/of somatische MMR-gen analyse verricht moet worden indien MMRd in een tumor wordt aangetoond en wat het beleid is indien de MMRd onverklaard (UMMRd) blijft.
Technieken om MMR-deficiëntie op te sporen
Voor de detectie van mismatch repair deficiëntie kan immunohistochemische kleuring (IHC) van de MMR-eiwitten MLH1, PMS2, MSH2 en MSH6 verricht worden. Omdat MLH1- en MSH2-deficiëntie praktisch altijd samengaat met respectievelijk PMS2- en MSH6-verlies, kan eerst PMS2- en MSH6-kleuring worden verricht. Bij een verlies van aankleuring volgt kleuring van MLH1 en MSH2, mede om te bepalen of MLH1-promoter-methylatieonderzoek nodig is en als aanwijzing welk gen meest waarschijnlijk aangedaan is. Een twee-antilichaam MMR-teststrategie leidt niet tot significante fouten in de identificatie van dMMR en bespaart kosten, zoals aangetoond in meerdere Nederlandse studies (Stello, 2017; Post, 2021; Vink-Börger, 2024). Ook Snowsill (2020) bevestigde de betrouwbaarheid en kosteneffectiviteit van deze aanpak.
IHC-evaluatie van MMR-eiwitten is niet altijd eenvoudig. Interpretatie is afhankelijk van de expertise van de patholoog en de kwaliteit van technieken en materialen. Uitdagend zijn de interpretatie van heterogene of subclonale kleuringspatronen, en zwakkere kleuring intensiteiten, die ten onrechte geduid kunnen worden als intacte MMR-expressie. De sensitiviteit van IHC-MMR kan afnemen door somatische of kiembaan missense-mutaties, die leiden tot een weliswaar stabiel eiwit met behouden antilichaambinding, maar zonder MMR-functie. Bou Farhat (2024) toonde aan dat bij 1% van de CRC/EC's dMMR wordt gemist bij gebruik van alleen IHC-MMR. Zij pleiten voor NGS in de diagnostiek, al lijken de resultaten overschat door methodologische beperkingen. Hechtman (2020) rapporteerde dat in 6% van de MMRd-patiënten (door kiembaan- of somatische mutaties) intacte MMR-kleuring aanwezig was, voor de praktijk zou dit dan kunnen betekenen dat rond de 0,5% van MMRd EC en CRC worden gemist met standaard IHC MMR in deze groepen tumoren. Deze 6% intacte kleuring in MMRd patiënten is echter mogelijk ook een overschatting. Singh et al. geven in antwoord op dit onderzoek aan dat niet alleen volledige afwezigheid van kleuring, maar bijvoorbeeld ook heterogene kleuring of ‘dot-like nuclear kleuring’ reden is tot verder onderzoek (Singh 2021).
Bij moleculaire MSI-analyse wordt gekeken naar microsatellieten, korte repetitieve DNA-sequenties. Bij een defect MMR-systeem zal de lengte van microsatellieten gaan variëren, wat leidt tot MSI (van Lier, 2010). Waar in het verleden gebruik werd gemaakt van fragmentlengte-analyse van vijf gestandaardiseerde markers (klassieke pentaplex-MSI), wordt MSI-analyse tegenwoordig doorgaans verricht via NGS-analyse van veel meer markers, zeker als het onderdeel is van een veel bredere moleculaire analyse van de tumor. Hoewel de MSI-test een belangrijk alternatief blijft voor IHC bij MMRd-detectie, is deze in algemene zin duurder, duurt de uitslag langer en is de test minder breed toepasbaar in pathologie laboratoria.
Conclusie: IHC-MMR en MSI-tests blijven relatief gevoelige, snelle en kosteneffectieve methoden voor MMRd-detectie en Lynch syndroom screening.
MLH1 promoter methylatie (PM) in tumor en kiembaan
MLH1-PM komt voor in 4-10% van alle CRC en 18-20% van de EC (Eikenboom, 2022, Ryan, 2019; Loong, 2024), neemt sterk toe met leeftijd en is sterk geassocieerd met een BRAF hotspot mutatie, rechtszijdig CRC, bepaalde histologische kenmerken en vrouwelijk geslacht (Advani, 2021; Christenson, 2023). Bijna altijd gaat het om een somatisch, nieuw ontstane verandering. Het is daarom wenselijk dat alvorens bij afwezige MLH1 (en PMS2) kleuring in een tumor kiembaan diagnostiek te adviseren, eerst MLH1-PM analyse wordt verricht.
In een klein deel van de met name jonge patiënten kan sprake zijn van kiembaan of post zygotisch MLH1-PM, vaak in mozaïek (Hitchins, 2023). Deze mensen hebben een verhoogde kans op nogmaals tumorvorming en het is daarom belangrijk om dit te weten voor vervolg controles. Vaak ontstaat deze kiembaan MLH1-PM de-novo als gevolg van een "primaire epimutatie" zonder duidelijke genetische basis; dragers hebben daarom meestal geen aangedane familiegeschiedenis. Er kan echter ook sprake zijn van een onderliggende variant in de MLH1 promoter regio, waarbij familieleden wel een verhoogde kans hebben op ook kiembaan MLH1-PM.
Het voorkomen MLH1-PM in de kiembaan (constitutional MLH1 promoter methylation) betreft in ongeselecteerde CRC-cohorten ongeveer 0,6-1,0% van alle mensen met een negatieve IHC MLH1, (Ward, 2013; Castillejo, 2015; Pearlman, 2017; Zyla, 2021; Niessen, 2009; Joo, 2023; Helderman, 2024). In cohorten geselecteerd op jonge leeftijd en familiegeschiedenis ligt dit tussen de 4 tot 16% (Castillejo, 2015; Niessen, 2009; van Roon, 2010; Pineda, 2012; Crucianelli, 2014 Goel, 2011) met diagnose-leeftijd variërend tussen de 18-60 jaar. De Amerikaanse studie uit 2023 van Hitchins (2023) beschrijft een analyse in 2 population-based, ongeselecteerde cohorten van respectievelijk 1,566 en 3310 CRC waar in 105 (6,7%) en 281 (8.5%) CRCs respectievelijk MLH1-PM was aangetoond in de tumor. In deze studie werd vervolgens, indien DNA beschikbaar was voor verder onderzoek, MLH1 kiembaan PM aangetoond in respectievelijk 4% (4/95) en 1.4% (4/281). Op één na (een man met CRC op 74 jaar) waren al deze patiënten met kiembaan MLH1-PM 55 jaar of jonger. Ondanks dat kiembaan (post-zygotisch) MLH1-PM zeldzaam is in het totale cohort, is het frequent aanwezig in CRC met MLH1 PM < 50 jaar. Een eerdere studie uit 2021 is onderdeel van dit cohort en wordt niet apart besproken (Pearlman, 2021). In het eerste cohort waren er 10 van de 105 (9,5%) gevallen met MLH1-PM < 60 jaar; In het tweede grotere cohort was dit 35 van de 281 (11%). Het testen van deze patiënten met MLH1-PM in de tumor op kiembaan MLH1-PM geeft een detectie ratio van 16% (7/45). In de leeftijdsgroep 55-59 jaar is dit echter slechts 1 op de 21. De auteurs stellen daarom voor om de leeftijdsgrens voor kiembaan analyse op 55 jaar te stellen. In een Nederlandse studie is ook maar één keer kanker op 60-jarige leeftijd gevonden (van Roon, 2010). Ook is beschreven dat MLH1-PM en kiembaan MMR-varianten elkaar niet uitsluiten (Helderman, 2024), dus dat MLH1-PM de 2e hit in de tumor is. Het lijkt daarom voor nu raadzaam om ook bij het detecteren van MLH1-PM in de tumor te verwijzen voor kiembaan MLH1-PM en MLH1 DNA-onderzoek bij darmkankerpatiënten met een leeftijd van diagnose <50 jaar naast andere factoren die wijzen op een mogelijk MMR-gendefect.
Er is meer onderzoek nodig om te zien of deze leeftijdgrens van <50 jaar sensitief genoeg is. Om die reden is hierover ook een kennisvraag geformuleerd.
Over het voorkomen van kiembaan MLH1-PM in geval van EC en andere tumoren zijn nog weinig data beschikbaar. Een kleinere studie beschrijft drie losse patiënten met EC met MLH1 kiembaan PM en daarnaast een EC-cohort studie (bestaande uit twee verschillende population-based cohorten van EC-patiënten met MLH1-PM) waarin één patiënt met een laag-niveau mozaïek MLH1-PM werd aangetoond. Dit komt neer op 2% van de patiënten onder 60 jaar (1/45 EC met MLH1-PM) en 17% (1/6 met MLH1-PM) van de patiënten onder/gelijk aan 50 jaar (Hitchins, 2023). Daarnaast is beschreven dat MLH1-PM ook bij EC voor kan komen bij patiënten met een onderliggend MMR-kiembaan defect (Helderman, 2024). Van belang is wel dat MLH1-PM veel vaker voorkomt bij EC dan bij CRC, ook onder de 60 jaar (Kahn, 2019, Loong, 2024; Andersson, 2024; Kaya, 2024). Voor andere tumoren is niet goed bekend hoe vaak MLH1-PM voorkomt en hoe vaak dit kiembaan methylatie betreft. Voor nu is, gezien ook de kleine aantallen en ten behoeve van de uniformiteit het voorstel om alle Lynch geassocieerde tumoren met MLH1-PM <50 jaar door te verwijzen voor aanvullend MLH1-PM onderzoek in kiembaan/ normaal weefsel en onderzoek naar een eventueel onderliggend MMR-defect, omdat MLH1-PM ook een second hit kan zijn of los kan staan van onderliggend MMR-kiembaan defect.
De opbrengst van tumoronderzoek naar Lynch syndroom bij patiënten met Lynch geassocieerde maligniteiten
MMRd-analyse in andere tumoren dan darm-/baarmoeder kanker voor de detectie van Lynch syndroom
In toenemende mate wordt MMRd-analyse verricht in het kader van het afstemmen van behandeling. Hierbij wordt met enige regelmaat MMRd gevonden in andere tumoren dan CRC en EC, waarbij de vraag rijst of er kiembaan diagnostiek verricht moet worden en of MMRd analyse niet ook routinematig in andere tumoren dan CRC en EC verricht zou moeten worden. Een studie door Latham (2019) heeft MSI en IHC-analyse verricht in 15,054 patiënten met meer dan 50 verschillende carcinomen en laat zien dat 50% van de gedetecteerde Lynch syndroom patiënten (met een bewezen kiembaan mutatie) carcinomen heeft anders dan colorectaal of endometriumcarcinoom.
In de literatuur werd gezocht naar prevalenties van MMRd of MSI-H en MMRd gerelateerd aan MLH1 promoter methylatie, kiembaan mutaties, somatische mutaties en onverklaarde MMRd van de diverse tumorsoorten. In eerste instantie werden per tumortype systematische reviews en meta-analyses geselecteerd. Dit is vervolgens aangevuld met cohortstudies voor de tumortypen waarvoor geen meta-analyses beschikbaar waren. Alle geselecteerde onderzoeken beschreven per tumortype, al dan niet volledig, de prevalentie van afwijkende IHC-MMR en/of MSI-H, MLH1-PM, Lynch syndroom en somatische varianten. Een samenvatting hiervan is gegeven in tabel 2.
Ovarium kanker:
Op basis van de meta-analyse van Mitric (2023) kan geconcludeerd worden dat 6% van de ovariumtumoren een MMRd profiel laat zien afhankelijk of immunohistochemie of moleculaire MSI is verricht. In endometroïd type ovariumcarcinoom, de meest voorkomende vorm van ovariumcarcinoom bij Lynch syndroom, toont zowel immunohistochemisch als moleculair 12% van de tumoren een MSI-profiel, in sereuze tumoren is dat 1% (Mitric 2023).
Afhankelijk van type ovariumtumor wordt in 1-3% Lynch syndroom aangetoond, waarbij het minst frequent in sereus type ovariumcarcinoom, wat de meest voorkomende vorm van ovariumcarcinoom is. Alle ovariumcarcinomen worden in het kader van tumor first diagnostiek voor een BRCA aanleg moleculair getest, Het meest doelmatig lijkt echter om alleen in niet-sereuze ovariumcarcinomen moleculaire MSI of IHC MMR analyse te verrichten. Omdat bij 76% van de MSI-H tumoren, met een MLH1-profiel, sprake was van methylatie van de MLH1 promoter moet MLH1-PM eerst uitgesloten worden, voordat de patiënt verwezen wordt naar de klinische genetica voor kiembaan diagnostiek, tenzij sprake is van MLH1-PM <50 jaar.
Pancreas, galblaas, galweg (inclusief papil van Vater) kanker:
Waarschijnlijk kan in ongeveer 5% van de pancreas en galweg-, galblaas adenocarcinomen IHC dMMR worden aangetoond, hoewel bij galblaaskanker zelden een MMRd profiel lijkt te worden gevonden. In totaal wordt bij ongeveer 1% Lynch syndroom aangetoond. Deze getallen stijgen naarmate leeftijd van diagnose jonger is; 2,8-4,5% bij diagnose <50 jaar. Routinematig verrichten van IHC dMMR lijkt daarom doelmatig bij diagnose <50 jaar. Hoewel data beperkt zijn, lijkt ook methylatie van de MLH1-promoter bij deze tumoren in de tumor verantwoordelijk te kunnen zijn voor een MLH1-profiel. Indien sprake is van MLH1-PM <50 jaar lijkt verwijzing naar de klinische genetica op dit moment wel zinvol.
Urotheel cel carcinoom hogere urinewegen en blaas:
Op basis van de huidige gegevens komt bij waarschijnlijk 5-10% van de hogere urineweg urotheelcel carcinomen (UTUC) dMMR en/of MSI voor en wordt in ongeveer de helft daarvan een kiembaan PV gevonden met name in MSH6 of MSH2. Omdat naar verwachting ook bij deze tumoren de kans op een kiembaan aanleg afneemt met toenemende leeftijd, lijkt het doelmatig om IHC MMR te verrichten in alle UTUC gediagnostiseerd <70 jaar. Methylatie van de MLH1-promoter is ook bij deze tumoren beschreven en dient uitgesloten te zijn bij een MLH1-profiel in de tumor, alvorens te verwijzen voor kiembaan diagnostiek, tenzij er sprake is van MLH1-PM <50 jaar.
Gezien het veel frequenter voorkomen van blaas urotheelcelcarcinoom (BUC), met een lage frequentie van MMRd (1,5%) en Lynch syndroom (1%), wordt routinematig MMRd analyse in BUC niet doelmatig geacht, tenzij dit op ongebruikelijk jonge leeftijd (<50 jaar) voor komt.
Dunne darmkanker en maagkanker:
Op basis van de beschikbare studies heeft 20-30% van de adenocarcinomen van de dunne darm een MMRd profiel met name die in het duodenum. In 5-10% wordt een kiembaan aanleg voor Lynch syndroom aangetoond. Gezien de zeldzaamheid van de tumor kan overwogen worden routinematig IHC MMR analyse te doen in alle dunne darmtumoren, maar dit dient in ieder geval bij diagnose <70 jaar te gebeuren. Omdat bij afwijkende aankleuring MLH1/PMS2 regelmatig methylatie van de MLH1-promoter gerapporteerd is, dient, alvorens te verwijzen naar de klinische genetica voor kiembaan diagnostiek eerst MLH1-PM in de tumor uitgesloten te zijn bij dit profiel, tenzij er sprake is van MLH1-PM <50 jaar.
Ongeveer 10% van de adenocarcinomen van de maag vertonen IHC dMMR. Echter, op basis van de beperkte data wordt slechts bij ongeveer 1% Lynch syndroom aangetoond. Routinematige MMRd-analyse voor de detectie van Lynch syndroom lijkt bij maagkanker daarom op dit moment alleen geïndiceerd wanneer deze op ongebruikelijk jonge leeftijd (<50 jaar) gediagnostiseerd wordt.
Hersentumoren:
Bij Lynch syndroom kunnen gliale hersentumoren voorkomen. Rodriguez-Hernández (2013) beschrijven dat IHC MMR eiwit expressie afwijkingen frequent (43%) voorkomen in laag- en hooggradige astrocytomen. Met moleculaire MSI-analyse wordt aanzienlijk minder MSI aangetoond. Ook Hadad (2023) vinden maar in 2% van glioblastomen MSI. Zowel IHC van de MMR-eiwitten als MSI is in de praktijk lastig te interpreteren. Het is ook bijzonder dat deze studie naast methylatie van de MLH1-promoter ook methylatie van de MSH2- en MSH6-promoter meldt, welke ook niet overeenkomt met het IHC-profiel van deze tumoren. Meest waarschijnlijk is de frequentie van 43% zoals gevonden door Rodriquez-Hernandez een sterke overschatting. In 1-5% van de gliale hersentumoren bleek sprake van Lynch syndroom. Op basis de huidige data is routinematig testen van gliale hersentumoren op MSI voor de detectie van Lynch syndroom niet aan de orde.
Talgkliertumoren:
Talgkliertumoren horen bij het tumor spectrum van Lynch syndroom, voorheen benoemd als Muir-Torre syndroom. In Nederland komen ongeveer 200 patiënten met een adnextumoren van de huid (waaronder talgkliertumoren) per jaar voor, waarvan 50 onder de 70 jaar (IKNL, 2022-2023). Takliertumoren worden ingedeeld in talgklieradenomen, talgklierepitheliomen (=sebaceoma) en talgkliercarcinomen. De ligging wordt verdeeld in (1) perioculair/ooglid en extraoculair (Ferreira, 2020).
Twee studies lieten zien dat MMR-deficiëntie in 30- 50% van de onderzochte talgkliertumoren voorkwam (Cook 2023, Kataapuram 2023). In de laatste studie werd in 15% Lynch syndroom aangetoond, maar in deze studie betrof dit wel een mogelijk geselecteerde groep aangezien maar in een deel verdere diagnostiek verricht werd.
Over het algemeen zijn talgklieradenomen en extra-oculaire laesies vaker MMR deficiënt, maar de frequentie van Lynch syndroom is in alle talgklier tumoren >10% (Gaskin, 2011; Sinson, 2023; Singh, 2008). Dit percentage zal waarschijnlijk lager liggen in de oudere leeftijdsgroepen, maar dit is niet uitgebreid onderzocht. Gezien de huidige data en omdat het een vrij zeldzame tumor betreft, lijkt het doelmatig in ieder geval routinematig IHC MMR te verrichten bij diagnose <70 jaar. Bij de casus boven de 70 jaar kan alsnog IHC MMR en/of kiembaan analyse ingezet worden in geval van een talgklieradenoom, een extra-oculaire ligging, een voorgeschiedenis van Lynch geassocieerde tumoren en/of belaste familieanamnese. Er zijn beperkt data in hoeverre methylatie van de MLH1-promoter voorkomt, maar dit lijkt zeldzaam; wel zijn enkele casus beschreven met kiembaan MLH1-PM (tussen 45-55 jaar) en dit moet overwogen worden in patiënten onder de 50 jaar (Walker, 2023; Zyla, 2021).
Prostaatkanker:
MSI is zeldzaam in prostaatkanker (3% of minder) en Lynch Syndroom nog zeldzamer. Mede gezien het een veel voorkomende tumor betreft, is er geen plaats voor routinematig testen van prostaatkanker op MMRd. Indien MMRd niet-routinematig wordt aangetoond, kan de leidraad voor verwijzing bij aanwijzing voor erfelijke aanleg voor kanker na tumor diagnostiek (www.artsengenetica.nl) gebruikt worden om te bepalen of verwijzing voor kiembaan diagnostiek geadviseerd wordt.
De opbrengst van tumoronderzoek naar Lynch syndroom bij patiënten met darmkanker en baarmoederkanker.
Colorectaal carcinoom (CRC)
Een Nederlandse studie van Vos (2020) liet de opbrengst zien van universeel IHC MMR en kiembaan analyse in een prospectief Nederlands cohort. In 3602 nieuw gediagnosticeerde CRCs jonger dan 70 jaar uit 19 ziekenhuizen, werd onderzoek gedaan naar MMR-deficiëntie (MMRd), MLH1-promoter methylatie (MLH1-PM), kiembaan en biallelische somatische MMR-events. De opbrengst werd geëvalueerd met behulp van gegevens uit het Nederlands Pathologie Register (PALGA) en twee regionale genetische centra. Pathologen pasten routinematig MMR-testen toe bij 84% van de CRCs en 10% bleek MMRd, waarvan 8% MLH1/PMS2 gerelateerd, grotendeels als gevolg van somatische MLH1-PM (66%). Van de patiënten met MMRd CRC zonder MLH1-PM werd 69% doorverwezen voor kiembaan diagnostiek, waarvan 55% verklaard werd door Lynch syndroom en 43% door somatische biallelisch MMR-events. De prevalentie van Lynch syndroom was 18% in CRC <40 jaar, 1,7% in CRC tussen de 40-64 jaar en 0,7% in CRC tussen de 65-69 jaar. De meeste gevallen met een dMMR tumor werden gevonden in de leeftijd 65-69 jaar, waarbij dMMR als gevolg van somatische oorzaken (13%) 20 keer vaker voorkwam dan het Lynch syndroom. In deze studie concluderen de auteurs dat tot de leeftijd van 65 jaar routinematige diagnostiek van (niet-) erfelijke oorzaken van dMMR CRCs effectief het Lynch syndroom identificeert en onverklaarde MMRD-diagnose vermindert. Boven de leeftijd van 64 jaar is de kans op Lynch syndroom echter klein.
Een meta-analysis in 2022 (Eikenboom, 2022) keek naar het aandeel patiënten met Lynch syndroom, met een sporadische MMRd tumor en onverklaarbare gevallen van MMRd-tumoren (UMMRd) in 58.580 niet-geselecteerde CRCs. Ongeveer 1 op de 10 CRCs had een MMR-deficiëntie, dit werd in ongeveer de helft verklaard door MLH1-promoter methylatie (MLH1-PM). Van alle patiënten met MMRd, die kiembaan genetisch onderzoek ondergingen, werd bij 38% een pathogene MMR-kiembaanvariant geïdentificeerd. In 2% van alle CRCs werd een kiembaan MMR pathogene variant (PV) aangetoond. Dit percentage steeg tot 7% en 5% van alle CRCs bij patiënten jonger dan respectievelijk 50 en 70 jaar. Verder werd duidelijk dat het percentage biallelische somatische events of kiembaanvarianten afhankelijk is van het specifieke MMR-eiwit dat ontbreekt bij kleuring. Voor MSH6 en PMS2 wordt de afwijkende eiwitkleuring vaker verklaard door een kiembaan PV voor dan voor MLH1 en MSH2. In 4% van het totale cohort werd geen verklaring voor de MMRd gevonden. Dit percentage bevat waarschijnlijk veel sporadische MMR-deficiënte CRCs omdat in meeste studies geen somatische test gedaan is. Volledige diagnostiek leidde tot een percentage onverklaarde MMRd (UMMRd) van slechts 0,6%.
Een retrospectieve cohortstudie van Christonson (2023) onderzocht 544 CRCs met next-generation sequencing en mismatch repair (MMR)-analyse. Ze vergeleken moleculaire en klinische kenmerken van 251 patiënten met CRC gediagnostiseerd tussen de 50-69 jaar en 60 patiënten met CRC >80 jaar. CRC kwam vaker rechtszijdig voor (82% vs. 35%), toonde vaker MMR-deficiëntie (35% vs. 8%) en BRAF p.V600E-mutaties (35% vs. 8%), maar toonde minder vaak stadium IV-ziekte (15% vs. 28%) en APC-mutaties (52% vs. 78%) in de groep >80 jaar. De toename van MMR-deficiëntie bij oudere patiënten werd vooral gezien bij vrouwen (48% vs. 10% bij mannen). BRAF p.V600E-mutaties kwamen vaker voor bij MMR-deficiënte CRC op latere leeftijd (67%). De conclusie van deze studie is dat CRC op latere leeftijd wordt gekenmerkt door een hogere frequentie van MMR-deficiëntie en BRAF p.V600E-mutaties, vooral bij vrouwen.
McRonald (2024) analyseerden de nationale dataset van de “National Cancer Registration and Analysis Service (NCRAS)”. Van alle patiënten met colorectale kanker (CRC), die in 2019 in Engeland werden gediagnosticeerd, werden uitslagen van dMMR-analyse (via immunohistochemie of microsatelliet instabiliteit), MLH1-promoter methylatie (MLH1-PM) en BRAF status, en kiembaan analyse van de MMR genen geïnventariseerd. Van de 8000 geteste CRC-patiënten waren 6000 bij diagnose ≤70 jaar oud. Slechts 44% van de CRCs werd getest op mismatch repair-deficiëntie (dMMR); 16% daarvan vertoonde dMMR of microsatellietinstabiliteit (MSI). Van deze dMMR/MSI-gevallen onderging slechts 51% verdere diagnostische tests. Van de MLH1-deficiënte tumoren had 67% (275/413) MLH1-PM. Slechts 1,3% van alle CRC-patiënten onderging een kiembaan (germline) MMR-genetische test; tot 37% van deze tests werd uitgevoerd buiten de NICE-richtlijnen. Van de dMMR-gevallen zonder MLH1-PM had 49% (103/210) een kiembaan pathogene variant (PV) in een MMR-gen, waarvan 25 in MLH1. Concluderend werd een aanzienlijk deel van de CRC-patiënten niet getest op dMMR of onderging geen verdere diagnostiek. Daarnaast werden veel kiembaan MMR-tests buiten richtlijnen uitgevoerd.
Abu-Ghazaleh (2022) deden een systematische review en meta-analyse van de literatuur in MEDLINE (Ovid), Embase en Web of Science om de prevalentie van Lynch syndroom in CRC-patiënten te berekenen. In totaal werden 51 studies geanalyseerd. De geschatte prevalentie van Lynch syndroom was 2,2% op basis van alle detectiemethoden. Studies die kiembaan (germline) tests uitvoerden op alle CRC-patiënten vonden een hogere prevalentie (5,1%) dan studies die alleen kiembaan tests deden bij patiënten met mismatch repair-deficiëntie (1,6%) of microsatellietinstabiliteit (1,1%). Echter, de hogere prevalentie bij directe kiembaan analyse is waarschijnlijk overschat omdat veel studies vooraf hoog-risicopatiënten selecteerden. Studies die CRC-patiënten ouder dan 75 jaar includeerden, rapporteerden een lagere Lynch syndroom-prevalentie (1,7% [95% CI = 1,3%–2,1%]) dan studies zonder deze oudere patiënten (4,4% [95% CI = 3,2%-5,5%]). Concluderend varieert de geschatte prevalentie van Lynch syndroom afhankelijk van de screeningsmethode en hebben oudere CRC-patiënten (>75 jaar) een lagere detectiekans van Lynch syndroom.
Kunnackal (2022) voerden een systematische review van alle publicaties in de MEDLINE-database tussen 2005 en 2017 uit om de effectiviteit van universele screening voor Lynch syndroom en de detectie van kiembaanmutaties in mismatch repair (MMR)-genen bij verschillende Lynch syndroom -gerelateerde tumoren te analyseren. De opbrengst van Lynch syndroom -screening en MMR-kiembaanmutaties verschilde significant per type tumor (p < 0.001) en was in endometriumkanker (EC) respectievelijk 22,65% en 2,6% en in colorectale kanker (CRC) 11,9% en 1,8%. Lynch syndroom -screening in EC detecteerde significant meer biallelische somatische events dan in CRC (16,94% vs. 5,23%, p < 0.0001). Routine Lynch syndroom -screening wordt aanbevolen voor meerdere tumortypen, maar er zijn geen gegevens over de invloed van leeftijd op de detectie van kiembaanmutaties.
West (2021) beschrijft een prospectieve studie, waarin alle nieuw gediagnosticeerde CRC van 50 jaar of ouder werd verwezen voor Lynch syndroom screening. De testen bestonden uit immunohistochemie voor MLH1, PMS2, MSH2 en MSH6, gevolgd door BRAF-mutatieanalyse ± MLH1-promotermethylatie (MLH1-PM) testen in gevallen die MLH1-verlies vertoonden. 15% van de patiënten vertoonden dMMR. 95% van de MLH1-deficiënte tumoren vertoonden MLH1-PM. Van de patiënten die in de uiteindelijke analyse waren opgenomen, hadden 81 (2,9%) een indicatie voor kiembaananalyse.
