Biologics
Uitgangsvraag
Welke biologics en JAK-remmers zijn aanbevolen voor kinderen/adolescenten met constitutioneel eczeem (CE)?
Welke screening en monitoring wordt aanbevolen bij kinderen en adolescenten met CE die met een biologic of JAK-remmer (gaan) worden behandeld?
Disclaimer Ten tijde van de ontwikkeling van deze module waren uitsluitend dupilumab, tralokinumab en upadacitinib geregistreerd en vergoed voor de behandeling van kinderen met constitutioneel eczeem. Inmiddels zijn ook lebrikizumab, baricitinib en abrocitinib voor specifieke leeftijdsgroepen geregistreerd en is hiervoor een vergoedingsstatus beschikbaar. Deze middelen zijn echter niet meegenomen in de huidige module. Standpunten van de NVDV met betrekking tot nieuwe geneesmiddelen die (nog) niet zijn opgenomen in de richtlijnen zijn te vinden via de volgende pagina: Standpunten en leidraden
Aanbeveling
Dupilumab en tralokinumab
Voorafgaand aan de behandeling
- Beoordeel het eczeem met de aanbevolen meetinstrumenten, zie hiervoor de module ‘Meetinstrumenten’.
- Anamnese: neem een anamnese af gericht op o.a. groei en ontwikkeling, medicatie gebruik, atopische comorbiditeiten (zoals voedselallergie, astma, rhinitis), pre-existente oogklachten ((allergische) conjunctivitis), parasitaire infecties en/of recent tropenbezoek, recidiverende (huid)infecties, gewrichtsklachten, doormaken waterpokken infectie, vaccinatiestatus en toekomstige reisplannen.
Vaccinaties:
-
-
- Check vaccinatiestatus volgens het Rijksvaccinatieprogramma (RVP), waarbij met name de levende (verzwakte) vaccins van belang zijn (BMR vaccinatie op leeftijd 14 maanden en 3 jaar). Overweeg de tweede BMR-vaccinatie naar voren te halen met een minimale tussenpoos van 28 dagen. Vaccineer niet met een levend (verzwakt) vaccin, inclusief de 1e BMR, tijdens behandeling met een biological. Een uitzondering hierop is de 2e BMR vaccinatie, die zonder problemen gegeven kan worden (expert opinie).
- Als de patiënt niet gevaccineerd wil worden heeft dupilumab/tralokinumab de voorkeur boven een JAK-remmer.
- Voor vaccinatieadviezen kan tabel 3 als handvat worden gebruikt. Voor aanvullende algemene informatie verwijzen wij naar de module ‘Vaccinaties bij juveniele idiopathische artritis (JIA)’. Vaccinatie bij kinderen met immunosuppressieve therapie is complex en vraagt om een individuele afweging. Daarbij spelen factoren zoals leeftijd, het type en de combinatie van immunosuppressiva en of een kind niet of onvolledig is gevaccineerd een rol. In complexe of uitzonderlijke situaties en bij twijfel wordt overleg met een kinderinfectioloog of kinderimmunoloog geadviseerd.
- Waterpokken/ varicella zoster: inventariseer of waterpokken zijn doorgemaakt ná de leeftijd van 1 jaar. Test bij twijfel of atypisch beloop varicella-antistoffen. Overweeg vaccinatie als antistoffen niet aantoonbaar zijn (expert opinie).
- Bespreek bij meisjes die seksueel actief zijn de kans op een zwangerschap en voer bij twijfel een zwangerschapstest (urine of bloed) uit. Adviseer aan deze patiëntgroep (vooralsnog) adequate anticonceptie tijdens en tot tenminste 3 maanden na de laatste dosis.
-
- Lichamelijk onderzoek:
- Beoordeel het eczeem met de aanbevolen meetinstrumenten, zie hiervoor de module ‘Meetinstrumenten’.
- Besteed aandacht aan: groei en ontwikkeling, aanwijzingen voor een onderliggende immuunstoornis of zeldzame genodermatose (zoals severe combined immunodeficiency (SCID), Netherton syndroom) in het bijzonder in het geval van erytrodermie, early onset dermatitis zonder atopische constitutie (vóór de leeftijd van 6 weken), forse aanhoudende exacerbatie, en/of therapieresistentie, secundaire (huid)infecties en oogklachten (zoals roodheid van oogleden en conjunctiva, tranende ogen).
- Aanvullend onderzoek:
- Routinematig laboratoriumonderzoek is niet nodig, ook niet voorafgaand aan de start van dupilumab en tralokinumab bij kinderen. Op indicatie kan laboratoriumonderzoek worden overwogen. Denk hierbij ook aan screening op varicella. Zie tabel 1. Indicaties kunnen zijn: klachten, bijwerkingen en/of (verdenking op) comorbiditeiten of uitsluiten van onderliggende aandoeningen.
- Overleg laagdrempelig met oogarts over indicatie voor oogheelkundig onderzoek bijvoorbeeld bij (anamnestisch) pre-existente oogklachten/oogheelkunde pathologie. Verwijs zo nodig.
- Overleg laagdrempelig met kinderarts/kinderallergoloog om (atopische) comorbiditeiten in kaart te brengen met aanvullend onderzoek.
- Definieer doormiddel van ‘samen beslissen’ behandeldoelen (zie module ‘Behandeldoelen’ in volwassenen richtlijn) en betrek daarbij afhankelijk van de leeftijd naast de ouders ook het kind.
- Bespreek met de patiënt om eventueel bij gecontroleerde ziekte of bijwerkingen in het geval van gecontroleerde ziekte, het interval tussen giften te verlengen (zie ‘Dosering’ bij ‘Overige zaken’ in de volwassenen richtlijn). Bij dupilumab betreft dosisverlaging en intervalverlenging off-label gebruik.
Tijdens de behandeling
- Beoordeel het eczeem met de aanbevolen meetinstrumenten, zie hiervoor de module ‘Meetinstrumenten’.
- Anamnese: neem een anamnese af gericht op eventuele bijwerkingen, zoals nieuw opgetreden of verergerende oogklachten (waarbij waakzaamheid is geboden dat (jonge) kinderen deze klachten minder goed kunnen uiten), nieuw ontstane huidafwijkingen anders dan CE, medicatie gebruik, aanwezigheid van infectie, check recent tropenbezoek, toestand en aanwezigheid van (atopische) comorbiditeiten, gewrichtsklachten en (actieve) kinderwens.
- Lichamelijk onderzoek:
- Beoordeel het eczeem met de aanbevolen meetinstrumenten, zie hiervoor de module ‘Meetinstrumenten’.
- Besteed aandacht aan: secundaire (huid)infecties, oogklachten (zoals roodheid van oogleden en conjunctiva, tranende ogen) en nieuw ontstane huidafwijkingen (head and neck dermatitis, psoriatiforme en lymfomatoïde huidafwijkingen).
- Aanvullend onderzoek:
- Laboratoriumonderzoek is op indicatie. Indicaties kunnen zijn: klachten, bijwerkingen en/of (verdenking op) comorbiditeiten, conform tabel 1.
- Overleg laagdrempelig met een oogarts over indicatie voor oogheelkundig onderzoek en/of intensievere behandeling bij (anamnestisch) optreden of verergeren van oogklachten/oogheelkundige pathologie. Bij patiënten die oogklachten ontwikkelen (al dan niet secundair aan dupilumab/tralokinumab) die zich niet met standaardbehandeling laat verhelpen, wordt geadviseerd hen te verwijzen naar een oogarts. Overweeg daarnaast intervalverlenging. Zie ‘Overige zaken’: ‘Ocular surface disease’ in volwassen richtlijn voor achtergrond en uitgebreid beleid hieromtrent.
- Check jaarlijks de vaccinatiestatus en adviseer het Rijkvaccinatieprogramma te volgen.
- Vaccineer niet met een levend (verzwakt) vaccin, inclusief de 1e BMR, tijdens de behandeling met een biologic of JAK remmer. Een uitzondering hierop is de 2e BMR vaccinatie, die zonder problemen gegeven kan worden.
- Voor vaccinatieadviezen kan tabel 3 als handvat worden gebruikt. Voor aanvullende algemene informatie verwijzen wij naar de module ‘Vaccinaties bij juveniele idiopathische artritis (JIA)’. In complexe of uitzonderlijke situaties en bij twijfel wordt overleg met een kinderimmunoloog of kinderinfectioloog geadviseerd.
- Overweeg de behandeling te staken indien na 16 weken geen initieel aanvaardbare respons is opgetreden. Bij een aanvankelijke gedeeltelijke respons kan bij doorbehandelen na 16 weken nog verbetering optreden
- Adviseer bij verbetering van het eczeem de topicale steroïden geleidelijk af te bouwen en niet abrupt te staken.
- Adviseer bij verbetering van het eczeem het emolliens blijvend te continueren.
- Overweeg bij gecontroleerde ziekte of bijwerkingen het interval tussen giften te verlengen (zie ‘Dosering’ bij ‘Overige zaken’ in de volwassenen richtlijn).
Na de behandeling
- Monitor de ziekteactiviteit en behandelwensen van patiënt en pas hier de toekomstige therapie op aan.
- Adviseer (vooralsnog) aan meisjes die zwanger kunnen worden de anticonceptie nog 3 maanden te continueren na staken van dupilumab/tralokinumab.
- Wijs patiënten met comorbide astma en allergische rhinoconjunctivitis op een mogelijke exacerbatie na staken dupilumab.
Overwegingen
Kwaliteit van het bewijs dupilumab en tralokinumab
Dupilumab
Voor de aanbevelingen over screening en monitoring is de literatuur niet systematisch beoordeeld op kwaliteit; er is geen GRADE-analyse gedaan. Voor het beantwoorden werd gebruik gemaakt van de NVDV-richtlijn constitutioneel eczeem (volwassenen). Indien de aanbevelingen overeenkwamen, werden deze overgenomen. Eventuele verschillen en/of onduidelijkheden werden besproken binnen de werkgroep, waarin verschillende disciplines vertegenwoordigd zijn (zie ook de samenstelling van de werkgroep onder ‘Verantwoording’).
Voor de aanbevelingen over effectiviteit en veiligheid werd voor alle uitkomstmaten (behalve de IDQOL welke alleen gerapporteerd werd door Paller et al., 2022) middels GRADE de kwaliteit beoordeeld op basis van een effectmaat afgeleid uit een meta-analyse (per uitkomstmaat) van 3 verschillende RCT’s (met verschillende populaties qua leeftijdsgroepen) (Paller et al., 2020; Paller et al., 2022; Simpson et al., 2020). Bij ‘Conclusies dupilumab (kwaliteit van bewijs)’ is per uitkomstmaat op basis van de GRADE-beoordeling, waarbij de kwaliteit van de RCT’s (risk of bias) en het bewijs invloed hebben op de uiteindelijke formulering, een conclusie geformuleerd.
De kwaliteit van het bewijs voor de superieure effectiviteit van dupilumab vs. placebo is over het algemeen redelijk (GRADE), waarbij de uitkomst betreft de verbetering van kwaliteit van leven voor kinderen van 6 maanden t/m 3 jaar (IDQOL-score) met enige voorzichtigheid moet worden geïnterpreteerd (GRADE: laag). Betreft de veiligheid kon er over het algemeen geen noemenswaardig verschil worden aangetoond tussen dupilumab en placebo, waarbij betreft serieuze interventie-afhankelijke AE’s en interventie-afhankelijke AE’s die resulteerden in discontinuatie de uitkomsten met enige voorzichtigheid moeten worden geïnterpreteerd (respectievelijk GRADE: laag; GRADE: zeer laag). In acht moet worden genomen dat het bewijs afkomstig is uit drie RCT’s (één per leeftijdscategorie) met een korte behandelduur (16 weken).
In de forest plots van de meta-analyses per uitkomstmaat zijn ook de effectmaten per leeftijdsgroep te raadplegen. Deze ‘alternatieve’ effectmaten per leeftijdsgroep zijn niet beoordeeld middels GRADE maar kunnen soms wel meer representatief zijn voor een individuele patiënt.
De kwaliteit van het bewijs is niet beoordeeld voor de open-label extensie (OLE) (Blauvelt et al., 2022; Cork et al., 2021), omdat deze niet placebo-gecontroleerd was. Deze studies hebben niet tot nieuwe bevindingen geleid t.o.v. de effectiviteits- en veiligheidsgegevens uit de RCT’s.
Tralokinumab
De kwaliteit van het bewijs voor de superieure effectiviteit van tralokinumab vs. placebo is over het algemeen hoog (GRADE). Betreft de veiligheid kon er over het algemeen geen noemenswaardig verschil worden aangetoond tussen tralokinumab en placebo, waarbij betreft serieuze interventie-afhankelijke AE’s de uitkomsten met enige voorzichtigheid moeten worden geïnterpreteerd (GRADE: laag). In acht moet worden genomen dat dit bewijs afkomstig is van één studie met een korte duur (16 weken).
Balans van gewenste en ongewenste effecten
Literatuuranalyse van de trials liet een gunstig effectiviteitsprofiel zien met een acceptabel veiligheidsprofiel zonder een significant meer aantal serieuze bijwerkingen betreft dupilumab en tralokinumab ten opzichte van placebo. Oogheelkundige bijwerkingen, zoals conjunctivitis, zijn ook gerapporteerd bij kinderen bij gebruik van dupilumab. Het effect van dupilumab en tralokinumab op type-2 geassocieerde comorbiditeiten, zoals eosinofiel astma, allergische rhinitis en voedselallergie, is bij kinderen nog onbekend. Jonge kinderen zullen bijwerkingen lastiger kunnen aangeven, waardoor deze mogelijk moeilijker te herkennen zijn. Het is belangrijk vanwege bovengenoemde redenen kinderen multidisciplinair te vervolgen. Er zijn nog geen langere termijn resultaten bekend over de effectiviteit en veiligheid van de behandeling van jonge kinderen met dupilumab en tralokinumab.
Professioneel perspectief
Effectiviteit en veiligheid
Dupilumab en tralokinumab zijn een effectieve behandeling voor kinderen en adolescenten met een matig tot ernstig constitutioneel eczeem die onvoldoende respons hebben op optimale topicale therapie (gepaard gaande met goede instructie en begeleiding), of wanneer het niet mogelijk is om de corticosteroïden af te bouwen naar een veilige onderhoudsdosering conform de Leidraad Dermatocorticosteroïden.
Er zijn op dit moment nog onvoldoende ‘real-world’ data bekend om de effectiviteit te vergelijken met die van de beschreven studies in deze richtlijn (REF). De groep kinderen die in aanmerking komt voor dupilumab en tralokinumab is immers klein.
Echter de ervaring in de praktijk leert dat dupilumab en tralokinumab effectief zijn. Door de inclusie van data in de landelijke registers (BioDay en TREAT NL/BE) kunnen effectiviteit data worden opgebouwd.
Door de inclusie van data in de landelijke registers (BioDay en TREAT NL/BE) kan ook data over de lange termijn bijwerkingen worden opgebouwd.
De meest evidente bijwerking die in de praktijk wordt gemeld is ooggerelateerde klachten, in het bijzonder bij dupilumab. Er zijn nog onvoldoende data bekend bij hoeveel patiënten er – ondanks behandeling en begeleiding door een oogarts – met dupilumab danwel tralokinumab gestopt moet worden.
Screening en monitoring
Groei en ontwikkeling: Het is belangrijk om de groei en ontwikkeling van kinderen in de gaten te houden. Dit kan door middel van de gegevens van de jeugdgezondheidszorg en groeicurves in het medisch patiëntendossier. Het is niet bekend of dupilumab of tralokinumab invloed hebben op de ontwikkeling en groei. Bij afwijkende groei, bijvoorbeeld door de ernst van het eczeem, is het belangrijk te verwijzen naar een kinderarts.
Vaccinaties: Het wordt aanbevolen om de vaccinatiestatus volgens het Rijksvaccinatieprogramma te checken omdat infecties ernstiger kunnen verlopen bij het gebruik van immunosuppressieve medicatie. Het wordt geadviseerd om niet te immuniseren met een levend (verzwakt) vaccin tijdens behandeling met een biologic. Indien toediening van een levend vaccin tijdens behandeling met een biologic toch noodzakelijk is, wordt geadviseerd te overleggen met een kinderimmunoloog/infectioloog over de te hanteren intervallen voor het staken, toedienen en hervatten van de behandeling.
Vanaf 1 januari 2025 krijgen kinderen binnen het rijksvaccinatieprogramma op de leeftijd van 3 jaar de tweede BMR-vaccinatie, waar dit voorheen pas bij 9 jaar was. In de overgangsfase naar deze aanpassing krijgt de groep kinderen geboren tussen 2016 tot en met 2021 de tweede BMR-vaccinatie op de leeftijd van 5 t/m 9 jaar. Bij kinderen die in deze groep vallen wordt aanbevolen om deze BMR-booster te vervroegen alvorens te starten met een biologic. Het minimale interval tussen de eerste en tweede BMR-vaccinatie is 28 dagen [Rijksvaccinatieprogramma, z.d.]. Maak voor meer diepgang en overwegingen gebruik van het hoofdstuk ‘Overige zaken’ in de modules ‘Dupilumab’ en ‘Tralokinumab’ in de CE richtlijn voor volwassenen.
