Criteria en tests per hoogrisicogroep bij PDO
Uitgangsvraag
- Welke criteria gelden ten aanzien van de aandoeningen die worden opgenomen in de dragerschapstests voor de verschillende hoogrisicogroepen?
- a) Welke genetische (inclusief biochemische) laboratorium technieken zijn beschikbaar voor de uitvoering van een preconceptie dragerschapstest en wat zijn hun eigenschappen (specificiteit, sensitiviteit)?
b) Hoe worden vervolgens gevonden varianten geïnterpreteerd en wat wordt in een uitslag gerapporteerd? - Welk type dragerschapstest en welke analyse is het meest geschikt voor welke hoogrisicogroep?
Aanbeveling
Streef bij het ontwikkelen van een dragerschapstest aandoeningen te includeren die voldoen aan de inclusiecriteria voor het opnemen van een aandoening in een preconceptie dragerschapstest. Hieronder worden aandoeningen verstaan die optreden op de (jonge) kinderleeftijd én:
- die gepaard gaan met een ernstige lichamelijke en/of verstandelijke beperking;
- en/of gepaard gaande met ernstige pijn;
- en/of (zeer) gepaard gaan frequente ziekenhuisbezoeken;
- en/of waarvoor geen genezende therapie beschikbaar is;
- en/of waarvoor in het algemeen een verkorte levensverwachting geldt.
Aanvrager van de dragerschapstest
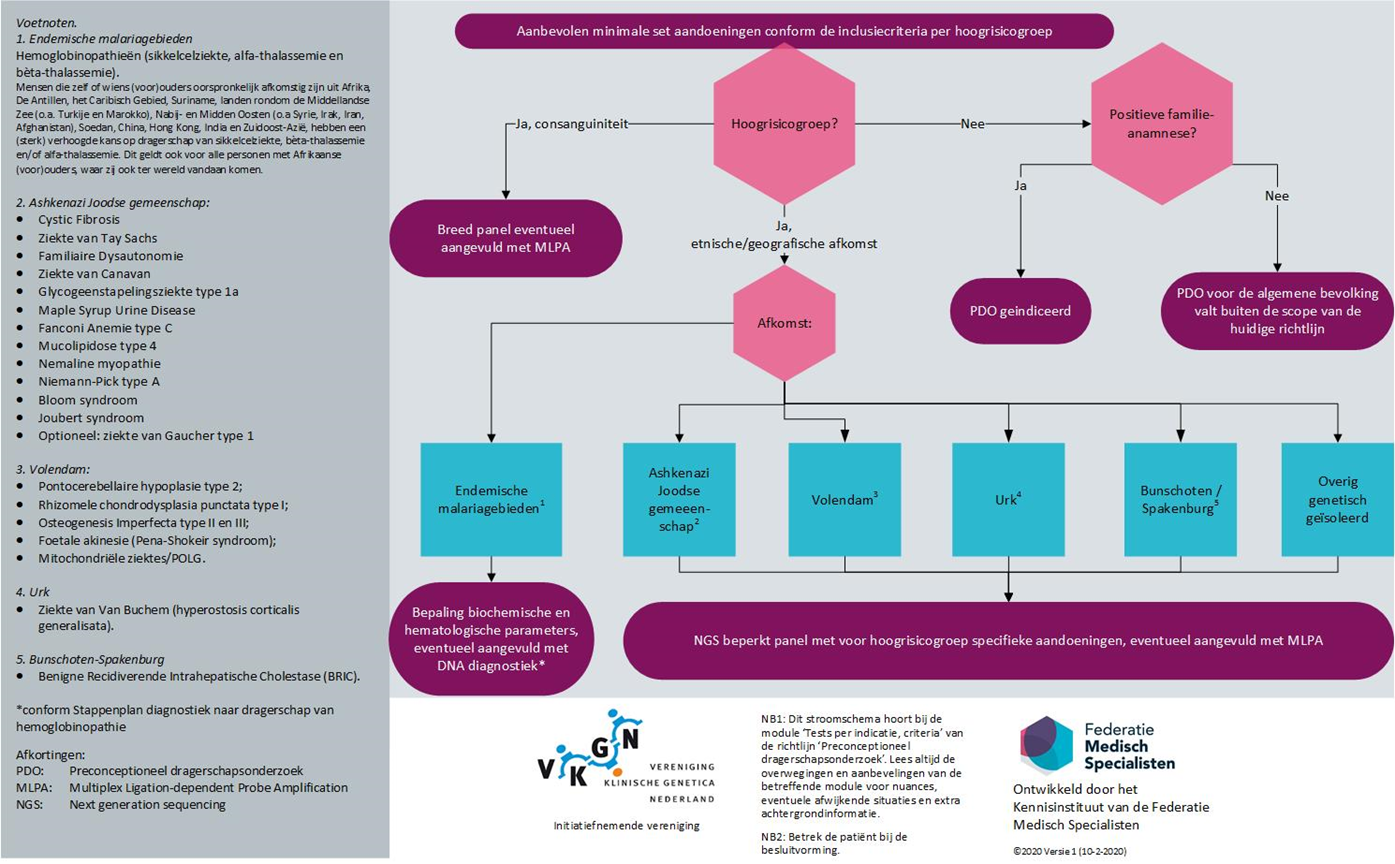
Bied paren (eventueel individuen) met kinderwens uit hoogrisicogroepen op basis van etnische en/of geografische afkomst de dragerschapstest aan die specifiek voor de desbetreffende groep beschikbaar is (zie het stroomschema bij de aanverwante producten).
Consanguine paren
Bied consanguine paren bij voorkeur een brede dragerschapstest aan en bespreek vooraf met het paar over welk soort aandoeningen dragerschap gerapporteerd zal (kunnen) worden.
Afdelingen Klinische Genetica
Streef naar een uniforme DNA-dragerschapstest per hoogrisicogroep. Indien meerdere centra een test voor eenzelfde hoogrisicogroep aan willen bieden, stem dan de inhoud van de test onderling af in de landelijke werkgroep WPCS VKGL/VKGN.
Update de testpanels aan de hand van toegenomen ervaring met de test en kennis uit de literatuur.
Overweeg bij nevenbevindingen een multidisciplinaire commissie in te richten. In deze commissie is bij voorkeur expertise aanwezig op de terreinen van de klinisch genetische laboratoriumdiagnostiek, de klinische genetica, het voor de uitslag relevante specialisme, alsmede het gezondheidsrecht en de medische ethiek, mogelijk aangevuld met een patiëntvertegenwoordiger (zie het Normatieve Kader).
Paren afkomstig uit endemische malaria gebieden met een verhoogd risico op dragerschap van hemoglobinopathieën
Voer PDO counseling uit bij individuen/ paren die afkomstig zijn uit deze hoogrisicogroep en volg het stappenplan van de diagnostiek naar dragerschap van hemoglobinopathie:
- Bepaal hematologische parameters (Hb, Ht, MCV, MCH, MCHC, erytrocyten) , ijzerstatus ( ferritine en ijzer) én Hb typering ( HPLC).
- Verricht aanvullend DNA diagnostiek naar alfa-thalassemie, indien er sprake is van een microcytair (hypochroom) bloedbeeld, zonder ijzerdeficiëntie, en zonder aanwijzingen voor dragerschap bèta-thalassemie.
- Verwijs het paar(individu) voor uitgebreid vervolgonderzoek met DNA diagnostiek naar een afdeling klinische genetica in Nederland als er bij stap 1 en 2 afwijkende uitslagen worden aangetoond passend bij dragerschap van sikkelcelziekte, bèta-thalassemie, andere Hb varianten of passend bij dragerschap van alfa-thalassemie, en indien er een wens is voor PGD of prenatale diagnostiek of het paar hierover geïnformeerd wil worden.
Meer informatie over beschikbare testen bij de
academische centra in Nederland zijn te vinden op: https://www.huisartsengenetica.nl/info/dragerschapstesten
Stroomschema: aanbevolen minimale set aandoeningen conform de inclusiecriteria per hoogrisicogroep (zie de module ‘Definitie van hoogrisicogroepen’) op basis van etnische/geografische afkomst (voor zover bekend ten tijde van de ontwikkeling van de richtlijn)
Overwegingen
1. Criteria voor inclusie van aandoeningen/genen/varianten
In de huidige situatie zijn er voor personen/paren die deel uitmaken van een specifieke hoogrisicogroep dragerschaptests beschikbaar waarin de voor die groep frequente autosomaal recessieve aandoening(en) zijn geïncludeerd. Criteria voor inclusie zijn in eerste instantie gebaseerd op de reeds aangegeven definitie van aandoeningen die optreden op de (jonge) kinderleeftijd, gepaard gaande met een ernstige lichamelijke en/of verstandelijke beperking, en/of gepaard gaande met ernstige pijn en/of (zeer) frequente ziekenhuisbezoeken, waarvoor geen genezende therapie beschikbaar is, en/of waarvoor in het algemeen een verkorte levensverwachting geldt (Henneman, 2016). De toepassing van deze criteria kan in de praktijk echter lastig zijn. De ernst van een aandoening is vaak moeilijk te definiëren en het criterium “niet behandelbaar” kan verschillend/subjectief geïnterpreteerd worden. Daarnaast kan de klinische expressie van verschillende varianten binnen één gen variëren van mild tot zeer ernstig. Zoals in de inleiding al is weergegeven wordt er bij een variabel klinisch beeld binnen één aandoening voor het bepalen van inclusie uitgegaan van de ernstigst mogelijke expressie. Hoewel er dus geen vastgelegde afspraken zijn, zijn in de literatuur wel een aantal handreikingen te vinden. Lazarin (2014), hebben in dit kader een voorstel gedaan voor een systematische classificatie van ziektebeelden met inachtneming van verschillende ziektebeeld karakteristieken (Korngiebel, 2016; Lazarin, 2014). Doel van deze studie was te komen tot een reproduceerbare tool om de ernst van een aandoening te classificeren. Overigens wordt in de praktijk de mate van ernst van een aandoening niet altijd hetzelfde ervaren door de patiënten als door artsen en is de weerspiegeling van de vraag naar reproductieve opties voor een aandoening niet altijd in lijn met de ernst van deze aandoening (zie normatieve kader). Deze factoren dienen in de post-testcounseling te worden meegenomen en besproken (zie de module ‘Pre- en post-test counseling bij PDO’).
