Verwijscriteria voor klinisch genetisch onderzoek bij adenomateuze polyposis
Uitgangsvraag
Welke endoscopische en histologische (verwijs)criteria zijn te gebruiken voor klinisch genetisch onderzoek (erfelijkheidsonderzoek) bij patiënten met adenomateuze polyposis?
Aanbeveling
Aanbeveling-1
Verwijs patiënten voor erfelijkheidsonderzoek bij:
- 10 of meer colorectale adenomen <60 jaar (cumulatief)
- 20 of meer colorectale adenomen <70 jaar (cumulatief)
Aanbeveling-2
Overweeg verwijzing voor erfelijkheidsonderzoek bij minder adenomen indien er sprake is van:
- Extra-colon manifestaties van polyposis syndromen, Lynch syndroom, CMMRD
- Een somatische KRAS c.34G>T-transversie
- Familiegeschiedenis van polyposis syndromen of Lynch syndroom geassocieerde tumoren, welke voldoet aan de verwijscriteria en waarbij geen geschikter familielid verwezen kan worden
- Reeds bekende familiaire pathogene variant(en) in een CRC-predispositie gen,
- Bij dominant overervende aandoeningen als cascadescreening
- Bij recessief overervende aandoening broers en zussen van de aangedane patiënt (homozygoot of compound heterozygoot) en bij dragerschapsfrequentie in bevolking>1/100 ook partner (of kinderen) van aangedane patiënt; bij consanguiniteit ook partner (of kinderen) van heterozygoten.
Overwegingen
Voor het beantwoorden van de vraag ‘Welke endoscopische en histologische (verwijs)criteria zijn te gebruiken voor erfelijkheidsonderzoek bij patiënten met 10-20 colorectale adenomen’ is geen systematische literatuursearch gedaan. Het antwoord op deze vragen is gezocht in internationale richtlijnen en studies die bekend zijn bij de werkgroep. De kwaliteit van bewijs is dan ook niet gegradeerd.
Verwijscriteria bij adenomateuze polyposis
In de studies van Stanlich (2019), Terlouw (2020) en Grover (2012) worden opsommingen gegeven van de uitkomsten van de studies naar opbrengst DNA-onderzoek bij (adenomateuze) polyposis patiënten, en de relatie met leeftijd. Op basis van deze drie studies lijkt DNA-onderzoek in elk geval geïndiceerd bij patiënten met ≥10 adenomen in de leeftijd <60 jaar en ≥20 adenomen in de leeftijd <70 jaar, zoals geadviseerd in de recente Europese richtlijnen.
Er is minder bekend over de opbrengst van DNA-onderzoek bij jonge patiënten (<50 jaar) met <10 adenomen of een oudere patiënt (>70 jaar) met meer dan 20 adenomen.
Bij jongere patiënten met enkele adenomen kan sprake zijn van (A)FAP, MAP, Lynch syndroom of CMMRD. Verwijzing dient daarom overwogen te worden indien er een positieve familiegeschiedenis is passend bij een van deze syndromen of er sprake is van andere kenmerken zoals FAP-gerelateerde extracolonische manifestaties.
De studie Terlouw (2020) laat zien dat er in de groep met 1-10 poliepen (totaal n=328, waarvan ongeveer 150 onder de 50 jaar) er 6 patiënten een kiembaan PV hebben (1 APC en 5 biallelische MUTYH) onder de 50 jaar, 2 daarvan hadden CRC 40-50 jaar, 1 had eerstegraads met polyposis en de andere 2 een eerstegraads met CRC <50 jaar). De 3% kans op PV in APC of MUTYH onder de 50 jaar wordt zeer waarschijnlijk hoger als je de afkap op 5 poliepen zet maar die informatie is helaas niet beschikbaar. Wel waren dus bij 5 van de 6 patiënt en ook andere kenmerken voor polyposis aanwezig zoals jong darmkanker of belast familieverhaal.
Ook indien sprake is van advanced adenomen op jongere leeftijd neemt de kans op een predispositie waarschijnlijk toe, maar hier zijn nog geen studies van bekend, al wordt in Lynch patiënten onder de 50 jaar wel in 5,4% een advanced adenoma gevonden (Alric 2024). In elk geval lijkt het van belang om een patiënt met advanced adenomen of met meerdere adenomen op jonge leeftijd (<30 jaar), indien besloten wordt vooralsnog niet te verwijzen voor erfelijkheidsonderzoek, wel controle coloscopieën aan te bieden (zie ook richtlijn coloscopie surveillance).
Op basis van de studies van Terlouw (2020) en Stanich (2019) is de kans op het vinden van een APC PV, biallelische MUTYH PVs of PV in een ander polyposis gen lager dan 1% in patiënten met minder dan 20 adenomen ouder dan 70 jaar. Bij patiënten met meer dan 20 adenomen in de groep ouder dan 70 is dit meer dan 1%, in de studie van Stanich (2019) zelfs 4,5% in de groep ouder dan 80 jaar. Echter, er is in deze studie alleen aangegeven dat het om een PV in de een van de MMR-genen, TP53, CHEK2 en CDH1 gaat, maar percentages per gen ontbreken. Mogelijk zit er dus een belangrijk deel CHEK2 PV tussen, die in deze groep geen klinische consequenties hebben. Waarschijnlijk zit er ook een belangrijk deel MMR PVs tussen, maar het is niet duidelijk of in deze groep mogelijk eerder al sprake was van een Lynch geassocieerde tumor in de index of een familielid.
Er is dus te weinig bewijs dat genetisch onderzoek bij patiënten met >20 adenomen >70 jaar zinvol is. Aan de andere kant is het aantal patiënten uit het bevolkingsonderzoek boven de 70 jaar met meer dan 20 adenomen zeer beperkt, zo’n 5 per jaar tussen de 70 en 80 jaar en daarboven waarschijnlijk nog minder (zie Figuur 2 (submodule Verwijscriteria voor klinisch genetisch onderzoek bij adenomateuze polyposis) op basis van RIVM-data van het bevolkingsonderzoek). In individuele gevallen, bij voorbeeld bij sterke wens van de patiënt, of in geval van gelijktijdig voorkomen duodenum poliepen kan daarom in overleg met de MDL-arts en Klinische Genetica soms toch verwijzing voor verder onderzoek overwogen worden.