Voor vaccinatieadviezen kan tabel 3 als handvat worden gebruikt. Voor aanvullende informatie verwijzen wij naar de module ‘Vaccinaties bij juveniele idiopathische artritis (JIA)’ voor algemene adviezen. Vaccinatie bij kinderen met immunosuppressieve therapie is complex en vraagt om een individuele afweging. Daarbij spelen factoren zoals leeftijd, het type en de combinatie van immunosuppressiva en of een kind niet of onvolledig is gevaccineerd een rol. In complexe of uitzonderlijke situaties en bij twijfel wordt overleg met een kinderimmunoloog of kinderinfectioloog geadviseerd.
Varicellazostervirus (VZV): Het is onbekend in hoeverre dupilumab en tralokinumab een immunosuppressieve uiting hebben op varicella (zoster) infecties. Immunosuppressieve medicatie kan infecties zoals VZV erger laten verlopen. Inventariseer of varicella (waterpokken) is doorgemaakt ná de leeftijd van 1 jaar. Test bij twijfel of atypisch beloop varicella-antistoffen. Als antistoffen niet aantoonbaar zijn overweeg vaccinatie tegen VZV.
Virale hepatitis en HIV: Immunosuppressieve medicatie kan infecties zoals hepatitis B/C en HIV erger doen laten verlopen. Het is onbekend in hoeverre dupilumab en tralokinumab een immunosuppressieve uiting hebben op hepatitis B/C en HIV-infectie.
- Routinematige screening op hepatitis B/C en HIV wordt bij kinderen en adolescenten niet aanbevolen. Screening is geïndiceerd enkel bij het bestaan van risicofactoren. Bovendien worden kinderen sinds 2011 gevaccineerd voor hepatitis B via het RVP.
- Overleg met een kinder MDL-arts bij het bestaan van een infectie met hepatitis B of C.
Tuberculose: Het is onbekend in hoeverre dupilumab en tralokinumab een immunosuppressieve uiting hebben op tuberculose infectie. Immunosuppressieve medicatie kan infecties zoals tuberculose erger doen verlopen en onder deze therapie kan latente tuberculose-infectie (LTBI) reactiveren. Het risico op activatie van LTBI is, op basis van het werkingsmechanisme van biologics, te verwaarlozen. Derhalve is de werkgroep van mening dat het niet noodzakelijk is om voorafgaand aan de behandeling screenend onderzoek te verrichten.
Kinderwens en zwangerschap: Bespreek bij meisjes die seksueel actief zijn de kans op een zwangerschap en voer bij twijfel een zwangerschapstest (urine of bloed) uit. Adviseer aan deze patiëntgroep adequate anticonceptie. Maak voor meer diepgang en overwegingen gebruik van het hoofdstuk ‘Overige zaken’ in de modules ‘Dupilumab’ en ‘Tralokinumab’ in de CE richtlijn voor volwassenen.
Atopische comorbiditeiten: Besteed aandacht aan de atopische comorbiditeiten bij het maken van een therapiekeuze, dosisverlaging, intervalverlenging en bij het staken van een behandeling met biologics.
Waarden en voorkeuren van patiënten
Hoewel er nog onvoldoende ‘real-world’ data bekend zijn, is er bekend dat bij dupilumab en tralokinumab een significante verbetering optreedt van het eczeem en de jeuk, waardoor uiteindelijk ook verbetering van de kwaliteit van leven.
Een nadeel van beide medicijnen kan zijn dat zij worden ingebracht middels een injectie. Kinderen kunnen aangeven dat dit pijnlijk is of angst hebben voor de injectie. Door verbinding te maken met het kind en vertrouwen te creëren, het kind te betrekken in diagnose, behandeling en follow-up, en daarnaast gebruik te maken van procedurele comfortzorg kan een kind hierin zo goed mogelijk worden begeleid.
Aanvaardbaarheid en haalbaarheid
CE is bij het merendeel van de kinderen goed te behandelen met lokale therapie, mits er voldoende tijd en energie geïnvesteerd wordt in voorlichting en begeleiding. Er is slechts een kleine groep kinderen die in aanmerking komt voor systemische immunosuppressieve/immunomodulerende behandeling. Bij voorkeur worden dupilumab en tralokinumab voorgeschreven door een gespecialiseerd centrum waar ervaring is met het gebruik van orale immunosuppressiva (conventionele middelen en JAK-remmers) en biologics bij kinderen en die deelnemen aan één van de eerdere genoemde landelijke registers. Mede gezien het feit dat de oogheelkundige bijwerkingen ook gerapporteerd zijn in de studies met dupilumab en tralokinumab bij kinderen en adolescenten met CE, is het raadzaam de behandeling ook in de gespecialiseerde centra te laten plaatsvinden waar kinderoogartsen aanwezig zijn. CE gaat verder vaak gepaard met atopische comorbiditeiten waarbij een multidisciplinair team met kinderartsen, kinderallergologen, kinderlongartsen, kinder KNO-artsen en kinderoogartsen onontbeerlijk is. Ook de dermatoloog dient actief uit vraag te doen naar de comorbiditeiten. Ook zijn deze disciplines van belang bij het herkennen en behandelen van eventuele bijwerkingen. Gespecialiseerde centra zijn gericht op deze multidisciplinaire samenwerking. Overweeg terug te verwijzen voor behandeling in de regio, indien er sprake is van gestabiliseerde ziekte.
Tabel 1. Te overwegen laboratoriumcontroles (dupilumab en tralokinumab)
|
Parameter |
Bij intake |
Tijdens de behandeling |
|
Volledig bloedbeeld incl. leukocyten differentiatie |
(X) |
(X) |
|
Serum kreatinine (incl. eGFR) |
(X) |
(X) |
|
ALAT |
(X) |
(X) |
|
Hepatitis B/C en HIV* |
(X) |
|
|
Screening op parasitaire infecties** |
(X) |
|
|
Screening op varicella-antistoffen*** |
(X) |
|
|
(X) = op indicatie; bij afwijkingen bij start dupilumab/tralokinumab herhaling van lab controle tijdens de behandeling. * Op indicatie: patiënten met risicofactoren voor een dergelijke infectie. HBV zit sinds 2011 in het Rijksvaccinatieprogramma. **Anamnestisch inventariseren (reisanamnese, klachten) en testen als daar een indicatie voor is op basis van klachten en/of risico op blootstelling. *** Test varicella-antistoffen bij twijfel over het doormaken van waterpokken ná leeftijd van 1 jaar, of een atypisch beloop in verband met het eventueel vaccineren voor waterpokken. |
||
Tabel 3. Handvat: overzicht van vaccinaties bij kinderen met CE die behandeld worden met dupilumab, tralokinumab of upadacitinib. Gebaseerd op de module Vaccinaties bij JIA (2025) - Richtlijn - Richtlijnendatabase en aangevuld met expert opinion.

Onderbouwing
Achtergrond
Achtergrond dupilimab (inleiding)
Dupilumab is het eerste volledig menselijke IgG4-monoklonale antilichaam dat op de markt werd gebracht voor de behandeling van CE en is in Nederland sinds 2018 beschikbaar voor de behandeling van volwassenen met CE, sinds september 2019 voor adolescenten 12-17 jaar, sinds december 2020 voor kinderen vanaf 6 jaar en sinds april 2023 is dupilumab ook geregistreerd voor kinderen vanaf de leeftijd van 6 maanden.
Dosering en toediening
De onderstaande informatie komt uit de SmPC, tenzij anders aangegeven. Dupilumab wordt toegediend middels subcutane injecties.
Adolescenten 12-17 jaar
Indien <60kg: subcutane oplaaddosis van 400 mg gevolgd door onderhoudsdosering van 200 mg Q2W
Indien ≥60kg: subcutane oplaaddosis van 600 mg gevolgd door onderhoudsdosering van 300 mg Q2W
Kinderen 6-12 jaar
Indien ≥15kg - <60kg: subcutane oplaaddosis van 300mg op dag 1 en dag 15 gevolgd door onderhoudsdosering 300mg Q4W (start 4 weken na dag 15 dosis)
Indien ≥60kg: subcutane oplaaddosis van 600 mg gevolgd door onderhoudsdosering van 300 mg Q2W
Kinderen 6 maanden-5 jaar
Indien ≥5kg - <15kg: 200 mg Q4W (zonder oplaaddosis)
Indien ≥15 - <30kg: 300 mg Q4W (zonder oplaaddosis)
De dupilumab voorgevulde pen is niet bedoeld voor gebruik bij kinderen jonger dan 12 jaar. Voor kinderen van 6 maanden tot 11 jaar met CE, is de voorgevulde spuit van dupilumab geschikt. Dupilumab wordt toegediend via subcutane injectie in de dij of de buik (bij voorkeur niet in het gebied 5 cm rond de navel). Als iemand anders de injectie toedient, kan ook de bovenarm worden gebruikt (niet geschikt voor kleine kinderen met een hele smalle bovenarm). Voor de oplaaddosis wordt aanbevolen om bij elke injectie de injectieplaats te wisselen. Dupilumab mag niet worden geïnjecteerd in een huid die gevoelig of beschadigd is of blauwe plekken of littekens vertoont. Een patiënt kan dupilumab zelf injecteren of de verzorger van de patiënt kan dupilumab toedienen, nadat zij goede instructies hebben gekregen. In de praktijk blijkt dat met name bij kinderen <12 jaar de behandeling veelal wordt opgestart op een (kinder)dagbehandeling met pedagogische en/of psygologische begeleiding.
Achtergrond tralokinumab (inleiding)
Tralokinumab is een volledig humaan monoklonaal IgG4 antilichaam gericht tegen interleukine-13, een belangrijke driver van inflammatie in de huid bij constitutioneel eczeem (CE) [Bieber, 2020]. Sinds oktober 2022 is tralokinumab door de EMA goedgekeurd voor de behandeling van adolescenten met ernstig eczeem (12-17 jaar) [SmPC Adtralza, 2022]. Sinds 1 december 2022 kan tralokinumab worden voorgeschreven als add-on geneesmiddel voor deze indicatie. De goedkeuring is gebaseerd op de resultaten van de Fase 3 ECZTRA 6 (ECZema TRAlokinumab trial No. 6) studie, waarin de effectiviteit en veiligheid van tralokinumab monotherapie (150 mg of 300 mg, elke 2 weken) wordt vergeleken met placebo bij adolescenten met matig tot ernstig CE [Paller, 2023; ECZTRA, 2017].
Achtergrond upadacitinib (inleiding)
Upadacitinib is een selectieve en reversibele JAK-remmer van voornamelijk JAK1, en in mindere mate JAK2 en JAK3, waardoor de fosforylering en activering van STAT’s verminderd wordt [FK upadacitinib, 2023]. Sinds augustus 2021 is upadacitinib goedgekeurd door de EMA voor de behandeling van volwassen patiënten en adolescenten vanaf 12 jaar met matig tot ernstig constitutioneel eczeem, die in aanmerking komen voor systemische therapie [SmPC Rinvoq]. De goedkeuring is gebaseerd op de resultaten van drie fase 3 studies: Measure Up 1 (MU1), Measure Up 2 (MU2) en AD Up (AU), waarin de effectiviteit en veiligheid van upadacitinib monotherapie (MU1 en MU2) en gecombineerd met topicale corticosteroïden (AU) wordt vergeleken met placebo bij adolescenten met matig tot ernstig CE [Guttman-Yassky 2021, Reick 2021, Silverberg 2021].
Conclusies / Summary of Findings
Conclusies dupilumab (kwaliteit van bewijs)
IGA
Literatuur: Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Dupilumab |
placebo |
Relatief |
Absoluut |
||
|
Proportie patiënten met IGA 0-1 |
||||||||||||
|
3 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
ernstiga,b |
niet ernstigc |
niet gevonden |
134/493 (27.2%) |
19/287 (6.6%) |
RR 3.99 |
198 meer per 1.000 |
⨁⨁⨁◯ |
CRUCIAAL |
|
Conclusie: dupilumab resulteert waarschijnlijk in een grote klinisch relevante toename in de proportie patiënten die IGA 0 of 1 bereikt t.o.v. placebo |
||||||||||||
CI: Confidence interval; RR: Risk ratio
Explanations
a. Paller et al. 2022 heeft ten opzichte van de andere twee trials maar 1 interventiegroep (200 of 300 mg afhankelijk van baseline gewicht), met een toedieningsregime van elke 4 weken, en dus geen groep met een toedieningsregime van elke 2 weken.
b. Absoluut gezien verschillen de doseringen van dupilumab enigzins per trial. Echter, alle studies hebben een interventiegroep met doseringen op basis van baseline gewicht. De rationale hierachter is vergelijkbare dupilumab-spiegels te krijgen in verschillende leeftijdsgroepen.
c. Power analyse: N=108
EASI
Literatuur: Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
|||||||||||||||||||||||||||||||||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
dupilimab |
placebo |
Relatief |
Absoluut |
|||||||||||||||||||||||||||||||||
|
Verandering in EASI-score t.o.v. baseline
Proportie patiënten met minstens 75% verbetering van EASI t.o.v. baseline (EASI-75) |
|||||||||||||||||||||||||||||||||||||||||||
|
3 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
ernstiga,b |
niet ernstigd |
niet gevonden |
277/493 (56.2%) |
48/287 (16.7%) |
RR 3.25 |
376 meer per 1.000 |
⨁⨁⨁◯ |
CRUCIAAL |
|||||||||||||||||||||||||||||||
|
Conclusie: dupilumab resulteert waarschijnlijk in een grote klinisch relevante verbetering van EASI-75 t.o.v. placebo |
|||||||||||||||||||||||||||||||||||||||||||
|
Proportie patiënten met minstens 90% verbetering van EASI t.o.v. baseline (EASI-90) |
|||||||||||||||||||||||||||||||||||||||||||
|
3 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
ernstiga,b |
niet ernstigc |
niet gevonden |
144/493 (29.2%) |
13/287 (4.5%) |
RR 6.19 |
235 meer per 1.000 |
⨁⨁⨁◯ |
CRUCIAAL |
|||||||||||||||||||||||||||||||
|
Conclusie: dupilumab resulteert waarschijnlijk in een grote klinisch relevante verbetering van EASI-90 t.o.v. placebo |
|||||||||||||||||||||||||||||||||||||||||||
CI: Confidence interval; RR: Risk ratio
Explanations
a. Paller et al. 2022 heeft ten opzichte van de andere twee trials maar 1 interventiegroep (200 of 300 mg afhankelijk van baseline gewicht), met een toedieningsregime van elke 4 weken, en dus geen groep met een toedieningsregime van elke 2 weken.
b. Absoluut gezien verschillen de doseringen van dupilumab enigzins per trial. Echter, alle studies hebben een interventiegroep met doseringen op basis van baseline gewicht. De rationale hierachter is vergelijkbare dupilumab-spiegels te krijgen in verschillende leeftijdsgroepen.
c. Power analyse: N=70
d. Power analyse: N=44
e. Power analyse: N=416
SCORAD
Literatuur: Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Dupilumab |
placebo |
Relatief |
Absoluut |
||
|
Gemiddelde van de percentuele verandering SCORAD t.o.v. baseline |
||||||||||||
|
3 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
ernstiga,b,c |
niet ernstigd |
niet gevonden |
493 |
287 |
- |
MD 32.89 lager |
⨁⨁⨁◯ |
CRUCIAAL |
|
Conclusie: dupilumab resulteert waarschijnlijk in een klinisch relevante afname van de SCORAD-score t.o.v. placebo |
||||||||||||
CI: Confidence interval; MD: Mean difference
Explanations
a. Paller et al. 2022 heeft ten opzichte van de andere twee trials maar 1 interventiegroep (200 of 300 mg afhankelijk van baseline gewicht), met een toedieningsregime van elke 4 weken, en dus geen groep met een toedieningsregime van elke 2 weken.
b. Absoluut gezien verschillen de doseringen van dupilumab enigzins per trial. Echter, alle studies hebben een interventiegroep met doseringen op basis van baseline gewicht. De rationale hierachter is vergelijkbare dupilumab-spiegels te krijgen in verschillende leeftijdsgroepen.
c. Het valt aan te nemen dat het de subjectieve symptomen (maximaal 20/103 punten) in de trial van Paller et al. 2022 gescoord zijn door de ouders/verzorgers. Dit is echter niet terug te vinden in de publicatie of het protocol van de trial.
d. Power analys: N=146
POEM
Literatuur: Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Dupilumab |
placebo |
Relatief |
Absoluut |
||
|
Gemiddelde verandering POEM t.o.v. baseline |
||||||||||||
|
3 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
ernstiga,b,c |
niet ernstig |
niet gevonden |
493 |
287 |
- |
MD 7.81 lager |
⨁⨁⨁◯ |
CRUCIAAL |
|
Conclusie: dupilumab resulteert waarschijnlijk in een klinisch relevante gemiddelde afname in POEM-score t.o.v. placebo. |
||||||||||||
CI: Confidence interval; MD: Mean difference
Explanations
a. Paller et al. 2022 heeft ten opzichte van de andere twee trials maar 1 interventiegroep (200 of 300 mg afhankelijk van baseline gewicht), met een toedieningsregime van elke 4 weken, en dus geen groep met een toedieningsregime van elke 2 weken.
b. Absoluut gezien verschillen de doseringen van dupilumab enigzins per trial. Echter, alle studies hebben een interventiegroep met doseringen op basis van baseline gewicht. De rationale hierachter is vergelijkbare dupilumab-spiegels te krijgen in verschillende leeftijdsgroepen.
c. Gescoord door patiënt: Paller et al. 2020 + Simpson et al. 2020. Gescoord door ouder/verzorger: Paller et al. 2022.