Zoals in de Algemene Inleiding beschreven heeft de richtlijnkerngroep de inclusiecriteria voor het opnemen van een aandoening in een preconceptie dragerschapstest als volgt geformuleerd: aandoeningen die optreden op de (jonge) kinderleeftijd en:
- die gepaard gaan met een ernstige lichamelijke en/of verstandelijke beperking;
- en/of gepaard gaande met ernstige pijn;
- en/of (zeer) gepaard gaan met frequente ziekenhuisbezoeken;
- en/of waarvoor geen genezende therapie beschikbaar is;
- en/of waarvoor in het algemeen een verkorte levensverwachting geldt.
In de richtlijn wordt geen exacte definitie uitgewerkt met betrekking tot het onderscheid tussen ernstige en niet-ernstige aandoeningen. Het regelmatig monitoren van actuele medisch-wetenschappelijke inzichten is daarbij echter van belang om te bezien of er wellicht aandoeningen danwel genen toegevoegd zouden moeten worden. De Werkgroep Preconceptie Dragerschapscreening (WPCS) van de VKGL en VKGN bespreekt dit twee keer per jaar.
2. Technische specificaties van de verschillende tests en variant interpretatie
2a. Welke genetische (inclusief biochemische) laboratorium technieken zijn beschikbaar?
PDO wordt aangeboden om (waarschijnlijk) pathogene varianten (klasse 4 en 5) in recessief overervende ziekteveroorzakende genen aan te tonen bij paren met een hoger risico om een dragerpaar te zijn ten opzichte van de algemene populatie (zie de module ‘Definitie van hoogrisicogroepen’). De (waarschijnlijk) pathogene veranderingen in het DNA kunnen berusten op veranderingen van één of enkele nucleotide(n), maar ook op grotere kopie veranderingen, namelijk verlies (deleties) of winst (duplicaties) van grotere delen van het DNA, die copy number variants (CNV’s) worden genoemd. Er wordt bij de beschikbare preconceptie dragerschapstests (parallel) gebruik gemaakt van verschillende genoomdiagnostische technieken omdat (nog) niet alle varianten met dezelfde techniek op te sporen zijn. De keuze van de techniek die wordt toegepast wordt bepaald door verschillende factoren zoals 1) de beoogde doelgroep, 2) het aantal te testen varianten of genen, 3) de gewenste sensitiviteit/specificiteit, 4) de snelheid en het gemak waarmee een test kan worden uitgevoerd en 5) de kosten.
Technische specificaties van gebruikte technieken
Sanger Sequencen
Sanger sequencen wordt gezien als de gouden standaard in variant analyse, aangezien nagenoeg alle nucleotidevarianten worden aangetoond. De sensitiviteit en specificiteit van hetgeen geanalyseerd wordt, is hoog. Het DNA van de adviesvrager wordt met specifieke, voor de vraagstelling ontworpen, essentiële target primers geamplificeerd, gelabeld en de volgordes worden bepaald met behulp van een capillaire sequencer. Resultaten worden vergeleken met het referentiegenoom. Met Sanger sequencen is het mogelijk om naar specifieke pathogene varianten te kijken (bijvoorbeeld foundermutaties) of naar een heel gen. Deze techniek is minder geschikt om naar een groot aantal genen tegelijk te kijken en is ook niet geschikt om exon-deleties en duplicaties of grotere CNV’s op te sporen.
Next Generation Sequencing
Met behulp van Next Generation Sequencen (NGS) is het mogelijk de nucleotide volgorde van een groot aantal genen tegelijk te bepalen. Dit kan een selectie (panel) zijn van een aantal specifieke varianten (bijvoorbeeld foundermutaties) in een beperkt aantal genen, een selectie van een aantal specifieke genen die in zijn geheel worden bekeken voor een specifieke risicogroep (gericht panel), of een selectie van de exonen van alle eiwitcoderende genen. Dit laatste wordt ook wel Whole Exome Sequencing (WES) genoemd. Na WES kan met hulp van software tools in de exoomdata vervolgens wel een selectie van de geamplificeerde targets worden gebruikt voor analyse (virtueel panel of filter). Belangrijk om te weten is dat bij een beperkte selectie voorafgaand aan de amplificatie (gericht panel) het aantal kopieën van de specifieke target sequenties hoger is dan wanneer de selectie het hele exoom betreft. Als gevolg hiervan geeft een gericht panel een hogere sensitiviteit en specificiteit dan wanneer er voor een virtueel panel wordt gekozen. Wel is alleen informatie beschikbaar over de specifieke set genen waarvoor het panel is ontworpen en is uitbreiding van het gerichte panel alleen mogelijk door een nieuw ontwerp. Een virtueel panel kan altijd aangepast worden en behoeft geen extra investeringen.
Een WES analyse is in staat om dragerschap voor een veel groter aantal aandoeningen te onderzoeken.
Wanneer er zonder selectie het hele genoom wordt gesequenced wordt er gesproken over Whole Genome Sequencing (WGS). WGS wordt in ieder geval in Nederland op dit moment nog niet aangeboden in een klinisch diagnostische setting.
Detectie van deleties en duplicaties (CNV analyse) met behulp van MLPA
Grotere (exon) deleties of duplicaties in een enkel gen kunnen worden opgespoord met de Multiplex Ligation-dependent Probe Amplification (MLPA) techniek. Bij MLPA wordt een specifieke multiplex PCR van target sequenties (meestal exonen van het betreffende gen) uitgevoerd. Het PCR product wordt vervolgens op lengte gescheiden op een capillaire sequencer en kwantitatief vergeleken met referentie DNA. De MLPA detecteert alleen exon deleties en duplicaties van vooraf gekozen target sequenties. Deze techniek wordt met name gebruikt bij genen waarin bekend is dat exondeleties/duplicaties voorkomen, zoals bijvoorbeeld de exon7-8 deletie in het SMN1 gen, oorzakelijk voor spinale musculaire atrofie (SMA)(Ogino, 2002).
Detectie van deleties en duplicaties (CNV analyse) met behulp van NGS
Het is mogelijk om grotere deleties en duplicaties met behulp van bio-informatische tools in NGS data te detecteren. Deze methode wordt in de meeste centra toegepast in het kader van de reguliere genetische diagnostiek. Daar waar een voldoende grote controle groep beschikbaar is, is de sensitiviteit en specificiteit vergelijkbaar met de MLPA techniek. Bij een gericht genpanel voor PDO is het mogelijk exondeleties/duplicaties te detecteren, gezien het hoge kopie aantal van de targetsequencies. De bredere WES techniek focust met name op de alle eiwitcoderende (exonen) regio’s in het genoom, waardoor de sensitiviteit lager is dan bij een gericht genpanel. In het kader van PDO is het gebruik van de WES techniek voor het opsporen van exondeleties/duplicaties derhalve (nog) niet voor de hand liggend.
Biochemische testen/metabole bepalingen
Voor specifieke aandoeningen is het mogelijk om biochemische of metabole bepalingen uit te voeren; zoals bij de hemoglobinopathieen verricht wordt (Traeger-Synodinos, 2015).
2b. Interpretatie en rapportage van genvarianten
Alle aangetoonde nucleotide veranderingen (volgordes die verschillen van het referentiegenoom) worden beoordeeld op technische eigenschappen, frequentie in de algemene populatie, conservering en (mogelijke) functionele effecten zoals beschreven in de ACMG guidelines (American College of Medical Genetics and Genomics) (Richards, 2015) . Volgens deze richtlijn worden varianten onderverdeeld in een 5-klassen systeem. Dit 5-klassen systeem varieert van goedaardig (benign, klasse 1) tot pathogeen (pathogenic, klasse 5). Bij voldoende bewijslast voor een biologisch ongunstig effect (op basis van literatuur, familie onderzoek, biologische eigenschappen, et cetera) wordt de variant beoordeeld als waarschijnlijk pathogeen (klasse 4) of pathogeen (klasse 5).