Endometriumcarcinoom (EC)
Een meta-analysis van Ryan (2019) bekeek 53 studies, waaronder 12.633 EC-patiënten. Het totale aandeel endometriumtumoren met microsatellietinstabiliteit of MMRd bij immunohistochemisch onderzoek was respectievelijk 0,27 (95% [CI] 0,25-0,28, I2: 71%) en 0,26 (95% CI 0,25-0,27, I2: 88%). Van de vrouwen met abnormale tumortesten had 0,29 (95% CI 0,25-0,33, I2: 83%) een kiembaan MMR-gen PV, zodat ongeveer 3% van de EC’s kan worden toegeschreven aan Lynch syndroom
Kunnackal John (2022) adviseert een verschillend beleid bij CRC en EC vanwege het feit dat MMR-deficiëntie bij EC veel vaker wordt verklaard door somatische events, namelijk om in die groep te beginnen met DNA-analyse in de tumor en niet in de kiembaan. Dit leidt echter tot minder eenduidige aanbevelingen met mogelijk juist weer averechts effect.
Kahn (2019) and Gordhandas (2020) identificeerden uit 29 geïncludeerde studies 6649 patiënten met endometriumkanker, waarvan 206 (3%) Lynch syndroom bleken te hebben. Van de patiënten had 28%-31% MMRd of MSI. De geschatte prevalentie van Lynch syndroom was 15% bij afwijkende IHC en 19% bij een MSI-high profiel; 13,7% van de MLH1-defiënte tumoren vertoonde geen promoter methylatie (PM), waarvan 32 (2.8% uit de gehele groep, 22,4% van de MLH1-deficiënte casus) een kiembaanvariant in MLH1 hadden. Screening op alleen familiegeschiedenis zou 43% van de Lynch syndroom gevallen hebben gemist. Patiënten met Lynch syndroom waren jonger (51,1 jaar), hadden een lager BMI en werden minder vaak in stadium I gediagnosticeerd. MLH1-PM patiënten waren gemiddeld ouder.
Jumaah (2021) heeft in een systematische review de prevalentie van MMRd bij endometriumkanker (EC) onderzocht in 25 studies met in totaal 7.459 patiënten. Een meta-analyse met een random-effect model berekende een totale MMRd frequentie van 24,5% (95% BI: 21,0%-28,1%). Bij Lynch syndroom was de MMRd frequentie 22,9% (95% BI: 14,9%-32,1%). Type I EC had een hogere MMRd (25,8%) dan type II (13,7%). MMRd was het hoogst bij stadium I-II (79,4%) en graad I-II (65,7%), en het laagst bij stadium III-IV (20,2%) en graad III (21,5%). Odds ratio’s toonden associaties met laag stadium EC (1,57), lymfovasculaire invasie (1,77) en diepe myometriuminvasie (1,27).
Loong (2024) verrichtten een retrospectieve, nationale, populatie-studie in de “English National Cancer Registration Dataset”. Ze includeerden alle endometriumcarcinomen (EC) gediagnostiseerd in 2019 en onderzochten frequentie en uitkomst van MMRd-analyse en kiembaan analyse van de MMR-genen. Van alle patiënten was 44% (3514/7928) ouder dan 70 jaar. Bij 17,8% (1408/7928) van de EC in 2019 werd immunohistochemie (IHC)- of microsatellietinstabiliteit (MSI)-analyse verricht; 31% (431/1405) van de geteste tumoren vertoonde MMRd; 80% (346/431) van de MMRd-tumoren was MLH1-negatief en/of MSI-positief; 43,1% (149/346) van de MLH1-negatieve/MSI-positieve tumoren onderging een MLH1-promoter methylatie (PM) test, waarvan 85% (126/149) methylatie vertoonde (MLH1-PM). Van de patiënten met EC die in aanmerking kwamen voor kiembaanonderzoek, kreeg 25% (26/104) een kiembaan MMR-test, waarbij 15 patiënten Lynch syndroom bleken te hebben. Concluderend laat deze studie zien dat slechts een klein percentage EC-patiënten in 2019 werd getest op MMRd en MLH1-PM.
Andersson (2024) screenden 221 EC gediagnostiseerd tussen 2008 en 2012 op het verlies van mismatch repair (MMR)-eiwitten via immunohistochemie (IHC) en het voorkomen van kiembaan MMR-gen pathogene varianten (PV); 24,4% (54/221) van de tumoren vertoonde verlies van ten minste één MMR-eiwit. De gemiddelde diagnoseleeftijd was 68,3 jaar in de MMR-deficiënte groep versus 65,3 jaar in de groep met normale MMR. Verdeling van het eiwitverlies: 83,3% (45/54): verlies van MLH1 en PMS2, 3,7% (2/54): verlies van MSH2 en MSH6, 11,1% (6/54): verlies van alleen MSH6, 1,9% (1/54): verlies van MLH1, PMS2 en MSH6, waarbij MSH2 niet beoordeeld kon worden. Van de 45 tumoren met MLH1/PMS2-verlies hadden 43 normale kiembaanresultaten. Er werden 5 kiembaan PV gevonden: 1 MLH1, 1 MSH2, 3 MSH6. Alle 5 kiembaan PV werden gedetecteerd bij patiënten jonger dan 70 jaar. Deze studie laat dus zien dat MMR-deficiëntie in EC vaak samenhangt met MLH1/PMS2-verlies. De meeste patiënten met MLH1/PMS2-verlies hadden geen erfelijke (kiembaan) PV en alle gedetecteerde kiembaan PV kwamen voor bij patiënten jonger dan 70 jaar, wat suggereert dat zeker op latere leeftijd MLH1-PM een belangrijke rol speelt.
Kaya (2024) beschrijft in een gecombineerde analyse van de PORTEC-1, -2, en -3 trials (n = 1254) dat 84 gevallen van MMRd endometriumkanker (EC) werden geïdentificeerd die niet gerelateerd waren aan MLH1-PM. Van deze gevallen was 37% geassocieerd met Lynch syndroom (MMRd EC), 38% was het gevolg van biallelisch somatische events (DS-MMRd EC), en 25% bleef onverklaard. MMRd EC vertoonde hogere percentages van MSH6-verlies (52% vs. 19%) of PMS2-verlies (29% vs. 3%) dan DS-MMRd EC, en vertoonde uitsluitend MMR-deficiënte klierfoci. DS-MMRd EC had hogere percentages van gecombineerd MSH2/MSH6-verlies (47% vs. 16%), verlies van meer dan 2 MMR-eiwitten (16% vs. 3%), en somatische POLE exonuclease domeinmutatie (25% vs. 3%) dan Lynch syndroom-MMRd.
Post (2021) rapporteert de uitkomsten van moleculair onderzoek in een studiecohort van (PORTEC-1, -2, en -3) 1336 EC's, waarvan 410 (30,7%) MMRd. In totaal waren er 380 (92,7%) volledig getriageerd: 275 (72,4%) waren MLH1-PM MMRd-EC's; 36 (9,5%) Lynch syndroom MMRd-EC's en 69 (18,2%) MMRd-EC's als gevolg van andere oorzaken. In totaal was de Lynch syndroom prevalentie in de onderzoekspopulatie 2,8% en onder MMRd-EC's 9,5%. Op verzoek werden extra gegevens verstrekt over het 70+ cohort. Het betrof in totaal 461 EC-patiënten, van wie er 108 MLH1-PM gerelateerde MMRd, 12 een andere vorm van somatisch MMRd en 6 Lynch-geassocieerde MMRd hadden. Lynch syndroom werd dus aangetoond in 1,3% (6/461) van het totale cohort en 33% (6/18) van de (niet-MLH1 PM) MMRd ECs. Een beperking was het ontbreken van kiembaan Lynch syndroom sequencing bij de hele onderzoekspopulatie, zodat de gevoeligheid van de op IHC gebaseerde triage om Lynch syndroom -patiënten te identificeren, niet kan worden beoordeeld.
In de studie van Hampel (2022) met 341 EC patiënten werd genetische testing uitgevoerd; 27% van de tumoren toonde MMRd, waarbij de meerderheid MLH1-PM vertoonde. Van de 22 (6,5%) gevallen met MMRd, die niet verklaard werden door MLH1-PM, werd bij 10 (2,9% van het totaal) Lynch syndroom vastgesteld (6 MSH6, 3 MSH2, 1 PMS2). Biallelische somatische MMR-events verklaarden de resterende 12 MMRd-gevallen (3,5% van het totaal). Tumoren in oudere leeftijdsgroepen vertoonden vaker MMRd en MLH1-PM, maar geen gevallen van Lynch syndroom. In de leeftijdsgroepen ouder dan 60 jaar werd opvallend geen Lynch syndroom aangetoond, van de tumoren tussen de 60 en 69 jaar (n=124) waren er 37 MMRd (30%), 31 met MLH1-PM. In de leeftijdsgroepen van 70-79 jaar (n=38) en 80-89 jaar (n=8) waren er 15 MMRd (33%), waarvan 14 met MLH1-PM.
Is er voor de detectie van Lynch syndroom reden voor uitbreiding van MMRd-analyse CRC en EC >70 jaar?
MMRd-analyse middels IHC MMR wordt sinds 2016 routinematig in Nederland verricht in CRC en EC gediagnostiseerd <70 jaar. MMRd-analyse wordt echter tegenwoordig bij CRC en EC vaker ook boven de 70 jaar uitgevoerd in het kader van de behandelkeuze. Er is twijfel of het doelmatig is patiënten met een MSI-high tumor in deze groep ook nadere diagnostiek aan te bieden naar Lynch syndroom, of dat het met name somatische afschakeling betreft in het bijzonder door methylatie van de MLH1 promoter (MLH1-PM). De groep met CRC > 70 is bijna net zo groot als het hele cohort onder de 70 (4300 vs. 5086, NKR-cohort 2020). Analyse van deze groep is moeilijk omdat er weinig studies zijn, die zich apart op de 70+ groep richten. Bovendien zijn de data binnen Nederland beperkt, omdat de vorige richtlijn zich tot de groep <70 beperkte. Zoals hierboven al vermeld, heeft Vos (2020) zich daarom gericht op de groep tussen 65 en 70. Van alle CRC-patiënten onder de 70, was 34% tussen de 65 en 70 jaar oud. 12% van deze groep had MLH1-PM in de tumor, 2% had MMRd zonder methylatie. 30% van die laatste groep had Lynch syndroom, maar opgemerkt moet worden dat de totale groep erg klein was (n=10, waarvan 3 met Lynch syndroom). Rond de 0,7% van de groep CRC 65-70 jaar had dus Lynch syndroom.
Eikenboom (2022) voerde een systematische review uit, waarin data werden verzameld van bijna 60.000 patiënten met CRC. 2% van de gehele groep had een kiembaanvariant (PV) in één van de MMR-genen. In de groep <50 jaar lag dit op 7,3%, en <70 op 5,0%. Boven de 70 kon dit niet worden bepaald. De Europese richtlijn van EHTG en ESCP adviseren iedere CRC-tumor te testen op MMRd/MSI (Seppala, 2021). Waarom de groep boven de 70 getest zou moet worden, wordt echter niet onderbouwd. Boven de 65 jaar vertonen veel tumoren met MLH1-afschakeling methylatie van de MLH1-promoter, wat de kosteneffectiviteit verlaagt omdat dit eerst moet worden uitgesloten. Christonson (2023) vergeleek de groep <70 met de groep >80. Het percentage MMRd was in de groep >80 veel hoger dan in de groep <70, maar dit was met name het geval bij vrouwen (50% vs. 10%). BRAF V600E, bij dMMR een marker voor MLH1- PM, was bij oudere vrouwen ook sterker toegenomen dan bij mannen (40% vs. 25%). 67% van alle (bij mannen en vrouwen) dMMR tumoren >80 en 63% van de MMRd tumoren tussen de 60 en 69 had een BRAF V600E mutatie (onder de 50 jaar ligt dat percentage onder de 25%). Daarbij moet worden aangemerkt dat slechts ~70% van alle CRCs met MLH1- PM de BRAF V600E mutatie hebben, terwijl deze praktisch afwezig is bij de niet-MLH1- PM MMRd tumoren (Deng, 2004; Parsons, 2012). Genoemde getallen zijn dus een onderschatting van het percentage MLH1- PM tumoren bij CRC op oudere leeftijd.
Dit wordt ondersteund door de Britse praktijk waar volgens de NICE guidelines alle CRCs getest moeten worden op MSI. Uit een recent overzicht (McRonald, 2024) blijkt dat 67% van de MLH1-deficiënte tumoren MLH1- PM vertoont. De groep 70-plussers maakte de helft van de totale groep uit (8000 van 16000, tabel 2). Het is al met al aannemelijk dat die het grootste deel van de MLH1- PM voor hun rekening nemen, al is dat niet aangegeven in dit artikel.
Om de kleine groep Lynch patiënten (volgens Vos (2020) maximaal 0,7%) uit alle MMRd tumoren te identificeren, zal dus in een significante groep eerst MLH1- PM moeten worden uitgesloten, hetgeen financiële en logistieke consequenties zal hebben. Aangezien dit probleem niet speelt bij MSH2/MSH6 of geïsoleerde PMS2-deficiënte tumoren, zou overwogen kunnen worden deze groep boven de 70 wel te verwijzen voor kiembaandiagnostiek. Echter, ook daar zal een groter deel van de MMRd tumoren verklaard worden door somatische varianten, vergeleken bij jongere patiënten (Elze, 2021).
Er is dus op grond van het vóórkomen van Lynch syndroom in deze groep geen reden routinematig IHC MMR/MSI in te zetten bij de groep CRC 70+. Als dit gedaan wordt ten bate van behandelkeuze, is het risico op Lynch syndroom bij de groep met afwijkende IHC van MLH1 en MSH2 waarschijnlijk klein, tenzij er bij patiënt of in de familie meer aanwijzing is voor Lynch syndroom.
Gezien de hogere gemiddelde leeftijd van CRC bij MSH6 en PMS2 PV dragers (zie submodule Colorectaal kankerrisico en surveillance bij het Lynch syndroom. risico CRC bij Lynch syndroom), kan in die situatie bij afwezige (geïsoleerde) MSH6 of PMS2 kleuring wel overwogen worden verwijzing voor kiembaan diagnostiek te adviseren, hoewel de kanker risico’s bij PMS2 wel veel lager liggen dan bij MSH6, zie ook submodule Colorectaal kankerrisico en surveillance bij het Lynch syndroom. Indien bij afwijkende PMS2 kleuring >70 jaar aanvullende MLH1 kleuring nodig is om te bepalen of sprake is van geïsoleerd PMS2 verlies, kan aanvullende kleuring en verwijzing wat betreft de werkgroep dan ook achterwege gelaten worden.
Wat betreft baarmoederkanker en de kans op Lynch syndroom bij diagnose >70 jaar, is nog minder bekend dan voor CRC. Wel lijkt het aandeel MLH1-PM nog groter dan bij CRC; Meerdere studies (Kahn, 2019; Loong, 2024; Andersson, 2024; Post, 2022) beschreven 25-30% MMRd/MSI in EC, waarbij rond de 3% sprake is van Lynch syndroom. Het overgrote deel van de MMRd/MSI lijkt in EC veroorzaakt te worden door MLH1-promotor methylatie (PM); >80% heeft afwijkende MLH1/PMS2 kleuring, waarvan >80% door MLH1-PM, terwijl deze onderzoeken geen puur 70+ groep beschrijven. Wel beschrijft Post (2022) dat 6 van de totaal 36 Lynch syndroom patiënten (17%) ouder dan 70 jaar waren, vijf hiervan hadden een MSH6 en één een PMS2 PV. Het is niet bekend of deze patiënten eerder al Lynch geassocieerde tumoren hadden (dit was het geval overigens in maar 11% van totaal Lynch syndroom -cohort) of een belast familieverhaal. In tegenstelling tot deze studie laat de studie van Hampel (2021) in de leeftijdsgroep 70-79 jaar (totaal n=38) en 80-89 jaar n= 8) zien dat er van de 15 met MMR-deficiëntie niemand Lynch syndroom had en 14 hadden MLH1-PM.
Van belang verder is nog dat uit kankerrisico-studies (zie ook submodule Colorectaal kankerrisico en surveillance bij het Lynch syndroom) voor met name MSH6 een toename blijkt in risico op EC vanaf 70 jaar (13% CR op 70 jaar naar 23% op 80 jaar, bij de PLSD is er van 70 naar 80 jaar een toename te zien (PLSD 2023, CR 41% op 70 jaar naar 46% op 80 jaar). Het is daarom mogelijk met name MSH6 PV draagsters met EC >70 jaar te missen bij routinematig tumor analyse <70 jaar. Aangezien in ieder geval een deel hiervan, op basis van een positieve voorgeschiedenis of familiegeschiedenis, Lynch syndroom -diagnostiek aangeboden zal krijgen, betreft dit echter waarschijnlijk een kleine groep. Vanuit praktische en doelmatige overwegingen lijkt het de commissie op dit moment verdedigbaar om ook bij EC vast te houden aan routinematige tumoranalyse op aanwijzing voor Lynch syndroom bij diagnose <70 jaar.
Als IHC MMR analyse gedaan wordt ten bate van behandelkeuze >70 jaar, is het risico op Lynch syndroom bij de groep met afwijkende IHC bij MLH1 en MSH2 waarschijnlijk net als bij CRC klein, tenzij er bij patiënt of in de familie meer aanwijzing is voor Lynch syndroom. Gezien de hogere gemiddelde leeftijd van CRC bij MSH6 en PMS2 (zie submodule Colorectaal kankerrisico en surveillance bij het Lynch syndroom) en de eenduidigheid is ook het advies bij bekend geïsoleerd MSH6 en PMS2 verlies te verwijzen voor kiembaan onderzoek. Wel kan, gezien de lagere tumor risico’s bij PMS2, indien MLH1 kleuring (nog) niet gedaan is, ervoor gekozen worden MLH1 kleuring en verwijzing achter wegen te laten.
Concluderend is het advies vooralsnog om routinematig IHC MMR/MSI in te zetten voor het opsporen van Lynch syndroom bij CRC en EC ≤70 jaar. Als >70 jaar analyse gedaan wordt ten bate van behandelkeuze, is het risico op Lynch syndroom bij de groep met afwijkende MLH1 of MSH2 IHC waarschijnlijk klein, tenzij er bij patiënt of in de familie aanwijzing is voor Lynch syndroom. Verdere analyse in de vorm van methylatie onderzoek van de MLH1-promoter en/of kiembaandiagnostiek dienen daarom te worden overwogen indien er bij patiënt of in de familie aanwijzing is voor Lynch syndroom zoals ook aangegeven in de flowchart en in submodule Verwijscriteria voor klinisch genetisch onderzoek bij CRC en genpanelanalyse. Gezien de latere gemiddelde leeftijd van diagnose bij MSH6 en PMS2 zou bij geïsoleerd afwijkende MSH6 of PMS2 IHC >70 jaar wel verwijzing voor kiembaan diagnostiek overwogen kunnen worden. Gezien de lagere tumor risico’s bij PMS2, dient aanvullende diagnostiek in de vorm van IHC MLH1 bij PMS2 beperkt te worden en kan als dit niet bekend is van verwijzing worden afgezien.
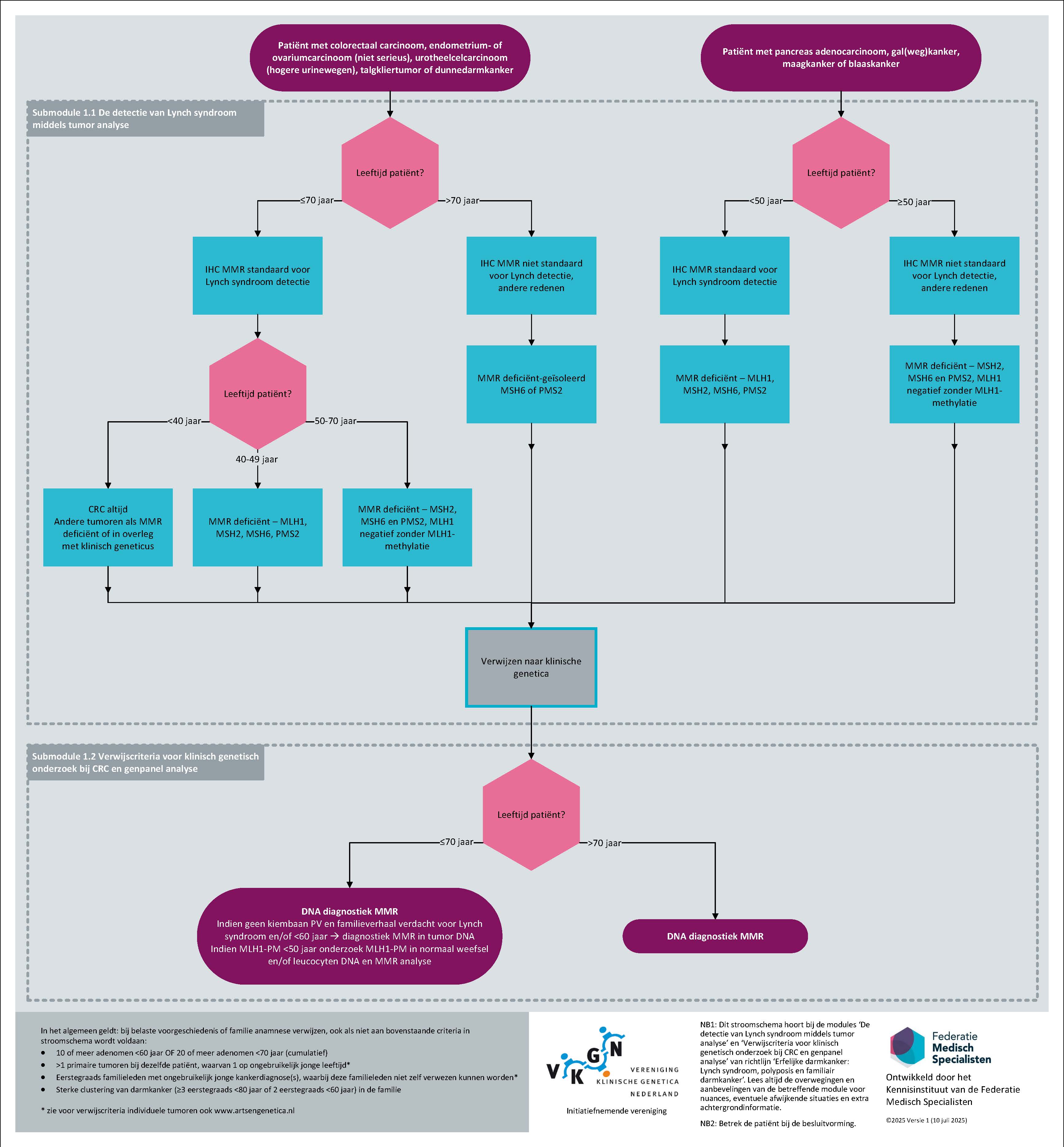
Figuur 1 flowchart (submodule De detectie van Lynch syndroom middels tumor analyse en submodule Verwijscriteria voor klinisch genetisch onderzoek bij CRC en genpanelanalyse); Beleid wanneer IHC MMR en DNA-diagnostiek vanwege verdenking Lynch syndroom
Beleid bij aanwijzing voor Lynch syndroom op basis van tumor analyse
Als er sprake is van een deficiëntie van het MMR-systeem, is er een kans op Lynch syndroom; deze kans verschilt sterk per betrokken gen en per type tumor. Met name afwijkende MLH1-(en PMS2) -kleuring wordt vaak veroorzaakt door methylatie van de MLH1-promoter (MLH1-PM). Belangrijk is ook te realiseren dat MMR-deficiëntie een andere vorm van erfelijke darmkanker niet uitsluit. Daarnaast is het zo dat niet alle tumoren bij een persoon met Lynch syndroom MMR deficiënt zijn en er kan sprake zijn van een sporadische tumor bij iemand met Lynch syndroom in de familie, een fenocopie. Bij blijvende verdenking is het daarom verstandig moleculaire MSI of IHC-analyse van een 2e tumor bij de patiënt of een aangedaan familielid te verrichten.
De betekenis van zowel een positieve als een negatieve test moet in de rapportage van de resultaten duidelijk worden verwoord. Hiervoor zijn ten bate van de patholoog/KMBP’er standaardformuleringen ontwikkeld (zie hiervoor: PALGA moleculaire diagnostiek).
Een flowchart voor verwijzing voor kiembaan diagnostiek naar de Klinische genetica bij MMRd in de tumor is bij aanbevelingen toegevoegd.
Tenslotte dient goede opvolging van patiënten met een MMRd tumor gewaarborgd te zijn en counseling voor kiembaan analyse aangeboden te worden aan alle patiënten en/of hun familieleden voor wie dit relevant kan zijn.
Welke adviezen zijn optimaal bij onverklaard MMRd (UMMRd)?
UMMRd Literatuur
Elze (2021) voerden een studie uit met twee cohorten. Cohort 1 bestond uit patiënten (N = 304) met een MMRd CRC of EC zonder MLH1-promotor methylatie (PM) verwezen naar de Klinische genetica voor verder onderzoek. Hiervan hadden 151 patiënten bewezen Lynch syndroom en 153 patiënten met MMRd CRC (n = 130) of EC (n = 23) geen kiembaan MMR-variant. Het tweede cohort bestond uit patiënten (N = 125) met MMRd CRC (n = 101) of EC (n = 28), waarbij Lynch syndroom en MLH1-PM elders waren uitgesloten. In totaal werd somatische MMRd geïdentificeerd in 88,8% van de geanalyseerde MMRd CRCs (182/205) en 80,9% van de EC's (38/47). Concluderend bleef na somatische analyse slecht een klein deel (2,7%; 32/252 tumoren) onverklaard (UMMRd).
De meta-analyse van Eikenboom (2022) keek naar het aandeel Lynch syndroom, sporadische MMRd en onverklaarde MMRd-gevallen in 58.580 niet-geselecteerde CRCs. In 4.42% van het totale cohort werd geen verklaring voor de MMRd gevonden. Dit percentage bevat waarschijnlijk veel sporadische MMRd CRCs omdat in meeste studies geen somatische test gedaan is. Volledige diagnostiek leidde tot een percentage onverklaarde MMRd van slechts 0,61%. In een literatuur review en analyse van een groot klinisch cohort vonden Eikenboom (2022) respectievelijk 14 en 7 onverklaarde MMRd (UMMRd) cases, waarbij de laatsten suspect bleven voor een gemist somatisch of kiembaan MMR-event. In 2 cases werd met longread sequencing alsnog een complexe MMR PV aangetoond.
Nugroho (2023) verrichtten een systematische review van studies, die kankerrisico's bij Lynch-like syndroom (LLS) onderzochten, waarin zes studies geïncludeerd zijn. De resultaten lieten zien dat de incidentie van CRC bij LLS-patiënten vergelijkbaar was met die van de algemene bevolking, maar lager dan bij Lynch syndroom-patiënten. De incidentie van CRC was hoger bij eerstegraadsverwanten van LLS-patiënten dan bij de algemene bevolking, maar lager dan bij Lynch syndroom-eerstegraadsverwanten. Ook EC kwam vaker voor bij LLS-patiënten dan bij de algemene bevolking, maar minder vaak dan bij Lynch syndroom -patiënten.
In een studie die gegevens van het Duitse Consortium voor Familiair Intestinaal Kanker analyseerde, werden 1448 patiënten prospectief gevolgd in een intensief coloscopisch screeningsprogramma (Bucksch 2022). De cumulatieve risico's voor adenomen en CRC werden vergeleken tussen drie risicogroepen: Lynch syndroom, Lynch-like syndroom (LLS) en familiaire colorectale kanker zonder de typische genetische kenmerken (FCCX). Het risico op CRC was het hoogst bij Lynch syndroom gevolgd door LLS, en het laagst bij FCCX. Er werd ook een verschil gevonden in de risico's voor eerste en tweede primaire CRC tussen de drie groepen, hoewel de risico's voor adenomen vergelijkbaar waren.
