Analgeticum bij analgosedatie
Uitgangsvraag
Welke analgeticum kan het beste worden gebruikt bij analgosedatie?
Aanbeveling
Gebruik remifentanil of een (middel-) langwerkend opioïde bij analgosedatie op de IC, afhankelijk van de ervaring en expertise met de beschikbare opioïden.
Overweeg remifentanil in plaats van (middel-) langwerkende opioïden bij de volgende patiëntengroepen:
- Patiënten met ernstige lever- en nierfunctiestoornissen;
- Patiënten met indicatie voor frequente neurologische controles.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Op basis van deze literatuursamenvatting is met een lage bewijskracht te concluderen dat remifentanil geen klinisch relevant voor- of nadeel oplevert in IC-opnameduur ten opzichte van langwerkende opioïden. Hetzelfde geldt voor het effect van remifentanil op mortaliteit, met eveneens zeer lage bewijskracht. De lage bewijskracht is met name te verklaren door de aanwezigheid van publicatiebias in de studies uit de review van Tan (2009) en het passeren van de grenzen van klinische relevantie. Ook wanneer er wordt gekeken naar de belangrijke uitkomstmaten hemodynamische adverse events, ventilatieduur en delier, is de bewijskracht voor het effect van remifentanil zeer laag. Met deze zeer lage bewijskracht was er alleen een klinisch relevant verschil in het optreden van delier, ten gunste van remifentanil (minder delier). Er waren geen data beschikbaar voor de uitkomstmaat autodetubaties.
Hoewel in de literatuur geen duidelijke aanwijzingen zijn gevonden voor een voordeel van remifentanil gebruik in de algemene IC-populatie, is op basis van het gunstige farmacokinetisch profiel te beredeneren dat bepaalde patiëntengroepen hier wel voordeel van kunnen hebben. Hierbij zou je kunnen denken aan patiënten met ernstige lever- of nierfunctiestoornissen, waarbij andere opioïden door verminderde afbraak snel in de weefsels kunnen opstapelen, waarbij dit door het lever- en nierfunctie onafhankelijke metabolisme van remifentanil niet het geval is. Daarnaast zou remifentanil, vanwege de ultrakorte werkingsduur, een meerwaarde kunnen hebben bij patiënten die meerdere keren per dag neurologische controles/evaluatie behoeven, omdat je het sedatieve effect snel “aan en uit” kunt zetten.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Goede analgesie is belangrijk voor het comfort en de stressreductie van de patiënt. Aangezien pijn veel voorkomt bij IC -patiënten is het gebruik maken van de sederende eigenschappen van analgetica zoals opioïden vaak een goede keuze. De stuurbaarheid van een ultrasnel en -kort werkend opioïde zoals remifentanil heeft als voordeel dat hierbij enerzijds snel en effectief kan worden geacteerd op pijnlijk interventies (bijvoorbeeld verzorging) en anderzijds hierna weer snel kan worden gestreefd naar een zo oppervlakkig mogelijk bewustzijnsniveau, wat de communicatie ten goede komt. In de gevonden studies is niet gekeken naar ervaring van patiënten en familieleden zelf. Hier is dus sprake van een kennislacune.
Kosten (middelenbeslag)
Er zijn geen grote prijsverschillen tussen remifentanil en de overige (middel-)langwerkende opioïden.
Aanvaardbaarheid, haalbaarheid en implementatie
Analgosedatie met remifentanil wordt op Nederlandse IC’s reeds veelvuldig toegepast en is veilig gebleken onder respiratoire en hemodynamische monitoring. Vanwege het ultra-snelwerkende effect volstaat het ophogen van de continue toedieningsstand voor een snel effect en wordt een bolus of oplaad-dosering afgeraden, dit dient bekend te zijn bij het personeel dat de analgesie bijstuurt. Wanneer wordt gekozen om remifentanil niet als standaard analgosedativum toe te passen, maar alleen incidenteel op indicatie (zie bij aanbevelingen), dan is het goed om te realiseren dat de effecten van dosisaanpassingen binnen enkele minuten merkbaar zijn. Het vergt enige ervaring in het gebruik om de stuurbaarheid optimaal te kunnen benutten.