Voor klasse 5 varianten geldt dat pathogeniciteit bewezen is. Dit zijn veelal varianten die meerdere keren beschreven zijn in meerdere afzonderlijke patienten, en/of functioneel getest zijn of die resulteren in het verlies van het eiwit waarbij dit verlies een bekend mechanisme is voor dat betreffende ziektebeeld. Voor klasse 4 varianten wordt aangehouden dat het zeer waarschijnlijk is dat de variant pathogeen is. Dit kunnen bijvoorbeeld varianten zijn die wel eerder beschreven zijn maar in een enkele of beperkt aantal patiënten, varianten die in mutatie hotspot gebieden liggen en qua voorspelling een ongunstig effect lijken te hebben of varianten die niet eerder zijn gevonden, maar op basis van kennis van andere varianten in het zelfde gen, een voorspeld gelijkwaardig effect zullen hebben. Aangezien de PDO resultaten leiden tot mogelijk ingrijpende reproductieve keuzes voor of tijdens de zwangerschap is er algemene consensus dat alleen klasse 4 en 5 varianten worden gemeld in de rapportage. Varianten waarvan de betekenis op basis van huidige kennis onduidelijk is (VUS, klasse 3 of lager) worden niet teruggekoppeld en functionele opvolging van dergelijke varianten valt niet binnen de scope van deze richtlijn.
De beoordeling van deze varianten en de beslissing welke gerapporteerd zullen worden in de uitslag is de verantwoordelijkheid van de laboratoriumspecialisten klinische genetica en de betrokken klinisch genetici. PDO is daarom ingebed in daarvoor ISO15189 geaccrediteerde laboratoria binnen de afdelingen Klinisch Genetica.
Interpretatie van nevenbevindingen
De Gezondheidsraad geeft als definitie voor nevenbevindingen: bevindingen die bij toeval worden gedaan en die los staan van de hulpvraag of de klachten waarmee de patiënt bij de dokter kwam en op grond waarvan de dokter tot diagnostisch onderzoek besloot (ook genaamd “bijvangst” of “incidental finding”) (Gezondheidsraad, 2014). Definitie van de VKGN/VKGL handreiking (in voorbereiding) is: pathogene varianten (klasse 4 en 5) in ziekte-veroorzakende genen die niet in relatie staan tot datgene waarvoor de test is aangevraagd.
In het kader van PDO wordt met nevenbevindingen bedoeld: (waarschijnlijk) pathogene DNA varianten in één van de onderzochte genen waar de dragerschapstest niet direct op gericht was, maar die niet afgeschermd konden worden bij de analyse van de testresultaten en die wel medische relevantie hebben of kunnen hebben voor de geteste persoon of haar/zijn nageslacht of overige familie. Het kan hier gaan om varianten die bij het (toekomstig) nageslacht een mildere klinische expressie (kunnen) hebben dan in deze richtlijn gedefinieerd als aandoeningen/genen/varianten die in een dragerschapstest horen. Ook kan het gaan om het aantonen van twee pathogene varianten bij een geteste persoon, waardoor deze persoon zelf een autosomaal recessieve aandoening blijkt te hebben (of kan krijgen). Het kan tevens gaan om (waarschijnlijk) pathogene varianten in genen die zowel een autosomaal dominante als autosomaal recessieve overerving kennen, waardoor ook een aanleg voor een autosomaal dominante aandoening ontdekt kan worden bij degene die getest wordt.
Afhankelijk van de gebruikte PDO tests is er meer of minder kans op nevenbevindingen (zie het Normatieve kader voor definitie nevenbevinding en uitleg kansen). In het algemeen geldt dat bij een breder panel de kans op nevenbevindingen groter wordt. In eerste instantie strekt het tot aanbeveling die test te gebruiken die voor een paar het meest voor de hand ligt, afhankelijk van de hoogrisicogroep waartoe zij behoren en het risico op nevenbevindingen hierbij in de counseling mee te nemen. Voor consanguine paren zal de test een bredere test (WES-gebaseerd) zijn, omdat er een verhoogde kans op gezamenlijk dragerschap voor één van de honderden zeer zeldzame aandoeningen is (zie de module ‘Definitie van hoogrisicogroepen’). Hierdoor zal de kans op nevenbevindingen groter zijn dan bij de kleinere panels. Echter, het detecteren van een nevenbevinding betekent niet vanzelfsprekend dat deze in een uitslag gerapporteerd moet worden. Zo kan bijvoorbeeld afgesproken worden met de bij de aandoening betrokken specialist dat varianten in het CFTR gen die leiden tot congenital bilateral absence of vas deferens (CBAVD) en niet tot cystic fibrosis niet gemeld worden in het rapport (Casals, 2000).
Het verdient aanbeveling hiervoor een lokale multidisciplinaire commissie in te stellen met als taak het beleid te helpen bepalen en te adviseren bij soms lastige besluitvorming in concrete gevallen. In deze commissie is bij voorkeur expertise aanwezig op de terreinen van de klinisch genetische laboratoriumdiagnostiek, de klinische genetica, het voor de uitslag relevante specialisme, alsmede het gezondheidsrecht en de medische ethiek, mogelijk aangevuld met een patiëntvertegenwoordiger (zie het Normatieve Kader).
3. Aanbevelingen per hoogrisicogroep
3a. Etnische of geografische afkomst
Wanneer een preconceptietest wordt uitgevoerd op basis van afkomst, is het van belang dat de aandoeningen waar een verhoogd risico op bestaan, zich in de test bevinden (zie het stroomschema bij de aanverwante producten). Voor deze paren bestaat in de context van deze richtlijn de voorkeur voor een beperkte, hoog sensitieve en specifieke test met alleen of in elk geval die aandoeningen erin waar een verhoogd risico op bestaat. Een discussie kan ontstaan wanneer een koppel tot meer dan één hoogrisicogroep behoort, bijvoorbeeld een consanguin paar van Ashkenazi-Joodse afkomst. Het is raadzaam in dergelijke situaties met het koppel te overleggen wat de voorkeur dient en wat de voor- en nadelen van elk van de tests zijn.
Paren afkomstig uit endemische malaria gebieden: hoogrisicogroep hemoglobinopathieën
Voor personen die een verhoogd risico hebben op dragerschap van hemoglobinopathieën, hoeft niet in eerste instantie een dragerschapstest middels DNA onderzoek te worden aangevraagd (Traeger-Synodinos, 2015). Dragers van sikkelcelziekte en bèta-thalassemie zijn eenvoudig op te sporen middels een Hb typering (HPLC test). Dragers van alfa-thalassemie hebben, net als dragers van bèta-thalassemie, meestal een microcytair (laag MCV), hypochroom (laag MCH) bloedbeeld, welke vaak, maar niet altijd, gepaard gaat met een milde anemie (laag Hb) (KNOV, 2010).
Een niet op DNA-onderzoek gebaseerde biochemische test voor het opsporen van hemoglobinopathie dragerschap omvat de volgende bepalingen: Hb, Ht, MCV, MCH, MCHC, erytrocyten, ferritine en ijzer, en een Hb typering (ook wel HPLC of Hb electrophorese genoemd). Deze testen kunnen worden aangevraagd door alle zorgverleners in de 1e, 2e en 3e lijn.
Aan de hand van de uitkomsten van deze laboratoriumuitslagen kan worden geconcludeerd of er sprake is van dragerschap van sikkelcelziekte of bèta-thalassemie (bij afwijkende Hb typering) en/of het waarschijnlijk is of iemand drager is van alfa-thalassemie (bij microcytaire hypochrome anemie zonder aanwijzingen voor ijzergebrek).
Bij een positieve (afwijkende) uitslag heeft de partner een indicatie om ook te worden onderzocht om na te gaan of er sprake is van een dragerpaar met verhoogd risico op het krijgen van een kind met een hemoglobinopathie.
Indien bij beide partners een afwijkende uitslag wordt aangetoond, dient in tweede instantie DNA diagnostiek te worden aangevraagd, als het paar overweegt gebruik te maken van prenatale diagnostiek (PND) of pre-implantatie genetische diagnostiek (PGD).
Als de partners echter afzien van PND of PGD is een bevestiging middels DNA diagnostiek niet noodzakelijk.
Voor meer uitgebreide informatie met betrekking tot de diagnostiek naar hemoglobinopathie dragerschap wordt verwezen naar Het rapport van de werkgroep hemoglobinopathieën van de Vereniging Hematologische Laboratoriumdiagnostiek (2017).
Kort samengevat is het stappenplan van de diagnostiek naar dragerschap van hemoglobinopathie als volgt:
- Hematologische parameters (Hb, Ht, MCV, MCH, MCHC, erytrocyten) , ijzerstatus ( ferritine en ijzer) én Hb typering ( HPLC).
- Indien microcytair (hypochroom) bloedbeeld, zonder ijzer deficiëntie, en zonder aanwijzingen voor dragerschap bèta-thalassemie: DNA onderzoek alfa thalassemie inzetten.
- Bij afwijkende uitslagen passend bij dragerschap van sikkelcelziekte, bèta-thalassemie of andere Hb varianten, is uitgebreid vervolgonderzoek met DNA diagnostiek alleen geïndiceerd indien er een wens is voor PGD of prenatale diagnostiek. Hiervoor dient het paar (of individu) verwezen te worden naar een afdeling Klinische Genetica in Nederland.
De individuele uitslagen worden door het referentielaboratorium geïnterpreteerd en kunnen worden nagelezen in het werkboek van de NHS Sickle Cell and Thalassemia werkgroep (NHS, 2012). Zie ook www.hbpinfo.com.
Let op: Het is belangrijk te weten dat de combinatie van dragerschap van sikkelcelziekte (HbS) met dragerschap bèta-thalassmie ook een verhoogd risico op een klinisch ernstig fenotype geeft. Zowel dragerschap van sikkelcelziekte als dragerschap van bèta-thalassemie wordt veroorzaakt door een pathogene genvariant in het HBB gen. Als de ene partner drager is van sikkelcelziekte en de andere partner drager is van bèta-thalassemie, geeft dit wel 25% kans op het krijgen van een kind met een ernstige vorm van hemoglobinopathie (gelijkend op sikkelcelziekte). Zie ook tabel 1.