In hoeverre moet somatische MMR-gen analyse verricht worden indien geen MLH1-PM of Lynch syndroom aangetoond wordt bij MMRd een tumor?
Patiënten worden aangeduid als patiënten met onverklaard MMRd (UMMRd) of met het Lynch-like syndroom (LLS). Beter is, zoals Katz eerder hebben opgemerkt, dat het gebruik van de term LLS wordt vermeden aangezien dit onterecht toch suggereert dat Lynch syndroom waarschijnlijk is, en dat UMMRd voortaan als volgt wordt gedefinieerd: tumoren die, bij MMR-deficiënte eiwitkleuring voor ten minste 1 MMR-eiwit, niet (genoeg) verklaard zijn door pathogene MMR, somatische dan wel kiembaan, events. Zie ook begrippenlijst met de definitie UMMRd.
Op dit moment zijn alleen voor CRC en EC-data beschikbaar ten aanzien van onverklaarde (UMMRd).
De meta-analyse van Eikenboom (2022) laat zien dat het percentage gevallen van UMMRd voor CRC sterk afhankelijk is van de volledigheid en het type diagnostiek dat wordt gebruikt. In studies die alle diagnostische stadia voltooiden, werden kiembaanvarianten gevonden in ongeveer 3% van de CRCs. Bovendien leidde volledige diagnostiek tot een percentage UMMRd van slechts 0,61%. Dit lage percentage UMMRd in CRC bij volledige diagnostiek wordt bevestigd in de literatuur review en retrospectieve studie van een klinisch cohort van dezelfde onderzoeksgroep, waarbij in een groot klinisch cohort slechts 7 patiënten met een UMMRd over blijven, waarvan bij 2 alsnog een complexe kiembaan variant wordt aangetoond met longread sequencing. Deze worden nader beschreven in de studie van te Paske (2022), waarin bij 8/32 (18,8%) patiënten met UMMRd een kiembaan PV in een niet-coderende sequentie van een MMR-gen wordt aangetoond.
Onder andere Elze (2021) en de Vos (2020) tonen aan dat het percentage somatisch verklaarde MMRd, zowel door MLH1-PM als biallelische MMR-gen events in de tumor, met de leeftijd toe neemt voor alle MMR-genen. De kans dat na uitsluiting van MLH1-PM in de tumor (indien relevant) en negatieve kiembaan diagnostiek toch sprake is van Lynch syndroom is dus afhankelijk van leeftijd van diagnose en uiteraard ook familiegeschiedenis (indien informatief) en neemt sterk af >65 jaar (Hitchins 2023). Het lijkt op dit moment het meest doelmatig om bij een MMRd tumor en negatieve kiembaan diagnostiek alleen bij blijvende verdenking op Lynch syndroom verdere diagnostiek naar (gemiste) somatische en kiembaan afwijkingen te verrichten. Deze grens kan arbitrair gelegd worden bij diagnose <60 jaar, tenzij de voor- of familiegeschiedenis verdacht is voor Lynch syndroom, waarbij ook eventueel onderzoek bij andere familieleden uitsluitsel kan geven of een gemiste MMR-gen kiembaan aanleg waarschijnlijk is of niet.
Wat is het klinisch beleid indien de MMRd onverklaard (UMMRd) blijft?
Enkele studies hebben naar het darmkanker risico in (eerstegraads familieleden (FDR) van) mensen met onverklaarde mismatch repair deficiënte (UMMRd) tumoren gekeken, in de meeste gevallen is overigens geen somatische tumor analyse gedaan. Nugroho (2023) beschrijft zes studies, waarbij het merendeel een hoger CRC-incidentie in “Lynch-like-syndroom” (LLS) FDRs vonden dan in de algemene bevolking maar lager dan bij Lynch syndroom FDRs. Ook was het risico op EC hoger in LLS-patiënten dan in de algemene bevolking, maar lager dan in Lynch syndroom patiënten. Ook de studie van Bucksch (2022) beschrijft het hoogste CRC-risico in Lynch syndroom, gevolgd door LLS het laagst in FCC.
Bij benadering zou er sprake zijn van een gemiddeld twee keer verhoogd risico op CRC en lijkt er geen onderbouwing voor een Lynch controle advies in de meerderheid van deze families. Afhankelijk van leeftijd en familieanamnese kan een advies gebaseerd op de familiair CRC (FCC)-criteria worden gegeven, als aan FCC-criteria wordt voldaan, zie submodule Incidentie, risico’s en surveillance bij familiair colorectaal carcinoom .
Bij een sterk belaste voor- of familiegeschiedenis of MMRd tumor bij een tweede familielid en geen kiembaan MMR-gen PV of somatische verklaring, kan intensievere darm- en eventueel baarmoeder surveillance (volgens Lynch syndroom protocol) worden overwogen.
Kwaliteit van bewijs
Niet van toepassing, want de gebruikte studies zijn niet met GRADE beoordeeld.
Waarden en voorkeuren van patiënten (en eventueel hun naasten/verzorgers)
Vanuit de patiënten wordt aangegeven dat zij het van groot belang achten dat iedere Lynch patiënt gediagnostiseerd wordt, niet alleen voor optimaal beleid voor de patiënt maar ook zodat familieleden van preventieve opties gebruik kunnen maken. Routinematig IHC-MMR/IHC analyse, ook bij andere Lynch syndroom geassocieerde tumoren, draagt hier in grote mate aan bij, hoewel goede opvolging bij een afwijkende tumor analyse nog wel een punt van zorg is. Ook dient een afweging gemaakt te worden wat betreft doelmatigheid, zodat moleculaire analyses en klinische genetische zorg beschikbaar blijven voor degenen met het hoogste risico.
Kostenaspecten
Verschillende studies, waaronder Nederlandse, hebben de kosteneffectiviteit van routinematige tumor analyse voor de detectie van Lynch syndroom in CRC en EC <70 jaar aangetoond. Gezien de opbrengst van tumor analyse en identificatie van Lynch syndroom in andere Lynch syndroom geassocieerde tumoren, is naar alle waarschijnlijkheid uitbreiding van indicatie van routinematige tumor analyse op MMd/ IHC bij diagnose <70 of <50 jaar bij andere Lynch syndroom geassocieerde tumoren ook kosteneffectief (Snowsill, 2020; Stinton, 2021; Leenen, 2016; Goverde, 2016).
Gelijkheid ((health) equity/equitable)
Aangezien de aanbevelingen het routinematig testen van tumoren op aanwijzing voor Lynch syndroom betreffen, leidt dit naar verwachting tot meer gelijkheid van zorg ten opzichte van de huidige praktijk, waarbij bij veel Lynch syndroom geassocieerde tumoren nog afgegaan wordt op familieanamnese, welke minder betrouwbaar kan zijn bij verminderde gezondheidsvaardigheden van patiënten en onvoldoende systematisch uitvragen van de familiegeschiedenis door de zorgverleners.
Aanvaardbaarheid:
Sinds de richtlijn van 2015 wordt routinematige tumor analyse op aanwijzing voor Lynch syndroom al landelijk toegepast voor alle CRC en EC-patiënten gediagnostiseerd <70 jaar. Bij afwijkende tumor analyse kunnen patiënten kiezen voor verdere kiembaan analyse naar Lynch syndroom of niet. De huidige adviezen wijken hier niet van af, alleen is de indicatie voor tumor-analyse verbreed.
Het opsporen van dragers van een aanleg voor Lynch syndroom en aanbieden van surveillance, leidt tot een afname van morbiditeit en mortaliteit in deze groep. Het voorkomt kanker diagnoses en behandeling, met name ook bij jonge mensen, die nog een belangrijke maatschappelijke rol hebben. De adviezen in deze richtlijn, worden daarom door de werkgroep als aanvaardbaar beoordeeld.
Haalbaarheid
In de aanbevelingen is nadrukkelijk rekening gehouden met de haalbaarheid van uitbreiding van analyse door bij frequente tumoren, waarbij opbrengst laag lijkt af te zien van routinematig IHC-MMR/MSI-testen of een leeftijdsgrens aan te houden, welke doelmatig lijkt.
Op basis van de ervaring met routinematige tumor analyse op Lynch syndroom bij alle CRC en EC <70 jaar, welke sinds 2015 in Nederland verricht wordt, worden de voorgestelde aanbevelingen haalbaar geacht.
Rationale van aanbeveling-1: weging van argumenten voor en tegen de interventie
Op basis van de prevalentie van Lynch syndroom bij andere Lynch syndroom geassocieerde tumoren dan CRC en EC, is het zinvol routinematig IHC-MMR/MSI analyse uit te breiden naar een groot deel van deze tumoren. Hierbij is per tumor op grond van de huidige gegevens afgewogen voor welke leeftijdscategorie deze analyse het meest doelmatig is.
Eindoordeel:
Sterke aanbeveling voor (Doen)
Rationale van aanbeveling-2: weging van argumenten voor en tegen de interventies
Ondanks de beperkingen blijven IHC-MMR en de traditionele moleculaire MSI-testen relatief gevoelige, snelle en kosteneffectieve methoden voor het detecteren van MMRd met als doel het opsporen van Lynch syndroom. Omdat bij een MLH1- en MSH2-aanleg ook afwijkende kleuring van respectievelijk PMS2 en MSH6 verwacht wordt, kan ervoor gekozen worden in eerste instantie MSH6 en PMS2 kleuring te verrichten en indien afwijkend ook kleuring van de andere 2 MMR eiwitten te verrichten, mede om te bepalen of verder onderzoek naar MLH1-promoter methylatie (PM) nodig is. MLH1-PM is vaak sporadisch, maar indien de diagnose <50 jaar is of er een belaste familieanamnese is, dan is er een reële kans dat er alsnog een kiembaan MLH1 pathogene variant, dus Lynch syndroom, aanwezig is.
Eindoordeel: Sterke aanbeveling voor (Doen)
Rationale van aanbeveling-3: weging van argumenten voor en tegen de interventies
Indien in geval van een MMRd tumor zonder MLH1-promoter methylatie (PM) geen kiembaan aanleg wordt aangetoond, hangt de kans dat alsnog sprake is van Lynch syndroom samen met de eigen voorgeschiedenis en familieanamnese van de patiënt. Hoe ouder de patiënt bij diagnose en hoe minder suspect de familie (indien informatief), des te kleiner de kans dat sprake is van Lynch syndroom. Dit dient meegenomen te worden in de afweging of verdere (dure) somatische analyse zinvol is en of familieleden blootgesteld moeten worden aan intensieve controles.
Bij benadering zou er sprake zijn van een gemiddeld twee keer verhoogd risico op CRC en lijkt er geen onderbouwing voor een Lynch controle advies in de meerderheid van deze families. Afhankelijk van leeftijd en familieanamnese kan een advies gebaseerd op de familiair CRC (FCC)-criteria worden gegeven, als aan FCC-criteria wordt voldaan, zie submodule Incidentie, risico’s en surveillance bij familiair colorectaal carcinoom. Bij een sterk belaste voor- of familiegeschiedenis of MMRd tumor bij een tweede familielid en geen kiembaan MMR-gen PV of somatische verklaring, kan intensievere darm- en eventueel baarmoeder surveillance (volgens Lynch syndroom protocol, submodule Colorectaal kankerrisico en surveillance bij het Lynch syndroom en submodule Extracolonische kankerrisico en surveillance bij het Lynch syndroom worden overwogen.
Eindoordeel:
Sterke aanbeveling voor (Doen)
Onderbouwing
Lynch Syndrome is the most common hereditary colorectal (CRC) and endometrial cancer (EC) syndrome, with an estimated prevalence of 2-3% of patients with CRC and EC. It is caused by pathogenic germline variants in one of the mismatch repair (MMR) genes MLH1, MSH2, MSH6, PMS2 or a deletion of the 3’ end of the EPCAM gene. Tumors with defective MMR display a microsatellite instability (MSI) phenotype. Since 2015, it has been standard policy in the Netherlands to perform immunohistochemical staining (IHC) for mismatch repair deficiency (MMRd) for every patient <70 years old with CRC or EC, to diagnose Lynch Syndrome. The question has risen whether this policy should be extended to CRC and EC patients> 70 years of age and to other Lynch-associated malignancies (ovarian cancer, pancreatic cancer, biliary tract and gall bladder cancer, urothelial cancer, small bowel cancer, stomach cancer, brain tumors, sebaceous gland tumors) and prostate cancer. Extension of the policy to other Lynch-associated malignancies will lead to better detection of Lynch syndrome and to reduced incidence and mortality from especially CRC through colonoscopy surveillance programs. However, this must be weighed against additional costs for IHC staining and possible unnecessary somatic and germline testing. In case no germline PV in one of the MMR genes is found in a patient with an MMRd tumor, somatic tumor analysis may exclude the diagnosis Lynch syndrome by finding biallelic somatic MMR gene events in the tumor. However, it should be weighted to what extent somatic analysis should be performed and what advice should be given in case the MMRd remains unexplained (UMMRd). These questions will be addressed in this chapter of the revised guideline.
Description of studies
A total of 35 studies were included in the analysis of the literature. Important study characteristics and results are summarized in Table 2. A description of each study categorized per cancer type, is provided below. The risk of bias of included studies was not assessed. Therefore, the risk of bias table was not included.
Epithelial ovarian cancer
Mitric (2023) performed a systematic review and meta-analysis in which the aim was to assess the prevalence of MMR deficiency (MMRd), microsatellite instability (MSI)-high, and Lynch syndrome, in epithelial ovarian cancer (EOC) of any histologic subtype. The search date was February 2nd, 2022. Related to the intervention and outcome, studies were included that met the following criteria: 1) IHC MMR testing, if tested for at least three MMR proteins (MLH1, MSH2, MSH6), 2) MSI testing using the national cancer institute NCI five markers and 3) germline sequencing if tested for at least three MMR proteins (MLH1, MSH2, MSH6). Included study designs were cohort, cross-sectional, and case-series. The outcomes relevant for this literature analysis included prevalence of MMRd, MSI-H, and Lynch syndrome.
Pancreatic cancer, biliary tract cancer and gallbladder cancer
Agaram (2010) performed a retrospective cohort study of 54 cases with ampullary carcinoma, to investigate the prevalence of MMRd by IHC. The tumor samples were resected at Memorial Sloan-Kettering Cancer Center, New York, NY, and were selected from the pathology database. IHC was assessed with antibodies against MLH1, MSH2, MSH6, and PMS2. Lack of nuclear staining in the invasive carcinoma cells was defined as abnormal. The outcomes relevant for this literature analysis were the prevalence of MMRd and prevalence of MMRd patients with lost MLH1/PMS2 expression.
Hu (2018) performed a retrospective cohort study in which the aim was to investigate the prevalence of MMRd in patients with pancreatic ductal adenocarcinoma (PDAC). Between January 2006 and July 2017, the MSKCC institutional tumor registry was searched for PDAC tumor samples. Finally, 833 patients were included. MMRd was tested using IHC, germline testing, or tumor and germline DNA sequencing using MSK-IMPACT. IHC was assessed with antibodies against MLH1, MSH2, MSH6, and PMS2. MSI analysis was performed using the five microsatellite loci by PCR or MSI sensor analysis. MSI-H was defined as microsatellite instability at ³2 loci or as >10% of the microsatellite loci showing microsatellite instability. NGS was conducted using the MSK-IMPACT assay. Outcomes relevant for this literature analysis included the proportion of patients with overall MMRd, with lost MLH1 and/or PMS2 expression, and with Lynch syndrome.
Christakis (2019) performed a study in which the aim was to determine the prevalence of microsatellite instability in upper gastrointestinal tract cancers and to screen for patients with Lynch syndrome. In total, 645 patients with upper gastrointestinal tract cancers were prospectively enrolled in an institutional cohort study at the Dana Farber Cancer Institute, Boston, and were sequenced using NGS. From these upper gastrointestinal tract cancers, pancreatic carcinoma (n=199), gastric carcinomas (n=97), bile duct carcinomas (n=60), small bowel carcinomas (n=29), and gallbladder carcinomas (n=19) were relevant for this literature analysis. Then, microsatellite instability was defined as >3 microsatellite indel events per megabase pair in the targeted genome. All samples with microsatellite instability detected by NGS, and with available pathology material, were assessed for MMRd by IHC for MLH1, PMS2, MSH2, and MSH6 expression using a case-control study design. Each case (microsatellite instable) was matched with ³2 controls (microsatellite stable). Depending on tissue availability, IHC was also performed on all samples greater than 2 but fewer than 3 microsatellite indel events per megabase pair. Tumor samples were additionally assessed for MLH1 promoter methylation status. Outcomes relevant for this literature analysis included the proportion of patients with overall MMRd, with lost MLH1 and/or PMS2 expression, with MLH1 promoter hypermethylation, with Lynch syndrome, and with somatic mutations.
Abrha (2020) performed a retrospective review of all patients with extracolonic gastrointestinal (GI) malignancies at Stanford Comprehensive Cancer Institute screened for MMRd via IHC between January 2016 and December 2017. A total of 1425 gastrointestinal malignancies of non-squamous etiology were screened. After exclusion of cases with insufficient medical information or unknown primary location, 1374 gastrointestinal cancer patients were left for analyses. Among those gastrointestinal malignancies, pancreatic carcinoma (n=244), gastric carcinoma (n=150), gallbladder carcinoma (n=41), and small bowel cancer (n=20) were relevant for this literature analysis. IHC was performed for the 4 MMR proteins (MLH1, MSH2, MSH6 and PMS2). Germline testing for Lynch syndrome was performed in tumor material deficient for MSH2/MSH6, MSH6 alone or PMS2 alone or if the following criteria were met: MLH1/PMS2 deficient, BRAF mutation negative, and/or MLH1 promoter methylation negative. In case of negative Lynch syndrome testing, specimens were sent for somatic tumor sequencing. Outcomes relevant for this literature analysis included the prevalence of overall MMRd, MLH1 promoter methylation, biallelic somatic pathogenic events, and Lynch syndrome.
Mettman (2020) performed a retrospective cohort study in which the aim was to determine the prevalence of MMRd in pancreatic adenocarcinoma in fine-needle aspiration (FNA) specimens. The medical record system of the University of Kansas Medical Center was reviewed to identify all FNA specimens with a diagnosis of pancreatic adenocarcinoma. IHC staining was conducted for the 4 MMR proteins (MLH1, MSH2, MSH6 and PMS2) to cases with sufficient tumor material only. In total, 184 pancreatic FNA specimens were identified, of which 65 contained sufficient material to perform IHC staining. Outcomes relevant for this literature analysis included the prevalence of overall MMRd.
Grant (2021) performed a retrospective cohort study in which the aim was to better understand MMRd pancreatic cancer. The Ontario Pancreas Cancer Study was used for this purpose, and this study prospectively collected epidemiological data and biospecimens from patients with pancreatic cancer in Ontario, Canada. IHC for MMR testing was conducted for the 4 MMR proteins (MLH1, MSH2, MSH6, and PMS2) as well as whole-genome and transcriptome sequencing. MMRd was defined as one of the following: somatic loss of a pair of MMR proteins by IHC or biallelic pathogenic events from whole-genome sequencing. MSI was detected using MSIsensor2. Outcomes relevant for this literature analysis included the prevalence of overall MMRd, lost MLH1/PMS2 expression, with a germline pathogenic variant, and of somatic mutations.
Kryklyva (2022) conducted a retrospective cohort study aiming to explore the prevalence of MMRd in early-onset carcinomas of duodenal (DC), ampullary (AC, and pancreatic (PC) origin. A nationwide retrospective search in the Nationwide Network and Registry of Histopathology and Cytopathology (PALGA) was performed to identify all patients with DC, AC and PC under the age of 50 in the Netherlands between January 2002 and December 2012. IHC MMR was performed using standard procedures. MSI status was assessed using five mononucleotide markers. Cases with more than one unstable marker were classified as having a high degree of MSI (MSI-H). MLH1 promoter methylation was detected using MS-MLPA on tumor DNA from MLH1- and PMS2-deficient cases. The outcomes relevant for this literature analysis were the proportion of patients with MMRd, MLH1 promoter methylation, and Lynch.
Kubo (2022) performed a retrospective cohort study in which the aim was to determine the prevalence of MSI-H status in hepato-biliary-pancreatic malignancies. In total, 283 patients from 7 hospitals in the Kansai Hepato-Biliary Oncology Group (KHBO) were included. Patients with MSI status detected by NGS were excluded from the study. Relevant malignancies for this literature analysis were intra- and extrahepatic cholangiocarcinoma (n=88), gallbladder carcinoma (n=18), and pancreatic carcinoma (n=144). MSI status was assessed by PCR fragment analysis, with MSI-H defined as ³2 unstable markers. The outcomes relevant for this literature analysis were the proportion of patients with overall MMRd, MLH1 promoter methylation, and Lynch syndrome.
Moy (2015) performed a retrospective cohort study in which the aim was to determine the prevalence of MMRd in patients with gallbladder carcinoma. The surgical pathology files of Massachusetts General Hospital in Boston were searched and cases from 1988 to 2012 were selected. Protein expression of 4 MMR proteins were evaluation by IHC staining. MSI was defined as complete loss of nuclear staining for ³1 of the 4 MMR proteins. Lynch syndrome was assessed in the group with MSI. Hot spot mutations were detected using SNaPshot, which is a mutation assay. The outcomes relevant for this literature analysis were the prevalence of overall MMRd, loss of expression of MLH1 and/or PMS2, and Lynch syndrome.
Goeppert (2019) performed a retrospective cohort study in which the aim was to determine the prevalence of MSI among gallbladder carcinomas. The cohort included tumor material from 69 patients who underwent hepatobiliary surgery at the University Hospital Heidelberg between 1995 and 2010. None of the patients received radio- and/or chemotherapy prior to surgery. All tumors classified as MSI were analyzed by IHC staining to determine the prevalence of MMRd proteins (MLH1, MSH2, MSH6, and PMS2). MMR-induced MSI status was assessed using the MSI marker panel. One outcome relevant for this literature analysis included the prevalence of overall MMRd.
Ju (2020) performed a retrospective cohort study in which the aim was to determine the prevalence of MMRd/MSI in patients with cholangiocarcinomas. In total, 96 cases diagnosed between 1993 and 2014 were identified after a search in the Citrix Copath and EPIC Hyperspace database. MMRd was assessed by IHC staining on 4 MMR genes. All cases with any MMR loss on IHC were tested for MSI status. MSI status was evaluated using the MSI Analysis System, analyzing 5 mononucleotide regions. MSI-H was defined as having novel allele peaks in the tumor sample with absence in the corresponding normal sample at 2 or more of the 5 mononucleotide repeat markers tested. MSI-L was defined as any cases with peak shifts in only 1 of the 5 regions tested. Cases were considered MMRd if the tumor sample had loss of expression of ³1 MMR proteins on IHC and MSI-L or -H status. Cases with lost MLH1/PMS2 expression were additionally tested for MLH1 promoter methylation. The outcomes relevant for this literature analysis included the prevalence of overall MMRd, loss of expression of MLH1 and/or PMS2 with MLH1 promoter methylation, and Lynch syndrome.
Ando (2022) performed a retrospective cohort study in which the aim was to determine the prevalence of MMRd/MSI-H among biliary tract carcinomas. In total, 116 patients diagnosed with biliary tract carcinoma and undergoing surgery between January 2008 and December 2017 at Kagawa University Hospital in Japan, were enrolled in the study. IHC staining of 4 MMR proteins was performed. Abnormal staining was defined as the absence of nuclear staining in the tumor cells while there is presence of nuclear staining of non-neoplastic cells. MSI status was performed in patients with MMRd and in patients with a family history of Lynch syndrome -associated cancers, using the MSI analysis system. MSI-H was defined as ³2 markers demonstrating altered numbers of repeats. The outcomes relevant for this literature analysis included the prevalence of overall MMRd and loss of expression of MLH1 and/or PMS2.
Upper tract urothelial cancer (UTUC) and bladder cancer
Rasmussen (2022) performed a systematic review and meta-analysis in which the aim was to study the effect of universal screening of newly diagnosed upper tract urothelial carcinomas (UTUC) for MMRd. The search date was December 1, 2021. Studies were eligible if (1) these were population-based screening cohorts, (2) screening using IHC staining of at least 2 MMR proteins, and (3) inclusion of UTUC cases (bladder cancers excluded). MMRd was defined as complete loss or MMR protein staining in tumor cells with positive internal control. For two studies, MMR protein loss was defined as <20% or <30% MMR-positive tumor cells, respectively. MSI-H was defined as instability of ³2 MSI markers, except for 1 study where it was defined as ³3 markers. The outcomes relevant for this literature analysis included the proportion of screened UTUC patients with loss of MMR protein expression, MSI status, and Lynch syndrome. A potential limitation of this meta-analysis was that it remains unknown whether some included studies are based on a true screening cohort of newly diagnosed UTUC.
Audenet (2019) performed an observational study in which the aim was to investigate genomic differences between UTUC and urothelial carcinoma of the bladder (UCB). In total, 195 UTUC patients and 454 UCB patients were included in the cohort. Tumor samples of these patients were analyzed using targeted NGS (MSK-IMPACT). Relevant for this literature analysis, MSI status was assessed using MSI-sensor algorithm (version 0.2). MSI-H was defined as an MSI-sensor score ³10. MMRd was further investigated by calculating the tumor mutational burden. The outcomes relevant for this literature analysis included the prevalence of overall MMRd, Lynch syndrome, somatic mutations, and Lynch-like syndrome.
Kagawa (2021) performed a retrospective cohort study in which the aim was to assess MMRd among bladder cancer patients. In total, 618 patients diagnosed with bladder cancer and undergoing surgery between June 1997 and February 2018 at Saitama Medical Center in Japan, were enrolled in the study. MMRd tumors were defined as tumors that showed total absence of nuclear staining. MSI status was performed using the MSI analysis system, analyzing 5 mononucleotide markers. MSI-H was defined as ³2 markers demonstrating altered numbers of repeats. The outcomes relevant for this literature analysis included the prevalence of overall MMRd, MSI-H, loss of expression of MLH1 and/or PMS2 with MLH1 promoter methylation, and Lynch syndrome.
Kullman (2023) performed a retrospective cohort study in which the aim was to compare the frequency of MSI in UTUC patients, determined using the Bethesda panel and the Idylla MSI assay. In total, 249 tumors collected from 1995 to 2017 from the renal pelvis or ureter were retrieved from the archives of 3 collaborating Institutes of Pathology: Málaga (Spain), Marburg and Erlangen (Germany). MSI status was analyzed using the Bethesda panel of 5 markers. Tumors were defined as MSI if at least 2 of the 5 markers are different with novel peaks. IHC staining was conducted to detect MMRd (MLH1, MSH2, MSH6, PMS2) to validate microsatellite testing. In addition to the Bethesda panel, MSI status was assessed using the Idylla MSI assay, which detects mutations in 7 new MSI biomarkers (ACVR2A, BTBD7, DIDO1, MRE11, RYR3, SEC31A, and SULF2). MSI-H was defined as detection of a mutation in ³2 biomarkers. The outcomes relevant for this literature analysis included the prevalence of overall MMRd, MSI status, loss of expression of MLH1 and/or PMS2.
Ma (2023) performed a retrospective cohort study in which the aim was to predict the efficacy of PD-1/PD-L1 inhibitors in Chinese patients with MMRd urothelial carcinoma. In total, 232 patients with UTUC and 374 patients with BUC who underwent surgical resection between 2018 and 2022 were identified through a search in the database of Shandong Provincial Hospital affiliated to Shandong First Medical University, Jinan, China. Patients who received chemoradiotherapy were excluded from the study. IHC staining was conducted to determine MMR protein expression of MLH1, MSH2, MSH6, and PMS2. MMRd was defined as the absence of nuclear staining of MMR proteins. MSI status was detected using MSI-sensor (V0.2) NGS. MSI-H was defined as MSI-sensor scores ³10. MSI status was also tested using PCR and was performed using the MSI Analysis System, version 1.2, which analyzed 5 mononucleotide markers. MSI-H was defined as the presence of novel alleles in the tumor sample, but absence in the corresponding normal sample at ³2 tested markers. The outcomes relevant for this literature analysis included the prevalence of overall MMRd, MSI-H, loss of expression of MLH1 and/or PMS2, Lynch syndrome, and somatic mutations.
