Behandeling influenza - opname
Uitgangsvraag
Wat is het beste beleid ten aanzien van keuze en duur (toedieningswijze, timing, dosis) van antivirale behandeling voor kinderen en volwassenen die in aanmerking zouden komen voor ziekenhuisopname met een influenza-like-illness?
De uitgangsvraag besteedt (indien mogelijk) specifieke aandacht aan de volgende risicogroepen:
- Ouderen
- Zwangeren
- Kinderen jonger dan 2 jaar, in het bijzonden zuigelingen
- Mensen met een chronische ziekte (comorbiditeit)
- Mensen met verminderde afweer en/of gebruik van immunosuppressiva/immunotherapie
Met patiënten wordt bedoeld:
- Opgenomen patiënt: patiënt opgenomen in een ziekenhuis.
Aanbeveling
Aanbeveling antivirale behandeling voor kinderen en volwassenen
Overweeg behandeling met oseltamivir voor 5 dagen bij kinderen en volwassenen die verdacht worden van influenza in afwachting van diagnostiek of bewezen influenza hebben en opgenomen worden in het ziekenhuis.
Start geen dubbeltherapie met NAIs en gebruik geen hogere doseringen dan standaard.
Zie kinderformularium en SWAB-richtlijn CAP voor dosering (https://www.kinderformularium.nl; https://swab.nl/nl/cap).
Overweeg Zanamivir iv indien orale toediening niet mogelijk is en/of indien er resistentie tegen oseltamivir is aangetoond.
Aanbeveling subgroep zwangeren
Overweeg behandeling met oseltamivir voor 5 dagen bij zwangere patiënten die verdacht worden van influenza in afwachting van diagnostiek of bewezen influenza hebben en opgenomen worden in het ziekenhuis.
Zie kinderformularium en SWAB-richtlijn CAP voor dosering
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Er zijn geen data beschikbaar voor de gedefinieerde cruciale uitkomstmaten in RCT’s, maar er is wel een observationele studie uitgevoerd (Muthuri, 2014). Er zijn geen RCTs met voldoende patiënten welke het effect van NAI op de relevante uitkomstmaten hebben bestudeerd of het effect ervan kon niet worden vastgesteld omdat te laat begonnen werd met antivirale therapie in deze trials. Voor bewijs van effectiviteit is men volledig afhankelijk van observationele data, waardoor er dus minder bewijskracht is.
Inherent aan het ontbreken van data uit RCTs is er dus ook geen bewijskracht voor gunstige dan wel ongunstige effecten van NAI behandeling voor patiënten met verdenking op influenza of een bewezen influenza infectie. De overall bewijskracht is in alle gevallen zeer laag of niet aanwezig. Vanwege het feit dat geadviseerd wordt om hoog risico patiënten (zwangeren, ouderen, kinderen en immuungecompromiteerden) die zich presenteren in het ziekenhuis met verdenking op of een bewezen influenza infectie direct te behandelen met een NAI zijn er geen placebo gecontroleerde studies met voldoende patiënten welke het effect van NAI op de relevante uitkomstmaten hebben bestudeerd.
De observationele studie van Muthuri (2014) met een meta-analyse op individueel patiënten niveau liet zien dat de behandeling met een NAI in vergelijking met geen behandeling bij volwassen patiënten geassocieerd was met een verminderde mortaliteit en dat deze associate in subgroepanalyses alleen significant was wanneer de behandeling vroeg (< 2 dagen) werd gestart en niet wanneer deze laat (na 2 tot 5 dagen) werd gestart (Muthuri, 2014).
Wel is er bewijs (zoals aangegeven in UV3) dat in gezonde volwassenen en kinderen met influenza het starten van een orale NAI de koorts en symptomen sneller doet verdwijnen (module 'Behandeling influenza - geen opname'). Deze data bieden een rationale om orale NAI voor te schrijven binnen 2 dagen na ontstaan van symptomen in een groep welke een hoog risico heeft op een gecompliceerd beloop (opname in ziekenhuis, IC opname en mortaliteit). Alhoewel er dus alleen observationele studies of RCT’s met te kleine aantallen zijn en hiervan meta-analyses bestaan wordt er in de praktijk NAI voorgeschreven aan vrijwel iedereen met een hoog risico die opgenomen wordt. De aanpak om hoog risico patiënten met influenza te behandelen met een NAI wordt ondersteund door een meta-analyse van 3085 patiënten met een bewezen influenza infectie en een hoog risico voor opname welke liet zien dat het gebruik van een orale NAI bij ambulante patiënten met influenza infectie het risico op opname verlaagd (adjusted odds ratio, 0.24; 95% confidence interval, 0.20 tot 0.30) (Venkatasan, 2017).
De huidige standard voor de behandeling van influenza is een NAI oraal of intraveneus. Oseltamivir is het meest gebruikte middel. Een fase 3b RCT met 626 patiënten met bewezen influenza heeft intraveneus zanamivir vergeleken met oraal oseltamivir en vond geen verschil in effectiviteit (Marty, 2017). Zanamivir was non inferieur aan oseltamivir. Er is geen bewijs en er zijn ook geen aanwijzingen vanuit observationele studies dat een behandelduur langer dan 5 dagen in specifieke gevallen nodig zou zijn of dat er in sommige gevallen zou moeten behandelen met een hogere dosering NAI.
Tijdens de zwangerschap kan oseltamivir waarschijnlijk veilig worden gebruikt. Er zijn geen aanwijzingen voor teratogeniciteit of een verhoogde risico op nadelige zwangerschapsuitkomsten. Met zanamivir is aanzienlijk minder ervaring. Terughoudendheid met het voorschrijven van dit middel is aangewezen.
Vooralsnog zijn er geen vergelijkende gerandomiseerde studies van voldoende grootte bij opgenomen patiënten naar andere antivirale therapieën, zoals baloxavir.
Waarden en voorkeuren van patiënten (en eventueel hun verzorgers)
Voor opgenomen patiënten is het doel allereerst hun ernstige ziekte te overleven. Zoals hierboven beschreven is er in dat licht een argument om alle potentieel bijdragende medicatie voor te schrijven. Anderzijds is het niet in het belang van de patiënt, en niet ethisch, een patiënt bloot te stellen aan potentiële bijwerkingen van medicatie die niet bijdraagt aan hun herstel. In dit kader is het belangrijk Samen Beslissen toe te passen waarbij eventueel ouders/naasten worden betrokken.
Welk aspect van bovengenoemde overwegingen het zwaarst weegt, zal per persoon verschillen. Omdat artsen hun patiënten wat deze behandeling betreft niet goed kunnen informeren is het belangrijk dat er een richtlijn is die hierin een keuze maakt. Omdat die keuze niet gebaseerd is op enig wetenschappelijk bewijs, adviseert de werkgroep patiënten, indien mogelijk, in studieverband te behandelen bij influenza bij opgenomen patiënten. Tot meer bewijs beschikbaar is adviseert de werkgroep de richtlijnen van het RIVM/LCI te volgen.
Kosten (middelenbeslag)
Op dit moment is er geen verschil in de kosten van verkrijgbare middelen. Wel is het maken van onnodige kosten in een maatschappelijk perspectief onwenselijk. Een aantal nieuwe middelen komt wellicht op korte termijn beschikbaar. De kosten daarvan zijn nog onbekend, maar potentieel hoog. De werkgroep adviseert om nieuwe middelen in opgenomen patiënten alleen in studie verband te gebruiken, om de toegevoegde waarde bovenop geen behandeling en de toegevoegde waarde bij verstrekking in plaats van, of additioneel aan, de huidige middelen, te bepalen.
Aanvaardbaarheid voor de overige relevante stakeholders
Op dit moment is er geen bewijs voor een belangrijk voordeel in de behandeling van pediatrische of volwassen opgenomen patiënten met antivirale middelen tegen influenza. Zoals hierboven beschreven is het wel begrijpelijk dat het middel in de huidige richtlijnen wordt geadviseerd, omdat opgenomen patiënten ernstig ziek zijn (zie “Voor- en nadelen van de interventie en de kwaliteit van het bewijs”).
Anderzijds kan een niet-bijdragende behandeling tijdens een grote uitbraak of pandemie de maatschappij op grote kosten jagen. Indien een behandeling in opgenomen patiënten niet effectief zou zijn, maar wel in een andere populatie, zou het behandelen van opgenomen patiënten tekorten (en dus onderbehandeling van andere patiëntgroepen) van het medicament kunnen veroorzaken.
Haalbaarheid en implementatie
Niet relevant.
Rationale/ balans tussen de argumenten voor en tegen de interventie
Aanbeveling antivirale behandeling voor kinderen en volwassenen
Omdat er geen bewijs is vanwege het ontbreken van RCT’s van voldoende grootte bij patiënten die worden opgenomen met influenza heeft de werkgroep argumenten verzameld uit observationele studies, RCT’s met te kleine aantallen en meta-analyses, ook hebben we de data overwogen van de effecten van NAI bij gezonde mensen met influenza welke aangeven dat behandeling de koorts en symptomen sneller kan doen verdwijnen.
Aanbeveling subgroep zwangeren
Zwangere patiënten met influenza hebben een verhoogd risico op complicaties. Alhoewel er geen gerandomiseerde prospectieve placebo gecontroleerde trials zijn verricht die het effect van antivirale therapie bij zwangeren met influenza hebben bestudeerd suggereert de PRIDE-studie (n=2166 zwangere patiënten met influenza) dat zwangere patiënten die werden opgenomen in het ziekenhuis en een vroege behandeling met NAI kregen (n=971) een gunstiger klinisch beloop hadden in vergelijking met zwangere patiënten die een late behandeling kregen met een NAI (Muthuri, 2014).