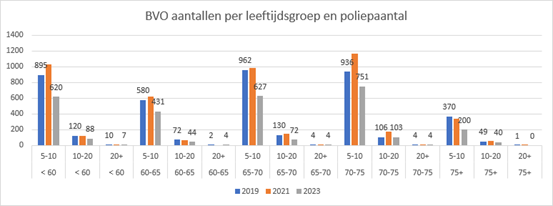
Figuur 2 (submodule Verwijscriteria voor klinisch genetisch onderzoek bij adenomateuze polyposis) Aantallen mensen met poliepen per leeftijdscategorie in 2019, 2021 en 2023 vanuit het bevolkingsonderzoek naar darmkanker (RIVM).
Tenslotte geeft het aanwezig zijn van de KRAS c.34G>T mutatie in darmkanker een sterke indicatie dat sprake kan zijn van MUTYH geassocieerde polyposis (MAP) (van Puijenbroek, 2012; Aime, 2015; Disel, 2024), waarbij MAP in 10 tot 25% van deze groep wordt teruggevonden, ook in patiënten met minder dan 10 poliepen en CRC. KRAS analyse vindt doorgaans plaats bij stadium IV CRCs in het kader van behandelingskeuzes. Indien hierbij de c.34G>T mutatie wordt aangetoond, valt verwijzing voor kiembaan MUTYH-analyse zeker te overwegen.
Bij voorkeur vindt genetische diagnostiek in eerste instantie plaats in een aangedaan persoon, die voldoet aan de verwijscriteria. Als deze persoon zelf echter niet kan of wil verwezen worden naar de Klinische genetica, dan kan ook een eerstegraads familielid verwezen worden, zeker als dit een jonger familielid betreft (onder 50 jaar).
Uiteraard komen ook personen bij wie in de familie een erfelijke aanleg voor een polyposis syndroom is aangetoond, in aanmerking voor (presymptomatisch) onderzoek. Voor dominante aandoeningen zoals bij APC, verdient het in het kader van doelmatigheid de voorkeur dat de persoon met het hoogste risico op de familiaire aanleg als eerste getest wordt (cascade-screening). Voor recessief overgeërfde syndromen wordt DNA-onderzoek aangeboden in eerste instantie alleen aan broers en zussen. DNA-onderzoek bij de andere ouder (tbv kinderen), is van belang als dragerschap in de populatie hoog is. Dit is het geval voor MUTYH, in veel geografische regio's is het dragerschap voor een MUTYH PV rond de 1 op 70 (~1,6%) op basis van gnomAD 4.1.0 database met Europese dragers (zonder Finland), voor de drie meest frequente varianten, is de allel frequentie ~0,8%. De kans op het erven van twee pathogene MUTYH-varianten voor kinderen van iemand met MAP is dan 0,5-1%. Voor kinderen van ouders met één MUTYH PV zakt deze kans naar 0,25-0,5%. Andere bekende recessieve polyposis syndromen, zoals NTHL1-, MLH3 en MSH3-geassocieerde polyposis, hebben lagere dragerschapspercentages (ongeveer 1 op de 300) (Terradas, 2019); Daarom wordt het risico op het overerven van twee pathogene varianten zeer laag (<1/600) en aanvullend testen van de andere ouder of de kinderen zelf wordt niet geadviseerd. Als ouders bloedverwant zijn, is de kans uiteraard groter en moet testen wel worden overwogen.
Voor het advies welke familieleden hoe en wanneer DNA-onderzoek aan te bieden in geval er sprake is van een bekende erfelijke aanleg, wordt verwezen naar de samenvatting in de VKGN/StOET richtlijnen www.artsengenetica.nl.
Tenslotte komen bij mensen met adenomateuze poliepen soms ook andere poliepen voor, met namen serrated poliepen. Voor de verwijscriteria in geval van serrated poliepen verwijzen we naar submodule Erfelijkheidsonderzoek en surveillance bij serrated polyposis syndroom.
Voor syndromen met poliepen met andere histologie, met name hamartomateuze poliepen, zijn de korte verwijsadviezen van de vorige richtlijn (2015) behouden. Voor meer achtergrond over deze andere en ook andere zeldzame (adenomateuze) polyposis syndromen wordt er verwezen naar de richtlijnen van de VKGN en StOET (zie ook: www.artsengenetica.nl).
Balans tussen gewenste en ongewenste effecten
Bij patiënten met polyposis en hun naasten kunnen verschillende overwegingen meespelen in de keuze voor kiembaanonderzoek. Kiembaanonderzoek biedt in de meeste gevallen een beperkte opbrengst, vooral bij patiënten met minder dan 20 adenomateuze poliepen en oudere leeftijd (>50 jaar). Dit kan voor patiënten een reden zijn om af te zien van dit onderzoek, evenals de kosten (eigen risico) en praktische of emotionele belasting. Het niet verwezen worden voor genetisch onderzoek kan onzekerheid en zorgen over genetische risico’s vermijden. Echter, het vinden van een kiembaan variant of kiembaan/postzygotisch mozaïek (afhankelijk van het aantal poliepen varieert deze kans tussen de 5 en 90%) kan daarentegen ook juist duidelijkheid geven voor patiënt en familieleden en de mogelijkheid voor presymptomatisch onderzoek bij familieleden en deelname aan preventieve programma’s. Surveillance bij patiënten met een hoog risico op poliep vorming en daarmee op CRC leidt tot een sterk verminderd risico op (laat stadium) CRC. Bij patiënten op oudere leeftijd en of met een slechte gezondheid zal DNA-onderzoek voor henzelf niet altijd meer van belang zijn, maar dit kan wel van belang zijn voor familieleden. Er zijn geen studies bekend die hebben gekeken naar lange termijn opbrengst in de zin van overlevingswinst of gezondheidswinst van het wel of geen erfelijkheid onderzoek doen bij patiënten met polyposis.
Kostenaspecten
Op dit moment worden er naar verwachting zo’n 300 tot 500 patiënten per jaar verwezen voor erfelijkheidsonderzoek vanwege darmpoliepen. De aanpassingen aan de criteria zullen naar verwachting niet leiden tot een toename van dit aantal. Bij beter toepassen van de criteria, welke grotendeels gelijk zijn aan die uit 2015, maar nu met meer onderbouwing, zal dit naar verwachting eerder leiden tot een afname van kosten.