Pivovarcikova (2023) performed a retrospective cohort study in which the aim was to investigate a cohort of UTUC patients for potential association with Lynch syndrome. In total, 180 patients diagnosed with UTUC between 2010 and 2022 were retrieved from the Šikl’s Department of Pathology, Czech Republic. Cases with extension of bladder cancer into the ureter were excluded. All cases were screened for MMRd by IHC staining using the antibodies MLH1, MSH2, MSH6 and PMS2. Cases with loss of both MLH1 and PMS2 staining were further tested for MLH1 promoter methylation. Patients with loss of MSH6, MSH2, PMS2 or MLH1 (without promoter methylation) were referred to genetic counseling and germline mutations testing. Outcomes relevant for this literature analysis included the prevalence of overall MMRd, loss of expression of MLH1 and/or PMS2 with MLH1 promoter hypermethylation, and with Lynch syndrome
Small bowel and gastric cancer
Christakis (2019): please read page 19 for the description of this study.
Abrha (2020): please read page 19 for the description of this study.
Aparacio (2021) performed a retrospective cohort study in which the aim was to assess the prevalence of MMRd among patients with small bowel cancer. In total, tumor blocks were used of 180 patients, recruited from January 2009 to December 2012 in the French NADEGE cohort with 74 participating institutions. IHC staining was conducted to assess the expression of the MMR proteins MLH1, MSH2, MSH6, and PMS2. Absence of staining of ³1 MMR protein was considered MMRd. Targeted NGS was used to investigate the presence of 739 somatic mutations. Outcomes relevant for this literature analysis included the prevalence of overall MMRd, loss of expression of MLH1 and/or PMS2, Lynch syndrome, and somatic mutations.
Latham (2021) performed a retrospective cohort study in which the aim was to determine the prevalence of MMRd among patients with small bowel cancer. In total, 100 patients diagnosed with primary small bowel cancer between 2006 and 2019 were identified at Memorial Sloan Kettering Cancer Center. Tumor DNA was sequenced using MSK-IMPACT. MSI status was conducted using MSI-sensor, which is an NGS-based bioinformatics platform that reports the percentage of unstable loci as a cumulative score. MSI-H is defined as MSI-sensor score ³10. IHC staining was conducted to determine the expression of MMR proteins. MMRd was defined as either lack of IHC staining or MSI-sensor score ³10. Germline analysis was conducted and only patients with (likely) pathogenic variants were considered germline positive. Outcomes relevant for this literature analysis included overall MMRd, with lost MLH1 and/or PMS2 and MLH1 promoter methylation, Lynch syndrome, and somatic mutations.
Sanchez (2021) performed a multicenter retrospective cohort study in which the aim was to determine the prevalence of Lynch syndrome among patients with MMRd small bowel cancer. In total, 94 patients diagnosed with small bowel cancer between 2004 and 2020 in 4 Spanish tertiary hospitals, were recruited. IHC staining was conducted to test the expression of the 4 MMR proteins (MLH1, MSH2, MSH6, and PMS2). All MMRd tumors underwent germline analysis. Lynch syndrome was identified if a germline pathogenic variant in one of the MMR genes was confirmed. Outcomes relevant for this literature analysis included the prevalence of overall MMRd, lost expression in MLH1 and/or PMS2, and Lynch syndrome
Suerink (2021) conducted a retrospective cohort study in which a large collection of small bowel cancers was used to investigate the prevalence of MMRd and Lynch syndrome. A nationwide search in the Nationwide Network and Registry of Histopathology and Cytopahtology (PALGA) was used to identify samples from patients with small bowel cancer. All neoplasms of the small bowel were extracted for the 5-year period, 2012-2016. Both resectable (n=332) and biopsied (n=68) samples were obtained. IHC staining was used to obtain information about PMS2 and MSH6 expression, followed by IHC for MLH1 and/or MSH2. Adenocarcinomas with aberrant expression of at least one of the MMR proteins in the absence of MLH1 promoter methylation underwent DNA variant analysis using an NGS panel. The outcomes relevant for this literature analysis included the proportion of patients with MMRd, MLH1 methylation, Lynch syndrome, and somatic mutations.
Kryklyva (2022): please read page 20 for the description of this study.
De Back (2024) performed a nationwide retrospective cohort study in which the aim was to investigate incidence, treatment and prognosis of small bowel cancer. In total, data of 2697 patients diagnosed with small bowel cancer between January 1999 and December 2019 were retrieved from the Netherlands Cancer Registry and Pathology Archive. MMR status had been done before, using IHC staining of the 4 MMR proteins. Outcomes relevant for this literature analysis included the prevalence of overall MMRd and Lynch syndrome.
Brain tumors
Rodriguez-Hernández (2013) performed a study in which the aim was to characterize the MMR system defects that could be involved in malignant astrocytoma pathogenesis. This study analyzed a study cohort and a validation cohort. The study comprised 96 patients diagnosed with astrocytoma grades II (n=20), III (n=19), and IV (n=57) who were recruited from June 2000 until March 2006 at the University Hospital of Salamanca, Spain. The validation cohort constituted 71 patients diagnosed with astrocytoma grades III (n=12) and IV (n=59) who were recruited from April 2004 until December 2010 at the University Hospital 12 de Octobre in Madrid. IHC staining was conducted to determine the expression of the MMR proteins MLH1, MSH2 and MSH6 (not PMS2). IHC staining of MSH6 was also performed in tumor sections from patients in the validation cohort. Aberrant methylation in MLH1, MSH2 and MSH6 was detected using the SALSA MS-MLPA Kit ME011. MSI status was assessed by PCR in tumor DNA obtained from 88 patients using a panel of 8 markers, as recommended by the National Cancer Institute. MSI-H was defined if ³30% markers demonstrated instability. In patients with negative IHC staining for MLH1, MSH2 and/or MSH6 and in those with MSI-H tumors, mutational analysis of MMR genes was performed by PCR. Outcomes relevant for this literature analysis included overall MMRd, MSI-H, MLH1 promoter methylation, germline variations, and somatic mutations.
Hadad (2023) performed a cohort study in which the aim was to investigate MMRd in a cohort of adults diagnosed with primary glioblastoma. In total, 459 glioblastoma patients aged ³25y who underwent surgical biopsy or resection between 2017 and 2022 at the University of California, San Francisco, were included. All tumors were evaluated using targeted NGS. Mutations in MMR genes MLH1, MSH2, MSH6 and PMS2, were identified using IHC staining. MSI status was performed using MSIsensor2 analysis of 5 markers, using the UCSF500 NGS Panel. MSI was present, if ³10% of the 86 microsatellites were unstable. Somatic tumor mutational burden (TMB) was determined by calculating the number of somatic mutations divided by the total coding footprint of the assay. Somatic hypermutation was defined as tumors TMB ³15 mutations per Mb. All genomic DNA were tested for methylation. The subgroup of tumors with hypermutation and biallelic inactivation of an MMR protein was called “De novo replication repair deficient (RRD) glioblastoma, IDH-wildtype”.
Outcomes relevant for this literature analysis included the prevalence of overall MMRd, Lynch syndrome, and somatic mutations.
Sebaceous gland tumors
Kunnackal John (2022) performed a systematic review and meta-analysis in which the aim was to assess the prevalence of Lynch syndrome in unselected colorectal carcinoma (CRC), endometrial carcinoma (EC), ovarian carcinoma (OC), urinary tract (UT) and sebaceous tumors (ST). The tumor types relevant for this literature analysis included ST only. Studies performing universal screening for Lynch syndrome using MSI testing or IHC staining published between 2005 and 2017 were included. Lynch syndrome was defined as missing ³1 protein on IHC staining or MSI-H cases. MLH1 promoter methylation was considered a somatic mutation. A diagnosis of Lynch syndrome was made by germline testing for pathogenic mutations in the mismatch repair genes MLH1, PMS2, MSH2, MSH6 or EPCAM. Outcomes relevant for this literature analysis included the prevalence of overall MMRd as a result of IHC or MSI testing.
Cook (2023) performed a population-based retrospective cohort study in which the aim was to investigate the proportion of sebaceous carcinomas (SC) screened for MMR and the proportion found to be MMRd. In total, data from 1077 patients diagnosed with SC during January 2008 and December 2018 from the National Cancer Registration and Analysis Service in England were used. IHC staining was conducted in 220 cases to test for the expression of 4 MMR proteins (MLH1, MSH2, MSH6, and PMS2). Germline DNA MMR testing was performed among 24 cases. Outcomes relevant for this literature analysis included the prevalence of overall MMRd, loss of MLH1 and/or PMS2 expression, and Lynch syndrome.
Kattapuram (2023) performed a retrospective cohort study in which the aim was to characterize patients with sebaceous lesions (SL). In total, 447 patients with 473 SLs diagnosed between January 2009 and December 2019 at the University of Michigan, were identified. SL encompassed sebaceous carcinoma (SC), sebaceous adenoma (SA), sebaceoma (SO), or sebaceous neoplasm not otherwise specified (SN). Patients with sebaceous hyperplasia but no other SL were excluded. IHC staining was conducted to test the expression of the MMR proteins MLH1, MSH2, MSH6, PMS2, and EPCAM. Abnormal staining was present if staining ³1 MMR protein was absent. None of the patients underwent testing for MLH1 promoter methylation. Germline genetic testing was based on the presence of MMRd. Outcomes relevant for this literature included the prevalence of overall MMRd, lost expression of MLH1 and/or PMS2, MLH1 promoter methylation, and Lynch syndrome.
Sinson (2023) performed a retrospective cohort study of non-selected sebaceous neoplasms (SN) to identify associations with Muir-Torre syndrome (MTS). MTS is a phenotypic variant of Lynch syndrome. In total, 123 tumor specimens were identified from archived samples collected from 2004 to 2017 in all pathological laboratories in the Poitou-Charentes region of France. IHC staining was conducted to test the expression of the 4 MMR proteins. MMRd was present if ³1 MMR protein lost 100% of its expression in tumor cells. MSI status was determined by microsatellite analysis and was assigned when ³2 microsatellites were unstable. MLH1 promoter methylation analysis was conducted among unstable tumors with MLH1/PMS2 loss. Patients with a known mutation in one of the genes of the MMR system were considered MTS-SL. Outcomes relevant for this literature analysis included the prevalence of overall MMRd, MSI status, MLH1 and/or PMS2 loss, and Lynch syndrome.
Prostate cancer
Abida (2019) performed a prospective analysis to determine the prevalence of MSI-H/dMMR prostate cancer and the clinical benefit of anti–PD-1/PD-L1 therapy in patients with prostate cancer. In this case series, 1551 tumors from 1346 patients were prospectively analyzed at Memorial Sloan Kettering Cancer Centre using the MSK-IMPACT targeted sequencing assay between January 2015 and January 2018. Tumors were also profiled with optional germline analysis and IHC for MMR proteins (MSH2, MSH6, MLH1, PMS2). Microsatellite instability was assessed with the MSI-sensor algorithm, and tumor mutation burden was evaluated. Pathogenic mutations in MMR genes were considered oncogenic. Statistical comparisons of MSI-H and dMMR frequencies were made across disease subsets. Outcomes relevant for this literature analysis included overall MMRd, MSI-H, germline mutation, and somatic alteration.
Pritzlaff (2019) conducted a clinical lab cohort to assess genetic predispositions in prostate cancer patients. A total of 1812 men underwent multigene panel testing between April 2012 and September 2017 at Ambry Genetics, USA. A separate analysis was done for 150 patients with prior genetic testing, including BRCA1/2 or Lynch syndrome screening, IHC for MMRd, and other somatic testing. Patients were tested for up to 67 cancer susceptibility genes using Sanger or next-generation sequencing, along with gross deletion/duplication analysis. Outcomes relevant for this literature analysis included isolated MMRd, MLH1 and positive testing for pathogenic MMR variants.
Kagawa (2021b) conducted a retrospective cohort study on 337 prostate cancer patients who underwent surgical resection at Saitama Medical Center in Japan between January 2001 and May 2016. IHC for MMR proteins (MLH1, MSH2, MSH6, PMS2) was performed on formalin-fixed paraffin-embedded tissue sections. MMRd was identified when tumor cells showed no nuclear staining compared to adjacent normal tissue. Genetic testing, including Sanger sequencing and MLPA analysis, was performed on blood and tumor samples to detect germline variants and copy number variations in MMR genes. Somatic alterations in the MSH2 gene, including promoter methylation, were also examined. Outcomes relevant for this literature analysis included MMRd by IHC, loss of MLH1/PMS2 and Lynch syndrome.
Oka (2023) conducted a retrospective study on 129 prostate cancer patients who underwent radical prostatectomy at Toranomon Hospital in Tokyo, Japan, between January 2012 and December 2015. The study aimed to investigate MMRd by analyzing tumor specimens using IHC to identify the MMR proteins MLH1, PMS2, MSH2, and MSH6. Tissue microarrays were constructed from formalin-fixed, paraffin-embedded tumor and control samples. Immunohistochemical staining was performed with standardized automated methods. MMRd was defined by the complete loss of nuclear staining in tumor cells, with adjacent normal tissue serving as a control. Tumors exhibiting MMRd were referred for genetic counseling regarding Lynch syndrome, and germline genetic testing was conducted using next-generation sequencing, with variant confirmation by Sanger sequencing. Outcomes relevant for this literature analysis included MMRd in IHC and germline mutation in MSH2.
Truong (2023) analyzed somatic and germline variants in prostate cancer patients who underwent MSK-IMPACT sequencing at Memorial Sloan Kettering Cancer Center between 2015 and 2020. The goal was to identify tumor variants that would trigger confirmatory germline testing, using a 10% germline probability threshold. The sequencing covered ≥341 somatic and ≥76 germline genes, focusing on 60 cancer risk genes. Germline variants were classified according to the ACMG/AMP criteria, and somatic variants were assessed for oncogenic potential using the OncoKB platform. The main objective was to identify variants in tumor-only sequencing that would warrant confirmatory germline testing due to their high probability of being germline in origin. A threshold of 10% germline probability was used. The study also investigated whether tumor location or other factors, like tumor mutation burden TMB or microsatellite instability MSI status, modified the likelihood of a germline variant. Additionally, the variant allele frequency VAF of variants was analyzed to distinguish between pathogenic germline and somatic variants. Outcomes relevant for this literature analysis included overall MMRd by MSI status, loss of MLH1 and germline MMR variant, Lynch syndrome overall and germline carries without MSI-H tumor.
Results
For each cancer type, please see Table 2 for a summary of the prevalences of overall MMRd and/or MSI, loss of MLH1/PMS2 and/or MLH1 promoter methylation (MLH1-PM), germline (likely) pathogenic variants (PV) diagnosing Lynch syndrome, somatic (L)PV/LOH, UMMRd, and age and sex.
|
Study characteristics |
Prevalence |
Age and Sex |
|||||
|
Author (year) |
Study design |
Overall MMRd (IHC MMR) or MSI |
Loss of MLH1/PMS2, MLH1-PM |
Germline (likely) PV MMR gene |
Somatic (likely) PV MMR gene and/or LOH |
UMMRd |
|
|
Epithelial ovarian cancer (EOC) |
|||||||
|
Mitric (2023) |
Meta-analysis |
EOC overall · MMRd: 6% (95% CI 5% to 8%), I2 of 80%, based on N=328/5814 patients from 27 studies · MSI-H: 13% (95% CI 12% to 15%), I2 of 0%, based on N=315/2375 patients from 14 studies Endometrioid · MMRd: 12% (95% CI 9% to 16%), I2 of 60%, based on N=193/1625 patients from 19 studies · MSI-H: 12% (95% CI 8% to 18%), I2 of 0%, based on N=19/158 patients from 5 studies Serous · MMRd: 1% (95% CI 0% to 2%), I2 of 56%, based on N=16/1903 patients from 12 studies · MSI-H: 9% (95% CI 4% to 19%), I2 of 0%, based on N=71/556 patients from 6 studies Other · MMRd: 3% (95% CI 1% to 14%), I2 of 0%, based on N=24/465 patients from 14 studies · MSI-H: 13% (95% CI 8% to 20%), I2 of 0%, based on N=16/125 patients from 5 studies |
· MLH1-PM: 76% (95% CI 64% to 84%), I2 of 0%, based on N=53/70 patients with MLH1 deficiency from 13 studies |
EOC overall · 2% (95% CI 1% to 3%), I2 of 88%, based on N=232/14809 patients from 14 studies Endometrioid · 3% (95% CI 2% to 5%), I2 of 58%, based on N=42/1198 patients from 6 studies Serous · 1% (95% CI 0% to 2%), I2 of 84%, based on N=43/6016 patients from 7 studies Other · 1% (95% CI 0% to 2%), I2 of 0%, based on N=11/1043 patients from 6 studies |
Not reported |
Not reported |
· Information about age at diagnosis not reported · 100% female |
|
Summary Range or estimate upon availability |
|
· MMRd: 1% (serous) - 12% (endometrioid) · MSI-H: 9% (serous) - 13% (other) Source: Mitric (2023) |
· MLH1-PM: 76% (95% CI 64% to 84%) based on patients with lost MLH1 Source: Mitric (2023) |
· 1% (95% CI 0% to 2%) Source: Mitric (2023) |
Not reported |
Not reported |
Not reported |
|
Pancreatic ductal cancer (PDAC), biliary tract cancer (BTC) and gallbladder cancer (GBC) |
|||||||
|
Agaram (2010) · Ampullary adenocarcinomas |
Retrospective cohort study · Setting: Pathology database of MSKCC, New York · Period: not reported |
· MMRd: N=3/54 (5.6%) |
· Lost MLH1/PMS2: N=1/3 (33.3%) MMRd patients |
Not reported |
Not reported |
Not reported |
Total cohort · Mean age of 64y · N=20/54 (37.0%) females |
|
Hu (2018) · Pancreatic adenocarcinoma |
Retrospective cohort study · Setting: MSKCC institutional tumor registry · Period: 2006-2017 |
· MMRd by IHC, germline testing or NGS: N=7/833 (0.8%) |
· Not reported |
· N=7/7 (100%) MMRd patients · Overall: N=7/833 (0.8%) |
Not reported |
Not reported |
MMRd tumours · Age range: 42-88y · Sex not reported |
|
Christakis (2019) · Upper gastrointestinal tract carcinomas |
Institutional cohort study · Setting: Dana Farber Cancer Institute, Boston, USA · Period: not reported |
Pancreas · MSI: N=2/199 (1.0%) Gallbladder · MSI: N=0/19 (0%) Bile duct · MSI: N=1/60 (2%) Ampullary · MSI: N=0/11 (0%)
|
Pancreas · Lost MLH1/PMS2: N=1/2 (50%) MSI patientsà data on MLH1PM not reported Gallbladder · Not reported Bile duct · Not available Ampullary · Not reported |
Pancreas · Overall: N=1/199 (0.5%) Gallbladder · Not reported Bile duct · Not reported Ampullary · Not reported
|
Pancreas · 1/199 (0.5%) Gallbladder · Not reported Bile duct · Not available Ampullary · Not reported
|
Pancreas · Not reported Gallbladder · Not reported Bile duct · Not reported Ampullary · Not reported
|
Total cohort · Median age: 65.3y (range, 19-93.6) · N=219/645 (34%) females LS patients · Median age: 69.6y (range, 49.8-74.6) · Sex not reported |
|
Abrha (2020) · Gastrointestinal malignancies |
Retrospective cohort study · Setting: SCCI · Period: January 2016 – December 2017 |
Pancreas · MMRd: N=13/244 (5.3%) à 3/13 MMRd cases were ampullary carcinomas Gallbladder · MMRd: N=1/41 (2.5%) Cholangiocarcinoma · MMRd: N=0/44 (0%) Ampullary · MMRd: N=3/14 (21.4%) |
Pancreas · MLH1-PM: N=3/13 (23.1%) MMRd patients (n=5 false positives) Gallbladder · Not reported Cholangiocarcinoma · Not reported Ampullary · MLH1-PM: N=2/3 (66.7%) MMRd patients |
Pancreas · N=3/13 (23.1%) MMRd patients (n=5 false positives) · Overall: N=3/244 (1.2%) Gallbladder · Not reported Cholangiocarcinoma · Not reported Ampullary · Not reported |
Pancreas · Double somatic: N=2/13 (15.4%) MMRd patients (n=5 false positives) · Possibly double somatic: N=3/13 (23.1%) MMRd patients (n=5 false positives) Gallbladder · Not reported Cholangiocarcinoma · Not reported Ampullary · Not reported |
Pancreas · N=2/13 (11.1%) MMRd patients (n=5 false positives) Gallbladder · Not reported Cholangiocarcinoma · Not reported Ampullary · Not reported |
Pancreatic MMRd patients · Median age at diagnosis: 66y (IQR, 57-72) · N=7/13 (54%) females Gallbladder · Not reported Cholangiocarcinoma · Not reported Ampullary · Not reported |
|
Mettman (2020) · Pancreatic adenocarcinoma |
Retrospective cohort study · Setting: Department of Pathology and Laboratory Medicine, University of Kansas Medical Center · Period: December 2017 – September 2019 |
· N=184 pancreatic fine-needle aspiration specimens · N=65/184 (35.3%) with sufficient material to perform IHC · IHC: N=5/65 (7.7%) absence of labeling for one or two of the MMR proteins, but results were invalid |
Not reported |
Not reported |
Not reported |
Not reported |
Cohort that underwent IHC MMR testing · Median age: 71y (range, 34-93) · N=30/65 (64.2%) females |
|
Grant (2021) · Pancreatic adenocarcinoma |
Retrospective cohort study · Setting: Ontario Pancreas Study, Canada · Period: not reported |
Clinical cohort · MMRd by IHC or WGS: N=12/1213 (1.0%), not all tested: MMRd by IHC: N=8/53 (7.5%) Genomics cohort · MSI-H by WGS: N=9/288 (3.1%) |
Genomics cohort · Lost MLH1/PMS2: N=2/288 (0.7%) MMRd patients
|
· N=14/519 (2.7%) patients that underwent germline MMR gene sequencing |
N=4/288 (1.4%) from the genomics cohort who had MMRd tumours |
Not reported |
MMRd tumours · Median age: 61.5y (IQR, 52.5-74.8) · N=8/12 (66.7%) females |
|
Kryklyva (2022) · Ampullary and pancreatic carcinoma, early onset (<50y) |
Retrospective cohort study · Setting: PALGA · Period: January 2002-December 2012 |
Pancreas · MSI: N=2/44 (4.5%) Ampullary · MSI: N=1/23 (4.3%) |
Pancreas · Lost MLH1/PMS2: N=0/2 (0%) MSI patients Ampullary · Lost MLH1/PMS2: N=1/1 (100%) MSI patients |
Pancreas · N=2/2 (100%) MSI patients · Overall: N=2/44 (4.5%) Ampullary · N=1/1 (100%) MSI-patients · Overall: N=1/23 (4.3%) |
Pancreas · N=0/2 (0%) MSI patients Ampullary · N=0/1 (0%) MSI patients |
Pancreas · N=0/2 (0%) MSI patients Ampullary · N=0/1 (0%) MSI patients |
Pancreas · Age: 43y (range, 30-49) · N=16/44 (36.4%) females Ampullary · Age: 44y (range, 33-49) · N=13/23 (56.5%) females |
|
Kubo (2022) · Hepato-biliary-pancreatic malignancies |
Retrospective cohort study · Setting: 7 hospitals in the KHBO Group · Period: January 2019-April 2021 |
Pancreas · MSI-H: N=2/144 (1.4%) Gallbladder · MSI-H: N=1/18 (5.6%) Cholangiocarcinoma · MSI-H: N=8/88 (9.1%) |
Pancreas · Not reported Gallbladder · Not reported Cholangiocarcinoma · Not reported
|
Pancreas · N=0/2 (0%) MSI-H patients Gallbladder · N=0/1 (0%) MSI-H patients Cholangiocarcinoma · N=0/8 (0%) MSI-H patients
LS overall: 0% |
Pancreas · Not reported Gallbladder · Not reported Cholangiocarcinoma · Not reported |
Pancreas · Not reported Gallbladder · Not reported Cholangiocarcinoma · Not reported |
Pancreatic MSI-H tumours · Age range: 54-67y · N=0/2 (0%) females Gallbladder MSI-H tumours · Age: 70y · N=1/1 (100%) males Cholangiocarcinoma MSI-H tumours · Age range: 51-77y · N=6/8 (75%) males |
|
Moy (2015) · Gallbladder cancer |
Retrospective cohort study · Setting: surgical pathology files of Massachusetts General Hospital, Boston · Period: 1988-2012 |
· MMRd in N=6/77 (7.8%) |
· Lost MLH1/PMS2: N=1/6 (16.7%)
|
· N=0/6 (0%) MSI-patients · Overall: N=0/77 (0%) |
Not reported |
Not reported |
MSI tumours · Mean age: 70.8y · N=2/6 (33.3%) males |
|
Goeppert (2019) · Gallbladder cancer |
Retrospective cohort study · Setting: University Hospital Heidelberg, Germany · Period: 1995-2010 |
· MSI-H: N=1/69 (1.4%) · IHC: “no loss of nuclear immunoreactivity for all tested repair proteins” |
Not detected |
Not reported |
Not reported |
Not reported |
MSI-H tumour · Age at diagnosis: 68y · N=0/1 (0%) male |
|
Ju (2020) · Intra- and extrahepatic cholangiocarcinomas |
