Behandeling influenza - geen opname
Uitgangsvraag
Wat is het beste beleid ten aanzien van keuze en duur (toedieningswijze, timing, dosis) van antivirale behandeling voor kinderen en volwassenen die niet in het ziekenhuis opgenomen worden met influenza-like-illness?
De uitgangsvraag besteedt (indien mogelijk) specifieke aandacht aan de volgende risicogroepen:
- Ouderen
- Zwangeren
- Kinderen jonger dan 2 jaar, in het bijzonden zuigelingen
- Mensen met een chronische ziekte (comorbiditeit)
- Mensen met verminderde afweer en/of gebruik van immunosuppressiva/immunotherapie
Met patiënten wordt bedoeld:
- Niet-opgenomen patiënt: poliklinische patiënten, patiënten op de SEH, patiënten in het verpleegtehuis. De patiëntenpopulatie in de huisartsenpraktijk valt vooralsnog buiten deze richtlijn.
Aanbeveling
Aanbeveling volwassenen
Bespreek met patiënten die zich binnen 48 uur na het begin van de symptomen melden de voordelen en nadelen van een behandeling met antivirale therapie.
Bespreek met patiënten uit de risicogroepen, of patiënten die ernstiger ziek zijn, die zich binnen 48 na de start van de symptomen van een ILI melden, dat behandeling met antivirale therapie* een verkorting van de ziekteduur met ongeveer een dag en een mogelijke verlaging van de kans op complicaties kan betekenen.
Patiënten uit een risicogroep zijn:
- ouderen ≥ 60 jaar;
- zwangeren;
- kinderen < 2 jaar in het bijzonder zuigelingen;
- mensen met een chronische ziekte;
- mensen met verminderde afweer en/of gebruik van immunosuppressiva.
* Behandeling met baloxavir is nog onvoldoende onderzocht bij zwangeren, kinderen <12 jaar en immuungecommpromitteerden.
Aanbeveling kinderen
Adviseer geen behandeling van influenza bij kinderen vanwege te lage bewijskracht voor effectiviteit, tenzij ze onderdeel zijn van een risicogroep.
Kinderen in een risicogroep zijn:
- kinderen < 2 jaar in het bijzonder zuigelingen;
- kinderen met een chronische ziekte;
- kinderen met verminderde afweer en/of gebruik van immunosuppressiva.
Overwegingen
De onderstaande overwegingen gelden in principe voor het overgrote deel van de patiëntenpopulatie waarop de uitgangsvraag betrekking heeft.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Wat betreft de cruciale uitkomstmaten ‘complicaties’ en ‘hospitalisatie’ is de effectiviteit van de interventie niet aangetoond. Het is echter ook niet uitgesloten dat de interventie leidt tot minder complicaties bij risicogroepen. De individuele patient data meta-analyse van Venkatasan (2017) suggereert die uitkomst (minder hospitalisatie van patiënten die antivirale therapie kregen tijdens de 2009-pandemie).
Wat betreft de belangrijke uitkomstmaten: alhoewel alle Grade-coderingen voor de bewijskracht hoogstens laag zouden zijn, lijkt er voor de uitkomsten ‘verlichting van symptomen’, ‘tijd tot verdwijnen koorts’ en ‘terugkeer tot normale gezondheid’ wel een significante verkorting van de duur bij volwassenen. Of deze verkorting van minder dan een dag relevant moet worden genoemd, is een onderwerp van discussie met de patiënt.
Na het uitvoeren van de literatuursearch werden nog enkele artikelen gepubliceerd, die hier kort worden beschreven.
Het onderzoek van Butler (2020) betrof een open-label, pragmatic, randomised controlled trial, in 15 Europese landen, met publieke financiering, van oseltamivir met gebruikelijke zorg versus alleen gebruikelijke zorg voor influenza-like illness in de huisartspraktijk. De gemiddelde verkorting van de ziekteduur was hier ook één dag. Opvallende uitkomst was dat oudere, ziekere patiënten met meer comorbiditeit en langere ziekteduur, 3 dagen eerder opknapten. In tegenstelling tot de andere bronnen, werd er geen verschil gevonden tussen wel of niet bevestigde influenza diagnose.
Zoals aangegeven in de introductie is kort voor de autorisatie van deze richtlijn het middel baloxavir, Xofluza®, goedgekeurd door de EMA voor gebruik van behandeling en preventie van van ongecompliceerde influenza (type A en B) bij volwassenen en kinderen vanaf 12 jaar. Uit de aanvullende search zijn de volgende artikelen gekomen die zijn meegenomen in de conclusies.
In een eerste gerandomiseerde dubbelblinde gecontroleerde studie met gezonde niet opgenomen patienten wordt beschreven dat patienten die behandeld werden met baloxavir klinisch herstelden na gemiddeld 54 uur vergeleken met 80 uur voor patienten die een placebo kregen. Dit effect was vergelijkbaar met de behandeling met oseltamivir (Hayden, 2018). In een subanalyse van deze studie is gekeken naar de adolescenten uit deze trial en worden vergelijkbare bevindingen beschreven als voor de hele patientengroep (Porthsmouth, 2020). In een andere gerandomiseerde placebo gecontroleerde fase III studie is gekeken naar het effect van behandeling met baloxavir in niet opgenomen patienten met ongecompliceerde influenza virus infectie die een hoog risico op complicaties hadden. Hierin werd aangetoond dat baloxavir-behandelde patienten klinisch verbeterden na 73 uur, vergeleken met 102 uur bij patienten behandeld met placebo en 81 uur bij behandeling met oseltamivir. (Ison, 2020) Beide studies toonden een snellere daling van de virale lading tijdens behandeling met baloxavir vergeleken met placebo en oseltamivir.
Er zijn ook klinische studies uitgevoerd die de effectiviteit en veiligheid beschrijven van het middel bij de behandeling van kinderen onder de leeftijd van 12 jaar. Baloxavir lijkt goed te worden verdragen en effectief te zijn en aanleiding te geven tot een snelle daling van de virale lading en klinisch herstel maar vergelijkingsgroepen ontbraken (Hirotsu, 2020; Baker, 2020; Yokoyama, 2020; Nakazawa, 2020).
Een belangrijke kanttekening bij de behandeling met baloxavir is het risico op ontwikkeling van antivirale resistentie. Tijdens behandeling met baloxavir ontstaat in een relatief hoog percentage patienten (2,2-9,7%) van patiënten resistentie, veroorzaakt door enkelvoudige mutaties in het targeteiwit PA. Het hoogste risico op resistentie-ontwikkeling wordt gezien bij kinderen en immuungecompromitteerden (Hirotsu, 2020). Ontwikkeling van resistentie tijdens behandeling kan gepaard gaan met een tijdelijke hernieuwde stijging in virale lading, langduriger virale uitscheiding, vertraging van het klinisch herstel en in enkele gevallen met terugkeer van symtomen (Hayden, 2018; Uehara, 2020). De implicaties op de behandeling bij specifieke patientengroepen zoals kinderen, opgenomen patienten, zwangeren en immuungecompromiteerde patienten vereisen aanvullend onderzoek (Uehara, 2020).
In de meta-analyse van Dobson (2015) werden alle gepubliceerde en ongepubliceerde Roche-gesponsorde RCT’s (dubbelblind) geïncludeerd waarbij tweemaal daags 75mg Oseltamivir werd voor geschreven bij volwassenen in vergelijking met placebo; deze studies werden ook geïncludeerd in Jefferson (2014) (zie literatuuranalyse). De studie van Dobson (2015) paste echter een individuele analyse op de patiëntdata toe waardoor gedetailleerde informatie wordt weergegeven. Tijd tot verlichting van symptomen, complicaties, ziekenhuisopname en veiligheidsuitkomsten werden gerapporteerd in de ITTI (intention-to-treat-infected) en ITT (intention-to-treat) populatie. De belangrijke uitkomstmaten waren vergelijkbaar met de eerdere gerefereerde literatuur De resultaten van de cruciale uitkomstmaten laten zien dat oseltamivir in volwassenen het risico op lage luchtweginfectie-complicaties en ziekenhuisopnames enigszins verlaagt. De resultaten van Dobson laten ook zien dat er een verhoogde kans is op misselijkheid en overgeven bij gebruik van oseltamivir.
Voor de cruciale uitkomstmaten is de bewijskracht laag. Alleen in de IPD meta-analyse van Dobson blijkt behandeling met oseltamivir in de ITTI-populatie een significant effect te hebben op secundaire luchtweginfecties en ziekenhuisopname. De gevonden ongunstige effecten (misselijkheid) zijn bekend en niet ernstig. De reden voor de lage bewijskracht is gelegen in de omstandigheid dat de producent van oseltamivir nooit een RCT heeft willen opzetten met de cruciale uitkomstmaten (en ook nauwelijks in risicopatiënten). Daarom eigenlijk alleen maar post hoc analyses en observationeel onderzoek.
De significante effecten van oseltamivir op belangrijke uitkomstmaten suggereren de mogelijkheid voor effecten op de cruciale uitkomsten. Aangezien die cruciale uitkomsten veel minder vaak voorkomen, zal het number to treat om die cruciale uitkomsten te voorkomen groot zijn. Bovendien zou iedereen met een ILI zich dan binnen 48 uur moeten melden. Dat past niet in de Nederlandse cultuur van ‘even aanzien’ bij luchtweginfecties.
In de Nederlandse huisartspraktijk wordt zelden antivirale therapie voorgeschreven. De ontwikkeling van resistentie komt derhalve ook niet vaak voor. Orale therapie met oseltamivir wordt vanwege het gebruiksgemak waarschijnlijk vaker voorgeschreven dan inhalatietherapie met zanamivir.
De bijwerkingen van antivirale therapie zijn gering. De positief voorspellende waarde van de huisartsdiagnose is tijdens de griepepidemie circa 60%. Indien een ILI toch geen influenza blijkt te zijn, zou de antivirale therapie gestaakt kunnen worden. Het risico voor de patiënt die onnodig antivirale therapie heeft gebruikt is niet groot. De meest frequent (zeer vaak en vaak) gemelde bijwerkingen van oseltamivir zijn misselijkheid of braken, maagpijn, maagklachten, hoofdpijn en pijn. Deze bijwerkingen treden meestal na de eerste dosis van het geneesmiddel op en zullen doorgaans stoppen als de behandeling voortgezet wordt. De frequentie van deze bijwerkingen neemt af wanneer het geneesmiddel met voedsel wordt ingenomen. Voorts zijn er nog een aantal zeldzame bijwerkingen, met name bij kinderen en ouderen, die moeilijk van de ziektesymptomen kunnen worden onderscheiden.
Waarden en voorkeuren van patiënten (en eventueel hun verzorgers)
Sommige patiënten zouden wellicht voor een verkorting van de ziekteduur met een dag of een mogelijk snellere terugkeer naar de normale gezondheid kiezen. Misschien geldt dat nog meer voor patiënten uit de bekende risicogroepen die vaker een complicatie krijgen na een virale luchtweginfecties.
Kosten (middelenbeslag)
De kosten van behandeling zijn ongeveer 30 euro; vermindering van de ziekteduur met ongeveer een dag kan voor sommige patiënten kosteneffectief zijn. De kosten voor het behandelen van iedereen met een ILI binnen 48 uur zullen hoog zijn en waarschijnlijk niet kosteneffectief.
Aanvaardbaarheid voor de overige relevante stakeholders
De morele of ethische bezwaren zijn misschien het medicaliserende effect wat zou uitgaan van het behandelen van een ILI binnen 48 uur. Dit kan onderdeel zijn van de bespreking met de patiënt.
Haalbaarheid en implementatie
Het advies om Samen Beslissen toe te passen bij het bespreken van de behandeling met antivirale middelen, zal nauwelijks tot weerstand leiden, omdat het goed medisch handelen is. De therapietrouw zal daar zeker door verbeterd worden. In de toekomst zal een goede sneltest voor influenza in de eerste lijn wel tot verhoging van de kosten leiden, maar ook tot verbeteren van de indicatiestelling. Gezien het lage percentage patiënten dat uiteindelijk wordt opgenomen in de studies zal er een grote groep patiënten behandeld moeten worden om een verschil te vinden tussen interventie en placebogroep.
Rationale/ balans tussen de argumenten voor en tegen de interventie
De meeste patiënten met ILI zullen pas na enkele dagen hulp zoeken; het behandelvenster van 48 uur zal dan meestal verstreken zijn. Indien een patiënt om hem of haar geldende redenen toch binnen 48 uur zich meldt en aangeeft dat voor hem een geringe verkorting van de duur van de ziekteperiode van belang is, valt antivirale therapie te overwegen. Het kan de duur van de ziekteverschijnselen verminderen, een snellere terugkeer naar de normale gezondheid en de besmettelijkheid wellicht ook door kortere viral shedding. De bijwerkingen zijn gering.
Onderbouwing
Nederlanders is altijd geleerd om virale luchtweginfecties ‘uit te zieken’. In de richtlijnen staat dat het niet nodig is om daarvoor een arts te bezoeken, behalve bij bepaalde risicocondities. Antivirale therapie voor influenza moet binnen 48 na het begin van de symptomen starten. Feit is dat antivirale therapie buiten het ziekenhuis niet of nauwelijks wordt voorgeschreven. Ook patiënten kennen de werking van antivirale therapie niet en kiezen er daarom ook niet voor. Meestal ligt daar geen gedeelde besluitvorming aan ten grondslag.
Sommige patiënten zouden wellicht voor een verkorting van de ziekteduur met een dag kiezen. Misschien geldt dat nog meer voor patiënten uit de bekende risicogroepen die vaker een complicatie krijgen na een virale luchtweginfecties. Het is echter onduidelijk of er risicogroepen zijn bij wie antivirale therapie complicaties kan voorkomen. Vandaar deze richtlijn.
Volwassenen
Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)
|
Laag GRADE |
Het is mogelijk dat de behandeling van antivirale medicatie het aantal nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis) waarbij antibioticagebruik nodig is, vermindert bij niet-opgenomen volwassen patiënten met influenza-like illness.
Bronnen: (Jefferson, 2014; Campion, 1998; Kohno, 2010; Treanor, 2000) |
|
Laag GRADE |
Behandeling van patiënten met bevestigde influenza met antivirale medicatie leidt mogelijk tot minder complicaties waarbij antibioticagebruik nodig is vergeleken met placebo.
Bronnen: (Hayden, 2018) |
Hospitalisatie
|
Laag GRADE |
Het is mogelijk dat de behandeling met antivirale medicatie bij aanvankelijk niet-opgenomen volwassen patiënten met influenza-like illness de kans op opname in het ziekenhuis vermindert.
Bronnen: (Jefferson, 2014) |
Verlichting van symptomen
|
Laag GRADE |
Behandeling van patiënten met influenza-like illness met antivirale medicatie resulteert mogelijk in een snellere verlichting van symptomen vergeleken met placebo.
Bronnen: (Campion, 1998; Hayden, 1997; Makela, 2000; Matsumoto, 1999; Monto, 1999; Puhakka, 2003; McLean, 2015; Nicholson, 2000; Treanor, 2000; Jefferson, 2016; Hayden, 2018) |
|
Laag GRADE |
Behandeling van patiënten met bevestigde influenza met antivirale medicatie resulteert mogelijk in een statistisch significant snellere verlichting van symptomen vergeleken met placebo.
Bronnen: (Boivin, 2000; Campion, 1998; Hayden, 1997; Makela, 2000; Matsumoto, 1999; Puhakka, 2003; Li, 2003; Nicholson, 2000; Treanor, 2000; Kohno, 2010; Whitley, 2015; Hayden, 2018) |
Verdwijnen van koorts
|
Laag GRADE |
Behandeling van patiënten met influenza-like illness met antivirale medicatie resulteert mogelijk in het sneller oplossen van koorts vergeleken met placebo.
Bronnen: (Campion, 1998; Monto, 1999; Nicholson, 2000) |
|
Laag GRADE |
Behandeling van patiënten met bevestigde influenza met antivirale medicatie resulteert mogelijk in het sneller oplossen van koorts vergeleken met placebo.
Bronnen: (Campion, 1998; Nicholson, 2000; Whitley, 2015; Hayden, 2018) |
Terugkeer tot normale gezondheid
|
Laag GRADE |
Behandeling van patiënten met influenza-like illness met antivirale medicatie zou terugkeer tot normale gezondheid kunnen versnellen vergeleken met placebo.
Bronnen: (Treanor, 2000) |
|
Redelijk GRADE |
Behandeling van patiënten met bevestigde influenza met antivirale medicatie versnelt waarschijnlijk de terugkeer tot normale gezondheid vergeleken met placebo.
Bronnen: (Treanor, 2000; Hayden, 2018) |
Kinderen
Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)
|
Laag GRADE |
Behandeling van niet-opgenomen kinderen met influenza-like illness met antivirale medicatie lijkt niet tot minder complicaties (mortaliteit, pneumonie, sepsis) te leiden.
Bronnen: (Jefferson, 2014; Johnston, 2005; Portsmouth, 2020) |
Complicaties waarbij antibioticagebruik nodig is
|
Zeer laag GRADE |
Er bestaat onzekerheid over het effect van een behandeling met antivirale medicatie bij niet-opgenomen kinderen met bevestigde influenza op complicaties waarbij antibioticagebruik nodig is in vergelijking met placebo.
Bronnen: (Portsmouth, 2020) |
Hospitalisatie
|
Zeer laag GRADE |
We zijn onzeker over het effect van een behandeling met antivirale medicatie bij niet-opgenomen kinderen met influenza-like illness op hospitalisatie in vergelijking met placebo of standaardzorg.
Bronnen: (Jefferson, 2014; Venkatesan, 2017; Johnston, 2005) |
Verlichting van symptomen
|
Laag GRADE |
Behandeling van patiënten met influenza-like illness met antivirale medicatie resulteert mogelijk in een snellere verlichting van symptomen in vergelijking met placebo.
Bronnen: (Jefferson, 2014; Heinonen, 2010; Johnston, 2005; Portsmouth, 2020) |
Tijd tot verdwijnen van koorts
|
Laag GRADE |
Behandeling van patiënten met influenza-like illness met antivirale medicatie resulteert mogelijk in een vermindering van het aantal dagen tot verdwijnen van koorts in vergelijking met placebo.
Bronnen: (Heinonen, 2010; Portsmouth, 2020) |
Terugkeer tot normale gezondheid
|
Laag GRADE |
Behandeling van kinderen met influenza-like illness met antivirale medicatie resulteert mogelijk tot snellere terugkeer tot normale gezondheid in vergelijking met placebo.
Bronnen: (Heinonen, 2010; Johnston, 2005) |
Hoog-risicopatiënten
Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)
|
Laag GRADE |
Het is mogelijk dat behandeling met antivirale medicatie het aantal nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis) waarbij antibioticagebruik nodig is, vermindert bij patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen met influenza-like illness vergeleken met placebo.
Bronnen: (Lin, 2006; Murphy, 2000; Ison, 2020) |
Hospitalisatie
|
Laag GRADE |
Het is mogelijk dat behandeling met antivirale medicatie de kans op opname in het ziekenhuis verlaagt bij niet-opgenomen patiënten met minimaal 1 hoog-risico conditie met influenza-like illness of bevestigde influenza.
Bronnen: (Jefferson, 2014) |
|
Laag GRADE |
Het is niet aangetoond dat behandeling met antivirale medicatie bij patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen met influenza-like illness tot minder opname in het ziekenhuis leidt.
Bronnen: (Lin, 2006) |
|
Laag GRADE |
Het is niet aangetoond dat behandeling met antivirale medicatie bij hoogrisicopatienten (long/hart/neurologische/nier/leveraandoeningen/diabetes/obesitas/ immuungecompromitteerd/jongeren met aspirinetherapie) met bevestigde influenza tot minder opname in het ziekenhuis leidt.
Bron: (Ison, 2020) |
Verlichting van symptomen
|
Laag GRADE |
Het is niet aangetoond dat behandeling met antivirale medicatie bij patiënten met multimorbiditeit of COPD/astma met influenza-like illness tot snellere verlichting van symptomen leidt vergeleken met placebo.
Bronnen: (Cooper, 2003; Murphy, 2000) |
|
Laag GRADE |
Het is mogelijk dat behandeling met antivirale medicatie bij hoogrisicopatiënten met bevestigde influenza resulteert in een snellere verlichting van symptomen vergeleken met placebo.
Bronnen: (Cooper, 2003; Lin, 2006; Murphy, 2000; Ison, 2020) |
Verdwijnen van koorts
|
Laag GRADE |
Het is mogelijk dat behandeling met antivirale medicatie bij patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen met influenza-like illness het aantal uur tot verdwijnen van koorts verminderd in vergelijking met placebo.
Bronnen: (Lin, 2006) |
|
Laag GRADE |
Het is mogelijk dat behandeling met antivirale medicatie bij hoogrisicopatienten (long/hart/neurologische/nier/leveraandoeningen/diabetes/obesitas/ Immuungecompromitteerd/jongeren met aspirinetherapie) met bevestigde influenza het aantal uur tot verdwijnen van koorts vermindert in vergelijking met placebo.
Bron: (Ison, 2020) |
Terugkeer tot normale gezondheid
|
Laag GRADE |
Het is mogelijk dat behandeling met antivirale medicatie bij patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen met influenza-like illness het aantal dagen voor terugkeer tot normale gezondheid vermindert in vergelijking met placebo.
Bronnen: (Lin, 2006) |
|
Laag GRADE |
Er bestaat onzekerheid over het effect van de behandeling met antivirale medicatie bij hoogrisicopatienten (long/hart/neurologische/nier/leveraandoeningen/diabetes/obesitas/ Immuungecompromitteerd/jongeren met aspirinetherapie) met bevestigde influenza op de tijd tot terugkeer tot normale gezondheid in vergelijking met placebo.