Onderbouwing
De ernst van een influenza infectie kan variëren van een milde zelflimiterende ziekte tot een levensbedreigende situatie op de intensive care. Op dit moment wordt geadviseerd dat therapie met antivirale middelen overwogen kan worden bij patiënten met influenza of verdenking op influenza welke een hoog risico hebben op complicaties, zoals bewoners van verpleeghuizen, ouderen, kinderen, zwangeren, mensen die opgenomen moeten worden en immuungecompromitteerden. De indicaties voor behandeling met antivirale therapie staan beschreven in de richtlijn Klinisch behandeling van antivirale therapie bij influenza en de Verenso richtlijn Influenzapreventie in verpleeg- en verzorgingshuizen (Verenso, 2004; Cools, 2006). Ook wordt beschreven dat er overwogen kan worden om immuungecompromiteerde patiënten langer te behandelen dan 5 dagen (Engelhard, 2013; Uyeki, 2019). De vraag blijft welke patiënten baat hebben bij de behandeling met antivirale middelen, wanneer antivirale therapie opgestart moet worden, met welk middel en wat de duur is van de therapie.
Volwassenen
IC-opname (cruciaal)
|
Laag GRADE |
Antivirale behandeling van volwassenen met influenza-like illness ≥ 2 dagen na start van symptomen lijkt geen effect te hebben op IC-opname in vergelijking met placebo of standaardzorg.
Bronnen: (De Jong, 2014; Ramirez, 2018; Ison, 2003) |
Opnameduur (cruciaal)
|
Laag GRADE |
Antivirale behandeling van volwassenen met influenza-like illness ≥2 dagen na start van symptomen lijkt geen effect te hebben op opnameduur in het ziekenhuis in vergelijking met placebo of standaardzorg.
Bronnen: (De Jong, 2014; Ramirez, 2018; Ison, 2003) |
Mortaliteit (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk wat het effect is van antivirale behandeling van volwassenen met influenza-like illness op mortaliteit in vergelijking met placebo of standaardzorg.
Bronnen: (De Jong, 2014; Ramirez, 2018; Ison, 2003; Muthuri, 2014) |
Adverse events (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk wat het effect is van antivirale behandeling van volwassenen met influenza-like illness op adverse events in vergelijking met placebo of standaardzorg.
Bronnen: (De Jong, 2014; Ramirez, 2018; Ison, 2003) |
Kinderen
IC-opname (cruciaal)
|
- GRADE |
Het effect op IC-opname van antivirale behandeling van kinderen met influenza-like illness in vergelijking met placebo is onbekend. Geen van de studies rapporteert deze uitkomstmaat. |
Opnameduur (cruciaal)
|
Laag GRADE |
Antivirale behandeling van kinderen (tot en met 9 jaar) met influenza-like illness lijkt geen effect te hebben op opnameduur in het ziekenhuis in vergelijking met placebo.
Bronnen: (Dawood, 2016) |
Mortaliteit (cruciaal)
|
Laag GRADE |
Antivirale behandeling van kinderen (< 16 jaar) met influenza-like illness lijkt geen effect te hebben op mortaliteit in vergelijking met placebo.
Bronnen: (Muthuri, 2014) |
Adverse events (belangrijk)
|
Laag GRADE |
Antivirale behandeling van kinderen (tot en met 9 jaar) met influenza-like illness lijkt geen effect te hebben op adverse events in vergelijking met placebo.
Bronnen: (Dawood, 2016) |
Zwangeren
Mortaliteit (cruciaal)
|
Laag GRADE |
Antivirale behandeling van opgenomen zwangeren leidt mogelijk tot minder mortaliteit in vergelijking met placebo of standaardzorg.
Bronnen: (Muthuri, 2014) |
|
- GRADE |
De effectiviteit van behandeling met antivirale medicatie bij opgenomen zwangeren met influenza-like illness op IC-opname, opnameduur en adverse events, is onbekend. Geen van de studies onderzocht deze uitkomstmaten bij zwangeren. |
Ouderen
|
- GRADE |
De effectiviteit van behandeling met antivirale medicatie bij opgenomen ouderen met influenza-like illness op IC-opname, opnameduur, mortaliteit en adverse events, is onbekend. Geen van de studies onderzocht deze uitkomstmaten bij ouderen. |
Mensen met een chronische ziekte (comorbiditeit)
|
- GRADE |
De effectiviteit van behandeling met antivirale medicatie bij opgenomen patiënten met comorbiditeit en influenza-like illness op IC-opname, opnameduur, mortaliteit en adverse events, is onbekend. Geen van de studies onderzocht deze uitkomstmaten bij opgenomen patiënten met comorbiditeit. |
Mensen met een verminderde afweer en/of gebruik van immunosuppressiva/ immunotherapie
|
- GRADE |
De effectiviteit van behandeling met antivirale medicatie bij opgenomen patiënten met een verminderde afweer en/of immunosuppressiva of immunotherapie gebruiken en influenza-like illness op IC-opname, opnameduur, mortaliteit en adverse events, is onbekend. Geen van de studies onderzocht deze uitkomstmaten bij patiënten met een verminderde afweer en/of immunosuppressiva of immunotherapie. |
Beschrijving studies
Ramirez (2018) verrichtte een gerandomiseerde ongeblindeerde studie naar de effectiviteit van een behandeling met oseltamivir bij volwassenen opgenomen in 9 ziekenhuizen met symptomen die overeenkomen met acute onderste luchtweginfectie (acute bronchitis, acute exacerbatie van chronische bronchitis, en CAP). Deze patiënten hadden mediaan al 5 dagen symptomen voordat zij zich in het ziekenhuis presenteerden. In totaal werden 1107 patiënten gediagnosticeerd met een luchtweginfectie gerandomiseerd naar de groep die binnen 24 uur na opname behandeld werd met 2-daags 75 mg voor 7 dagen oseltamivir (N=551) of de controlegroep die standaard zorg kreeg (N=556). In de interventiegroep was de gemiddelde leeftijd 62 (SD 18) jaar en was 55% man. In de controlegroep was de gemiddelde leeftijd 62 (SD 20) jaar en was 56% man. Tweederde van de patiënten was gevaccineerd tegen influenza. De resultaten werden beschreven voor een intention-to-treat sample (ITT), dit was de totale gerandomiseerde groep en een per protocol sample (ITTI) waarbij influenza was bevestigd met laboratoria testen (respectievelijk 45 zonder oseltamivir en 29 met oseltamivir behandeld). Beperking daarbij is dat in de analyse van deze laatste groep alleen de patiënten werden meegenomen die de gerandomiseerde behandeling hadden ondergaan en geen dosis oseltamivir hadden gemist.
Dawood (2016) verrichtte een dubbelblinde gerandomiseerde studie naar de veiligheid van vroege oseltamivir behandeling van kinderen van 0 tot 9 jaar opgenomen minder dan 7 dagen na ontstaan van symptomen in 5 ziekenhuizen in Panama en El Salvador met influenza. Deelnemers kregen in totaal 10 doses, elke 12 uur 1 dosis, oseltamivir (dosis 0 tot 11 maanden: 3 mg/kg; ≥ 12 maanden: 30 mg/dosis voor kinderen ≤ 15 kg, 45 mg voor kinderen 15 tot 23 kg, 60 mg voor kinderen 23 tot 40 kg en 75 mg voor kinderen ≥ 40 kg) of placebo, waarbij de eerste dosis 2 uur na inclusie gegeven werd. In totaal werden er 683 kinderen gerandomiseerd (ITT safety groep), waarvan 345 kinderen naar de oseltamivir groep (50% was jonger dan 1 jaar) en 343 kinderen naar de placebo groep (53% was jonger dan 1 jaar). Kinderen hadden mediaan 63 uur symptomen voordat zij oseltamivir kregen en 67 uur voordat zij placebo kregen. Kinderen die uiteindelijk laboratorium-bevestigde influenza hadden werden gedefinieerd als de Intention tot Treat efficacy (ITTI) groep. De primaire uitkomsten waren opnameduur en tijd tot oplossen hypoxie en verhoogde ademhaling bij kinderen in de ITTI groep. Als secundaire uitkomstmaat werden adverse events gerapporteerd.
De Jong (2014) verrichtte een dubbelblinde gerandomiseerde studie in 323 ziekenhuizen van 21 landen naar de effectiviteit en veiligheid van intraveneuze peramivir bij volwassenen en kinderen met influenza. In totaal werden 405 patiënten gerandomiseerd en 398 opgenomen in de intention-to-treat infected (ITTI) populatie. Vervolgens werd een onderverdeling gemaakt in 2 groepen, 1 groep die een neuraminidase inhibitor (NAI) ontving als onderdeel van de standaard zorg (99% kreeg oseltamivir) en 1 groep die geen NAI ontving als onderdeel van de standaard zorg. Deze laatste groep was de populatie voor de primaire effectiviteit analyse (N=121). In deze groep werden 78 patiënten gerandomiseerd naar de groep die een behandeling onderging met dagelijks eenmalig intraveneuze peramivir (dosis 10 mg/kg, tot maximaal 600 mg/dag) voor 5 dagen boven op de standaard influenza behandeling van het desbetreffende ziekenhuis. Er werden 43 patiënten gerandomiseerd naar de groep die alleen de standaard influenza behandeling ontving. In de peramivir groep was de gemiddelde leeftijd 44 (range 19 tot 86) jaar en was 53% man. In de controlegroep was de gemiddelde leeftijd 40 (range 13 tot 72) jaar en was 53% man. Over de 4 verschillende groepen (met of zonder NAI in de SoC en met of zonder peramivir) hadden 52-74% van de patiënten symptomen korter dan 48 uur bij randomisatie.