Bij langdurig gebruik van remifentanil, maar ook van andere opioïden, dient bij het staken rekening te worden gehouden met onttrekking. Dit kan een reden zijn om na detubatie de opioïde niet direct te stoppen, maar langzaam af te bouwen. Een nadeel van remifentanil is, dat het niet is opgenomen in de opioïde conversie tabellen, waardoor het lastig kan zijn om van remifentanil naar een andere opioïde te switchen.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Remifentanil levert geen klinisch relevant voor- of nadeel op in IC-opnameduur ten opzichte van langwerkende opioïden. Gezien het gunstige farmacokinetische profiel (ultrakorte werkingsduur en lever- en nierfunctie onafhankelijke metabolisme) is te beredeneren dat bepaalde patiëntengroepen voordeel kunnen hebben van remifentanil.
Onderbouwing
Achtergrond
Analgosedatie bij IC-patiënten is gebaseerd op het principe dat eerst discomfort, waaronder pijn, dient te worden bestreden met analgetica die vaak intrinsiek ook een sederende werking hebben, alvorens ‘pure’ sedativa toe te passen. De achterliggende gedachte is dat bij adequate pijnbehandeling een minder diepe sedatie nodig is met als voordeel minder nadelige effecten van sedativa, zoals sterk verlaagd bewustzijn, hemodynamische en respiratoire depressie. De meest gebruikte vorm van analgosedatie is de continue toediening van opioïden waarbij er keuze is uit (middel-)langwerkende opioïden als morfine, fentanyl, sufentanil en alfentanil of het ultra kortwerkende remifentanil. Naast de korte eliminatiehalfwaardetijd onderscheidt remifentanil zich van de andere opioïden doordat het metabolisme (nagenoeg) onafhankelijk is van de lever- en nierfunctie, omdat het wordt gemetaboliseerd door niet-specifieke bloed- en weefsel esterasen. Dit is relevant, omdat bij veel IC-patiënten sprake is van lever- en nierfunctiestoornissen. Door dit metabolisme en de ultrakorte halfwaardetijd ontstaat geen stapeling van het medicament in de weefsels. De dosering is daardoor makkelijk en snel te titreren op het gewenste effect. Remifentanil heeft in verhouding tot andere opioïden mogelijk wel meer hemodynamische effecten en ademdepressie.
Op veel Nederlandse IC’s is remifentanil inmiddels het middel van eerste keus voor analgosedatie bij geïntubeerde patiënten. Het is de vraag of het gunstige farmacologische profiel van remifentanil daadwerkelijk leidt tot een ‘stuurbaarder’ analgosedatie beleid ten opzichte van de (middel-)langwerkende opioïden; en of dit opweegt tegen de eventuele nadelige effecten van remifentanil.
Conclusies / Summary of Findings
1. ICU length of stay (crucial)
|
Low GRADE |
Remifentanil may result in little to no difference in ICU length of stay when compared with long-acting opioids in ICU patients.
Sources: Tan, 2009 (Baillard, 2005; Dahaba, 2004; Breen, 2005; Muellejans, 2004; Muellejans, 2006); Cevik, 2011; Liu, 2017; Spies, 2011. |
2. Mortality (crucial)
|
Very low GRADE |
The evidence is very uncertain about the effect of remifentanil on mortality when compared with long-acting opioids in ICU patients.
Sources: Tan, 2009 (Baillard, 2005; Dahaba, 2004; Breen, 2005); Liu, 2017. |
3. Hemodynamic adverse events (important)
|
Very low GRADE |
The evidence is very uncertain about the effect of remifentanil on hemodynamic adverse events when compared with long-acting opioids in ICU patients.
Sources: Tan, 2009 (Dahaba, 2004; Breen, 2005; Muellejans, 2004; Muellejans, 2006); Cevik, 2011. |
4. Delirium (important)
|
Very low GRADE |
The evidence is very uncertain about the effect of remifentanil on delirium when compared with long-acting opioids in ICU patients.