PP-NRS
Literatuur: Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Dupilumab |
placebo |
Relatief |
Absoluut |
||
|
Gemiddelde percentuele afname van het wekelijks gemiddelde van de dagelijkse PP-NRS score t.o.v. baseline |
||||||||||||
|
3 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
ernstiga,b,c |
niet ernstig |
niet gevonden |
493 |
287 |
- |
MD 31.54 lager |
⨁⨁⨁◯ |
CRUCIAAL |
|
Conclusie: dupilumab resulteert waarschijnlijk in een klinisch relevante percentuele afname van het wekelijks gemiddelde van de dagelijkse PP-NRS score t.o.v. placebo |
||||||||||||
CI: Confidence interval; MD: Mean difference
Explanations
a. Paller et al. 2022 heeft ten opzichte van de andere twee trials maar 1 interventiegroep (200 of 300 mg afhankelijk van baseline gewicht), met een toedieningsregime van elke 4 weken, en dus geen groep met een toedieningsregime van elke 2 weken.
b. Absoluut gezien verschillen de doseringen van dupilumab enigzins per trial. Echter, alle studies hebben een interventiegroep met doseringen op basis van baseline gewicht. De rationale hierachter is vergelijkbare dupilumab-spiegels te krijgen in verschillende leeftijdsgroepen.
c. In tegenstelling tot Paller et al. 2020 en Simpson et al. 2020, werd deze uitkomstmaat in de trial van Paller et al. 2022 door de ouders/verzorgers gescoord.
CDLQI en IDQOL
Literatuur: Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Dupilumab |
placebo |
Relatief |
Absoluut |
||
|
Gemiddelde verandering in CDLQI score t.o.v. baseline |
||||||||||||
|
3 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
ernstiga,b |
niet ernstig |
niet gevonden |
457 |
246 |
- |
MD 4.08 lager |
⨁⨁⨁◯ |
BELANGRIJK |
|
Conclusie: dupilumab resulteert waarschijnlijk in een afname van de CDLQI-score t.o.v placebo; deze verbetering is niet klinisch relevant. |
||||||||||||
|
Gemiddelde verandering in IDQOL score t.o.v. baseline (Paller et al. 2022) |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
zeer ernstigc |
niet gevonden |
36 |
41 |
- |
MD 8.9 lager |
⨁⨁◯◯ |
BELANGRIJK |
|
Conclusie: dupilumab zou kunnen resulteren in een afname van de IDQOL-score t.o.v. placebo; deze verbetering is waarschijnlijk klinisch relevant. |
||||||||||||
CI: Confidence interval; MD: Mean difference
Explanations
a. Paller et al. 2022 heeft ten opzichte van de andere twee trials maar 1 interventiegroep (200 of 300 mg afhankelijk van baseline gewicht), met een toedieningsregime van elke 4 weken, en dus geen groep met een toedieningsregime van elke 2 weken.
b. Absoluut gezien verschillen de doseringen van dupilumab enigzins per trial. Echter, alle studies hebben een interventiegroep met doseringen op basis van baseline gewicht. De rationale hierachter is vergelijkbare dupilumab-spiegels te krijgen in verschillende leeftijdsgroepen.
c. Kleine steekproef: N=77; De bovengrens van het CI van de MD is lager dan de ondergrens van het MCID (6 punten).
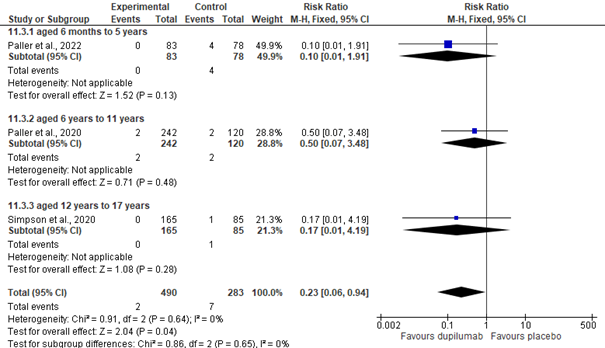
Veiligheid – interventie-gerelateerde (serieuze) AE’s
Literatuur: Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Dupilumab |
placebo |
Relatief |
Absoluut |
||
|
Proportie patiënten met ≥1 TEAE |
||||||||||||
|
3 |
gerandomiseerde trials |
ernstiga |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
325/490 (66.3%) |
205/283 (72.4%) |
RR 0.91 |
65 minder per 1.000 |
⨁⨁⨁◯ |
BELANGRIJK |
|
Conclusie: dupilumab heeft waarschijnlijk niet of nauwelijks effect op het aantal patiënten met ≥1 TEAE t.o.v. placebo. |
||||||||||||
|
Proportie patiënten met ≥1 serious TEAE |
||||||||||||
|
3 |
gerandomiseerde trials |
ernstiga |
niet ernstig |
niet ernstig |
ernstigb |
niet gevonden |
2/490 (0.4%) |
7/283 (2.5%) |
RR 0.23 |
19 minder per 1.000 |
⨁⨁◯◯ |
BELANGRIJK |
|
Conclusie: dupilumab zou kunnen resulteren in een grote vermindering van het aantal patiënten met ≥1 serious TEAE t.o.v. placebo; dit verschil is mogelijk niet klinisch relevant. |
||||||||||||
|
Proportie patiënten met ≥1 TEAE resulterende in permanente discontinuatie van trial |
||||||||||||
|
3 |
gerandomiseerde trials |
ernstiga |
niet ernstig |
niet ernstig |
zeer ernstigc |
niet gevonden |
3/490 (0.6%) |
4/283 (1.4%) |
RR 0.46 |
8 minder per 1.000 |
⨁◯◯◯ |
BELANGRIJK |
|
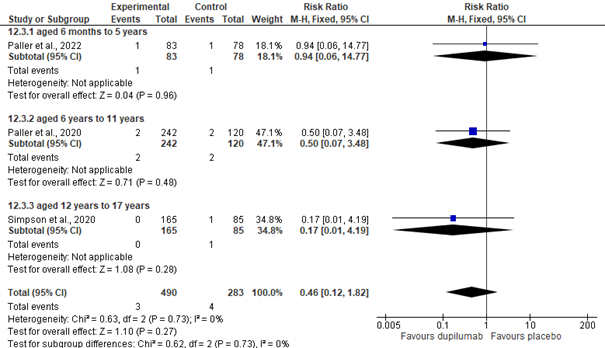
Conclusie: de werkgroep is onzeker over het effect van dupilumab op het aantal patiënten met ≥1 TEAE die resulteren in permanente discontinuatie t.o.v. placebo. |
||||||||||||
CI: Confidence interval; RR: Risk ratio
Explanations
a. Een hoog aantal patiënten in de placebogroep van Paller et al. 2022 (n=49 vs. n=16 in dupilmab-groep) had 'rescue therapy' nodig. Ondanks dat hier uitvoerig voor gecorrigeerd is bij andere uitkomstmaten (zie risk of bias analyse, is te vinden onder conclusies dupilumab (kwaliteit van bewijs)), levert dat betreft de bijwerkingen wel onnauwkeurigheid op. Het risico op bias bestaat dat daardoor minder TEAE's, serious TEA's en TEAE waardoor uitval, gerelateerd aan CE opvlamming, voorkwamen in de placebogroep.
b. Hoewel significant verschil, mogelijk geen klinisch relevant verschil (bovengrens CI=0.94).
c. Zowel klinisch relevant voordeel als nadeel mogelijk op basis van spreiding CI.
Onderbouwing tralokinumab
Onderstaande kopjes vallen onder de onderbouwing en worden op de Richtlijnendatabase als bijlagen weergegeven.
Conclusies tralokinumab (kwaliteit van bewijs)
IGA
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Tralokinumab |
placebo |
Relatief |
Absoluut |
||
|
Proportie patiënten met IGA 0-1 |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
38/195 (19.5%) |
4/94 (4.3%) |
RR 4.58 |
152 meer per 1.000 |
⨁⨁⨁⨁ |
CRUCIAAL |
|
Conclusie: tralokinumab resulteert in een grote, klinisch relevante, toename in de proportie patiënten die IGA 0 of 1 bereikt ten opzichte van placebo |
||||||||||||
EASI
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Tralokinumab |
placebo |
Relatief |
Absoluut |
||
|
Proportie patiënten dat 75% verbetering van EASI behaalt (EASI-75) |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
55/195 (28.2%) |
6/94 (6.4%) |
RR 4.42 |
218 meer per 1.000 |
⨁⨁⨁⨁ |
CRUCIAAL |
|
Conclusie: tralokinumab resulteert in een grote, klinisch relevante, verbetering d.m.v. de proportie patiënten die EASI-75 bereikt ten opzichte van placebo |
||||||||||||
|
Proportie patiënten dat 90% verbetering van EASI behaalt (EASI-90) |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
36/195 (18.5%) |
4/94 (4.3%) |
RR 4.34 |
142 meer per 1.000 |
⨁⨁⨁⨁ |
CRUCIAAL |
|
Conclusie: tralokinumab resulteert in een grote, klinisch relevante, verbetering d.m.v. de proportie patiënten die EASI-90 bereikt ten opzichte van placebo |
||||||||||||
|
Gemiddelde percentuele verandering (afname) EASI-score |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
zeer ernstiga, b |
niet gevonden |
187 |
87 |
- |
MD 7.13 lager |
⨁⨁◯◯ |
CRUCIAAL |
|
Conclusie: tralokinumab zou kunnen resulteren in een verbetering in de percentuele afname van EASI t.o.v. placebo, dit is mogelijk niet klinisch relevant. |
||||||||||||
CI: Confidence interval; MD: Mean difference; RR: Risk ratio
Explanations
- placebogroep van n=36 na exclusie van patiënten die noodmedicatie kregen, volgens powerberekening, voor 80%, minimaal n=50 nodig per groep.
- Als de het percentuele MD en het CI daarvan wordt omgerekend naar een absoluut verschil overlapt dit de grenzen van een klinisch relevante afname. (REF)
SCORAD
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Tralokinumab |
placebo |
Relatief |
Absoluut |
||
|
Gemiddelde percentuele verandering in SCORAD |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
195 |
94 |
- |
MD 18.8 lager |
⨁⨁⨁⨁ |
CRUCIAAL |
|
Conclusie: tralokinumab resulteert in een klinisch relevante verbetering (afname) in SCORAD ten opzichte van placebo. |
||||||||||||
CI: Confidence interval; MD: Mean difference; RR: Risk ratio
POEM
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Tralokinumab |
placebo |
Relatief |
Absoluut |
||
|
Proportie patiënten met ≥6 punten afname in POEM-score |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
80/187 (42.8%) |
9/86 (10.5%) |
RR 4.09 |
323 meer per 1.000 |
⨁⨁⨁⨁ |
CRUCIAAL |
|
Conclusie: tralokinumab resulteert in een grote, klinisch relevante, verbetering (afname) in de POEM-score ten opzichte van placebo. |
||||||||||||
CI: Confidence interval; MD: Mean difference; RR: Risk ratio
Jeuk - Adolescent Worst Pruritus NRS
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Tralokinumab |
placebo |
Relatief |
Absoluut |
||
|
Proportie patiënten met ≥4 punten afname in wekelijks gemiddelde van de AWP-NRS-score |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
46/191 (24.1%) |
3/90 (3.3%) |
RR 7.23 |
208 meer per 1.000 |
⨁⨁⨁⨁ |
CRUCIAAL |
|
Conclusie: tralokinumab resulteert in een grote, klinisch relevante, verbetering in de AWP-NRS ten opzichte van placebo. |
||||||||||||
CI: Confidence interval; MD: Mean difference; RR: Risk ratio
Kwaliteit van leven – CDLQI
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Tralokinumab |
placebo |
Relatief |
Absoluut |
||
|
Gemiddelde verandering in CDLQI |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
niet ernstig |
niet gevonden |
195 |
94 |
- |
MD 2.3 lager |
⨁⨁⨁⨁ |
BELANGRIJK |
|
Conclusie: tralokinumab resulteert in een verbetering van de CDLQI-score t.o.v. placebo; dit is geen klinisch relevante afname. |
||||||||||||
CI: Confidence interval; MD: Mean difference; RR: Risk ratio
Veiligheid – AE’s en serious TEAE’s
|
Certainty assessment |
Aantal patiënten |
Effect |
Certainty |
Importantie |
||||||||
|
Aantal studies |
Studieopzet |
Risk of bias |
Inconsistentie |
Indirect bewijs |
Onnauwkeurigheid |
Andere factoren |
Tralokinumab |
placebo |
Relatief |
Absoluut |
||
|
Proportie patiënten met met ≥1 AE |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
ernstiga |
niet gevonden |
129/195 (66.2%) |
58/94 (61.7%) |
RR 1.07 |
43 meer per 1.000 |
⨁⨁⨁◯ |
BELANGRIJK |
|
Conclusie: tralokinumab heeft waarschijnlijk niet of nauwelijks effect op het aantal patiënten met ≥1 adverse event t.o.v. placebo; dit verschil is waarschijnlijk niet klinisch relevant. |
||||||||||||
|
Proportie patiënten met ≥1 serious TEAE |
||||||||||||
|
1 |
gerandomiseerde trials |
niet ernstig |
niet ernstig |
niet ernstig |
zeer ernstigb |
niet gevonden |
4/195 (2.1%) |
5/94 (5.3%) |
RR 0.39 |
32 minder per 1.000 |
⨁⨁◯◯ |
BELANGRIJK |
|
Conclusie: tralokinumab zou kunnen resulteren in een klinisch relevante vermindering van het aantal patiënten met ≥1 serious treatment-emergent adverse event ten opzichte van placebo. |
||||||||||||
CI: Confidence interval; MD: Mean difference; RR: Risk ratio
Explanations
a. Klinisch relevant voordeel mogelijk (bovengrens CI=1.29)
b. Zowel klinisch relevant voordeel als nadeel mogelijk op basis van spreiding CI.
Samenvatting literatuur
Dupilumab (resultaten)
Beschrijving van de studies
De drie RCT’s onderzochten allen dubbelblind, placebo-gecontroleerd en gerandomiseerd interventie met dupilumab als behandeling voor matig tot ernstig CE. Bij het opzetten en uitvoeren van de trials werd in de (of één van de 2) interventiegroep(en) rekening gehouden met dosering van dupilumab in relatie tot baseline gewicht, met een toedieningsregime van elke 4 weken (q4w) [Paller et al. 2022; Paller et al. 2020; Simpson et al. 2020]. In 2 trials [Paller et al. 2020; Simpson et al. 2020] werd tevens een tweede interventiegroep toegevoegd (ratio 1:1:1) waarin een vaste dosering werd gegeven zonder rekening te houden met baseline gewicht, met een toedieningsfrequentie van elke 2 weken (q2w).
LIBERTY AD PRESCHOOL
Paller et al. (2022) voerden een dubbelblinde, 16 weken, placebo-gecontroleerde RCT uit in 31 centra, onder 162 kinderen in de leeftijdscategorie 6 maanden t/m 5 jaar met matig tot ernstig CE (inclusie criteria: IGA ≥3; EASI ≥16; BSA ≥10%; wekelijks gemiddelde Worst Itch (WI) NRS intensiteit ≥4; binnen 6 maanden voor het screeningsbezoek een onvoldoende respons op topicale corticosteroïden). Patiënten werden gerandomiseerd (1:1) in de combinatie subcutaan a) placebo [n=83] of b) dupilumab [n=79] (lichaamsgewicht ≥5 kg tot <15 kg: 200 mg; lichaamsgewicht ≥15 kg tot <30 kg: 300 mg) elke 4 weken plus eens per dag laag-potente topicale corticosteroïden (hydrocortison acetaat 1% crème).