Retrospective cohort study · Setting: institutional database (Citrix Copath and EPIC Hyperspace) · Period: 1993-2014 |
· Possible MMRd by IHC: N=9/96 (9.4%) · MSI: N=6/96 (6.3%) of which N=4 with MSI-H and N=2 with MSI-L. These 6 cases were grouped into the MMRd group |
· Lost MLH1/PMS2: N=5/6 (83.3%), none showed MLH1-PM |
· Not tested |
Not reported |
Not reported |
MMRd tumours · Mean age: 68y · N=3/6 (50%) males |
|
Ando (2022) · Biliary tract carcinomas |
Retrospective cohort study · Setting: Kagawa University Hospital, Japan · Period: January 2008-December 2017 |
· MMRd: N=5/116 (4.3%) · MSI-H: 2/5 (40%) MMRd patients which was determined by IHC |
· Lost MLH1/PMS2: N=1/5 (20%) cases of intrahepatic cholangiocarcinoma
|
Not reported |
Not reported |
Not reported |
Total cohort · Median age at diagnosis: 73y (range, 38-93) · N=73/116 (62.9%) males MMRd patients · Age range: 55-93 years · N=3/5 (60%) males |
|
Summary Range or estimate upon availability |
- |
Pancreas · MMRd: 0.8-7.5% Sources: Hu (2018), Abrha (2020), Grant (2021) · MSI: 1.0-4.5% Sources: Christakis (2019), Grant (2021), Kryklyva (2022), Kubo (2022) Gallbladder · MMRd: 2.5% Source: Abrha (2020) · MSI: 0-7.8% Sources: Christakis (2019), Kubo (2022), Moy (2015), Goeppert (2019) Bile duct · MMRd: 0-9.4% Sources: Abrha (2020), Ju (2020), Ando (2022) · MSI: 1.7-9.1% Sources: Christakis (2019), Kubo (2022), Ju (2020), Ando (2022) Ampullary · MMRd: 5.6-21.4% Sources: Agaram (2010), Abrha (2020) · MSI: 0-4.3% Sources: Christakis (2019), Kryklyva (2022)
|
Pancreas · MLH1/PMS2: 0-50% MMRd/MSI patients Sources: Christakis (2019), Grant (2021), Kryklyva (2022) · MLH1-PM: 23.1% Source: Abrha (2020) · Isolated lost MLH1: 0.7% Source: Grant (2021) Gallbladder · MLH1/PMS2: 16.7% Source: Moy (2015) Bile duct · MLH1/PMS2: 20-83.3% (MMRd cases) Sources: Ju (2020), Ando (2022) · MLH1-PM: 0% Source: Ju (2020) Ampullary · MLH1/PMS2: 33.3-100% MMRd/MSI patients Sources: Agaram (2010), Kryklyva (2022) · MLH1-PMhm: 66.7% Source: Abrha (2020) |
Pancreas · Overall: 0.5-4.5% Sources: Hu (2018), Christakis (2019), Abrha (2020), Grant (2021), Kryklyva (2022) Gallbladder · Overall: 0% Source: Moy (2015) Bile duct · Overall: 0% Source: Kubo (2022) Ampullary · Overall: 4.3% Source: Kryklyva (2022)
|
Pancreas · 0-15.4% Sources: Christakis (2019), Abrha (2020), Grant (2021), Kryklyva (2022) Gallbladder Not reported Bile duct Not reported Ampullary · 0% Source: Kryklyva (2022) |
Pancreas · 0 (MSI-patients) -11.1% (MMRd patients) Sources: Abrha (2020), Kryklyva (2022) Gallbladder Not reported Bile duct Not reported Ampullary · 0% Source: Kryklyva (2022) |
Pancreas (total cohort) · Age: 43-71y · 34-64.2% females Sources: Christakis (2019), Mettman (2020), Kryklyva (2022) Gallbladder (MSI-H tumours) · Age: 68-70.8y · 0-100% males Sources: Kubo (2022), Moy (2015), Goeppert (2019) Bile duct (MSI-H tumours) · Age: 51-77y · 75% males Source: Kubo (2022) Ampullary · Age: 44-64y · 37-56.5% females Sources: Agaram (2010), Kryklyva (2022)
|
|
Upper tract urothelial cancer (UTUC) and urothelial bladder cancer (UBC) |
|||||||
|
Rasmussen (2022) · UTUC excluding UBC |
Meta-analysis
|
· MMRd: 9% (range, 2.4% to 39.0%), based on N=140/1559 patients from 11 studies · MSI-H: 3.9% (range, 1.7% to 13%), based on N=903 patients from 4 studies |
· Lost MLH1: 4.1%, based on N=67/1628 patients from 12 studies |
· Suspected and verified LS: 4.7% (range, 0.8% to 13.9%), based on N=51/1087 patients from 8 studies
|
Not reported |
Not reported |
LS patients · Median age at onset: 38-64y (range, 38-86) · UTUC in females: 45% (range, 0% to 100%) among verified LS patients |
|
Audenet (2019) · UTUC and UBC
|
Both retrospective and prospective cohort study · 111 samples were prospectively profiled and 84 samples retrospectively |
UTUC · MSI-H: N=12/194 (6.2%) · MSI-H (MSI sensore score ³10): N=10/12 (83.3%) Lynch patients; 2 with LS indeterminate MSI sensor scores (3-10) UBC · MSI-H: N=4/454 (0.9%)
|
UTUC Not reported UBC Not reported |
UTUC · From N=47 patients consent to assess for germline mutations (MSI-H and based on family) · N=12/47 (25.5%) patients with (likely) pathogenic germline mutation · Overall: N=12/194 (6.2%) UBC · N=1/4 (25%) MSI patients · Overall: N=1/454 (0.2%) |
UTUC · N=1/12 (8.3%) UBC · Not reported
|
UTUC · N=1/12 (8.3%) UBC · Not reported
|
UTUC · Median age: 67.1y (IQR, 58.1-74.5) · N=74/195 (38%) females UBC · Median age: 67.5y (IQR, 60.1-74.4) · N=367/454 (81%) males |
|
Kagawa (2021) · UBC only |
Retrospective cohort study · Setting: Saitama Medical Center, Japan · Period: June 1997-February 2018 |
· MMRd: N=9/618 (1.5%) à the 9 MMRd cases by IHC were tested for MSI · MSI-H: N=7/9 (78%) MMRd cases
|
· Lost MLH1/PMS2: N=3/9 (33.3%) IHC MMRd cases · MLH1-PM: N=0/3 (0%)
|
· Minimal: N=2/618 (0.6%, 95% CI: 0.1, 1.2%) · Maximal: N=7/618 (1.1%, 95% CI: 0.5, 2.3%) |
Not reported |
Not reported |
MMRd patients · Median age: 68y (range, 63-79) · N=9/9 (100%) males |
|
Kullmann (2023) · UTUC excluding UBC |
Retrospective cohort study · Setting: archives of three collaborating institutes of Pathology: Málaga, Marburg and Erlangen · Period: 1995-2017 |
· MSI using Bethesda panel and Idylla assay: N=4/243 (1.6%) · MMRd: N=5/156 (3.2%)
|
Not reported |
Not reported |
Not reported |
Not reported |
MSI-H tumours · Age range 52 to 83y · N=1/4 (25%) females |
|
Ma (2023) · UTUC and UBC |
Retrospective cohort study · Setting: search in database of Shandong Provincial Hospital, Jinan, China · Period: 2018-2022 |
UTUC · MMRd: N=9/219 (3.9%) UBC · MMRd: N=1/361 (0.3%) UTUC + UBC · MMRd: N=5/13 (38.4%)
|
UTUC + UBC · “No cases with MLH1 and/or PMS2 loss were identified”
|
UTUC · N=5/9 (55.6%) had mutation in MSH2 gene UBC · Not reported UTUC + UBC · N=2/5 (40%) (1 no data) |
Not reported |
Not reported |
UTUC · Age of MMRd cases: 64y (SD 13.5) · N=7/14 (50.0%) females UBC · Age of MMRd cases: 62.3y (SD 16.7) · N=3/6 (50.0%) males |
|
Pivovarcikova (2023) · UTUC excluding UBC |
Retrospective cohort study · Setting: Šikl’s Department of Pathology, Czech Republic · Period: 2010-2022 |
· MMRd: N=15/180 (8.3%)
|
· Lost MLH1/PMS2: N=3/15 (20%) · MLH1-PM: N=1/3 (33.3%) |
· Overall: N=5/180 (2.8%), including 4 mutations in MSH6 and 1 mutation in MSH2 |
Not reported |
Not reported |
· Mean age of 66.2y among 5 LS cases with UTUC · N=3/5 (60%) females |
|
Summary Range or estimate upon availability |
- |
UTUC · MMRd: 3.2-9% Sources: Rasmussen (2022), Kullmann (2023), Ma (2023), Pivovarcikova (2023) · MSI: 1.6-6.2% Sources: Rasmussen (2022), Audenet (2019), Kullmann (2023) UBC · MMRd: 0.3-1.5% Sources: Kagawa (2021), Ma (2023) · MSI: 0.9% Source: Audenet (2019) |
UTUC · Lost MLH1/PMS2: 0-20% Sources: Rasmussen (2022), Pivovarcicova (2023) · MLH1-PM: 33.3% Source: Pivovarcicova (2023) UBC · Lost MLH1/PMS2: 0% Sources: Kagawa (2021) · MLH1-PM: 0% Source: Kagawa (2021) |
UTUC · Overall: 2.3-6.2% Sources: Rasmussen (2022), Audenet (2019), Pivovarcicova (2023) UBC · Overall: 0.2-1.1% Sources: Audenet (2019), Kagawa (2021) |
UTUC · 8.3% Source: Audenet (2019) UBC Not reported |
UTUC · 8.3% Source: Audenet (2019) UBC Not reported |
UTUC · Age: 52-83y (MSI tumours), 64y (MMRd tumours), 67.1y (all patients) · 25% females (MSI tumours), 50% females (MMRd tumours), 38% females (all patients) Sources: Audenet (2019), Kullmann (2023), Ma (2023) UBC · Age: 62.3-68y (MMRd tumours), 67.5y (all patients) · 50-100% males (MMRd tumours), 81% males (all patients) Sources: Audenet (2019), Kagawa (2021), Ma (2023) |
|
Small bowel cancer (SBA) and gastric cancer (GC) |
|||||||
|
Christakis (2019) · Upper gastrointestinal tract cancers |
Institutional cohort study · Setting: Dana Farber Cancer Institute, Boston, USA · Period: not reported
|
Small bowel · MSI: N=8/29 (28%) Gastric · MSI: N=9/97 (9%) |
Small bowel · Lost MLH1/PMS2: N=4/8 (50%) MMRd patients · MLH1-PM: N=2/4 (50%) Gastric · Lost MLH1/PMS2: N=7/9 (77.8%) MMRd patients · MLH1PMm: N=4/7 (57.1%) |
Small bowel · Overall: 4/29 (13.8%) Gastric · Overall: 1/97 (1.0%)
|
Small bowel · N=2/8 (25%) Gastric · N=1/9 (11.1%)
|
Small bowel · N=0/8 (0%) Gastric · N=3/9 (33.3%) |
Total cohort · Median age: 65.3y (range, 19-93.6) · N=219/645 (34%) females LS patients · Median age: 69.6y (range, 49.8-74.6) · Sex not reported |
|
Abrha (2020) · Gastrointestinal malignancies |
Retrospective cohort study · Setting: SCCI · Period: January 2016 – December 2017 |
Small bowel · MMRd: N=1/20patients (5.0%) Gastric · MMRd: N=15/150 (10%)
|
Small bowel · Not reported Gastric · MLH1-PM: N=11/15 (73.3%) MMRd patients |
Small bowel · Not reported Gastric · N=0/15 (0%) MMRd patients (1 not tested) |
Small bowel · Not reported Gastric · Double somatic: N=1/15 (6.7%) MMRd patients · Possibly double somatic: N=2/15 (13.3%) MMRd patients
|
Small bowel · Not reported Gastric · N=1/15 (6.7%) MMRd patients |
Small bowel; MMRd patients · Age and sex not reported Gastric; MMRd patients · Median age at diagnosis: 77y (IQR, 71-86) · N=10/15 (67%) males |
|
Aparicio (2021) · Small bowel adenocarcinoma (SBA) |
Retrospective cohort study · Setting: France · Period: January 2009-December 2012 |
· MMRd: N=50/180 (28%) patients (66% duodenum, 22% jejunum, 12% ileum)
|
· Lost MLH1/PMS2: N=21/50 (42%) MMRd patients |
· N=17/50 (34%) MMRd patients · Overall: N=17/180 (9.4%) |
· N=113/125 (90.4%) at least one genomic alteration |
Not reported |
MMRd tumours · Median age: 58y · N=28/50 (56%) males |
|
Latham (2021) · SBA |
Retrospective cohort study · Setting: patients diagnosed with SBA at MSKCC from 2006-2019 |
· MMRd: N=26/100 (26%) |
· Lost MLH1 and/or PMS2 (IHC): N=17/26 (65.4%) MMRd patients · MLH1-PM: N=7/9 (77.8%) LS negative SBA patients |
· N=10/26 (38.5%) · Overall: N=10/100 (10%)
|
Among n=16 LS-negative SBA patients · N=2 patients declined additional work-up · N=9/14 (64.3%) had a somatic driver |
Not reported |
Total cohort · Median age: 60y · N=59/95 (62%) males MMRd LS cohort · Median age: 47.5y · N=5/10 (50%) males |
|
Sanchez (2021) · SBA |
Retrospective cohort study · Setting: 4 Spanish tertiary hospitals · Period: 2004-2020 |
· MMRd: N=20/94 (21.3%) |
· Lost MLH1/PMS2: N=7/20 (35%) MMRd patients · Isolated lost MLH1: N=2/20 (10%) MMRd patients
|
· N=9/15 (60%) with MMRd tumours that had germline testing Overall: N=9/94 (10.1%) SBAs |
Not reported |
Not reported |
MMRd tumours · Median age at diagnosis: 58y (IQR, 44.5-69) · N=12/20 (60%) males |
|
Suerink (2021) · Small bowel cancer |
Retrospective cohort study · Setting: PALGA · Period: 2012 – 2016 |
Resections · MMRd: N=74/332 (22.3%) Biopsies · MMRd: N=3/68 (4.4%) |
Resections · MLH1-PM: N=30/74 (40.5%) MMRd patients Biopsies · MLH1-PM: N=2/3 (66.7%) MMRd patients |
Resections · N=20/74 (27.0%) MMRd patients · Overall: N=20/332 (6.0%) Biopsies · N=0/3 (0%) MMRd patients · Overall: N=0/68 (0%) |
Resections, 2 somatic hits · N=10/74 (13.5%) MMRd patients Biopsies, 2 somatic hits · N=0/3 (0%) MMRd patients |
Resections · N=8/74 (10.8%) MMRd patients Biopsies · N=1/3 (33.3%) MMRd patients |
LS · Mean age of 54.6y · N=13/20 (65.0%) males Sporadic MMRd cancers · Mean age of 68.8y · N=23/44 (52.3%) males |
|
Kryklyva (2022) · Duodenal carcinoma |
Retrospective cohort study · Setting: PALGA · Period: January 2002-December 2012 |
<50y · MMRd: N=11/23 (47.8%) ³50y · MMRd: N=1/18 (5.6%)
|
<50y · Lost MLH1/PMS2: N=3/11 (27.3%) MMRd patients ³50y · Lost MLH1/PMS2: N=1/1 (100%) MMRd patients |
<50y · N=5/9 (55.6%) MMRd patients (2 failed) · Overall: N=5-7/23 (22-30%) ³50y · N=0/1 (0%) MMRd patients |
<50y · N=2/9 (22.2%) ³50y · N=1/1 (100%) |
Not reported |
<50y (n=23) · Age: 46y (range, 17-49) · N=15/23 (65.2%) males ³50y (n=18) · Age: 70y (range, 57-77) · N=13/18 (72.2%) males |
|
de Back (2024) · Small bowel cancer |
Retrospective cohort study · Setting: Netherlands Cancer Registry and PALGA · Period: 1999-2019 |
· MMRd: N=194/635 (30.6%) |
Not reported |
· N=107/194 (55%) · Overall: N=107/2697 (4%) to N=107/635 (16.9%) |
Not reported |
Not reported |
Total cohort · Median age: 69y (IQR, 60-77) · N=1410/2697 (52.3%) males |
|
Summary Range or estimate upon availability |
- |
Small bowel · MMRd: 4.4-47.8% Sources: Abrha (2020), Aparicio (2021), Latham (2021), Sanchez (2021), Suerink (2021), Kryklyva (2022), de Back (2024) · MSI: 28% Source: Christakis (2019) Gastric · MMRd: 10% Source: Abrha (2020) · MSI: 9% Source: Christakis (2019) |
Small bowel · Lost MLH1/PMS2: 27.3-65.4% (MMRd cases) Sources: Aparicio (2021), Latham (2021), Sanchez (2021), Kryklyva (2022) · MLH1-PM: 40.5% (resections, MMRd cases), 50%, 66.7% (biopsies, MMRd cases), 77.8% (LS negative cases) Sources: Christakis (2019), Latham (2021), Suerink (2021) · Isolated lost MLH1: 10% Source: Sanchez (2021) Gastric · Lost MLH1/PMS2: 77.8% (MMRd cases) Source: Christakis (2019) · MLH1-PM: 57.1%, 73.3% (MMRd cases) Sources: Christakis (2019), Abrha (2020) |
Small bowel · Overall: 0-30% Sources: Christakis (2019), Aparicio (2021), Latham (2021), Sanchez (2021), Suerink (2021), Kryklyva (2022), de Back (2024) Gastric · Overall: 0-1% Sources: Christakis (2019), Abrha (2020)
|
Small bowel · 0% (biopsies, MMRd cases), 13.5% (resections, MMRd cases), 22.2% (MMRd cases), 25% (MSI cases), 64.3% (LS negative cases) Sources: Christakis (2019), Latham (2021), Suerink (2021), Kryklyva (2022) Gastric · 6.7% (MMRd cases), 11.1% (MSI cases) Sources: Christakis (2019), Abrha (2020)
|
Small bowel · 0% (MSI cases), 10.8% (resections, MMRd cases), 33.3% (biopsies, MMRd cases) Sources: Christakis (2019), Suerink (2021) Gastric · 6.7% (MMRd cases), 33.3% (MSI cases) Sources: Christakis (2019), Abrha (2020)
|
Small bowel · Age: 46y (total cohort <50y), 47.5y (MMRd LS cases), 54.6y (LS cases), 58y (MMRd cases), · 50% males (MMRd LS cases), 56-60% males (MMRd cases), 65% males (LS cases), 65.2% males (total cohort <50y) Sources: Aparicio (2021), Latham (2021), Sanchez (2021), Suerink (2021), Kryklyva (2022) Gastric · Age: 77y (MMRd cases) · 67% males (MMRd cases) Source: Abrha (2020) |
|
Brain tumors (BT) |
|||||||
|
Rodriguez-Hernandez (2013) · Astrocytomas (N=20 low-grade, N=19 anaplastic, and N=57 glioblastoma) |
Study cohort · Setting: University Hospital of Salamanca, Spain · Recruitment period: June 2000-March 2006
|
· MMRd: N=41/96 (43%) · MSI-H: N=4/88 (5%) à all four glioblastomas |
· MLH1-PM: N=11/92 (12.0%) · Also, MSH2/MSH6 methylation and not corresponding with IHC staining |
· N=5/44 (11.4%) with MMR germline mutation · Overall: N=5/96 (5.2%)
“Only one glioblastoma was associated with LS” |
N=1/44 (2.3%) with MMR somatic mutation |
N=27/44 (61.4%) |
Low-grade · Median age: 35y (IQR, 30.3-46.0) · N=11/20 (55%) males Anaplastic · Median age: 57y (IQR, 47.0-66.0) · N=12/19 (63%) males Glioblastoma · Median age: 63y (IQR, 54.5-69.0) · N=36/57 (63%) males |
|
Hadad (2023) · Glioblastoma |
· Setting: University of California, San Francisco · Period: 2017-2022 |
· MSI-H by NGS: N= 9/459 (2.0%) |
Not reported |
· N=4/9 (44.4%) MSI-H patients · Overall: N=4/459 (0.9%) |
N=3/9 (33.3%) or N=3/459 (0.65%) with somatic mutations |
Not reported |
De novo RRD GBM, IDH-wildtype · Mean age: 50y (IQR, 40-57) · N=3/9 (33.3%) males |
|
Summary Range or estimate upon availability |
- |
Astrocytomas · MSI: 5% Source: Rodriguez-Hernandez (2013) Glioblastoma · MSI: 2% Source: Hadad (2023)
|
Astrocytomas · MLH1-PM: 12% Sources: Rodriguez-Hernandez (2013) Glioblastoma Not reported |
Astrocytomas · Overall: 5.2% Source: Rodriguez-Hernandez (2013) Glioblastoma · Overall: 0.9% Source: Hadad (2023)
|
Astrocytomas · 2.3% Source: Rodriguez-Hernandez (2013) Glioblastoma · 0.65% Source: Hadad (2023) |
Astrocytomas · 61.4% Source: Rodriguez-Hernandez (2013) Glioblastoma Not reported |
Glioblastoma · Age: 50-63y · 33.3% males (MSI cases), 63% males (total cohort) Sources: Rodriguez-Hernandez (2013), Hadad (2023) |
|
Sebaceous gland tumor (SGT) |
|||||||
|
Kunnackal John (2022) · Sebaceous tumors |
Meta-analysis |
52.5% (95% CI 48.74% to 56.26%), based on N=355/676 patients from 10 studies |
Not reported |
18.8% (95% CI 13.03% to 24.57%), based on N=33/176 patients from 2 studies |
Not reported |
Not reported |
Age and sex not reported |
|
Cook (2023) · Sebaceous tumors |
Retrospective cohort study · Setting: National Cancer Registration and Analysis Service, England · Period: |
· MMR IHC performed in N=220/1077 (20%) of tumours · MMRd: N=70/220 (32%) |
· Lost MLH1/PMS2: N=15/70 (21.4%) MMRd patients
|
· N=12/24 (50%) tested patients · Overall: N=12/220 (5.5%) |
Not reported |
Not reported |
Total cohort · Median age at diagnosis: 76y (IQR: 17) · N=632/1077 (58.7%) males |
|
Kattapuram (2023) · Sebaceous lesions |
Retrospective cohort study · Setting: University of Michigan · Period: January 2009-December 2019 |
· IHC conducted in N=173/427 (41%)patients · N=92/173 (53%) with abnormal staining (n=20 known LS cases excluded) |
· Lost MLH1/PMS2: N=13/92 (14.1%) patients with abnormal staining · “No patient underwent MLH1-PM testing”
|
· N=69 abnormal IHC staining referred to genetics · Overall: N=6 newly diagnosed à N=6/69 (9%); N=20 with existing LS à N=26/69 (37.7%) |
Not reported |
Not reported |
Total cohort · Mean age at diagnosis: 69.3y (SD 12.4) · N=281/447 (63%) males LS patients · Mean age at diagnosis: 59.7y (SD 10.9) · N=15/27 (56%) males |
|
Sinson (2023) · Sebaceous lesions |
Retrospective cohort study · Setting: pathological laboratories in the Poitou-Charentes region of France · Period: 2004-2017 |
· MMRd: N=87/123 (70.7%) of tumours · MSI: N=59/123 (47.8%) |
· Lost MLH1/PMS2: N=12/123 (9.8%) sebaceous lesions · MLH1-PM: N=0/12 (0%) |
· Overall: N=25/123 (20.3%) tumours (not all tumours tested) |
Not reported |
Not reported |
All histological subtypes · Mean age: 71.1y (range, 30-96) · Sex ratio (M:W): 1.7 |
|
Summary Range or estimate upon availability |
- |
· MMRd: 32-70.7% Sources: Kunnackal John (2022), Cook (2023), Kattapuram (2023), Sinson (2023) · MSI: 47.8% Source: Sinson (2023)
|
· Lost MLH1/PMS2: 14.1%, 21.4% (MMRd cases), 9.8% (all cases) Sources: Cook (2023), Kattapuram (2023), Sinson (2023) · MLH1-PM: 0% Source: Sinson (2023) |
· Overall: 5.5%, 9%, 18.8% Sources: Kunnackal John (2021), Cook (2023), Kattapuram (2023) |
Not reported |
Not reported |
· Age: 69.3-76y · 58.7-63% males Sources: Cook (2023), Kattapuram (2023), Sinson (2023) |
|
Prostate cancer (PRC) |
|||||||
|
Abida (2019) · Prostate cancer |
Case series · Setting: prospective analysis of patients with prostate cancer undergoing treatment at MSKCC, New York · Period: January 2015-Januari 2018 |
· MSI-H: N=32/1033 (3.1%) |
Not reported |
· N=7/32 (21.9%) MSI-H patients · Overall: N=7/1033 (0.68%) |
· N=6/1033 (0.6%) with somatic alteration, but without evidence of MSI or hypermutation |
Not reported |
MSI-H cohort · Median age at diagnosis: 64.5y (range, 39-85) · 100% males |
|
Pritzlaff (2020) · Prostate cancer |
Clinical lab cohort · Setting: Ambry genetics, USA · Period: April 2012-September 2017 |
· MMRd: 2.8% |
No prior genetic testing · Isolated MLH1 loss: N=2/1501 (0.1%) tested patients
|
No prior genetic testing · N=26/1501 (1.7%)
|
Not reported |
Not reported |
Total cohort · Median age at diagnosis: 60y (IQR 54-66) · 100% males |
|
Kagawa (2021) · Prostate cancer |
Retrospective cohort study · Setting: Saitama Medical Center and Saitama Medical University, Japan · Period: Januari 2001-May 2016 |
· MMRd: N=4/337 (1.2%) |
Not present |
· N=0/2 (0%) that were tested· Overall: N=0/337 (0%) |
· N=2/4 at least one somatic alteration inactivating MSH2 without MSH2 hypermethylation was identified |
· N=2/337 (0.6%) with supposed Lynch-like syndrome |
Total cohort · Median age: 67y (range, 52-81) · 100% males
MMRd patients · Age range: 64-75y · 100% males
|
|
Oka (2023) · Prostate cancer |
Retrospective cohort study · Setting: Toranomon Hospital, Tokyo, Japan · Period: 2012-2015 |
· MMRd: N=1/129 (0.8%) |
Not present |
· N=1/129 (0.8%) |
Not reported |
Not reported |
Total cohort · Median age at diagnosis: 68y (range, 52-79) · 100% males
MMRd patient · Age: 68y · 100% male |
|
Truong (2023) · Prostate cancer |
Retrospective cohort study · Setting: MSKCC · Period: 2015-2020 |
· N=1883 patients included in study · MSI status available in n=1603 tumours · MSI-H: N=35/1603 (2.2%) |
· Lost MLH1: N=0/3 (0%)
|
· N=6/35 (17%) MSI-H patients · Overall: N=6/1605 (0.4%) · N=8 germline carriers without MSI-H tumour |
Not reported |
Not reported |
Total cohort · Median age at diagnosis: 62y (IQR, 56-68) · 100% males |
|
Summary Range or estimate upon availability |
- |
· MMRd: 0.8-2.8% Sources: Pritzlaff (2020), Kagawa (2021), Oka (2023) · MSI: 2.2-3.1% Sources: Abida (2019), Truong (2023) |
· Isolated lost MLH1: 0-0.1% Sources: Pritzlaff (2020), Truong (2023) |
· Overall: 0-1.7% Sources: Abida (2019), Pritzlaff (2020), Kagawa (2021), Oka (2023), Truong (2023) |
· 0.59-0.6%, Sources: Abida (2019), Kagawa (2021) |
· 0.6% Source: Kagawa (2021) |
· Age: 60-68y (all cases), 64.5y (MSI-H cases), 64-75y (MMRd cases) · 100% males Sources: Abida (2019), Pritzlaff (2020), Kagawa (2021), Oka (2023), Truong (2023) |
Abbreviations: MMRd, mismatch repair deficiency; IHC, immunohistochemistry; UMMRd, unexplained mismatch repair deficiency; MLH1-PM, MLH1 promoter methylation; LS, Lynch syndrome; MSI(-H), microsatellite instability (- high); EOC, epithelial ovarian carcinoma; PDAC, Pancreatic ductal cancer, BTC, biliary tract cancer; GBC, gallbladder cancer; UTUC, upper tract urothelial cancer; UBC, urothelial bladder cancer; SBA, small bowel adenocarcinoma; GC, gastric Cancer; BT, brain tumor; SGT, sebaceous gland tumor; PRC, prostate cancer; NGS, next generation sequencing; WGS, whole generation sequencing; IQR, interquartile range; SD, standard deviation; MSKCC, Memorial Sloan-Kettering Cancer Center; USA, United States of America; SCCI, Stanford Comprehensive Cancer Institute; KHBO, Kansai Hepato-Biliary Oncology Group; PALGA, Nationwide Network and Registry of Histopathology and Cytopathology in the Netherlands; De novo RRD GBM, de novo replication repair deficient glioblastoma.
A systematic review of the literature was performed to answer the following questions: What is the yield of standard tumor testing (IHC MMR) for MMRd and Lynch syndrome in tumors (ovarian cancer, pancreatic cancer, biliary tract and gall bladder cancer, urothelial cancer, small bowel cancer, stomach cancer, brain tumors, sebaceous gland tumors and prostate cancer) and germline DNA analysis at the population level?
Table 1. PICO
|
Patients |
Patients with ovarian cancer, pancreatic cancer, biliary tract and gall bladder cancer, urothelial cancer, small bowel cancer, stomach cancer, brain tumors, sebaceous gland tumors, prostate cancer |
|
Intervention |
MMRd tests using immunohistochemistry (IHC) followed by germline tests in case of MMRd |
|
Control |
Not applicable |
|
Outcomes |
Prevalence of overall MMRd, MLH1 methylation, germline mutations, somatic mutations, unexplained MMRd |
|
Other selection criteria |
n/a |
Relevant outcome measures
The guideline panel considered the prevalence of overall MMRd, MLH1 promoter methylation, germline and somatic MMR (likely) pathogenic variants or LOH and UMMRd as critical outcome measures for decision making.
A priori, the guideline panel did not define the outcome measures listed above but used the definitions in the studies.
No minimal clinically (patient) important differences were defined since the aim of this guideline chapter was to provide an overview of reported prevalences and not to assess the clinical important differences in outcomes.
Search and select (Methods)
A systematic literature search was performed by a medical information specialist using the following bibliographic databases: Embase.com and Ovid/Medline. Both databases were searched from 2010 to 2024 for systematic reviews and observational studies (both comparative and non-comparative research). Systematic searches were completed using a combination of controlled vocabulary/subject headings (e.g., Emtree-terms, MeSH) wherever they were available and natural language keywords. The overall search strategy was derived from three primary search concepts: (1) Lynch syndrome and/or UMMRd; (2) types of cancer; (3) (molecular) tumor testing (IHC, MMRd, MSI) and/or germline MMR gene analysis. Duplicates were removed using EndNote software. After deduplication a total of 1469 records were imported for title/abstract screening.