Ison (2003) verrichtte een dubbelblinde gerandomiseerde studie in 7 Amerikaanse ziekenhuizen naar de effectiviteit en veiligheid van vernevelde zanamivir of placebo en altijd rimantadine bij volwassenen opgenomen met een onderste luchtweginfectie door een bevestigde influenza A of B virus infectie met symptomen van 4 dagen of minder. Wat de uiteindelijke duur van symptomen was bij start van therapie, is niet gegeven. In totaal werden 41 geïnfecteerde patiënten opgenomen in de trial, waarvan er 2 negatief bleken voor influenza op basis van kweek en serologie en werden geëxcludeerd uit de effectiviteit analyse. Er werden 20 patiënten gerandomiseerd naar de zanamivir groep (mediaan leeftijd 67; range 24 tot 93; 90% man) en 21 patiënten naar de placebo groep (mediaan leeftijd 61; range 22 tot 80; 67% man). De follow-up duur was 28 dagen. Primaire uitkomstmaat was de afwezigheid van faryngeale influenza virale uitscheiding op dag 3 van de behandeling. Secundaire uitkomstmaten waren duur van de koorts, hospitalisatie en frequentie van complicaties.
Muthuri (2014) verrichtte een meta-analyse van individuele patiënt data om de associatie tussen NAI en mortaliteit bij patiënten die opgenomen werden in het ziekenhuis met een pandemische influenza A HiN1pdm09 virusinfectie. Er werd een systematische search gedaan in 11 databases om potentiële databronnen op te sporen in april 2012 voor observationele studies en RCT’s uitgevoerd tussen maart-april 2009 en augustus 2010 (einde van pandemie). De primaire uitkomstmaat was mortaliteit, gedefinieerd als overlijden binnen 30 dagen van ziekte. Gebruik van NAI (onafhankelijk van timing) werd vergeleken met geen gebruik van NAI, en als vroege behandeling (start ≤ 2 dagen na eerste symptomen) versus late behandeling (start > 2 dagen na eerste symptomen). Er werden propensity scores berekend voor de waarschijnlijkheid van NAI behandeling voor elke patiënt binnen elke individuele dataset door middel van multivariabele logistische regressie analyse voor binaire variabelen en gegeneraliseerde propensity score schattingen voor de continue tijd tot behandeling variabele. Covariaten waren leeftijd, geslacht, comorbiditeit (ja of nee), een proxy-indicator van ernstige ziekte (ja of nee). Subgroepanalyses werden gedaan voor onder andere kinderen (onder de 16 jaar), zwangere vrouwen en IC-patiënten. De gegevens van in totaal 29.234 patiënten uit 78 verschillende centra kon in de analyse worden geïncludeerd.
Resultaten
Volwassenen
1. IC-opname (cruciaal)
In de studie van Ramirez (2018) was er in de ITT sample geen verschil in IC-opname tussen de groepen die werden gerandomiseerd naar oseltamivir (N=14/551) of standaard zorg (N=22/556). Het RR op IC-opname was 0,65 (95% BI 0,34 tot 1,26) in het voordeel van interventie, maar dit verschil was niet statistisch significant. In de ITTI sample (met laboratorium bevestigde influenza) was er ook geen verschil in IC-opname tussen de groepen die werden gerandomiseerd naar oseltamivir (N=1/29) of standaard zorg (N=1/45). Het RR op IC-opname was in de ITTI sample 1,55 (95% BI 0,10 tot 23,85).
In de RCT van De Jong (2014) was er bij patiënten met een bevestigde diagnose voor influenza geen verschil in IC-opname tussen de groepen die werden gerandomiseerd naar intraveneuze peramivir (N=0/78) of standaard zorg zonder NAI (N=1/43). Het RR op IC-opname was 0,19 (95% BI 0,01 tot 4,56) in het voordeel van de interventie, maar dit verschil was niet statistisch significant.
IC-opname werd niet gerapporteerd in de studie van Ison (2003).
2. Opnameduur (cruciaal)
In de studie van Ramirez (2018) was er in de ITT sample geen verschil (P=0,52) in opnameduur tussen de groepen die werden gerandomiseerd naar oseltamivir (mediaan 4 dagen; IQR 3) of standaard zorg (mediaan 4 dagen; IQR 4). In de ITTI sample (met laboratorium bevestigde influenza) was er ook geen verschil (P=0,35) in opnameduur tussen de twee groepen, met een mediane opnameduur van 4 dagen (IQR 4) in de oseltamivir groep en een mediane opnameduur van 4 dagen (IQR 5) in de controlegroep.
In de studie van Ison (2003) was er geen verschil (P=0,52) in opnameduur tussen de groepen die werden gerandomiseerd naar zanamivir (gemiddelde 4,7 dagen; SD 2,3) of placebo (gemiddelde 5,2 dagen; SD 2,3).
Opnameduur werd niet gerapporteerd in de studie van De Jong (2014).
3. Mortaliteit (cruciaal)
In de studie van Ramirez (2018) was er in de ITT sample geen verschil in mortaliteit na 1 jaar tussen de groepen die werden gerandomiseerd naar oseltamivir (N=84/551) of standaard zorg (N=94/556). Het RR op mortaliteit was 0,90 (95% BI 0,69 tot 1,18) in het voordeel van interventie, maar dit verschil was niet statistisch significant. In de ITTI sample (met laboratorium bevestigde influenza) was er ook geen verschil in mortaliteit na 1 jaar tussen de twee groepen (RR 0,52; 95% BI 0,11 tot 2,29).
In de RCT van De Jong (2014) was er bij patiënten met een bevestigde diagnose voor influenza geen verschil in mortaliteit na 28 dagen tussen de groepen die werden gerandomiseerd naar intraveneuze peramivir (N=0/78) of standaard zorg (N=2/43). Het RR op mortaliteit was 0,11 (95% BI 0,006 tot 2,27) in het voordeel van de interventie, maar dit verschil was niet statistisch significant.
In de RCT van Ison (2003) was er bij patiënten met influenza geen verschil in mortaliteit na 28 dagen tussen de groepen die werden gerandomiseerd naar zanamivir (N=2/20) en de placebo (N=1/21) groep. Het RR op mortaliteit was 2,10 (95% BI 0,21 tot 21,39) in het voordeel van de placebogroep, maar dit verschil was niet statistisch significant.
Een meta-analyse met de resultaten van deze drie studies liet ook geen verschil zien tussen de groepen, met een RR van 0,58 (95% BI 0,15 tot 2,21) (figuur 1), waarbij uiteindelijk 3% in de antivirale arm overleed versus 8% in de arm met placebo of standaardzorg.
Figuur 1 Uitkomstmaat mortaliteit vergelijking antivirale behandeling versus placebo of standaard zorg
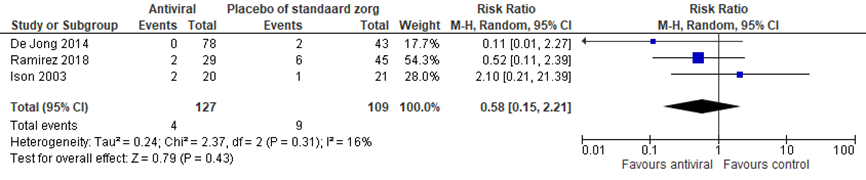
In de gepoolde meta-analyse van Muthuri (2014) op individueel patiënten niveau met 19.816 volwassen patiënten (≥ 16 jaar) met bevestigde of gediagnosticeerde influenza was de waarschijnlijkheid van mortaliteit bij patiënten die behandeld werden met NAI lager vergeleken met geen NAI (OR 0,75; 95% BI 0,64 tot 0,87). Bij een vergelijking tussen vroege NAI behandeling (≤ 2 dagen na eerste symptomen) en late behandeling (na 2 tot 5 dagen) hadden volwassen patiënten met een vroege behandeling een betere overleving vergeleken met late behandeling (OR 0,45; 95% BI 0,38 tot 0,54). Wanneer langer dan 2 dagen na start van de symptomen gestart werd met de behandeling was er geen verschil in overleving vergeleken met de groep zonder behandeling (OR 1,01; 95% BI 0,76 tot 1,33). In de subgroep met alleen laboratorium bevestigde influenza (alle leeftijden) was de waarschijnlijkheid van mortaliteit lager bij patiënten die behandeld werden met NAI vergeleken met geen NAI (OR 0,82; 95% BI 0,70 tot 0,95), en ook lager bij patiënten die vroeg behandeld werden met NAI vergeleken met late NAI (OR 0,48; 95% BI 0,38 tot 0,54). Ook in deze groep was er geen verschil in overleving als er later dan 2 dagen na start van de symptomen met behandeling werd gestart vergeleken met geen behandeling (OR 1,17; 95% BI 0,92 tot 1,51).
In alle analyses werd gecorrigeerd voor behandeling propensity (bij quintiel), corticosteroïdengebruik en antibioticagebruik.
3.1 Subgroep zwangere vrouwen: uitkomst mortaliteit
Muthuri (2014) verrichtte een subgroep analyse op individueel patiënten met 2166 zwangeren met bevestigde of gediagnosticeerde influenza. De waarschijnlijkheid van mortaliteit bij zwangeren die behandeld werden met NAI was lager vergeleken met zwangeren die geen NAI ontvingen (OR 0,46; 95% BI 0,23 tot 0,89; P=0,02).
4. Adverse events (belangrijk)
In de studie van Ramirez (2018) was er in de ITT sample geen verschil in adverse events, gedefinieerd als heropname binnen 30 dagen, tussen de groepen die werden gerandomiseerd naar oseltamivir (N=75/551) of standaard zorg (N=76/556). Het RR op adverse events was 0,996 (95% BI 0,74 tot 1,34) in het voordeel van interventie, maar dit verschil was niet statistisch significant. In de ITTI sample (met laboratorium bevestigde influenza) was er ook geen verschil in adverse events tussen de twee groepen (RR 0,93; 95% BI 0,21 tot 3,60).