Bron: (Ison, 2020) |
|
- GRADE |
De effectiviteit van behandeling met antivirale medicatie bij ouderen met influenza-like illness op het aantal nieuw opgetreden complicaties waarbij antibioticagebruik nodig is, hospitalisatie, verlichting van symptomen, verdwijnen van koorts, en terugkeer tot normale gezondheid, is onbekend. Geen van de studies onderzocht deze uitkomstmaten bij ouderen. |
Beschrijving studies
Ten eerste is een RCT geïncludeerd (McLean, 2015) die de effectiviteit van oseltamavir (n=114, mediaan leeftijd 18 jaar, 43% man) vergeleek met placebobehandeling (n=51, mediaan leeftijd 17 jaar, 33% man) bij 134 volwassenen en kinderen (leeftijd 1 tot 79 jaar) met PCR-bevestigde griep, met de tijd tot verdwijnen van de symptomen, de kans dat na 4 of 7 dagen de symptomen waren verdwenen, kans op positieve RT-PCR, en ernst van de symptomen als uitkomstmaten.
Taieb (2019) combineerde in een netwerk meta-analyse 22 bronnen die de effectiviteit van antivirale middelen bij griepverschijnselen beschreven. Daarin is op 14 november 2016 gezocht in de MEDLINE (vanaf 1946), EMBASE (vanaf 1974), Cochrane Central Register of Controlled Trials (CENTRAL) en Igaku Chuo Zasshi (ICHUSHI) databases op RCTs van antivirale middelen (oseltamivir, tamiflu, gs4071, zanamivir, relenza, laninamivir, CS-8958, inavir, peramivir, rapvab, rapiacta, neuramidase inhibitor, cap-dependent endonuclease inhibitor, cap-snatching inhibitor en PA endonuclease inhibitor) bij humane influenza. Een aantal in deze review geïncludeerde studies is geen RCT, aangezien er ook congres abstracts en stukken van regulatory websites (EMA, FDA, PMDA) werden geïncludeerd. Daarnaast zijn de karakteristieken van de individuele studies zeer beperkt beschreven, waardoor geen GRADE-waardering mogelijk zou zijn. In de analyse werd enkel Baloxavir vergeleken met andere behandelingen en placebo. Om antwoord te kunnen geven op de uitgangsvraag en de informatie te waarderen volgens de GRADE methode, zijn daarom alle fulltext artikelen uit de review geanalyseerd die (1) RCT en (2) Engelstalig waren, en (3) een antiviraal middel vergeleken met placebo. Dertien RCT’s uit de analyse van Taieb (2019) zijn geanalyseerd; 7 bestudeerden Zanamivir (Boivin, 2000; Campion, 1998; Hayden, 1997; Makela, 2000; Matsumoto, 1999; Monto, 1999; Puhakka, 2003), 3 Oseltamivir (Li, 2003; Nicholson, 2000; Treanor, 2000), 2 Peramivir (Kohno, 2010; Whitley, 2015), waarmee het totaal aantal geanalyseerde RCT’s uitkwam op 12. In de studie van Matsumoto (1999) werd overigens een brede onderzoekspopulatie gehanteerd, waarbij 6% (7/116) van de patiënten gehospitaliseerd was bij aanvang van de studie.
Jefferson (2014) voerde een systematische review uit met niet-gepubliceerde gerandomiseerde, placebo gecontroleerde trials onder volwassenen en kinderen met bevestigde of vermoedelijke blootstelling aan van nature voorkomende griep. De literatuursearch werd uitgevoerd tot 22 juli 2013 in Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, Pubmed, de online GlaxoSmithKline Clinical Trials Register (GSK), Database of Reviews of Effect (DARE), de NHS Economic Evaluation Database (NHSEED), de Health Economic Evaluation Database (HEED) en de referentielijsten van artikelen. RCT’s werden geïncludeerd als de effecten van neuraminidase-remmers voor profylaxe, profylaxe na blootstelling (PEP) en behandeling van influenza werden onderzocht. Verder werden alleen studies geïncludeerd indien de studiepopulatie bestond uit voorheen gezonde personen (kinderen en volwassenen), waarbij personen met chronische ziekten (zoals astma, diabetes, hypertensie) werden geïncludeerd, maar personen met ziekten die een significant effect hadden op het immuunsysteem (zoals maligniteiten of HIV-infecties) werden uitgesloten. Daarnaast werden alleen studies meegenomen met personen die waren blootgesteld aan van nature voorkomende griep met of zonder symptomen.
In deze literatuursamenvatting werden alleen de trials meegenomen die de, door de werkgroep vastgelegde uitkomstmaten, rapporteerden. In totaal hadden 28 ongepubliceerde trials één of meerdere cruciale of belangrijke uitkomstmaten gerapporteerd. Hiervan gingen acht studies over oseltamivir bij volwassen en drie over oseltamivir bij kinderen. Vijftien van de 28 studies onderzochten de effectiviteit van zanamivir bij volwassen en twee de effectiviteit van zanamivir bij kinderen.
Heinonen (2010) voerde een gerandomiseerd dubbelblind, placebo gecontroleerde studie uit onder kinderen tussen de 1 en 3 jaar oud met laboratorium bevestigde influenza tijdens de seizoenen 2007 en 2008 en 2008 en 2009. Patiënten werden geïncludeerd als ze een leeftijd hadden tussen de 1 en 3 jaar, minder dan 24 uur koorts had (een orale, rectale of axillaire temperatuur van ≥ 38.0°C) en 1 of meer tekenen of symptomen had van een luchtweginfectie (hoesten, rhinitis of zere keel) of een positief resultaat van de influenza sneltest. Patiënten werden geëxcludeerd als ze een virologische infectie hadden met een andere infectie dan influenza, als er verdenking was op een ernstige invasieve bacteriële infectie waarbij onmiddellijke ziekenhuisopname noodzakelijk was, slecht gecontroleerde onderliggende medische aandoening, bekende immunosuppressie, allergie voor oseltamivir, een behandeling ondergaan van oseltamivir in de voorafgaande 4 weken of deelname aan een andere klinische studie met een onderzoeksgeneesmiddel.
De interventie bestond uit een behandeling met oseltamivir tweemaal daags voor 5 dagen (met een totaal van 10 doses). De dosering van oseltamivir was 30 mg tweemaal daags voor kinderen die minder dan 15 kilogram wogen en 45 mg tweemaal daags voor kinderen die tussen de 15,1 en 23 kilogram wogen. De controle behandeling was een placebo.
In totaal werden er 37 patiënten geïncludeerd in de interventie groep en 61 in de controlegroep. De gemiddelde leeftijd in de interventie en controlegroep waren 2,3 jaar (SD=0,8) en 2,5 jaar (SD=0,8), respectievelijk. Verder was het percentage man in de interventie en controlegroep 62,2% en 62,3%, respectievelijk. Er werden geen prognostische factoren meegenomen om te corrigeren voor mogelijke confounding.
Venkatasan (2017) verrichtte een meta-analyse met individuele patiënt data naar de associatie tussen neuraminidase inhibitors (NAI) en ziekenhuisopname bij niet-opgenomen patiënten met laboratorium-bevestigde of klinisch gediagnosticeerde influenza A H1N1pdm09. Data werd verzameld in 9 centra (Argentinië, Canada, Frankrijk, Duitsland, Israël, Saudi-Arabië, Singapore, Slovenië en UK). De primaire uitkomstmaat was influenza-gerelateerde ziekenhuisopname. Gebruik van NAI (onafhankelijk van timing en setting) werd vergeleken met geen gebruik van NAI. Indien de data beschikbaar was werd bovendien een vergelijking gemaakt tussen vroege behandeling (start ≤ 2 dagen na eerste symptomen) en late behandeling (start > 2 dagen na eerste symptomen) met NAI. Er werden propensity scores berekend voor de waarschijnlijkheid van NAI behandeling voor elke patiënt binnen elke individuele dataset door middel van multivariabele logistische regressie analyse voor binaire variabelen en gegeneraliseerde propensity score schattingen voor de continue tijd tot behandeling variabele. Covariaten waren leeftijd, geslacht, comorbiditeit (ja of nee), indicator voor ziekte ernst (gedocumenteerde ernstige ademnood of kortademigheid). Subgroepanalyses werden gedaan voor onder andere patiënten met een hoog-risico conditie, wat gedefinieerd werd als het hebben van minstens 1 chronische ziekte met indicatie voor seizoen griepvaccinatie of 65 jaar of ouder waren. Ook werd een subgroep analyse uitgevoerd voor kinderen (< 16 jaar). De gegevens van in totaal 3376 patiënten werden geïncludeerd, waarvan 1671 (49,9%) niet werden opgenomen. In totaal waren er 1019 niet-opgenomen patiënten (30,1%) met minstens 1 hoog-risico conditie.
Lin (2006) voerde een RCT uit onder patiënten met chronische ademhalingsaandoeningen (astma of COPD) of chronische hartziekten (coronaire hartziekte of chronisch hartfalen), die zich presenteerden in het ziekenhuis binnen 48 uur na de start van de symptomen voor influenza. Patiënten werden gerandomiseerd naar een behandeling met 75 mg orale Oseltamivir twee keer per dag voor vijf achtereenvolgende dagen (interventiegroep) of een routinematige symptomatische behandeling (controlegroep). Alle patiënten werden 21 dagen gevolgd of totdat ze herstelden van hun infectie. In totaal werden er 58 patiënten geïncludeerd in de interventiegroep en 60 in de controlegroep. Influenza was gediagnosticeerd in 47% (27/58) van de interventiegroep en 48% (29/60) van de controlegroep. Deze groep werd gedefinieerd als de ITTI populatie en meegenomen in de analyse. De gemiddelde leeftijd was 48,1 jaar (SD 0,8) in de interventie en 52,3 jaar (SD 16,0) in de controlegroep. In de interventiegroep was 63% man en in de controlegroep 55%.
Murphy (2000) verrichtte een gerandomiseerd dubbelblind placebo gecontroleerde studie naar de behandeling van influenza bij patiënten van 12 jaar of ouder met astma of COPD. Patiënten met onderliggende astma en/of COPD met acute influenza-like-ilness van minder dan 36 uur werden geworven voor de studie wanneer het bekend was dat influenza circuleerde in de gemeenschap. Patiënten werden gerandomiseerd naar een behandeling met 2 inhalaties van Zanamivir (5 mg per inhalatie) per dag of 2 inhalaties met een placebo (lactose poeder) per dag voor 5 dagen. De follow-up periode was 28 dagen. In totaal werden 525 patiënten met astma en/of COPD en met influenza-like-ilness geïncludeerd, waarvan er 262 naar de Zanamivir groep en 263 naar de placebogroep werden gerandomiseerd. De gemiddelde leeftijd was 36 jaar (range 12 tot 88) in de interventie en 40 jaar (range 12 tot 85) in de controlegroep. In de interventiegroep was 42% man en in de controlegroep 43%. Uiteindelijk hadden 313 patiënten (60%) laboratorium bevestigde influenza.
Johnston, 2005 voerde een gerandomiseerd dubbelblind placebo gecontroleerde studie uit in drie landen (Argentinië, Spanje, Verenigd Koninkrijk) naar de behandeling van influenza bij kinderen van 6 tot 12 jaar oud met astma, gedurende de griepseizoenen 1998 en 1999. Patiënten werden gerandomiseerd naar een behandeling met Oseltamivir (2mg/kg) twee keer per dag door middel van een orale siroop (6mg/mL) of een placebo. De follow-up periode betrof vier weken. Inclusiecriteria waren kinderen van 6 tot 12 jaar met astma (zodanige astma dat medische follow-up/ ziekenhuiszorg nodig was) en influenza symptomen (temperatuur > 38.6°C) inclusief één respiratoir symptoom die zich binnen 48u na de start van de symptomen meldden. In totaal werden 170 patiënten met ILI geïncludeerd in de Oseltamivirgroep waarvan 84 patiënten bewezen influenza hadden (ITTI); 164 met ILI werden in de placebogroep geïncludeerd (ILI) waarvan 84 bewezen influenza hadden (ITTI).
Het onderzoek van Hayden, 2018 verrichtte twee gerandomiseerde dubbelblinde placebo gecontroleerde studies naar de behandeling van patiënten met ILI (fase 2 en fase 3 trial).
In de fase 2 trial ontvingen patiënten in de leeftijd van 20-64 jaar met acute influenza (op basis van een positieve antigeen sneltest) een enkele dosis baloxavir van 10, 20 of 40 mg of een placebo. In de fase 3 trial (bekend als CAPSTONE-1) ontvingen ILI patiënten in de leeftijd van 12-64 jaar een enkele dosis baloxavir of placebo (40 mg bij patiënten <80 kg lichaamsgewicht; 80 mg bij patiënten ≥80 kg) in combinatie met een oseltamivir-placebo tweedagelijks voor 5 dagen (interventie) of een single-dosis placebo met een oseltamivir placebo, tweedagelijks voor 5 dagen. Kinderen in de leeftijd van 12-19 jaar ontvingen alleen baloxavir of een placebo, gedurende één dag. De studie werd gesponsord door de farmacie, waarbij de farmaceut betrokken was bij alle onderdelen van het onderzoek.
Portsmouth, 2020 verrichtte subanalyse op de studiedata van Hayden, 2018 voor de groep patiënten tussen de 12 t/m 17 jaar. In de baloxavirgroep zaten 63 patiënten en in de placebogroep 27 patiënten, beide met bewezen influenza (ITTI populatie). Voor achtergrondinformatie over de dosis etc., zie de tekst hierboven.
Ison, 2020 verrichtte een gerandomiseerd dubbelblind placebo gecontroleerde studie naar de behandeling van patiënten vanaf 12 jaar met ILI en met een hoog risico op het ontwikkelen van influenza-gerelateerde complicaties. Risicogroepen die werden geincludeerd waren patiënten ouder dan 64 jaar; met een chronische longaandoening; neurologische aandoeningen; hartziekten; diabetes mellitus; lever of nieraandoeningen; immuungecomprommitteerden; BMI≥40 kg/m2 of jongeren tot 19 jaar die langdurige aspirinetherapie ondergingen. De studie werd uitgevoerd in 17 landen met in totaal 551 locaties waar patiënten werden geincludeerd (CAPSTONE-2) waarbij een deel van geïncludeerde patiënten uiteindelijk afviel vanwege non-compliance met de Good Clinical Practice guidelines op sommige studielocaties. Patiënten met bevestigde influenza (RT-PCR getest) werden gerandomiseerd naar een behandeling met een enkele orale dosis baloxavir (40 mg bij patiënten <80 kg lichaamsgewicht; 80 mg bij patiënten ≥80 kg) in combinatie met een oseltamivir-placebo tweedagelijks voor 5 dagen (interventie) of een single-dosis placebo met een oseltamivir placebo, tweedagelijks voor 5 dagen. De follow-up betrof 22 dagen (waarvan 14 dagen follow-up voor influenza symptomen en 22 dagen voor laboratorium tests). In de interventiegroep werden 388 patiënten met ILI geincludeerd, waarvan 50% man was en een gemiddelde leeftijd van 52 jaar. In de controlegroep werden 386 patiënten geincludeerd, waarvan 47% man en een gemiddelde leeftijd van 52 jaar. De studie werd gesponsord door de farmacie, waarbij de farmaceut betrokken was bij alle onderdelen van het onderzoek.
Resultaten
Volwassenen
Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis) (cruciaal)
In de literatuur werd alleen de uitkomstmaat “nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)” beschreven in de systematische literatuur review van Jefferson (2014). Er waren zes studies die oseltamivir in volwassen beschreven met in totaal 2179 patiënten in de interventiegroep en 1496 patiënten in de controlegroep. Er werd een risk ratio gevonden van 0,91 in het voordeel van de antivirale medicatie Oseltamivir, met een 95% BI van 0,40 tot 2,06 (M76001, WV15671, WV156707, WV15812/WV15872, WV15819/WV15876/WV15978, WV16277). Dit verschil is niet statistisch significant (figuur 1).
Figuur 1 Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis) bij antivirale therapie van oseltamivir versus placebo in volwassenen
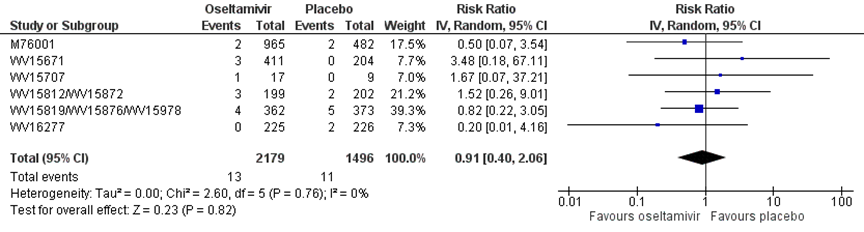
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Nieuw ontstane complicaties waarbij antibiotica behandeling nodig was, werd beschreven in 3 studies voor een ITT populatie van 668 versus 686 patiënten (Campion, 1998; Kohno, 2010; Treanor, 2000). Het relatieve risico (RR) op complicaties was 0,68 (95% BI 0,52 tot 0,90) in het voordeel van de interventie, een verschil dat statistisch significant is (figuur 2). Slechts 1 studie (Hayden, 2018) beschreef de complicaties in een ITTI populatie, en vond geen significante verschillen tussen 456 patiënten met antivirale medicatie en 231 patiënten met placebobehandeling (RR 0,81; 95% BI van 0,37 tot 1,76). Slechts 1 studie (Hayden, 2018) beschreef de complicaties in een ITTI populatie, en vond geen significante verschillen tussen 456 patiënten met antivirale medicatie en 231 patiënten met placebobehandeling (RR 0,81; 95% BI van 0,37 tot 1,76).
Figuur 2 Complicaties geassocieerd met antibioticagebruik (relatief risico) in ITT populatie bij antivirale therapie versus placebo
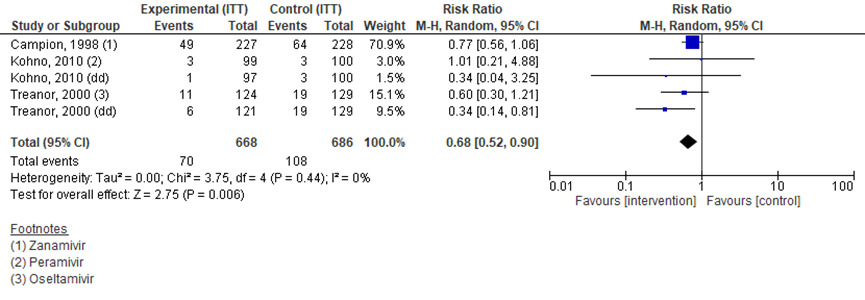
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Er waren tien studies die Zanamivir in volwassen beschreven met in totaal 2301 patiënten in de interventiegroep en 2087 patiënten in de controlegroep. Er werd een risk ratio gevonden van 0,86 in het voordeel van de antivirale medicatie Zanamivir, met een 95% BI van 0,49 tot 1,50 (JNAI-07, NAI30008n NAI30010, NAI30011, NAI30012, NAI30015, NAIA2005, NAIA3002, NAIB3001, NAIB3002). Dit verschil is niet statistisch significant (figuur 3).
Figuur 3 Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis) bij antivirale therapie van zanamivir versus placebo in volwassenen
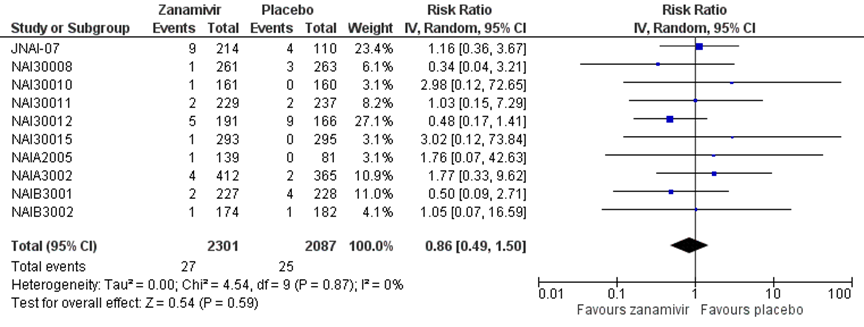
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Hospitalisatie (cruciaal)
De uitkomstmaat “hospitalisatie” werd beschreven in de systematische literatuur review van Jefferson (2014). Er waren zeven studies die oseltamivir in volwassen beschreven met in totaal 2663 patiënten in de interventiegroep en 1731 patiënten in de controlegroep. Er werd een risk ratio gevonden van 0,92 in het voordeel van de antivirale medicatie Oseltamivir, met een 95% BI van 0,57 tot 1,50 (M76001, WV15670, WV15671, WV156707, WV15812/WV15872, WV15819/WV15876/WV15978, WV16277). Dit verschil is niet statistisch significant (figuur 4).
Figuur 4 Hospitalisatie bij antivirale therapie van oseltamivir versus placebo in volwassenen
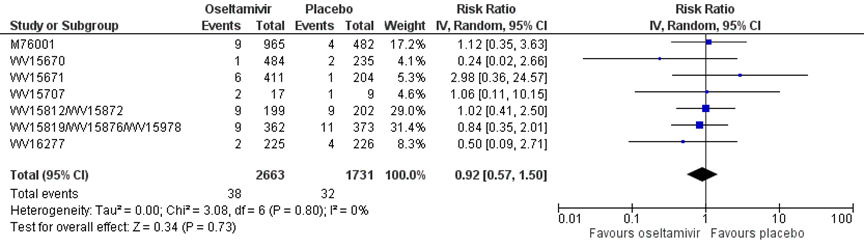
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Venkatesan (2017) verrichtte een meta-analyse met individuele patiënt data naar de associatie tussen NAI en hospitalisatie bij niet-opgenomen patiënten. De waarschijnlijkheid van hospitalisatie bij patiënten met bevestigde of gediagnosticeerde influenza die behandeld werden met NAI was lager vergeleken met patiënten die niet behandeld werden met NAI (gecorrigeerde OR 0,24; 95% BI 0,20 tot 0,30; P<0,001).
In de subgroep met alleen laboratorium bevestigde influenza (alle leeftijden, N=3085) was de waarschijnlijkheid van hospitalisatie lager bij patiënten die behandeld werden met NAI vergeleken met geen NAI (gecorrigeerde OR 0,24; 95% BI 0,19 tot 0,29; P<0,001).
Bij een vergelijking tussen vroege NAI behandeling (≤ 2 dagen na eerste symptomen) en late behandeling (na 2 tot 5 dagen) met 473 patiënten hadden patiënten met een vroege behandeling een lagere waarschijnlijkheid tot hospitalisatie (gecorrigeerde OR 0,44; 95% BI 0,23 tot 0,86; P=0,16). In alle analyses werd gecorrigeerd voor behandeling propensity (bij quintiel) en antibioticagebruik.