De primaire uitkomstmaat na 16 weken was het percentage patiënten met een IGA-score van 0-1 (‘clear’ of ‘almost clear’). Het belangrijkste secundaire eindpunt na 16 weken was het percentage patiënten met minstens 75% verbetering (t.o.v. baseline) in de Eczema Area and Severity Index (EASI-75).
LIBERTY AD PEDS
Paller et al. (2020) voerden een dubbelblinde, 16 weken, placebo-gecontroleerde RCT uit in 61 centra, onder 367 kinderen van 6 t/m 11 jaar met matig tot ernstig CE (inclusie criteria: IGA =4; EASI ≥21; BSA ≥15%; diagnose ten minste 1 jaar voor screeningsbezoek; binnen 6 maanden voor het baseline bezoek een onvoldoende respons op topicale corticosteroïden [TCS]). Patiënten werden gerandomiseerd (1:1:1) in 3 groepen: a) dupilumab 300mg, q4w [n=122] (600 mg oplaaddosis) of dupilumab, q2w [n=122] met voor baseline gewicht: 15 tot <30 kg, 100 mg (200 mg oplaaddosis); en baseline gewicht ≥30 kg, 200 mg (400 mg oplaaddosis), of c) placebo [n=123], plus eens per dag laag-potente topicale corticosteroïden (in alle groepen). De placebogroep kreeg gerandomiseerd de hoeveelheid (in ml) en frequentie (q2w of q4w) corresponderende met interventiegroep A dan wel interventiegroep B toegediend. De primaire uitkomstmaat was het percentage patiënten met een IGA-score van 0 of 1 na 16 weken; een verbetering van 75% in de Eczema Area and Severity Index (EASI-75) vanaf baseline tot week 16. Belangrijke secundaire eindpunten omvatten het percentuele verschil in EASI en het wekelijks gemiddelde van de Peak Pruritus (PP) NRS vanaf baseline t.o.v. week 16.
LIBERTY AD ADOL
Simpson et al. (2020) voerden een dubbelblinde, 16 weken, placebo-gecontroleerde RCT uit in 45 centra, onder 251 adolescenten (12 t/m 17 jaar oud) met matig tot ernstig CE inadequaat gecontroleerd met topicale medicatie of voor wie topicale therapie niet geadviseerd was (aanvullende inclusie criteria: IGA ≥3; EASI ≥16; BSA ≥10%; gemiddelde PP NRS ≥4). De patiënten werden willekeurig toegewezen (1:1:1, gestratificeerd op ernst en lichaamsgewicht) aan een behandeling van 16 weken met dupilumab, 200 mg, na een initiële oplaaddosis van 400 mg (n = 43; baseline gewicht <60 kg), of dupilumab, 300 mg, na een initiële oplaaddosis van 600 mg (n = 39; baseline gewicht ≥60 kg), elke 2 weken; dupilumab, 300 mg (na een initiële oplaaddosis van 600 mg), elke 4 weken (n = 84); of placebo (n = 85). Primaire uitkomstmaten: het percentage patiënten met EASI-75 en IGA-score van 0-1 na 16 behandelweken.
LIBERTY AD PED-OLE
Studieresultaten werden meegenomen ter overweging. Gezien geen vergelijkend onderzoek werd er geen GRADE- en RoB-analyse gedaan.
Cork et al. (2021) [Open-label extension]
Kinderen (leeftijd ≥ 6 tot < 12 jaar) met ernstig CE werden ingeschreven in een wereldwijde, multicentrische, fase IIa, open-label, oplopende dosis, opeenvolgende cohortstudie en daaropvolgende open-label extension studie. Patiënten kregen een enkele dosis dupilumab van 2 of 4 mg/kg, gevolgd door een 8-weken durende farmacokinetische sampeling, en vervolgens wekelijks 2 of 4 mg/kg gedurende 4 weken (fase IIa), gevolgd door dezelfde wekelijkse behandeling (OLE). Primaire eindpunten waren het concentratie-tijdprofiel van dupilumab en treatment-emergent adverse events (TEAEs). Secundaire uitkomstmaten omvatten de EASI en de PP NRS.
Blauvelt et al. (2022) [Open-label extension, ongoing]
Deze open-label extension studie onderzocht de lange termijn veiligheid, werkzaamheid en farmacokinetiek van dupilumab bij adolescenten met matig tot ernstig CE die hebben deelgenomen aan eerdere dupilumab-studies. Patiënten die waren ingeschreven volgens het oorspronkelijke studieprotocol kregen subcutane dupilumab volgens een gewichtsafhankelijk schema (2 of 4 mg/kg per week). Na wijziging van het protocol werden patiënten overgezet op subcutane dupilumab 300 mg elke 4 weken (q4w), ongeacht het gewicht, en nieuw ingeschreven patiënten begonnen met dupilumab 300 mg q4w. Patiënten met een ontoereikende klinische respons (IGA- score van 0 of 1 werd niet bereikt) op het q4w-schema konden worden opgehoogd naar de goedgekeurde dupilumab-doseringsschema's van 200 of 300 mg elke 2 weken (lichaamsgewicht < 60 of ≥ 60 kg, respectievelijk). Bij patiënten van wie de IGA-score van 0 of 1 werd gehandhaafd gedurende een periode van 12 weken na week 40, werd gestopt met dupilumab, gecontroleerd op terugval, en indien nodig opnieuw gestart met dupilumab.
Risk of bias
Paller et al. (2022)
De studie is van redelijke kwaliteit, met een laag risico op selection bias, detection bias, performance bias en reporting bias. Er is echter een aanzienlijk risico op ‘attrition bias’ omdat n=49 patiënten in de placebogroep ‘noodmedicatie’ ontvingen (ten opzichte van n=16 in de dupilumab-groep). Aan hen werd voor de missende data, data toegerekend (imputed). De methode daarvoor is helder beschreven. Er is bij de GRADE-analyse niet afgewaardeerd voor ‘risk of bias’, behalve bij de veiligheidsuikomstmaten waar dit wel vertekening geeft van de resultaten (zie ook GRADE-conclusies). Een studieprotocol is beschikbaar op ClinicalTrials.gov. De Risk of bias analyse is te vinden onder conclusies dupilumab (kwaliteit van bewijs) .
Paller et al. (2020)
De studie is van redelijke kwaliteit, met een laag risico op selection bias, detection bias, attrition bias en reporting bias. Wegens een operationele fout waren 68 patiënten mogelijk niet geblindeerd, wat een hoog risico vermoedt op ‘performance bias’. Een gemodificeerde analyse (te beschouwen als sensitiviteitsanalyse), waarbij deze patiënten werden geëxcludeerd, gaf vergelijkbare resultaten. Door de auteurs werd gesteld dat hierdoor derhalve geen (performance) bias is opgetreden. Er is bij de GRADE-analyse niet afgewaardeerd voor ‘risk of bias’. Een studieprotocol is beschikbaar op ClinicalTrials.gov. De Risk of bias analyse is te vinden onder conclusies dupilumab (kwaliteit van bewijs).
Simpson et al. (2020)
De studie is van goede kwaliteit. Er werd door de werkgroep geen potentiële bias geïdentificeerd. Een studieprotocol is beschikbaar op ClinicalTrials.gov. De Risk of bias analyse is te vinden onder conclusies dupilumab (kwaliteit van bewijs) .
Beschrijving van de resultaten
Meta-analyse
Een meta-analyse van de 3 geïncludeerde trials werd verricht per uitkomstmaat.
De studies zijn beoordeeld op Risk of Bias volgens het Cochrane Handbook for Systematic Reviews of Interventions [Higgins, 2011] en de zekerheid van het bewijs middels de Grading of Recommendations Assessment, Development and Evaluation methodiek [Schünemann, 2013]. Alle studies konden op basis van de uitkomstmaten worden gepoold voor een meta-analyse met ReviewManager (RevMan versie 5.4), op de volgende uitkomstmaten:
- Proportie patiënten met IGA response 0 of 1 (0=clear; 1=almost clear) (cruciaal)
- Verandering in EASI score t.o.v. baseline na 16 behandelweken (cruciaal); proportie patiëntenmet minstens 75% verbetering van EASI t.o.v. baseline (EASI-75) (cruciaal); proportie patiënten met minstens 90% verbetering van EASI t.o.v. baseline (EASI-90) (cruciaal)
- Verandering in aangedane oppervlakte (gemeten met SCORAD) score t.o.v. baseline (cruciaal)
- Verandering in Patient-Oriented Outcome Measurement (POEM) score t.o.v. baseline (cruciaal)
- Verandering in wekelijks gemiddelde van de dagelijkse peak pruritus numeric rating scale (PP-NRS) scores t.o.v. baseline (cruciaal)
- Verandering in CDLQI (Children’s Dermatology Life Quality Index) of de IDQOL (Infants’ Dermatitis Quality of Life Index) scores t.o.v. baseline (belangrijk)
- Proportie patiënten met ≥1 treatment-emergent adverse event (TEAE); proportie patiënten ≥1 serious TEAE; proportie patiënten ≥1 TEAE resulterende in permanente discontinuatie van therapie (belangrijk)
Voor continue uitkomstmaten is gekozen voor het gemiddelde verschil (mean difference [MD]) tussen de dupilumab-groep en de placebogroep; voor dichotome uitkomstmaten de risk ratio (RR). In het geval van uitkomstmaten met verschillende meetmethoden is gekozen voor de standardized mean difference (SMD), echter dit was niet aan de orde.
Vanwege de klinische heterogeniteit in leeftijdsgroepen werden de effectmaten tevens weergegeven per subgroep (ingedeeld naar leeftijd). Voor de 2 trials met twee dupilumab-interventiegroepen (Paller et al. 2020; Simpson et al. 2020), met elk een verschillend toedieningsregime (q2w vs. q4w), zijn deze interventiegroepen gepoold vergeleken met de placebogroep. Per uitkomstmaat werd tevens een alternatieve analyse verricht per toedieningsregime vs. placebo (q2w vs. placebo; q4w vs. placebo).
In het geval van verhoogde statistische heterogeniteit (I2 > 50%) is middels alternatieve analyse gepoogd te bezien welke studie dit mogelijk veroorzaakt. De kwaliteit van het bewijs niet verlaagd op basis van een verhoogde heterogeniteit (>50%), maar deze heterogeniteit werd wel verklaard zodat duidelijk is voor welke subgroep(en) het algehele bewijs mogelijk minder of meer toepasselijk is. Zie ook ‘Conclusies (kwaliteit van bewijs)’ voor de effectmaat van het algehele bewijs per uitkomstmaat en de kwaliteit daarvan. Zie ook de ‘forest plots’ van de meta-analyses per uitkomstmaat voor tevens de effectmaat per leeftijdscategorie.
IGA
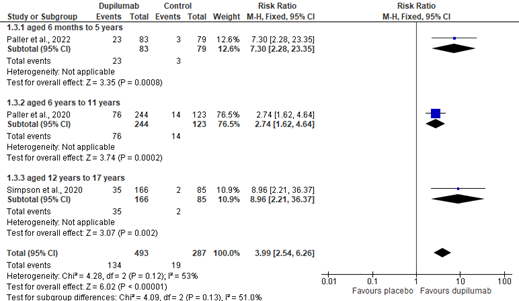
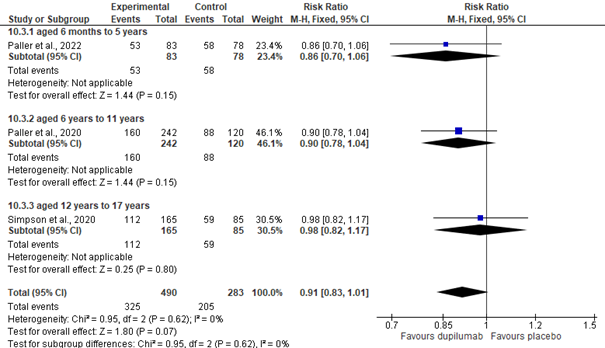
Meta-analyse liet zien dat in week 16 significant meer kinderen (6 maanden t/m 17 jaar) die dupilumab kregen, ten opzichte van placebo, een IGA van 0 of 1 hadden (summary RR= 3.99 [95% CI: 2.54, 6.26]) (zie figuur 1a). De alternatieve analyse gaf vergelijkbare significante uitkomsten. De statistische heterogeniteit tussen de trials was hoger dan op basis van kans verwacht mag worden (I2=53%). Mogelijk is dit te wijten aan de trial van Paller et al. 2020 (leeftijdsgroep: 6 t/m 11 jaar). Het gepoolde effect is mogelijk een onderschatting voor de jongste (Paller et al. 2022) en oudste (Simpson et al. 2020) leeftijdsgroepen; het excluderen van Paller et al. 2020 resulteerde in een significant overall effect van summary RR= 8.07 [95% CI 3.26, 19.95], met een lage heterogeniteit (I2=0%). Dit zou verklaard kunnen worden door een verschil in inclusie criteria ten opzichte van de andere trials met (IGA 4 vs. IGA 3-4).
EASI
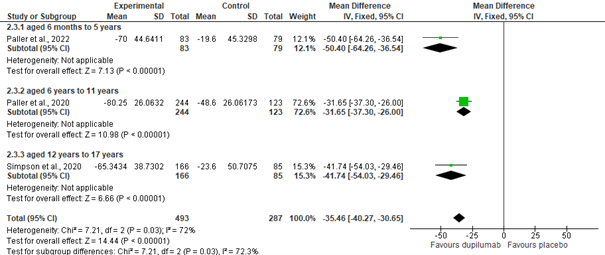
Meta-analyse liet zien dat, ten opzichte van baseline, in week 16 de het gemiddelde van de percentuele verandering (afname in EASI) significant groter was bij kinderen die dupilumab kregen vs. placebo (summary MD= -35.46 [95% CI: -40.27, -30.65])(zie figuur 2a). De alternatieve analyse gaf vergelijkbare significante uitkomsten. De statistische heterogeniteit tussen de trials was hoger dan op basis van kans verwacht mag worden (I2=72%). Mogelijk is dit te wijten aan de trial van Paller et al. 2020 (leeftijdsgroep: 6 t/m 11 jaar). Het gepoolde effect is mogelijk een onderschatting voor de jongste (Paller et al. 2022) en oudste (Simpson et al. 2020) leeftijdsgroepen; het excluderen van Paller et al. 2020 resulteerde in een significant overall effect van MD= 45.55 [95% CI -54.75, -36.36], met een lage heterogeniteit (I2=0%). Dit zou verklaard kunnen worden door een verschil in inclusie criteria ten opzichte van de andere trials (EASI ≥21 vs. EASI ≥16), met als resultaat absoluut hogere baseline waarden in interventie- en placebogroep.
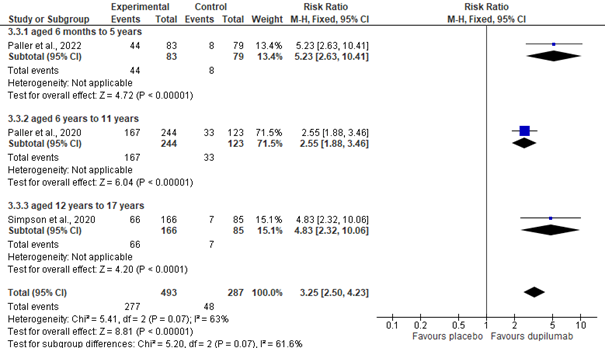
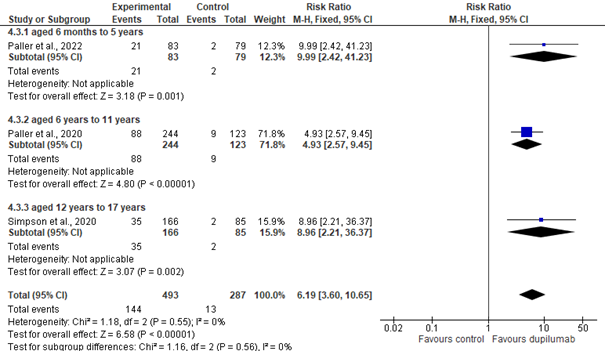
In week 16 hadden significant meer kinderen (6 maanden t/m 17 jaar) die dupilumab kregen, ten opzichte van placebo, een reductie van meer of gelijk aan 75% en 90% in EASI (EASI-75, EASI-90), respectievelijk; summary RR= 3.25 [95% CI 2.50, 4.23], en summary RR= 6.19 [95% CI: 3.60, 10.65](zie figuur 3a en 4a). Voor EASI-75 was de statistische heterogeniteit tussen de trials hoger dan op basis van kans verwacht mag worden (I2=63%). Mogelijk is dit te wijten aan de trial van Paller et al. 2020 (leeftijdsgroep: 6 t/m 11 jaar). In tegenstelling tot EASI-90, is het gepoolde effect voor EASI-75 mogelijk een onderschatting voor de jongste (Paller et al. 2022) en oudste (Simpson et al. 2020) leeftijdsgroepen; het excluderen van Paller et al. 2020 resulteerde in een significant overall effect van summary RR 5.02 [95% CI: 3.03, 8.31], met een lage heterogeniteit (I2=0%).