In de RCT van De Jong (2014) was er bij patiënten met een bevestigde diagnose voor influenza geen verschil in adverse event, gedefinieerd als incidentie van de complicaties otitis, sinusitis, bronchitis of pneumonia, tussen de groepen die werden gerandomiseerd naar intraveneuze peramivir (N=15/78) of standaard zorg (N=9/43). Het RR op adverse events was 0,88 (95% BI 0,42 tot 1,83) in het voordeel van de interventie, maar dit verschil was niet statistisch significant.
In de RCT van Ison (2003) was er bij patiënten met influenza geen verschil in serious adverse events tussen de groepen die werden gerandomiseerd naar zanamivir (N=1/20) en de placebo (N=3/21) groep. Het RR op adverse events was 0,35 (95% BI 0,04 tot 3,09) in het voordeel van de zanamivir groep, maar dit verschil was niet statistisch significant.
Gezien het verschil in definities voor adverse events werd er geen meta-analyse uitgevoerd.
Kinderen
1. IC-opname (cruciaal)
IC-opname werd niet gerapporteerd in de studie van Dawood (2016).
2. Opnameduur (cruciaal)
In de studie van Dawood (2016) was er in de ITTI sample geen verschil (P=0,22) in opnameduur tussen de groepen die werden gerandomiseerd naar oseltamivir (mediaan 3 dagen; IQR 2 tot 4) of placebo (mediaan 5 dagen; IQR 3 tot 7).
3. Mortaliteit (cruciaal)
Mortaliteit werd niet gerapporteerd in de studie van Dawood (2016).
In de gepoolde meta-analyse van Muthuri (2014) op individueel patiënten niveau met 9.218 kinderen (< 16 jaar) met bevestigde of gediagnosticeerde influenza was er geen verschil in de waarschijnlijkheid van mortaliteit bij kinderen die behandeld werden met NAI vergeleken met geen NAI (OR 0,82 (95% BI 0,58 tot 1,17). Ook in de vergelijking van vroege versus late behandeling met NAI was er geen verschil in mortaliteit bij de kinderen (OR 0,67; 95% BI 0,44 tot 1,03).
4. Adverse events (belangrijk)
In de RCT van Dawood (2016) was er in de groep kinderen die opgenomen werden met influenza-like illness geen verschil in adverse events tussen de groepen gerandomiseerd naar oseltamivir of placebo. Adverse events werden gedefinieerd als een ongunstig effect opgemerkt door een arts tijdens ziekenhuisopname of supervisoren na ontslag tijdens deelname aan het onderzoek. In de leeftijdsgroep jonger dan 1 jaar (N=351) waren er 24 (van de 170) adverse events met een ernst minder dan graad 3 in de oseltamivir groep en 39 (van de 181) met een ernst minder dan graad 3 in de in de placebogroep (RR 0,65; 95% BI 0,41 tot 1,04). In de leeftijdsgroep van 1 jaar of ouder waren deze aantallen respectievelijk 34 (van de 171) en 32 (van de 161) (RR 0,99; 95% BI 0,65 tot 1,53). In de leeftijdsgroep jonger dan 1 jaar waren er 6 (van de 170) adverse events met een ernst van graad 3 of meer in de oseltamivir groep en 8 (van de 181) met een ernst van graad 3 of meer in de in de placebogroep (RR 0,80; 95% BI 0,28 tot 2,25). In de leeftijdsgroep van 1 jaar of ouder waren deze aantallen respectievelijk 1 (van de 171) en 4 (van de 161) (RR 0,24; 95% BI 0,27 tot 2,08).
Bewijskracht van de literatuur
De bewijskracht voor alle beschreven uitkomstmaten is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog.
Volwassenen
Voor de uitkomstmaat IC-opname bij volwassenen is met 2 niveaus afgewaardeerd tot laag gezien beperkingen in de onderzoeksopzet (risk of bias vanwege het niet houden aan intention-to treat-principe in ITTI populatie); en het geringe aantal patiënten en brede betrouwbaarheidsinterval (imprecisie).
Voor de uitkomstmaat opnameduur bij volwassenen is met 2 niveaus afgewaardeerd tot laag gezien beperkingen in de onderzoeksopzet (risk of bias vanwege het niet houden aan intention-to-treat principe in ITTI populatie); en het geringe aantal patiënten en brede betrouwbaarheidsinterval (imprecisie).
Voor de uitkomstmaat mortaliteit bij volwassenen is met 3 niveaus afgewaardeerd tot zeer laag gezien beperkingen in de onderzoeksopzet (risk of bias vanwege het niet houden aan intention-to-treat principe in ITTI populatie); inconsistentie in resultaten en het geringe aantal patiënten en brede betrouwbaarheidsinterval (imprecisie).
Voor de uitkomstmaat adverse events bij volwassenen is met 3 niveaus afgewaardeerd tot zeer laag gezien beperkingen in de onderzoeksopzet (risk of bias vanwege het niet houden aan intention-to-treat principe in ITTI populatie); verschillende uitkomstmaten (indirectheid) en het geringe aantal patiënten en brede betrouwbaarheidsinterval (imprecisie).
Kinderen
Vanwege het ontbreken van die studies die de uitkomsten IC-opname rapporteren bij kinderen is de uitkomst IC-opname niet beoordeeld.
Voor de uitkomstmaat opnameduur is met 2 niveaus afgewaardeerd tot laag gezien het geringe aantal patiënten (imprecisie: -2).
De bewijskracht voor de uitkomstmaat mortaliteit is gebaseerd op observationeel onderzoek en start derhalve laag. De studiepopulatie was van voldoende grootte en er werd gecorrigeerd voor eventueel verstorende variabelen. Derhalve blijft de bewijskracht op laag.
De bewijskracht voor de uitkomstmaat adverse events is met 2 niveaus verlaagd gezien het geringe aantal patiënten (imprecisie: -2).
Zwangere vrouwen
De bewijskracht voor de uitkomstmaat mortaliteit is gebaseerd op observationeel onderzoek en start derhalve laag. De studiepopulatie was van voldoende grootte en er werd gecorrigeerd voor eventueel verstorende variabelen. Derhalve blijft de bewijskracht op laag.
Vanwege het ontbreken van studies die de uitkomsten IC-opname, opnameduur en adverse events rapporteren bij zwangere vrouwen is de bewijskracht van deze uitkomsten niet beoordeeld.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat is de effectiviteit van antivirale behandeling bij kinderen en volwassenen die opgenomen worden voor verdenking op influenza-like-illness?
P: kinderen en volwassenen die opgenomen worden met verdenking op influenza-like-illness;
I: antivirale behandeling;
C: geen behandeling of placebo, paracetamol;
O: IC-opname, opnameduur, mortaliteit, adverse events van de behandeling.
Relevante uitkomstmaten
De werkgroep achtte IC-opname, opnameduur en mortaliteit 30 dagen, voor de besluitvorming cruciale uitkomstmaten; en adverse events een voor de besluitvorming belangrijke uitkomstmaat.
De werkgroep definieerde niet a priori de genoemde uitkomstmaten van de PICO, maar hanteerde de in de studies gebruikte definities.
De werkgroep heeft ervoor gekozen geen vooraf gestelde afkapwaarden voor klinische (patiënt) relevante verschillen te definiëren, maar statistisch significante verschillen per uitkomstmaat te rapporteren. Hiervoor is gekozen, omdat de grens voor klinische relevantie verschillend zal zijn voor bepaalde risicogroepen en deze grenzen waarschijnlijk in overleg met de patiënt gekozen moeten worden. Voor de conclusies wordt daarom uitgegaan van statistische significantie voor de effectiviteit van antivirale middelen.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID), Embase (via Embase.com) en de Cochrane Library (via Wiley) is op 3 mei 2019 met relevante zoektermen gezocht naar systematische reviews en gerandomiseerd onderzoek vanaf 1996 voor de drie uitgangsvragen over behandeling met antivirale middelen. Een separate search is gedaan naar specifieke risicogroepen voor de behandeling van influenza. Beide zoekstrategieën worden in de module 'Behandeling influenza - geen opname' beschreven. De algemene literatuurzoekactie leverde 517 treffers op en de specifieke zoekactie voor risicogroepen 755 treffers. Studies werden geselecteerd op grond van de volgende selectiecriteria: vergelijkend (observationeel of gerandomiseerd) onderzoek naar de antivirale behandeling van patiënten die in het ziekenhuis waren opgenomen. Op basis van titel en abstract werden uit de algemene literatuurzoekactie voor het beantwoorden van de uitgangsvraag over opgenomen patiënten in eerste instantie 33 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werd voor de algemene zoekvraag 29 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording) en vier studies definitief geselecteerd. Voor de specifieke risicogroepen werd aanvullend één studie definitief geselecteerd. De aanvullende literatuurzoekactie naar baloxavir (zie module 4) leverde geen extra resultaten op.
Resultaten
Vier RCT’s en één observationele studie zijn opgenomen in de literatuuranalyse. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk-of-biastabellen.
- Cools H, Kroes L. Update 2006 van de NVVA richtlijn influenzapreventie in verpleeghuizen en verzorgingshuizen Tijdschrift voor Verpleeghuisgeneeskunde 2006;31:148-151 doi10.1007/BF04075176
- Dawood, F. S., Jara, J., Gonzalez, R., Castillo, J. M., De León, T., Estripeaut, D.,... & Lawson, A. M. (2016). A randomized, double-blind, placebo-controlled trial evaluating the safety of early oseltamivir treatment among children 0–9 years of age hospitalized with influenza in El Salvador and Panama. Antiviral research, 133, 85-94.