De uitkomstmaat “hospitalisatie” werd niet gerapporteerd in de studies voor het antivirale middel zanamivir dan wel baloxavir in volwassenen.
Tijd tot verlichting van symptomen (belangrijk)
De meest gangbare uitkomstmaat beschreven in de geselecteerde artikelen is de “tijd tot verlichting van symptomen”, waarbij sommige studies een variant gebruikten, zoals alleviation of “major symptoms”, “clinically important symptoms”, “5 symptoms” of “3 symptoms”. De studies van Nicholson (2000) en Treanor (2000) hanteerden “duration of illness” en “illness duration”, welke meer overeenkomen met “alleviation of symptoms” dan met “return to usual health”. Voor de analyse zijn deze allen samengenomen als 1 uitkomstmaat en in de figuren zijn de verschillende uitkomstmaten gespecificeerd. De resultaten werden beschreven op basis van een intention-to-treat analyse (ITT) en/of op basis van een intention-to-treat infected populatie (ITTI).
In de gepubliceerde RCT’s van Taieb (2019) werden in totaal 932 patiënten geïncludeerd in de interventiegroep en 459 patiënten in de controlegroep. Er werd een gemiddeld verschil gevonden van -0,64 in het voordeel van Oseltamivir, met een 95% BI van -1,07 tot -0,22 (McLean, 2015; Nicholson, 2000; Treanor, 2000).
De systematische literatuur review van Jefferson (2014) beschreef ook de uitkomstmaat “tijd tot verlichting van symptomen” in ongepubliceerde RCT’s. Er waren acht studies die Oseltamivir in volwassenen beschreven met in totaal 2208 patiënten in de interventiegroep en 1746 patiënten in de controlegroep. Er werd een verschil gevonden van -0,67 in het voordeel van de antivirale medicatie Oseltamivir, met een 95% BI van -1,01 tot -0,32 (M76001, WV15670, WV15671, WV156707, WV15730, WV15812/WV15872, WV15819/WV15876/WV15978, WV16277). Dit verschil is statistisch significant figuur 5).
Figuur 5 Tijd tot verlichting van symptomen (in dagen) bij antivirale therapie van Oseltamivir versus placebo in volwassenen
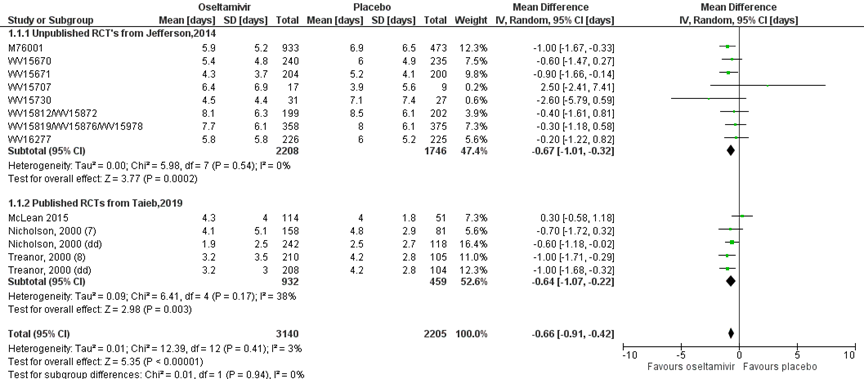
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Er waren dertien studies die Zanamivir in volwassen beschreven met in totaal 3031 patiënten in de interventiegroep en 2380 patiënten in de controlegroep. Er werd een verschil gevonden van -0,60 in het voordeel van de antivirale medicatie Zanamivir, met een 95% BI van -0,81 tot -0,39 (JNAI-01, JNAI-04, JNAI-07, NAI30008, NAI30010, NAI30012, NAI30015, NAIA/B2008, NAIA2005, NAIA3002, NAIB2005, NAIB3001, NAIB3002). Dit verschil is statistisch significant.
In de gepubliceerde RCT’s van Taieb (2019) werden in totaal 1870 patiënten geïncludeerd in de interventiegroep en 1309 patiënten in de controlegroep. Er werd een gemiddeld verschil gevonden van -0,79 in het voordeel van Zanamivir, met een 95% BI van -1,11 tot -0,47 (Campion, 1998; Hayden, 1997; Makela, 2000; Matsumo, 1999; Monto, 1999; Puhakka, 2003) (figuur 6).
Figuur 6 Tijd tot verlichting van symptomen (in dagen) bij antivirale therapie van Zanamivir versus placebo in volwassenen
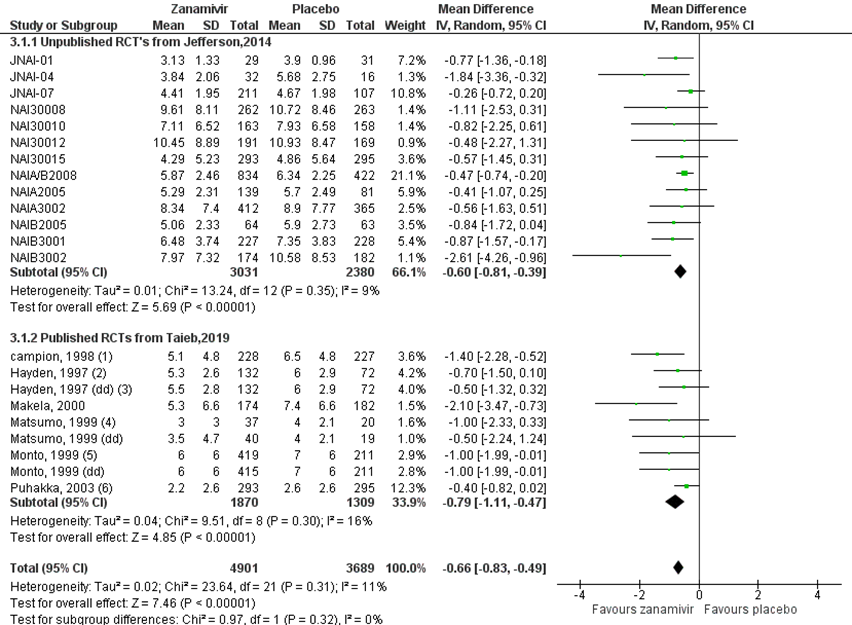
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Slechts één studie (Hayden, 2018) beschreef de tijd tot verlichting van symptomen bij antivirale therapie van Baloxavir in volwassenen. Er werd een verschil gevonden van -1 (95% BI van -1,41 tot -0,58) in het voordeel van de interventie, bij in totaal 919 patiënten. Dit verschil is statistisch significant.
In de ITTI populatie werd eenzelfde beeld gevonden: een gemiddeld verschil van -0,90 (95% BI -1,11 tot -0,70) in het voordeel van de interventie, in 12 studies met in totaal 2358 patiënten in de interventiegroep en 1536 patiënten in de placebogroep (Boivin, 2000; Campion, 1998; Hayden, 1997; Makela, 2000; Matsumoto, 1999; Puhakka, 2003; Li, 2003; Nicholson, 2000; Treanor, 2000; Kohno, 2010; Whitley, 2015; Hayden, 2018). Dit verschil is statistisch significant. Voor Zanamivir werd een verschil gevonden van -1,30 (95% BI -2,04 tot -0,56), voor Oseltamivir -1,03 (95% BI -1,57 tot -0,49), voor Peramivir -0,70 (95% BI -0,96 tot -0,45) en voor Baloxavir -1,00 (95% BI -1,35 tot -0,65) (figuur 7).
Figuur 7 Tijd tot verlichting van symptomen (in dagen) in ITTI populatie bij antivirale therapie versus placebo
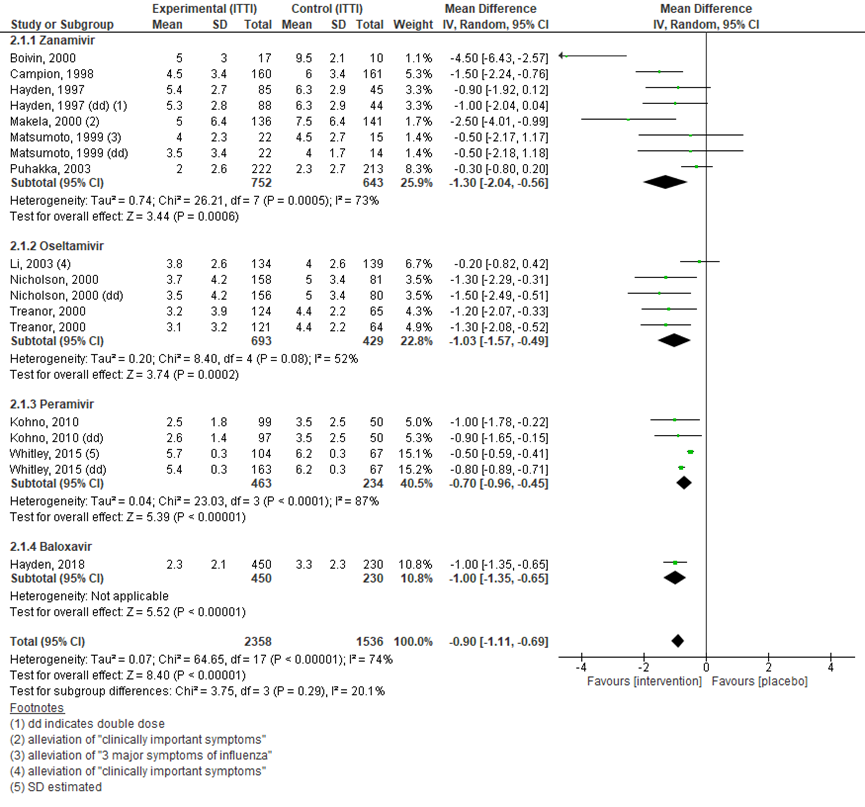
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Tijd tot verdwijnen van koorts (belangrijk)
Drie studies uit de review van Taieb (2019), waarvan 2 met 2 verschillende doseringen, beschreven tijd tot verdwijnen van koorts in een ITT populatie (figuur 7). Bij 1125 patiënten in de interventiegroep en 605 in de controlegroep (Campion, 1998; Monto, 1999; Nicholson, 2000) werd een gemiddeld verschil gevonden van -0,74 (95% BI van -1,36 tot -0,12), een statistisch significant verschil in het voordeel van de interventie. In de ITTI populatie werd in 4 studies met in totaal 1766 patiënten (Campion, 1998; Nicholson, 2000; Whitley, 2015; Hayden, 2018) een verschil gevonden van -2,05 (95% BI -3,22 tot -0,33), een significant verschil in het voordeel van antivirale medicatie (figuur 8).
De uitkomstmaat “tijd tot verdwijnen van koorts” werd niet gerapporteerd in de studie van Jefferson (2014).
Figuur 8 Tijd tot verdwijnen van koorts (in dagen) in ITT populatie bij antivirale therapie versus placebo
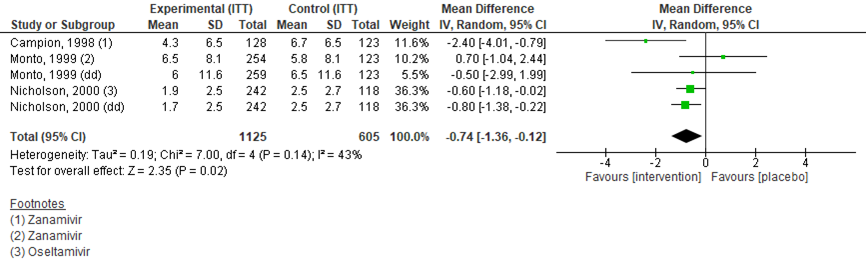
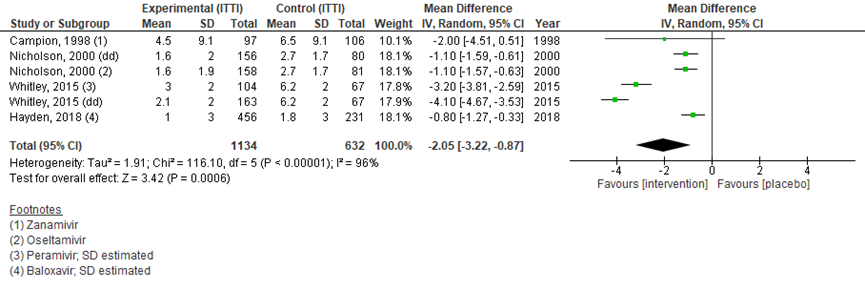
dd: double dose, de hoge dosis in het geval van een studie met lage en hoge dosis; Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Tijd tot terugkeer normale gezondheid (belangrijk)
Slechts één studie (Treanor, 2000) uit de review van Taieb (2019) beschreef de tijd tot terugkeer normale gezondheid in een ITT populatie en vond een verschil van -3,05 (95% BI van -4,81 tot -1,29) in het voordeel van de interventie, bij in totaal 627 patiënten. Dit verschil is statistisch significant. Deze studie beschreef deze uitkomstmaat ook in een ITTI populatie, en gecombineerd met een andere studie (Hayden, 2018) werd bij 1061 patiënten (701 interventie, 360 placebo) een gemiddeld verschil gevonden van -2,12 (95% BI -3,14 tot -1,09), eveneens een statistisch significant verschil.
De uitkomstmaat “tijd tot terugkeer normale gezondheid” werd niet gerapporteerd in de studie van Jefferson (2014).
Kinderen
Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis) (cruciaal)
Er waren drie studies die Oseltamivir in kinderen beschreven met in totaal 682patiënten in de interventiegroep en 681 patiënten in de controlegroep. Er werd een risk ratio gevonden van 1,96 in het voordeel van de placebo, met een 95% BI van 0,67 tot 5,74 (Johnston, 2005, WV15758, WV15759/WV15871). Dit verschil is niet statistisch significant (figuur 9).
Figuur 9 Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis) bij antivirale therapie van Oseltamivir versus placebo in kinderen
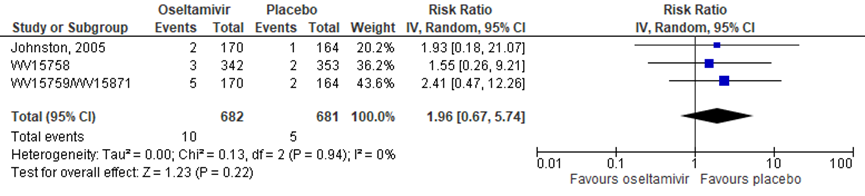
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
De uitkomstmaat “nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)” werd niet gerapporteerd in de studies voor het antivirale middel Zanamivir in kinderen.
Portsmouth (2020) rapporteerde complicaties die resulteerden in antibiocabehandeling voor het middel baloxavir. In de baloxavirgroep (ITTI populatie) werd geen complicatie gemeld (0/63) en in de placebogroep één (1/27) complicatie (bronchitis).
Hospitalisatie (cruciaal)
Er waren drie studies die Oseltamivir in kinderen beschreven met in totaal 677 patiënten in de interventiegroep en 682 patiënten in de controlegroep. Er werd een risk ratio gevonden van 1,92 in het voordeel van de placebo, met een 95% BI van 0,70 tot 5,23 (NV16871, WV15758, WV15759/WV15871). Dit verschil is niet statistisch significant (figuur 10).
Figuur 10 Hospitalisatie bij antivirale therapie van oseltamivir versus placebo in kinderen
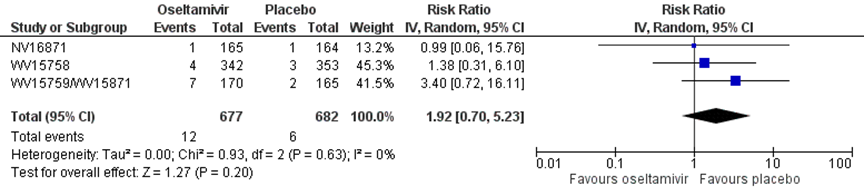
Johnston (2005) rapporteerde een gecombineerde uitkomstmaat waarbij het onduidelijk is of dit de ILI of ITTI populatie betreft. Daarom zijn de resultaten niet toegevoegd aan de meta-analyse in figuur 11. In de Oseltamivirgroep was sprake van 27% ongeplande ziekenhuisbezoeken of ziekenhuisopnames; in de placebogroep betrof dit 23%. Er werd geen p-waarde gerapporteerd. Deze resultaten zijn in lijn met de gevonden richting van het effect in figuur 11.
Venkatesan (2017) verrichtte een subgroep analyse op individueel patiënten met 1747 niet-opgenomen kinderen jonger dan 16 jaar. Daarbij werd geen onderscheid gemaakt tussen kinderen met en zonder laboratorium bevestigde influenza. De waarschijnlijkheid van hospitalisatie bij de kinderen die behandeld werden met NAI was lager vergeleken met de kinderen die geen NAI ontvingen (OR 0,25; 95% BI 0,18 tot 0,34; P<0,001).
De uitkomstmaat “hospitalisatie” werd niet gerapporteerd in de studies voor het antivirale middel Zanamivir in kinderen.
De uitkomstmaat “hospitalisatie” werd niet gerapporteerd in de studie voor het antivirale middel baloxavir bij kinderen.
Tijd tot verlichting van symptomen (belangrijk)
Slechts één ongepubliceerde studie in de review van Jefferson (2014) (WV15758) beschreef de tijd tot verlichting van symptomen bij antivirale therapie van Oseltamivir in kinderen. Er werd een verschil gevonden van -29,4 uur (95% BI van -47,04 tot -11,76) in het voordeel van de interventie, bij in totaal 669 patiënten. Dit verschil is statistisch significant.
Er was twee studies (Heinonen (2010) en Johnston (2005)) die de tijd tot verlichting van symptomen beschreef bij antivirale therapie van Oseltamivir in kinderen. Heinonen rapporteerde in de interventie en controle groep een mediane aantal dagen tot verlichting van symptomen gevonden van 10,4 dagen (IQR= 4,6 tot 12,4) en 13,3 dagen (IQR= 10,3 to 17,1), en een verschil tussen de 2 groepen van 2,8 dagen in het voordeel van de interventiegroep (P<0,001).
Johnston (2005) rapporteerde de tijd tot verlichting van symptomen in de ITTI populatie bij kinderen met astma. Er werd een mediane tijdsduur gerapporteerd zonder IQR. De interventiegroep had een mediane tijd van 90,4 uur en de placebogroep 115,6 uur met een verschil van 25,3 uur in het voordeel van de interventiegroep (p=0,1197).
Er waren twee studies die Zanamivir in kinderen beschreven met in totaal 396 patiënten in de interventiegroep en 327 patiënten in de controlegroep. Er werd een gepoold verschil gevonden van -1,08 (95% BI van -2,32 tot 0,15), in het voordeel van de interventie. Dit verschil is niet statistisch significant (figuur 11).
Figuur 11 Tijd tot verlichting van symptomen (in dagen) bij antivirale therapie van Zanamivir versus placebo in kinderen
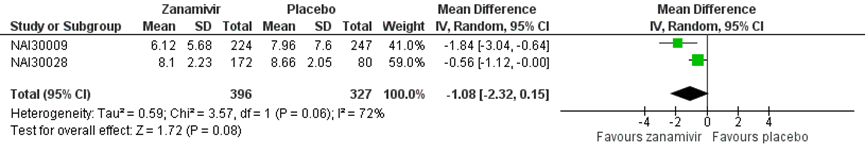
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Voor het middel baloxavir rapporteerde de studie van Portsmouth (2020) de tijd tot verlichting van symptomen in de ITTI populatie. Dit was in de interventiegroep 54,1u en in de controlegroep 92,7u met een mediaan verschil van 38,6u (p=0,0055, significant).
Tijd tot verdwijnen van koorts (belangrijk)
Er was één studie (Heinonen,2010) die de tijd tot verdwijnen van koorts beschreef bij antivirale therapie van Oseltamivir in kinderen. Er werd in de interventie en controlegroep een mediane aantal dagen tot verdwijnen van koorts gevonden van 1,7 dagen (IQR= 0,9 tot 2,9) en 2,9 dagen (IQR= 1,2 tot 4,7), en een verschil tussen de 2 groepen van 1,2 dagen in het voordeel van de interventiegroep (P=0,004).
De uitkomstmaat “tijd tot verdwijnen van koorts” werd niet gerapporteerd in de studies voor het antivirale middel Zanamivir in kinderen.
Voor het middel baloxavir rapporteerde de studie van Portsmouth (2020) de tijd tot verlichting van koorts in de ITTI populatie. De mediane tijd was in de interventiegroep 27,1 (95%BI van 22,0 – 35,0) uur en in de controlegroep 43,1 uur (95%BI: 26,9 tot 50,8).
Tijd tot terugkeer normale gezondheid (belangrijk)
Er was één studie (Heinonen,2010) die de tijd tot terugkeer van normale gezondheid beschreef bij antivirale therapie van Oseltamivir in kinderen. Er werd in de interventie en controlegroep een mediane aantal dagen tot terugkeer van normale gezondheid gevonden van 4,3 dagen (IQR= 2,2 tot 5,9) en 5,7 dagen (IQR= 4,2 tot 10,3), en een verschil tussen de twee groepen van 1,4 dagen in het voordeel van de interventiegroep (P=0,004).
Johnston (2005) rapporteerde de tijd tot normale gezondheid in de ITTI populatie bij kinderen met astma. Er werd een mediane tijdsduur gerapporteerd zonder IQR.
De interventiegroep had een mediane tijd tot terugkeer naar normale gezondheid van 101,4 uur en de placebogroep 114 uur met een verschil van 12,6 uur in het voordeel van de interventiegroep (p=0,4555).
De uitkomstmaat “tijd tot terugkeer van normale gezondheid” werd niet gerapporteerd in de studies voor de antivirale middellen Zanamivir en baloxavir in kinderen.
Hoog-risicopatiënten
Nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)
Lin (2006) beschreef in de ITTI populatie van patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen het aantal patiënten waar complicaties ontstonden gedurende de behandeling. 3 van de 27 (11%) patiënten in de interventiegroep van de ITTI populatie en 13 van de 29 (45%) in de controlegroep ontwikkelden complicaties. Dit komt neer op een RR van 0,15 (95% BI 0,04 tot 0,63; P=0,009) ten gunste van Oseltamivir. De complicaties die optraden waren tracheitis, bronchitis, pneumonie, nasosinusitis en pharyngitis, dit werd niet gespecificeerd naar groep.