Tabel 1 Risicocombinaties bij dragers van α en β defecten
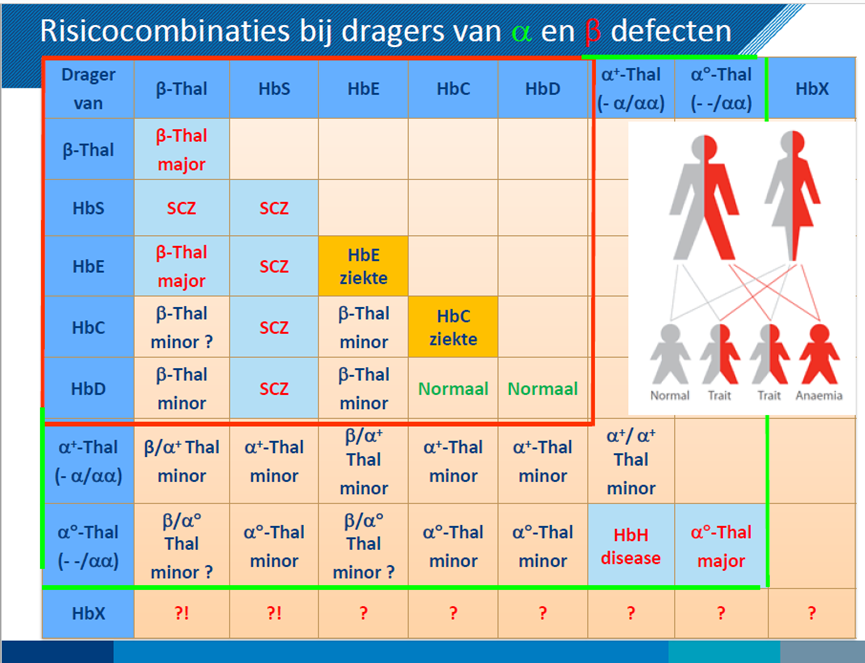
Bron: Dr. C.L. Harteveld, LUMC
Hoogrisicogroep Ashkenazi-Joodse gemeenschap
Paren met kinderwens, waarvan beide partners of één van beide partners van oorspronkelijk Ashkenazi-Joodse afkomst zijn, kunnen worden verwezen naar een afdeling Klinische Genetica in Nederland. Afhankelijk van de familieanamnese en afkomst, kunnen verschillende tests worden overwogen.
Personen die oorspronkelijk afkomstig zijn uit de Ashkenazi-Joodse gemeenschap, hebben een hogere kans op dragerschap van een aantal (ernstige) autosomaal recessieve ziekten: de ziekte van Tay-Sachs, familiaire dysautonomie (Riley-Day syndroom), de ziekte van Canavan, Bloom syndroom, Fanconi anemie type C, glycogeenstapelingsziekte type 1a, Maple Syrup Urine Disease, mucolipidose type 4, Niemann-Pick type A, nemaline myopathie, Joubert syndroom type 2, cystic fibrosis en Gaucher type 1 (Scott, 2010; ACOG, 2017a). Deze ziekten zijn, op de ziekte van Gaucher type 1 na, zijn dan ook standaard opgenomen in de beschikbare dragerschapstesten in Nederland voor paren (of individuen) van Ashkenazi-Joodse afkomst. Men kan zich optioneel laten testen op dragerschap van de ziekte Gaucher type 1. Deze aandoening is niet standaard toegevoegd aan de specifieke dragerschapstest voor de Ashkenazi-Joodse populatie, omdat deze aandoening milder is qua ernst en hiermee niet voldoet aan de inclusie criteria voor het opnemen van een aandoening in een preconceptiedragerschapstest.
Hoogrisicogroep genetisch geïsoleerde gemeenschappen
Paren met een kinderwens, waarvan beide partners of één van beide partners afkomstig zijn uit een genetisch geïsoleerde gemeenschap met een verhoogde kans op dragerschap van een specifieke autosomaal recessieve aandoening, kunnen worden verwezen naar een afdeling Klinische Genetica in Nederland. Afhankelijk van familieanamnese en afkomst, kunnen verschillende tests worden overwogen. De beschikbare testen verschillen ten aanzien van het aantal aandoeningen en de wijze waarop de uitslagen worden gegenereerd en gerapporteerd (individuele uitslagen versus parenuitslagen).
In Volendam is destijds door de afdeling Klinische Genetica van Amsterdam UMC (AMC en VUmc) samen met de Verloskundigenpraktijk Waterland Oost een spreekuur opgericht voor mensen met een kinderwens (https://verloskundigenwaterlandoost.nl/). Hier kunnen paren met een kinderwens uit Volendam terecht voor vragen en erfelijkheidsvoorlichting. Paren afkomstig uit Volendam kunnen naar dit spreekuur verwezen worden, maar ook, indien gewenst naar iedere andere afdeling Klinische Genetica in Nederland. De tests worden aangevraagd door of onder supervisie van een klinisch geneticus.
3b. Consanguine paren
Consanguine paren hebben een hoger risico om een dragerpaar te zijn van één of meerdere autosomaal recessieve aandoeningen ten opzichte het algemene bevolkingsrisico (zie de module ‘Definitie van hoogrisicogroepen’). Zij zijn vaker een dragerpaar van zéér zeldzame autosomaal recessieve aandoeningen die bij niet-consanguine paren vrijwel nooit voor komen. Een benadering met een te beperkt genpanel is, met het oog op het mogelijk missen van een gezamenlijk dragerschap van een (zeer) zeldzame ernstige autosomaal recessieve aandoening, derhalve voor consanguine paren minder geschikt (Sallevelt, 2017). Een brede dragerschapstest verdient daarom de voorkeur, waarbij bij de rapportage van de uitslagen rekening gehouden dient te worden met de hierboven genoemde inclusiecriteria van de richtlijnkerngroep. Hiermee wordt bedoeld dat het aanbod van PDO primair gericht is op het aantonen van dragerschap van ernstige, op de kinderleeftijd optredende en niet goed behandelbare aandoeningen. Het beschikbaar zijn van deskundigen voor een multidisciplinaire commissievoor eventuele toetsing van gevonden varianten/genen is aan te raden.
In deze commissie is bij voorkeur expertise aanwezig op de terreinen van de klinisch genetische laboratoriumdiagnostiek, de klinische genetica, het voor de uitslag relevante specialisme, alsmede het gezondheidsrecht en de medische ethiek, mogelijk aangevuld met een patiëntvertegenwoordiger (zie het Normatieve Kader).
Algemeen (zowel geldend voor a als voor b)
Uiteraard is elk centrum dat (een) preconceptietest(s) voor (een) hoogrisicogroep(en) aanbiedt, verantwoordelijk voor de inhoud en het updaten van de inhoud hiervan. Bij voorkeur is er voor elke specifieke hoogrisicogroep één zelfde test beschikbaar in het land. Wanneer een centrum de intentie heeft een nieuwe of reeds bestaande PDO test aan te gaan bieden, is de afspraak dit vooraf in de WPCS te bespreken en te overleggen om uniformiteit te bewaken.
De PDO tests zouden zo moeten worden samengesteld dat er een maximale klinische toepasbaarheid is met minimale belasting van de adviesvrager, laboratorium en medisch specialist, tegen zo laag mogelijke kosten (Stevens, 2017).
Onderbouwing
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen uit de 1e, 2e en 3e lijn die betrokken zijn bij de medische zorg voor patiënten/paren in de fertiele leeftijd en/of hun nageslacht uit de hiervoor omschreven hoogrisicogroepen. Daarnaast is de richtlijn geschreven voor laboratoriumspecialisten klinische genetica van de klinisch genetische laboratoria die zich bezighouden met het inrichten en uitvoeren van een (preconceptie) dragerschapstest voor hoogrisicogroepen. Hieronder volgen de criteria ten aanzien van de aandoeningen die worden opgenomen in de dragerschapstests voor de verschillende risicogroepen, alsmede de aanbevelingen welke test voor welke hoogrisicogroep het meest geschikt is en dan ook de voorkeur heeft.
Voor paren uit een aantal bekende hoogrisicogroepen (zie de module ‘Definitie van hoogrisicogroepen’) wordt al preconceptie dragerschapsonderzoek (PDO) aangeboden. Hoewel er brede consensus lijkt te zijn ontbreken echter (landelijke) afspraken over inhoud, uitvoering en aanbod.
Er werd geen literatuuronderzoek uitgevoerd naar uitgangsvragen in deze module omdat de criteria afhankelijk zijn van de test-eigenschappen (resultaten uitgangsvraag 2a van deze module) en klinische overwegingen. Deze zijn hieronder beschreven.
Er is wel een literatuur search verricht naar diagnostische accuratesse van Whole Exome Sequencing (WES) analyse ten opzichte van analyse met een beperkt panel om inzicht te krijgen in de sensitiviteit en specificiteit van de gebruikte dragerschaptests. De Sangermethode is hier als referentiestandaard aangehouden.
P (Patiënten): de laboratoriumtesten die gebruikt worden om op (gezamenlijk) dragerschap van recessieve aandoeningen te screenen;
I (Interventie): whole exome sequencing;
C (comparison): targeted panel sequencing;
R (referentiestandaard): sanger Sequencen gekozen;
O (Outcome): variant class III/IV/V;VUS (Variant of Unknown Significance) (likely) pathogenic variant(s), variant class 3,4,5; targeted enrichment, NGS coverage, unsolicited findings, late onset disease, sensitivity, specificity.