De kosten van genetisch onderzoek (genetische counseling en gen-panel analyse) zijn zo’n 3000 euro. Deze kosten wegen doorgaans wel op tegen de opbrengsten – in geval er een kiembaan variant wordt gevonden kan gericht surveillance en preventieve ingrepen worden aangeboden aan familieleden met de aanleg en kosten voor eventuele kankerbehandeling worden voorkomen. Ook is er een afname van preventieve coloscopieën bij familieleden die een familiare aanleg niet hebben of die ten onrechte (frequent) colonoscopie ondergaan.
Gelijkheid ((health) equity/equitable)
De interventie draagt bij aan gelijkheid in de zorg, omdat het duidelijke indicaties stelt voor genetisch onderzoek bij polyposis patiënten. Waar specifiek aanvullend onderzoek nodig is, wordt dit bepaald op basis van duidelijke klinische indicaties, waardoor een eerlijke en consistente aanpak wordt gehanteerd.
Erfelijkheidsonderzoek valt onder de basis verzekerde zorg, maar mensen zijn wel hun eigen bijdrage en eventueel eigen risico kwijt als zij geen andere medische kosten hebben.
Voor sommige mensen kan het moeten betalen van eventueel eigen risico een reden zijn om af te zien van erfelijkheidsonderzoek, of uit te stellen tot een jaar dat zij meer medische kosten hebben, wat wel ongelijkheid van zorg kan veroorzaken.
Aanvaardbaarheid:
Ethische aanvaardbaarheid
De diagnostiek lijkt aanvaardbaar voor de betrokkenen. Er zijn geen grote ethische bezwaren. Het genetisch onderzoek kan in een klein aantal gevallen tot extra onzekerheid leiden als er sprake is van een variant van onbekende betekenis (VUS). Het huidige beleid in Nederland is echter dat een VUS alleen gemeld wordt indien deze zeer verdacht is en/of nader onderzoek tot her-classificatie kan leiden. In praktijk gaat dit om een zeer klein deel van de gevallen.
Het kan zijn dat een of meerdere familieleden geen DNA-onderzoek wil en andere wel. Dit is een algemeen probleem bij genetisch onderzoek. Patiënten kunnen nooit worden verplicht genetisch onderzoek te doen maar soms kan een uitslag bij een kind ook een uitslag voor een ouder betekenen. In principe gaat het recht in deze gevallen op het wel willen weten voor op niet willen weten.
Duurzaamheid
Bij de interventie spelen de duurzaamheidsaspecten een rol aangezien het doen van genetisch onderzoek een energie intensief onderzoek is. Goede criteria zorgen voor het doelmatig gebruik van genetisch onderzoek. Ook kan de uitslag hiervan leiden tot een verminderde noodzaak van coloscopieën bij familieleden of juist heel gericht aanbieden van controle en preventie en daarmee het voorkomen van kanker en daarbij horende behandelingen.
Haalbaarheid
De geformuleerde verwijscriteria zullen naar verwachting niet leiden tot extra belasting voor de klinisch genetische centra en de gen-panel diagnostiek is over het algemeen al standaard zorg in de klinisch genetische laboratoria. De toevoeging van onderzoek naar APC-mozaïek leidt tot meer werk en kosten, maar moleculaire diagnostiek wordt in toenemende mate al verricht in tumoren, waaronder darmkanker. Ook leidt deze diagnostiek tot een reductie van onnodige coloscopieën bij familieleden als een mozaïek bij een polyposis patiënt wordt aangetoond. Naar verwachting zijn de voorgestelde adviezen in de praktijk dus haalbaar.
Rationale van aanbeveling-1: weging van argumenten voor en tegen de interventie
Eindoordeel: Sterke aanbeveling voor (Doen)
Rationale van aanbeveling-2: weging van argumenten voor en tegen de interventie
Eindoordeel: Zwakke aanbeveling voor (Doen)
Onderbouwing
Achtergrond
About half of Western individuals will be diagnosed with at least one or more adenomatous polyps of the colon during their lives (van Hees, 2015). When ten or more adenomatous polyps are present, they suffer from what is termed polyposis. A UK-based study showed that ten or more adenomas were detected in about 1.1% of patients who underwent a colonoscopy as part of the population screening for CRC (Alexander, 2021).
In older guidelines (from before 2015), the recommendation was that people with ten or more polyps undergo genetic testing. Often, however, no (hereditary) predisposition is detected. The percentage of people in whom no (germline) pathogenic variant (PV) is found, so-called polyposis e.c.i. or colonic polyposis of unknown etiology (CPUE), even seems to increase (Terlouw, 2020), which seems partly due to more accurate endoscopic techniques. The Dutch guideline in 2015 introduced stricter criteria for referral to Clinical Genetics for genetic testing being >10 adenomas under the age of 60 years or >20 adenomas under the age of 70 years.
The question is, however, whether the criteria from the 2015 guideline are efficient. What are the findings in studies in patients with fewer than 10 adenomas, and is the cut-off at 70 years justified?
Samenvatting literatuur
Description of the Guidelines
The 2015 Dutch guideline on hereditary colorectal cancer (CRC) recommended offering DNA testing to patients with 10 or more adenomas under 60 years of age and 20 or more adenomas under 70 years of age, or a personal history of desmoid tumors, hepatoblastoma, cribriform-morular papillary thyroid cancer, and CHRPE.
The European Hereditary Tumour Group (EHTG) guideline 2024 recommends a multigene germline panel in patients with >20 cumulative colorectal adenomas. The threshold may be reduced to 10 cumulative adenomas if diagnosed under the age of 60 years, or a family history of polyposis or CRC, or an extracolonic manifestation of FAP. This guideline also recommends that multi-gene germline testing (for CRC and polyposis syndromes) is performed in patients with gastrointestinal cancers under 50. Finally, they advise that somatic mutation testing for APC mosaic should be considered in patients with unexplained polyposis, who meet the criteria for testing mentioned above.
The British guideline from 2020 (Monahan, 2020) recommends gene panel testing in cases with a total of ≥10 adenomas <60 years of age, or patients ≥60 years of age with a ≥20 adenomas, or ≥10 with a family history of multiple adenomas or CRC.
The NCCN guideline 2024 (Hodan, 2024) advises testing with a personal history of ≥1 of the following criteria:
Recommend testing if ≥1 of the following criteria: ≥20 adenomas; known pathogenic variant in family; multifocal/bilateral congenital hypertrophy of retinal pigment epithelium (CHRPE); cribriform-morular variant of papillary thyroid cancer; family history of polyposis and family unwilling/unable to have testing.