Lin (2006) rapporteerde het aantal patiënten dat antibiotica ontving gedurende de influenza bij patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen. Er was een statistisch significant verschil in het antibioticagebruik tussen de Oseltamivir groep (10 van de 27; 37%) en de controlegroep (20 van de 29; 69%). Dit komt neer op een RR van 0,26 (95% BI 0,09 tot 0,80) in het voordeel van de interventiegroep.
Murphy (2000) rapporteerde het aantal patiënten dat antibiotica nodig had voor influenza-gerelateerde complicaties. Er was geen statistisch significant verschil (P=0,064) tussen het antibioticagebruik (RR 0,42; 95% BI 0,18 tot 0,96), met 7 van de 160 (4%) personen in de Zanamivir groep en 16 van de 153 (10%) personen in de controlegroep die antibiotica nodig hadden.
Ison (2020) rapporteerde het aantal influenza-gerelateerde complicaties in de ITTI popluatie. In de baloxavirgroep werden 11/388 (2,8%) complicaties geregistreerd, in de controlegroep 40/386 (10.4%) met een p-waarde <0,0001). Dit komt neer op een RR van 0,27 (95%BI van 0,14 tot 0,53) in het voordeel van de baloxavirgroep (statistisch significant). Het aantal complicaties wat resulteerde in antibioticabehandeling was 13/388 in de baloxavirgroep en 29/386 in de placebogroep. Dit komt neer op een RR van 0,45 (95%BI van 0,24 tot 0,84) in het voordeel van de baloxavirgroep (statistisch significant).
Hospitalisatie
Venkatesan (2017) verrichtte een subgroep analyse op individueel patiënten met 1019 niet-opgenomen patiënten met minimaal 1 hoog-risico conditie. Daarbij werd geen onderscheid gemaakt patiënten met en zonder laboratorium bevestigde influenza. De waarschijnlijkheid van hospitalisatie bij de hoog risicogroep die behandeld werden met NAI was lager vergeleken met de hoog risicogroep die geen NAI ontvingen (OR 0,27; 95% BI 0,19 tot 0,38; P<0,001).
Lin (2006) rapporteerde hoeveel patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen per groep tijdens de studie in het ziekenhuis werden opgenomen voor influenza. Er was geen statistisch significant verschil tussen de groepen in het aantal ziekenhuisopnamen met 2 opnames (7%) in de interventiegroep en 5 opnames in de controlegroep (RR 0,38; 95% BI 0,07 tot 2,17).
Ison (2020) rapporteerde het aantal ziekenhuisopnames in de ITTI populatie. Er was geen significant verschil tussen beide groepen: er waren 3 ziekenhuisopnames in de baloxavirgroep (0,8%) en 5 in de placebogroep (1,3%). Dit komt neer op een RR van 0,6 (95%BI van 0,14 tot 2,48).
Tijd tot verlichting van symptomen
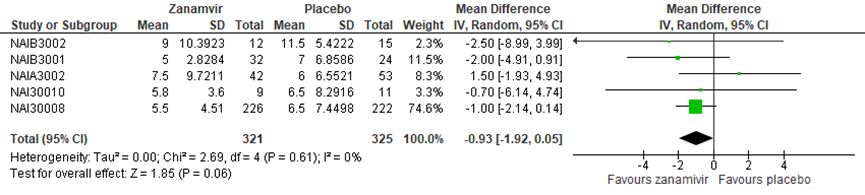
Jefferson verrichtte geen subgroep analyse naar patiënten met multimorbiditeit. Cooper (2003) deed dit wel, in deze analyse werden vijf van de ongepubliceerde studies met in totaal 646 patiënten geïncludeerd, die ook in Jefferson werden beschreven. Mogelijk waren er nog andere studies in de review van Jefferson die ook een subgroep met multimorbiditeit beschreven, maar dat is niet te controleren. Hoog risicopatiënten werden gedefinieerd als diegene van 65 jaar en ouder of diegene met bepaalde chronische medische condities, zoals ademhalingsziekte, hartziekte en pulmonaire afwijkingen. In de ITTI populatie met multimorbiditeit werd geen verschil gevonden tussen Zanamivir en placebo in de tijd tot verlichting van symptomen, met een gepoold gemiddeld verschil van -0,93 (95% BI -1,92 tot 0,05; figuur 12). In de ITT populatie met multimorbiditeit werd een verschil gevonden in het voordeel van Zanamivir met een gepoold gemiddeld verschil van -1,99 (95% BI -3,04 tot -0,93; figuur 13).
Figuur 12 Tijd tot verlichting van symptomen (in dagen) in ITTI populatie subgroep hoog risicopatiënten zanamivir versus placebo (Bron: Cooper, 2003)
Figuur 13 Tijd tot verlichting van symptomen (in dagen) in ITT populatie subgroep hoog risicopatiënten Zanamivir versus placebo (Bron: Cooper, 2003)
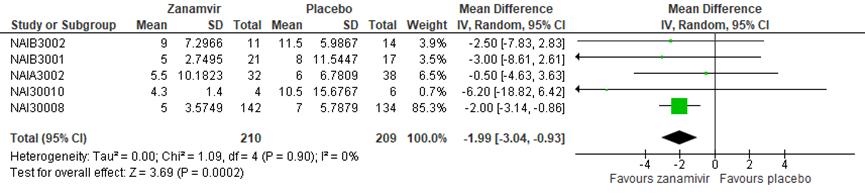
Lin (2006) rapporteerde de tijd tot het verdwijnen van alle influenza symptomen. Er was een statistisch significant verschil (P=0,048) in de tijd tot het verdwijnen van symptomen tussen de Oseltamivir groep (gemiddeld 110,4 uur; SD 125,9) en de controlegroep (gemiddeld 174,4 uur; SD 93,4).
Murphy (2000) rapporteerde de mediane tijd tot verdwijnen van influenza symptomen in de populatie met COPD of astma. Er was een statistisch significant verschil (P=0.009) in de mediane tijd tot verdwijnen van symptomen in de ITTI populatie met laboratorium bevestigde influenza (mediane verschil 1,5; 95% BI 0,50 tot 3,25) tussen de Zanamivir-groep (mediaan 5,5) en de placebogroep (mediaan 7,0). In de ITT populatie was er geen statistisch significant verschil (P=0,123) tussen de Zanamivir-groep (mediaan 6,0) en de controlegroep (mediaan 7,0).
Ison (2020) beschreef de tijd tot verlichting van symptomen in uren. In de baloxavirgroep betrof dit gemiddeld 72,3u (95%BI van 67,2 tot 85,1) en in de placebogroep 102,3 u (95%BI van 14,6 tot 42,8). Dit geeft een gemiddeld verschil van 29,1u (95%BI van 14,6 tot 42,8) in het voordeel van de baloxavirgroep (statistisch significant, p<0,0001).
Tijd tot verdwijnen van koorts (belangrijk)
De studie van Lin (2006) beschreef de tijd tot verdwijnen van koorts bij patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen. Er was een statistisch significant verschil in tijd tussen de Oseltamivir groep (gemiddeld 57,2 uur; SD 45,4) en de controlegroep (gemiddeld 104,4 uur; SD 76,9). Dit was een gemiddeld verschil van 47,2 uren (SD 45,2; P=0,005) in het voordeel van de interventiegroep.
Ison (2020) beschreef de tijd tot het verdwijnen van de koorts in uren. In de baloxavirgroep betrof dit gemiddeld 30,8u (95%BI van 28,2 tot 35,4) en in de placebogroep 50,7 u (95%BI van 44,6 tot 58,8). Dit geeft een gemiddeld verschil van 19,8u (statistisch significant, p<0,001) in het voordeel van de baloxavirgroep.
Tijd tot terugkeer normale gezondheid (belangrijk)
Lin (2006) rapporteerde de tijd tot herstel naar de basis gezondheidsstatus bij patiënten met astma/COPD of coronaire hartziekte/chronisch hartfalen. Er was een statistisch significant verschil in tijd tussen de Oseltamivir-groep (gemiddeld 6,2 dagen; SD 3,7) en de controlegroep (gemiddeld 11,2 dagen; SD 6,3). Dit was een gemiddeld verschil van 5 dagen (SD 44,6; P=0,001) in het voordeel van de interventiegroep.
Ison (2020) beschreef de tijd tot herstel naar de basis gezondheidsstatus in uren. In de baloxavirgroep betrof dit gemiddeld 126,4u (95%BI van 104,6 tot 153,4) en in de placebogroep 149,8 u (95%BI van 124,7 tot 175,7). Dit geeft een gemiddeld verschil van 23,4u (95%BI van -21,8 tot 52,2) in het voordeel van de baloxavirgroep (niet significant, p=0,46). Dit werd gerapporteerd voor een deel van de patiënten (n=274 baloxavirgroep; n=274 in de placebogroep).
Bewijskracht van de literatuur
De bewijskracht voor alle beschreven uitkomstmaten is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog.
Volwassenen
Voor de uitkomstmaat “nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (gebrek aan randomisering, toewijzing verbergen, incomplete uitkomsten en blindering) en brede betrouwbaarheidsintervallen (imprecisie).
Voor de uitkomstmaat “hospitalisatie” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (gebrek aan randomiseren, toewijzing verbergen, incomplete uitkomsten en blindering) en brede betrouwbaarheidsintervallen (imprecisie).
Voor de uitkomstmaat “tijd tot verlichting van symptomen” is zowel bij de ITT populatie (influenza-like illness) als bij de ITTI populatie (bevestigde influenza) met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (gebrek aan randomisering, toewijzing verbergen, incomplete uitkomsten en blindering) en brede betrouwbaarheidsintervallen (imprecisie).
Voor de uitkomstmaat “tijd tot verdwijnen van koorts” is zowel in de ITT als in de ITTI populatie met 2 niveaus afgewaardeerd tot laag vanwege het geringe aantal patiënten (imprecisie) en brede betrouwbaarheidsintervallen (imprecisie).
Voor de uitkomstmaat “tijd tot terugkeer normale gezondheid” is in de ITT populatie met 2 niveaus afgewaardeerd tot laag vanwege brede betrouwbaarheidsintervallen (imprecisie) en het geringe aantal patiënten (imprecisie). In de ITTI populatie is afgewaardeerd met 1 niveau tot redelijk vanwege het geringe aantal patiënten (imprecisie).
Kinderen
Voor de uitkomstmaat “nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (gebrek aan randomisering, incomplete uitkomsten en blindering) en brede betrouwbaarheidsintervallen (imprecisie).
Voor de uitkomstmaat “hospitalisatie” is met 3 niveaus afgewaardeerd tot zeer laag vanwege beperkingen in de onderzoeksopzet (gebrek aan randomisering, incomplete uitkomsten en blindering), inconsistentie in studieresultaten en brede betrouwbaarheidsintervallen (imprecisie).
Voor de uitkomstmaat “tijd tot verlichting van symptomen” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (gebrek aan randomisering, incomplete uitkomsten en blindering) en het geringe aantal patiënten (imprecisie).
Voor de uitkomstmaat “tijd tot verdwijnen van koorts” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (risico op selectief rapporteren en loss to follow-up) en het geringe aantal patiënten (imprecisie).
Voor de uitkomstmaat “tijd tot terugkeer normale gezondheid” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (risico op selectief rapporteren en loss to follow-up) en het geringe aantal patiënten (imprecisie).
Hoog-risicopatiënten
Voor de uitkomstmaat “nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis)” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (onduidelijke randomisering en blindering) en het geringe aantal patiënten (imprecisie).
De bewijskracht voor de uitkomstmaat “hospitalisatie” is afkomstig uit een observationele studie en een RCT en begint zodoende op laag. De studiepopulatie was van voldoende grootte en er werd gecorrigeerd voor eventueel verstorende variabelen. Derhalve blijft de bewijskracht op laag.
Voor de uitkomstmaat “tijd tot verlichting van symptomen” is zowel bij de ITT populatie (influenza-like illness) als bij de ITTI populatie (bevestigde influenza) met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (gebrek aan randomiseren, toewijzing verbergen, incomplete uitkomsten en blindering) en brede betrouwbaarheidsintervallen (imprecisie).
Voor de uitkomstmaat “tijd tot verdwijnen van koorts” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (onduidelijke randomisering en blindering) en het geringe aantal patiënten (imprecisie).
Voor de uitkomstmaat “tijd tot terugkeer normale gezondheid” is met 2 niveaus afgewaardeerd tot laag vanwege beperkingen in de onderzoeksopzet (onduidelijke randomisering en blindering) en het geringe aantal patiënten (imprecisie).
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat is de effectiviteit van antivirale behandeling vergeleken met geen behandeling of placebo met betrekking tot de uitkomsten mortaliteit, complicaties, ziekenhuisopname en ziektedagen?
P: kinderen en volwassenen die niet opgenomen worden met influenza-like illness;
I: antivirale middelen;
C: Geen behandeling, placebo, paracetamol;
O: tijd tot verlichting van symptomen, tijd tot verdwijnen van koorts, tijd tot terugkeer normale gezondheid, nieuw opgetreden complicaties (mortaliteit, pneumonie, sepsis), hospitalisatie, complicaties waarbij antibioticagebruik nodig is.
Relevante uitkomstmaten
De werkgroep achtte nieuw opgetreden complicaties waarbij antibioticagebruik nodig is (mortaliteit, pneumonie, sepsis) en hospitalisatie voor de besluitvorming cruciale uitkomstmaten; en tijd tot verdwijnen van koorts, tijd tot verlichting van symptomen, en tijd tot terugkeer normale gezondheid voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde niet a priori de genoemde uitkomstmaten, maar hanteerde de in de studies gebruikte definities.
De werkgroep heeft ervoor gekozen geen grenzen voor klinische (patiënt) relevante verschillen te definiëren, maar statistisch significante verschillen te rapporteren. Hiervoor is gekozen, omdat de grens voor klinische relevantie verschillend zal zijn voor bepaalde subgroepen en deze grenzen waarschijnlijk in overleg met de patiënt gekozen moeten worden. Voor de conclusies wordt daarom uitgegaan van statistische significantie voor de effectiviteit van antivirale middelen.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID), Embase (via Embase.com) en de Cochrane Library (via Wiley) is op 3 mei 2019 met relevante zoektermen gezocht naar systematische reviews en gerandomiseerd onderzoek vanaf 1996 voor de drie uitgangsvragen (modules 'Behandeling influenza; geen opname, opname en IC opname') over behandeling met antivirale middelen. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 517 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: vergelijkend (observationeel of gerandomiseerd) onderzoek naar de antivirale behandeling van patiënten die niet in het ziekenhuis waren opgenomen. Voor observationeel onderzoek was de extra vereiste dat er was gecorrigeerd voor confounding. Op basis van titel en abstract werden in eerste instantie 62 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens één netwerk meta-analyse, één systematische review, één meta-analyse met individuele patiënt data en drie RCT’s definitief geselecteerd (zie exclusietabel onder het tabblad Verantwoording) en 56 studies geëxcludeerd.
Een aparte search werd verricht naar de effectiviteit van antivirale middelen bij de volgende risicogroepen:
- Ouderen.
- Zwangeren.
- Kinderen < 2jaar en in het bijzonder zuigelingen.
- Mensen met een chronische ziekte (comorbiditeit).
- Mensen met een verminderde afweer en/of gebruik van immunosuppressiva/ immunotherapie.
In de databases Medline (via OVID), Embase (via Embase.com) en de Cochrane Library (via Wiley) is op 2 oktober 2019 met relevante zoektermen gezocht naar systematische reviews, gerandomiseerd en observationeel onderzoek vanaf 1996 voor de hierboven genoemde risicogroepen over behandeling met antivirale middelen. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 755 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: vergelijkend (observationeel of gerandomiseerd) onderzoek naar de antivirale behandeling van patiënten die in één van de risicogroepengroepen kon worden ingedeeld: ouderen, zwangeren, kinderen <2jaar en in het bijzonder zuigelingen, mensen met een chronische ziekte (comorbiditeit), mensen met een verminderde afweer en/of gebruik van immunosuppressiva/ immunotherapie. Voor observationeel onderzoek was de extra vereiste dat er was gecorrigeerd voor confounding. Op basis van titel en abstract werden in eerste instantie 37 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden 34 studies geëxcludeerd. Vervolgens werden aan bovenstaande searchresultaten nog twee RCT’s toegevoegd en één studie werd toegevoegd aan de module over behandeling van opgenomen patiënten. In totaal werden daarmee 5 studies opgenomen in deze module over hoogrisicopatiënten en één studie in de module 'Opname' over hoogrisicopatienten.
Aanvullende literatuurzoekactie baloxavir:
In de databases Medline (via OVID) en Embase (via Embase.com) is op 19 februari 2021 met relevante zoektermen gezocht naar systematische reviews, gerandomiseerd en observationeel onderzoek vanaf oktober 2019 over behandeling met baloxavir. Deze literatuurzoekactie is een aanvulling op hierboven beschreven literatuurzoekacties. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 58 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: vergelijkend (observationeel of gerandomiseerd) onderzoek naar de antivirale behandeling van patiënten die niet in het ziekenhuis waren opgenomen. Voor observationeel onderzoek was de extra vereiste dat er was gecorrigeerd voor confounding. Op basis van titel en abstract werden in eerste instantie 16 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens twee RCT’s definitief geselecteerd (zie exclusietabel onder het tabblad Verantwoording) en 14 studies geëxcludeerd.
De eerdere algemene search naar antivirale middelen én de search naar de subgroepen werden opnieuw doorgenomen om te checken of hier nog studies over baloxavir konden worden geidentificeerd. Dit leverde uit de algemene search één studie op die al was geincludeerd via de netwerk meta-analyse van Taieb en uit de subgroep search leverde dit 2 treffers op die na het lezen van de volledige tekst alsnog werden geexcludeerd (geen relevante uitkomstmaten en beschrijving van een al geïncludeerde studie).
Resultaten
Een netwerk meta-analyse, een systematische review, een meta-analyse met individuele patiënt data en vijf RCT’s vormden de basis voor de literatuuranalyse. Bij nadere bestudering bleek het noodzakelijk de oorspronkelijke RCT’s uit de netwerk meta-analyse te gebruiken. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk-of-biastabellen.
- Baker, J., Block, S. L., Matharu, B., Macutkiewicz, L. B., Wildum, S., Dimonaco, S., ... & Piedra, P. A. (2020). Baloxavir marboxil single-dose treatment in influenza-infected children: a randomized, double-blind, active controlled phase 3 safety and efficacy trial (miniSTONE-2). The Pediatric infectious disease journal, 39(8), 700.
- Boivin, G., Goyette, N., Hardy, I., Aoki, F., Wagner, A., & Trottier, S. (2000). Rapid antiviral effect of inhaled zanamivir in the treatment of naturally occurring influenza in otherwise healthy adults. The Journal of infectious diseases, 181(4), 1471-1474.
- Butler, C. C., van der Velden, A. W., Bongard, E., Saville, B. R., Holmes, J., Coenen, S., ... & Llor, C. (2020). Oseltamivir plus usual care versus usual care for influenza-like illness in primary care: an open-label, pragmatic, randomised controlled trial. The Lancet, 395(10217), 42-52.
- Campion, K., Silagy, C., Keene, O., & Cooper, C. (1998). Randomised trial of efficacy and safety of inhaled zanamivir in treatment of influenza A and B virus infections. The Lancet, 352(9144), 1877.
- Cooper, N. J., Sutton, A. J., Abrams, K. R., Wailoo, A., Turner, D., & Nicholson, K. G. (2003). Effectiveness of neuraminidase inhibitors in treatment and prevention of influenza A and B: systematic review and meta-analyses of randomised controlled trials. Bmj, 326(7401), 1235.
- Hayden, F. G., Osterhaus, A. D., Treanor, J. J., Fleming, D. M., Aoki, F. Y., Nicholson, K. G., ... & Wightman, K. (1997). Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenzavirus infections. New England Journal of Medicine, 337(13), 874-880.
- Hayden, F. G., Sugaya, N., Hirotsu, N., Lee, N., de Jong, M. D., Hurt, A. C., ... & Kawaguchi, K. (2018). Baloxavir marboxil for uncomplicated influenza in adults and adolescents. New England Journal of Medicine, 379(10), 913-923.
- Heinonen, S., Silvennoinen, H., Lehtinen, P., Vainionpää, R., Vahlberg, T., Ziegler, T., ... & Heikkinen, T. (2010). Early oseltamivir treatment of influenza in children 1–3 years of age: a randomized controlled trial. Clinical Infectious Diseases, 51(8), 887-894.
- Hirotsu, N., Sakaguchi, H., Sato, C., Ishibashi, T., Baba, K., Omoto, S., ... & Watanabe, A. (2020). Baloxavir marboxil in Japanese pediatric patients with influenza: safety and clinical and virologic outcomes. Clinical Infectious Diseases, 71(4), 971-981.
- Ison, M. G., Portsmouth, S., Yoshida, Y., Shishido, T., Mitchener, M., Tsuchiya, K., ... & Hayden, F. G. (2020). Early treatment with baloxavir marboxil in high-risk adolescent and adult outpatients with uncomplicated influenza (CAPSTONE-2): a randomised, placebo-controlled, phase 3 trial. The Lancet Infectious Diseases.
- Jefferson, T., Jones, M. A., Doshi, P., Del Mar, C. B., Hama, R., Thompson, M. J., ... & Howick, J. (2014). Neuraminidase inhibitors for preventing and treating influenza in adults and children. Cochrane database of systematic reviews, (4).
- Johnston, S. L., Ferrero, F., Garcia, M. L., & Dutkowski, R. (2005). Oral oseltamivir improves pulmonary function and reduces exacerbation frequency for influenza-infected children with asthma. The Pediatric infectious disease journal, 24(3), 225-232.
- Kohno, S., Kida, H., Mizuguchi, M., & Shimada, J. (2010). Efficacy and safety of intravenous peramivir for treatment of seasonal influenza virus infection. Antimicrobial agents and chemotherapy, 54(11), 4568-4574.
- Li, L., Cai, B., Wang, M., & Zhu, Y. (2003). A double-blind, randomized, placebo-controlled multicenter study of oseltamivir phosphate for treatment of influenza infection in China. Chinese medical journal, 116(1), 44-48.