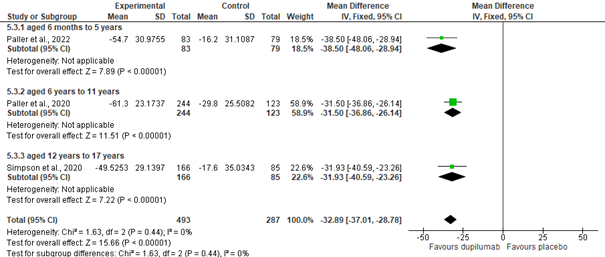
SCORAD
Meta-analyse liet zien dat, ten opzichte van baseline, in week 16 de het gemiddelde van de percentuele verandering (afname in SCORAD) significant groter was bij kinderen die dupilumab kregen vs. placebo (summary MD= -32.89 [95% CI: -37.01, -28.78])(zie figuur 5a). De alternatieve analyse gaf vergelijkbare significante uitkomsten. De statistische heterogeniteit was zeer laag (I2=0%).
POEM
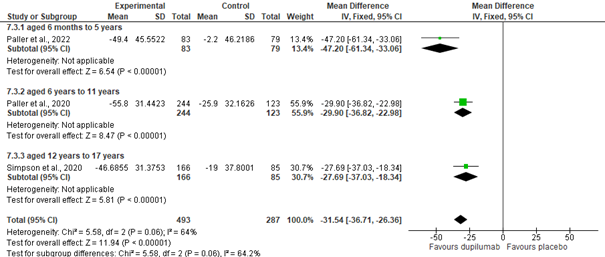
POEM werd in de trial van Paller et al. 2022 (6 maanden t/m 5 jaar) gescoord door de ouders/verzorgers. In Paller et al. 2020 (6 t/m 11 jaar) en Simpson et al. 2020 (12 t/m 17 jaar) werd deze score ingevuld door de patiënt. Meta-analyse liet zien dat, ten opzichte van baseline, in week 16 de gemiddelde verandering (afname in POEM) significant groter was bij kinderen die dupilumab kregen vs. placebo (summary MD= -7.81 [95% CI: -9.00, -6.62])(zie figuur 6a). De alternatieve analyse gaf vergelijkbare significante uitkomsten. De statistische heterogeniteit tussen de trials was hoger dan op basis van kans verwacht mag worden (I2=45%). Mogelijk is dit te wijten aan de trial van Simpson et al. 2020; het excluderen van deze trial resulteerde in een significant overall effect van MD= -8.48 [95% CI: -9.87, -7.09], met een zeer lage heterogeniteit (I2= 0%). Het is mogelijk dat adolescenten (12 t/m 17 jaar) de items van de POEM beter kunnen beoordelen dan kinderen van 6 t/m 11 jaar en ouders van kinderen met de leeftijd 6 maanden t/m 5 jaar oud, en deze daardoor hoger beoordelen (waardoor lagere gemiddelde verandering t.o.v. baseline).
Jeuk – Peak Pruritus NRS
Ten opzichte van baseline, in week 16, was de gemiddelde percentuele afname van het wekelijks gemiddelde van de dagelijkse PP-NRS score significant groter bij kinderen die dupilumab kregen vs. placebo (summary MD= -31.54 [95% CI:-36.71, -26.36]. De alternatieve analyse gaf vergelijkbare significante uitkomsten. De statistische heterogeniteit tussen de trials was hoger dan op basis van kans verwacht mag worden (I2=64%) (zie figuur 6a). Exclusie van Paller et al. 2022 gaf een summary MD van -29.12 [95% CI: -34.68, -23.56], met een zeer lage statistische heterogeniteit (I2= 0%). Mogelijk is de reden hiervan dat de PP-NRS werd in de trial van Paller et al. 2022 (6 maanden t/m 5 jaar) gescoord door de ouders/verzorgers. In Paller et al. (6 t/m 11 jaar) en Simpson et al. (12 t/m 17 jaar) werd deze score ingevuld door de patiënt. De schaal was hetzelfde (11-punts Likertschaal).
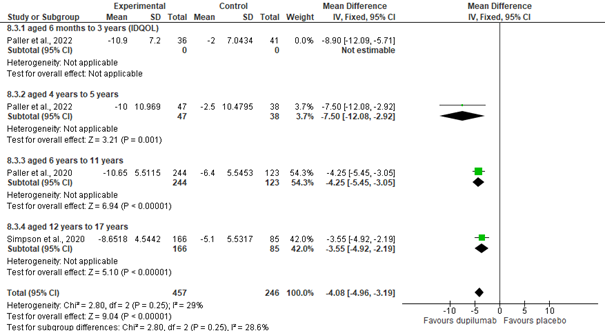
Kwaliteit van leven
In week 16 was de gemiddelde afname van de CDLQI score significant groter bij kinderen die dupilumab kregen vs. placebo (summary MD= -4.08 [95% CI:-4.08, -3.19]), met een redelijk lage, waarschijnlijk niet relevante, heterogeniteit (I2= 29%), zie figuur 7a. De alternatieve analyse gaf vergelijkbare significante uitkomsten.
CDLQI werd in alle trials gescoord door de participanten. Echter, voor de leeftijdsgroep 6 maanden t/m 3 jaar (deelpopulatie van Paller et al. 2022) werd voor de beoordeling van de kwaliteit van leven de IDQOL gescoord door de ouders/verzorgers. Derhalve werd deze subgroep niet meegenomen in de meta-analyse. In deze subgroep werd een significante gemiddelde afname van de IDQOL gevonden (MD= -8.90 [95% CI: -12.09, -5.71], p<.00001), zie figuur 7a.
Veiligheid
Voor de TEAE’s zijn, in week 16, de proporties per groep (dupilumab vs. placebo) vergeleken voor: patiënten met A. ≥1 TEAE, B. ≥1 serious TEAE, en C. ≥1 TEAE resulterende in permanente discontinuatie van therapie. Ten aanzien van A. liet meta-analyse geen significant verschil zien (summary RR= 0.91 [0.83, 1.01]), zie figuur 8a. Alternatieve subanalyse van het ‘elke 4 weken toedieningsregime’ liet wel een significant verschil zien ten voordele van de dupilumab-groep (summary RR= 0.89 [95% CI: 0.79, 0.99]). Voor B. werd een significant overall verschil gezien ten voordele van dupilumab (summary RR= 0.23 [95%CI:0.06, 0.94], zie figuur 9a. Sub-analyses per toedieningsregime (elke 4 weken; elke 2 weken) waren niet significant verschillend voor dupilumab-groep vs. placebogroep. Voor C. werd geen significant verschil gevonden (summary RR= 0.46 [95% CI: 0.12, 1.82]), zie figuur 10a. Tabel 2 (in bijlagen) geeft een overzicht van de bijwerkingen bij de bovenstaande uitkomstmaten, per studie.
Aanvullende veiligheidsgegevens uit trials AD PRESCHOOL, AD PEDS en AD ADOL (16 weken)
Virale gastro-enteritis en cariës kwamen meer voor in de dupilumab-groep dan in de placebo-groep. Echter, het aantal patiënten met deze AE’s was te klein of hier conclusies op te baseren (Paller, Simpson, et al., 2022), de andere trials kwamen nog niet eerder tot deze bevinding.
Evenals in de RCT met de leeftijdsgroep 6 maanden t/m 5 jaar (Paller, Simpson, et al., 2022), vond een gepoolde sub-analyse (Paller, Beck, et al., 2022) van de trials in kinderen/adolescenten van 6 t/m 11 jaar en 12 t/m 17 jaar met matig tot ernstig eczeem (Paller et al., 2020; Simpson et al., 2020), dat dupilumab niet het totale risico op infectie verhoogde, en in de dupilumab-groepen werd een lagere incidentie gezien van huidinfecties vergeleken met placebo. Eveneens deden zich geen serieuze infecties voor.
De incidentie van conjunctivitis (van verschillende etiologie) van mild tot matige ernst was hoger in de dupilumab-groepen van alle RCT’s, consistent met dupilumab trials in trials met volwassen (Bansal et al., 2021; Paller et al., 2020; Paller, Simpson, et al., 2022; Simpson et al., 2020). Enkel eenmalig resulteerde (bacteriële) conjunctivitis in tijdelijke discontinuatie van therapie (Paller et al., 2020). Injectieplaats reacties waren meer prevalent in de dupilumab-groepen van kinderen en adolescenten in de leeftijdsgroep 6 t/m 17 jaar (Paller et al., 2020; Paller, Simpson, et al., 2022), in de trial met de jongste groep kinderen werd ten aanzien van injectieplaats reacties geen verschil waargenomen en was de incidentie zeer laag (Paller, Simpson, et al., 2022)
Een lichte verhoging in ‘mean eosinophil count’ werd gezien in de dupilimab-groep, zonder klinische relevantie (Paller, Simpson, et al., 2022), vergelijkbaar als ook gerapporteerd op basis van de andere geïncludeerde trials met kinderen en adolescenten (Paller et al., 2021; Siegfried et al., 2021).
Lange termijn gegevens - open-label extension trials (52 weken)
AD PED-OLE (52 weken)
Cork et al. (2021) [6 t/m 11 jaar]
Van de 38 geïncludeerde kinderen voltooiden 37 de IIa-fase en 33 gingen door naar de OLE-studie. TEAEs waren meestal mild tot matig en van voorbijgaande aard; geen enkele resulteerde tot het stopzetten van de behandeling. De meest gemelde TEAEs waren neus-keelholteontsteking (2 mg/kg, 47%; 4 mg/kg, 56%) en verergering van AD (respectievelijk 29% en 13%). Dupilumab elke 4 weken verbeterde AD met verdere verbeteringen tot week 52.
In de OLE-studie gebruikte 82% (2 mg/kg) en 94% (4 mg/kg) van de kinderen gelijktijdige topicale medicatie, waarbij de meerderheid (65% en 69%) gebruik maakte van hoog-potente (groep III) TCS.
Het gemiddelde EASI- en PP-NRS (Peak Pruritus Numeric Rating Scale)-score verbeterden respectievelijk met -37%/-33% en -17%/-20% na week 2 (fase IIa) en met -92%/-84% en -70%/-58% na week 52 (OLE).
Het percentage patiënten dat EASI-75 of IGA 0-1 behaalde bij patiënten die dupilumab kregen met doses van 2 mg/kg en 4 mg/kg was respectievelijk 94% en 75% bij week 52 van de OLE-studie voor EASI-75, en voor IGA 0-1 respectievelijk 76% en 25% bij week 52 van de OLE-studie.
Blijvende verbeteringen werden ook waargenomen in EASI-50, EASI-90 en SCORAD (en het percentage lichaamsoppervlakte aangetast door CE) in de OLE-studie tot week 52. CE-symptomen en de kwaliteit van leven zoals beoordeeld door POEM (Patient-Oriented Eczema Measure) en CDLQI (Children's Dermatology Life Quality Index) toonden verbetering vanaf baseline tot week 48 van de OLE-studie.
De werkzaamheid en veiligheid Paller et al. 2020 (gerandomiseerde, placebogecontroleerde fase III-studie bij kinderen van ≥ 6 tot < 12 jaar met ernstig CE die dupilumab kregen in combinatie met gelijktijdig gebruik van TCS) met een grotere sample size van deze patiëntenpopulatie dan in deze studie, waren over het algemeen vergelijkbaar.
Er waren geen bijwerkingen die resulteerden in het stopzetten van de behandeling, en geen van de gemelde ernstige TEAEs werd beschouwd als gerelateerd aan dupilumab. Huidinfecties deden zich voornamelijk voor bij patiënten die op het moment van het optreden van TEAE nog niet met dupilumab werden behandeld. De incidentie van conjunctivitis in de OLE-studie bij kinderen van ≥ 6 tot < 12 jaar (vijf tot 21 patiënten per 100 patiëntjaren) was vergelijkbaar met die bij adolescenten (negen tot 10 patiënten per 100 PYs) en volwassenen (12 patiënten per 100 PYs).
Blauvelt et al. (2022) [12 t/m 17 jaar]
Gegevens van 294 patiënten (gemiddelde leeftijd 14,7 jaar) werden geanalyseerd, waarvan 102 (34,7%) de 52-weken follow-up hadden voltooid op het moment van de databasevergrendeling.
De gemiddelde procentuele veranderingen in EASI toonden aanzienlijke verbetering vanaf de start van de studie tot week 52, met een gemiddelde procentuele verandering van -83,5% ± 23,5 in EASI. Respectievelijk 93,1%, 81,2% en 56,4% vertoonden ten minste een verbetering van 50%, 75% of 90% de EASI. De gemiddelde procentuele veranderingen in SCORAD vertoonden aanzienlijke verbetering vanaf de start van de studie tot week 52, met een gemiddelde procentuele verandering van -65,0% ± 21,3 in SCORAD bij week 52. De gemiddelde verandering ± SD van de CDLQI vanaf de start van de studie was -11,8 ± 6,7 bij week 52.
Bij de meeste patiënten (70,9%) moest de dosis van dupilumab opgewaardeerd worden volgens het goedgekeurde behandelingsregime. Het percentage patiënten (na dosisverhoging) dat een IGA-score van 0 of 1 of een verbetering van 75% in EASI vertoonde, nam in de loop van de tijd toe (respectievelijk 35,7% en 51,9% 48 weken na de eerste dosisverhoging). In week 52 had 29,4% van de patiënten gedurende 12 weken IGA-score 0 of 1 behouden en was gestopt met medicatie; 56,7% had een terugval en werd vervolgens opnieuw behandeld, met een gemiddelde tijd tot herstart van 17,5 (± SD 17,3) weken.
Het hoge percentage patiënten dat dosisverhoging nodig had vanwege een onvoldoende respons op de dosering van 'elke 4 weken', ondersteunt het 'elke 2 weken' regime als optimaal voor deze leeftijdsgroep. Tevens ervoer de meerderheid van de patiënten die de medicatie stopten nadat ze gedurende 12 weken IGA-score 0 of 1 hadden behouden, een terugkeer van de ziekte, wat suggereert dat het voortzetten van de dupilumab-behandeling nodig is om de werkzaamheid te handhaven.
Het lange termijn veiligheidsprofiel van dupilumab was vergelijkbaar met dat bij volwassenen en consistent met het bekende veiligheidsprofiel. De meeste behandeling-gerelateerde bijwerkingen waren mild tot matig. Bij week 52 had 42,7% van de patiënten een IGA-score van 0 of 1.
Concluderend, in lijn met de resultaten van korte termijnbehandeling, toonde lange termijn behandeling met dupilumab een acceptabel veiligheidsprofiel en resulteerde in toenemende klinische voordelen bij voortgezette behandeling in de loop van de tijd.
Dupilumab – Forest plots
Figuur 1a. Meta-analyse IGA 0 of 1 (week 16)
Figuur 2a. Meta-analyse percentuele gemiddelde verandering in EASI (week 16 t.o.v. baseline)
Figuur 3a. Meta-analyse proportie patiënten met EASI-75 (week 16)
Figuur 4a. Meta-analyse proportie patiënten met EASI-90 (week 16)
Figuur 5a. Meta-analyse percentuele gemiddelde verandering in SCORAD (week 16 t.o.v. baseline)
Figuur 6a. Meta-analyse percentuele gemiddelde verandering in POEM (week 16 t.o.v. baseline)

Figuur 6a. Meta-analyse: gemiddelde percentuele verandering in wekelijks gemiddelde van dagelijkse PP-NRS score (week 16 t.o.v. baseline)
Figuur 7a. Meta-analyse gemiddelde verandering in CDLQI en uitkomst + subgroep analyse IDQOL (week 16 t.o.v. baseline)
Tabel 2. Bijwerkingen (week 16)
|
|
|
≥1 TEAE |
≥1 serious TEAE |
≥1 TEAE → discontinuation |
|
Paller 2022 |
Dupilumab |
64% |
0 |
1 (1%)
1. atopic dermatitis flare |
|
Placebo |
74% |
4 (5%)
1. atopic dermatitis + infected dermatitis 2. hypersensitivityone 3. staphylococcal bacteraemia 4. staphylococcal cellulitis. |
1 (1%)
1. nightmares due to blood draws |
|
|
Paller 2020 |
Dupilumab |
66.1%
|
2 (0.8%)
Dupilumab 100/200 mg every 2 weeks + TCS: 1. 1 event of food allergy 2. 1 event of conjunctivitis bacterial |
2 (0.8%)
Dupilumab 300 mg every 4 weeks + TCS: 1. 1 event of food allergy 2. 1 event of urinary tract infection |
|
Placebo |
73.3% |
2 (1.7%)
1. 1 event of asthma 2. 1 event of dermatitis atopic |
2 (1.7%)
1. 1 event of asthma 2. 1 event of dermatitis atopic |
|
|
Simpson 2020 |
Dupilumab |
67.9% |
0
|
0 |
|
Placebo |
69.4% |
1 (1.2%)
1. appendicitis
|
1 (1.2%)
1. AD exacerbation |
Figuur 8a. Meta-analyse: proportie patiënten met ≥1 TEAE’s (week 16)
Figuur 9a. Meta-analyse: proportie patiënten met ≥1 serious TEAE’s (week 16)
Figuur 10a. Meta-analyse: proportie patiënten met ≥1 TEAE’s resulterende in permanente discontinuatie (week 16)
Samenvatting literatuur tralokinumab (resultaten)
Beschrijving van de studies
ECZTRA 6
Paller et al. (2023) voerden een dubbelblinde,16 weken, placebo-gecontroleerde RCT uit in 72 centra, onder 301 adolescenten in de leeftijdscategorie 12 t/m 17 jaar met matig tot ernstig constitutioneel eczeem (IGA ≥3; EASI ≥16; BSA ≥10%; Adolescent Worst Pruritus Numeric Rating Scale (NRS) gemiddelde van ≥ 4 in de week voorafgaande aan baseline; voorgeschiedenis van onvoldoende respons op TCS en/of TCI, of medisch niet mogelijk).