Relevante uitkomstmaten
Binnen de kerngroep werd de gevoeligheid van de techniek (sensitiviteit, specificiteit) als belangrijke uitkomstmaat beschouwd. Idealiter geldt: alle bekende (waarschijnlijk) pathogene varianten (klasse 4 en 5) worden gevonden en er zijn geen fout positieve resultaten.
Doel van deze search was de technische verschillen en verschillen in uitkomst van de gebruikte technieken in kaart te brengen om zo een zo goed mogelijk advies te kunnen geven over de te adviseren test per hoogrisicogroep.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID), Embase (via Embase.com) is op 19 april 2019 met relevante zoektermen gezocht naar vergelijkend onderzoek naar whole exome sequencing en targeted panel sequencing met Sanger sequencing als gouden standaard. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 511 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: diagnostisch onderzoek waarbij WES werd vergeleken met targeted panel sequencing met Sanger sequencing als referentiestandaard. Op basis van titel en abstract werden in eerste instantie 22 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 22 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), en 0 studies definitief geselecteerd.
Er zijn geen studies opgenomen in de literatuuranalyse.
- ACOG (2017a). ACOG Committee Opinion No. 690. Carrier screening in the age of genomic medicine. Obstet Gynecol. 2017 Mar;129(3):e35-e40.
- Casals T (2000). Heterogeneity for mutations in the CFTR gene and clinical correlation in patients with congenital absence of the vas deferens. Hum Reprod, 15(7):1476-83.
- Henneman L, Borry P, Chokoshvili D, et al. (2016). Responsible implementation of expanded carrier screening. Eur J Hum Genet; 24(6): e1-e12.
- Korngiebel, D.M., McMullen C.K., Amendola L.M. (2016). Generating a taxonomy for genetic conditions relevant to reproductive planning. Am J Med Genet A. 170(3): 565-573.
- KNOV (2010). Anemie in de verloskundige praktijk. Website bezocht 1-9-2019: https://www.knov.nl/serve/file/knov.nl/knov_downloads/669/file/KNOV-Standaard%20Anemie%20in%20de%20verloskundige%20praktijk.pdf
- Lazarin G (2014). Systematic classification of disease severity for evaluation of expanded carrier screening panels. Plos One, 9(12):e114391.
- NHS Sickle Cell and Thalassaemia Screening Programme (2012). ISBN 13: 978-0-9565846-8-7.
- Ogino S. (2002). SMN dosage analysis and risk assessment for spinal muscular atrophy. Am J Hum Genet, 70(6):1596-8.
- Richards S (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetic in Medicine 17(5):405-24.
- Sallevelt S (2017). A comprehensive strategy for exome-based preconception carrier screening. Genet Med, 19(5):583-592.
- Scott SA, Edelmann L, Liu L et al. (2010). Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum Mutat; 31: 1240–1250.
- Stevens B (2017). Finding middle ground in constructing a clinically useful expanded carrier screening panel. Obstet Gynecol, 130(2):279-284.
- Thomas E (2005). Two pragmatic trials of treatment for shoulder disorders in primary care: generalisability, course, and prognostic indicators. Annals of the rheumatic diseases, 64(7), 1056-1061.
- Traeger-Synodinos J, Harteveld CL, Old JM et al. (2015). EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur J Hum Genet; 23, 426–437.
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
diSibio, 2017 |
Voldoet niet aan PICO: geen vergelijking met Sanger |
|
Sheppard, 2018 |
Voldoet niet aan PICO: geen vergelijking met Sanger, ES ipv WES |
|
Dillon, 2018 |
Voldoet niet aan PICO: geen vergelijking |
|
LaDuca, 2017 |
Voldoet niet aan PICO: verkeerde vergelijking (NGS versus WES) |
|
Fujita, 2017 |
Voldoet niet aan PICO: verkeerde vergelijking (NGS versus SNV's ) |
|
Cirino, 2017 |
Voldoet niet aan PICO: geen vergelijking van WES en panel met Sanger |
|
Chang, 2017 |
WGS versus targeted NGS ne Sanger als R gebruikt. Ik mis echter de P: er wordt geen dragerschap opgespoord voor recessieve aandoeningen. |
|
Sun, 2015 |
Voldoet niet aan PICO: verkeerde vergelijking (WGS versus WES) |
|
Pereira, 2015 |
Voldoet niet aan PICO: verkeerde vergelijking (Sanger versus WES) |
|
Guo, 2015 |
Voldoet niet aan PICO: verkeerde vergelijking (Sanger versus WES) |
|
Wang, 2018 |
WES versus targeted enrichment NGS, maar geen Sanger gebruikt als R. als ik het goed lees. Er werden enkele patienten die Sanger kregen naast WES/NGS en het lijkt erop dat die varianten met Sanger zijn opgepikt en niet met WES/NGS. |
|
Montaut, 2018 |
Voldoet niet aan PICO: geen vergelijking met Sanger |
|
Saudi Mendeliome Group, 2015 |
Er werd geen dragerschap van AR aandoeningen van gezonde individuen opgespoord, dus er werd niet voldaan aan P. |
|
Al-Herz, 2018 |
Voldoet niet aan PICO: geen vergelijking |
|
Haer-Wigman, 2017 |
Voldoet niet aan PICO: niet gekeken naar diagnostische accuratesse |
|
Balicza, 2016 |
Voldoet niet aan PICO: geen vergelijking |
|
Comander, 2017 |
Voldoet niet aan PICO: alleen panel based vergeleken met Sanger |
|
Keser, 2017 |
Voldoet niet aan PICO: geen vergelijking |
|
Consugar, 2015 |
NGS met targeted enrichment vergeleken met WES. Echter er wordt niet gezocht naar recessieve aandoeningen, dus voldoet niet aan P. en geen Sanger gebruikt als referentie |
|
Monies, 2016 |
Voldoet niet aan PICO: geen vergelijking |
|
Bauer, 2019 |
Voldoet niet aan PICO: geen panel |
|
Palmer, 2018 |
trio ES (panel) werd vergeleken met Sanger, voldoet niet aan P, geen vergelijking om (gezamenlijk) dragerschap op te sporen |
Beoordelingsdatum en geldigheid
Publicatiedatum : 25-06-2020
Beoordeeld op geldigheid : 02-03-2020
Voor het beoordelen van de actualiteit van deze richtlijn is de richtlijnkerngroep niet in stand gehouden. Uiterlijk in 2025 bepaalt het bestuur van de VKGN of de modules van deze richtlijn nog actueel zijn. Op modulair niveau is een onderhoudsplan geschreven. Bij het opstellen van de richtlijn heeft de kerngroep per module een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten. In een artikel van Burke (2019) wordt uiteengezet dat een evidence-based onderbouwing vaak erg lastig is voor genetische geneeskunde gezien de snelle veranderingen in genpanels en tests. Een andere aanpak voor het schrijven van evidence-based richtlijnmodules is daarom om de CPAD methode te volgen (Clinical practice advisory document) zoals beschreven in het artikel van Burke (2019). De kerngroep adviseert deze methode als uitgangspunt te nemen bij een herziening of aanvulling van deze richtlijn. Voor de ontwikkeling van deze richtlijn (gestart in januari 2018) kon deze methode niet als uitgangspunt genomen worden vanwege de verschijningsdatum.
De VKGN is regiehouder van deze richtlijn en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
|
Module[1] |
Regie-houder(s)[2] |
Jaar van autorisatie |
Eerst-volgende beoordeling actualiteit richtlijn[3] |
Frequentie van beoordeling op actualiteit[4] |
Wie houdt er toezicht op actualiteit[5] |
Relevante factoren voor wijzigingen in aanbeveling[6] |
|
Tests per indicatie, criteria |
VKGN |
2020 |
2025 |
Elke 5 jaar |
VKGN |
Niet van toepassing |
[1] Naam van de module
[2] Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders)
[3] Maximaal na vijf jaar
[4] (half)Jaarlijks, eens in twee jaar, eens in vijf jaar
[5] regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) kerngroep die in stand blijft
[6] Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen
Algemene gegevens
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijn.
Doel en doelgroep
Doel
De richtlijn zal handvatten bieden voor de medische indicatiestelling en dus het, al dan niet actief, aanbieden van en verwijzen voor preconceptie dragerschapsonderzoek (PDO) aan hoogrisicogroepen. Dit stelt paren met kinderwens uit deze hoogrisicogroepen in staat om autonome reproductieve beslissingen te nemen bij voorkeur voorafgaande aan een zwangerschap (zie reproductieve opties in de Algemene inleiding).
Doelgroep
Deze richtlijn gaat over preconceptie dragerschapsonderzoek (PDO) voor hoogrisicogroepen. Met hoogrisicogroepen wordt bedoeld: paren (en individuen) die op basis van etniciteit of geografische afkomst en/of consanguïniteit een hogere kans hebben om een drager of dragerpaar te zijn van één of meerdere (specifieke) autosomaal recessieve aandoeningen ten opzichte van het algemene populatierisico van de gehele bevolking om drager of dragerpaar te zijn van die aandoening(en).