- Lin, J. T., Yu, X. Z., Cui, D. J., Chen, X. Y., Zhu, J. H., Wang, Y. Z., & Wu, X. D. (2006). A multicentre, randomized, controlled trial of oseltamivir in the treatment of influenza in a high-risk Chinese population. Current medical research and opinion, 22(1), 75-82.
- Mäkelä, M. J., Pauksens, K., Rostila, T. A. A., Fleming, D. M., Man, C. Y., Keene, O. N., & Webster, A. (2000). Clinical efficacy and safety of the orally inhaled neuraminidase inhibitor zanamivir in the treatment of influenza: a randomized, double-blind, placebo-controlled European study. Journal of Infection, 40(1), 42-48.
- McLean, H. Q., Belongia, E. A., Kieke, B. A., Meece, J. K., & Fry, A. M. (2015, September). Impact of Late Oseltamivir Treatment on Influenza Symptoms in the Outpatient Setting: Results of a Randomized Trial. In Open forum infectious diseases (Vol. 2, No. 3). Oxford University Press.
- Monto, A. S., Robinson, D. P., Herlocher, M. L., Hinson Jr, J. M., Elliott, M. J., & Crisp, A. (1999). Zanamivir in the prevention of influenza among healthy adults: a randomized controlled trial. Jama, 282(1), 31-35.
- Murphy, K. R., Eivindson, A., Pauksens, K., Stein, W. J., Tellier, G., Watts, R., ... & Loeschel, E. (2000). Efficacy and safety of inhaled zanamivir for the treatment of influenza in patients with asthma or chronic obstructive pulmonary disease. Clinical Drug Investigation, 20(5), 337-349.
- Muthuri, S. G., Venkatesan, S., Myles, P. R., Leonardi-Bee, J., Al Khuwaitir, T. S., Al Mamun, A., ... & Beovic, B. (2014). Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: a meta-analysis of individual participant data. The lancet Respiratory medicine, 2(5), 395-404.
- Nakazawa, M., Hara, K., Komeda, T., & Ogura, E. (2020). Safety and effectiveness of baloxavir marboxil for the treatment of influenza in Japanese clinical practice: A postmarketing surveillance of more than 3000 patients. Journal of Infection and Chemotherapy, 26(7), 729-735
- Nicholson, K. G., Aoki, F. Y., Osterhaus, A. D. M. E., Trottier, S., Carewicz, O., Mercier, C. H., ... & Ward, P. (2000). Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. The Lancet, 355(9218), 1845-1850.
- Portsmouth, S., Hayden, F. G., Kawaguchi, K., Ishibashi, T., Kinoshita, M., Shishido, T., ... & Uehara, T. (2021). Baloxavir Treatment in Adolescents With Acute Influenza: Subgroup Analysis From the CAPSTONE-1 Trial. Journal of the Pediatric Infectious Diseases Society, 10(4), 477-484.
- Puhakka, T., Lehti, H., Vainionpää, R., Jormanainen, V., Pulkkinen, M., Sharp, S., ... & Tisdale, M. (2003). Zanamivir: a significant reduction in viral load during treatment in military conscripts with influenza. Scandinavian journal of infectious diseases, 35(1), 52-58.
- Taieb, V., Ikeoka, H., Ma, F. F., Borkowska, K., Aballéa, S., Tone, K., & Hirotsu, N. (2019). A network meta-analysis of the efficacy and safety of baloxavir marboxil versus neuraminidase inhibitors for the treatment of influenza in otherwise healthy patients. Current medical research and opinion, 1-10.
- Treanor, J. J., Hayden, F. G., Vrooman, P. S., Barbarash, R., Bettis, R., Riff, D., ... & US Oral Neuraminidase Study Group. (2000). Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. Jama, 283(8), 1016-1024.
- Uehara, T., Hayden, F. G., Kawaguchi, K., Omoto, S., Hurt, A. C., De Jong, M. D., ... & Kida, H. (2020). Treatment-emergent influenza variant viruses with reduced baloxavir susceptibility: impact on clinical and virologic outcomes in uncomplicated influenza. The Journal of infectious diseases, 221(3), 346-355.
- Venkatesan, S., Myles, P. R., Leonardi-Bee, J., Muthuri, S. G., Al Masri, M., Andrews, N., ... & Loh, T. P. (2017). Impact of outpatient neuraminidase inhibitor treatment in patients infected with influenza A (H1N1) pdm09 at high risk of hospitalization: an individual participant data metaanalysis. Clinical Infectious Diseases, 64(10), 1328-1334.
- Whitley, R., Laughlin, A., Carson, S., Mitha, E., Tellier, G., Stich, M., ... & Sheridan, W. P. (2015). Single dose peramivir for the treatment of acute seasonal influenza: integrated analysis of efficacy and safety from two placebo-controlled trials. Antiviral therapy, 20(7), 709-19.
- Yokoyama, T., Sakaguchi, H., Ishibashi, T., Shishido, T., Piedra, P. A., Sato, C., ... & Uehara, T. (2020). Baloxavir marboxil 2% granules in Japanese children with influenza: an open-label phase 3 study. The Pediatric infectious disease journal, 39(8), 706.
Evidence table for systematic review of RCTs and observational studies (intervention studies)
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Taieb, 2019 |
SR and network meta-analysis of RCTs
Literature search up to November 2016
A: Hayden 2018, phase 2 trial B: Hayden, 2018, phase 3 trial C: Kohno, 2010 D: Treanor, 2000 E: Nicholson, 200 F: Matsumoto, 1999 G: Li, 2003 H: Puhakka, 2003 I: Kohno, 2011 J: Duval, 2010 K: Watanabe, 2010 L: Boivin, 2000 M: Makela, 2000 N: Monto, 1999 O: Hayden, 1977 P: Campion, 1998 Q: Whitley, 2015 (NCT00419263) R: Ikematsu, 2011 S: Kashiwagi, 2000 T: PMDA zanamivir 2017 U: PMDA Laninamivir, 2010 V: FDA Oseltamavir, 2012
Study design: RCT (parallel / cross-over), cohort (prospective / retrospective), case-control
Setting and Country: Creativ-Ceutical, London, UK; Hirotsu Clinic, Kanagawa, Japan.
Source of funding: This study (design and conduct, data collection, management, analysis and interpretation of the data, review and approval of the manuscript) was funded by Shionogi & Co. Ltd.
Conflicts of interest: V.T., F.M., K.B. and S.A. have disclosed that they have received grant funding from Shionogi & Co. Ltd during the conduct of the study and outside of the submitted work. K.T. has disclosed that she is an employee of Shionogi Limited. H.I. has disclosed that he is an employee of Shionogi & Co. Ltd. N.H. has disclosed that he has received personal fees and other from Shionogi & Co. Ltd, outside the submitted work. |
Inclusion criteria SR: RCTs; patients aged 12 to 80 years with flu symptoms, otherwise healthy;
Exclusion criteria SR: Case reports, letters and historical articles; High-risk patients; studies focusing on patients with risk factors.
22 studies included
Study characteristics: (CI: confirmed influenza; ILI: influenza-like illness) A: CI, age 20-64 B: CI, 12-64 C: CI, 20-64 D: ILI, 18-65 E: ILI, 18-65 F: ILI, 16-65 G: ILI, 18-65 H: ILI, 17-29 I: CI, 20-80 J: CI, 18-85 K: CI, 20-77 L: ILI, >12 years old M: ILI, 12-81 N: ILI, >13 O: ILI, >13 P: ILI, >12 Q: CI, >18 R: CI, children + adults S: ILI, 16-80 T: CI, adults U: N/A, adults V: CI, >13
N, sex and mean age of individual studies were not reported in SR.
|
A: Baloxavir 10/20/40 mg single dose B: Baloxavir 40/80 mg single dose C: Peramivir 300 mg D: Oseltamivir 75 mg 2x/d E: Oseltamivir 75 mg 2x/d F: Zanamivir 10 mg 2x/d G: Oseltamivir 75 mg 2x/d H: Zanamivir 10 mg 2x/d I: Peramivir 300 mg once daily J: Zanamivir 10 mg 2x/d + placebo K: Laninamivir 40 mg L: Zanamivir 10 mg 2x/d M: Zanamivir 10 mg 2x/d N: Zanamivir 10 mg 2x/d O: Zanamivir 10 mg 2x/d P: Zanamivir 10 mg 2x/d Q: Peramivir 300 mg R: Laninamivir 40 mg S: Oseltamivir 75 mg 2x/d T: Zanamivir 20 mg U: Laninamivir 40 mg V: Oseltamivir 150 mg
|
A: placebo B: Oseltamivir 75 mg 2x/d + placebo C: placebo D: placebo E: placebo F: placebo G: placebo H: placebo I: Oseltamivir 75 mg 2x/d J: Oseltamivir 75 mg 2x/d + placebo K: Oseltamivir 75 mg 2x/d L: Placebo M: Placebo N: Placebo O: Placebo P: Placebo Q: Placebo R: Zanamivir 10 mg 2x/d S: Placebo T: Placebo U: Oseltamivir 150 mg V: Placebo
|
End-point of follow-up: Not reported in SR
For how many participants were no complete outcome data available? Not reported in SR
|
1. Time (h) to alleviation of all symptoms Median difference (95% CI) (* indicates significant difference, random effects model) Baloxavir versus… - Placebo 29.36 (15.34; 45.82)* - Zanamivir 20 mg 19.96 (3.23; 39.07)* - Laninamivir 40 mg 6.41 (-15.48; 32.10) - Oseltamivir 150 mg 6.33 (-6.89; 19.54) - Peramivir 300 mg 7.60 (-8.49; 24.78)
14 studies, 5403 patients, ITTI analysis
2. Time (h) to resolution of fever Median difference (95% CI) (* indicates significant difference, random effects model) Baloxavir versus… - Placebo 19.12 (6.44; 33.25)* - Zanamivir 20 mg 4.65 (-20.06; 41.56) - Laninamivir 40 mg 0.41 (-18.82; 24.20) - Oseltamivir 150 mg -1.14 (-14.16; 10.33) - Peramivir 300 mg -1.69 (-17.50; 14.48)
9 studies, 3772 patients, ITTI analysis
3. Time (h) to return to pre-influenza health status: Median difference (95% CI) (no significant differences, random effects model) Baloxavir versus… - Placebo 33.25 (-24.07; 90.96) - Zanamivir 20 mg 18.77 (-61.58; 107.20) - Laninamivir 40 mg Not reported - Oseltamivir 150 mg -6.88 (-70.17; 54.81) - Peramivir 300 mg 0.84 (-77.97; 77.66)
4. Pneumonia OR (95% CI) Baloxavir versus placebo 0.91 (0.07; 10.18) Not significant
3 studies, 3758 patients, ITTI analysis |
|
Evidence table for intervention studies (randomized controlled trials and non-randomized observational studies (cohort studies, case-control studies, case series))1
This table is also suitable for diagnostic studies (screening studies) that compare the effectiveness of two or more tests. This only applies if the test is included as part of a test-and-treat strategy otherwise the evidence table for studies of diagnostic test accuracy should be used.
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3 |
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
McLean, 2015 |
Type of study: Double-blind RCT
Setting and country: Marshfield Clinic in Marshfield, Wisconsin, USA.
Funding: This work was supported by the Centers for Disease Control and Prevention
Conflicts of interest: H. Q. M., J. K. M., E. A. B., and B. A. K. report research grant support from MedImmune |
Inclusion criteria: Age 1-72; Patients were eligible for influenza testing if they reported feverishness, chills, or cough <120 hours (5 days) in duration; Influenza confirmed by RT-PCR.
Exclusion criteria: Treatment initiation ≥120 hours after illness onset
N total at baseline: Intervention: n=114, median age 18 (IQR 8, 36), 43% male. Control: n=51, median age 17 (IQR 7, 38), 33% male.
Influenza type: Both intervention and control: 75% type A, 25% type B
Groups were comparable at baseline |
Intervention: 75 mg oseltamivir twice daily for 5 days (patients >88 pounds)
Children ≤88 pounds received a liquid form of oseltamivir at a concentration of 15 mg/mL |
Control: Placebo capsule/liquid |
Length of follow-up: At least 7 days after initiation of study or until symptoms had resolved (up to 14 days).
Loss-to-follow-up: Not reported
Incomplete outcome data: Not reported
|
1. Time from treatment initiation to symptom resolution Median days (95% CI): I: 4 (3.5, 5) C: 4 (3.5, 4.5), P=0.4
2. Probability of symptom resolution on day 3–4 of treatment (95% CI) I: 0.39 (0.30, 0.48) C: 0.35 (0.21, 0.48), P=0.6
3. Probability of symptom resolution on day 7 of treatment (95% CI) I: 0.82 (0.75, 0.89) C: 0.89 (0.80, 0.98), P=0.2
Only ITT population.
Results are grouped by “Late treatment (48–119 hours)” and “Early and late treatment (<120 hours)”, but groups were not different. Only combined group is reported here. |
|
|
Boivin, 2000 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter study in Canada
Funding: Glaxo Wellcome Canada.
Conflicts of interest: G.B. is a fellow of the Medical Research Council of Canada. |
Inclusion criteria: Healthy persons >12 years old and high-risk subjects (defined as elderly patients or those with chronic respiratory, cardiovascular, or renal diseases) were enrolled if they were able to take the first dose of study medication on the first or second day of their influenza-like symptoms.
Exclusion criteria: Not reported
N ITTI at baseline: Intervention: n=17 Control: n=10.
No patient characteristics specified. |
Intervention: Zanamivir 10 mg twice daily for 5 days |
Control: Placebo |
Length of follow-up: 14 days
Loss-to-follow-up: Not reported
Incomplete outcome data: Not reported
|
Time to alleviation of all symptoms (days) ITTI I: 5.0 (no SD) C: 9.5 (no SD) P=0.35 |
|
|
Campion, 1998 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter study in Australia, New Zealand and South Africa
Funding: This study was supported by Glaxo Wellcome Australia Ltd
Conflicts of interest: Not reported |
Inclusion criteria: Individuals aged 12 years or older who presented with influenza-like illness of 36 h duration or less were eligible for entry into the study. Patients had to present with fever (>37·8ºC), feverishness, or both, and at least two of myalgia, cough, headache, or sore throat. Exclusion criteria: Suspected bacterial infection, recent use of influenza antiviral therapy, recent participation in a trial of another investigational product, or a history of drug misuse; (possible) pregnancy, breastfeeding.
Patient characteristics: Intervention: n=227, 59% male, age 36±13, influenza A 46%, influenza B 35%.
Control: n=228, 46% male, age 38±14, influenzaA 48%, influenza B 32%.
Groups were comparable at baseline |
Intervention: 10 mg inhaled Zanamivir twice daily for 5 days |
Control: Placebo |
Length of follow-up: 28 days
Loss-to-follow-up: 31 patients withdrew from the study early because of adverse events (four on placebo), withdrawn consent (three on placebo, five on zanamivir), lost to follow-up (ten on placebo, seven on zanamivir), and two because of other reasons (one on placebo and one on zanamivir because of protocol violations)
Incomplete outcome data: Not reported
|
1. Median time (days) to alleviation of clinically important symptoms ITT population I: 5.0 (n=228) C: 6.5 (n=227) Difference (95% CI) 1.5 (0.5, 2.25) P=0.011
ITTI population I: 4.5 (n=160) C: 6.0 (n=161) Difference (95% CI) 1.5 (0.5, 2.25) P=0.004
2. Resolution of fever (days) ITT I: 4.5 (n=128) C: 6.5 (n=123) Difference (95% CI) 2.0 (1.0, 4.0) favouring zana P<0.001
ITTI I: 4.5 (n=97) C: 6.5 (n=106) Difference (95% CI) 2.0 (1.25, 4.5) P<0.001
3. Complications and associated antibiotic use in intention-to-treat and high-risk patients Complications I: 49/227 (22%) C: 64/228 (28%) RR 0.77 (0.56, 1.06) favouring zana P= 0.135
Associated AB use I: 45/227 (20%) C: 52/228 (23%) RR 0.87 (0.61, 1.24) favouring zana P=0.508 |
|
|
Hayden, 1997 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: 2 parallel multicentre trials in Europe and North America
Funding: Not reported
Conflicts of interest: Drs. Hayden, Treanor, Aoki, and Nicholson have served as ad hoc consultants to Glaxo Wellcome. |
Inclusion criteria: Age >18 (Europe) or >13 (North America); acute influenza-like illness Of 48 hours; presence of fever and at least two other symptoms (headache, myalgia, cough, and sore throat.
Exclusion criteria: Suspected bacterial infection or recent use of antimicrobial drugs, immunization with influenza vaccine for the current season, pregnancy or breastfeeding, concurrent use of intranasal or inhaled medications, and underlying conditions for which influenza immunization is currently recommended.
Patient characteristics: Inhaled zanamivir: n=85, age 31±11, 59% male, influenza A 52%, influenza B 48% Inhaled+intranasal zanamivir: n=88, age 32±12, 57% male, influenza A 57%, influenza B 43% Placebo: n=89, age 33±12, 52% male, influenza A 60%, influenza B 40%
Groups were comparable at baseline |
Intervention: 10 mg of zanamivir by inhalation by mouth plus 6.4 mg by intranasal spray
Or
10 mg of zanamivir by inhalation by mouth plus intranasal placebo
Twice daily for 5 days |
Control: Placebo inhalation plus nasal sprays twice daily for 5 days |
Length of follow-up: 28 days
Loss-to-follow-up: One patient randomly assigned to the placebo group failed to take the study drug. Eight other patients in the placebo group withdrew, as did 10 assigned to inhaled zanamivir and 10 assigned to inhaled and intranasal zanamivir. The most common reason for withdrawal was failure to return for the scheduled study visits.
Incomplete outcome data: Not reported
|
1. Median time (days) to alleviation of major symptoms ITT: N, median, mean±SD Placebo 144, 5, 6.0±2.9 Inhaled 132, 5, 5.3±2.6 Difference -0.7 (-1.4 to 0) P=0.04 Inhaled+nasal 141, 4, 5.5±2.8 Difference -0.6 (-1.2 to 0.1)
ITTI: N, median, mean±SD Placebo 89, 5, 6.3±2.9 Inhaled 85, 4, 5.4±2.7 Difference -0.8 (-1.7 to 0) P=0.05
Inhaled+nasal 88, 4, 5.3±2.8 Difference -1.0 (-1.8 to -0.1) P=0.02
2. Adverse events 18% of 144 patients assigned to placebo, 23% of 132 patients assigned to inhaled zanamivir and 25% of 141 patients assigned to intranasal and inhaled zanamivir. During drug administration, adverse events related to the upper respiratory tract (9 percent of patients given placebo, 7 percent of those given inhaled zanamivir, and 11 percent of those given intranasal and inhaled zanamivir) or gastrointestinal tract (5 percent, 7 percent, and 9 percent, respectively) were the most common, but these reactions were difficult to distinguish from symptoms due to the underlying illness. The frequencies of local irritation of the nose (5 to 6 percent) or eyes (<1 percent) were similar in the three groups. |
|
|
Hayden, 2018
JapicCTI-153090 (phase 2)
And
NCT 02954354 (phase 3) |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Phase 2 and phase 3 trial in Japan and the USA
Funding: Funded by Shionogi & Co., Ltd. (Osaka, Japan).
Conflicts of interest: Many authors were received consulting fees, lecture fees, travel support and/or served on an advisory board for one or several companies. |
Inclusion criteria: Presence of fever (axillary temperature, ≥38.0°C), at least one systemic symptom and at least one respiratory symptom of at least moderate severity, and a symptom duration of no more than 48 hours.
Exclusion criteria: Pregnant women, patients weighing less than 40 kg, and those with illness resulting in hospitalization
Patient characteristics: Baloxavir: ITT n=610; ITTI n=456, mean age 32, 51% male, Influenza A 88%, Influenza B 8%.
Control: ITT n=309; ITTI n=231, mean age 33, 52% male, influenza A 88%, influenza B 9%
Groups were comparable at baseline. |
Intervention: Baloxavir Marboxil, single oral dose (40 mg for patients weighing <80 kg or 80 mg for those weighing ≥80 kg) for 5 days |
Control: Placebo |
Length of follow-up: 28 days
Loss-to-follow-up: Intervention: 34 withdrawn: 2 for adverse events 1 inclusion/exclusion criteria 17 withdrew 12 lost to follow-up 2 other reason
Control: 34 withdrawn: 2 for adverse events 2 lack of efficacy per investigator 9 withdrew 5 lost to follow-up 2 other reason
Incomplete outcome data: Not reported |
1. Time (h) to alleviation of symptoms ITTI Intervention (n=456) 53.7 Control (n=231) 80.2 Mean difference 26.5 (95% CI 17.8 to 35.8)
ITT Intervention (n=610): 65.4 Control (n=309): 88.6 Mean difference 23.2 (14.0 to 34.2)
2. Time (h) to resolution of fever ITTI? ITT? Intervention 24.5 Control 42. No SD provided.
3. Time (h) to return to usual health ITTI? ITT? Intervention: 129.2 Control: 168.8. No SD provided.
4. Complications that resulted in antibiotic treatment ITTI? ITT? Intervention: 16/456 (3.5%) Control: 10/231 (4.3%) |
|
|
Ison, 2020 NCT02949011
|
Type of study: double-blind, placebo-controlled and oseltamivir-controlled trial in
Setting and country: 551 sites in 17 countries and territories
Funding: This study was sponsored by Shionogi, manufacturer of baloxavir Marboxil. The sponsor (Shionogi) designed the trial, and the authors’ access to the data was not restricted by confidentiality agreements.
The sponsor of the study was involved in the study design, data collection, data analysis, and preparation of the manuscript.