Consider testing if personal history of ≥1 of the following criteria: between 10–19 cumulative adenomas, desmoid tumor, hepatoblastoma, unilateral CHRPE, or individual meets criteria for serrated polyposis (SPS) with at least some adenomas.
Description of studies
An overview of the outcome of genetic testing in adenomatous polyposis patients is given in Table 1.
Stanich (2019)
Stanich (2019) reported the outcome of constitutional DNA testing in 3789 patients with 10 or more colorectal polyps (75 with hamartomatous polyps). All patients had genetic testing of at least 14 colorectal cancer and polyposis genes (APC, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, TP53). Three additional polyposis genes (GREM1, POLD1, and POLE) were evaluated in only a subset of patients, as they were added to the multigene panel assay during the study period. In addition, 1867 patients with between 20–99 colorectal polyps and 270 100+ colorectal polyps were tested. The adenomatous polyposis cohort was further stratified by age at testing. It was decided a priori to exclude any age groups with <10 patients within this cohort. Also, the low-risk variants APC p.I1307K and CHEK2 p.I1517T, as well as monoallelic MUTYH mutations, were not considered as mutation carriers in this analysis as they are not known to result in a polyposis phenotype.
Of the 1342 patients with between 10–19 adenomatous colorectal polyps, a total of 31 of these patients (2,1%) had a PV in one of the adenomatous polyposis genes, (10 in APC (1%), 10 MUTYH biallelic (1%),1 in POLE (0,1%) and 1 patient had CMMRD (0,1%)), 13 (1%) had a PV in a hamartomatous polyposis gene (2 in SMAD4 (0,1%), 5 in BMPR1A, 3 in PTEN (0,3%), 1 in STK11, 2 in GREM1) and 41 had a PV in one of the MMR genes (3,3%), 17 in CHEK2 (1,3%) and 1 in CDH1.
In the 1637 patients with between 20-100 adenomatous polyps a PV in adenomatous polyp gene was found in 139 patients (8.5%), a PV in a hamartomatous polyposis gene in 40 (2.4%) and 19 in an MMR gene (1.1%), 3 in TP53 (0.2%), 22 in CHEK2 (1.3%) and 5 in CDH1 (0.3%),
In 219 patients with more than 100 adenomatous polyps, 90 patients (41%) had PV in adenomatous polyp genes, 9 (4.1%) in a hamartomatous gene, and only 2 PVs were found in an MMR gene, 1 in TP53 and 4 in CHEK2.
Authors conclude that the diagnostic yield of pathogenic variants remained above 5% in all ages and cohorts, despite a decrease with age. In the multiple adenoma cohort, the yield was higher in those patients with a personal or family history of cancer. In patients with 10–19 adenomas, though only 2.1% had a PV in the “traditional” polyposis predisposition gene, another 1,1% had a PV in a hamartomatous polyposis gene, 3.3% had a PV in one of the MMR genes, and 1.3% had a PV in the CHEK2-gene. The question is whether or not these MMR gene variants would also have been found through CRC tumor testing. Interestingly, in the supplemental files, the percentages of patients with polyps and no history of CRC are mentioned, in the group with 10-19 adenomas, the prevalence of PV in the MMR genes is almost two times lower, around 1.7%, (12/685), but still considerable high and therefore convincing evidence to include the MMR genes also in the polyposis panel. Although in most panels these genes are already included because of also the possibility of biallelic MMR mutation, causing a severe tumor phenotype with adenomatous polyposis, CMMRD.
In this study CHEK2 was also included in the analysis, explaining about 20% of all the PVs found. Still, the prevalence of around 1-1.5% in most groups is slightly more often than in the general (western) populations; in gnomAD (4.1.0 nonUKB), 0.23% is a carrier (https://gnomad.broadinstitute.org/variant/22-28695868-AG-A?dataset=gnomad_r4), in the Netherlands this is estimated to be > 0.6 % (personal note from molecular geneticist). Also, the clinical value of the findings of a CHEK2 variant is questionable since there are no clinical consequences if there is not a family history of breast cancer, according to the Dutch guidelines for breast cancer.
Unfortunately, the table showing the prevalence in the Adenoma Cohorts by Age does not specify which nonpolyposis colorectal cancer gene variant was found per age group, so although the almost 8% PV found in the age group above 80 could concern variant in moderate penetrant risk genes such as CHEK2 or it could highly selected cases based on family history or other cancer history. Notable, the younger age groups have a lower percentage of found variants.
Grover (2012)
A study in 2012 describes the rate of APC and biallelic MUTYH pathogenic variants (PVs) found in 7225 individuals with colorectal polyposis. PVs in APC or common European founder biallelic PVs in MUTYH were identified in 87/970 (9%) individuals with 10–19 adenomas and 559/3253 (17%) individuals with 20–99 adenomas. (Grover 2012) There was an incremental increase in the odds of a PV with an increasing number of adenomas and earlier age at adenoma diagnosis, particularly under the age of 50 years.
Terlouw (2020)
Terlouw (2020) studied the outcomes of DNA testing in 2082 polyposis patients tested between 1992 and 2017. Contrary to the other studies, Terlouw et al, also included individuals without adenomas (n = 336) but with extracolonic manifestations, CRC with KRAS codon 12 G>T transversion, a first-degree relative >10 adenomas and/or multiple primary CRCs. In 4% APC and 1% biallelic MUTYH variants were detected in these 0-adenoma patients.
Also, a relatively large cohort of patients with 1–9 adenomas was included (n = 328; APC tested n = 217 and MUTYH tested n = 309), one APC and six biallelic MUTYH variants were identified (2% variant detection rate). In this group, the APC PV carrier had already developed adenomas by age 20 and had an FDR with >100 polyps. Of the MAP patients, four were affected with CRC between the ages of 39 and 53. Information on KRAS status in tumor DNA was available for one patient, showing a somatic KRAS c.34G > T transversion.
In the group with 10–19 adenomas (n = 406; APC tested n = 324 and MUTYH tested n = 401), three FAP (1%) and six MAP patients (1,5%) were diagnosed who all developed adenomas aged under 60. Approximately 9% APC and 10% MUTYH biallelic PV mutations were found in patients with between 20-50 adenomas; this number is 12% and 18%, respectively, for 50-99 polyps and more than 70% and 7% in case someone has more than 100 adenomas.
No MUTYH or APC variants were found in patients with fewer than 20 adenomas aged over 70 (n = 82). One MAP patient was found in the patients with over 20 adenomas aged over 70 (1/90, 1.1%).