Beoogde gebruikers van de richtlijn
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen uit de 1e, 2e en 3e lijn die betrokken zijn bij de medische zorg voor patiënten/paren in de reproductieve leeftijd en/of hun nageslacht, uit de hiervoor omschreven hoogrisicogroepen. Daarnaast is de richtlijn geschreven voor klinisch moleculair genetici en hun collega’s werkzaam in de laboratoria voor genoomdiagnostiek van de afdelingen Genetica van de UMC’s die zich bezighouden met het inrichten en uitvoeren van (preconceptie) dragerschapstests voor hoogrisicogroepen (zie de module ‘Criteria en tests per hoogrisicogroep’).
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2018 een multidisciplinaire richtlijnkerngroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de samenstelling van de kerngroep en de klankbordgroep).
De richtlijnkerngroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname. De richtlijnkerngroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
De klankbordgroep (onder meer bestaande uit afgevaardigden van de VKGN, VKGL, KNOV, NVOG, NVK, VvF, VSOP, en NHG) heeft de conceptteksten gelezen en hier feedback op gegeven vóórdat de richtlijn in commentaar is gegaan. De richtlijnkerngroep heeft de feedback vervolgens doorgenomen en indien nodig aanpassingen gedaan. De richtlijnkerngroep bepaalt de inhoud van de versie die aan externe partijen wordt voorgelegd tijdens de commentaarfase.
Kerngroep
- Prof. dr. I.M. (Irene) van Langen, klinisch geneticus & hoogleraar Klinische Genetica, werkzaam in het UMCG, Groningen, VKGN (voorzitter)
- Dr. P. (Phillis) Lakeman, klinisch geneticus werkzaam in het Amsterdam UMC, locatie AMC, Amsterdam, VKGN (vice-voorzitter)
- Dr. L. (Lidewij) Henneman, senior onderzoeker en universitair hoofddocent afdeling Klinische Genetica, werkzaam in het Amsterdam UMC, locatie VUmc, Amsterdam
- Dr. Ir. A.D.C. (Aimee) Paulussen, laboratoriumspecialist klinische genetica, werkzaam in het Maastricht UMC+, Maastricht, VKGL
- Prof. dr. W.J. (Wybo) Dondorp, Socrates hoogleraar Humanisme & ethiek van reproductieve genetica; universitair hoofddocent Biomedische ethiek, afdeling Health, Ethics & Society, Universiteit Maastricht
- Dr. S. (Sanne) van der Hout, assistant professor Biomedische ethiek, afdeling Health, Ethics & Society, Universiteit Maastricht
- Dr. I. (Ilse) Feenstra, klinisch geneticus, werkzaam in het Radboudumc, Nijmegen, VKGN
- Dr. S.C.E.H. (Suzanne) Sallevelt, klinisch geneticus, werkzaam in het Maastricht UMC+, Maastricht, VKGN
- Drs. J.J.T. (Jeske) van Harssel, klinisch geneticus, werkzaam in het UMC Utrecht, Utrecht, VKGN
- Drs. K.E. (Kyra) Stuurman, klinisch geneticus, werkzaam in het Erasmus MC, Rotterdam, VKGN
- Dr. N.S. (Nicolette) den Hollander, klinisch geneticus, werkzaam in het LUMC, Leiden, VKGN
- Dr. S. (Silvana) van Koningsbruggen, laboratoriumspecialist klinische genetica, werkzaam in Amsterdam UMC, locatie AMC, Amsterdam, VKGL
- Dr. T. (Trijnie) Dijkhuizen, laboratoriumspecialist klinische genetica, werkzaam in het UMCG, Groningen, VKGL
- Drs. E. (Elsbeth) van Vliet-Lachotzki, patiëntvertegenwoordiger, Vereniging Samenwerkende Ouder- en Patiëntenorganisaties, VSOP
- Dr. M. (Mirjam) Plantinga, senior onderzoeker afdeling Genetica, werkzaam in het UMCG, Groningen
Klankbordgroep
- Dr. K. (Kristin) Abbott, laboratoriumspecialist klinische genetica, werkzaam in het UMCG, Groningen, VKGL
- Dr. H.T. (Hennie) Brüggenwirth, laboratoriumspecialist klinische genetica, werkzaam in het Erasmus MC, Rotterdam, VKGL
- Dr. K. (Karin) Huijsdens- van Amsterdam, laboratoriumspecialist klinische genetica, werkzaam in het UMCU, Utrecht, VKGL
- Dr. C.L. (Kees) Harteveld, laboratoriumspecialist klinische genetica, werkzaam in het LUMC, Leiden, VKGL
- Dr. I. (Irene) Homminga, IVF-arts, werkzaam in het UMCG, Groningen, VvF
- Dr. L.C. (Lieke) de Jong - Potjer, huisarts, werkzaam bij Huisarts, werkzaam bij Huisartsenpraktijk het Kompas , NHG
- Dr. R.J.T. (Ron) van Golde, gynaecoloog, werkzaam in het Maastricht UMC+, Maastricht, NVOG
- Dr. M.F.C.M. (Maarten) Knapen, gynaecoloog-perinatoloog, werkzaam in het Erasmus MC, Rotterdam, NVOG
- Prof. mr. J.G. (Jaap) Sijmons, hoogleraar Gezondheidsrecht, werkzaam bij het UMCU, Utrecht
- Dr. C.R. (Carsten) Lincke, kinderarts, werkzaam in het ErasmusMC, Rotterdam, NVK
- Dr. M. (Marjolein) Peters, kinderarts, voorheen werkzaam in het Amsterdam UMC, Amsterdam, NVK
- Dr. S. (Suze) Jans, wetenschapsredacteur, Utrecht; onderzoeker, afdeling Child health TNO Leiden, KNOvV
Met ondersteuning van
- E.A. (Ester) Rake, MSc, adviseur Kennisinstituut van de Federatie Medisch Specialisten
- M. (Margriet) Moret-Hartman, senior adviseur Kennisinstituut van de Federatie Medisch Specialisten
- M. (Marjolein) de Weerd, senior adviseur Kennisinstituut van de Federatie Medisch Specialisten (tot 1 augustus 2019)
Met dank aan
- Dr. M.E. (Marleen) Jansen, postdoc onderzoeker afdeling Klinische Genetica, werkzaam in het Amsterdam UMC, locatie VUmc, Amsterdam
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle kerngroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van de kerngroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de federatie Medisch Specialisten.
|
Kerngroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Dijkhuizen |
Laboratoriumspecialist Klinische Genetica, UMCG |
Lid WPCS VKGN/VKGL (onbetaald) |
Betrokken bij aanbod dragerschapstesten via instelling. |
Geen |
|
Dondorp |
Bijzonder hoogleraar Humanisme & ethiek van reproductieve genetica (Stichting Socrates – Humanistisch Verbond); Universitair hoofddocent Faculteit Health, Medicine & Life Sciences, afd Health, Ethics & Society, Universiteit Maastricht, 0,8 fte |
Lid WPCS VKGN/VKGL (onbetaald)
|
Was co-auteur van position document over de verantwoorde introductie van preconceptionele dragerschapsscreening: Henneman L et al. Responsible implementation of expanded carrier screening. Eur J Hum Genet. 2016;24(6):e1-e12.
Was secretaris van Gezondheidsraadcommissie 'Screening en de rol van de overheid'. Schreef in die hoedanigheid het advies 'Screening: tussen hoop en hype' (GR 2008).
Was lid van Gezondheidsraadcommissie Prenatale screening (advies in 2016).
Als ethicus betrokken bij onderzoek naar verantwoord (toekomstig) aanbod van dragerschapstest/screening in het MUMC, Maastricht
Was medeaanvrager van het door ZonMw gehonoreerde (inmiddels afgeronde) POM-project (preconceptionele dragerschapsscreening op maat). |
Geen |
|
Feenstra |
Klinisch geneticus, Radboudumc |
Lid WPCS VKGN/VKGL (onbetaald) -Voorzitter werkgroep prenatale genetica (onbetaald) -Lid Raad van Advies stichting prenatale screening Nijmegen (vacatie) |
Geen |
Geen |
|
Harssel |
Klinisch Geneticus, UMC Utrecht |
Lid WPCS VKGN/VKGL (onbetaald) -Lid werkgroep prenatale genetica (onbetaald) -Lid commissie Kwaliteit VKGN (onbetaald) |
Geen |
Geen |
|
Henneman |
Senior onderzoeker (Associate professor) en Universitair Hoofddocent, afdeling Klinische Genetica VUmc (vakgebied community genetics en public health genomics) |
Voorzitter wetenschappelijke vereniging Nederlandse Associatie voor Community Genetics en Public Health Genomics (NACGG) (onbetaald). -Lid bestuur Nederlandse Vereniging voor Humane Genetica (NVHG) (onbetaald) -Lid Forum Biotechnologie en Genetica (FBG), (onbetaald). -Secretaris Stichting Preconceptiezorg Nederland, (onbetaald). -Lid WPCS VKGN/VKGL (onbetaald) -Lid Kerngroep Niet Invasieve Prenatale Testen (NIPT) Consortium, (onbetaald -Website redactielid: www.huisartsengenetica.nl (onbetaald). - Lid Gezondheidsraad commissie Screening rond zwangerschap en geboorte (advies screening op niet-behandelbare aandoeningen) (vacatie). |
Hoofdaanvrager ZonMw- gefinancierde onderzoeksprojecten:- POM studie (Preconceptionele dragerschapscreening Op Maat); project rond de ontwikkeling en verdere implementatie van preconceptionele dragerschapscreening en opzet website www.benikdrager.nl (i.s.m. Erfocentrum) (afgerond, looptijd 2015-2017).