Initially, 48 studies were selected based on title and abstract screening. After reading the full text, 13 studies were excluded (see the exclusion table under the tab ‘Evidence tabellen’), and 35 studies were included.
- Abida W, Cheng ML, Armenia J, Middha S, Autio KA, Vargas HA, Rathkopf D, Morris MJ, Danila DC, Slovin SF, Carbone E, Barnett ES, Hullings M, Hechtman JF, Zehir A, Shia J, Jonsson P, Stadler ZK, Srinivasan P, Laudone VP, Reuter V, Wolchok JD, Socci ND, Taylor BS, Berger MF, Kantoff PW, Sawyers CL, Schultz N, Solit DB, Gopalan A, Scher HI. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019 Apr 1;5(4):471-478. doi: 10.1001/jamaoncol.2018.5801. PMID: 30589920; PMCID: PMC6459218.
- Abrha A, Shukla ND, Hodan R, Longacre T, Raghavan S, Pritchard CC, Fisher G, Ford J, Haraldsdottir S. Universal Screening of Gastrointestinal Malignancies for Mismatch Repair Deficiency at Stanford. JNCI Cancer Spectr. 2020 Jun 19;4(5):pkaa054. doi: 10.1093/jncics/pkaa054. PMID: 33225206; PMCID: PMC7667994.
- Agaram NP, Shia J, Tang LH, Klimstra DS. DNA mismatch repair deficiency in ampullary carcinoma: a morphologic and immunohistochemical study of 54 cases. Am J Clin Pathol. 2010 May;133(5):772-80. doi: 10.1309/AJCPGDDE8PLLDRCC. PMID: 20395525.
- Ando Y, Kumamoto K, Matsukawa H, Ishikawa R, Suto H, Oshima M, Kamada H, Morishita A, Kobara H, Matsunaga T, Haba R, Masaki T, Suzuki Y, Okano K. Low prevalence of biliary tract cancer with defective mismatch repair genes in a Japanese hospital-based population. Oncol Lett. 2022 Jan;23(1):4. doi: 10.3892/ol.2021.13122. Epub 2021 Nov 4. PMID: 34820003; PMCID: PMC8607234.
- Aparicio T, Svrcek M, Henriques J, Afchain P, Lièvre A, Tougeron D, Gagniere J, Terrebonne E, Piessen G, Legoux JL, Lecaille C, Pocard M, Gornet JM, Zaanan A, Lavau-Denes S, Lecomte T, Deutsch D, Vernerey D, Puig PL. Panel gene profiling of small bowel adenocarcinoma: Results from the NADEGE prospective cohort. Int J Cancer. 2021 Apr 1;148(7):1731-1742. doi: 10.1002/ijc.33392. Epub 2021 Jan 4. PMID: 33186471.
- Audenet F, Isharwal S, Cha EK, Donoghue MTA, Drill EN, Ostrovnaya I, Pietzak EJ, Sfakianos JP, Bagrodia A, Murugan P, Dalbagni G, Donahue TF, Rosenberg JE, Bajorin DF, Arcila ME, Hechtman JF, Berger MF, Taylor BS, Al-Ahmadie H, Iyer G, Bochner BH, Coleman JA, Solit DB. Clonal Relatedness and Mutational Differences between Upper Tract and Bladder Urothelial Carcinoma. Clin Cancer Res. 2019 Feb 1;25(3):967-976. doi: 10.1158/1078-0432.CCR-18-2039. Epub 2018 Oct 23. PMID: 30352907; PMCID: PMC6359971.
- de Back TR, Linssen JDG, van Erning FN, Verbakel CSE, Schafrat PJM, Vermeulen L, de Hingh I, Sommeijer DW. Incidence, clinical management and prognosis of patients with small intestinal adenocarcinomas from 1999 through 2019: A nationwide Dutch cohort study. Eur J Cancer. 2024 Mar;199:113529. doi: 10.1016/j.ejca.2024.113529. Epub 2024 Jan 9. PMID: 38232410.
- Christakis AG, Papke DJ, Nowak JA, Yurgelun MB, Agoston AT, Lindeman NI, MacConaill LE, Sholl LM, Dong F. Targeted Cancer Next-Generation Sequencing as a Primary Screening Tool for Microsatellite Instability and Lynch Syndrome in Upper Gastrointestinal Tract Cancers. Cancer Epidemiol Biomarkers Prev. 2019 Jul;28(7):1246-1251. doi: 10.1158/1055-9965.EPI-18-1250. Epub 2019 Apr 26. PMID: 31028081.
- Cook S, Pethick J, Kibbi N, Hollestein L, Lavelle K, de Vere Hunt I, Turnbull C, Rous B, Husain A, Burn J, Lüchtenborg M, Santaniello F, McRonald F, Hardy S, Linos E, Venables Z, Rajan N. Sebaceous carcinoma epidemiology, associated malignancies and Lynch/Muir-Torre syndrome screening in England from 2008 to 2018. J Am Acad Dermatol. 2023 Dec;89(6):1129-1135. doi: 10.1016/j.jaad.2023.03.046. Epub 2023 Apr 7. PMID: 37031776.
- Goeppert B, Roessler S, Renner M, Loeffler M, Singer S, Rausch M, Albrecht T, Mehrabi A, Vogel MN, Pathil A, Czink E, Köhler B, Springfeld C, Rupp C, Weiss KH, Schirmacher P, von Knebel Doeberitz M, Kloor M. Low frequency of mismatch repair deficiency in gallbladder cancer. Diagn Pathol. 2019 May 8;14(1):36. doi: 10.1186/s13000-019-0813-5. PMID: 31068195; PMCID: PMC6506936.
- Grant RC, Denroche R, Jang GH, Nowak KM, Zhang A, Borgida A, Holter S, Topham JT, Wilson J, Dodd A, Jang R, Prince R, Karasinska JM, Schaeffer DF, Wang Y, Zogopoulos G, Berry S, Simeone D, Renouf DJ, Notta F, O'Kane G, Knox J, Fischer S, Gallinger S. Clinical and genomic characterisation of mismatch repair deficient pancreatic adenocarcinoma. Gut. 2021 Oct;70(10):1894-1903. doi: 10.1136/gutjnl-2020-320730. Epub 2020 Sep 15. PMID: 32933947.
- Hadad S, Gupta R, Oberheim Bush NA, Taylor JW, Villanueva-Meyer JE, Young JS, Wu J, Ravindranathan A, Zhang Y, Warrier G, McCoy L, Shai A, Pekmezci M, Perry A, Bollen AW, Phillips JJ, Braunstein SE, Raleigh DR, Theodosopoulos P, Aghi MK, Chang EF, Hervey-Jumper SL, Costello JF, de Groot J, Butowski NA, Clarke JL, Chang SM, Berger MS, Molinaro AM, Solomon DA. "De novo replication repair deficient glioblastoma, IDH-wildtype" is a distinct glioblastoma subtype in adults that may benefit from immune checkpoint blockade. Acta Neuropathol. 2023 Dec 11;147(1):3. doi: 10.1007/s00401-023-02654-1. PMID: 38079020; PMCID: PMC10713691.
- Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, Lowery MA, Diaz LA Jr, Mandelker D, Yu KH, Zervoudakis A, Kelsen DP, Iacobuzio-Donahue CA, Klimstra DS, Saltz LB, Sahin IH, O'Reilly EM. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clin Cancer Res. 2018 Mar 15;24(6):1326-1336. doi: 10.1158/1078-0432.CCR-17-3099. Epub 2018 Jan 24. PMID: 29367431; PMCID: PMC5856632.
- Ju JY, Dibbern ME, Mahadevan MS, Fan J, Kunk PR, Stelow EB. Mismatch Repair Protein Deficiency/Microsatellite Instability Is Rare in Cholangiocarcinomas and Associated With Distinctive Morphologies. Am J Clin Pathol. 2020 Apr 15;153(5):598-604. doi: 10.1093/ajcp/aqz199. PMID: 31844887.
- Kagawa M, Kawakami S, Yamamoto A, Suzuki O, Kamae N, Eguchi H, Okazaki Y, Yamamoto G, Akagi K, Tamaru JI, Yamaguchi T, Arai T, Ishida H. Identification of Lynch syndrome-associated DNA mismatch repair-deficient bladder cancer in a Japanese hospital-based population. Int J Clin Oncol. 2021 Aug;26(8):1524-1532. doi: 10.1007/s10147-021-01922-y. Epub 2021 Jul 2. PMID: 34213665.
- Kagawa M, Kawakami S, Yamamoto A, Suzuki O, Eguchi H, Okazaki Y, Akagi K, Tamaru JI, Arai T, Yamaguchi T, Ishida H. Prevalence and clinicopathological/molecular characteristics of mismatch repair protein-deficient tumours among surgically treated patients with prostate cancer in a Japanese hospital-based population. Jpn J Clin Oncol. 2021 Apr 1;51(4):639-645. doi: 10.1093/jjco/hyaa207. Erratum in: Jpn J Clin Oncol. 2022 Apr 6;52(4):402. doi: 10.1093/jjco/hyac021. PMID: 33244609.
- Kattapuram M, Shabet C, Austin S, Jacobs MF, Koeppe E, Smith EH, Lowe L, Else T, Cha KB. A retrospective cohort study of genetic referral and diagnosis of Lynch syndrome in patients with cutaneous sebaceous lesions. Fam Cancer. 2023 Jul;22(3):295-301. doi: 10.1007/s10689-022-00322-z. Epub 2022 Nov 28. PMID: 36437392.
- Kryklyva V, Brosens LA, Marijnissen-van Zanten MA, Ligtenberg MJ, Nagtegaal ID. Mismatch repair deficiency in early-onset duodenal, ampullary, and pancreatic carcinomas is a strong indicator for a hereditary defect. J Pathol Clin Res. 2022 Mar;8(2):181-190. doi: 10.1002/cjp2.252. Epub 2021 Dec 6. PMID: 34873870; PMCID: PMC8822371.
- Kubo S, Nagano H, Tsujie M, Seo S, Gotoh K, Wada H, Nakashima S, Ioka T. Microsatellite instability in patients with hepato-biliary-pancreatic malignancies in clinical practice (KHBO 1903). Int J Clin Oncol. 2022 Aug;27(8):1340-1347. doi: 10.1007/s10147-022-02187-9. Epub 2022 Jun 19. PMID: 35718824.
- Kullmann F, Strissel PL, Strick R, Stoehr R, Eckstein M, Bertz S, Wullich B, Sikic D, Wach S, Taubert H, Olbert P, Heers H, Lara MF, Macias ML, Matas-Rico E, Lozano MJ, Prieto D, Hierro I, van Doeveren T, Bieche I, Masliah-Planchon J, Beaurepere R, Boormans JL, Allory Y, Herrera-Imbroda B, Hartmann A, Weyerer V. Frequency of microsatellite instability (MSI) in upper tract urothelial carcinoma: comparison of the Bethesda panel and the Idylla MSI assay in a consecutively collected, multi-institutional cohort. J Clin Pathol. 2023 Feb;76(2):126-132. doi: 10.1136/jclinpath-2021-207855. Epub 2021 Sep 28. PMID: 34583948; PMCID: PMC9887356.
- Kunnackal John G, Das Villgran V, Caufield-Noll C, Giardiello FM. Comparison of universal screening in major Lynch-associated tumors: a systematic review of literature. Fam Cancer. 2022 Jan;21(1):57-67. doi: 10.1007/s10689-020-00226-w. Epub 2021 Jan 11. PMID: 33426601.
- Latham A, Shia J, Patel Z, Reidy-Lagunes DL, Segal NH, Yaeger R, Ganesh K, Connell L, Kemeny NE, Kelsen DP, Hechtman JF, Nash GM, Paty PB, Zehir A, Tkachuk KA, Sheikh R, Markowitz AJ, Mandelker D, Offit K, Berger MF, Cercek A, Garcia-Aguilar J, Saltz LB, Weiser MR, Stadler ZK. Characterization and Clinical Outcomes of DNA Mismatch Repair-deficient Small Bowel Adenocarcinoma. Clin Cancer Res. 2021 Mar 1;27(5):1429-1437. doi: 10.1158/1078-0432.CCR-20-2892. Epub 2020 Nov 16. PMID: 33199489; PMCID: PMC7925361.
- Ma YT, Hua F, Zhong XM, Xue YJ, Li J, Nie YC, Zhang XD, Ma JW, Lin CH, Zhang HZ, He W, Sha D, Zhao MQ, Yao ZG. Clinicopathological characteristics, molecular landscape, and biomarker landscape for predicting the efficacy of PD-1/PD-L1 inhibitors in Chinese population with mismatch repair deficient urothelial carcinoma: a real-world study. Front Immunol. 2023 Nov 6;14:1269097. doi: 10.3389/fimmu.2023.1269097. PMID: 38022513; PMCID: PMC10657814.
- Mettman D, Haer E, Olyaee M, Rastogi A, Madan R, O'Neil M, Kelting S, Dennis K, Fan F. The utility of immunohistochemical testing for mismatch repair proteins in fine needle aspiration specimens of pancreatic adenocarcinoma. Ann Diagn Pathol. 2020 Aug;47:151552. doi: 10.1016/j.anndiagpath.2020.151552. Epub 2020 Jun 17. PMID: 32570025.
- Mitric C, Salman L, Abrahamyan L, Kim SR, Pechlivanoglou P, Chan KKW, Gien LT, Ferguson SE. Mismatch-repair deficiency, microsatellite instability, and Lynch syndrome in ovarian cancer: A systematic review and meta-analysis. Gynecol Oncol. 2023 Mar;170:133-142. doi: 10.1016/j.ygyno.2022.12.008. Epub 2023 Jan 20. PMID: 36682091.
- Moy AP, Shahid M, Ferrone CR, Borger DR, Zhu AX, Ting D, Deshpande V. Microsatellite instability in gallbladder carcinoma. Virchows Arch. 2015 Apr;466(4):393-402. doi: 10.1007/s00428-015-1720-0. Epub 2015 Feb 14. PMID: 25680569.
- Oka S, Urakami S, Hagiwara K, Hayashida M, Sakaguchi K, Miura Y, Inoshita N, Arai M. The prevalence of Lynch syndrome (DNA mismatch repair protein deficiency) in patients with primary localized prostate cancer using immunohistochemistry screening. Hered Cancer Clin Pract. 2023 Oct 12;21(1):20. doi: 10.1186/s13053-023-00265-1. PMID: 37828628; PMCID: PMC10568829.
- Pivovarcikova K, Pitra T, Alaghehbandan R, Buchova K, Steiner P, Hajkova V, Ptakova N, Subrt I, Skopal J, Svajdler P, Farcas M, Slisarenko M, Michalova K, Strakova Peterikova A, Hora M, Michal M, Daum O, Svajdler M, Hes O. Lynch syndrome-associated upper tract urothelial carcinoma frequently occurs in patients older than 60 years: an opportunity to revisit urology clinical guidelines. Virchows Arch. 2023 Oct;483(4):517-526. doi: 10.1007/s00428-023-03626-2. Epub 2023 Aug 23. PMID: 37612527.
- Pritzlaff M, Tian Y, Reineke P, Stuenkel AJ, Allen K, Gutierrez S, Jackson M, Dolinsky JS, LaDuca H, Xu J, Black MH, Helfand BT. Diagnosing hereditary cancer predisposition in men with prostate cancer. Genet Med. 2020 Sep;22(9):1517-1523. doi: 10.1038/s41436-020-0830-5. Epub 2020 May 22. PMID: 32439974; PMCID: PMC7462744.
- Rasmussen M, Madsen MG, Therkildsen C. Immunohistochemical Screening of Upper Tract Urothelial Carcinomas for Lynch Syndrome Diagnostics: A Systematic Review. Urology. 2022 Jul;165:44-53. doi: 10.1016/j.urology.2022.02.006. Epub 2022 Feb 22. PMID: 35217028.
- Rodríguez-Hernández I, Garcia JL, Santos-Briz A, Hernández-Laín A, González-Valero JM, Gómez-Moreta JA, Toldos-González O, Cruz JJ, Martin-Vallejo J, González-Sarmiento R. Integrated analysis of mismatch repair system in malignant astrocytomas. PLoS One. 2013 Sep 20;8(9):e76401. doi: 10.1371/journal.pone.0076401. PMID: 24073290; PMCID: PMC3779191.
- Sánchez A, Bujanda L, Cuatrecasas M, Bofill A, Alvarez-Urturi C, Hernandez G, Aguilera L, Carballal S, Llach J, Herrera-Pariente C, Iglesias M, Rivero-Sánchez L, Jung G, Moreno L, Ocaña T, Bayarri C, Pellise M, Castells A, Castellví-Bel S, Balaguer F, Moreira L. Identification of Lynch Syndrome Carriers among Patients with Small Bowel Adenocarcinoma. Cancers (Basel). 2021 Dec 20;13(24):6378. doi: 10.3390/cancers13246378. PMID: 34944998; PMCID: PMC8699558.
- Sinson H, Karayan-Tapon L, Godet J, Rivet P, Alleyrat C, Battistella M, Pierron H, Morel F, Lecron JC, Favot L, Frouin E. Immunohistochemistry, Molecular Biology, and Clinical Scoring for the Detection of Muir-Torre Syndrome in Cutaneous Sebaceous Tumors: Which Strategy? Dermatology. 2023;239(6):889-897. doi: 10.1159/000534126. Epub 2023 Sep 15. PMID: 37717564.
- Suerink M, Kilinç G, Terlouw D, Hristova H, Sensuk L, van Egmond D, Farina Sarasqueta A, Langers AMJ, van Wezel T, Morreau H, Nielsen M; PALGA-group collaborators. Prevalence of mismatch repair deficiency and Lynch syndrome in a cohort of unselected small bowel adenocarcinomas. J Clin Pathol. 2021 Nov;74(11):724-729. doi: 10.1136/jclinpath-2020-207040. Epub 2020 Oct 12. PMID: 33046565; PMCID: PMC8543220.
- Truong H, Breen K, Nandakumar S, Sjoberg DD, Kemel Y, Mehta N, Lenis AT, Reisz PA, Carruthers J, Benfante N, Joseph V, Khurram A, Gopalan A, Fine SW, Reuter VE, Vickers AJ, Birsoy O, Liu Y, Walsh M, Latham A, Mandelker D, Stadler ZK, Pietzak E, Ehdaie B, Touijer KA, Laudone VP, Slovin SF, Autio KA, Danila DC, Rathkopf DE, Eastham JA, Chen Y, Morris MJ, Offit K, Solit DB, Scher HI, Abida W, Robson ME, Carlo MI. Gene-based Confirmatory Germline Testing Following Tumor-only Sequencing of Prostate Cancer. Eur Urol. 2023 Jan;83(1):29-38. doi: 10.1016/j.eururo.2022.08.028. Epub 2022 Sep 15. PMID: 36115772; PMCID: PMC10208030.
- In overwegingen
- Advani SM, Swartz MD, Loree J, Davis JS, Sarsashek AM, Lam M, Lee MS, Bressler J, Lopez DS, Daniel CR, Morris V, Shureqi I, Kee B, Dasari A, Vilar E, Overman M, Hamilton S, Maru D, Braithwaite D, Kopetz S. Epidemiology and Molecular-Pathologic Characteristics of CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer. Clin Colorectal Cancer. 2021 Jun;20(2):137-147.e1. doi: 10.1016/j.clcc.2020.09.007. Epub 2020 Oct 12. PMID: 33229221.
- Andersson E, Keränen A, Lagerstedt-Robinson K, Ghazi S, Lindblom A, Tham E, Mints M. Universal testing in endometrial cancer in Sweden. Hered Cancer Clin Pract. 2024 Aug 22;22(1):14. doi: 10.1186/s13053-024-00288-2. PMID: 39175077; PMCID: PMC11342736.
- Bou Farhat E, Adib E, Daou M, Naqash AR, Matulonis U, Ng K, Kwiatkowski DJ, Sholl LM, Nassar AH. Benchmarking mismatch repair testing for patients with cancer receiving immunotherapy. Cancer Cell. 2024 Jan 8;42(1):6-7. doi: 10.1016/j.ccell.2023.12.001. Epub 2023 Dec 28. Erratum in: Cancer Cell. 2024 Feb 12;42(2):323. doi: 10.1016/j.ccell.2024.01.009. PMID: 38157866.
- Bucksch K, Zachariae S, Ahadova A, Aretz S, Büttner R, Görgens H, Holinski-Feder E, Hüneburg R, Kloor M, von Knebel Doeberitz M, Ladigan-Badura S, Moeslein G, Morak M, Nattermann J, Nguyen HP, Perne C, Redler S, Schmetz A, Steinke-Lange V, Surowy H, Vangala DB, Weitz J, Loeffler M, Engel C; German Consortium for Familial Intestinal Cancer. Adenoma and colorectal cancer risks in Lynch syndrome, Lynch-like syndrome and familial colorectal cancer type X. Int J Cancer. 2022 Jan 1;150(1):56-66. doi: 10.1002/ijc.33790. Epub 2021 Sep 14. PMID: 34469588.
- Castillejo A, Hernández-Illán E, Rodriguez-Soler M, Pérez-Carbonell L, Egoavil C, Barberá VM, Castillejo MI, Guarinos C, Martínez-de-Dueñas E, Juan MJ, Sánchez-Heras AB, García-Casado Z, Ruiz-Ponte C, Brea-Fernández A, Juárez M, Bujanda L, Clofent J, Llor X, Andreu M, Castells A, Carracedo A, Alenda C, Payá A, Jover R, Soto JL. Prevalence of MLH1 constitutional epimutations as a cause of Lynch syndrome in unselected versus selected consecutive series of patients with colorectal cancer. J Med Genet. 2015 Jul;52(7):498-502. doi: 10.1136/jmedgenet-2015-103076. Epub 2015 Apr 23. PMID: 25908759.
- Christenson ES, Tsai HL, Le DT, Jaffee EM, Dudley J, Xian RR, Gocke CD, Eshleman JR, Lin MT. Colorectal cancer in patients of advanced age is associated with increased incidence of BRAF p.V600E mutation and mismatch repair deficiency. Front Oncol. 2023 Jun 7;13:1193259. doi: 10.3389/fonc.2023.1193259. PMID: 37350948; PMCID: PMC10284017.
- Crucianelli F, Tricarico R, Turchetti D, Gorelli G, Gensini F, Sestini R, Giunti L, Pedroni M, Ponz de Leon M, Civitelli S, Genuardi M. MLH1 constitutional and somatic methylation in patients with MLH1 negative tumors fulfilling the revised Bethesda criteria. Epigenetics. 2014 Oct;9(10):1431-8. doi: 10.4161/15592294.2014.970080. PMID: 25437057; PMCID: PMC4622593.
- Dabir PD, Bruggeling CE, van der Post RS, Dutilh BE, Hoogerbrugge N, Ligtenberg MJL, Boleij A, Nagtegaal ID. Microsatellite instability screening in colorectal adenomas to detect Lynch syndrome patients? A systematic review and meta-analysis. Eur J Hum Genet. 2020 Mar;28(3):277-286. doi: 10.1038/s41431-019-0538-7. Epub 2019 Nov 6. PMID: 31695176; PMCID: PMC7028913.
- Deng G, Bell I, Crawley S, Gum J, Terdiman JP, Allen BA, Truta B, Sleisenger MH, Kim YS. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004 Jan 1;10(1 Pt 1):191-5. doi: 10.1158/1078-0432.ccr-1118-3. PMID: 14734469.
- Eikenboom EL, van der Werf-'t Lam AS, Rodríguez-Girondo M, Van Asperen CJ, Dinjens WNM, Hofstra RMW, Van Leerdam ME, Morreau H, Spaander MCW, Wagner A, Nielsen M. Universal Immunohistochemistry for Lynch Syndrome: A Systematic Review and Meta-analysis of 58,580 Colorectal Carcinomas. Clin Gastroenterol Hepatol. 2022 Mar;20(3):e496-e507. doi: 10.1016/j.cgh.2021.04.021. Epub 2021 Apr 19. PMID: 33887476.
- Elze L, Mensenkamp AR, Nagtegaal ID, van Zelst-Stams WAG; Dutch LS-Like Study Group; de Voer RM, Ligtenberg MJL. Somatic Nonepigenetic Mismatch Repair Gene Aberrations Underly Most Mismatch Repair-Deficient Lynch-Like Tumors. Gastroenterology. 2021 Mar;160(4):1414-1416.e3. doi: 10.1053/j.gastro.2020.11.042. Epub 2020 Nov 28. PMID: 33253688.
- Ferreira I, Wiedemeyer K, Demetter P, Adams DJ, Arends MJ, Brenn T. Update on the pathology, genetics and somatic landscape of sebaceous tumours. Histopathology. 2020 Apr;76(5):640-649. doi: 10.1111/his.14044. Epub 2020 Mar 17. PMID: 31821583.
- Gaskin BJ, Fernando BS, Sullivan CA, Whitehead K, Sullivan TJ. The significance of DNA mismatch repair genes in the diagnosis and management of periocular sebaceous cell carcinoma and Muir-Torre syndrome. Br J Ophthalmol. 2011 Dec;95(12):1686-90. doi: 10.1136/bjophthalmol-2011-300612. Epub 2011 Oct 6. PMID: 21979897.
- Gordhandas S, Kahn RM, Gamble C, Talukdar N, Maddy B, Baltich Nelson B, Askin G, Christos PJ, Holcomb K, Caputo TA, Chapman-Davis E, Frey MK. Clinicopathologic features of endometrial cancer with mismatch repair deficiency. Ecancermedicalscience. 2020 Jun 18;14:1061. doi: 10.3332/ecancer.2020.1061. PMID: 32582376; PMCID: PMC7302890.
- Goel A, Nguyen TP, Leung HC, Nagasaka T, Rhees J, Hotchkiss E, Arnold M, Banerji P, Koi M, Kwok CT, Packham D, Lipton L, Boland CR, Ward RL, Hitchins MP. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. 2011 Feb 15;128(4):869-78. doi: 10.1002/ijc.25422. PMID: 20473912; PMCID: PMC3794437.
- Goverde A, Spaander MC, van Doorn HC, Dubbink HJ, van den Ouweland AM, Tops CM, Kooi SG, de Waard J, Hoedemaeker RF, Bruno MJ, Hofstra RM, de Bekker-Grob EW, Dinjens WN, Steyerberg EW, Wagner A; LIMO study group. Cost-effectiveness of routine screening for Lynch syndrome in endometrial cancer patients up to 70years of age. Gynecol Oncol. 2016 Dec;143(3):453-459. doi: 10.1016/j.ygyno.2016.10.008. Epub 2016 Oct 24. PMID: 27789085.
- Hampel H, Pearlman R, de la Chapelle A, Pritchard CC, Zhao W, Jones D, Yilmaz A, Chen W, Frankel WL, Suarez AA, Cosgrove C, Backes F, Copeland L, Fowler J, O'Malley D, Salani R, McElroy JP, Stanich PP, Goodfellow P, Cohn DE. Double somatic mismatch repair gene pathogenic variants as common as Lynch syndrome among endometrial cancer patients. Gynecol Oncol. 2021 Jan;160(1):161-168. doi: 10.1016/j.ygyno.2020.10.012. Epub 2020 Oct 21. PMID: 33393477; PMCID: PMC7783191.
- Hechtman JF, Rana S, Middha S, Stadler ZK, Latham A, Benayed R, Soslow R, Ladanyi M, Yaeger R, Zehir A, Shia J. Retained mismatch repair protein expression occurs in approximately 6% of microsatellite instability-high cancers and is associated with missense mutations in mismatch repair genes. Mod Pathol. 2020 May;33(5):871-879. doi: 10.1038/s41379-019-0414-6. Epub 2019 Dec 19. PMID: 31857677; PMCID: PMC7195218.