- De Jong, M. D., Ison, M. G., Monto, A. S., Metev, H., Clark, C., O'Neil, B.,... & Sheridan, W. P. (2014). Evaluation of intravenous peramivir for treatment of influenza in hospitalized patients. Clinical Infectious Diseases, 59(12), e172-e185.
- Engelhard, D., Mohty, B., De La Camara, R., Cordonnier, C., & Ljungman, P. (2013). European guidelines for prevention and management of influenza in hematopoietic stem cell transplantation and leukemia patients: summary of ECIL‐4 (2011), on behalf of ECIL, a joint venture of EBMT, EORTC, ICHS, and ELN. Transplant Infectious Disease, 15(3), 219-232.
- Ison, M. G., Gnann Jr, J. W., Nagy-Agren, S., Treannor, J., Paya, C., Steigbigel, R.,... & NIAID Collaborative Antiviral Study Group. (2003). Safety and efficacy of nebulized zanamivir in hospitalized patients with serious influenza. Antiviral therapy, 8(3), 183-190.
- Muthuri, S. G., Venkatesan, S., Myles, P. R., Leonardi-Bee, J., Al Khuwaitir, T. S., Al Mamun, A.,... & Beovic, B. (2014). Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: a meta-analysis of individual participant data. The lancet Respiratory medicine, 2(5), 395-404.
- Ramirez, J., Peyrani, P., Wiemken, T., Chaves, S. S., & Fry, A. M. (2018). A randomized study evaluating the effectiveness of oseltamivir initiated at the time of hospital admission in adults hospitalized with influenza-associated lower respiratory tract infections. Clinical Infectious Diseases, 67(5), 736-742., course, and prognostic indicators. Annals of the rheumatic diseases, 64(7), 1056-1061.
- Uyeki, T. M., Bernstein, H. H., Bradley, J. S., Englund, J. A., File Jr, T. M., Fry, A. M.,... & Ison, M. G. (2019). Clinical practice guidelines by the Infectious Diseases Society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenza. Clinical Infectious Diseases, 68(6), e1-e47.
- Verenso (2004). Richtlijn Influenzapreventie. https://www.verenso.nl/_asset/_public/Richtlijnen_kwaliteit/richtlijnen/database/RL-Influenza-2004.pdf
- Wiersinga, W. J., Bonten, M. J., Boersma, W. G., Jonkers, R. E., Aleva, R. M., Kullberg, B. J.,... & Sachs, A. P. E. (2018). Management of community-acquired pneumonia in adults: 2016 guideline update from the Dutch Working Party on Antibiotic Policy (SWAB) and Dutch Association of Chest Physicians (NVALT). Netherlands journal of medicine,
Evidence table for intervention studies (randomized controlled trials and non-randomized observational studies (cohort studies, case-control studies, case series))1
This table is also suitable for diagnostic studies (screening studies) that compare the effectiveness of two or more tests. This only applies if the test is included as part of a test-and-treat strategy - otherwise the evidence table for studies of diagnostic test accuracy should be used
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3 |
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Ramirez, 2018 |
Type of study: RCT, unblinded
Setting and country: multicentre, USA
Funding and conflicts of interest: ‘’This work was supported by an award to the University of Louisville by the CDC (cooperative agreement IP000420-01). Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed. |
Inclusion criteria: Adult patients hospitalized at 1 of the 9 hospitals in Louisville, Kentucky, were eligible to participate if they -had symptoms consistent with acute LRTIs over 3 consecutive influenza seasons (2010–2011, 2011–2012, and 2012–2013). -did not have an order for oseltamivir or zanamivir from the admitting clinician written on their admission orders, had no known allergies to oseltamivir, and were able to give informed consent or had a proxy to give consent within 48 hours of hospitalization
Exclusion criteria: pregnant or incarcerated.
N total at baseline: Intervention ITT: 551 Intervention ITTI: 41 Control ITT: 556 Control ITTI 55
Important prognostic factors: age ± SD: I (ITT): 62±18 yrs C (ITT): 62±20 yrs
Sex: I (ITT): 55% M C (ITT): 56% M
Nursing home resident I (ITT): 5% C (ITT): 5%
Groups comparable at baseline? yes |
Standard of care plus oral oseltamivir administered as early as possible but within 24 hours of enrollment. Oseltamivir was dosed at 75 mg twice daily for 7 days. Seven days was chosen to standardize treatment time across all levels of severity. The oseltamivir dose was adjusted in patients with renal insufficiency according to the package insert. |
Standard of care: Empiric antibiotic therapy was given according to standard clinical practice. Patients randomized to standard of care where a clinician later suspected or confirmed influenza virus infection could be started on oseltamivir at the discretion of the clinician caring for the patient. |
Length of follow-up: 1 year
Loss-to-follow-up, N (%): Intervention, ITTI: 4 (9.8) Reasons: voluntary withdrawn
Control, ITTI: 1 (1.8) Reasons: voluntary withdrawn
|
1. ICU-admission I (ITT): 14/551 C (ITT): 22/556 RR (ITT): 0.65 (0.34 to 1.26)
I (ITTI): 1/29 C (ITTI): 1/45 RR (ITTI): 1.55 (0.10 to 23.85)
2. Length of hospital stay, median days (IQR) I (ITT): 4 (3) C (ITT): 4 (4) P=0.52
I (ITTI): 3 (4) C (ITTI): 4 (5) P=0.35
3. Mortality (1 year) I (ITT): 84/551 C (ITT): 94/556 RR (ITT): 0.90 (0.69 to 1.18)
I (ITTI): 2/29 C (ITTI): 6/45 RR (ITTI): 0.52 (0.11 to 2.39)
4. Adverse events Defined as rehospitalization within 30 days I (ITT): 75/551 C (ITT): 76/556 RR (ITT): 0.996 (0.74 to 1.34)
I (ITTI): 3/29 C (ITTI): 5/45 RR (ITTI): 0.93 (0.21 to 3.60)
|
ITT: Intention-to treat ITTI: Intention-to treat infected LRTI: lower respiratory tract infection
Acute LRTI was defined as the presence of 2 respiratory signs or symptoms and 1 sign of acute infection at the time of admission. Respiratory signs and symptoms included new or increased cough, change in sputum production (color or quantity), evidence for reduced oxygenation (O2 saturation <90% breathing on room air or, for patients on home oxygen therapy, a 1-L increase in their oxygen requirement), new onset shortness of breath, rapid respiratory rate (≥24 breaths per minute), or a record of new auscultatory findings (rales, rhonchi, wheezing). Signs or symptoms of acute infection included fever (temperature >38°C), hypothermia (temperature <35.6°C), subjective fever, or a report of chills or myalgias; changes in white blood cells (leukocytosis, leukopenia, abnormal differential (left shift, change in lymphocyte number or proportion)); or altered mental status. This was a composite definition of standard criteria used for diagnosis of acute bronchitis, acute exacerbation of chronic bronchitis, and CAP. |
|
Dawood, 2016 |
Type of study: RCT, double blind
Setting and country: tertiary care hospitals, Panama (N=3) and El Salvador (N=2)
Funding and conflicts of interest: This study was funded by the US Centers for Disease Control and Prevention through Cooperative Agreement number RFA-GH-13-00102CONT14 with the Universidad del Valle de Guatemala. The authors have no conflicts of interest to declare. |
Inclusion criteria: children had to be aged ≤9 years and hospitalized <7 days after symptom onset with symptoms meeting a modified version of the World Health Organization criteria for severe acute respiratory infection (cough or sore throat plus age-specific tachypnea
Exclusion criteria: history of prematurity (born at <37 weeks gestation) or birth weight <2,500 grams in children aged <3 months; history of renal dysfunction; history of gastrointestinal resection that might hinder medication absorption; concomitant severe vomiting (defined as >3 episodes in the past 24 hours); prior serious adverse reaction to oseltamivir; receipt of oseltamivir during the 5 days prior to presentation at the admitting hospital; and previous enrollment in this study during a hospitalization that ended less than 14 days prior to the current admission.
N total at baseline: Intervention ITT: 345 Intervention ITTI: 19 Control ITT: 343 Control ITTI: 11
Important prognostic factors: age < 1 yr, 1-2 yr, 3-4 yr (%): I (ITT): 50, 39, 6 I (ITTI): 47, 26, 11 C (ITT): 53, 36, 6 C (ITTI): 36, 55, 0
Sex not reported
Groups comparable at baseline? yes |
Active drug (oseltamivir) every 12 h for 10 doses. The first dose was given within 2 h of enrollment. For children aged 0e11 months, study drug was dosed at 3 mg/kg/dose. For children aged ≥12 months, study drug was dosed based on standard unit dosing: 30 mg/dose for children ≤15 kg, 45 mg for children >15-23 kg, 60 mg for children >23-40 kg, and 75 mg for children >40 kg. Oseltamivir was prepared according to package insert guidelines for emergency compounding of an oral suspension from 75 mg TAMIFLU® capsules modified to yield a concentration of 15mg/5 mL.
|
Placebo every 12 h for 10 doses. The first dose was given within 2 h of enrollment. Placebo was prepared using corn starch and suspension vehicle to produce a suspension with the appearance and taste of the active suspension
|
Length of follow-up: 7-9 days after discharge
Incomplete outcome data: Intervention: 36 (10%) Reasons: lost to follow up (N=25), did not meet eligibility criteria (N=3), voluntary withdrawal (N=5), transferred (N=2), death (N=1)
Control: 29 (8%) N (%) lost to follow up (N=20), did not meet eligibility criteria (N=2), voluntary withdrawal (N=3), transferred (N=4)
|
1. ICU-admission Not reported
2. Length of hospital stay, median days (IQR) I (ITTI): 3 (2-4) C (ITTI): 5 (3-7) P=0.22
3. Mortality Not reported
4. Adverse events Defined as unfavourable effect noted by a physician during hospitalization or guardians after discharge during trial participation
Subgroup age < 1 year (N=351) - Any event <grade 3 severity I (ITT): 24/170 C (ITT): 39/181 RR (ITT): 0.65 (0.41 to 1.04) - Any event ≥grade 3 severity I (ITT): 6/170 C (ITT): 8/181 RR (ITT): 0.80 (0.28 to 2.25)
Subgroup age ≥ 1 year (N=332) - Any event <grade 3 severity I (ITT): 34/171 C (ITT): 32/161 RR (ITT): 0.99 (0.65 to 1.53) - Any event ≥grade 3 severity I (ITT): 1/171 C (ITT): 4/161 RR (ITT): 0.24 (0.27 to 2.08) |
Conducted during 2012-2013 Study sites included three hospitals in Panama and two in El Salvador that provided specialized pediatric care with intensive care units and capacity to provide mechanical ventilation.