ZonMw VIMP (Verspreidings- en lmplementatieplan) Preconceptie dragerschapsscreening Op Maat (POM) (looptijd 2018-2019). Werkzaam en verbonden aan een instelling (Amsterdam UMC) dat preconceptie dragerschapstesten aanbiedt (zonder winstoogmerk) www.dragerschapstesten.nl.
Betrokken (geweest) als onderzoeker bij een (onbetaald onderzoeksproject) naar de evaluatie van een kinderwensspreekuur in een genetisch geïsoleerde populatie, het aanbod via www.dragerschapstest.nl en (ZonMw gefinancieerd) www.vumc.nl/CFtest.
Was eerste auteur van het PPPC European Society of Human Genetics position document over de verantwoorde introductie van preconceptionele dragerschapsscreening: Henneman L et al. Responsible implementation of expanded carrier screening. Eur J Hum Genet. 2016;24(6):e1-e12.
Was lid van Gezondheidsraadcommissie Prenatale screening (advies in 2016). |
Geen |
|
Van Koningsbruggen |
Laboratorium specialist klinische genetica, AMC |
Lid WPCS VKGN/VKGL (onbetaald) |
Betrokken bij aanbod dragerschapstesten via instelling. |
Geen |
|
Lakeman |
Klinisch geneticus te AMC Amsterdam |
Lid WPCS VKGN/VKGL (onbetaald) -Lid werkgroep PIL (vacatie) |
Sinds mei 2016 biedt Amsterdam UMC, locatie AMC een preconceptie dragerschaptest aan voor paren met kinderwens aan hoogrisicogroepen en aan koppels zonder a priori verhoogd risico (www.dragerschapstesten.nl). Door de richtlijn zal meer aandacht komen voor deze en ander in NL beschikbare testen. Hierdoor zal er meer aanndacht zijn voor de wetenschappelijke studies over het aanbod van deze test.
Was co-auteur van onder andere het PPPC ESHC position document over de verantwoorde introductie van preconceptionele dragerschapsscreening: Henneman L et al. Responsible implementation of expanded carrier screening. Eur J Hum Genet. 2016 ;24(6):e1-e12.
Was medeaanvrager van het door ZonMw gehonoreerde (inmiddels afgeronde) POM-project (preconceptionele dragerschapsscreening op maat). |
Geen |
|
Den Hollander |
Klinisch geneticus LUMC 80% |
geen (Commissie kwaliteit VKGN onbetaald) lid |
Betrokken bij aanbod dragerschapstesten via instelling. |
Geen |
|
Paulussen |
Laboratorium Specialist Klinische Genetica (LSKG) MUMC+ |
Bestuurslid Nederlandse Vereniging Humane Genetica (NVHG, penningmeester) (onbetaald), Lid Registratie Commissie VKGL (onbetaald), Lid WPCS VKGN/VKGL (onbetaald) |
Klinisch 'kartrekker' van de dragerschapstest die op dit moment geïmplementeerd is (niet-commercieel) door de afdeling klinische genetica van het MUMC+ |
Geen |
|
Plantinga |
senior onderzoeker afdeling genetica UMCG (0,6 FTE) programma manager leidende coalitie patientenparticipatie UMCG (0,3 FTE) |
Visiting research fellow CELS Southampton (onbetaald) Secretaris landelijke PCS werkgroep (onbetaald) Secretaris NACGG (onbetaald) |
Betrokken bij UMCG onderzoek naar implementatie van dragerschapsscreening via de huisarts |
Geen |
|
Sallevelt |
klinisch geneticus MUMC+ |
Lid WPCS VKGN/VKGL (onbetaald) |
Klinisch 'kartrekker' van de PCS test die op dit moment geimplementeerd is (niet-commercieel) door de afdeling klinische genetica van het MUMC+ |
Geen |
|
Stuurman |
klinisch geneticus Erasmus MC |
Geen |
Geen |
Geen |
|
Van Langen |
Hoogleraar Klinische Genetica, UMCG, Groningen |
Lid van de vaste commissie bevolkingsonderzoek Gezondheidsraad. (vacatie) - Lid WPCS VKGN/VKGL (onbetaald) |
In het UMCG behoren wij tot de voorlopers met betrekking tot aanbod van en wetenschappelijk onderzoek op het gebied van (universele) dragerschapsscreeningstests. Ik heb een joint PhD (samen met collega A.Lucassen in Southampton) die de evaluatie van de pilot in 1e lijn doet (en deze pilot heeft uitgevoerd), dit is dus op ander vlak. Ook doet zij nu onderzoek naar verschillen (ethisch) naar aanbod in 1e lijn (alle paren met kinderwens) en in 2e/3e lijn binnen fertiliteitspopulatie. Tot stand komen/brengen van richtlijn zal haar/ons geen voordeel geven, voor zover ik kan inschatten. Promotie volgt in 2020. |
Geen |
|
Van Vliet |
Beleidsmedewerker VSOP patiënten perspectief ethiek, genetica en perinatale zorg. 18 uur Vanuit de VSOP gedetacheerd voor 45% bij Stichting Erfocentrum als project leider ZwangerWijzer.nl en medisch adviseur. |
Lid van de Beraadsgroep Gezondheidsraad (vacatiegeld). - Lid Programma commissie Prenatale Screening downsyndroom en SEO. Tevens lid werkgroep kwaliteit, voorlichting en onderzoek. Centrum voor bevolkingsonderzoek, RIVM, (vacatiegeld en reiskosten) - Lid werkgroep Preconceptie Indicatielijst (PIL) , College perinatale Zorg (vacatiegeld), in eindfase - Lid van het TRIDENT Consortium en TRIDENT 2 studie: onderzoek naar mening zwangeren over ervaringen NIPT in Nederland VUMC (project financiering ZONMW). - Lid begeleidingscommissie studie eerste trimester virtual reality echoscopie in de verloskundige zorg (project financiering) - Project lid Studie A loyalty program to motivate vulnerable women to engage in preconception care: ‘from voucher to tablet’”. (project financiering ZONMW) - Project lid APROPOS-II. A Stepped Wedge Cluster Randomized Controlled Trial to evaluate the effect of a locally tailored approach for Preconception Care (project financiering ZONMW) - Adviseur VIMP Preconceptionele dragerschap screening op maat (POM) (project financiering ZONMW) (afgerond) |
VSOP is een vereniging van circa 80 ouder- en patiëntenorganisaties betrokken bij zeldzame, vaak erfelijke, aangeboren aandoeningen |
Geen |
|
Van der Hout |
Universitair docent Biomedische Ethiek, Universiteit Maastricht |
Lid WPCS VKGN/VKGL (onbetaald) -Lid werkgroep Pre-implantatie Genetische Diagnostiek, MUMC Maastricht. (Onbetaald). -Lid Commissie Medisch-Ethische en -Juridische Aangelegenheden (CMEJA), MUMC Maastricht. (Onbetaald). |
Als ethicus betrokken bij onderzoek naar verantwoord (toekomstig) aanbod van dragerschapstest/screening in het MUMC, Maastricht
Co-auteur van richtlijn van de European Society of Human Embryology and Reproduction (ESHRE) over de ethiek van preconceptionele dragerschapsscreening aan paren die in aanmerking komen voor medisch geassisteerde voortplanting: De Wert et al. Ethics of Preconception Carrier Screening in Assisted Reproduction (work in progress). |
Geen |
|
Klankbord-groeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Abbott |
laboratoriumspecialist (buitengewoon), UMCG |
diagnostiek NGS analyse pipeline coördineren, plus laboratoriumspecialist taken |
Geen |
Geen |
|
Bruggenwirth |
Laboratoriumspecialist Klinische genetica |
Geen |
Geen |
Geen |
|
Harteveld |
Senior-Laboratorium Specialist Klinische Genetica LDGA, Leids Universitair Medisch |
Associate-Editor International Journal of Laboratory hematology (onbetaald) |
Geen |
Geen |
|
Homminga |
Fertiliteitsarts, UMCG |
Geen |
Geen |
Geen |
|
Huijsdens |
Laboratoriumspecialist klinische genetica, UMCU |
Geen |
Geen |
Geen |
|
Jans |
Wetenschappelijk Redacteur Tijd schrift voor verloskundigen (KNOV) 0,33 FTE Beleidsadviseur KNOV 0, 1 FTE |
Geen |
Geen |
Geen |
|
Jong-Potjer |
Vrijgevestigde huisarts te Zoetermeer (3 dagen/ week) |
Project kwetsbare zwangere in Zoetermeer, betaald vanuit subsidie gemeente |
Geen |
Geen |
|
Knapen |
gynaecoloog perinatoloog ErasmusMC, 0.6 fte |
gynaecoloog star/shl, gynaecologische consulten, betaald. |
Geen |
Geen |
|
Lincke |
kinderarts-erfelijke en aangeboren aandoeningen |
voorzitter Klinische Adviesraad en vaccincommissie Lareb (onbetaald) |
Geen |
Geen |
|
Peters |
Kinderarts-hematoloog |
Geen |
Geen |
Geen |
|
Sijmons |
partner Nysingh advocaten - notarissen NV, hoogleraar gezondheidsrecht Universiteit Utrecht (0,2) |
voorzitter voering voor gezondheidsrecht (onbetaald), raadsheer plaatsvervanger hof amsterdam (onbetaald), lid gezondheidsraad (onbetaald) lid redactie tijdschrift voor gezondeheidsrecht (onbetaald) hoofddocent Grotius academy opleiding gezondheidrecht (betaald) BV besturen binnen de zorg (onbetaald) |
Geen |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde van de patiëntenvereniging in de kerngroep (VSOP). De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Vereniging Samenwerkende Ouder- en Patiëntenorganisaties en de Patiëntenfederatie Nederland.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn (module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden bij de aanverwante producten.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van Medisch Specialisten.