Patiënten werden gerandomiseerd (1:1:1) in de groepen a) tralokinumab 150 mg [n=98] of b) tralokinumab 300 mg [n=97] of c) placebo [n=94], elke 2 weken na een initiële oplaaddosis (dubbele interventie dosering) voor 16 weken (initiële fase).
Patiënten die in week 16 geen IGA-score van 0 of 1 behaalden, geen EASI-75 bereikten, of die noodmedicatie kregen tussen week 2 en 16, werden overgeschakeld naar een behandeling met ‘open-label’ tralokinumab, 300 mg, elke 2 weken. Het bereiken van IGA-score 0 of 1 of EASI-75 waren de primaire uitkomstmaten van de initiële fase en vormden beiden tevens de enige uitkomstmaten, naast ‘adverse events’, van interesse die werden gemeten tijdens zowel de onderhoudsfase als de open-label arm, die beide 52 weken duurden.
Noodmedicatie werd gebruikt in de initiële behandelfase door, respectievelijk, 33.7% en 29.9% van de patiënten die tralokinumab, 150 mg, en tralokinumab, 300 mg, kregen in vergelijking met 56.4% in de placebogroep (voornamelijk TCS van elke sterkte). Noodmedicatie werd sneller geïnitieerd bij patiënten die tralokinumab, 150 mg, kregen, in vergelijking met tralokinumab, 300 mg.
Voor de onderhoudsfase (=week 16 t/m week 52), werden patiënten uit de tralokinumab groepen die wel IGA 0 of 1 en/of EASI-75 hadden in week 16 [n=50] gererandomiseerd (1:1) met hun originele dosering (150 mg of 300 mg) in de groepen ‘elke 2 weken’ en ‘elke 4 weken’. In de ‘elke 4 weken’ groep kregen patiënten tussendoor elke 4 weken placebo toegediend, ten behoeve van blindering. IGA en EASI werden gescoord t/m week 52. Patiënten die in deze fase noodmedicatie kregen (na week 2) en/of permanent uitvielen of die om een andere reden dan noodmedicatie werden geswitcht naar open-label, werden als non-responder beschouwd. Twintig patiënten werden pas in week 52 als non-responder beschouwt en overgezet naar open-label; zij werden niet meegenomen in de open-label analyses. In tegenstelling tot het initiële protocol, was er in de onderhoudsfase geen placebogroep.
In totaal werden 214 patiënten, aan het einde van week 16, geswitcht naar open-label, 300 mg, elke 2 weken (met de optie tot gebruik van zwak- of matig potente TCS of TCI). IGA en EASI werden gescoord t/m week 52.
Risk of bias
De risk of bias beoordeling heeft betrekking op de initiële fase van deze trial (week 0-16).
De studie is van goede kwaliteit. Er werd door de werkgroep geen potentiële bias geïdentificeerd. Een studieprotocol is beschikbaar op ClinicalTrials.gov. De Risk of bias analyse is te vinden onder conclusies dupilumab (kwaliteit van bewijs).
Beschrijving van de resultaten
De studie is beoordeeld op Risk of Bias volgens het Cochrane Handbook for Systematic Reviews of Interventions [Higgins, 2011] en de zekerheid van het bewijs middels de Grading of Recommendations Assessment, Development and Evaluation methodiek [Schünemann, 2013]. GRADE beoordeling werd gedaan voor de uitkomstmaten vastgesteld in de PICO (als mogelijk):
- Proportie patiënten die IGA response 0 of 1 (0=clear; 1=almost clear) behaalt (cruciaal)
- Verandering in EASI-score t.o.v. baseline na 16 behandelweken (cruciaal); proportie patiënten dat 75% verbetering van EASI t.o.v. baseline behaalt (EASI-75) (cruciaal); proportie patiënten dat 90% verbetering van EASI t.o.v. baseline behaalt (EASI-90) (cruciaal)
- Verandering in aangedane oppervlakte (gemeten met SCORAD) score t.o.v. baseline (cruciaal)
- Verandering in Patient-Oriented Outcome Measurement (POEM) score t.o.v. baseline (cruciaal)
- Verandering in wekelijks gemiddelde van de Adolescent Worst Pruritus (AWP)- NRS (numeric rating scale) score t.o.v. baseline (cruciaal)
- Verandering in CDLQI (Children’s Dermatology Life Quality Index) score t.o.v. baseline (belangrijk)
- Proportie patiënten met ≥1 treatment-emergent adverse event (TEAE); proportie patiënten ≥1 serious TEAE (belangrijk)
Voor continue uitkomstmaten is gekozen voor het gemiddelde verschil (mean difference [MD]) tussen de tralokinumab-groep en de placebogroep; voor dichotome uitkomstmaten de risk ratio (RR). In het geval van uitkomstmaten met verschillende meetmethoden is gekozen voor de standardized mean difference (SMD), echter dit was niet aan de orde.
IGA
Initiële fase
Significant meer adolescenten die tralokinumab 150 mg (21.4% [n=21/98]; t.o.v. placebo: RR=5.05 [95% CI: 1,80, 14.12], P=0.002) en 300 mg (17.5% [n=17/97]; t.o.v. placebo: RR= 4.12 [95%CI: 1.22, 11.79], P=0.008) elke 2 weken kregen, ten opzichte van placebo (4.3% [n=4/94]) hadden in week 16 een IGA van 0 of 1 zonder het gebruik van noodmedicatie. Voor de tralokinumab-groepen gepooled was dit 20,5% [n=38/195] (t.o.v. placebo: RR = 4.58 [95% CI: 1.68, 12.45], P=0.003).
Onderhoudsfase
In week 52, werd een IGA-score van 0 of 1 behouden zonder het gebruik van noodmedicatie bij 62.9% (n=22/35) van de patiënten in alle tralokinumab-groepen (gepoold).
Open-label arm
In week 52 werd alsnog een IGA-score van 0 of 1 bereikt, zonder gebruik van noodmedicatie, door respectievelijk 35,3% (n=23/65) van de patiënten die overstapten van de tralokinumab, 150 mg, elke 2 weken groep; 31,4 % (n=22/70) van de tralokinumab, 300 mg, elke 2 weken groep; en 27.8% (n=22/79) van de placebogroep.
EASI
Initiële fase
Significant meer adolescenten die tralokinumab 150 mg (28.6% [n=28/98]; t.o.v. placebo: RR=4.48 [95% CI: 1,80, 14.12], P=0.002) en 300 mg (27.8% [n=27/97]; t.o.v. placebo: RR= 4.36 [95% CI: 1.89, 10.08], P=0.0006) elke 2 weken kregen, ten opzichte van placebo (6.4% [n=6/94]), hadden in week 16 een reductie van meer of gelijk aan 75% (EASI-75) zonder gebruik van noodmedicatie. Voor de tralokinumab-groepen gepoold was dit 28.2% [n=55/195] (t.o.v. placebo: RR = 4.42 [95% CI: 1.97, 9.89], P=0.0003).
EASI-90 werd tevens door significant meer adolescenten die tralokinumab 150 mg (19,4% [n=19/98]; t.o.v. placebo: RR= 4.56 [95% CI: 1.61, 12.90], P=0.004) en 300 mg (17.5% [n=17/97]; t.o.v. placebo: RR= 4.12 [95% CI: 1.44, 11.79], P= 0.008) elke 2 weken kregen bereikt, vergeleken met placebo (4.3% [n=4/94]) zonder het gebruik van noodmedicatie. Voor de tralokinumab-groepen gepoold was dit 18.5% [n=36/195] (t.o.v. placebo: RR 4.34 [95% CI: 1.59,11.83], P= 0.004).
Ten opzichte van baseline, in week 16, was het gemiddelde van de percentuele verandering (afname in EASI) significant groter voor adolescenten die tralokinumab kregen vs. placebo (150 mg-groep: MD= -7.21 [95% CI:-10.95, -3.47], P= 0.0002; 300 mg-groep: MD= -7.05 [95% CI: -10.87, -3.23], P= 0.0003; gepoold: MD= -7.13 [95% CI:-10.42, -3.84], P<0.0001). Patiënten die noodmedicatie kregen werden bij bovenstaande analyses niet geëxcludeerd, het ging hier overwegend om het gebruik van TCS (elke sterkte). Na exclusie van patiënten die noodmedicatie kregen (n=115) werd tevens een significante afname gezien ten voordele van de tralokinumab-groepen (gepoold: MD= -9.76 [95% CI: -14.46, -5.06], P<0.0001), hoewel dat resulteert in een redelijk kleine groep patiënten (met name placebogroep van n=36).
Onderhoudsfase
In week 52, werd EASI-75 behouden zonder het gebruik van noodmedicatie bij 53.2% (n=25/47) van de patiënten in alle tralokinumab-groepen (gepoold).
Open-label arm
In week 52, werd EASI-75 alsnog behaald, zonder gebruik van noodmedicatie, door 63.1% (n=41/65) van de patiënten die overstapten van de tralokinumab, 150 mg, elke 2 weken groep; 52.9% uit de tralokinumab, 300 mg, elke 2 weken groep; en 65.8% (n=52/79) uit de placebogroep.
SCORAD
Ten opzichte van baseline, in week 16, was het gemiddelde van de percentuele verandering (afname) in SCORAD significant groter voor adolescenten die tralokinumab kregen vs. placebo (150 mg-groep: MD= -18.00 [95% CI: -25.53, -10.47], P<0.00001; 300 mg-groep: MD= -19.60 [95% CI: -27.13, -12.07], P<0.00001; gepoold: MD= -18.80 [95% CI: -25.55, -12.04], P<0.00001).
POEM
Het percentage patiënten dat in week 16 ten opzichte van baseline klinisch significante verbeteringen ervoer in de Patient Oriented Eczema Measure (een afname van ≥6 punten) was hoger bij de groep die tralokinumab, 150 mg kreeg (38.7% [n=44/94]; t.o.v. placebo: RR= 3.70 [95% CI: 1.89, 7.22], P= 0.0001), en in de groep die tralokinumab, 300 mg kreeg (46.8% [n=44/94]; t.o.v. placebo: RR= 4.47 [95% CI: 2.32, 8.61], P<0.00001), dan in de placebogroep (10.5% [n=9/86]). Voor de tralokinumab-groepen gepoold was dit 42.8% [n=80/187] (t.o.v. placebo: RR= 4.09 [95% CI: 2.16, 7.75], P< 0.0001).
Jeuk - Adolescent Worst Pruritus NRS
Het percentage patiënten dat in week 16 ten opzichte van baseline klinisch significante verbeteringen ervoer in het wekelijks gemiddelde van de Adolescent Worst Pruritus NRS (een afname van ≥4 punten) was hoger bij de groep die tralokinumab, 150 mg kreeg (23.2% [n=22/95]; t.o.v. placebo: RR= 6.95 [95% CI: 2.15, 22.41], P= 0.001), en in de groep die tralokinumab, 300 mg kreeg (25.0% [n=24/96]; t.o.v. placebo: RR= 7.50 [95% CI: 2.34, 24.05], P= 0.0007), dan in de placebogroep (3.3% [n=3/90). Voor de tralokinumab-groepen gepoold was dit 24.1% [n=46/191] (t.o.v. placebo: RR= 7.23 [95% CI: 2.31, 22.61], P= 0.0007).
Kwaliteit van leven
Ten opzichte van baseline, in week 16, was het gemiddelde verandering (afname) in CDLQI significant groter voor adolescenten die tralokinumab kregen vs. placebo (150 mg-groep: MD= -2.00 [95% CI: -3.81, -0.19], P= 0.03; 300 mg-groep: MD= -2.60 [95% CI: -4.41, -0.79], P= 0.005; gepoold: MD= -2.30 [95% CI: -3.90, -0.69], P= 0.005).
Veiligheid
Initiële fase
Tralokinumab werd goed verdragen, waarbij het merendeel van de bijwerkingen mild tot matig van ernst was. De meest voorkomende bijwerkingen waren bovenste luchtweginfecties, atopische dermatitis (verergering van de ziekte), reactie op de injectieplaats, astma en hoofdpijn. In de initiële behandelperiode waren de percentages patiënten met 1 of meer bijwerkingen (≥1 AE) vergelijkbaar bij degenen die tralokinumab, 150 mg, elke 2 weken kregen (67,3%); tralokinumab, 300 mg, elke 2 weken (64,9%); en placebo (61,7%). Ten opzichte van placebo: 150 mg-groep: RR= 1.09 [95% CI: 0.88, 1.35], P= 0.42; 300 mg-groep: RR= 1.05 [95% CI: 0.85, 1.31], P= 0.64; gepoold: RR= 1.07 [95% CI: 0.89, 1.29], P= 0.47. Er waren weinig ernstige bijwerkingen die gerelateerd werden aan de behandeling (serious treatment-emergent adverse events [TEAE’s]), Respectievelijk had 3.1%, 1.0% en 5.3% ≥1 serious TEAE (gepoolde dosis: RR= 0.39 [95% CI: 0.11, 1.40], P= 0.15) en slechts 1 bijwerking die leidde tot het stoppen van de behandeling (niet beschouwd als gerelateerd aan de behandeling).
Aanvullende veiligheidsgegevens initiële fase
Het percentage patiënten met conjunctivitis (als bijwerking van speciaal belang), inclusief de 4 ‘preferred terms’ conjunctivitis, bacteriële conjunctivitis, virale conjunctivitis en allergische conjunctivitis, was laag en vergelijkbaar tussen de twee tralokinumab-groepen vs. de placebogroep (150 mg-groep: 4.1%; 300 mg-groep: 3.1%; en placebogroep: 2.1%). Keratitis werd gerapporteerd bij 1 patiënt in de tralokinumab 300 mg-groep.
De frequenties van andere bijwerkingen van speciaal belang, zoals eczema herpeticum en huidinfecties die systemische behandeling vereisten, waren laag in alle behandelingsgroepen. Er werd geen toename van acne waargenomen bij patiënten die tralokinumab kregen (gepoolde doses: 1,5%) in vergelijking met degenen die placebo kregen (4,5%). Het percentage patiënten met virale en bacteriële huidinfecties (1,5% vs. 0%), misselijkheid (2% vs. 0%) en herpes simplex-infecties (1,0% vs. 2,1%) was laag in de initiële fase.
In de initiële fase werd bij enkele patiënten die tralokinumab kregen, tijdelijk een verhoging van eosinofielen waargenomen. Voortgezette behandeling in de open-label fase liet geen verdere toenames in de loop van de tijd zien en er werden geen klinische gevolgen, gerelateerd aan eosinofilie, gerapporteerd. Gedurende de gehele studie werden er geen bijwerkingen gerelateerd aan eosinofilie gemeld.
Onderhoudsfase en open-label arm
De soorten en frequenties van bijwerkingen die werden waargenomen tijdens de onderhoudsfase in de open-label arm (t/m 52 weken) waren vergelijkbaar met die in de initiële fase.