One weakness of this study was that not all patients with low adenoma counts were tested for both APC and MUTYH. Moreover, variants in other genes were not taken into account. Many of the patients were tested for POLE/D, MSH3, and NTHL1 on a research basis, the proven variant carriers were excluded in this study. Possible variants in other genes, such as SMAD4, BMPR1A, and PTEN, might be present, albeit likely only in a small percentage.
Based on this study, in contrast with the outcome of Stanich 2019, testing patients with adenomas above the age of 70 should be taken cautiously since the variant detection rate was 1%. The higher percentage found in the study of Stanich in older age groups (>70 and >80 yrs) is likely explained by also including MMR genes in the panel, which unexpectedly showed germline PVs up to 6-7% likely (exact percentage not given, it was grouped together with CHEK2, CDH1 en TP53 germline variants). More research is needed to see whether this finding of MMR PVs in older aged patients is because of selection bias (polyposis patients with also CRC who did not undergo IHC MMR testing before, for example). Notably, the same group published in 2022 a study (Jain et al.) on adenoma counts in known Lynch syndrome patients, showing that 6% of patients with Lynch syndrome had 10 or more cumulative adenomatous polyps. Authors note that this is similar to rates of multiple adenomas seen in the general population in a recent study by Sullivan et 2022.
For now, it seems too early to propose MMR testing in patients with >10 adenomas, but in case of a history of advanced colonic neoplasia or Lynch related tumor or an affected family history of Lynch related tumors this should be considered.
Also, in general, other more specific circumstances might warrant panel testing for polyposis genes, such as polyps below age 20 and numerous primary CRCs (≥2) and between 5-10 polyps depending on age and in consultation with clinical geneticist.
The 2% heterozygote MUTYH carriers detected in this study is higher than expected based on the 1% prevalence reported in the Exome Aggregation Consortium database but similar to what Grover (2012) found in patients with 20 adenomas [Grover et al]. Although this could mean that MUTYH heterozygotes might possibly have a higher risk for developing adenomas, this has no clinical consequences because the total number is still low, and CRC risk is not significantly increased for MUTYH (Nielsen, 2012).
In patients with <10 adenomas and CRC younger than 50, the detection rate of MAP may also be above 5% (see also submodule Verwijscriteria voor klinisch genetisch onderzoek bij CRC en genpanelanalyse), specifically when a hot spot KRAS G>T is present. Referral and DNA analysis in patients with CRC under the age of 50 are covered in submodule Verwijscriteria voor klinisch genetisch onderzoek bij CRC en genpanelanalyse and will not be discussed separately in this submodule.
Table 1 a-c: outcomes of DNA testing in polyposis patients
Table 1a. Prevalence gene variants in patients with 1-10 adenomas
|
|
APC |
MUTYH biallelic |
Polyposis gene other |
|
Terlouw
|
0,3% 1/328 |
1.8% (6/328) |
ND |
|
Terlouw > 60 yrs |
0/61 |
0/103 |
ND |
|
Grover |
44/1147 (4%) |
19/1147 (1.7%) |
ND |
1b. Prevalence gene variants in patients with 10-20 adenomas
|
|
APC |
MUTYH biallelic |
Polyposis gene other |
Hamar-tomateus |
MMR |
Other (TP53, CDH1) |
|
Terlouw
|
3 /402 (0.7%) |
6 /402 (1.5%) |
ND |
ND |
ND |
ND |
|
Terlouw >70 |
0/82 |
0/82 |
ND |
ND |
ND |
ND |
|
Stanich |
14/1342 (1%) |
15/1342 (1.1%) |
2/1342 (0.2%) |
13/1342 (1%) |
41/1342 (3.3%) includes chek2
|
1 (0.1%) |
|
Stanich 70-80 yrs
|
1/151 (0.7%) |
0/151 |
0/151 |
|
7/151 (4.6%) includes chek2
|
|
|
Stanich >80 yrs |
0/39 |
0/39 |
0/39 |
|
3/93 (7.7%), includes chek2 |
|
|
Grover |
50/970 (5%) |
37/970 (3.8%) |
ND |
ND |
ND |
ND |
Table 1c. Prevalence gene variant in patients with 20-100 adenomas
|
|
APC |
MUTYH biallelisch |
Polyposis gene other |
Hamar-tomatous |
MMR |
Other (TP53, CDH1) |
|
Terlouw
|
65/712(9%) |
82/712 (12%) |
ND |
ND |
ND |
ND |
|
Terlouw >70 yrs
|
0/90 |
1/90 (1.1%) |
ND |
ND |
ND |
ND |
|
Stanich |
65/1637 (4%) |
73/1673 (4.5%) |
1/1673 (0.1%) |
40/1673 (2.4%) |
19/1637 (1.1%) includes chek2 |
8/1637 (0.5%) |
|
Stanich >70 yrs |
8/297 2.7% (APC and MUTYH combined) |
See APC |
See APC |
2/297 0.7% |
11/297 (3.7%) includes chek2 |
Not specified |
|
Stanich > 80 |
2/44 4.5% (APC and MUTYH combined) |
See APC |
See APC |
0/44 0% |
3/44 6.8% includes chek2 |
Not specified |
|
Grover
|
326/3253 (10%)
|
233/3253 (7%) |
|
|
|
|
Van Puijenbroek (2012)
In a Dutch study the KRAS c.34G > T mutation was detected in 10 of 210 colon tumors, in one patent biallelic MUTYH PVS were detected (10%). This was in a 61-year-old patient with a cecum carcinoma and three adenomas. MUTYH hotspot mutation screening in 182 patients without the somatic c.34G>T KRAS mutation led to the detection of three monoallelic germline and no biallelic MUTYH mutation carriers.
Aimé (2015)
A French study compared 86 adenomas and 19 CRCs of 30 MAP patients to 135 adenomas and five CRCs of 47 familial adenomatous polyposis (FAP) patients. Secondly, the germline MUTYH gene sequence was analyzed in patients carrying the KRAS c.34G>T in CRCs diagnosed between 2008 and 2012. The c.34G>T was present in 39.7% of MAP adenomas versus 1.6% of FAP adenomas (P < 0.01). Sensitivity and specificity for detecting MAP were 39.7% and 98%, respectively. Sensitivity increased with the number of adenomas tested (P = 0.039). KRAS exon 2 analysis was performed on 2239 CRC and 2.2% harbored the c.34G>T transversion. Among 28 carriers of the KRAS c.34G>T mutation, biallelic MUTYH mutations were detected in seven patients (25%). One patient did not have any polyp or family history and did not fulfill criteria for MUTYH testing.