Sources: Tan, 2009 (Muellejans, 2006); Liu, 2017; Spies, 2011. |
5. Duration of mechanical ventilation (important)
|
Low GRADE |
Remifentanil may result in little to no difference in duration of mechanical ventilation when compared with long-acting opioids in ICU patients.
Sources: Tan, 2009 (Dahaba, 2004; Breen, 2005; Muellejans, 2004); Cevik, 2011; Liu, 2017; Spies, 2011. |
6. Selfextubations (important)
|
- GRADE |
The outcome measure selfextubations was not reported in the included studies.
Sources: - |
Samenvatting literatuur
Description of studies
Tan (2009) performed a meta-analysis about the effects of remifentanil as a sedative agent in critically ill adult patients. An electronic database search was performed in EMBASE (from 1988) and MEDLINE (from 1966) until August 2009 by two independent researchers. Studies were included if they were randomised controlled trials; compared remifentanil with another opioid, hypnotic or sedative agent; and included adult patients (>18 years old). In total, 11 trials involving 1067 patients were included for the meta-analysis. To answer our clinical question, we selected six studies from this review (Baillard, 2005; Dahaba, 2004; Breen, 2005; Muellejans, 2006; Muellejans, 2004). The five other studies did not fulfil our selection criteria in terms of language, comparison, or outcome measures. The effects of remifentanil were evaluated on length of ICU stay, hospital mortality and the duration of mechanical ventilation.
Cevik (2011) performed a RCT to examine the effect of fentanyl and remifentanil on sedation and analgesia at the ICU. The trial enrolled 32 adult patients (mean age 51.2y; 53% men) requiring mechanical ventilation and sedation and were randomly allocated to the groups. The remifentanil group (n=16) received remifentanil at an initial dose of 0.05 μg/kg/min (mean daily dose 4.97mg (SD 1.31) and the fentanyl group (n=16) received fentanyl at an initial dose of 0.015 μg/kg/min (mean daily dose 18.41mg (SD 2.87). Both groups received midazolam infusion at an initial dose of 0.03mg/kg/h. None of the patients received adjuvant analgesics or rescue pain therapy. The effects were evaluated on length of ICU stay, hemodynamic adverse events, and duration of mechanical ventilation.
Liu (2017) performed a single-center RCT about the effect of analgesic-based sedation protocols on delirium and outcomes in critically ill patients at a surgical ICU undergoing mechanical ventilation for longer than 24 hours. A total of 105 patients (mean age 64.2y; 52.4% men) were enrolled in the trial and were randomly allocated to one of the three groups. The remifentanil group (n=35) received 1μg/kg/hr remifentanil and midazolam; the fentanyl group (n=35) received 1μg/ kg/hr fentanyl and midazolam; and the control group received 1μg/kg/hr saline and midazolam. Midazolam was administered with a loading dose of 0.05 mg/kg followed by 0.02±0.1 mg/kg/hr. To answer our clinical question, we compared the effects of the remifentanil group with the effects of the fentanyl group. The effects were evaluated on length of ICU stay, 28th day all-cause mortality, hemodynamic adverse events, delirium, and duration of mechanical ventilation. Patients were followed until death or the 28th day from the admission to the ICU.
Spies (2011) performed a two-centre RCT about the effect of remifentanil and fentanyl in mechanically ventilated patients. A total of 60 patients who were expected to receive ventilation between >24h and <48h (mean age 63.5y; 78,3% men) were enrolled in the trial. Patients were randomly allocated to receive remifentanil (n=28), infused at 0.1–0.4 lg/kg ideal body weight/min; or fentanyl (n=32) infused at 0.02–0.08 lg/kg ideal body weight/min. The effects were evaluated on length of ICU stay, hospital mortality and the duration of mechanical ventilation. Patients were followed for a maximum of 30 days from the moment of inclusion.
Results
1. ICU length of stay (crucial)
ICU length of stay was described in eight RCTs (Baillard, 2005; Dahaba, 2004; Breen, 2005; Cevik, 2011; Liu, 2017; Muellejans, 2004; Muellejans, 2006; Spies, 2011), comprising 579 patients. Different drugs were used as a comparator, such as sufentanil (Baillard, 2005), morphine (Dahaba, 2004), fentanyl or morphine (Breen, 2005) and fentanyl (Cevik, 2011; Liu, 2017; Muellejans, 2004; Muellejans, 2006; Spies, 2011). Pooled data resulted in a mean difference (MD) of -0.36 days (95% Confidence Interval (CI) -0.94 to 0.21) in favour of remifentanil (Figure 3.2). This difference is not clinically relevant.