- Helderman NC, Andini KD, van Leerdam ME, van Hest LP, Hoekman DR, Ahadova A, Bajwa-Ten Broeke SW, Bosse T, van der Logt EMJ, Imhann F, Kloor M, Langers AMJ, Smit VTHBM, Terlouw D, van Wezel T, Morreau H, Nielsen M. MLH1 Promoter Hypermethylation in Colorectal and Endometrial Carcinomas from Patients with Lynch Syndrome. J Mol Diagn. 2024 Feb;26(2):106-114. doi: 10.1016/j.jmoldx.2023.10.005. Epub 2023 Dec 5. PMID: 38061582.
- Hitchins MP, Dámaso E, Alvarez R, Zhou L, Hu Y, Diniz MA, Pineda M, Capella G, Pearlman R, Hampel H. Constitutional MLH1 Methylation Is a Major Contributor to Mismatch Repair-Deficient, MLH1-Methylated Colorectal Cancer in Patients Aged 55 Years and Younger. J Natl Compr Canc Netw. 2023 Jul;21(7):743-752.e11. doi: 10.6004/jnccn.2023.7020. PMID: 37433431; PMCID: PMC11578100.
- Hitchins MP, Alvarez R, Zhou L, Aguirre F, Dámaso E, Pineda M, Capella G, Wong JJ, Yuan X, Ryan SR, Sathe DS, Baxter MD, Cannon T, Biswas R, DeMarco T, Grzelak D, Hampel H, Pearlman R. MLH1-methylated endometrial cancer under 60 years of age as the "sentinel" cancer in female carriers of high-risk constitutional MLH1 epimutation. Gynecol Oncol. 2023 Apr;171:129-140. doi: 10.1016/j.ygyno.2023.02.017. Epub 2023 Mar 8. PMID: 36893489; PMCID: PMC10153467.
- Joo JE, Mahmood K, Walker R, Georgeson P, Candiloro I, Clendenning M, Como J, Joseland S, Preston S, Graversen L, Wilding M, Field M, Lemon M, Wakeling J, Marfan H, Susman R, Isbister J, Edwards E, Bowman M, Kirk J, Ip E, McKay L, Antill Y, Hopper JL, Boussioutas A, Macrae FA, Dobrovic A, Jenkins MA, Rosty C, Winship IM, Buchanan DD. Identifying primary and secondary MLH1 epimutation carriers displaying low-level constitutional MLH1 methylation using droplet digital PCR and genome-wide DNA methylation profiling of colorectal cancers. Clin Epigenetics. 2023 Jun 3;15(1):95. doi: 10.1186/s13148-023-01511-y. PMID: 37270516; PMCID: PMC10239107.
- Jumaah AS, Al-Haddad HS, Salem MM, McAllister KA, Yasseen AA. Mismatch repair deficiency and clinicopathological characteristics in endometrial carcinoma: a systematic review and meta-analysis. J Pathol Transl Med. 2021 May;55(3):202-211. doi: 10.4132/jptm.2021.02.19. Epub 2021 Apr 14. PMID: 33845554; PMCID: PMC8141969.
- Kahn RM, Gordhandas S, Maddy BP, Baltich Nelson B, Askin G, Christos PJ, Caputo TA, Chapman-Davis E, Holcomb K, Frey MK. Universal endometrial cancer tumor typing: How much has immunohistochemistry, microsatellite instability, and MLH1 methylation improved the diagnosis of Lynch syndrome across the population? Cancer. 2019 Sep 15;125(18):3172-3183. doi: 10.1002/cncr.32203. Epub 2019 May 31. PMID: 31150123.
- Kaya M, Post CCB, Tops CM, Nielsen M, Crosbie EJ, Leary A, Mileshkin LR, Han K, Bessette P, de Boer SM, Jürgenliemk-Schulz IM, Lutgens L, Jobsen JJ, Haverkort MAD, Nout RA, Kroep J, Creutzberg CL, Smit VTHBM, Horeweg N, van Wezel T, Bosse T. Molecular and Clinicopathologic Characterization of Mismatch Repair-Deficient Endometrial Carcinoma Not Related to MLH1 Promoter Hypermethylation. Mod Pathol. 2024 Mar;37(3):100423. doi: 10.1016/j.modpat.2024.100423. Epub 2024 Jan 6. PMID: 38191122.
- Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, Middha S, Hechtman J, Zehir A, Dubard-Gault M, Tran C, Stewart C, Sheehan M, Penson A, DeLair D, Yaeger R, Vijai J, Mukherjee S, Galle J, Dickson MA, Janjigian Y, O'Reilly EM, Segal N, Saltz LB, Reidy-Lagunes D, Varghese AM, Bajorin D, Carlo MI, Cadoo K, Walsh MF, Weiser M, Aguilar JG, Klimstra DS, Diaz LA Jr, Baselga J, Zhang L, Ladanyi M, Hyman DM, Solit DB, Robson ME, Taylor BS, Offit K, Berger MF, Stadler ZK. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol. 2019 Feb 1;37(4):286-295. doi: 10.1200/JCO.18.00283. Epub 2018 Oct 30. Erratum in: J Clin Oncol. 2019 Apr 10;37(11):942. doi: 10.1200/JCO.19.00517. PMID: 30376427; PMCID: PMC6553803.
- Leenen CH, Goverde A, de Bekker-Grob EW, Wagner A, van Lier MG, Spaander MC, Bruno MJ, Tops CM, van den Ouweland AM, Dubbink HJ, Kuipers EJ, Dinjens WN, van Leerdam ME, Steyerberg EW. Cost-effectiveness of routine screening for Lynch syndrome in colorectal cancer patients up to 70 years of age. Genet Med. 2016 Oct;18(10):966-73. doi: 10.1038/gim.2015.206. Epub 2016 Mar 3. PMID: 26938782.
- Loong L, Huntley C, Pethick J, McRonald F, Santaniello F, Shand B, Tulloch O, Goel S, Lüchtenborg M, Allen S, Torr B, Snape K, George A, Lalloo F, Norbury G, Eccles DM, Tischkowitz M, Antoniou AC, Pharoah P, Shaw A, Morris E, Burn J, Monahan K, Hardy S, Turnbull C. Lynch syndrome diagnostic testing pathways in endometrial cancers: a nationwide English registry-based study. J Med Genet. 2024 Nov 25;61(12):1080-1088. doi: 10.1136/jmg-2024-110231. PMID: 39433398; PMCID: PMC11671912.
- McRonald FE, Pethick J, Santaniello F, Shand B, Tyson A, Tulloch O, Goel S, Lüchtenborg M, Borthwick GM, Turnbull C, Shaw AC, Monahan KJ, Frayling IM, Hardy S, Burn J. Identification of people with Lynch syndrome from those presenting with colorectal cancer in England: baseline analysis of the diagnostic pathway. Eur J Hum Genet. 2024 May;32(5):529-538. doi: 10.1038/s41431-024-01550-w. Epub 2024 Feb 15. PMID: 38355963; PMCID: PMC11061113.
- Niessen RC, Hofstra RM, Westers H, Ligtenberg MJ, Kooi K, Jager PO, de Groote ML, Dijkhuizen T, Olderode-Berends MJ, Hollema H, Kleibeuker JH, Sijmons RH. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009 Aug;48(8):737-44. doi: 10.1002/gcc.20678. PMID: 19455606.
- Nugroho PP, Ghozali SAS, Buchanan DD, Pisano MI, Reece JC. Risk of cancer in individuals with Lynch-like syndrome and their families: a systematic review. J Cancer Res Clin Oncol. 2023 Jan;149(1):25-46. doi: 10.1007/s00432-022-04397-0. Epub 2022 Oct 17. PMID: 36251064; PMCID: PMC9889410.
- Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012 Mar;49(3):151-7. doi: 10.1136/jmedgenet-2011-100714. PMID: 22368298.
- Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, Goldberg RM, Paskett E, Shields PG, Freudenheim JL, Stanich PP, Lattimer I, Arnold M, Liyanarachchi S, Kalady M, Heald B, Greenwood C, Paquette I, Prues M, Draper DJ, Lindeman C, Kuebler JP, Reynolds K, Brell JM, Shaper AA, Mahesh S, Buie N, Weeman K, Shine K, Haut M, Edwards J, Bastola S, Wickham K, Khanduja KS, Zacks R, Pritchard CC, Shirts BH, Jacobson A, Allen B, de la Chapelle A, Hampel H; Ohio Colorectal Cancer Prevention Initiative Study Group. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017 Apr 1;3(4):464-471. doi: 10.1001/jamaoncol.2016.5194. PMID: 27978560; PMCID: PMC5564179.
- Pearlman R, Frankel WL, Swanson BJ, Jones D, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, Goldberg RM, Paskett E, Shields PG, Freudenheim JL, Stanich PP, Lattimer I, Arnold M, Prior TW, Haut M, Kalady MF, Heald B, Paquette I, Draper DJ, Brell JM, Mahesh S, Weeman K, Bastola S, Zangmeister J, Gowda A, Kencana F, Malcolm A, Liu Y, Cole S, Bane C, Li C, Rehmus E, Pritchard CC, Shirts BH, Jacobson A, Cummings SA, de la Chapelle A, Hampel H. Prospective Statewide Study of Universal Screening for Hereditary Colorectal Cancer: The Ohio Colorectal Cancer Prevention Initiative. JCO Precis Oncol. 2021 May 5;5:PO.20.00525. doi: 10.1200/PO.20.00525. PMID: 34250417; PMCID: PMC8232834.
- Pineda M, Mur P, Iniesta MD, Borràs E, Campos O, Vargas G, Iglesias S, Fernández A, Gruber SB, Lázaro C, Brunet J, Navarro M, Blanco I, Capellá G. MLH1 methylation screening is effective in identifying epimutation carriers. Eur J Hum Genet. 2012 Dec;20(12):1256-64. doi: 10.1038/ejhg.2012.136. Epub 2012 Jul 4. PMID: 22763379; PMCID: PMC3499751.
- Post CCB, Stelloo E, Smit VTHBM, Ruano D, Tops CM, Vermij L, Rutten TA, Jürgenliemk-Schulz IM, Lutgens LCHW, Jobsen JJ, Nout RA, Crosbie EJ, Powell ME, Mileshkin L, Leary A, Bessette P, Putter H, de Boer SM, Horeweg N, Nielsen M, Wezel TV, Bosse T, Creutzberg CL. Prevalence and Prognosis of Lynch Syndrome and Sporadic Mismatch Repair Deficiency in Endometrial Cancer. J Natl Cancer Inst. 2021 Sep 4;113(9):1212-1220. doi: 10.1093/jnci/djab029. PMID: 33693762; PMCID: PMC8418420.
- Ryan NAJ, Glaire MA, Blake D, Cabrera-Dandy M, Evans DG, Crosbie EJ. The proportion of endometrial cancers associated with Lynch syndrome: a systematic review of the literature and meta-analysis. Genet Med. 2019 Oct;21(10):2167-2180. doi: 10.1038/s41436-019-0536-8. Epub 2019 May 14. PMID: 31086306; PMCID: PMC8076013.
- Seppälä TT, Latchford A, Negoi I, Sampaio Soares A, Jimenez-Rodriguez R, Sánchez-Guillén L, Evans DG, Ryan N, Crosbie EJ, Dominguez-Valentin M, Burn J, Kloor M, Knebel Doeberitz MV, Duijnhoven FJBV, Quirke P, Sampson JR, Møller P, Möslein G; European Hereditary Tumour Group (EHTG) and European Society of Coloproctology (ESCP). European guidelines from the EHTG and ESCP for Lynch syndrome: an updated third edition of the Mallorca guidelines based on gene and gender. Br J Surg. 2021 May 27;108(5):484-498. doi: 10.1002/bjs.11902. PMID: 34043773; PMCID: PMC10364896.
- Singh RS, Grayson W, Redston M, Diwan AH, Warneke CL, McKee PH, Lev D, Lyle S, Calonje E, Lazar AJ. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol. 2008 Jun;32(6):936-42. doi: 10.1097/pas.0b013e31815b0cc2. PMID: 18551751.
- N. Singh, R. Wong, N. Tchrakian, S.G. Allen, B. Clarke, C.B. Gilks, Interpretation of mismatch repair protein expression using obsolete criteria results in discrepancies with microsatellite instability and mutational testing results. Comment on Hechtman et al. Mod Pathol 2020; 33:871-879, Mod Pathol, 34 (2021), pp. 1031-1032
- Sinson H, Karayan-Tapon L, Godet J, Rivet P, Alleyrat C, Battistella M, Pierron H, Morel F, Lecron JC, Favot L, Frouin E. Immunohistochemistry, Molecular Biology, and Clinical Scoring for the Detection of Muir-Torre Syndrome in Cutaneous Sebaceous Tumors: Which Strategy? Dermatology. 2023;239(6):889-897. doi: 10.1159/000534126. Epub 2023 Sep 15. PMID: 37717564.
- Snowsill TM, Ryan NAJ, Crosbie EJ. Cost-Effectiveness of the Manchester Approach to Identifying Lynch Syndrome in Women with Endometrial Cancer. J Clin Med. 2020 Jun 1;9(6):1664. doi: 10.3390/jcm9061664. PMID: 32492863; PMCID: PMC7356917.
- Stelloo E, Jansen AML, Osse EM, Nout RA, Creutzberg CL, Ruano D, Church DN, Morreau H, Smit VTHBM, van Wezel T, Bosse T. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann Oncol. 2017 Jan 1;28(1):96-102. doi: 10.1093/annonc/mdw542. PMID: 27742654.
- Stinton C, Jordan M, Fraser H, Auguste P, Court R, Al-Khudairy L, Madan J, Grammatopoulos D, Taylor-Phillips S. Testing strategies for Lynch syndrome in people with endometrial cancer: systematic reviews and economic evaluation. Health Technol Assess. 2021 Jun;25(42):1-216. doi: 10.3310/hta25420. PMID: 34169821; PMCID: PMC8273681.
- Te Paske IBAW, Mensenkamp AR, Neveling K; ERN-GENTURIS Lynch-Like Working Group; Hoogerbrugge N, Ligtenberg MJL, De Voer RM. Noncoding Aberrations in Mismatch Repair Genes Underlie a Substantial Part of the Missing Heritability in Lynch Syndrome. Gastroenterology. 2022 Dec;163(6):1691-1694.e7. doi: 10.1053/j.gastro.2022.08.041. Epub 2022 Aug 28. PMID: 36037994.
- van Lier MG, Wagner A, van Leerdam ME, Biermann K, Kuipers EJ, Steyerberg EW, Dubbink HJ, Dinjens WN. A review on the molecular diagnostics of Lynch syndrome: a central role for the pathology laboratory. J Cell Mol Med. 2010 Jan;14(1-2):181-97. doi: 10.1111/j.1582-4934.2009.00977.x. Epub 2009 Nov 19. PMID: 19929944; PMCID: PMC3837620.
- van Roon EH, van Puijenbroek M, Middeldorp A, van Eijk R, de Meijer EJ, Erasmus D, Wouters KA, van Engeland M, Oosting J, Hes FJ, Tops CM, van Wezel T, Boer JM, Morreau H. Early onset MSI-H colon cancer with MLH1 promoter methylation, is there a genetic predisposition? BMC Cancer. 2010 May 5;10:180. doi: 10.1186/1471-2407-10-180. PMID: 20444249; PMCID: PMC2880297.
- Vink-Börger E, den Bakker M, Voorham R, van Nederveen F, Nagtegaal I. Mismatch repair deficiency: how reliable is the two-antibody approach? A national real-life study. Histopathology. 2024 Oct;85(4):639-648. doi: 10.1111/his.15236. Epub 2024 Jun 11. PMID: 38859771.
- Vos JR, Fakkert IE, Spruijt L, Willems RW, Langenveld S, Mensenkamp AR, Leter EM, Nagtegaal ID, Ligtenberg MJL, Hoogerbrugge N; FINAL Group. Evaluation of yield and experiences of age-related molecular investigation for heritable and nonheritable causes of mismatch repair deficient colorectal cancer to identify Lynch syndrome. Int J Cancer. 2020 Oct 15;147(8):2150-2158. doi: 10.1002/ijc.33117. Epub 2020 Jun 8. PMID: 32510614; PMCID: PMC7496272.
- Walker R, Mahmood K, Joo JE, Clendenning M, Georgeson P, Como J, Joseland S, Preston SG, Antill Y, Austin R, Boussioutas A, Bowman M, Burke J, Campbell A, Daneshvar S, Edwards E, Gleeson M, Goodwin A, Harris MT, Henderson A, Higgins M, Hopper JL, Hutchinson RA, Ip E, Isbister J, Kasem K, Marfan H, Milnes D, Ng A, Nichols C, O'Connell S, Pachter N, Pope BJ, Poplawski N, Ragunathan A, Smyth C, Spigelman A, Storey K, Susman R, Taylor JA, Warwick L, Wilding M, Williams R, Win AK, Walsh MD, Macrae FA, Jenkins MA, Rosty C, Winship IM, Buchanan DD; Family Cancer Clinics of Australia. A tumor focused approach to resolving the etiology of DNA mismatch repair deficient tumors classified as suspected Lynch syndrome. J Transl Med. 2023 Apr 26;21(1):282. doi: 10.1186/s12967-023-04143-1. PMID: 37101184; PMCID: PMC10134620.
- Ward RL, Dobbins T, Lindor NM, Rapkins RW, Hitchins MP. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet Med. 2013 Jan;15(1):25-35. doi: 10.1038/gim.2012.91. Epub 2012 Aug 9. PMID: 22878509; PMCID: PMC3908650.
- West NP, Gallop N, Kaye D, Glover A, Young C, Hutchins GGA, Brockmoeller SF, Westwood AC, Rossington H, Quirke P; Yorkshire Cancer Research Bowel Cancer Improvement Programme Group. Lynch syndrome screening in colorectal cancer: results of a prospective 2-year regional programme validating the NICE diagnostics guidance pathway throughout a 5.2-million population. Histopathology. 2021 Nov;79(5):690-699. doi: 10.1111/his.14390. Epub 2021 Jun 21. PMID: 33872400.
- Win AK, Jenkins MA, Dowty JG, Antoniou AC, Lee A, Giles GG, Buchanan DD, Clendenning M, Rosty C, Ahnen DJ, Thibodeau SN, Casey G, Gallinger S, Le Marchand L, Haile RW, Potter JD, Zheng Y, Lindor NM, Newcomb PA, Hopper JL, MacInnis RJ. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol Biomarkers Prev. 2017 Mar;26(3):404-412. doi: 10.1158/1055-9965.EPI-16-0693. Epub 2016 Oct 31. PMID: 27799157; PMCID: PMC5336409.
- Zyla R, Graham T, Aronson M, Velsher L, Mrkonjic M, Turashvili G. MLH1 epimutation is a rare mechanism for Lynch syndrome: A case report and review of the literature. Genes Chromosomes Cancer. 2021 Sep;60(9):635-639. doi: 10.1002/gcc.22957. Epub 2021 May 15. PMID: 33934415.
Risk of Bias tables
Not applicable
Table of excluded studies
|
Reference |
Reason for exclusion |
|
Abu-Ghazaleh N, Kaushik V, Gorelik A, Jenkins M, Macrae F. Worldwide prevalence of Lynch syndrome in patients with colorectal cancer: Systematic review and meta-analysis. Genet Med. 2022 May;24(5):971-985. doi: 10.1016/j.gim.2022.01.014. Epub 2022 Feb 15. PMID: 35177335. |
Wrong population: CRC patients |
|
Dabir PD, Bruggeling CE, van der Post RS, Dutilh BE, Hoogerbrugge N, Ligtenberg MJL, Boleij A, Nagtegaal ID. Microsatellite instability screening in colorectal adenomas to detect Lynch syndrome patients? A systematic review and meta-analysis. Eur J Hum Genet. 2020 Mar;28(3):277-286. doi: 10.1038/s41431-019-0538-7. Epub 2019 Nov 6. PMID: 31695176; PMCID: PMC7028913. |
Wrong population: colorectal adenomas |
|
Dal Buono A, Poliani L, Greco L, Bianchi P, Barile M, Giatti V, Bonifacio C, Carrara S, Malesci A, Laghi L. Prevalence of Germline Mutations in Cancer Predisposition Genes in Patients with Pancreatic Cancer or Suspected Related Hereditary Syndromes: Historical Prospective Analysis. Cancers (Basel). 2023 Mar 20;15(6):1852. doi: 10.3390/cancers15061852. PMID: 36980738; PMCID: PMC10047356. |
No population-based study (ascertainment bias) |
|
Eikenboom EL, Moen S, van Leeuwen L, Geurts-Giele WRR, Tops CMJ, van Ham TJ, Dinjens WNM, Dubbink HJ, Spaander MCW, Wagner A. Unexplained mismatch repair deficiency: Case closed. HGG Adv. 2022 Dec 14;4(1):100167. doi: 10.1016/j.xhgg.2022.100167. PMID: 36624813; PMCID: PMC9823207. |
Wrong population: CRC patients |
|
Eikenboom EL, van der Werf-'t Lam AS, Rodríguez-Girondo M, Van Asperen CJ, Dinjens WNM, Hofstra RMW, Van Leerdam ME, Morreau H, Spaander MCW, Wagner A, Nielsen M. Universal Immunohistochemistry for Lynch Syndrome: A Systematic Review and Meta-analysis of 58,580 Colorectal Carcinomas. Clin Gastroenterol Hepatol. 2022 Mar;20(3):e496-e507. doi: 10.1016/j.cgh.2021.04.021. Epub 2021 Apr 19. PMID: 33887476. |
Wrong population: CRC patients |
|
Gordhandas S, Kahn RM, Gamble C, Talukdar N, Maddy B, Baltich Nelson B, Askin G, Christos PJ, Holcomb K, Caputo TA, Chapman-Davis E, Frey MK. Clinicopathologic features of endometrial cancer with mismatch repair deficiency. Ecancermedicalscience. 2020 Jun 18;14:1061. doi: 10.3332/ecancer.2020.1061. PMID: 32582376; PMCID: PMC7302890. |
Wrong population: EC patients Wrong outcomes: association with age, cancer stage, histology and BMI |
|
Jumaah AS, Al-Haddad HS, Salem MM, McAllister KA, Yasseen AA. Mismatch repair deficiency and clinicopathological characteristics in endometrial carcinoma: a systematic review and meta-analysis. J Pathol Transl Med. 2021 May;55(3):202-211. doi: 10.4132/jptm.2021.02.19. Epub 2021 Apr 14. PMID: 33845554; PMCID: PMC8141969. |
Wrong population: EC patients |
|
Kahn RM, Gordhandas S, Maddy BP, Baltich Nelson B, Askin G, Christos PJ, Caputo TA, Chapman-Davis E, Holcomb K, Frey MK. Universal endometrial cancer tumor typing: How much has immunohistochemistry, microsatellite instability, and MLH1 methylation improved the diagnosis of Lynch syndrome across the population? Cancer. 2019 Sep 15;125(18):3172-3183. doi: 10.1002/cncr.32203. Epub 2019 May 31. PMID: 31150123. |
Wrong population: EC patients |
|
Kunnackal John G, Das Villgran V, Caufield-Noll C, Giardiello F. Worldwide variation in Lynch syndrome screening: case for universal screening in low colorectal cancer prevalence areas. Fam Cancer. 2021 Apr;20(2):145-156. doi: 10.1007/s10689-020-00206-0. Epub 2020 Sep 11. PMID: 32914371. |
Wrong population: CRC patients |
|
Mendelsohn RB, Herzog K, Shia J, Rahaman N, Stadler ZK, Shike M. Molecular Screening for Lynch Syndrome in Young Patients With Colorectal Adenomas. Clin Colorectal Cancer. 2017 Sep;16(3):173-177. doi: 10.1016/j.clcc.2017.01.002. Epub 2017 Feb 2. PMID: 28242162. |
Wrong population: colorectal adenoma patients |
|
Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014 Mar;23(3):437-49. doi: 10.1158/1055-9965.EPI-13-1165. Epub 2014 Jan 14. PMID: 24425144. |
Wrong population: risk of prostate cancer in LS patients |
|
Ryan NAJ, Glaire MA, Blake D, Cabrera-Dandy M, Evans DG, Crosbie EJ. The proportion of endometrial cancers associated with Lynch syndrome: a systematic review of the literature and meta-analysis. Genet Med. 2019 Oct;21(10):2167-2180. doi: 10.1038/s41436-019-0536-8. Epub 2019 May 14. PMID: 31086306; PMCID: PMC8076013. |
Wrong population: EC patients |
|
Stinton C, Jordan M, Fraser H, Auguste P, Court R, Al-Khudairy L, Madan J, Grammatopoulos D, Taylor-Phillips S. Testing strategies for Lynch syndrome in people with endometrial cancer: systematic reviews and economic evaluation. Health Technol Assess. 2021 Jun;25(42):1-216. doi: 10.3310/hta25420. PMID: 34169821; PMCID: PMC8273681. |
Wrong population: EC patients Wrong outcomes: sensitivity and specificity of different molecular tests |
Beoordelingsdatum en geldigheid
Publicatiedatum : 29-09-2025
Beoordeeld op geldigheid : 15-09-2025
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Belangrijkste wijzigingen t.o.v. vorige versie:
|
Onderwerp |
Wijzigingen meest recente versie |
|
Module 1: Diagnostiek en verwijzing |
|
|
Module 2: Familiair colorectaal carcinoom |
|
|
Module 3: Lynch syndroom |
|
|
Module 4: Adenomateuze polyposis |
|
|
Module 5: Serrated polyposis en overige vormen van polyposis |
|
|
Andere aanpassingen |
|
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2022 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de zorg voor patiënten met Erfelijke darmkanker.