Conflicts of interest: Many authors were received consulting fees, lecture fees, travel support and/or served on an advisory board for one or several companies. |
Inclusion criteria: outpatients aged 12 years or older with suspected influenza A or influenza B virus infection (clinically diagnosed influenza-like illness as defined by a fever (axillary temperature ≥38·0°C), at least one systemic symptom and at least one respiratory symptom of at least moderate severity, and a symptom duration of 48 h or less) who were considered at high risk of developing influenza-associated complications (including asthma or chronic lung disease, endocrine disorders, heart disease, age 65 years or older, and metabolic disorders). Patients included in the analysis had RT_PCR confirmed influenza (ITTI population)
Exclusion criteria: Some patients at high risk were excluded, including pregnant women or women who were breastfeeding, patients with hepatic impairment, patients with cancer within 5 years, patients with untreated HIV or a CD4 count of less than 350 cells per μL in the past 6 months, patients on immunosuppressive treatment for organ or bone marrow transplantation, and patients receiving at least 20 mg prednisolone. Patients with severe influenza requiring inpatient treatment and patients with a known allergy to oseltamivir were also excluded.
N total at baseline: Baloxavir: mITT n=388; mean age 52,3, 50% male
Control: mITT n=386; mean age 51,9, 47% male
Groups were comparable at baseline |
Intervention: single oral dose of baloxavir at baseline (40 mg for patients weighing <80 kg and 80 mg for those weighing ≥80 kg) and oseltamivir matched placebo twice daily for 5 days. |
Control: single oral dose of baloxavir-matched placebo at baseline and oseltamivir-matched placebo twice daily for 5 days |
Length of follow-up: 22 days (14 days for influenza symptoms, 22 for safety laboratory tests)
Loss-to-follow-up: Intervention: 33 did not complete study: 6 adverse events 5 protocol deviation 13 withdrew 7 lost to follow-up 2 other reasons
Control: 34 did not complete study: 7 adverse events 3 protocol deviation 2 lack of efficacy 13 withdrew 5 lost to follow-up 4 other reasons
Incomplete outcome data: Not reported |
1. Time (h) to alleviation of symptoms Intervention: 73,2 (95%CI 67,2-85,1) Control: 102,3 (95%CI 92,7-113,1) Mean difference 29,1 (95% CI 14,6 to 42,8)
2. Time (h) to resolution of fever Intervention (n=380): 30,8 (95%CI 28,2 – 35,4) Control (n=385): 50,7 (95%CI 44,6 – 58,8) Median difference: 19,8, p<0,001
3. Time (h) to return to usual health Intervention (n=274): 126,4 (95%CI 104,6 – 153,4) Control (n=274): 149,8 (95%CI 124,7−175,7) Mean difference: 23,4 (95%CI –21,8 – 52,2; p=0·46)
4. Influenza-related complications Intervention (n=388): 11 (2,8%) Control (n=386): 40 (10,4%) RR: 0.27 [0.14 to 0.53] p<0,0001* * Due to differences in sinusitis or bronchitis or requiring antibiotics for suspected or proven secondary infections
5. Hospitalisation Intervention (n=388): 3 (0,8%) Control (n=386): 5 (1,3%) P=0.5047 RR: 0.6 [0.14 to 2.48]
6. Complications that resulted in antibiotic treatment Intervention (n=388): 13 (3,4%) Control (n=386): 29 (7,5%) P=0,0112 RR: 0.45 [0.24 to 0.84]
Adverse events were reported in 183 (25%) of 730 patients who received baloxavir, 216 (30%) of 727 patients who received placebo (ILI group). |
|
|
Johnston, 2005
|
Type of study: Randomized double blind placebo-controlled trial.
Setting and country: UK, Argentinia, Spain (multinational, multicentre study)
Funding and conflicts of interest: Dr. Johnston has received speakers’ fees and remuneration for sitting on advisory boards for Hoffmann-La Roche and GlaxoSmithKline. Supported by F. Hoffman-La Roche (Basel, Switzerland). |
Inclusion criteria: children 6-12 years with asthma severe enough to require regular medical follow-up monitoring or hospital care if they met the following criteria: presented with influenza symptoms (recorded temperature of ≥100.0°F (≥38.7°C) plus 1 respiratory symptom (cough or coryza)), presented within 48 hours after symptom onset and were able to perform the pulmonary function tests. Exclusion criteria: Subjects were excluded if they fulfilled any of the following criteria: tested positive for respiratory syncytial virus (rapid diagnostic test); were taking immunosuppressive medication (except inhaled or orally administered steroids for asthma treatment); had known HIV infection; had uncontrolled (requiring change of therapy or hospitalization within 4 weeks before the first dose of the study drug) renal, vascular, neurologic, metabolic or pulmonary (excluding asthma) disease; were transplant recipients (except corneal transplants); had an allergy to the test medication (or paracetamol/acetaminophen); had undergone antiviral treatment of influenza in the previous 2 weeks; had active cancer at any site; were female subjects of childbearing potential or had participated in a clinical trial with an investigational drug within 4 weeks before study day 1.
N total at baseline: Influenza like illness (called: ‘safety population’ by the authors) I: 170 C: 164
Intention to treat infected population (ITTI): Intervention: 84 Control: 95
Important prognostic factors2: age: median (range) ILI I: 9 (5-12); C: 9 (5-12) ITTI: I: 9 (6-12); C: 9 (5-12))
Sex: ILI: I: 65%M; C: 62%M ITTI I: 70% M; C: 63% M
Race Caucasian ILI: I: 88%; C: 87% ITTI: I: 87%; C: 90%
Asthma severity Mild ILI: I: 43%; C: 46% ITTI: I: 49%; C: 55% Moderate ILI: I: 49%; C: 49% ITTI: I: 48%; C: 41% Severe ILI: 8%; C: 5% ITTI: I: 3%; C4%
Current vaccination yes ILI: I: 18%; C: 21% ITTI: I: 17%; C: 12%
Time from onset of symptoms to first dose Total (h), mean ± SD ILI: I: 27.5 ± 12.1 C: 26.9 ±12.1 ITTI: I: 27.9 ± 11.6 C: 26.8 ± 11.5
<24 h, % ILI: I: 38%; C: 42% ITTI: I: 37%; C: 43%
≥24 h, % ILI: I: 62%; C: 58% ITTI: I: 63%; C: 57%
Predicted % of peak flow at baseline ILI: I: 73.2 ± 19.2 C: 72.6 ± 18.1 ITTI: I: 71.9 ± 19.8 C: 71 ± 17
Predicted % of FEV1 at baseline ILI: I: 77.4 ± 23.2 C: 77.8 ± 21.4 ITTI: I: 75.6 ± 21.4 C: 81 ± 20.1
Groups comparable at baseline? In the ILI population, the 2 treatment arms were broadly comparable. In the ITTI population, a greater proportion of patients in the oseltamivir group had moderate/ severe asthma and were recruited into the study >24 hours after the onset of symptoms. According to the authors, these imbalances were associated with a greater degree of respiratory compromise at baseline, as evidenced by the lower percent predicted FEV1 in the oseltamivir group |
Describe intervention (treatment/procedure/test): oseltamivir (2 mg/kg) twice daily, as orally administered syrup (6 mg/mL)
|
Describe control (treatment/procedure/test): Placebo twice daily, as orally administered syrup (6 mg/mL) |
Length of follow-up: 4 weeks
Loss-to-follow-up: None
Incomplete outcome data: None; missing values for asthma exacerbation were linearly interpolated.
|
Outcome measures and effect size (include 95%CI and p-value if available):
New complications (mortality, pneumonia, sepsis) ILI: Deaths: I: 0/170 C: 0/164 Pneumonia: I: 2/170 C: 1/164
Hospitalisation Defined as unplanned physician visits or hospitalizations because of influenza or secondary illnesses ILI/ITTI? I: 27% C: 23% P value not reported Not clearly reported whether these values were based on the whole ILI population.
Time to alleviation of symptoms, median hours ITTI population I: 90.4 C: 115.6 Difference: 25.3, p=0.1197
Time to resolution of fever Not reported
Time to return to usual health, median hours ITTI population I: 101.4 C: 114 Difference: 12.6; p=0.4555 |
Timeframe: 2 consecutive influenza seasons (1998-1999) in the northern and southern hemispheres.
The primary efficacy endpoint was the time to freedom from illness and was defined as the time when all of the following conditions were met for the first time for a period of 21.5 hours (24 hours ± 10%): alleviation of symptoms (score of 0 (no problem) or 1 (minor problem) on the symptom questionnaire), return to normal health and activity (i.e., return to school or normal style of play behavior, indicated by a yes response from the parent/guardian) and return to an afebrile state (≤98.9°F or ≤37.2°C).
Return to normal health and activity was defined as the time taken to return to preinfluenza health and activity status for the first time for a minimal period of 21.5 hours, as assessed by the parent/guardian with a simple yes/no questionnaire. The duration of symptoms was defined as the time to the alleviation of all 18 symptoms from the first dose of the study drug.
Asthma exacerbation was defined as a ⬎20% reduction from the highest peak flow reading recorded up to day 28 |
|
Kohno, 2010 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter study at Nagasaki University Hospital, Nagasaki, and 74 other centers in Japan.
Funding: Funded by Shionogi & Co., Ltd. (Osaka, Japan).
Conflicts of interest: Not reported |
Inclusion criteria: Previously healthy adults aged 20-64 years reporting onset of influenza-like illness within the previous 48 h. The time of onset of influenza-like illness was defined as either when the body temperature first rose to ≥1°C above normal or when the subject experienced at least 2 of the 7 influenza symptoms: headache, aches or pains in muscles or joints, feverishness, fatigue, cough, sore throat, and nasal congestion. At enrolment, a diagnosis of influenza was required based on a positive rapid antigen test for influenza virus, fever of ≥38°C, and the presence of at least 2 of the 7 symptoms at moderate to high severity, based on subjects’ self-reporting.
Exclusion criteria: Respiratory dysfunction or chronic respiratory disorders requiring pharmacotherapy, convulsions or other neurological symptoms, active clinically important chronic illness or known infection with human immunodeficiency virus, renal impairment requiring hemodialysis, suspected bacterial infection, treatment with steroids or other immunosuppressants, use of anti-influenza virus drugs within the past 7 days; and a history of hypersensitivity, allergy, or serious adverse drug reactions to anti-influenza virus drugs or acetaminophen. Women who were (likely to be) pregnant or breast feeding were also excluded.
Patient characteristics: Peramivir 300mg: n=99, age 34±10, 47% male, Influenza A/H1 75%, Influenza A/H3 21%. Peramivir 600mg: n=97, Age 34±10, 55% male, Influenza A/H1 71%, Influenza A/H3 26%. Control: n=100, age 34±10, 51% male, Influenza A/H1 72%, Influenza A/H3 24%.
Groups were comparable at baseline |
Intervention: Single intravenous infusion of 300 mg Peramivir
Or
Single intravenous infusion of 600 mg Peramivir |
Control: Placebo infusion |
Length of follow-up: 14 days
Loss-to-follow-up: A total of 300 subjects were randomly assigned to one of the treatment groups. Of these, one subject did not have laboratory-confirmed influenza virus infection, two subjects withdrew after randomization but before treatment, and one subject did not have any symptom assessment data after randomization.
Incomplete outcome data: Not reported
|
1. Median time (hours) to alleviation of symptoms Only ITTI 300mg: median 59.1 (50.9 to 68.1), hazard ratio 0.681 (0.511 to 0.909), P=0.0092 600mg: 59.9 (54.4 to 68.1), hazard ratio 0.666 (0.499 to 0.890) Placebo: 81.8 (68.0 to 101.5)
2. Complications Physician-diagnosed secondary complications (pneumonia, bronchitis, sinusitis, and otitis media) occurred in three recipients of 300 mg peramivir (3 cases of bronchitis; 3.0%), one recipient of 600 mg peramivir (1 case of otitis media; 1.0%), and three placebo recipients (3 cases of bronchitis; 3.0%). |
|
|
Li, 2003 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter study in China
Funding: not reported.
Conflicts of interest: not reported. |
Inclusion criteria: Patients without other medical history, aged 18 to 65 years old, displaying symptoms consistent with influenza, received care no more than 36 hours after onset of feeling unwell, willing to use contraception during the trial.
Exclusion criteria: Pregnancy, high likelihood of bacterial infection, steroid or immune-suppressant therapy, influenza vaccination less than 12 months before.
Patient characteristics: Intervention: n=216. ITTI n=134, 47% male, age 32±12, influenza A 61%, influenza B 37%. Control: n=235. ITTI n=139, 53% male, age 30±11, influenza A 63%, influenza B 36%.
Groups were comparable at baseline. |
Intervention: Oseltamivir phosphate capsule, 75 mg twice daily for 5 days |
Control: Placebo |
Length of follow-up: 21 days
Loss-to-follow-up: Not reported
Incomplete outcome data: Not reported
|
1. Median time (hours) to alleviation of clinically important symptoms ITTI Intervention: n=134, 91.6 hours (80.2 to 101.3) Control: 139, 95.0 (84.5 to 105.3)
ITT Intervention (n=216): 83.5 h (72.7 to 94.0) Control (n=235): 87.7 h (77.1 to 97.0)
2. Secondary complications (bronchitis, pneumonia, sinusitis, tonsillitis, otitis media) Intervention: 7/134 (5.2%) Control: 7/139 (5.0%)
Antibiotic use Intervention: 5/134 (3.7%) Control: 5/139 (3.6%)
3. Adverse events Adverse events were similarly reported in both the oseltamivir group and the placebo group. The main adverse events following test drug were gastrointestinal symptoms, neurological symptoms and rashes. |
|
|
Makela, 2000 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter study in Finland, Sweden and UK?
Funding: This study was supported by Glaxo Wellcome Research and Development, Greenford, U.K.
Conflicts of interest: Not reported |
Inclusion criteria: Patients were required to have a fever (>37.8°C for patients aged less than 65 years, ³ 37.2°C for patients aged 65 years or more) and at least two of the following symptoms: headache, myalgia, cough and sore throat. They had to start therapy within 2 days of symptom onset.
Exclusion criteria: Women who were pregnant or at risk of pregnancy were excluded.
Patient characteristics: Total n=356, 96% influenza A (H3N2), mean age 37.2 years. Patient data were not specified per group. |
Intervention: Zanamivir 10 mg inhaled orally twice daily for 5 days |
Control: Placebo |
Length of follow-up: 28 days
Loss-to-follow-up: Placebo: 3 discontinued trial medication (2 for adverse events; 1 for other reasons). 3 lost to follow–up. Intervention: 4 discontinued trial medication (2 for adverse events; 2 for other reasons). 4 discontinued trial procedures (2 for adverse events; 1 lost to follow-up; 1 for other reasons)
Incomplete outcome data: Not reported
|
1. Median time (days) to alleviation of clinically important symptoms ITTI Zanamivir, n=136, 5.0 days Placebo, n=141, 7.5 days Difference 2.5 (1.0 to 4.0) P<0.001 Hazard ratio 1.60 (1.22 to 2.09)
ITT Zanamivir, n=174, 5.0 days Placebo, n=182, 7.5 days Difference 2.5 (0.75 to 3.5) P<0.001 Hazard ratio 1.50 (1.18 to 1.89)
2. Incidence of complications ITT Placebo 61/182 (34%) Intervention 40/174 (23%) P=0.037 |
|
|
Matsumoto, 1999 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: multicentre study in Japan
Funding: Not reported
Conflicts of interest: Data analysis was performed by Nippon Glaxo Ltd. |
Inclusion criteria: Aged between 16 and 65 years without pre-existing complications or concurrent disease, within 36h of the onset of influenza-like symptoms; an axillary temperature of at least 37.5ºC and had at least two of the following four symptoms: headache, sore throat, myalgia and cough.
Exclusion criteria: Pregnant or lactating women.
Patient characteristics: Zanamivir inhalation: n=37, age 29.9±9.7, 35% male, confirmed influenza 59.5%, influenza A 40.9%, influenza B 59.1%. Zanamivir double: n=40, age 30.3±9.6, 38% male, confirmed influenza 55.0%, influenza A 45.5%, influenza B 54.5%. Control: n=39, age 29.0±10.6, 38% male, confirmed influenza 74.4%, influenza A 48.3%, influenza B 51.7%.
Groups were comparable at baseline |
Intervention: 10 mg inhaled zanamivir (two inhalations) plus intranasal placebo
or
10 mg inhaled zanamivir plus 6.4 mg intranasal zanamivir (two sprays per nostril) |
Control: Inhaled placebo plus intranasal placebo |
Length of follow-up: 28 days
Loss-to-follow-up: Not reported
Incomplete outcome data: Not reported
|
1. Median time (days) to alleviation of three major symptoms of influenza ITTI Placebo, n=29: 4 (IQR 3, >5) Zanamivir, n=22: 3.5 (IQR 3, 5) Zanamivir double, n=22: 4 (IQR 2, 5)
ITT Placebo, n=39: 4 (IQR 3, >5) Zanamivir, n=37: 3 (IQR 2, 4) Zanamivir double, n=40: 3.5 (IQR 2, >5)
2. Alleviation of 5 influenza symptoms At day 3 (ITT): Placebo 18.0% Zanamivir (combined groups) 45.5%, P=0.006.
At day 5 (ITT): Placebo 51.3% Zanamivir (combined groups) 68.8%, P=0.005.
3. Adverse events that were considered by the investigator to be at least possibly related to study medication 22.5% in the zanamivir inhalation plus intranasal group, 16.2% in the zanamivir inhalation group and 20.5% in the placebo group; 19.5% in the combined zanamivir group. In the zanamivir group, three patients reported hoarse voice, two patients reported headache and two patients diarrhoea. In the placebo group, two patients reported diarrhoea. These were the only events that were reported by more than one patient. There were no serious adverse events. |
|
|
Monto, 1999 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter study in North America and Europe.
Funding: GlaxoWellcome Research and Development
Conflicts of interest: Not reported. |
Inclusion criteria: Otherwise healthy persons ages ≥13 years who presented with symptoms of influenza of ≤48 h duration. Subjects ages >65 years or with the following chronic illnesses were included and regarded as being at high risk for developing complications or having more severe or prolonged illness: cardiovascular conditions (excluding hypertension), respiratory conditions, and endocrine or metabolic conditions.
Exclusion criteria: Persons with any unstable chronic illness, those who had taken other antiviral agents in the previous 7 days, and those with known or suspected hypersensitivity to any component of the study medication. Pregnant or breast-feeding women and women at risk of becoming pregnant during the study were also excluded
Patient characteristics: placebo: n=422, 44% male, age 36±14, 57% influenza positive. Zanamivir 2x: n=419, 43% male, age 35±14, 58% influenza positive. Zanamivir 4x n=415, 45% male, age 34±13, 58% influenza positive.
Groups were comparable at baseline. |
Intervention: Zanamivir, 10 mg 2x/day by oral inhalation plus 6.4 mg 2x/day by nasal spray
Or
Zanamivir, 10 mg 4x/day by oral inhalation plus 6.4 mg 4x/day by nasal spray |
Control: Placebo 2x or 4x/day, group combined for analysis |
Length of follow-up: 21 days
Loss-to-follow-up: Placebo: withdrawn 24 (6%), 12 adverse events 8 lost to follow-up 4 other reasons
Zanamivir 2x/d: withdrawn 24 (6%) 13 adverse events 9 lost to follow-up 2 other reasons
Zanamivir 4x/d: withdrawn 26 (6%) 10 adverse events 12 lost to follow-up 4 other reasons
Incomplete outcome data: Placebo 24 (6%) Zanamivir 2x/d 30 (7%) Zanamivir 4x/d 21 (5%) |
1. Median time (days) to alleviation of major symptoms ITT Placebo 7.0 Zana 2x 6.0, difference 1.0 (0.0, 2.0), P=0.012 Zana 4x 6.0, difference 1.0 (0.0, 2.0), P=0.014
2. Resolution of fever (days) ITT Placebo 6.5 (n=246) Zana 2x 5.75 (n=254), difference 0.75 (-1.0, 2.0), P=0.049 Zana 4x 6.0 (n=259), difference 0.5 (-1.0, 2.0), P=0.274
3. Adverse events Placebo: 59 (14%); Zanamivir 2x/day: 46 (11%); zanamivir 4x/day: 53 (13%)
The most commonly reported adverse events during treatment were nasal signs and symptoms, headaches, bronchitis, and gastrointestinal events.
Withdrawal due to possible adverse events: Placebo: 12 (3%); Zanamivir 2x/day: 13 (3%); zanamivir 4x/day: 10 (2%) |
|
|
Nicholson, 2000 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter trial in 51 centres in Europe, 11 in Canada, and one in China
Funding: This study was supported by grants from F Hoffmann-La Roche.
Conflicts of interest: Not reported |
Inclusion criteria: aged 18–65 years and presented within 36h of onset of influenza-like illness with fever of at least 38°C, with at least one respiratory symptom (cough, sore throat, or nasal symptom) and at least one constitutional symptom (headache, malaise, myalgia, sweats or chills, or fatigue).
Exclusion criteria: Women with a positive urine pregnancy test, patients who had been vaccinated against influenza in the previous 12 months, had active clinically important chronic illness or known HIV-1 infection, were receiving steroids or other immune-suppressants, and who had a history of drug or alcohol abuse.
Patient characteristics: Oseltamivir 75 mg: n=242, age 38.2±11.1, 50% male, influenza confirmed 66%, of which influenza A 97%. Oseltamivir 150 mg: n=242, age 36.7±11.8, 53% male, influenza confirmed 69% of which influenza A 97%. Placebo: n=235, age 37.4±11.9, 50% male, influenza confirmed 69% of which influenza A 96%.
Groups were comparable at baseline. |
Intervention: Oseltamivir 75 mg, twice daily for 5 days
or
Oseltamivir 150 mg, twice daily for 5 days |
Control: Placebo |
Length of follow-up: 21 days
Loss-to-follow-up: Seven patients took no study medication (confirmed by capsule counts) and were excluded from all analyses. 38 patients (5%) withdrew from the study—15 because of adverse events (six placebo, three oseltamivir 75 mg, six oseltamivir 150 mg), and 23 were lost to follow-up (three oseltamivir 150 mg), withdrew for personal reasons (five placebo, one oseltamivir 75 mg, four oseltamivir 150 mg), or were withdrawn for protocol violations (four placebo, four oseltamivir 75 mg, one oseltamivir 150 mg) or for early improvement (one oseltamivir 150 mg).
Incomplete outcome data: Not reported |
1. Median time (hours) to alleviation of symptoms: duration of illness ITTI Placebo (n=161): 116.5 (95% CI 101.5 to 137.8) Oseltamivir 75 mg (n=158): 87.4 (73.3 to 104.7), P=0.02. Oseltamivir 150 mg (n=156): 81.8 (68.2 to 100.0), P=0.01.