The level of evidence in the literature
The level of evidence regarding the outcome ICU length of stay started at high because it was based on randomized controlled trials but was downgraded by two levels due to publication bias (risk of bias, -1) and heterogeneity (inconsistency, -1). The final level is low.
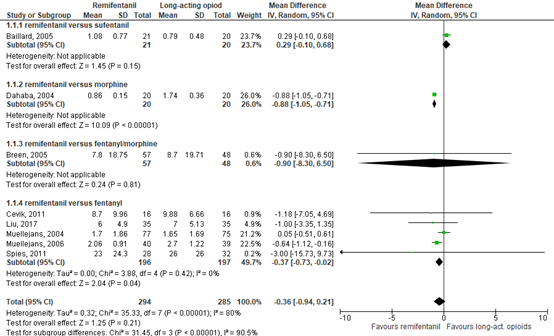
Figure 2. Forest plot of remifentanil versus long-acting opioids on ICU length of stay
2. Mortality (crucial)
Mortality was described by four RCTs (Baillard, 2005; Dahaba, 2004; Breen, 2005; Liu, 2017), comprising 256 patients. Pooled data resulted in a risk ratio (RR) of 0.99 (95% CI 0.66 to 1.48) in favour of remifentanil (Figure 3.3). This difference was not clinically relevant.
The level of evidence in the literature
The level of evidence regarding the outcome mortality started at high because it was based on randomized controlled trials but was downgraded by two levels due to publication bias (risk of bias, -1) and crossing the borders of clinical relevance on both sides (imprecision, -2). The final level is very low.
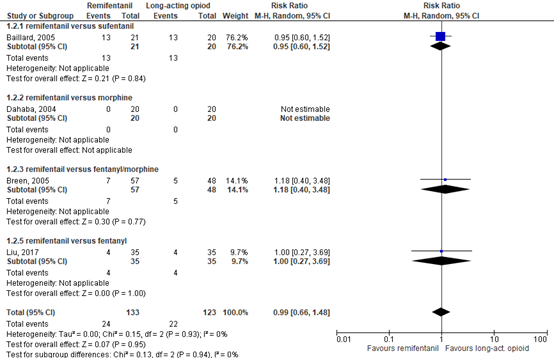
Figure 3. Forest plot of remifentanil versus long-acting opioids on mortality
3. Hemodynamic adverse events (important)
Hemodynamic adverse events were described by five RCTs by the incidence of hypotension (Dahaba, 2004; Breen, 2005; Cevik, 2011; Muellejans, 2004; Muellejans, 2006), comprising 452 patients. Data resulted in a RR of 1.13 (95% CI 0.55 to 2.33) in favour of long-acting opioids (Figure 3.4). This difference was not clinically relevant.
The level of evidence in the literature
The level of evidence regarding the outcome hemodynamic adverse events started at high because it was based on randomized controlled trials but was downgraded by two levels due to publication bias (risk of bias, -1) and crossing the borders of clinical relevance on both sides (imprecision, -2). The final level is very low.
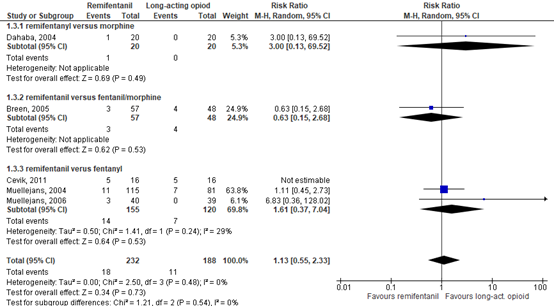
Figure 4. Forest plot of remifentanil versus long-acting opioids on hemodynamic adverse events
4. Delirium (important)
Delirium was described by three RCTs (Liu, 2017; Muellejans, 2006; Spies, 2011), comprising 209 patients. Data resulted in a RR of 0.79 (95% CI 0.49 to 1.27) in favour of remifentanil (Figure 3.5). This difference was clinically relevant.