The study was terminated early primarily based on lower than anticipated rates of accrual of participants with laboratory confirmed influenza and additionally due to the observation at some sites that length of hospitalization may at times have been influenced by factors independent of the children's clinical courses such as social reasons and variation in physician practices. |
|
De Jong, 2014 |
Type of study: Double blind RCT
Setting and country: multicentre, multi country
Funding and conflicts of interest: This work was supported in whole or in part by the Biomedical Advanced Research and Development Authority, Office of the Assistant Secretary for Preparedness and Response, US Department of Health and Human Services, Washington DC (contract HHSO100200 700032C). Additional funding was provided by BioCryst Pharmaceuticals.
Potential conflicts of interest. M. D. d. J. has received research support from Crucell, is a consultant to Crucell and AIMM Therapeutics, and is a member of Data and Safety Monitoring Board for studies sponsored by GlaxoSmithKline, for which fees were paid to the Academic Medical Center. M. G. I. has received research support, paid to orthwestern University, from BioCryst, Cellex, Chimerix, Crucell, GlaxoSmithKline, and NexBio; has been a paid consultant for Alios, Abbott, Crucell, Genentech/Roche, and Vistera and an unpaid consultant for Adamas, BioCryst, Biota, Cellex, Clarassance, GlaxoSmithKline, Gen-MarkDx, Romark, Toyama/MediVector, NexBio, Theraclone, and Vertex; and has been a paid member of the Data Monitoring Boards for studies sponsored by Biota and NexBio. A. M. has been a paid consultant for BioCryst and Roche. H. M. has received research support from BioCryst. C. C. has received research support from BioCryst, GlaxoSmithKline, and NexBio. B. O has received research support from BioCryst, has been paid a consultant to Zoll Circulation, and has received grant support from the DMC Foundation and |
Inclusion criteria: -male or nonpregnant female (pregnant women were eligible in the USA) adults, adolescents, or children (aged 6–11 years) with positive influenza rapid antigen test results.
Exclusion criteria: hospitalization >24 hours at screening, prior NAI or adamantane treatment, or confirmed bacterial infection.
N total at baseline: Intervention: 78 Control: 43
Important prognostic factors2: For example Mean age (range): I: 44 (19-86) C: 40 (13-72)
Sex: I: 53% M C: 53% M
Groups comparable at baseline? yes |
Intravenous peramivir once daily for 5 days. Children and adolescents received peramivir at a dosage of 10 mg/kg once daily, to a maximum of 600 mg per day. Peramivir doses were lowered to 150 or 100 mg once daily in subjects with creatinine clearance of 30–49 or 10–29 mL/min/1.73 m2, respectively. The study drug was added to the institution’s SOC influenza treatment. At some study sites, the SOC consisted of treatment with oseltamivir (n=216) or zanamivir (n=1).
|
Intravenous placebo once daily for 5 days with standard of care influenza treatment. At some study sites, the SOC consisted of treatment with oseltamivir (n=216) or zanamivir (n=1). |
Length of follow-up: 14 days
Loss-to-follow-up: not reported
|
1. ICU-admission* I: 0/78 C: 1/43 RR: 0.19 (0.01 to 4.56)
2. Length of hospital stay Not reported
3. Mortality (28 days) I: 0/78 C: 2/43 RR: 0.11 (0.006 to 2.27)
4. Adverse events Defined as incidence as complications (otitis, sinusitis, bronchitis, pneumonia) I: 15/78 C: 9/43 RR: 0.88 (0.42 to 1.83) |
Study was conducted between September 2009 and November 2012.
NAI: neuraminidase inhibitor
(*)Part of the population was already admitted to the ICU at baseline. |
|
Ison, 2003 |
Type of study: Double blind RCT
Setting and country: 7 centres, USA
Funding and conflicts of interest: Not reported |
Inclusion criteria: - Male or female patients ≥10 years of age that were hospitalized with influenza A or B virus infection with symptoms of 4 days duration or less - at least one of the following lower respiratory tract signs: new infiltrate on chest radiograph; new onset of respiratory distress (dyspnea, severe cough) - 15 mmHg or greater decrease in alveolar-arterial oxygen gradient compared to the patient’s known or expected baseline gradient; and/or arterial oxygen saturation ≤90% by transcutaneous (fingertip) oximetry on room air. - Female patients were required to have a negative urine or serum pregnancy test
Exclusion criteria: Patients with known hypersensitivity to neuraminidase inhibitors, pregnant or breastfeeding patients, patients who were intubated or had an expectation of imminent demise.
N total at baseline: Intervention: 20 Control: 21
Important prognostic factors2: For example Median age (range): I: 67 (24-93) C: 61 (22-80)
Sex: I: 90% M C: 67% M
Groups comparable at baseline? yes |
Zanamivir (ZNV) 16 mg (as 16 mg/ml in normal saline) administered by the Medic-Aid Sidestream Disposable Nebulizer (Medic-Aid, West Sussex, UK) with a mouthpiece attachment at an airflow of 6–7 l/min and completed within 10 min. Dosis: 4 times/day for 5 days. Patients discharged early were continued on the same treatment schedule with dry powder-inhaled ZNV plus rimantadine (n=3). All patients with influenza A virus infection (40/41) also received rimantadine orally for 5 days.
|
Placebo (normal saline) administered by the Medic-Aid Sidestream Disposable Nebulizer (Medic-Aid, West Sussex, UK) with a mouthpiece attachment at an airflow of 6–7 l/min and completed within 10 min. Dosis: 4 times/day for 5 days. Patients discharged early were continued on the same treatment schedule with placebo plus rimantadine (n=2). All patients with influenza A virus infection (40/41) also received rimantadine orally for 5 days. |
Length of follow-up: 28 days
Incomplete outcome data: Intervention: 9 (45%) Reasons: left the hospital prior to completion 5-day dosing regime because they were clinically improved (N=4), discharge early (N=5)
Control: 7 (33%) Reasons: left the hospital prior to completion 5-day dosing regime because they were clinically improved (N=2), dischare early (N=5)
|
1. ICU-admission Not reported
2. Length of hospital stay, mean±SD I: 4.7±2.3 C: 5.2±2.3 P=0.52
3. Mortality I: 2/20 C: 1/21 RR: 2.1 (0.21 to 21.39)
4. Adverse events Defined as serious adverse events I: 1/20 C: 3/21 RR: 0.35 (0.04 to 3.09) |
Study was conducted between January 1998 and April 1999.
High proportion of patients lost to follow-up. |
|
Muthuri, 2014 |
Type of study: individual participant data meta-analysis
Setting and country: admitted patients in 78 studies
Funding and conflicts of interest: funded by Hoffmann- La Roche. |
Inclusion criteria: described in an appendix (not online available)
Exclusion criteria:
N total at baseline: 29234 Any NAI treatment: 18803 No NAI treatment: 10431
Important prognostic factors: For example Age, median (IQR): 26 (11-44) Sex: 49.4% M Pregnant women: 22.8% Any comorbidity: 37.7%
Groups comparable at baseline? Not calculated |
Neuraminidase inhibitor treatment: - yes versus no - early NAI (starting treatment ≤2 days after symptom onset) versus later (initiation >2 days after symptom onset) - early NAI versus none - later NAI versus none.
|
No NAI or late NAI
|
Length of follow-up: within 30 days
Loss-to-follow-up: 0 (only those with complete data included)
|
3. Mortality NAI treatment versus None
Early NAI treatment versus late
*Adjusted for treatment propensity (by quintile), corticosteroid use and antibiotic use |
|
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures.
- Provide data per treatment group on the most important prognostic factors ((potential) confounders).
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls.
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders.
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Ramirez, 2018 |
A computer-generated randomizer was used to assign all patients into block sizes of 8. |
Unlikely |
Likely, no blinding was performed |
Unlikely, blinding was not possible but unlikely that this influenced the outcomes |
Unclear, blinding of outcome assessors not reported |
Unlikely |
Unlikely |
Likely, per protocol analysis (patients with lab-confirmed I-LRTI) only performed in those who underwent assigned treatment |
|
Dawood, 2016 |
Enrolled children eligible for randomization were assigned 1:1 to receive oseltamivir or placebo using site-stratified randomization sheets with randomly permuted blocks of 10 four digit numbers paired with treatment assignments (5 oseltamivir and 5 placebo per block). Randomization sheets were generated by a statistician without other study involvement. |
Unlikely |
Unlikely, as participants were blinded for treatment allocation |
Unlikely, as care providers were blinded for treatment allocation |
Unlikely, as outcome assessors were blinded for treatment allocation |
Unlikely |
Unlikely |
Unlikely, intention-to-treat analysis was performed |
|
De Jong, 2014 |
Randomization was stratified by duration of illness (≤48 versus >48–72 hours), SOC treatment (NAI versus non-NAI versus no antiviral therapy), influenza subtype (influenza Aversus B versus not determined), and intensive care unit (ICU) admission status at randomization. |
Unlikely |
Unlikely |
Unlikely, not reported but unlikely that this influenced the outcomes |
Unclear, blinding of outcome assessors not reported |
Unlikely |
Unlikely |
Unlikely, intention-to-treat analysis was performed |
|
Ison, 2003 |
A computer-generated block randomization scheme was utilized and the sites were provided with the study drug and randomization envelopes. |
Unlikely |
Unlikely, as participants were blinded for treatment allocation. |
Unlikely, as care providers were blinded for treatment allocation |
Unlikely, as investigators were blinded for treatment allocation |
Unlikely |
Likely, as 45% of the intervention and 33% of the control group were lost to follow-up. |
Unlikely, intention-to-treat analysis was performed |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear.