ITT Placebo: 116 (99.8 to 129.5) Oseltamivir 75 mg: 97.6 (79.1 to 115.3), P=0.05 Oseltamivir 150 mg: 89.4 (79.1 to 103.7), P=0.03
2. Resolution of fever (hours, ≤37.2˚C) ITTI Placebo: 67 (95% CI 56 to 74) Oseltamivir 75 mg: 39 (31 to 45), P=0.002. Oseltamivir 150 mg: 37 (31 to 44), P<0.001
ITT Placebo: 59 (48 to 68) Oseltamivir 75 mg: 45 (38 to 52), P=0.2 Oseltamivir 150 mg: 41 (33 to 46), P=0.003
3. Tolerability Transient upper-gastrointestinal events generally occurred at the start of treatment and resolved within 1–2 days and were more frequent in the oseltamivir groups than in the placebo group (nausea: 75 mg group mean frequency 12% (range 2·9–12·6%), 150 mg group 12% (2·5–12·1%), placebo group 4%; vomiting: 10% (2·6–11·3%), 9% (1·9–10·3%), and 3%). |
|
|
Puhakka, 2003 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Turku University Hospital, Finland
Funding: Funding for this study (protocol number NAI30015) was provided by GlaxoSmithKline Research and Development, Greenford, UK.
Conflicts of interest: Not reported |
Inclusion criteria: Patients (conscripts of the Finnish Defence Forces) were required to have an influenza-like illness of less than 48 h duration, defined as the presence of fever (temperature ≥37.8°C) and at least 2 of the following symptoms: headache, muscle/joint aches and pain, sore throat and cough.
Exclusion criteria: Patients who were hypersensitive to any component of the study medication or paracetamol, immunocompromised, currently using antibiotics for respiratory tract infection, had used influenza or any other antiviral therapy within 7 d of the study. Women of childbearing potential were required to use acceptable methods of contraception and to have a negative urine test for pregnancy.
Patient characteristics: Intervention: n=293, age 19.2±1.0, 99% male, influenza positive 76%, >99% influenza A. Control: n=295, age 19.3±1.3, >99% male, influenza positive 72%, >99% influenza A.
Groups were comparable at baseline |
Intervention: Zanamivir inhalation 10 mg twice daily for 5 days |
Control: Placebo |
Length of follow-up:
Loss-to-follow-up: Zanamivir: 5 (2%) discontinued prematurely: 1 withdrew consent 1 lost to follow-up 3 other
Placebo: 9 (3%) discontinued prematurely: 1 adverse event 3 withdrew consent 4 lost to follow-up 1 other
Incomplete outcome data: Not reported
|
1. Median time (days) to alleviation of clinically important symptoms ITTI Zanamivir (n=222) 2.0 Placebo (n=213) 2.33 Difference 0.33 (-0.17 to 1.00) P=0.080
ITT Zanamivir (n=293) 2.17 Placebo (n=295) 2.67 Difference 0.50 (0.00 to 0.83) P=0.166
2. Return to normal health ITTI Zanamivir 4.50 Placebo 6.33 Difference 1.83 (no CI reported), P=0.101
3. Adverse effects Inhaled zanamivir was well tolerated. The incidence of adverse events reported during treatment, most of which were mild or moderate intensity, was similar in the zanamivir (77/293, 26%) and placebo (80/295, 27%) groups. The most common adverse events during treatment (zanamivir versus placebo) were sinusitis (6% versus 10%), tonsillitis (4% versus 3%), pneumonia (3% versus 1%), diarrhoea (2% versus 2%), ear nose and throat haemorrhage (2% versus 2%), musculoskeletal pain (2% versus 0%), fever (2% versus 0%) and cough (1% versus 2%). |
|
|
Treanor, 2000 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Sixty primary care and university health centers throughout the United States.
Funding and conflicts of interest: Dr Treanor has received grant support and serves as a paid consultant for Roche, Gilead, Glaxo, Robert Wood Johnson Pharmaceutical Research Institute, and BioChem Pharma. He is also a consultant or on the speaker bureau’s of Wyeth Lederle, Aviron, Pateur Merleaux Connaught (Aventis), Novartis, Acquila Biopharmaceuticals and Biota. Dr. Hayden has received grant support and/or consultation payments from Glaxo Wellcome, Abbott, Johnson & Johnson, PRI, Aviron, and Viropharma. |
Inclusion criteria: Previously healthy adults aged 18 to 65 years who presented within 36 hours of onset of influenza symptoms and who had documented oral temperature of 38°C or higher at enrolment plus 1 or more respiratory symptom (cough, sore throat, or nasal symptoms) and 1 or more constitutional symptom (headache, malaise, myalgia, sweats and/or chills, or fatigue).
Exclusion criteria: Influenza vaccination in the 12 months prior to the beginning of the study; active, clinically significant chronic illness or hiv disease; were receiving systemic steroids or other immunosuppressants; or had a history of alcohol or drug abuse.
Patient characteristics: Placebo: n=209, age 32.6±10.2, 46% male, 62% infected, 95% influenza A. Oseltamivir 75 mg: n=210, age 32.2±10.8, 47% male, 59% infected, 90% influenza A. Oseltamivir 150 mg: n=208, age 33.1±9.8, 55% male, 58% infected, 90% influenza A. Groups were comparable at baseline |
Intervention: Oseltamivir 75 mg 2x/d for 5 days
Or
Oseltamivir 150 mg 2x/d for 5 days |
Control: Placebo |
Length of follow-up: 21 days
Loss-to-follow-up: Placebo: 11 withdrawals (5%): 7 loss to follow-up 3 refused drug 1 adverse event (<1%)
Oseltamivir 75 mg 16 withdrawals (8%): 8 loss to follow-up 4 refused drug 1 adverse event (<1%) 2 protocol violation 1 early improvement
Oseltamivir 150 mg 19 withdrawals (9%): 6 loss to follow-up 5 refused drug 6 adverse event (3%) 2 protocol violation
Incomplete outcome data: Not reported |
1. Median time (hours) to alleviation of symptoms (illness duration) ITTI Placebo (n=129): 103.3 (95% CI 92.6 to 118.7) Oseltamivir 75 mg (n=124): 71.5 (60.0 to 93.2), P<0.001. Oseltamivir 150 mg (n=121): 69.9 (60.0 to 87.9), P=0.006
ITT Placebo (n=209): 97.0 (86.3 to 113.6) Oseltamivir 75 mg (n=210): 76.3 (66.3 to 89.2), P<0.004. Oseltamivir 150 mg (n=208): 74.3 (68.1 to 87.5), P=0.004.
2. Return to normal health (h) ITTI Placebo: 178 (156 to 273) Osel 75mg: 132 (123 to 152) Osel 150mg: 154 (131 to 157)
ITT Placebo: 178 (156 to 273) Osel 75mg: 134 (128 to 155) Osel 150mg: 155 (133 to 157)
3. Complications and associated antibiotic use Complications: otitis media, sinusitis, bronchitis, pneumonia. Total complications Placebo: 19/129 (15%) Osel 75mg: 11/124 (9%) Osel 150mg: 6/121 (5%) Combined intervention versus placebo P=0.03
Antibiotic use Placebo: 14/129 (11%) Osel 75mg: 8/124 (6%) Osel 150mg: 4/121 (3%) Combined intervention versus placebo P=0.05 |
|
|
Whitley, 2015
NCT 00419263
NCT 00610935 |
Type of study: Double-blind placebo-controlled RCT
Setting and country: Multicenter study at locations in USA, Canada, Australia, New Zealand and South Africa.
Funding: The conduct of these studies was supported in whole or in part with funding from the Office of Public Health Emergency Preparedness, Office of Public Health Emergency Countermeasures, Washington, DC, USA. Additional funding was provided by the study Sponsor, BioCryst Pharmaceuticals, Durham, NC, USA.
Conflicts of interest: RW is a Director of Gilead Sciences. AL, SC, EM, GT and MS received research support from BioCryst. JE has been a paid consultant for BioCryst, Biota, Gilead Sciences and Tobira Therapeutics. WJA, SD, PC and WPS are previous or current employees of BioCryst and received salary, stock and stock options. |
Inclusion criteria: Previously healthy males and non-pregnant females aged ≥18 years who presented within 48 h of onset of influenza symptoms with a positive rapid antigen test (RAT) for influenza A or influenza B performed at the clinic site and who had documented fever ≥38.0°C (oral), one or more respiratory symptoms (cough, sore throat or nasal symptoms) and one or more constitutional symptoms (headache, myalgia, feverishness or fatigue) were considered for enrolment.
Exclusion criteria: receipt of any influenza antiviral treatment during the previous 7 days or influenza immunization within 21 days prior to enrolment; active, clinically significant chronic illness including hepatitis B or C, or HIV infection with CD4+ T-cell count <350 cells/mm3; congestive heart failure meeting NYHA Class II or greater severity; a clinically significant abnormal ECG; severe COPD or severe persistent asthma; clinical findings suggesting the presence of complications of influenza (for example, sinusitis, otitis, bronchitis, pneumonia); current therapy with systemic corticosteroids, other immunosuppressants or anticoagulants; or a history of alcohol or drug abuse.
Patient characteristics: Peramivir 300: n=172, age 35±13, 48% male, 94% infected, 74% influenza A. Peramivir 150: n=114, age 37±16, 41% male, 72% infected, 74% influenza A. Placebo: n=141, age 34±12, 50% male, 94% infected, 73% influenza A.
Groups were comparable at baseline. |
Intervention: Peramivir 300 mg
Or
Peramivir 150 mg
as bilateral 2-ml intramuscular injections in each gluteal muscle |
Control: Placebo injections |
Length of follow-up: 14 days
Loss-to-follow-up: Discontinued study: Peramivir 300: 4 1 adverse event 1 lost to follow-up 2 other
Peramivir 150: 1 1 lost to follow-up
Placebo: 4 3 lost to follow-up 1 other
Incomplete outcome data: Not reported
|
1. Median time (hours) to alleviation of symptoms ITTI (no ITT reported) Placebo (n=134): 148.2±7.25 Peramivir 300 (n=163): 130.6±6.89 Difference -21.6, P=0.161 HR 0.838 (95% CI 0.648 to 1.085) Peramivir 150 (n=104): 136.2±8.15 Difference -20.7, no statistics reported.
2. Time to resolution of fever (h) ITTI Placebo: 79.9±5.66 Peramivir 300: 54.3±3.81 Difference -24.0, P=0.004 HR 0.648 (0.472 to 0.797) Peramivir 150: 71.5±6.50 Difference -15.1
3. Adverse events Peramivir 300: 70/172 (41%) Peramivir 150: 43/113 (38%) Placebo: 62/139 (45%)
|
|
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures.
- Provide data per treatment group on the most important prognostic factors ((potential) confounders).
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls.
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders.
Table of quality assessment for systematic reviews of RCTs and observational studies
Based on AMSTAR checklist (Shea, 2007; BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher, 2009; PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/NA |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Taieb, 2019 |
Yes: “The objective of this study was to assess the relative efficacy and safety of baloxavir in otherwise healthy influenza patients, compared to placebo, oseltamivir, zanamivir, laninamivir and peramivir using a network meta-analysis based on all published randomized controlled trials. |
Yes |
Yes |
No |
N/A |
No |
unclear |
No |
unclear |
- Research question (PICO) and inclusion criteria should be appropriate and predefined.
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched.
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons.
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported.
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs).
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table et cetera).
- Clinical and statistical heterogeneity should be assessed; clinical: enough similarities in patient characteristics, intervention and definition of outcome measure to allow pooling? For pooled data: assessment of statistical heterogeneity using appropriate statistical tests (for example Chi-square, I2)?
- An assessment of publication bias should include a combination of graphical aids (for example funnel plot, other available tests) and/or statistical tests (for example Egger regression test, Hedges-Olken). Note: If no test values or funnel plot included, score “no”. Score “yes” if mentions that publication bias could not be assessed because there were fewer than 10 included studies.
- Sources of support (including commercial co-authorship) should be reported in both the systematic review and the included studies. Note: To get a “yes,” source of funding or support must be indicated for the systematic review AND for each of the included studies.
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome accessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
McLean, 2015 |
Patients were randomized to receive oseltamivir or placebo in a 2:1 ratio |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
|
Boivin, 2000 |
Not reported |
unclear |
unlikely |
unlikely |
unclear |
unclear |
unclear |
unclear |
|
Campion, 1998 |
The randomisation code was generated by an internal computer programme in blocks of six. Individual computer-generated randomisation envelopes were kept in one sealed envelope at each site. These remained unopened for all patients. The randomisation code was broken once the database was closed. |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
|
Hayden, 1997 |
Not reported |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
unlikely |
|
Hayden, 2018 / Portsmouth, 2020 |
Not reported |
unlikely |
unlikely |
unlikely |
unlikely |
likely |
unlikely |
unlikely |
| Ison, 2020
|
An interactive web response system |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
|
|
Johnston, 2005 |
Not described |
unclear |
unclear |
unclear |
unclear |
unlikely |
unlikely |
Unlikely |
|
Kohno, 2010 |
Computer-generated randomization was conducted by a central randomization facility with sole access to the code, using a minimization method to balance current smoking behavior at screening and composite symptom scores at screening among the three groups. Each center dispensed the study drug, which was unrecognizable without a drug number, according to the instructions of the randomization center, using assigned randomization numbers. |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
unlikely |
|
Li, 2003 |
Eligible patients were assigned sequentially randomized numbers. The randomization code was designed by Roche Basal. |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
unlikely |
|
Makela, 2000 |
Each patient was randomized independently, with no stratification for centre. The randomization code was generated using an internal Glaxo Wellcome program. The study medication was packed in individually numbered treatment. |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
unlikely |
|
Matsumoto, 1999 |
Not reported |
unlikely |
unlikely |
unlikely |
unclear |
likely |
unlikely |
unlikely |
|
Monto, 1999 |
Not reported |
unlikely |
unlikely |
unlikely |
unclear |
unclear |
unlikely |
unlikely |
|
Nicholson, 2000 |
Randomisation was computer generated by a central randomisation facility, which had sole access to the code. Each centre provided its own medication in individually numbered packs, according to the instructions of the randomisation centre. |
unlikely |
unlikely |
unlikely |
unclear |
unclear |
unlikely |
unlikely |
|
Puhakka, 2003 |
A computer-generated randomization schedule in blocks of 6 |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
unlikely |
|
Treanor, 2000 |
Randomization occurred at the time of study entry by telephone contact with an automated service that had sole access to the code key and was stratified by study site and smoking behavior. |
Unlikely: Participants and staff remained blinded to allocation status throughout the study. |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
unlikely |
|
Whitley, 2015 |
Participants were centrally randomized |
Unlikely: Participants, clinic staff and sponsor personnel remained blinded to treatment allocation throughout conduct of both studies |
unlikely |
unlikely |
unlikely |
unclear |
unlikely |
unlikely |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Tabel Exclusie na het lezen van het volledige artikel (algemene search)
|
Auteur en jaartal |
Redenen van exclusie |
|
Nakamura 2017 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Rafalsky 2016 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Dixit, 2015 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Yang 2019 |
Geen systematische search en studieselectie uitgevoerd |
|
Higashigu, 2018 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Doll 2017 |
Verkeerd design, systematic review van systematic reviews |
|
Boikos 2017 |
Includeert ook niet-gerandomiseerde studie-designs waarvan in 95% van de gevallen niet gecorrigeerd is voor confounding |
|
Lehnert 2016 |
Verkeerd design, systematic review van systematic reviews |
|
Heneghan 2016 |
Review van Taieb, 2019 is recenter |
|
Balasingam 2016 |
Studie d.m.v. opzettelijke infectie, voldoet niet aan PICO. |
|
Vargas-Sandoval 2015 |
Conference abstract |
|
Qiu 2015 |
Review van Taieb, 2019 is recenter; ook vergelijking met andere middelen en traditionele Chinese medicatie (niet uitgesplitst) |
|
Nguyen-Van-Tam 2015 |
Overview artikel, geen systematische search en studieselectie |
|
Michiels 2015 |
Review van Taieb, 2019 is recenter |
|
Dobson 2015 |
Alle studies worden geïncludeerd in Jefferson, 2014 |
|
Geneesmiddelenbulletin, 2015 |
Nieuwe analyse op basis van studies geïncludeerd in Jefferson, 2014, te gebruiken in overwegingen. |
|
Jefferson 2014b |
Duplicatie van Jefferson, 2014 (oude titel, aangepast: healthy verwijderd uit de titel) |
|
Jefferson 2014c |
Studies worden geïncludeerd in Jefferson, 2014 |
|
Heneghan 2014 |
Studies worden geïncludeerd in Jefferson, 2014 |
|
Alves Galvao, 2014 |
Amantadine en rimantidine worden niet (meer) in Nederland gebruikt als antiviraal middel |
|
Michiels, 2013 |
Jefferson, 2014 is een recentere systematische review |
|
Ceyhan, 2012 |
Geen relevante uitkomstmaten |
|
Santesso, 2013 |
Samenvatting van Hsu, 2012 |
|
Muthuri, 2013 |
Analyse uitgebreider in Muthuri 2014, zie inclusie in module ‘opgenomen patiënten’ |
|
Michiels, 2013 |
Systematische review van systematische reviews: verkeerd studiedesign |
|
Ebell, 2013 |
studies geincludeerd in Jefferson, 2014 |
|
Beck, 2013 |
Geen systmatische reveiw |
|
Wang, 2012 |
Cochrane review children. Jefferson 2014 more recent. |
|
Jefferson, 2012 |
Update in Jefferson 2014 |
|
Hsu, 2012 |
Eén geïncludeerde studie (Blumentals) heeft gecorrigeerd voor confounding d.m.v. propensity scoring maar rapporteert alleen uitkomsten gerelateerd aan central nerve system. |
|
Cayley Jr, 2012 |
Commentaar |
|
Hernain, 2011 |
Geen systematische review |
|
Ceyhan, 2012 |
Therapie niet beschikbaar in Nederland (Enfluvir) |
|
Mancuso, 2010 |
Geen transparante search en selectiecriteria |
|
Jefferson, 2010. |
Update in Jefferson 2014 |
|
Gibbs, 2010 |
Heinonen, 2010 is de originele RCT en werd geïncludeerd |
|
Falagas, 2010 |
Taieb, 2019 is recenter; inclusie overlapt |
|
Cayley Jr, 2010 |
Commentaar |
|
Wang, 2011 |
Therapie niet beschikbaar in Nederland |
|
Shun-Shin, 2009 |
Jefferson, 2014 is een recentere systematische review |
|
Jefferson and Jones, 2009 |
Update in Jefferson 2014 |
|
Jefferson, 2009 |
Update in Jefferson 2014 |
|
Burch, 2009 |
Jefferson, 2014 is een recentere systematische review |
|
Matheson, 2007 |
Jefferson, 2014 is een recentere systematische review |
|
Kohno, 2010 |
Inclusie in Taieb, 2019 |
|
Duval, 2010 |
Inclusie in Taieb, 2019 |
|
Jefferson, 2006 (Cohcrane) |
Update in Jefferson 2014 |
|
Jefferson, 2006 (Lancet) |
Update in Jefferson 2014 |
|
Bettis, 2006 |
Inclusie in Taieb, 2019 |
|
van der Wouden, 2005 |
Verkeerde interventie (vacccinatie) |
|
Turner, 2003 |
Verkeerde uitkomstmaten |
|
Matheson, 2003 |
Jefferson, 2014 is een recentere systematische review |
|
McNicholl, 2001 |
Geen transparante search en selectiecriteria |
|
Kaiser, 2003 |
Geïncludeerd in Jefferson, 2014 |
|
Ison, 2003 |
Geïncludeerd in uitgangsvraag over gehospitaliseerde patiënten |
|
Makela, 2000 |
Inclusie in Taieb, 2019 |
|
Unknown, 2001 (Prespcrire International) |
Overzichtsartikel, geen systematische review |
|
Kaiser, 2000 |
Geïncludeerd in Jefferson, 2014 |
|
Jefferson, 2000 |
Update in Jefferson 2014 |
|
Demicheli, 2000 |
Verkeerde populatie (healthy adults) |
|
Monto, 1999 |
Geen systematische review |
|
Bardsley-Elliot, 1999 |
Overzicht van farmakinetiek van Oseltamivir, voldoet niet aan PICO |
Tabel Exclusie na het lezen van het volledige artikel (search risicogroepen)
Vanwege de grote overeenkomst met searchresultaten van de algemene search, is er geen exclusietabel van de risicogroepen opgenomen. Deze is op te vragen bij het Kennisinstituut.