Zoeken en selecteren
dupilumab
PICO bij alle uitgangsvragen
Om de uitgangsvraag te beantwoorden is een systematische literatuuranalyse uitgevoerd. Voor dit onderzoek is de volgende PICO opgesteld:
(P) Kinderen met constitutioneel eczeem (leeftijd: 0 t/m 18 jaar)
(I) JAK-remmer of biologic (zie onder)
(C) Geen behandeling, placebo, conventionele systemische therapie, biologic/JAK-remmer
(O) Zie uitkomstmaten
Biologics: dupilumab, tralokinumab
JAK-remmers: baricitinib, upadacitinib, abrocitinib
Uitkomstmaten bij alle uitgangsvragen
De werkgroep definieerde de uitkomstmaten als volgt en hanteerde de in de studies gebruikte definities.
Primair (cruciaal):
- EASI-score, EASI-75, EASI-90
- SCORAD-index
- POEM-score
- Jeuk (PP-NRS of zoals gerapporteerd in studie)
Secundair (belangrijk):
- Kwaliteit van leven: (C)DLQI
- Veiligheid (o.a. serious adverse events [ziekenhuis opname, mortaliteit], andere bijwerkingen)
- Kosteneffectiviteit
Zoeken en selecteren voor alle uitgansvragen
Er werd een systematische zoekstrategie uitgevoerd in de elektronische databases Embase en Medline. De zoekstrategie is toegevoegd onder verantwoording.
Studies werden geïncludeerd wanneer deze overeenkwamen met de elementen van de PICO en aan de volgende in- en exclusiecriteria voldeden:
Inclusie:
- Vergelijkend onderzoek
- Systematische reviews o.b.v. vergelijkend onderzoek
- Full-tekst geschreven in het Nederlands of Engels
- Controlegroep: placebo, andere behandeling of geen behandeling
- Niet vergelijkend onderzoek wordt op hoofdlijnen beschreven bij de overwegingen als er geen RCT’s zijn, of als aanvulling als er wel RCT’s zijn en dit van toegevoegde waarde wordt geacht (NB: in dit geval geen analyse middels GRADE)
Exclusie:
- Interventies die niet in Nederland beschikbaar voor kinderen of waarvan dat niet verwacht wordt dat ze in de nabije toekomst beschikbaar komen
- Studies waarin verschillende dosis of behandelfrequenties zijn onderzocht (zonder controle groep)
- Publicaties die analyses presenteren over subgroepen op basis van een co-morbiditeit naast constitutioneel eczeem, mits deze studies geen aanvullende effectiviteit- en veiligheidsanalyses presenteren.
Resultaten
Er werden in totaal 158 studies/zoekresultaten geïncludeerd op basis van beoordeling van titel en abstract, waarvan 84 zoekresultaten over dupilumab. Een groot deel hiervan werd geëxcludeerd wegens dat dit abstract-/posterpresentaties waren van een conferentie/meeting. Voor al deze resultaten werd achteraf gekeken of de gepubliceerde trial was geïncludeerd. Als dit niet zo was en er werd een volledige publicatie gevonden werd deze alsnog geïncludeerd en aangeboden voor full-tekstscreening.
De gevonden systematische reviews (specifiek over dupilumab [=4], en overige systematische reviews over meerdere middelen/middelen groepen [=6]) bleken na full-tekst beoordeling niet bijdragende voor het beantwoorden van de onderzoeksvraag inzake dupilumab.
Op basis van PICO en in- en exclusiecriteria werden er uiteindelijk 3 RCT’s geïncludeerd. Publicaties op basis van deze trials die rapporteren over gepoolde en/of post-hoc analyses, of relevante reviews werden meegenomen voor de overwegingen. Daarnaast werd een open-label extensie (OLE) trial meegenomen voor de overwegingen.
Referenties
- Referenties dupilumab
- Bansal, A., Simpson, E. L., Paller, A. S., Siegfried, E. C., Blauvelt, A., de Bruin-Weller, M., . . . Ardeleanu, M. (2021). Conjunctivitis in Dupilumab Clinical Trials for Adolescents with Atopic Dermatitis or Asthma. American Journal of Clinical Dermatology, 22(1), 101-115. doi:doi:10.1007/s40257-020-00577-1
- Blauvelt, A., Guttman-Yassky, E., Paller, A. S., Simpson, E. L., Cork, M. J., Weisman, J., . . . Bansal, A. (2022). Long-Term Efficacy and Safety of Dupilumab in Adolescents with Moderate-to-Severe Atopic Dermatitis: Results Through Week 52 from a Phase III Open-Label Extension Trial (LIBERTY AD PED-OLE). Am J Clin Dermatol, 23(3), 365-383. doi:10.1007/s40257-022-00683-2
- Cork, M. J., Thaçi, D., Eichenfield, L. F., Arkwright, P. D., Sun, X., Chen, Z., . . . Bansal, A. (2021). Dupilumab provides favourable long-term safety and efficacy in children aged ≥ 6 to < 12 years with uncontrolled severe atopic dermatitis: results from an open-label phase IIa study and subsequent phase III open-label extension study. Br J Dermatol, 184(5), 857-870. doi:10.1111/bjd.19460
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
- Paller, A. S., Beck, L. A., Blauvelt, A., Siegfried, E. C., Cork, M. J., Wollenberg, A., . . . Cyr, S. L. (2022). Infections in children and adolescents treated with dupilumab in pediatric clinical trials for atopic dermatitis—A pooled analysis of trial data. Pediatric Dermatology, 39(2), 187-196. doi:doi:10.1111/pde.14909
- Paller, A. S., Siegfried, E. C., Thaçi, D., Wollenberg, A., Cork, M. J., Arkwright, P. D., . . . Shumel, B. (2020). Efficacy and safety of dupilumab with concomitant topical corticosteroids in children 6 to 11 years old with severe atopic dermatitis: A randomized, double-blinded, placebo-controlled phase 3 trial. Journal of the American Academy of Dermatology, 83(5), 1282-1293. doi:doi:10.1016/j.jaad.2020.06.054
- Paller, A. S., Simpson, E. L., Siegfried, E. C., Cork, M. J., Wollenberg, A., Arkwright, P. D., . . . Bansal, A. (2022). Dupilumab in children aged 6 months to younger than 6 years with uncontrolled atopic dermatitis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London, England), 400(10356), 908-919. doi:doi:10.1016/S0140-6736(22)01539-2
- Paller, A. S., Wollenberg, A., Siegfried, E., Thaçi, D., Cork, M. J., Arkwright, P. D., . . . Levit, N. A. (2021). Laboratory Safety of Dupilumab in Patients Aged 6-11 Years with Severe Atopic Dermatitis: Results from a Phase III Clinical Trial. Paediatr Drugs, 23(5), 515-527. doi:10.1007/s40272-021-00459-x
- Rijksvaccinatieprogramma. (z.d.). Tijdstip van Vaccinaties. Geraadpleegd op 10 december 2024, van https://rijksvaccinatieprogramma.nl/professionals/richtlijnen/uitvoering/7-tijdstip-van-vaccinaties
- Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html
- Siegfried, E. C., Bieber, T., Simpson, E. L., Paller, A. S., Beck, L. A., Boguniewicz, M., . . . Bansal, A. (2021). Effect of Dupilumab on Laboratory Parameters in Adolescents with Atopic Dermatitis: Results from a Randomized, Placebo-Controlled, Phase 3 Clinical Trial. American Journal of Clinical Dermatology, 22(2), 243-255. doi:doi:10.1007/s40257-020-00583-3
- Simpson, E. L., Paller, A. S., Siegfried, E. C., Boguniewicz, M., Sher, L., Gooderham, M. J., . . . Bansal, A. (2020). Efficacy and Safety of Dupilumab in Adolescents with Uncontrolled Moderate to Severe Atopic Dermatitis: A Phase 3 Randomized Clinical Trial. JAMA Dermatology, 156(1), 44-56. doi:doi:10.1001/jamadermatol.2019.3336
- Referenties tralokinumab
- Adtralza® (tralokinumab) EU Product Information. LEO Pharma; October 2022.
- Bieber T. Interleukin-13: targeting an underestimated cytokine in atopic dermatitis. Allergy 2020; 75:54-62
- Blauvelt A. Long-term 2-year safety and efficacy of tralokinumab in adults with moderate-to-severe atopic dermatitis: Interim analysis of the ECZTEND open-label extension trial. J Am Acad Dermatol. 2022 Oct;87(4):815-824. doi: 10.1016/j.jaad.2022.07.019. Epub 2022 Jul 19. PMID: 35863467
- ClinicalTrialsRegister.eu. Tralokinumab Monotherapy for Adolescent Subjects With Moderate to Severe Atopic Dermatitis - ECZTRA 6 (ECZema TRAlokinumab Trial no. 6). Identifier: 2017-005143-33.
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
- Paller AS, Flohr C, Cork M, Bewley A, Blauvelt A, Hong HC, Imafuku S, Schuttelaar MLA, Simpson EL, Soong W, Arlert P, Lophaven KW, Kurbasic A, Soldbro L, Vest NS, Wollenberg A. Efficacy and Safety of Tralokinumab in Adolescents With Moderate to Severe Atopic Dermatitis: The Phase 3 ECZTRA 6 Randomized Clinical Trial. JAMA Dermatol. 2023 Apr 19:e230627. doi: 10.1001/jamadermatol.2023.0627. Epub ahead of print. PMID: 37074705; PMCID: PMC10116386.
- Paller A. Efficacy and safety of tralokinumab in adolescents with moderate-to-severe atopic dermatitis: results of the phase 3 ECZTRA 6 Randomized Clinical Trial. JAMA dermatology, e230627. Advance online publication. https://doi.org/10.1001/jamadermatol.2023.0627
- Rijksvaccinatieprogramma. (z.d.). Tijdstip van Vaccinaties. Geraadpleegd op 10 december 2024, van https://rijksvaccinatieprogramma.nl/professionals/richtlijnen/uitvoering/7-tijdstip-van-vaccinaties
- Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html
- Wollenberg A. European guideline (EuroGuiDerm) on atopic eczema: part I - systemic therapy. J Eur Acad Dermatol Venereol. 2022 Sep;36(9):1409-1431. doi: 10.1111/jdv.18345. PMID: 35980214.
Verantwoording
Beoordelingsdatum en geldigheid
Publicatiedatum : 12-12-2025
Beoordeeld op geldigheid : 12-12-2025
De Nederlandse Vereniging voor Dermatologie en Venereologie (NVDV) is regiehouder van deze richtlijn en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van het richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van deze richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
Algemene gegevens
Aanleiding en afbakening onderwerp
Op initiatief van de Nederlandse Vereniging voor Dermatologie en Venereologie (NVDV) is de richtlijn Constitutioneel Eczeem (CE) in de periode 2022-2025 gedeeltelijk herzien. Deze herziening betreft de toevoeging van modules over de ‘nieuwe systemische middelen’: biologics en JAK-remmers, in deze verantwoording gaat het specifiek om de module biologics en JAK-remmers bij kinderen en adolescenten met CE.
CE is een veel voorkomende aandoening met een grote impact op het leven van patiënten. Ondanks goede therapeutische opties hebben nog steeds veel patiënten suboptimale behandeling van hun CE. In een in Europa uitgevoerde enquête onder 1189 patiënten met ernstig CE, geven tot wel 1 op de 4 patiënten aan niet goed om te kunnen gaan met hun eczeem en het gevoel te hebben de aandoening niet onder controle te kunnen houden. Daarnaast geeft 23% aan géén positieve kijk te hebben op hun leven (met eczeem). Patiënten ervaren tevens veel problemen als gevolg van onbeheersbare en persisterende jeuk, die een groot deel (57%) drijft tot wanhoop.[EFA, 2018]
Het Nationaal CE Project [NCEP (nvdv.nl)], opgesteld door meerdere partijen, heeft als doel het verbeteren van de CE-zorg in en voor de gehele keten; onder meer door het verschaffen van goede voorlichting die aansluit bij de behoefte van de patiënt, verbeterde voorlichting over de behandelmogelijkheden en goede voorlichting en begeleiding tijdens de behandeling. Dit zijn bij uitstek doelen die aan bod dienen te komen in medisch specialistische richtlijnen (als ook die van de NVDV), te meer sinds de komst van relatief nieuwe medicamenten.
Met de komst van biologics en Januskinase (JAK)-remmers voor patiënten met CE is het arsenaal aan behandelmogelijkheden uitgebreid, hetgeen bijdraagt om patiënten met CE die niet uitkomen met conventioneel systemische middelen controle van ziekte te bieden, met inherent een betere kwaliteit van leven.
Derhalve zijn de modules over de herziene en toegevoegde onderwerpen (biologics, JAK-remmers) zo opgesteld dat ze relevante en praktische aanbevelingen bieden voor de verschillende momenten (indicatiestelling en keuze van middel, vóór start behandeling/screening, tijdens de behandeling/monitoring en bij het stoppen van de behandeling) van de specialistische dermatologische zorg in de tweede/derde lijn.
De module over dupilumab is herzien t.o.v. de richtlijn uit 2019. De overige modules in deze richtlijn zijn toegevoegd. Eind 2024 volgt naar verwachting een toevoeging over de behandeling van kinderen met CE middels biologics en JAK-remmers.
Doel en doelgroep
Doel
Deze modulaire herziening is een document met aanbevelingen ter ondersteuning van de dagelijkse praktijkvoering. De richtlijn berust op de resultaten van wetenschappelijk onderzoek en aansluitende meningsvorming (expert opinion) gericht op het vaststellen van goed medisch handelen. Deze modulaire herziening geeft aanbevelingen over de begeleiding en behandeling van patiënten met CE die in aanmerking komen voor een biologic of JAK-remmer.
Doelgroep
De richtlijn is primair bestemd voor dermatologen. Alhoewel de indicatiestelling en het voorschrijven van biologics en JAK-remmers bij CE primair door de dermatoloog plaatsvindt, zijn verpleegkundig specialisten en physician assistants nauw betrokken bij de zorg voor patiënten met CE. Derhalve is de richtlijn ook relevant voor deze doelgroep. Secundair is de richtlijn eventueel nuttig voor oogartsen, internisten, (ziekenhuis)apothekers, allergologen en gynaecologen. Voor patiënten werd informatie ontwikkeld voor www.thuisarts.nl. Voor huisartsen geldt primair de NHG-Standaard. Vooralsnog zijn biologics en JAK-remmers voor de behandeling van CE middelen die in de 2de, maar met name in de 3de lijn worden voorgeschreven.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn werd een multidisciplinaire werkgroep ingesteld. Bij het samenstellen van de werkgroep werd rekening gehouden met de geografische spreiding van de werkgroepleden en met een evenredige vertegenwoordiging van academische en niet-academische achtergrond. De werkgroepleden hebben onafhankelijk gehandeld en geen enkel lid ontving gunsten met het doel de richtlijnen te beïnvloeden.
Tabel 2: Overzicht werkgroepleden - Herziening NVDV-richtlijn CE 2022-2025 – specifiek voor de Kindermodule
|
Werkgroepleden – kindermodule |
Vereniging/affiliatie |
|
S. van den Ham-Stewart, dermatoloog (voorzitter) |
NVDV |
|
S.Pasmans, (kinder)dermatoloog |
NVDV |
|
P.A. Middelkamp Hup, (kinder)dermatoloog |
NVDV |
|
E. Mendels, (kinder)dermatoloog |
NVDV |
|
M. de Graaf, (kinder)dermatoloog |
NVDV |
|
M.A.J. van Rossum, kinderarts-reumatoloog |
NVK |
|
Ondersteuning – kindermodule |
Vereniging/affiliatie |
|
M.R. Masselink, arts-onderzoeker |
NVDV |
|
M.O. Hoogeveen, arts-onderzoeker |
NVDV |
|
W.A. van Enst, klinisch epidemioloog |
NVDV |
Belangenverklaringen
De KNMG-Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of ze in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatie management, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel (tabel 3). De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van de Nederlandse Vereniging voor Dermatologie en Venereologie.
Tabel 3: Overzicht belangenverklaringen werkgroepleden - Herziening NVDV-richtlijn CE 2022-2024
|
Werkgroep lid |
Hoofdfunct- ie(s) |
Nevenfunctie(s) |
Persoonlijke financiële belangen |
Persoonlijke relaties |
Extern gefinancierd onderzoek |
Intellectuele belangen en reputatie |
Overige belangen |
Getekend op |
Acties |
|
drs. S. van den Ham-Stewart |
Dermatoloog IJsselland ziekenhuis |
Project Huid in de Klas – Huidpatiënten Nederland: 1 sessie betaald, de rest onbetaald |
Geen |
Geen |
IJsselland ziekenhuis neemt deel aan Bioday Registry |
Geen |
Geen |
13 maart 2022 |
- |
|
dr. F.M. Garritsen |
Dermatoloog Haga ziekenhuis |
Lid domeingroep kinderdermatologie NVDV |
In 2021 deelname aan adviesraad over eczeem bij Abbvie en LeoPharma |
Geen |
Geen |
Geen |
Geen |
17 maart 2022 |
- |
|
dr. J.M. Oldhoff |
Dermatoloog UMC Groningen |
Voorzitter domeingroep SOA en huidinfecties Lid MDR SOA Lid leidraad PeIN Lid richtlijn alopecia
Allen onbetaald |
Geen |
Geen |
Geen |
Geen |
Geen |
29 maart 2022 |
- |
|
dr. L.A.A. Gerbens |
Dermatoloog Amsterdam UMC en Huid Medisch Centrum |
- Co-auteur European Dermatology Forum (EDF) EuroGuiDerm Atopic Dermatitis guideline, geleid doorprof. dr. A. Wollenberg en prof. dr. C. Flohr. Onbetaald.