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Popov, 2018 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Nakamura, 2017 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Marty, 2017 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Lee, 2017 |
Geen behandeling met antivirale middelen |
|
Hung, 2017 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Kakeya, 2014 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling |
|
Ison, 2014. |
Behandeling enkele versus. dubbele dosis intravenous peramivir |
|
Venkatesan, 2017 |
Geen gerandomiseerde studie |
|
Lee, 2017 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Doll, 2017 |
2 studies die over gehospitaliseerde patiënten gaat, zie Muthuri, 2014; Muthuri, 2013 |
|
Muthuri, 2016 |
Geen gerandomiseerde studie |
|
Vargas-Sandoval,2015 |
Conference abstract |
|
Nguyen-Van-Tam, 2015 |
Overview artikel, geen systematische search en studieselectie |
|
Sedyaningsih, 2013 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Lee, 2013 |
Geen gerandomiseerde studie |
|
Ison, 2013 |
Voldoet niet aan de PICO (geen vergelijking met placebo/geen behandeling) |
|
Muthuri, 2014 |
Geen gerandomiseerde studie |
|
Santesso, 2013 |
Geen systematische review op basis van gerandomiseerde studies |
|
Muthuri, 2013 |
Includeert ook niet gerandomiseerde studies (resultaten RCT’s niet te extraheren) |
|
Ebell, 2013 |
Betreft geen gehospitaliseerde patiënten, studies geincludeerd in Jefferson, 2012 (Literatuuranalyse UV3) |
|
Beck,2013 |
Betreft geen gehospitaliseerde patiënten |
|
Wang, 2012 |
Betreft geen gehospitaliseerde patiënten |
|
Ceyhan, 2012 |
Betreft geen gehospitaliseerde patiënten |
|
Mancuso, 2010 |
Geen systematische search en studieselectie, geen transparante methodologie |
|
Gibbs, 2010 |
Betreft niet-opgenomen patiënten |
|
Shun-Shin, 2009 |
Wang, 2013 is hier een update van |
|
Matheson, 2007 |
Wang, 2013 is hier een update van |
|
No author, 2006 |
Betreft geen gehospitaliseerde patiënten |
|
Makela, |
Niet-opgenomen patiënten, studie geincludeerd in Taieb, 2019 (literatuuranalyse UV3) |
Beoordelingsdatum en geldigheid
Publicatiedatum : 15-10-2021
Beoordeeld op geldigheid : 01-07-2021
|
Module[1] |
Regie-houder(s)[2] |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn[3] |
Frequentie van beoordeling op actualiteit[4] |
Wie houdt er toezicht op actualiteit[5] |
Relevante factoren voor wijzigingen in aanbeveling[6] |
|
Behandeling opgenomen patiënten |
NVMM |
2021 |
2023 |
Elke 2 jaar |
NVMM |
Nieuw beschikbare antivirale middelen |
[1] Naam van de module
[2] Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders)
[3] Maximaal na vijf jaar
[4] (half)Jaarlijks, eens in twee jaar, eens in vijf jaar
[5] regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) kerngroep die in stand blijft
[6] Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
De richtlijn is ontwikkeld in samenwerking met:
- Nederlandse Internisten Vereniging
- Nederlandse Vereniging van Artsen voor Longziekten en Tuberculose
- Nederlandse Vereniging voor Kindergeneeskunde
- Nederlandse Vereniging voor Klinische Geriatrie
- Nederlandse Vereniging voor Obstetrie en Gynaecologie
- Nederlandse Vereniging voor Intensive Care
- Vereniging van specialisten ouderengeneeskunde
- Nederlands Huisartsen Genootschap
- Rijksinstituut voor Volksgezondheid en Milieu
- Patiëntenfederatie Nederland
Doel en doelgroep
Doel
Het project was het herzien en uitbreiden van de richtlijn klinische behandeling met antivirale therapie van opgenomen patiënt met influenza. Bij de oorspronkelijke richtlijn waren niet alle betrokken wetenschappelijke verenigingen en patiënten meegenomen in het opstellen hiervan en misten aanbevelingen voor specifieke patiëntengroepen. Verder is bij de herziening de inhoud uitgebreid met de behandeling van niet-opgenomen patiënten, de nieuwste ontwikkelingen in diagnostiek en wijzigingen. Hierdoor zal de patiëntenzorg in overeenstemming gebracht worden met de laatste stand van zaken.
Doelgroep
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen die betrokken zijn bij de zorg voor patiënten met Influenza.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2018 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor de behandeling van patiënten met (verdenking) op influenza.
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname.
Werkgroep
- Dr. A. (Annelies) Riezebos-Brilman, arts-microbioloog, werkzaam in het UMC Utrecht (NVMM), t/m april 2021 werkzaam in het UMC Utrecht, sinds mei 2021 werkzaam in LabMicta, Hengelo (NVMM) (voorzitter)
- Prof. dr. Menno de Jong, hoogleraar/afdelingshoofd Medische Microbiologie & Infectiepreventie, werkzaam in het Amsterdam UMC te Amsterdam (NVMM)
- Drs. K.A.S. (Karen) Couderé, AIOS Medische microbiologie, werkzaam in Elisabeth TweeSteden ziekenhuis Tilburg (NVMM)
- Dr. F.L. (Frank) van de Veerdonk, internist-infectioloog, werkzaam in het Radboudumc te Nijmegen (NIV)
- Dr. A.W.J. (Aik) Bossink, longarts, werkzaam in het Diakonessenhuis te Utrecht (NVALT)
- Dr. P.L.A. (Pieter) Fraaij, kinderarts-infectioloog/immunoloog, werkzaam in het Erasmus MC-Sophia te Rotterdam (NVK)
- Drs. M. (Marieke) Zeeman, internist-ouderengeneeskunde en klinisch farmacoloog, werkzaam in Deventer ziekenhuis te Deventer (NVKG)
- Dr. W.J. (Wouter) Meijer, gynaecoloog, werkzaam in Gelre Ziekenhuizen te Zutphen (NVOG)
- Dr. L.P.G. (Lennie) Derde, infectioloog-intensivist, werkzaam in het UMC Utrecht (NVIC)
- Dr. P. (Paul) van Houten, specialist ouderengeneeskunde, werkzaam bij Zonnehuisgroep Amstelland te Amstelveen (Verenso)
- Mevr. K. (Klaartje) Spijkers (Patiëntenfederatie Nederland)
- Dr. G.A. (Ted) van Essen, huisarts, niet praktiserend (NHG)
- Dr. A.M. (Albert) Vollaard, infectioloog, werkzaam bij het RIVM te Bilthoven (RIVM)
Meelezer
- Stichting Kind en Ziekenhuis
Met ondersteuning van
- Dr. J. (Janneke) Hoogervorst-Schilp, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- E.A (Ester) Rake, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Actie |
|
Riezebos-Brilman (voorzitter) |
Arts-microbioloog met aandachtsgebied virologie, UMCU |
Counsilmember ESCV (European Society of Clinical Virology) |
geen |
Geen actie |
|
Meijer |
Gynaecoloog, Coöperatie medische Specialisten Gelre u.a. werkzaam in Gelre ziekenhuis locatie Zutphen |
geen |
geen |
Geen actie |
|
Bossink |
Longarts, vrijgevestigd medisch specialist Diakonessenhuis Utrecht |
geen |
Patent crossspot |
Geen actie |
|
Van Essen |
Huisarts, gepensioneerd |
Onbezoldigd voorzitter Nederlandse Influenza Stichting (https://influenzastichting.nl/) |
Een paar keer een vergoeding als spreker, van Sanofi Pasteur en van MSD. |
Geen actie |
|
Van de Veerdonk |
Internist-infectioloog werkzaam Radboudumc |
Onderzoeker betaald vanuit subsidies. |
Ik heb een VIDI subsidie gekregen waarbij uitgezocht wordt of neuraminidase remmers de kans op dodelijke schimmelinfecties kunnen verhogen. Ik heb geen baat bij welke uitkomst dan ook van de richtlijn. |
Geen actie |
|
De Jong |
Hoogleraar/afdelingshoofd Medische Microbiologie & Infectiepreventie, Amsterdam UMC, |
geen |
In afgelopen 5 jaar lid (ad-hoc) geweest van Scientific Advisory Boards voor Janssen, Crucell, Shionogi, MedImmune, Celltrion; allen op gebied van preklinisch en klinisch onderzoek van nieuwe antivirale middelen tegen influenza. Momenteel geen actief adviseurschap. |
Geen actie, want momenteel geen actief adviseurschap, en onafhankelijke beoordelen van nieuwe middelen (dus geen belangenverstrengeling) |
|
Derde |
Intensivist UMC Utrecht (0.45 FTE) |
Voorzitter taskforce NVIC: Draaiboek (influenza-) pandemie (onbetaald) |
Coördinerend onderzoeker Europa voor REMAP-CAP studie. Deze studie is een internationale 'adaptive platform trial' naar de beste behandeling van CAP op de IC, waarbij in het seizoen of tijdens een pandemie de behandeling van influenza daar deel van zou kunnen uitmaken. Coördinerend onderzoeker Europa voor REMAP-CAP studie. |
Geen actie, want de uitkomst van het genoemde onderzoek en de richtlijn hebben geen invloed op elkaar |
|
Zeeman |
Internist ouderengeneeskunde Deventer Ziekenhuis |
Klinisch Farmacoloog Deventer Ziekenhuis (binnen functie internist-og) |
geen |
Geen actie |
|
Vollaard |
Internist-infectioloog, Landelijke Coördinatie Infectieziektebestrijding (LCI) |
Arts-vrijwilliger bij Dokters van de Wereld: consultatie van ongedocumenteerde migranten |