Tabel Exclusie na het lezen van het volledige artikel (Baloxavir search)
|
Auteur en jaartal |
Redenen van exclusie |
|
Kakayu, 2019 |
Andere vergelijking. prospectief observationeel onderzoek naar BM versus NAI (oseltamivir, zanamivir, peramivir, laninamivir) voor fever duration. (ook vergelijking van BM <48h na onset vs >48h) |
|
Komeda, 2020 |
Andere vergelijking. retrospectief observationeel onderzoek naar BM versus NAI (oseltamivir, zanamivir, laninamivir) voor hospitalization incidence |
|
Sha, 2020 |
Andere vergelijking. retrospectief observationeel onderzoek naar BM versus oseltamivir |
|
Baker, 2020 |
Andere vergelijking. RCT naar BM versus oseltamivir |
|
Dziewiatkowski, 2019 |
Geen systematische review. |
|
Hayden, 2019 |
Geen systematische review |
|
Ng, 2019 |
Geen systematische review |
|
Saito, 2020 |
Andere vergelijking. prospectief observationeel onderzoek naar BM versus oseltamivir |
|
Shirley, 2020 |
Geen systematische review |
|
Watanabe, 2019 |
Subgroep-analyses voor uitkomsten per virus type/subtype. Niet relevant voor deze module. |
|
Yoshii, 2020 |
Andere vergelijking. prospectief observationeel onderzoek naar BM versus NAI |
|
Yoshino, 2020 |
Andere vergelijking. prospectief observationeel onderzoek naar BM versus oseltamivir of laninamivir |
|
Taieb, 2019 |
Voor BM vs placebo zijn 2 trials van Shionogi (farmaceut) geïncludeerd. Dit betreffen beiden de CAPSTONE-1 studie beschreven in Hayden 2018 |
|
Taieb, 2021 |
Zie Taieb, 2019, zelfde studies geincludeerd. |
Beoordelingsdatum en geldigheid
Publicatiedatum : 15-10-2021
Beoordeeld op geldigheid : 01-07-2021
|
Module[1] |
Regie-houder(s)[2] |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn[3] |
Frequentie van beoordeling op actualiteit[4] |
Wie houdt er toezicht op actualiteit[5] |
Relevante factoren voor wijzigingen in aanbeveling[6] |
|
Behandeling niet-opgenomen patiënten |
NVMM |
2021 |
2026 |
Elke 5 jaar |
NVMM |
Niet van toepassing |
[1] Naam van de module
[2] Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders)
[3] Maximaal na vijf jaar
[4] (half)Jaarlijks, eens in twee jaar, eens in vijf jaar
[5] regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) kerngroep die in stand blijft
[6] Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
De richtlijn is ontwikkeld in samenwerking met:
- Nederlandse Internisten Vereniging
- Nederlandse Vereniging van Artsen voor Longziekten en Tuberculose
- Nederlandse Vereniging voor Kindergeneeskunde
- Nederlandse Vereniging voor Klinische Geriatrie
- Nederlandse Vereniging voor Obstetrie en Gynaecologie
- Nederlandse Vereniging voor Intensive Care
- Vereniging van specialisten ouderengeneeskunde
- Nederlands Huisartsen Genootschap
- Rijksinstituut voor Volksgezondheid en Milieu
- Patiëntenfederatie Nederland
Doel en doelgroep
Doel
Het project was het herzien en uitbreiden van de richtlijn klinische behandeling met antivirale therapie van opgenomen patiënt met influenza. Bij de oorspronkelijke richtlijn waren niet alle betrokken wetenschappelijke verenigingen en patiënten meegenomen in het opstellen hiervan en misten aanbevelingen voor specifieke patiëntengroepen. Verder is bij de herziening de inhoud uitgebreid met de behandeling van niet-opgenomen patiënten, de nieuwste ontwikkelingen in diagnostiek en wijzigingen. Hierdoor zal de patiëntenzorg in overeenstemming gebracht worden met de laatste stand van zaken.
Doelgroep
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen die betrokken zijn bij de zorg voor patiënten met Influenza.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2018 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor de behandeling van patiënten met (verdenking) op influenza.
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname.
Werkgroep
- Dr. A. (Annelies) Riezebos-Brilman, arts-microbioloog, werkzaam in het UMC Utrecht (NVMM), t/m april 2021 werkzaam in het UMC Utrecht, sinds mei 2021 werkzaam in LabMicta, Hengelo (NVMM) (voorzitter)
- Prof. dr. Menno de Jong, hoogleraar/afdelingshoofd Medische Microbiologie & Infectiepreventie, werkzaam in het Amsterdam UMC te Amsterdam (NVMM)
- Drs. K.A.S. (Karen) Couderé, AIOS Medische microbiologie, werkzaam in Elisabeth TweeSteden ziekenhuis Tilburg (NVMM)
- Dr. F.L. (Frank) van de Veerdonk, internist-infectioloog, werkzaam in het Radboudumc te Nijmegen (NIV)
- Dr. A.W.J. (Aik) Bossink, longarts, werkzaam in het Diakonessenhuis te Utrecht (NVALT)
- Dr. P.L.A. (Pieter) Fraaij, kinderarts-infectioloog/immunoloog, werkzaam in het Erasmus MC-Sophia te Rotterdam (NVK)
- Drs. M. (Marieke) Zeeman, internist-ouderengeneeskunde en klinisch farmacoloog, werkzaam in Deventer ziekenhuis te Deventer (NVKG)
- Dr. W.J. (Wouter) Meijer, gynaecoloog, werkzaam in Gelre Ziekenhuizen te Zutphen (NVOG)
- Dr. L.P.G. (Lennie) Derde, infectioloog-intensivist, werkzaam in het UMC Utrecht (NVIC)
- Dr. P. (Paul) van Houten, specialist ouderengeneeskunde, werkzaam bij Zonnehuisgroep Amstelland te Amstelveen (Verenso)
- Mevr. K. (Klaartje) Spijkers (Patiëntenfederatie Nederland)
- Dr. G.A. (Ted) van Essen, huisarts, niet praktiserend (NHG)
- Dr. A.M. (Albert) Vollaard, infectioloog, werkzaam bij het RIVM te Bilthoven (RIVM)
Meelezer
- Stichting Kind en Ziekenhuis
Met ondersteuning van
- Dr. J. (Janneke) Hoogervorst-Schilp, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- E.A (Ester) Rake, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Actie |
|
Riezebos-Brilman (voorzitter) |
Arts-microbioloog met aandachtsgebied virologie, UMCU |
Counsilmember ESCV (European Society of Clinical Virology) |
geen |
Geen actie |
|
Meijer |
Gynaecoloog, Coöperatie medische Specialisten Gelre u.a. werkzaam in Gelre ziekenhuis locatie Zutphen |
geen |
geen |
Geen actie |
|
Bossink |
Longarts, vrijgevestigd medisch specialist Diakonessenhuis Utrecht |
geen |
Patent crossspot |
Geen actie |
|
Van Essen |
Huisarts, gepensioneerd |
Onbezoldigd voorzitter Nederlandse Influenza Stichting (https://influenzastichting.nl/) |
Een paar keer een vergoeding als spreker, van Sanofi Pasteur en van MSD. |
Geen actie |
|
Van de Veerdonk |
Internist-infectioloog werkzaam Radboudumc |
Onderzoeker betaald vanuit subsidies. |
Ik heb een VIDI subsidie gekregen waarbij uitgezocht wordt of neuraminidase remmers de kans op dodelijke schimmelinfecties kunnen verhogen. Ik heb geen baat bij welke uitkomst dan ook van de richtlijn. |
Geen actie |
|
De Jong |
Hoogleraar/afdelingshoofd Medische Microbiologie & Infectiepreventie, Amsterdam UMC, |
geen |
In afgelopen 5 jaar lid (ad-hoc) geweest van Scientific Advisory Boards voor Janssen, Crucell, Shionogi, MedImmune, Celltrion; allen op gebied van preklinisch en klinisch onderzoek van nieuwe antivirale middelen tegen influenza. Momenteel geen actief adviseurschap. |
Geen actie, want momenteel geen actief adviseurschap, en onafhankelijke beoordelen van nieuwe middelen (dus geen belangenverstrengeling) |
|
Derde |
Intensivist UMC Utrecht (0.45 FTE) |
Voorzitter taskforce NVIC: Draaiboek (influenza-) pandemie (onbetaald) |
Coördinerend onderzoeker Europa voor REMAP-CAP studie. Deze studie is een internationale 'adaptive platform trial' naar de beste behandeling van CAP op de IC, waarbij in het seizoen of tijdens een pandemie de behandeling van influenza daar deel van zou kunnen uitmaken. Coördinerend onderzoeker Europa voor REMAP-CAP studie. |
Geen actie, want de uitkomst van het genoemde onderzoek en de richtlijn hebben geen invloed op elkaar |
|
Zeeman |
Internist ouderengeneeskunde Deventer Ziekenhuis |
Klinisch Farmacoloog Deventer Ziekenhuis (binnen functie internist-og) |
geen |
Geen actie |
|
Vollaard |
Internist-infectioloog, Landelijke Coördinatie Infectieziektebestrijding (LCI) |
Arts-vrijwilliger bij Dokters van de Wereld: consultatie van ongedocumenteerde migranten |
geen |
Geen actie |
|
Fraaij |
Kinderarts Infectioloog en immunoloog, hoofd subspecialisme Kinderimmunologie en infectieziekten, ErasmusMC-Sophie, Rotterdam |
geen |
PREPARE Europe (EU FP? grant no. 602525) Ik ben bestuurslid bij Stichting Infecties bij Kinderen (INKI) Ik organiseer de cursus 'Antibiotica bij kinderen' |
Geen actie, want de uitkomst van het genoemde onderzoek en de richtlijn hebben geen invloed op elkaar |
|
Spijkers |
Senior adviseur patiëntenbelang |
Voorzitter Stichting Samen voor Duchenne (onbetaald) |
geen |
Geen actie |
|
Coudere |
AIOS Medische microbiologie, LMMI, ETZ, Tilburg |
Consulent dienst Gastro-enterologie en Hepatologie, UZA, Antwerpen: medewerker onderzoeksproject Hepatitis B en C screening bij sub-Saharaans Afrikaanse Migranten (funding Gilead) - betaald - einde 31 me 2019 |
geen |
Geen actie |
|
Van Houten |
Specialist ouderengeneeskunde, hoofd medische dienst Zonnehuisgroep Amstelland |
Lid Goed gebruik Hulpmiddelen bij ZonMw |
Voorzitter werkgroep Handreiking hygiëne en infectiepreventie voor specialisten ouderengeneeskunde bij Verenso. Adviseur Behandeladvies COVID-19 acute fase en nazorg bij Verenso |
Geen actie |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een werkgroeplid af te vaardigen namens de Patiëntenfederatie Nederland. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn (module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden in de bijlagen. In het licht van de bevindingen van de Kwaliteits- & Doelmatigheidsagenda over aantallen beschikbare indicatoren en de moeilijkheid van het ontwikkelen van toepasselijke indicatoren, is er besloten (vooralsnog) geen indicatoren te ontwikkelen.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. Tevens zijn er knelpunten aangedragen door afgevaardigden van de Nederlandse Vereniging voor Medische Microbiologie, Inspectie Gezondheidszorg & Jeugd, de Nederlandse Associatie Physician Assistants, het Nederlands Huisartsen Genootschap, de Nederlandse Vereniging voor Klinische Geriatrie, Rijksinstituut van Volksgezondheid en Milieu, Verpleegkundigen en Verzorgenden Nederland, Verenso, Vereniging Innovatieve Geneesmiddelen, Zorginstituut Nederland, en de Nederlandse Vereniging Artsen voor Longziekten en Tuberculose tijdens de invitational conference. Een verslag hiervan is opgenomen in de bijlagen.
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Voor de afzonderlijke uitgangsvragen werd aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie voor de oriënterende zoekactie en patiëntenperspectief zijn opgenomen in de zoekverantwoording.
Gedurende het zoekproces, heeft de werkgroep de internationale richtlijn van de Infectious Diseases Society of America (IDSA) beoordeeld op geschiktheid (Uyeki, 2019). Daarbij werd het adviesrapport ‘Adapteren van internationale richtlijnen naar de Nederlandse praktijk’ gevolgd van de Federatie Medisch Specialisten, welke is vastgesteld in de Raad Kwaliteit in 2017 (NVALT, NVU 2016). Er werd besloten om de IDSA Guideline niet als uitgangspunt te nemen om de richtlijn te updaten. Enerzijds zijn er geen transparante zoekstrategie en selectiecriteria in de IDSA Guideline beschikbaar; anderzijds is er geen duidelijke scheiding weergegeven tussen literatuuranalyse en Overwegingen in de IDSA Guideline. Daarmee voldoet de IDSA Guideline niet aan afspraken die wv-en onderling hebben afgesproken met betrekking tot het gebruiken en adapteren van internationale richtlijnen.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag.
Commentaar- en autorisatiefase [na autorisatiefase definitief maken]
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Aanvullingen module baloxavir - autorisatiefase
De aanvullingen in deze richtlijn betreffende het antivirale middel baloxavir werd toegevoegd aan de geautoriseerde richtlijn. Deze aanvullingen en wijzigingen zijn ter autorisatie dan wel accordering voorgelegd aan de deelnemende (wetenschappelijke) verenigingen en (patient) organisaties. Er werd geen aparte commentaarfase georganiseerd voor deze teksten, omdat er geen wijzigingen zijn aangebracht in de aanbevelingen.
Literatuur
- Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
- Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
- Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
- Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
- Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
- Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
- Nederlandse Vereniging voor Urologie en de Nederlandse Vereniging van Artsen voor Longziekten en Tuberculose. Adapteren van internationale richtlijnen naar de Nederlandse praktijk. Utrecht, 2016
- Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
- Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
- Schünemann HJ, Oxman AD, Brozek J, Glasziou P, Jaeschke R, Vist GE, Williams JW Jr, Kunz R, Craig J, Montori VM, Bossuyt P, Guyatt GH; GRADE Working Group. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008 May 17;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008 May 24;336(7654). doi: 10.1136/bmj.a139.
- Schünemann, A Holger J (corrected to Schünemann, Holger J). PubMed PMID: 18483053; PubMed Central PMCID: PMC2386626.
- Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
- Uyeki, T. M., Bernstein, H. H., Bradley, J. S., Englund, J. A., File Jr, T. M., Fry, A. M.,... & Ison, M. G. (2019). Clinical practice guidelines by the Infectious Diseases Society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenza. Clinical Infectious Diseases, 68(6), e1-e47.
Zoekverantwoording
Zoekverantwoording algemene search
|
Database(s): Medline, Embase |
Datum: 3 mei 2019 |
|
Periode: > 1996 |
Talen: Engels |
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
1996 – mei 2019 |
1 exp *Influenza, Human/ or exp influenzavirus a/ or exp influenzavirus b/ or exp influenza b virus/ or influenza*.ti. or flu.ti. or flue.ti. or h1n1*.ti. or h2n3*.ti. (89133) 2 exp *Antiviral Agents/ or exp Oseltamivir/ or exp Zanamivir/ or exp Rimantadine/ or exp Amantadine/ or (oseltamivir or zanamivir or baloxavir or peramivir or rimantadine or amantadine or lasinavir or moroxydine or umifenovir or antiviral therap* or antivirus agent*).ti. (200227) 3 exp Hospitalization/ or (hospital* or outpatient* or medical centre* or emergency room* or intensive care or icu or ic).ti,ab,kw. (1497555) 4 1 and 2 and 3 (904) 5 limit 4 to (english language and yr="1996 - 2019") (808) 6 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (391445) 7 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1852581) 8 5 and 7 (163) 9 1 and 2 (7302) 10 limit 9 to (english language and yr="1996 - 2019") (5419) 11 6 and 10 (122) (aantal systematische reviews) 12 8 not 11 (130) (aantal RCT’s) 13 11 or 12 (252)
= 252 |
518 |
|
Embase (Elsevier) |
('influenza'/exp/mj OR influenza*:ti OR flu:ti OR flue:ti OR h1n1*:ti OR h2n3*:ti OR 'influenza virus'/exp/mj) AND ('antivirus agent'/exp/mj OR 'oseltamivir'/exp OR 'zanamivir'/exp OR 'baloxavir'/exp OR 'baloxavir marboxil'/exp OR 'peramivir'/exp OR 'rimantadine'/exp OR 'amantadine'/exp OR 'lasinavir'/exp OR 'moroxydine'/exp OR 'umifenovir'/exp OR oseltamivir:ti OR zanamivir:ti OR baloxavir:ti OR peramivir:ti OR rimantadine:ti OR amantadine:ti OR lasinavir:ti OR moroxydine:ti OR umifenovir:ti OR 'antiviral therap*':ti OR 'antivirus agent*':ti) AND ('hospital patient'/exp OR hospital*:ti,ab OR outpatient*:ti,ab OR 'medical centre*':ti,ab OR 'emergency room*':ti,ab OR 'intensive care':ti,ab OR icu:ti,ab OR ic:ti,ab) AND (1996-2019)/py AND (english)/lim NOT 'conference abstract':it
Gebruikte filters: RCT’s: ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it = 262
('influenza'/exp/mj OR influenza*:ti OR flu:ti OR flue:ti OR h1n1*:ti OR h2n3*:ti OR 'influenza virus'/exp/mj) AND ('antivirus agent'/exp/mj OR 'oseltamivir'/exp OR 'zanamivir'/exp OR 'baloxavir'/exp OR 'baloxavir marboxil'/exp OR 'peramivir'/exp OR 'rimantadine'/exp OR 'amantadine'/exp OR 'lasinavir'/exp OR 'moroxydine'/exp OR 'umifenovir'/exp OR oseltamivir:ti OR zanamivir:ti OR baloxavir:ti OR peramivir:ti OR rimantadine:ti OR amantadine:ti OR lasinavir:ti OR moroxydine:ti OR umifenovir:ti OR 'antiviral therap*':ti OR 'antivirus agent*':ti) AND (1996-2019)/py AND (english)/lim NOT 'conference abstract':it
Gebruikte filters: Systematische reviews: ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) = 187
= 449 |
Zoekverantwoording risicogroepen
|
Database(s): Medline, Embase |
Datum: 2 oktober 2019 |
|
Periode: > 1996 |
Talen: Engels |
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
1996 – sep 2019
|
1 exp *Influenza, Human/ or exp influenzavirus a/ or exp influenzavirus b/ or exp influenza b virus/ or influenza*.ti. or flu.ti. or flue.ti. or h1n1*.ti. or h2n3*.ti. (90865) 2 exp Antiviral Agents/ or exp Oseltamivir/ or exp Zanamivir/ or exp Rimantadine/ or exp Amantadine/ or (oseltamivir or zanamivir or marboxil or peramivir or rimantadine or amantadine or lasinavir or moroxydine or umifenovir or antiviral* or antivirus*).ti,ab,kw. (387189) 3 exp *Pregnancy/ or exp *Chronic Disease/ or exp Immunocompromised Host/ or exp *Lung Diseases/ or exp Asthma/ or exp Immunosuppressive Agents/ or (pregnan* or post-partum or asthma or copd or immunocompromis* or immunosuppress*).ti,ab,kw. or (chronic adj4 (disease* or condition*)).ti,ab,kw. or ((lung or pulmonary or heart or renal or liver or cerebrovascular or neurological) adj3 disease*).ti,ab,kw. or ((organ or stem cell) adj3 transplant*).ti,ab,kw. or (immune adj3 (therap* or treatment*)).ti,ab,kw. (2362472) 4 1 and 2 and 3 (1500) 5 limit 4 to (english language and yr="1996 - 2019") (1261) 6 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (413444) 7 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1901849) 8 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ (Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies) (3274082) 9 5 and 6 (42) 10 (5 and 7) not 9 (130) 11 (5 and 8) not (9 or 10) (251) 12 9 or 10 or 11 (423)
= 423 |
755 |
|
Embase (Elsevier) |
('influenza'/exp/mj OR influenza*:ti OR flu:ti OR flue:ti OR h1n1*:ti OR h2n3*:ti OR 'influenza virus'/exp/mj)
AND ('antiviral therapy'/exp/mj OR 'oseltamivir'/exp/mj OR 'zanamivir'/exp/mj OR 'baloxavir marboxil'/exp/mj OR 'peramivir'/exp/mj OR 'rimantadine'/exp/mj OR 'amantadine'/exp/mj OR 'lasinavir'/exp/mj OR 'moroxydine'/exp/mj OR 'umifenovir'/exp/mj OR oseltamivir:ti,ab OR zanamivir:ti,ab OR marboxil:ti,ab OR peramivir:ti,ab OR rimantadine:ti,ab OR amantadine:ti,ab OR lasinavir:ti,ab OR moroxydine:ti,ab OR umifenovir:ti,ab OR antiviral*:ti,ab OR antivirus*:ti,ab)
AND ('pregnancy'/exp/mj OR 'chronic disease'/exp/mj OR 'immunocompromised patient'/exp OR 'lung disease'/exp/mj OR 'asthma'/exp OR 'immunosuppressive agent'/exp/mj OR pregnan*:ti,ab OR 'post partum':ti,ab OR ((chronic NEAR/4 (disease* OR condition*)):ti,ab) OR (((lung OR pulmonary OR heart OR renal OR liver OR cerebrovascular OR neurological) NEAR/3 disease*):ti,ab) OR asthma:ti,ab OR copd:ti,ab OR immunocompromis*:ti,ab OR (((organ OR 'stem cell*') NEAR/3 transplant*):ti,ab) OR ((immune NEAR/3 (therap* OR treatment*)):ti,ab) OR immunosuppress*:ti,ab OR 'high risk':ti,ab)
AND (english)/lim AND (1996-2019)/py NOT 'conference abstract':it
Gebruikte filters:
Systematische reviews: ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp)
RCT’s: ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it
Observationeel onderzoek: ‘major clinical study’/exp OR 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR ('prospective study'/de NOT 'randomized controlled trial'/de) OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (case:ab,ti AND ((control NEAR/1 (study OR studies)):ab,ti)) OR (follow:ab,ti AND ((up NEAR/1 (study OR studies)):ab,ti)) OR ((observational NEAR/1 (study OR studies)):ab,ti) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti)
= 591 |
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
2019 – feb 2021
|
1 exp *Influenza, Human/ or exp influenzavirus a/ or exp influenzavirus b/ or exp influenza b virus/ or influenza*.ti. or flu.ti. or flue.ti. or h1n1*.ti. or h2n3*.ti. (97402) 2 (baloxavir or marboxil or xofluza).ti,ab,kf. (179) 3 1 and 2 (152) 5 limit 3 to (english language and yr="2019 - 2021") (141) 6 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (496037) 7 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (2093390) 8 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ [Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies] (3659541) 9 5 and 6 (5) 10 (5 and 7) not 9 (31) 11 (5 and 8) not (9 or 10) (8) 12 9 or 10 or 11 (44)
= 44 |
58 |
|
Embase (Elsevier) |
('influenza'/exp/mj OR influenza*:ti OR flu:ti OR flue:ti OR h1n1*:ti OR h2n3*:ti OR 'influenza virus'/exp/mj)
AND ('baloxavir marboxil'/exp OR marboxil:ti,ab OR baloxavir*:ti,ab OR xofluza:ti,ab,kw)
AND [english]/lim AND [2019-2021]/py NOT 'conference abstract':it
Gebruikte filters:
Systematische reviews: ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp)
RCT’s: ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it
Observationeel onderzoek: ‘major clinical study’/exp OR 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR ('prospective study'/de NOT 'randomized controlled trial'/de) OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (case:ab,ti AND ((control NEAR/1 (study OR studies)):ab,ti)) OR (follow:ab,ti AND ((up NEAR/1 (study OR studies)):ab,ti)) OR ((observational NEAR/1 (study OR studies)):ab,ti) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti)
= 54 |