Werkgroep
- dr. M. (Maartje) Nielsen (voorzitter), Klinisch geneticus, Leids Universitair Medisch Centrum, Leiden, VKGN
- prof. dr. N. (Nicoline) Hoogerbrugge, internist, Radboud UMC, Nijmegen, VKGN
- dr. A. (Anja) Wagner, Klinisch geneticus, Erasmus MC Kanker Instituut, Universitair Medisch Centrum Rotterdam, Rotterdam, VKGN
- dr. S.W. (Sanne) Bajwa – ten Broeke, Klinisch geneticus, Universitair Medisch Centrum Groningen, Groningen, VKGN
- prof. Dr. M.E. (Monique) van Leerdam, MDL-arts, Nederlands Kanker Instituut, Amsterdam, Leiden University Medical Center, NVMDL
- dr. T.M. (Tanya) Bisseling, MDL-arts, Radboud UMC, Nijmegen, NVMDL
- dr. M.C.A. (Mariëtte) van Kouwen, MDL-arts, Radboud UMC, Nijmegen, NVMDL
- prof. Dr. E. (Evelien) Dekker, MDL-arts, Amsterdam UMC, Amsterdam, NVMDL
- drs. H. (Hicham) Bouchiba, Arts-onderzoeker MDL, Amsterdam UMC, Amsterdam, persoonlijke titel
- dr. C.J. (Charlotte) Verberne, Chirurg, Ziekenhuis Amstelland, Amstelveen
- dr. J.M. (Jorien) Woolderink, Gynaecoloog, Martini Ziekenhuis, Groningen, NVOG
- dr. R.S. (Chella) van der Post, Patholoog, Radboud UMC, Nijmegen, NVVP
- dr. J.E. (Jurgen) Seppen, Patiëntvertegenwoordiger, Stichting Lynch Polyposis
- dr. A.R. (Arjen) Mensenkamp, laboratoriumspecialist Klinische genetica, Radboud UMC, Nijmegen, VKGL
- dr. C.M.J. (Carli) Tops, laboratoriumspecialist Klinische genetica, Leids Universitair Medisch Centrum, Leiden, VKGL
- I.J.H. (Ivonne) Schoenaker, Verpleegkundig specialist MDL, Isala Ziekenhuis, Zwolle, V&VN oncologie
- L.J.(Lisette) Saveur, Verpleegkundig specialist MDL, Nederlands Kanker Instituut, Amsterdam, V&VN oncologie
Klankbordgroep:
- Dr. F.J.B. (Fränzel) van Duijnhoven, Universitair hoofddocent voeding, genen en kanker WUR, Wageningen, persoonlijke titel
- Dr. J.A.J. (Job) Verdonschot, AIOS klinische Genetica, Maastricht Universitair Medisch Centrum, Maastricht, persoonlijke titel
- Dr. A.G. (Toine) van der Heijden, Oncologisch uroloog, Radboud UMC, Nijmegen, persoonlijke titel
Met ondersteuning van
- dr. J. (Joppe) Tra, Senior-Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. D. (Dagmar) Nieboer, Senior-Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. J. (Josefien) Buddeke, Senior-Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. M. (Mirre) den Ouden-Vierwind, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. M. (Merel) Wassenaar, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. A.C. (Anniek) van Westing, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. L. (Leanne) Küpers, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. J. (Jing) de Haan- Du, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. S.N. (Sarah) van Duijn, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. M. (Majke) van Bommel, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- drs. E. (Evie) Verweg, Junior Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- D.P. (Diana) Gutierrez, projectsecretaresse, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een (sub-) module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Werkgroepleden |
||||
|
Nielsen |
Klinisch geneticus, volledig betaald door de afdeling. |
2012 - present: the International Society for Gastrointestinal Hereditary Tumors (InSiGHT)
Betaling per bijeenkomst (vacation money for expenses made)
Lid adviesraad Stichting Lynch- Polyposis (onbetaald) |
Lopende subsidiegelden – waar ik bij betrokken bij ben:
|
Geen |
|
Hoogerbrugge – van der Linden |
Medisch specialist en hoogleraar erfelijke kanker
|
Geen |
Geen extern gefinancierd onderzoek in relatie tot deze functie |
Geen |
|
Wagner |
Klinisch geneticus |
Bestuurslid Stichting Opsporing Erfelijke tumoren (onbetaald); |
Extern gefinancierd onderzoek. WP3 leider KWF-project nr: 14976 (Smart measurement of circulating tumour DNA: a tumour-agnostic computational tool to improve CRC-care) |
Geen |
|
Bajwa – ten Broeke |
Klinische geneticus UMCG PI, betaald (vanuit KWF-subsidie) |
Lid European Hereditary Tumor Group (onbetaald), Bestuurslid DCCG |
Ja, subsidie voor onderzoek door KWF (young investigator grant), 2021-2025 |
Geen |
|
Van Leerdam |
MDL-arts Antoni van Leeuwenhoek, Amsterdam (0,6 FTE) betaald
|
Medisch directeur stichting opsporing erfelijke tumoren, Leiden, onbetaald |
Extern gefinancierd onderzoek |
Geen |
|
Bisseling |
MDL-arts Radboud UMC 0.9 fte |
Geen |
Geen |
Geen |
|
Van Kouwen |
Maag-Darm-Leverarts Radboud UMC Nijmegen |
Geen |
Geen |
Geen |
|
Dekker |
Maag-Darm-Leverarts, FT, Amsterdam UMC (betaald)
|
Lid raad van Commissarissen The eNose Company (betaald)
|
I have received honorarium for consultancy* from FujiFilm, Olympus, GI Supply, PAION and Ambu, and speakers' fees from Olympus, GI Supply, Norgine, IPSEN, PAION and FujiFilm. -Studies gefinancierd door KWF, MLDS, TKI, ZonMW, Celtic, FujiFilm. * Single meeting about development, innovation and research |
Geen
Geen deelname aan adviesraden gedurende de richtlijnontwikkeling |
|
Bouchiba |
PhD kandidaat/Arts-onderzoeker maag-darm-leverziekten Amsterdam UMC. |
Geen |
Geen |
Geen |
|
Verberne |
Chirurg in het ziekenhuis Amstelland, Amstelveen |
Geen |
Geen |
Geen |
|
Woolderink |
Gynaecoloog, Martini Ziekenhuis Groningen |
Lid adviesraad Stichting Lynch- Polyposis (onbetaald) |
Geen |
Geen |
|
Van der Post |
Patholoog, Radboud UMC
|
Bestuurslid Stichting Opsporing Erfelijke tumoren (onbetaald) |
Geen extern gefinancierd onderzoek in relatie tot deze functie |
Geen |
|
Seppen |
Universitair hoofddocent, Amsterdam UMC |
Bestuurslid Stichting Lynch Polyposis |
Geen |
Geen |
|
Mensenkamp |
Laboratoriumspecialist klinische genetica, Radboud UMC Nijmegen |
Voorzitter landelijk overleg erfelijke borstkankerdiagnostiek (LOB)
|
-Medewerking verleend aan workshops variantclassificatie en betrokken als assessor bij kwaliteitsrondzendingen BRCA-diagnostiek op tumorweefsel (EMQN/GenQA, gesponsord door AstraZeneca, betaald aan de afdeling Genetica) -VCo-applicant CRAFT project bij KWF. |
Geen |
|
Tops |
Laboratorium specialist klinische genetica, KG, LUMC
|
Geen |
KWF 14469 - Functionele test Lynch genen - Geen projectleider |
Geen |
|
Schoenaker |
Verpleegkundige specialist AGZ (MDL Oncologie) Isala, betaald
|
Geen |
EASIER study; Electronic nose for breath Analysis after curative Surgery to detect dIstant mEtastases or locoregional Recurrence of colon cancer |
Geen |
|
Saveur |
Verpleegkundig specialist Antoni van Leeuwenhoek |
Geen |
Geen |
Geen |
|
|
||||
|
Klankbordgroepleden |
||||
|
Van Duijnhoven |
Associate Professor
|
Geen |
Onderzoeksproject gefinancierd door World Cancer Research Fund/ Wereld Kanker Onderzoek Fonds |
Geen |
|
Verdonschot |
AIOS Klinische Genetica, MUMC+ |
Geen |
Geen |
Geen |
|
Van der Heijden |
Oncologisch uroloog
|
Geen |
ZonMW; BladParadigm; RCT mpMRI versus TURT.Projectleider ja |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiënten perspectief door afvaardiging vanuit de patiëntenvereniging Stichting Lynch Polyposis in de werkgroep. De afgevaardigde heeft meebeslist bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen.
De conceptrichtlijn is tevens voor commentaar voorgelegd aan Stichting Lynch Polyposis de Nederlandse Federatie van Kankerpatiëntenorganisaties (NFK). De eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijnmodule is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd om te beoordelen of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling is de richtlijnmodule op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
|
Module |
Uitkomst raming |
Toelichting |
|
Submodule De detectie van Lynch syndroom middels tumor analyse |
Geen mogelijk financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en daarom naar verwachting geen substantiële financiële gevolgen zullen hebben voor de collectieve uitgaven. |
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep schriftelijk de knelpunten in de zorg voor patiënten met Erfelijke darmkanker. Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. Indien mogelijk werd de data uit verschillende studies gepoold in een random-effects model. Review Manager 5.4 werd gebruikt voor de statistische analyses. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
er is hoge zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; het is zeer onwaarschijnlijk dat de literatuurconclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Redelijk |
er is redelijke zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; het is mogelijk dat de conclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Laag |
er is lage zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; er is een reële kans dat de conclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Zeer laag |
er is zeer lage zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; de literatuurconclusie is zeer onzeker. |
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello, 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE-gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
Voor- en nadelen van interventies dienen goed met de patiënt te worden doorgenomen.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Zoekverantwoording
Literature search strategy
Algemene informatie
|
Cluster/richtlijn: VKGN Erfelijke darmkanker |
|
|
Uitgangsvraag/submodules: UV1 Wat is het optimale beleid voor moleculair onderzoek naar unexplained MMR-deficiëntie op populatieniveau? |
|
|
Database(s): Embase.com, Ovid/Medline |
Datum: 1 maart 2024 |
|
Periode: vanaf 2010 |
Talen: geen restrictie |
|
Literatuurspecialist: Alies Oost |
Rayyan review: https://rayyan.ai/reviews/949215 |
|
BMI-zoekblokken: voor verschillende opdrachten wordt (deels) gebruik gemaakt van de zoekblokken van BMI-Online https://blocks.bmi-online.nl/ Deduplication: voor het ontdubbelen is gebruik gemaakt van http://dedupendnote.nl:9777/ |
|
|
Toelichting: Voor deze vraag is gezocht op de elementen:
De sleutelartikelen worden gevonden met deze search, m.u.v. PMID 25560109. Deze bevat in titel/ abstract/ trefwoord geen termen voor moleculair tumoronderzoek en Lynch syndroom/ UMMRD. PMID 36037994 wordt op zich wel gevonden maar valt niet binnen de SRs en observationele studies. |
|
|
Te gebruiken voor richtlijntekst: In de databases Embase.com en Ovid/Medline is op 1 maart 2024 systematisch gezocht naar systematische reviews en observationele studies over moleculair tumoronderzoek naar Lynch (like) syndroom en/of unexplained Mismatch repair deficiëntie (UMMRD) bij bepaalde kankersoorten. De literatuurzoekactie leverde 1469 unieke treffers op. |
|
Zoekopbrengst
|
|
EMBASE |
OVID/MEDLINE |
Ontdubbeld |
|
SR |
123 |
83 |
137 |
|
Observationele studies |
1203 |
741 |
1332 |
|
Totaal |
|
|
1469* |
*in Rayyan
Zoekstrategie
Embase.com
|
No. |
Query |
Results |
|
#1 |
'Lynch syndrome'/exp OR 'Lynch syndrome ii'/exp OR ((Lynch NEAR/3 (syndrome* OR like)):ti,ab,kw) OR 'ls related':ti,ab,kw OR lls:ti,ab,kw OR 'muir torre syndrome*':ti,ab,kw OR (((mmrd OR 'mmr d' OR 'mmr deficien*' OR 'mismatch repair deficien*') NEAR/4 (unexplain* OR 'no identifiable cause')):ti,ab,kw) OR ummrd:ti,ab,kw |
11619 |
|
#2 |
'hereditary nonpolyposis colorectal cancer'/de OR 'familial colorectal cancer type x'/exp OR (((hereditary OR familial OR 'type x') NEAR/3 ('colorectal cancer*' OR 'colorectal neoplasm*' OR 'colorectal carcinoma*' OR 'colon cancer*' OR 'colon neoplasm*' OR 'colon carcinoma*')):ti,ab,kw) OR hnpcc:ti,ab,kw OR fcctx:ti,ab,kw OR 'ovary cancer'/exp OR ((ovar* NEAR/3 (adenocarcinom* OR cancer* OR carcinogenesis OR carcino* OR lymphom* OR malignan* OR neoplasm* OR sarcom* OR teratom* OR tumor* OR tumour*)):ti,ab,kw) OR 'urothelial tumor'/exp OR (((urothelial OR urothelium OR 'transitional cell' OR 'transitional epithelium') NEAR/3 (cancer* OR carcinogenesis OR carcinoma* OR malignan* OR neoplas* OR papilloma* OR tumor* OR tumour*)):ti,ab,kw) OR (((bladder OR 'renal pelvis' OR 'renal pelvic' OR ureter* OR urethra*) NEAR/3 (cancer OR carcinoma* OR malignan* OR neoplasm* OR papilloma* OR tcc)):ti,ab,kw) OR 'small intestine tumor'/exp OR ((('small bowel' OR 'small intestin*' OR duodenal OR duodenum OR ileal OR ileum OR jejunum OR jejunal) NEAR/3 (adenocarcinom* OR cancer* OR carcin* OR malignan* OR neoplas* OR sarcoma* OR tumor* OR tumour*)):ti,ab,kw) OR 'pancreas tumor'/exp OR (((pancreas OR pancreatic) NEAR/3 (adenocarcinom* OR cancer* OR carcinom* OR incidentalom* OR malignan* OR neoplas* OR sarcom* OR tumor* OR tumour*)):ti,ab,kw) OR pancreatoblastom*:ti,ab,kw OR 'prostate tumor'/exp OR ((prostat* NEAR/3 (adenocarcinom* OR adenoma* OR cancer* OR carcinom* OR cystadenom* OR lymphoma* OR malignan* OR neoplas* OR sarcom* OR tumor* OR tumour*)):ti,ab,kw) OR 'stomach tumor'/exp OR (((stomach OR gastric) NEAR/3 (adenocarcinom* OR cancer* OR carcin* OR choriocarcin* OR lymphom* OR malignan* OR melanom* OR neoplas* OR tumor* OR tumour*)):ti,ab,kw) OR 'biliary tract tumor'/exp OR (((biliary OR 'bile duct' OR gallbladder OR 'gall bladder') NEAR/3 (adenoma* OR adenocarcinom* OR adenomyomatos* OR cancer* OR carcinom* OR cystadeno* OR malignan* OR neoplas* OR papillomatos* OR sarcom* OR tumor* OR tumour*)):ti,ab,kw) OR 'sebaceous gland tumor'/exp OR ((sebaceous NEAR/3 (adenocarcinom* OR adeocarcinom* OR adenom* OR cancer* OR carcinom* OR malignan* OR neoplas* OR tumor* OR tumour*)):ti,ab,kw) OR sebaceoma*:ti,ab,kw OR 'brain tumor'/exp OR (((brain OR cerebellum OR cerebral OR cerebri OR cerebrum OR intracerebrum OR intracerebral OR intracranial) NEAR/3 (angiomatos* OR cancer* OR hemangio* OR lymphom* OR malignan* OR melanom* OR neoplas* OR tumor* OR tumour*)):ti,ab,kw) |
1424054 |
|
#3 |
'mismatch repair'/exp OR 'mismatch* repair':ti,ab,kw OR 'mismatch* dna repair':ti,ab,kw OR 'mis match* repair':ti,ab,kw OR 'mis match* dna repair':ti,ab,kw OR mmr:ti,ab,kw OR dmmr:ti,ab,kw OR mmrd:ti,ab,kw OR 'immunohistochemistry'/exp OR ihc:ti,ab,kw OR immunohistochemistry:ti,ab,kw OR immunohistocytochemistry:ti,ab,kw OR 'immunohistochemical analys*':ti,ab,kw OR 'antigen staining':ti,ab,kw OR immunostaining:ti,ab,kw OR immunocytochemistry:ti,ab,kw OR (((immunogold OR immunolabel*) NEAR/33 techni*):ti,ab,kw) OR 'microsatellite instability'/exp OR 'microsatellite instability':ti,ab,kw OR msi:ti,ab,kw OR 'replication error phenotyp*':ti,ab,kw OR 'germline testing'/exp OR (('genetic screening'/de OR 'genetic counseling'/de OR 'genetic profile'/de OR 'cancer genetics'/de) AND ('germline mutation'/exp OR 'germ line'/exp)) OR ((('germ line' OR germline) NEAR/3 (test* OR mmr OR mutation*)):ti,ab,kw) |
1021893 |
|
#4 |
#1 AND #2 AND #3 |
5408 |
|
#5 |
#4 NOT ('conference abstract'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) NOT (('animal'/exp OR 'animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
3001 |
|
#6 |
#5 AND [2010-2024]/py |
2559 |
|
#7 |
'meta analysis'/exp OR 'meta analysis (topic)'/exp OR metaanaly*:ti,ab OR 'meta analy*':ti,ab OR metanaly*:ti,ab OR 'systematic review'/de OR 'cochrane database of systematic reviews'/jt OR prisma:ti,ab OR prospero:ti,ab OR (((systemati* OR scoping OR umbrella OR 'structured literature') NEAR/3 (review* OR overview*)):ti,ab) OR ((systemic* NEAR/1 review*):ti,ab) OR (((systemati* OR literature OR database* OR 'data base*') NEAR/10 search*):ti,ab) OR (((structured OR comprehensive* OR systemic*) NEAR/3 search*):ti,ab) OR (((literature NEAR/3 review*):ti,ab) AND (search*:ti,ab OR database*:ti,ab OR 'data base*':ti,ab)) OR (('data extraction':ti,ab OR 'data source*':ti,ab) AND 'study selection':ti,ab) OR ('search strategy':ti,ab AND 'selection criteria':ti,ab) OR ('data source*':ti,ab AND 'data synthesis':ti,ab) OR medline:ab OR pubmed:ab OR embase:ab OR cochrane:ab OR (((critical OR rapid) NEAR/2 (review* OR overview* OR synthes*)):ti) OR ((((critical* OR rapid*) NEAR/3 (review* OR overview* OR synthes*)):ab) AND (search*:ab OR database*:ab OR 'data base*':ab)) OR metasynthes*:ti,ab OR 'meta synthes*':ti,ab |
1006035 |
|
#8 |
'major clinical study'/de OR 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR 'prospective study'/de OR 'comparative study'/de OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (('case control' NEAR/1 (study OR studies)):ab,ti) OR (('follow up' NEAR/1 (study OR studies)):ab,ti) OR (observational NEAR/1 (study OR studies)) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti) |
8098097 |
|
#9 |
'case control study'/de OR 'comparative study'/exp OR 'control group'/de OR 'controlled study'/de OR 'controlled clinical trial'/de OR 'crossover procedure'/de OR 'double blind procedure'/de OR 'phase 2 clinical trial'/de OR 'phase 3 clinical trial'/de OR 'phase 4 clinical trial'/de OR 'pretest posttest design'/de OR 'pretest posttest control group design'/de OR 'quasi experimental study'/de OR 'single blind procedure'/de OR 'triple blind procedure'/de OR (((control OR controlled) NEAR/6 trial):ti,ab,kw) OR (((control OR controlled) NEAR/6 (study OR studies)):ti,ab,kw) OR (((control OR controlled) NEAR/1 active):ti,ab,kw) OR 'open label*':ti,ab,kw OR (((double OR two OR three OR multi OR trial) NEAR/1 (arm OR arms)):ti,ab,kw) OR ((allocat* NEAR/10 (arm OR arms)):ti,ab,kw) OR placebo*:ti,ab,kw OR 'sham-control*':ti,ab,kw OR (((single OR double OR triple OR assessor) NEAR/1 (blind* OR masked)):ti,ab,kw) OR nonrandom*:ti,ab,kw OR 'non-random*':ti,ab,kw OR 'quasi-experiment*':ti,ab,kw OR crossover:ti,ab,kw OR 'cross over':ti,ab,kw OR 'parallel group*':ti,ab,kw OR 'factorial trial':ti,ab,kw OR ((phase NEAR/5 (study OR trial)):ti,ab,kw) OR ((case* NEAR/6 (matched OR control*)):ti,ab,kw) OR ((match* NEAR/6 (pair OR pairs OR cohort* OR control* OR group* OR healthy OR age OR sex OR gender OR patiënt * OR subject* OR participant*)):ti,ab,kw) OR ((propensity NEAR/6 (scor* OR match*)):ti,ab,kw) OR versus:ti OR vs:ti OR compar*:ti OR ((compar* NEAR/1 study):ti,ab,kw) OR (('major clinical study'/de OR 'clinical study'/de OR 'cohort analysis'/de OR 'observational study'/de OR 'cross-sectional study'/de OR 'multicenter study'/de OR 'correlational study'/de OR 'follow up'/de OR cohort*:ti,ab,kw OR 'follow up':ti,ab,kw OR followup:ti,ab,kw OR longitudinal*:ti,ab,kw OR prospective*:ti,ab,kw OR retrospective*:ti,ab,kw OR observational*:ti,ab,kw OR 'cross sectional*':ti,ab,kw OR cross?ectional*:ti,ab,kw OR multicent*:ti,ab,kw OR 'multi-cent*':ti,ab,kw OR consecutive*:ti,ab,kw) AND (group:ti,ab,kw OR groups:ti,ab,kw OR subgroup*:ti,ab,kw OR versus:ti,ab,kw OR vs:ti,ab,kw OR compar*:ti,ab,kw OR 'odds ratio*':ab OR 'relative odds':ab OR 'risk ratio*':ab OR 'relative risk*':ab OR 'rate ratio':ab OR aor:ab OR arr:ab OR rrr:ab OR ((('or' OR 'rr') NEAR/6 ci):ab))) |
14864900 |
|
#10 |
#6 AND #7 - SR |
123 |
|
#11 |
#6 AND (#8 OR #9) NOT #10 - observationeel |
1203 |
Ovid/Medline
|
# |
Searches |
Results |
|
1 |
exp Lynch Syndrome II/ or (Lynch adj2 (syndrome* or like)).ti,ab,kf. or 'ls related'.ti,ab,kf. or lls.ti,ab,kf. or 'Muir Torre syndrome*'.ti,ab,kf. or ((mmrd or 'mmr d' or 'mmr deficien*' or 'Mismatch repair deficien*') adj4 (unexplain* or 'no identifiable cause')).ti,ab,kf. or ummrd.ti,ab,kf. |
6088 |
|
2 |
exp Colorectal Neoplasms, Hereditary Nonpolyposis/ or ((hereditary or familial or 'type x') adj3 ('colorectal cancer*' or 'colorectal neoplasm*' or 'colorectal carcinoma*' or 'colon cancer*' or 'colon neoplasm*' or 'colon carcinoma*')).ti,ab,kf. or hnpcc.ti,ab,kf. or fcctx.ti,ab,kf. or exp Ovarian Neoplasms/ or (ovar* adj3 (adenocarcinom* or cancer* or carcinogenesis or carcino* or lymphom* or malignan* or neoplasm* or sarcom* or teratom* or tumor* or tumour*)).ti,ab,kf. or exp Urinary Bladder Neoplasms/ or exp Carcinoma, Transitional Cell/ or exp Ureteral Neoplasms/ or exp Urethral Neoplasms/ or ((urothelial or urothelium or 'transitional cell' or 'transitional epithelium') adj3 (cancer* or carcinogenesis or carcinoma* or malignan* or neoplas* or papilloma* or tumor* or tumour*)).ti,ab,kf. or ((bladder or 'renal pelvis' or 'renal pelvic' or ureter* or urethra*) adj3 (cancer or carcinoma* or malignan* or neoplasm* or papilloma* or tcc)).ti,ab,kf. or exp Duodenal Neoplasms/ or exp Ileal Neoplasms/ or exp Jejunal Neoplasms/ or (('small bowel' or 'small intestin*' or duodenal or duodenum or ileal or ileum or jejunum or jejunal) adj3 (adenocarcinom* or cancer* or carcin* or malignan* or neoplas* or sarcoma* or tumor* or tumour*)).ti,ab,kf. or exp Pancreatic Neoplasms/ or ((pancreas or pancreatic) adj3 (adenocarcinom* or cancer* or carcinom* or incidentalom* or malignan* or neoplas* or sarcom* or tumor* or tumour*)).ti,ab,kf. or pancreatoblastom*.ti,ab,kf. or exp Prostatic Neoplasms/ or (prostat* adj3 (adenocarcinom* or adenoma* or cancer* or carcinom* or cystadenom* or lymphoma* or malignan* or neoplas* or sarcom* or tumor* or tumour*)).ti,ab,kf. or exp Stomach Neoplasms/ or ((stomach or gastric) adj3 (adenocarcinom* or cancer* or carcin* or choriocarcin* or lymphom* or malignan* or melanom* or neoplas* or tumor* or tumour*)).ti,ab,kf. or exp Biliary Tract Neoplasms/ or ((biliary or 'bile duct' or gallbladder or 'gall bladder') adj3 (adenoma* or adenocarcinom* or adenomyomatos* or cancer* or carcinom* or cystadeno* or malignan* or neoplas* or papillomatos* or sarcom* or tumor* or tumour*)).ti,ab,kf. or exp Sebaceous Gland Neoplasms/ or (sebaceous adj3 (adenocarcinom* or adeocarcinom* or adenom* or cancer* or carcinom* or malignan* or neoplas* or tumor* or tumour*)).ti,ab,kf. or sebaceoma*.ti,ab,kf. or exp Brain Neoplasms/ or ((brain or cerebellum or cerebral or cerebri or cerebrum or intracerebrum or intracerebral or intracranial) adj3 (angiomatos* or cancer* or hemangio* or lymphom* or malignan* or melanom* or neoplas* or tumor* or tumour*)).ti,ab,kf. |
946327 |
|
3 |
exp DNA Mismatch Repair/ or 'mismatch* repair'.ti,ab,kf. or 'mismatch* dna repair'.ti,ab,kf. or 'mis match* repair'.ti,ab,kf. or 'mis match* dna repair'.ti,ab,kf. or mmr.ti,ab,kf. or dmmr.ti,ab,kf. or mmrd.ti,ab,kf. or exp Immunohistochemistry/ or ihc.ti,ab,kf. or immunohistochemistry.ti,ab,kf. or immunohistocytochemistry.ti,ab,kf. or 'immunohistochemical analys*'.ti,ab,kf. or 'antigen staining'.ti,ab,kf. or immunostaining.ti,ab,kf. or immunocytochemistry.ti,ab,kf. or ((immunogold or immunolabel*) adj3 techni*).ti,ab,kf. or exp Microsatellite Instability/ or 'microsatellite instability'.ti,ab,kf. or msi.ti,ab,kf. or 'replication error phenotyp*'.ti,ab,kf. or ((exp Genetic Testing/ or exp Genetic Carrier Screening/ or exp Genetic Counseling/ or exp Genetic Services/ or exp Genetic Profile/) and exp Germ-Line Mutation/) or (('germ line' or germline) adj3 (test* or mmr or mutation*)).ti,ab,kf. |
882282 |
|
4 |
1 and 2 and 3 |
2671 |
|
5 |
4 not (comment/ or editorial/ or letter/) not ((exp animals/ or exp models, animal/) not humans/) |
2548 |
|
6 |
limit 5 to yr="2010 -Current" |
2143 |
|
7 |
meta-analysis/ or meta-analysis as topic/ or (metaanaly* or meta-analy* or metanaly*).ti,ab,kf. or systematic review/ or cochrane.jw. or (prisma or prospero).ti,ab,kf. or ((systemati* or scoping or umbrella or "structured literature") adj3 (review* or overview*)).ti,ab,kf. or (systemic* adj1 review*).ti,ab,kf. or ((systemati* or literature or database* or data-base*) adj10 search*).ti,ab,kf. or ((structured or comprehensive* or systemic*) adj3 search*).ti,ab,kf. or ((literature adj3 review*) and (search* or database* or data-base*)).ti,ab,kf. or (("data extraction" or "data source*") and "study selection").ti,ab,kf. or ("search strategy" and "selection criteria").ti,ab,kf. or ("data source*" and "data synthesis").ti,ab,kf. or (medline or pubmed or embase or cochrane).ab. or ((critical or rapid) adj2 (review* or overview* or synthes*)).ti. or (((critical* or rapid*) adj3 (review* or overview* or synthes*)) and (search* or database* or data-base*)).ab. or (metasynthes* or meta-synthes*).ti,ab,kf. |
720199 |
|
8 |
Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or cohort.tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ [Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies] |
4633555 |
|
9 |
Case-control Studies/ or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or comparative study/ or control groups/ or controlled before-after studies/ or controlled clinical trial/ or double-blind method/ or historically controlled study/ or matched-pair analysis/ or single-blind method/ or (((control or controlled) adj6 (study or studies or trial)) or (compar* adj (study or studies)) or ((control or controlled) adj1 active) or "open label*" or ((double or two or three or multi or trial) adj (arm or arms)) or (allocat* adj10 (arm or arms)) or placebo* or "sham-control*" or ((single or double or triple or assessor) adj1 (blind* or masked)) or nonrandom* or "non-random*" or "quasi-experiment*" or "parallel group*" or "factorial trial" or "pretest posttest" or (phase adj5 (study or trial)) or (case* adj6 (matched or control*)) or (match* adj6 (pair or pairs or cohort* or control* or group* or healthy or age or sex or gender or patiënt * or subject* or participant*)) or (propensity adj6 (scor* or match*))).ti,ab,kf. or (confounding adj6 adjust*).ti,ab. or (versus or vs or compar*).ti. or ((exp cohort studies/ or epidemiologic studies/ or multicenter study/ or observational study/ or seroepidemiologic studies/ or (cohort* or 'follow up' or followup or longitudinal* or prospective* or retrospective* or observational* or multicent* or 'multi-cent*' or consecutive*).ti,ab,kf.) and ((group or groups or subgroup* or versus or vs or compar*).ti,ab,kf. or ('odds ratio*' or 'relative odds' or 'risk ratio*' or 'relative risk*' or aor or arr or rrr).ab. or (("OR" or "RR") adj6 CI).ab.)) |
5603459 |
|
10 |
6 and 7 - SR |
83 |
|
11 |
(6 and (8 or 9)) not 10 - observationeel |
741 |