- Projectleider UPDATE trial, beurs ZonMw. Mijn functie is in-kind.
- Data Monitoring Board (DMC) van A-STAR registry UK/Ireland. Onbetaald.
- Lid Board of Directors C3: The CHORD COUSIN Collaboration for Dermatology Outcomes. Onbetaald.
- Lid Scientific Committee Skin Inflammation & Psoriasis International Network (SPIN). Onbetaald.
- Advisory journal editor of Dutch Amsterdam Medical Student journal (AMSj). Onbetaald.
- Lid Executive Committee Harmonising Outcome Measurements for Eczema (HOME) initiative. Onbetaald.
- Lid steering committee of the international TREatment of ATopic eczema (TREAT) Registry Taskforce and of the Dutch TREAT NL registry. Onbetaald. |
Geen |
Geen |
Geen. Wel projectleider UPDATE trial; NB-UVB bij atopisch eczeem. Geen systemische medicatie is hierbij betrokken. |
Geen |
Geen |
11 maart 2022 |
- |
|
dr. M. de Graaf |
Dermatoloog, met aandachtsgebied kinderen (kinderdermatoloog) UMC Utrecht |
Geen |
Geen financieel belang Wel lid van adviesraad over systemische medicatie voor Constitutioneel Eczeem (CE) van Sanofi Genzyme, Eli Lilly en Leo Pharma. |
Geen |
Betrokken bij CE onderzoek naar: - JAK remmers (Eli Lilly, Abbvie, Pfizer) - Biologicals (Sanofi Genzyme, Leo Pharma) - anti bacteriële verbandmiddelen (ZonMW) |
Geen |
Geen |
11 maart 2022 |
- |
|
dr. I.M. Haeck |
Dermatoloog Reinier de Graaf ziekenhuis |
Adviesraad/consultant voor: Lilly, Abbvie, Sonofi/Regeneron, LEO Pharma
M18-891 studie Abbvie |
Geen |
Geen |
Geen |
Geen |
Geen |
29 maart 2022 |
- |
|
dr. J.L. Thijs |
AIOS dermatologie en post-doctoraal onderzoeker UMC Utrecht |
Spreker voor Sanofi, LeoPharma, Almirall (betaald) |
Geen |
Geen |
Medewerking aan wetenschappelijk onderzoek gefinancierd door Sanofi-Regeneron en LeoPharma |
Geen |
Geen |
16 maart 2022 |
- |
|
AIOS interne geneeskunde, fellow allergologie/klinische immunologie UMC Groningen |
Geen |
Geen |
Geen |
Geen |
Geen |
Geen |
29 maart 2022 |
- |
|
|
dr. C.M. van Luijk |
Oogarts UMC Utrecht.
Subspecialisme voorsegment: chirurgisch en medisch cornea, conjunctiva en complexe cataract operaties. |
Eenmalig betaalde nascholing door Santen (4-2021)
Eenmalig deelname adviescommissie bijwerkingen dupilumab waarvoor vergoeding door Regeneron (12-2019) |
Geen |
Geen |
Beurs voor onderzoek te besteden aan ciclosporine door Santen. Onderzoek nog niet gestart. Onafhankelijkheid verklaard in overeenkomst. |
Bijzondere en unieke expertise in atopische oogpathologie binnen multidisciplinair eczeem zorg team in UMC Utrecht |
Geen |
29 maart 2022 |
- |
|
dr. M.A.J. van Rossum |
Kinderarts-reumatoloog/immunoloog Amsterdam UMC en gedetacheerd naar Reade Reumatologie Amsterdam |
Geen |
Geen |
Geen |
Geen |
Geen |
Geen |
17 maart 2022 |
- |
|
drs. F. Korving |
Poliklinisch ziekenhuis apotheker Tergooi MC |
Werkgroeplid NVDV-richtlijn psoriasis Beoordeling NVDV-richtlijn Acne Vulgaris |
Geen |
Geen |
Geen |
Geen |
Geen |
23 maart 2022 |
- |
|
mw. K. Sterkenburg |
Beleidsmedewerker onderzoek & kwaliteit bij de Nederlandse Vereniging van Huidtherapeuten |
Projectlid 'Eczeemvriendelijk Veenendaal'. Betaald. |
Geen |
Geen |
Geen |
Geen |
Geen |
17 maart 2022 |
- |
|
mw. F. Schussler-Raymakers |
Verpleegkundig specialsist UMC Utrecht |
Adviesraad Sanofi genzyme (betaald) Adviesraad Leo Pharma (betaald) |
Geen |
Geen |
Geen |
Geen |
Geen |
10 juni 2022 |
- |
|
dhr. W. Kouwenhoven |
Patiëntvertegenwoordiger |
Geen |
Geen |
Geen |
Geen |
Geen |
Geen |
14 maart 2022 |
- |
Inbreng patiëntenperspectief
Er is aandacht besteed aan het patiëntenperspectief door de zitting neming van een patiëntvertegenwoordiger in de werkgroep en verwerking van het patiëntperspectief in de richtlijnmodules. Het conceptrichtlijn is tevens voor commentaar voorgelegd aan de Vereniging voor Mensen met Constitutioneel Eczeem (VMCE).
Wkkgz & Kwalitatieve raming van mogelijke substantiële financiële gevolgen
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (gebaseerd op het stroomschema ontwikkeld door FMS).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
|
Module |
Uitkomst Raming |
Toelichting |
|
Biologics en JAK-remmers bij kinderen en adolescenten met CE |
Geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen(en) niet breed toepasbaar zijn (<5.000 patiënten) en zal daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven |
Implementatie
In de verschillende fasen van de ontwikkeling van het richtlijn is rekening gehouden met de implementatie van het richtlijn(modules) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De richtlijn wordt via het internet verspreid onder alle relevante beroepsgroepen en ziekenhuizen en er zal in verschillende specifieke vaktijdschriften aandacht worden besteed aan deze richtlijn. Tevens zal een samenvatting worden gemaakt.
Het implementatieplan is te vinden als bijlage per module.
Werkwijze
Knelpuntenanalyse
In de voorbereidingsfase heeft een bijeenkomst plaatsgevonden waarvoor alle belanghebbenden zijn uitgenodigd. In deze bijeenkomst zijn knelpunten aangedragen door de werkgroepleden en gemandateerden van verschillende wetenschappelijke verenigingen en stakeholders. Voor een volledig overzicht wordt verwezen naar tabel 1.
Uitgangsvragen en uitkomstmaten
De uitgangsvragen van de EDF-guideline werden geadapteerd naar de Nederlandse situatie. De modules over de herziene onderwerpen (biologics, JAK-remmers,) zo opgesteld dat ze relevante en praktische aanbevelingen bieden voor de verschillende momenten (indicatiestelling en keuze van middel, vóór start behandeling/screening, tijdens de behandeling/monitoring en bij stoppen van de behandeling) van de specialistische dermatologische zorg in de tweede/derde lijn.
Indicatorontwikkeling
Er werden geen nieuwe indicatoren ontwikkeld voor deze richtlijn. De huidige indicatoren volstaan.
Kennislacunes
Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, als uitgangsvraag beschreven, is te vinden in bijlage 3.
Juridische betekenis van richtlijnen
Richtlijnen zijn geen wettelijke voorschriften maar wetenschappelijk onderbouwde en breed gedragen inzichten en aanbevelingen waaraan zorgverleners zouden moeten voldoen om kwalitatief goede zorg te verlenen. Aangezien richtlijnen uitgaan van ‘gemiddelde patiënten’, kunnen zorgverleners in individuele gevallen zo nodig afwijken van de aanbevelingen in de richtlijn. Afwijken van richtlijnen is, als de situatie van de patiënt dat vereist en na overleg met de patiënt, soms zelfs noodzakelijk. Een richtlijn beschrijft wat goede zorg is, ongeacht de financieringsbron (Zorgverzekeringswet (Zvw), Wet langdurige zorg (Wlz), Wet maatschappelijke ondersteuning (Wmo), aanvullende verzekering of eigen betaling door de cliënt/patiënt). Opname van een richtlijn in een register betekent dus niet noodzakelijkerwijs dat de in de richtlijn beschreven zorg verzekerde zorg is.
Commentaar- en autorisatiefase
De conceptrichtlijn is medio september tot november 2025 aan de betrokken (wetenschappelijke) verenigingen, (patiënt) organisaties en stakeholders voorgelegd ter commentaar.
- Drucker AM, Morra DE, Prieto-Merino D, Ellis AG, Yiu ZZN, Rochwerg B, et al. Systemic Immunomodulatory Treatments for Atopic Dermatitis: Update of a Living Systematic Review and Network Meta-analysis. JAMA Dermatol. 2022;158; 523-532.
- Drucker AM. Systemic immunomodulatory treatments for atopic dermatitis: a living systematic review and network meta-analysis. 2022. https://eczematherapies.com/research/.
- EFA: Giuseppe de Carlo, Sofia Romagosa, Isabel Proaño, Susanna Palkonen. Itching for Life: Quality of Life and costs for people with severe atopic eczema in Europe. EFA, European Federation of Allergy and Airways Diseases Patients’ Associations. 2018
Zoekverantwoording
Zoekstrategie
EMBASE (datum 16-02-2023)
Zoektermen
- #53. #22 AND #52 449
- #52. #31 OR #35 OR #41 OR #47 OR #51 5,500,155
- #51. #48 OR #49 OR #50 2,665,162
- #50. selection:ti,ab AND criteria:ti,ab 90,476
- #49. data:ti,ab AND extraction:ti,ab 94,751
- #48. review:ti,ab 2,552,805
- #47. #42 OR #43 OR #44 OR #45 OR #46 85,788
- #46. relevant:ti,ab AND journals:ti,ab 9,237
- #45. manual:ti,ab AND search*:ti,ab 15,030
- #44. hand:ti,ab AND search*:ti,ab 23,035
- #43. bibliograph*:ti,ab 36,217
- #42. reference:ti,ab AND list:ti,ab 7,912
- #41. #36 OR #37 OR #38 OR #39 OR #40 257,881
- #40. cinahl:ti,ab OR cinhal:ti,ab 50,644
- #39. bids:ti,ab 974
- #38. science:ti,ab AND citation:ti,ab 7,954
- #37. embase:ti,ab 180,463
- #36. cochrane:ti,ab 163,608
- #35. #32 OR #33 OR #34 847,118
- #34. systematic:ti,ab 624,016
- #33. meta:ti,ab AND analy*:ti,ab OR metaanaly*:ti,ab 341,493
- #32. meta AND 'analysis'/exp 324,430
- #31. #23 OR #24 OR #25 OR #26 OR #27 OR 2,773,441
#28 OR #29 OR #30
- #30. random*:ti,ab 1,889,947
- #29. randomized:ti,ab AND controlled:ti,ab AND 242,138
trial:ti,ab
- #28. trial:ti,ab 1,076,310
- #27. placebo*:ti,ab 357,228
- #26. (crossover*:ti,ab OR cross:ti,ab) AND over*:ti,ab 361,895
- #25. 'single blind':ti,ab AND procedure:ti,ab 1,014
- #24. 'double blind':ti,ab AND procedure:ti,ab 5,844
- #23. crossover:ti,ab AND procedure:ti,ab 3,917
- #22. #6 AND #10 AND #21 846
- #21. #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR 4,783
#18 OR #19 OR #20
- #20. cibinqo:ti,ab 2
- #19. abrocitinib:ti,ab 219
- #18. rinvoq:ti,ab 12
- #17. upadacitinib:ti,ab 1,097
- #16. olumiant:ti,ab 26
- #15. barcitinib:ti,ab 14
- #14. adtralza:ti,ab 6
- #13. tralokinumab:ti,ab 265
- #12. dupixent:ti,ab 58 16
- #11. dupilumab:ti,ab 3,423
- #10. #7 OR #8 OR #9 6,145,496
- #9. child* OR infant* OR babies* OR boy* OR girl* OR 5,926,382
pediatric* OR paediatric* OR adolescen*
- #8. 'adolescent'/exp 1,882,081
- #7. 'child'/exp 3,292,966
- #6. #1 OR #2 OR #3 OR #4 OR #5 233,597
- #5. 'atopic dermatitis'/exp 55,147
- #4. dermatitis:ti,ab 96,872
- #2. eczema:ti,ab 29,000
- #1. 'eczema'/exp 40,900
Resultaten = [449]
MEDLINE in Ovid (datum 16-02-2023)
Zoektermen
- 1 exp Dermatitis/ 113978
- 2 dermatitis.ti,ab. 65161
- 3 exp Eczema/ 12554
- 4 eczema.ti,ab. 18160
- 5 exp Dermatitis, Atopic/ 23970
- 6 or/1-5 140521
- 7 exp Child/ or exp Infant/ or exp Adolescent/ 3925316
- 8 (child* or infant* or babies* or boy* or girl* or pediatric* or paediatric* or adolescen*).tw. 2383196
- 9 or/7-8 4524919
- 10 dupilumab.ti,ab. 1945
- 11 Dupixent.ti,ab. 39
- 12 tralokinumab.ti,ab. 134
- 13 adtralza.ti,ab. 3
- 14 baricitinib.ti,ab. 991
- 15 olumiant.ti,ab. 20
- 16 upadacitinib.ti,ab. 413
- 17 rinvoq.ti,ab. 5
- 18 abrocitinib.ti,ab. 117
- 19 cibinqo.ti,ab. 1
- 20 or/10-19 3281
- 21 6 and 9 and 20 304
- 22 Meta-Analysis as Topic/ 22114
- 23 meta analy$.tw. 258028
- 24 metaanaly$.tw. 2514
- 25 Meta-Analysis/ 175760
- 26 (systematic adj (review$1 or overview$1)).tw. 274769
- 27 exp Review Literature as Topic/ 21784
- 28 or/22-27 435343
- 29 cochrane.ab. 127248
- 30 embase.ab. 145464
- 31 (psychlit or psyclit).ab. 917
- 32 (psychinfo or psycinfo).ab. 55430
- 33 (cinahl or cinhal).ab. 43492
- 34 science citation index.ab. 3697
- 35 bids.ab. 651
- 36 cancerlit.ab. 638
- 37 or/29-36 232497
- 38 reference list$.ab. 21631
- 39 bibliograph$.ab. 21950
- 40 hand-search$.ab. 8370
- 41 relevant journals.ab. 1329
- 42 manual search$.ab. 5833
- 43 or/38-42 53103
- 44 selection criteria.ab. 35272
- 45 data extraction.ab. 30849
- 46 or/44-45 63518
- 47 review/ 3116195
- 48 46 and 47 33947
- 49 Comment/ 996296
- 50 Letter/ 1207425
- 51 Editorial/ 636446
- 52 animal/ 7234887
- 53 human/ 21059072
- 54 52 not (52 and 53) 5059741
- 55 or/49-51,54 7117409
- 56 28 or 37 or 43 or 48 520149
- 57 56 not 55 494929
- 58 randomized controlled trial.pt. 586677
- 59 controlled clinical trial.pt. 95190
- 60 randomized.ab. 592581
- 61 placebo.ab. 235671
- 62 clinical trials as topic.sh. 200866
- 63 randomly.ab. 401908
- 64 trial.ti. 279285
- 65 or/58-64 1503720
- 66 57 or 65 1880213
- 67 21 and 66 86
- Resultaten = [86]
Alle resultaten
|
Database |
Datum |
# hits |
|
EMBASE |
16-02-2023 |
449 |
|
MEDLINE |
16-02-2023 |
86 |
|
Cochrane |
|
|
|
Totaal |
535 |
|
|
Duplicates |
143 |
|
|
Netto aantal |
392 |
|
Resultaten tralokinumab
Er werden in totaal 158 studies/zoekresultaten geïncludeerd op basis van beoordeling van titel en abstract, waarvan 5 zoekresultaten over talokinumab. Er werd geen systematische review gevonden waarin tralokinumab voor kinderen/adolescenten met CE werd besproken, en overige systematische reviews over meerdere middelen/middelen groepen [=6]) bleken na full-tekst beoordeling niet bijdragende voor het beantwoorden van de onderzoeksvraag inzake tralokinumab. Op basis van PICO en in- en exclusiecriteria werd er uiteindelijk 1 RCT geïncludeerd [Paller, 2023].