Disel (2024)
A recent study analyzed a large pan-cancer comprehensive genomic dataset from patients profiled (tissue next-generation sequencing) during routine clinical care. Pathogenic MUTYH alterations were identified in over 2% of 229 120 solid tumors in 30 tumor types, including germ cell (3.6%), adrenal gland (3.1%), colorectal tumors (2.7%), and small intestinal tumors (2.6%). Across tumor types, a majority of the MUTYH alterations were predicted to be monoallelic. A minority was biallelic, but this also included germline monoallelic cases with loss of heterozygosity. No difference was made in this study for biallelic germline PV carriers. So, it was not possible to calculate sensitivity or specificity for KRAS testing in this study to find MAP.
Terlouw (2025)
The rate of APC mosaicism was 9.4% in 541 patients with a broad spectrum of polyposis phenotypes. In patients who did not meet the scope of Dutch guidelines, the detection rate was 2.3% (5/219). In patients with ≥20 adenomas before the age of 60 or ≥30 adenomas before the age of 70, the detection rate was ≥10%. Of 34 mosaic patients who underwent an esophagogastroduodenoscopy, 26% were diagnosed with gastroduodenal polyps. In one patient, the mosaic variant was detected in semen, but none of the children tested in this cohort inherited the mosaic variant.
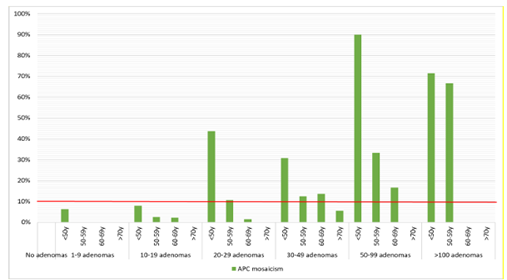
Figuur 1 (submodule Verwijscriteria voor klinisch genetisch onderzoek bij adenomateuze polyposis): Detection rates of APC mosaicism subdivided in adenoma count groups and stratified for age at first adenoma.
400 polyposis patients tested somatically for APC mosaicism, a recurrent splice variant in APC c.835-8A>G was detected in a significant proportion of colorectal lesions. This variant fits a mutation signature caused by the genotoxin colibactin. Further analysis of all APC mutations showed that at least one APC variant was detected in approximately 30% of patients that matched a colibactin-associated mutation signature.
Georgeson 2024 (preprint)
Another recent study of a large cohort of CRCs also describes that occurrence of the signature SBS88 is strongly associated with the presence of the APC splice variant c.835-8A>G and thus further confirmed the association found by Terlouw, in 7.5% (398/5,292) of the intestinal CRCs examined.
Zoeken en selecteren
No systematic review of the literature was performed. To answer the following question, “What are the endoscopic and histologic referral criteria to perform genetic testing in patients with adenomatous polyposis?”, recent international guidelines were used; the EHTG, British and NCCN guideline (Zaffaroni, 2024; Monahan, 2020; Hodan, 2024). The EHTG guideline is based on a recent structured literature search (Zaffaroni, 2024). Articles of interest that were published after the publication of this guidelines or were otherwise relevant were also included. These include Terlouw (2025), Georgeson (2025), Disel (2024), Terlouw (2020), Stanich (2019), Aimé (2015), Grover (2012), and van Puijenbroek (2012).
Referenties
- Aimé A, Coulet F, Lefevre JH, Colas C, Cervera P, Flejou JF, Lascols O, Soubrier F, Parc Y. Somatic c.34G>T KRAS mutation: a new prescreening test for MUTYH-associated polyposis? Cancer Genet. 2015 Jul-Aug;208(7-8):390-5. doi: 10.1016/j.cancergen.2015.04.005. Epub 2015 Apr 23. PMID: 26056087.
- Alexander JL, Johnston BJ, Smith TJ, Yong KK, Marshall SM, Fawkes JDC, Martin JP, Seward EW, Saunders B, Monahan KJ. Low Referral Rates for Genetic Assessment of Patients With Multiple Adenomas in United Kingdom Bowel Cancer Screening Programs. Dis Colon Rectum. 2021 Sep 1;64(9):1058-1063. doi: 10.1097/DCR.0000000000001972. PMID: 34039904.
- Disel U, Sivakumar S, Pham T, Fleischmann Z, Anu RI, Sokol ES, Kurzrock R. Increased KRAS G12C Prevalence, High Tumor Mutational Burden, and Specific Mutational Signatures Are Associated With MUTYH Mutations: A Pan-Cancer Analysis. Oncologist. 2024 Feb 2;29(2):e213-e223. doi: 10.1093/oncolo/oyad230. PMID: 37589222; PMCID: PMC10836311.
- Georgeson P, Steinfelder RS, Harrison TA, Pope BJ, Zaidi SH, Qu C, Lin Y, Joo JE, Mahmood K, Clendenning M, Walker R, Aglago EK, Berndt SI, Brenner H, Campbell PT, Cao Y, Chan AT, Chang-Claude J, Dimou N, Doheny KF, Drew DA, Figueiredo JC, French AJ, Gallinger S, Giannakis M, Giles GG, Goode EL, Gruber SB, Gsur A, Gunter MJ, Harlid S, Hoffmeister M, Hsu L, Huang WY, Huyghe JR, Manson JE, Moreno V, Murphy N, Nassir R, Newton CC, Nowak JA, Obón-Santacana M, Ogino S, Pai RK, Papadimitrou N, Potter JD, Schoen RE, Song M, Sun W, Toland AE, Trinh QM, Tsilidis K, Ugai T, Um CY, Macrae FA, Rosty C, Hudson TJ, Winship IM, Phipps AI, Jenkins MA, Peters U, Buchanan DD. Genotoxic colibactin mutational signature in colorectal cancer is associated with clinicopathological features, specific genomic alterations and better survival. medRxiv [Preprint]. 2024 Jan 30:2023.03.10.23287127. doi: 10.1101/2023.03.10.23287127. PMID: 37090539; PMCID: PMC10120801.
- Grover S, Kastrinos F, Steyerberg EW, Cook EF, Dewanwala A, Burbidge LA, Wenstrup RJ, Syngal S. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012 Aug 1;308(5):485-492. doi: 10.1001/jama.2012.8780. PMID: 22851115; PMCID: PMC3770297.