geen |
Geen actie |
|
Fraaij |
Kinderarts Infectioloog en immunoloog, hoofd subspecialisme Kinderimmunologie en infectieziekten, ErasmusMC-Sophie, Rotterdam |
geen |
PREPARE Europe (EU FP? grant no. 602525) Ik ben bestuurslid bij Stichting Infecties bij Kinderen (INKI) Ik organiseer de cursus 'Antibiotica bij kinderen' |
Geen actie, want de uitkomst van het genoemde onderzoek en de richtlijn hebben geen invloed op elkaar |
|
Spijkers |
Senior adviseur patiëntenbelang |
Voorzitter Stichting Samen voor Duchenne (onbetaald) |
geen |
Geen actie |
|
Coudere |
AIOS Medische microbiologie, LMMI, ETZ, Tilburg |
Consulent dienst Gastro-enterologie en Hepatologie, UZA, Antwerpen: medewerker onderzoeksproject Hepatitis B en C screening bij sub-Saharaans Afrikaanse Migranten (funding Gilead) - betaald - einde 31 me 2019 |
geen |
Geen actie |
|
Van Houten |
Specialist ouderengeneeskunde, hoofd medische dienst Zonnehuisgroep Amstelland |
Lid Goed gebruik Hulpmiddelen bij ZonMw |
Voorzitter werkgroep Handreiking hygiëne en infectiepreventie voor specialisten ouderengeneeskunde bij Verenso. Adviseur Behandeladvies COVID-19 acute fase en nazorg bij Verenso |
Geen actie |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een werkgroeplid af te vaardigen namens de Patiëntenfederatie Nederland. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn (module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden in de bijlagen. In het licht van de bevindingen van de Kwaliteits- & Doelmatigheidsagenda over aantallen beschikbare indicatoren en de moeilijkheid van het ontwikkelen van toepasselijke indicatoren, is er besloten (vooralsnog) geen indicatoren te ontwikkelen.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. Tevens zijn er knelpunten aangedragen door afgevaardigden van de Nederlandse Vereniging voor Medische Microbiologie, Inspectie Gezondheidszorg & Jeugd, de Nederlandse Associatie Physician Assistants, het Nederlands Huisartsen Genootschap, de Nederlandse Vereniging voor Klinische Geriatrie, Rijksinstituut van Volksgezondheid en Milieu, Verpleegkundigen en Verzorgenden Nederland, Verenso, Vereniging Innovatieve Geneesmiddelen, Zorginstituut Nederland, en de Nederlandse Vereniging Artsen voor Longziekten en Tuberculose tijdens de invitational conference. Een verslag hiervan is opgenomen in de bijlagen.
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Voor de afzonderlijke uitgangsvragen werd aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie voor de oriënterende zoekactie en patiëntenperspectief zijn opgenomen in de zoekverantwoording.
Gedurende het zoekproces, heeft de werkgroep de internationale richtlijn van de Infectious Diseases Society of America (IDSA) beoordeeld op geschiktheid (Uyeki, 2019). Daarbij werd het adviesrapport ‘Adapteren van internationale richtlijnen naar de Nederlandse praktijk’ gevolgd van de Federatie Medisch Specialisten, welke is vastgesteld in de Raad Kwaliteit in 2017 (NVALT, NVU 2016). Er werd besloten om de IDSA Guideline niet als uitgangspunt te nemen om de richtlijn te updaten. Enerzijds zijn er geen transparante zoekstrategie en selectiecriteria in de IDSA Guideline beschikbaar; anderzijds is er geen duidelijke scheiding weergegeven tussen literatuuranalyse en Overwegingen in de IDSA Guideline. Daarmee voldoet de IDSA Guideline niet aan afspraken die wv-en onderling hebben afgesproken met betrekking tot het gebruiken en adapteren van internationale richtlijnen.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag.
Commentaar- en autorisatiefase [na autorisatiefase definitief maken]
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Aanvullingen module baloxavir - autorisatiefase
De aanvullingen in deze richtlijn betreffende het antivirale middel baloxavir werd toegevoegd aan de geautoriseerde richtlijn. Deze aanvullingen en wijzigingen zijn ter autorisatie dan wel accordering voorgelegd aan de deelnemende (wetenschappelijke) verenigingen en (patient) organisaties. Er werd geen aparte commentaarfase georganiseerd voor deze teksten, omdat er geen wijzigingen zijn aangebracht in de aanbevelingen.
Literatuur
- Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
- Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
- Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
- Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
- Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
- Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
- Nederlandse Vereniging voor Urologie en de Nederlandse Vereniging van Artsen voor Longziekten en Tuberculose. Adapteren van internationale richtlijnen naar de Nederlandse praktijk. Utrecht, 2016
- Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
- Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
- Schünemann HJ, Oxman AD, Brozek J, Glasziou P, Jaeschke R, Vist GE, Williams JW Jr, Kunz R, Craig J, Montori VM, Bossuyt P, Guyatt GH; GRADE Working Group. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008 May 17;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008 May 24;336(7654). doi: 10.1136/bmj.a139.
- Schünemann, A Holger J (corrected to Schünemann, Holger J). PubMed PMID: 18483053; PubMed Central PMCID: PMC2386626.
- Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
- Uyeki, T. M., Bernstein, H. H., Bradley, J. S., Englund, J. A., File Jr, T. M., Fry, A. M.,... & Ison, M. G. (2019). Clinical practice guidelines by the Infectious Diseases Society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenza. Clinical Infectious Diseases, 68(6), e1-e47.
Zoekverantwoording
|
Database(s): Medline, Embase |
Datum: 3 mei 2019 |
|
Periode: > 1996 |
Talen: Engels |
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
1996 – mei 2019 |
1 exp *Influenza, Human/ or exp influenzavirus a/ or exp influenzavirus b/ or exp influenza b virus/ or influenza*.ti. or flu.ti. or flue.ti. or h1n1*.ti. or h2n3*.ti. (89133) 2 exp *Antiviral Agents/ or exp Oseltamivir/ or exp Zanamivir/ or exp Rimantadine/ or exp Amantadine/ or (oseltamivir or zanamivir or baloxavir or peramivir or rimantadine or amantadine or lasinavir or moroxydine or umifenovir or antiviral therap* or antivirus agent*).ti. (200227) 3 exp Hospitalization/ or (hospital* or outpatient* or medical centre* or emergency room* or intensive care or icu or ic).ti,ab,kw. (1497555) 4 1 and 2 and 3 (904) 5 limit 4 to (english language and yr="1996 - 2019") (808) 6 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (391445) 7 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1852581) 8 5 and 7 (163) 9 1 and 2 (7302) 10 limit 9 to (english language and yr="1996 - 2019") (5419) 11 6 and 10 (122) (aantal systematische reviews) 12 8 not 11 (130) (aantal RCT’s) 13 11 or 12 (252)
= 252 |
518 |
|
Embase (Elsevier) |
('influenza'/exp/mj OR influenza*:ti OR flu:ti OR flue:ti OR h1n1*:ti OR h2n3*:ti OR 'influenza virus'/exp/mj) AND ('antivirus agent'/exp/mj OR 'oseltamivir'/exp OR 'zanamivir'/exp OR 'baloxavir'/exp OR 'baloxavir marboxil'/exp OR 'peramivir'/exp OR 'rimantadine'/exp OR 'amantadine'/exp OR 'lasinavir'/exp OR 'moroxydine'/exp OR 'umifenovir'/exp OR oseltamivir:ti OR zanamivir:ti OR baloxavir:ti OR peramivir:ti OR rimantadine:ti OR amantadine:ti OR lasinavir:ti OR moroxydine:ti OR umifenovir:ti OR 'antiviral therap*':ti OR 'antivirus agent*':ti) AND ('hospital patient'/exp OR hospital*:ti,ab OR outpatient*:ti,ab OR 'medical centre*':ti,ab OR 'emergency room*':ti,ab OR 'intensive care':ti,ab OR icu:ti,ab OR ic:ti,ab) AND (1996-2019)/py AND (english)/lim NOT 'conference abstract':it
Gebruikte filters: RCT’s: ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it = 262
('influenza'/exp/mj OR influenza*:ti OR flu:ti OR flue:ti OR h1n1*:ti OR h2n3*:ti OR 'influenza virus'/exp/mj) AND ('antivirus agent'/exp/mj OR 'oseltamivir'/exp OR 'zanamivir'/exp OR 'baloxavir'/exp OR 'baloxavir marboxil'/exp OR 'peramivir'/exp OR 'rimantadine'/exp OR 'amantadine'/exp OR 'lasinavir'/exp OR 'moroxydine'/exp OR 'umifenovir'/exp OR oseltamivir:ti OR zanamivir:ti OR baloxavir:ti OR peramivir:ti OR rimantadine:ti OR amantadine:ti OR lasinavir:ti OR moroxydine:ti OR umifenovir:ti OR 'antiviral therap*':ti OR 'antivirus agent*':ti) AND (1996-2019)/py AND (english)/lim NOT 'conference abstract':it
Gebruikte filters: Systematische reviews: ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) = 187
= 449 |