In een artikel van Burke (2019) wordt uiteengezet dat een evidence-based onderbouwing vaak erg lastig is voor genetische geneeskunde gezien de snelle veranderingen in genpanels en tests. Een andere aanpak voor het schrijven van evidence-based richtlijnmodules is daarom om de CPADmethode te volgen (Clinical practice advisory document) zoals beschreven in het artikel van Burke. De kerngroep adviseert deze methode als uitgangspunt te nemen bij een herziening of aanvulling van deze richtlijn.
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de voorzitter van de kerngroep en de adviseur de knelpunten, mede op basis van 6 interviews met stakeholders. Tevens zijn er knelpunten aangedragen door de VKGL, VVF, KNOV, Stichting CZB, NVOG, VKGN, VSOP en diverse externe experts op persoonlijke titel via de invitational conference op 21 september 2018, waar ook een samenvatting van de resultaten van de interviews werd gepresenteerd. Een verslag hiervan is opgenomen onder de aanverwante producten.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de voorzitter en de adviseur concept-uitgangsvragen opgesteld. Deze zijn met de kerngroep besproken waarna de kerngroep de definitieve uitgangsvragen heeft vastgesteld. Vervolgens inventariseerde de kerngroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De kerngroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de kerngroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
De definiëring van hoogrisicogroepen (module 'Definitie van hoogrisicogroepen') is ontstaan door voortschrijdend inzicht. Er werd op 9 april 2019 oriënterend gezocht naar bestaande buitenlandse richtlijnen en systematische reviews via Medline (OVID) en de Cochrane library. Er is breed gezocht naar hoogrisicogroepen en (expanded) dragerschapsonderzoek voor recessieve aandoeningen. Hierbij is naast preconceptionele screening ook prenatale screening meegenomen, omdat mensen buiten Nederland zich vaak pas tijdens de zwangerschap op dragerschap laten testen en deze literatuur ook relevant is voor deze richtlijn. Cascadescreening (familiescreening naar aanleiding van uitkomsten van een individuele dragerschapstest of aandoening in de familie) wordt buiten beschouwing gelaten. Omdat dit een oriënterende search is, werd alleen de P (Patiënten) gedefinieerd.
- P (Patiënten): (expanded) carrier screening, universal carrier screening, ancestry based carrier screening, preconception carrier screening, reproductive carrier screening, screening for autosomal recessive disorders, screening for (severe) childhood disorders, ethnicity based screening, targeted carrier screening.
Hoe breder de bevolking (de patiëntengroep) wordt ingedeeld, hoe meer mensen uit de bevolking kunnen worden gerelateerd aan een hoogrisicogroep. Echter, een beperkt aantal hoogrisicogroepen zijn door wetenschappelijk onderzoek in Nederland bekend.
Vervolgens werd voor een aantal afzonderlijke uitgangsvragen (zie de modules ‘Criteria en tests per hoogrisicogroep’ en 'Individuele testuitslag versus parenuitslag’) aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De kerngroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR -voor systematische reviews; Cochrane - voor gerandomiseerd gecontroleerd onderzoek; Newcastle-Ottowa - voor observationeel onderzoek; QUADAS II - voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A. Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk* |
|
|
Laag |
|
|
Zeer laag |
|
*in 2017 heeft het Dutch GRADE Network bepaald dat de voorkeursformulering voor de op een na hoogste gradering ‘redelijk’ is in plaats van ‘matig’
B. Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008), en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in één of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De kerngroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De gehele bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de cruciale uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de kerngroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’. In deze richtlijn bleek uiteindelijk het opstellen van PICO’s en het selecteren van literatuur voor de analyse niet goed van toepassing (zie artikel Burke, 2019). Daarom is de gebruikte literatuur (veelal niet-systematisch literatuuronderzoek) beschreven in de Overwegingen en tezamen met alle argumenten (consensus based en expert based) meegenomen in het wegen en opstellen van de aanbevelingen.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de kerngroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag.
Ontwikkeling van kwaliteitsindicatoren
De kerngroep heeft besloten geen indicatoren te ontwikkelen bij deze richtlijn omdat de UMC’s verschillende soorten tests aanbieden en de overige aanbevelingen volgens de kerngroep niet relevant zijn om een indicator op te stellen zonder onnodige verhoging van de administratielast.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de kerngroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de kerngroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de kerngroep. De definitieve richtlijn werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd respectievelijk geaccordeerd.
Literatuur
Brouwers, M. C., Kho, M. E., Browman, G. P., Burgers, J. S., Cluzeau, F., Feder, G., ... & Littlejohns, P. (2010). AGREE II: advancing guideline development, reporting and evaluation in health care. Cmaj, 182(18), E839-E842.
Burke, W., Clayton, E. W., Wolf, S. M., Berry, S. A., Evans, B. J., Evans, J. P., ... & McGuire, A. L. (2019). Improving recommendations for genomic medicine: building an evolutionary process from clinical practice advisory documents to guidelines. Genetics in Medicine. doi: 10.1038/s41436-019-0549-3.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van Medisch Specialisten.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html
Schünemann, H. J., Oxman, A. D., Brozek, J., Glasziou, P., Jaeschke, R., Vist, G. E., ... & Bossuyt, P. (2008). Rating Quality of Evidence and Strength of Recommendations: GRADE: Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ, 336(7653), 1106.
Zoekverantwoording
Searchdatum: 19 april 2019
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
2015 – april 2019
|
1. exp Whole Exome Sequencing/or 'whole exome'.ti,ab,kw. (8769) 2. (sanger or panel or (parallel adj3 sequenc*)).ti,ab,kw. (132038) 3. 1 and 2 (1642) 4. limit 3 to (english language and yr="2015-Current") (1300) 5. (meta-analysis/or meta-analysis as topic/or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/or Editorial/or Letter/or (animals/not humans/)) (389660) 6. (exp clinical trial/or randomized controlled trial/or exp clinical trials as topic/or randomized controlled trials as topic/or Random Allocation/or Double-Blind Method/or Single-Blind Method/or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/or placebo*.tw.) not (animals/not humans/) (1847769) 7. 4 and 5 (18) 8. (4 and 6) not 7 (43) 9. 4 not (7 or 8) (1239) 10. Epidemiologic studies/or case control studies/or exp cohort studies/or Controlled Before-After Studies/or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/or historically controlled study/or interrupted time series analysis/(Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies) (3161873) 11. 4 and 10 (151) 12. 11 not (7 or 8) (137) 13. 7 or 8 or 12 (198)
= 198 (197 uniek) |
511 |
|
Embase (Elsevier) |
('whole exome sequencing'/exp/mj OR 'whole exome':ab,ti)
AND ('sanger sequencing'/exp OR 'targeted exome sequencing'/exp OR sanger:ab,ti OR panel:ti,ab OR ((parallel NEAR/3 sequenc*):ab,ti))
AND (english)/lim AND (2015-2019)/py NOT 'conference abstract':it
Gebruikte filters:
Systematische reviews: ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp)
RCT’s: ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it
Observationeel onderzoek: ‘major clinical study’/exp OR 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR ('prospective study'/de NOT 'randomized controlled trial'/de) OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (case:ab,ti AND ((control NEAR/1 (study OR studies)):ab,ti)) OR (follow:ab,ti AND ((up NEAR/1 (study OR studies)):ab,ti)) OR ((observational NEAR/1 (study OR studies)):ab,ti) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti)
= 431 (428 uniek) |
Orienterende search
Datum: 19 april 2019
|
Database |
Zoektermen |
|
Medline (OVID)
Richtlijnen
2008 – april 2019 |
1. exp Genetic Carrier Screening/or (carrier adj3 (screen* or test*)).ti,ab,kw. (10406) 2. (screening adj4 disorder*).ti,ab,kw. and (exp Genetic Testing/or (genetic adj3 (screen* or test*)).ti,ab,kw. or prenatal.ti,ab,kw.) (336) 3. 1 or 2 (10661) 4. limit 3 to (english language and yr="2008 -Current") (949) 5. limit 4 to (guideline or practice guideline) (5) 6. guideline*.ti. (70476) 7. 4 and 6 (8) 8. 5 or 7 (13)
= 13 (11 uniek) |
|
Medline (OVID)
Systematische reviews (Cochrane)
2008 – april 2019 |
1. exp Genetic Carrier Screening/or (carrier adj3 (screen* or test*)).ti,ab,kw. (10406) 2. (screening adj4 disorder*).ti,ab,kw. and (exp Genetic Testing/or (genetic adj3 (screen* or test*)).ti,ab,kw. or prenatal.ti,ab,kw.) (336) 3. 1 or 2 (10661) 4. limit 3 to (english language and yr="2008 -Current") (949) 5. (meta-analysis/or meta-analysis as topic/or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/or Editorial/or Letter/or (animals/not humans/)) (388404) 6. 4 and 5 (56) 7. cochrane.jw. (14189) 8. 4 and 7 (3)
= 56 (55 uniek) SR
= 3 (3 uniek) Cochrane reviews |