- Monahan KJ, Bradshaw N, Dolwani S, Desouza B, Dunlop MG, East JE, Ilyas M, Kaur A, Lalloo F, Latchford A, Rutter MD, Tomlinson I, Thomas HJW, Hill J; Hereditary CRC guidelines eDelphi consensus group. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020 Mar;69(3):411-444. doi: 10.1136/gutjnl-2019-319915. Epub 2019 Nov 28. PMID: 31780574; PMCID: PMC7034349.
- Stanich PP, Pearlman R, Hinton A, Gutierrez S, LaDuca H, Hampel H, Jasperson K. Prevalence of Germline Mutations in Polyposis and Colorectal Cancer-Associated Genes in Patients With Multiple Colorectal Polyps. Clin Gastroenterol Hepatol. 2019 Sep;17(10):2008-2015.e3. doi: 10.1016/j.cgh.2018.12.008. Epub 2018 Dec 14. PMID: 30557735.
- Terradas M, Munoz-Torres PM, Belhadj S, Aiza G, Navarro M, Brunet J, Capellá G, Valle L. Contribution to colonic polyposis of recently proposed predisposing genes and assessment of the prevalence of NTHL1- and MSH3-associated polyposes. Hum Mutat. 2019 Nov;40(11):1910-1923. doi: 10.1002/humu.23853. Epub 2019 Jul 29. PMID: 31243857.
- van Hees F, Saini SD, Lansdorp-Vogelaar I, Vijan S, Meester RG, de Koning HJ, Zauber AG, van Ballegooijen M. Personalizing colonoscopy screening for elderly individuals based on screening history, cancer risk, and comorbidity status could increase cost effectiveness. Gastroenterology. 2015 Nov;149(6):1425-37. doi: 10.1053/j.gastro.2015.07.042. Epub 2015 Aug 4. PMID: 26253304; PMCID: PMC4631390.
- van Puijenbroek M, Nielsen M, Tops CM, Halfwerk H, Vasen HF, Weiss MM, van Wezel T, Hes FJ, Morreau H. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res. 2008 Jan 1;14(1):139-42. doi: 10.1158/1078-0432.CCR-07-1705. PMID: 18172263.
Verantwoording
Beoordelingsdatum en geldigheid
Publicatiedatum : 29-09-2025
Beoordeeld op geldigheid : 15-09-2025
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Belangrijkste wijzigingen t.o.v. vorige versie:
|
Onderwerp |
Wijzigingen meest recente versie |
|
Module 1: Diagnostiek en verwijzing |
|
|
Module 2: Familiair colorectaal carcinoom |
|
|
Module 3: Lynch syndroom |
|
|
Module 4: Adenomateuze polyposis |
|
|
Module 5: Serrated polyposis en overige vormen van polyposis |
|
|
Andere aanpassingen |
|
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2022 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de zorg voor patiënten met Erfelijke darmkanker.
Werkgroep
- dr. M. (Maartje) Nielsen (voorzitter), Klinisch geneticus, Leids Universitair Medisch Centrum, Leiden, VKGN
- prof. dr. N. (Nicoline) Hoogerbrugge, internist, Radboud UMC, Nijmegen, VKGN
- dr. A. (Anja) Wagner, Klinisch geneticus, Erasmus MC Kanker Instituut, Universitair Medisch Centrum Rotterdam, Rotterdam, VKGN
- dr. S.W. (Sanne) Bajwa – ten Broeke, Klinisch geneticus, Universitair Medisch Centrum Groningen, Groningen, VKGN
- prof. Dr. M.E. (Monique) van Leerdam, MDL-arts, Nederlands Kanker Instituut, Amsterdam, Leiden University Medical Center, NVMDL
- dr. T.M. (Tanya) Bisseling, MDL-arts, Radboud UMC, Nijmegen, NVMDL
- dr. M.C.A. (Mariëtte) van Kouwen, MDL-arts, Radboud UMC, Nijmegen, NVMDL
- prof. Dr. E. (Evelien) Dekker, MDL-arts, Amsterdam UMC, Amsterdam, NVMDL
- drs. H. (Hicham) Bouchiba, Arts-onderzoeker MDL, Amsterdam UMC, Amsterdam, persoonlijke titel
- dr. C.J. (Charlotte) Verberne, Chirurg, Ziekenhuis Amstelland, Amstelveen
- dr. J.M. (Jorien) Woolderink, Gynaecoloog, Martini Ziekenhuis, Groningen, NVOG
- dr. R.S. (Chella) van der Post, Patholoog, Radboud UMC, Nijmegen, NVVP
- dr. J.E. (Jurgen) Seppen, Patiëntvertegenwoordiger, Stichting Lynch Polyposis
- dr. A.R. (Arjen) Mensenkamp, laboratoriumspecialist Klinische genetica, Radboud UMC, Nijmegen, VKGL
- dr. C.M.J. (Carli) Tops, laboratoriumspecialist Klinische genetica, Leids Universitair Medisch Centrum, Leiden, VKGL
- I.J.H. (Ivonne) Schoenaker, Verpleegkundig specialist MDL, Isala Ziekenhuis, Zwolle, V&VN oncologie
- L.J.(Lisette) Saveur, Verpleegkundig specialist MDL, Nederlands Kanker Instituut, Amsterdam, V&VN oncologie
Klankbordgroep:
- Dr. F.J.B. (Fränzel) van Duijnhoven, Universitair hoofddocent voeding, genen en kanker WUR, Wageningen, persoonlijke titel
- Dr. J.A.J. (Job) Verdonschot, AIOS klinische Genetica, Maastricht Universitair Medisch Centrum, Maastricht, persoonlijke titel
- Dr. A.G. (Toine) van der Heijden, Oncologisch uroloog, Radboud UMC, Nijmegen, persoonlijke titel
Met ondersteuning van
- dr. J. (Joppe) Tra, Senior-Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. D. (Dagmar) Nieboer, Senior-Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. J. (Josefien) Buddeke, Senior-Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. M. (Mirre) den Ouden-Vierwind, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. M. (Merel) Wassenaar, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. A.C. (Anniek) van Westing, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. L. (Leanne) Küpers, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. J. (Jing) de Haan- Du, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. S.N. (Sarah) van Duijn, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- dr. M. (Majke) van Bommel, Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- drs. E. (Evie) Verweg, Junior Adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- D.P. (Diana) Gutierrez, projectsecretaresse, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een (sub-) module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Werkgroepleden |
||||
|
Nielsen |
Klinisch geneticus, volledig betaald door de afdeling. |
2012 - present: the International Society for Gastrointestinal Hereditary Tumors (InSiGHT)
Betaling per bijeenkomst (vacation money for expenses made)
Lid adviesraad Stichting Lynch- Polyposis (onbetaald) |
Lopende subsidiegelden – waar ik bij betrokken bij ben:
|
Geen |
|
Hoogerbrugge – van der Linden |
Medisch specialist en hoogleraar erfelijke kanker
|
Geen |
Geen extern gefinancierd onderzoek in relatie tot deze functie |
Geen |
|
Wagner |
Klinisch geneticus |
Bestuurslid Stichting Opsporing Erfelijke tumoren (onbetaald); |
Extern gefinancierd onderzoek. WP3 leider KWF-project nr: 14976 (Smart measurement of circulating tumour DNA: a tumour-agnostic computational tool to improve CRC-care) |
Geen |
|
Bajwa – ten Broeke |
Klinische geneticus UMCG PI, betaald (vanuit KWF-subsidie) |
Lid European Hereditary Tumor Group (onbetaald), Bestuurslid DCCG |
Ja, subsidie voor onderzoek door KWF (young investigator grant), 2021-2025 |
Geen |
|
Van Leerdam |
MDL-arts Antoni van Leeuwenhoek, Amsterdam (0,6 FTE) betaald
|
Medisch directeur stichting opsporing erfelijke tumoren, Leiden, onbetaald |
Extern gefinancierd onderzoek |
Geen |
|
Bisseling |
MDL-arts Radboud UMC 0.9 fte |
Geen |
Geen |
Geen |
|
Van Kouwen |
Maag-Darm-Leverarts Radboud UMC Nijmegen |
Geen |
Geen |
Geen |
|
Dekker |
Maag-Darm-Leverarts, FT, Amsterdam UMC (betaald)
|
Lid raad van Commissarissen The eNose Company (betaald)
|
I have received honorarium for consultancy* from FujiFilm, Olympus, GI Supply, PAION and Ambu, and speakers' fees from Olympus, GI Supply, Norgine, IPSEN, PAION and FujiFilm. -Studies gefinancierd door KWF, MLDS, TKI, ZonMW, Celtic, FujiFilm. * Single meeting about development, innovation and research |
Geen
Geen deelname aan adviesraden gedurende de richtlijnontwikkeling |
|
Bouchiba |
PhD kandidaat/Arts-onderzoeker maag-darm-leverziekten Amsterdam UMC. |
Geen |
Geen |
Geen |
|
Verberne |
Chirurg in het ziekenhuis Amstelland, Amstelveen |
Geen |
Geen |
Geen |
|
Woolderink |
Gynaecoloog, Martini Ziekenhuis Groningen |
Lid adviesraad Stichting Lynch- Polyposis (onbetaald) |
Geen |
Geen |
|
Van der Post |
Patholoog, Radboud UMC
|
Bestuurslid Stichting Opsporing Erfelijke tumoren (onbetaald) |
Geen extern gefinancierd onderzoek in relatie tot deze functie |
Geen |
|
Seppen |
Universitair hoofddocent, Amsterdam UMC |
Bestuurslid Stichting Lynch Polyposis |
Geen |
Geen |
|
Mensenkamp |
Laboratoriumspecialist klinische genetica, Radboud UMC Nijmegen |
Voorzitter landelijk overleg erfelijke borstkankerdiagnostiek (LOB)
|
-Medewerking verleend aan workshops variantclassificatie en betrokken als assessor bij kwaliteitsrondzendingen BRCA-diagnostiek op tumorweefsel (EMQN/GenQA, gesponsord door AstraZeneca, betaald aan de afdeling Genetica) -VCo-applicant CRAFT project bij KWF. |
Geen |
|
Tops |
Laboratorium specialist klinische genetica, KG, LUMC
|
Geen |
KWF 14469 - Functionele test Lynch genen - Geen projectleider |
Geen |
|
Schoenaker |
Verpleegkundige specialist AGZ (MDL Oncologie) Isala, betaald
|
Geen |
EASIER study; Electronic nose for breath Analysis after curative Surgery to detect dIstant mEtastases or locoregional Recurrence of colon cancer |
Geen |
|
Saveur |
Verpleegkundig specialist Antoni van Leeuwenhoek |
Geen |
Geen |
Geen |
|
|
||||
|
Klankbordgroepleden |
||||
|
Van Duijnhoven |
Associate Professor
|
Geen |
Onderzoeksproject gefinancierd door World Cancer Research Fund/ Wereld Kanker Onderzoek Fonds |
Geen |
|
Verdonschot |
AIOS Klinische Genetica, MUMC+ |
Geen |
Geen |
Geen |
|
Van der Heijden |
Oncologisch uroloog
|
Geen |
ZonMW; BladParadigm; RCT mpMRI versus TURT.Projectleider ja |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiënten perspectief door afvaardiging vanuit de patiëntenvereniging Stichting Lynch Polyposis in de werkgroep. De afgevaardigde heeft meebeslist bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen.
De conceptrichtlijn is tevens voor commentaar voorgelegd aan Stichting Lynch Polyposis de Nederlandse Federatie van Kankerpatiëntenorganisaties (NFK). De eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijnmodule is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd om te beoordelen of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling is de richtlijnmodule op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
|
Module |
Uitkomst raming |
Toelichting |
|
Submodule Verwijscriteria voor klinisch genetisch onderzoek bij adenomateuze polyposis |
Geen mogelijk financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en daarom naar verwachting geen substantiële financiële gevolgen zullen hebben voor de collectieve uitgaven. |
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep schriftelijk de knelpunten in de zorg voor patiënten met Erfelijke darmkanker. Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. Indien mogelijk werd de data uit verschillende studies gepoold in een random-effects model. Review Manager 5.4 werd gebruikt voor de statistische analyses. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
er is hoge zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; het is zeer onwaarschijnlijk dat de literatuurconclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Redelijk |
er is redelijke zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; het is mogelijk dat de conclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Laag |
er is lage zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; er is een reële kans dat de conclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Zeer laag |
er is zeer lage zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; de literatuurconclusie is zeer onzeker. |
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello, 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE-gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
Voor- en nadelen van interventies dienen goed met de patiënt te worden doorgenomen.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.