The level of evidence in the literature
The level of evidence regarding the outcome delirium started at high because it was based on randomized controlled trials but was downgraded by two levels due to publication bias (risk of bias, -1) and crossing the borders of clinical relevance on both sides (imprecision, -2). The final level is very low.
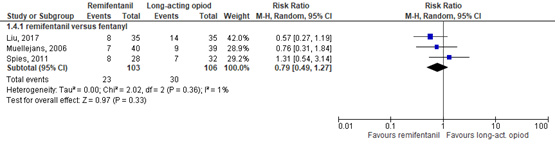
Figure 5. Forest plot of remifentanil versus long-acting opioids on delirium
5. Duration of mechanical ventilation (important)
Duration of mechanical ventilation was described by six RCTs (Dahaba, 2004; Breen, 2005; Cevik, 2011; Liu, 2017; Muellejans, 2004; Spies, 2011), comprising 386 patients. Pooled data resulted in a MD of -0.49 days (94% CI -1.11 to 0.12) in favour of remifentanil (Figure 3.6). This difference was not clinically relevant.
The level of evidence in the literature
The level of evidence regarding the outcome duration of mechanical ventilation started at high because it was based on randomized controlled trials but was downgraded by three levels due to publication bias (risk of bias, -1) and crossing the borders of clinical relevance on one side (imprecision, -1). The final level is low.
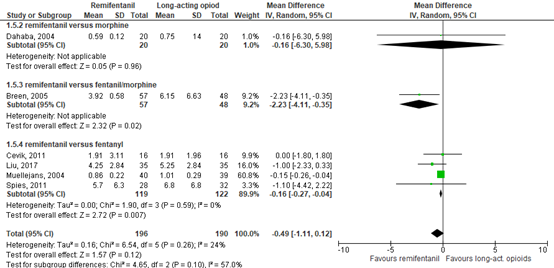
Figure 6. Forest plot of remifentanil versus long-acting opioids on duration of mechanical ventilation.
6. Selfextubations (important)
Selfextubations were not described as an outcome in the included studies.
Zoeken en selecteren
A systematic review of the literature was performed to answer the following question:
What are the (dis)advantages of remifentanil when compared to long-acting opioids in an analgosedation strategy on length of ICU stay, mortality, hemodynamic adverse events, delirium, duration of mechanical ventilation and selfextubations?
P: Adult Intensive Care patients in an analgosedation strategy;
I: Remifentanil;
C: Longer-acting opioids: Morphine, fentanyl, sufentanil, alfentanil;
O: Length of ICU stay, mortality, hemodynamic adverse events, delirium, duration of mechanical ventilation and selfextubations.
Relevant outcome measures
The guideline development group considered length of ICU stay and mortality as critical outcome measures for decision making; and hemodynamic adverse events, delirium, duration of mechanical ventilation and selfextubations as important outcome measures for decision making.
The working group defined the outcome measures as follows:
- Length of ICU stay: The number of days spent on the ICU.
- Mortality: The number of patients who died during IC/hospital stay.
- Hemodynamic adverse events: Hypotension/fluid requirement/vasopressor use/vasoactive medication.
- Delirium: The number of participants who were diagnosed with delirium during IC/hospital stay.
- Duration of mechanical ventilation: The number of days.
- Selfextubations: Percentage of unplanned detubations.
The working group defined the following differences as minimal clinically (patient) important differences:
- ICU length of stay: A difference of one day.
- Mortality: A difference of 3%, based on the SDD-trial (Smet, 2009) (RR ≤ 0.97, RR ≥ 1.03).
- Hemodynamic adverse events: A difference of25% in relative risk (RR ≤ 0.80, RR ≥ 1.25).
- Delirium: A difference of 5% in delirium incidence (RR ≤ 0.95 RR ≥ 1.05).
- Duration of mechanical ventilation: A difference of one day.
- Selfextubations: A difference of 50% in relative risk (RR <0.67, RR>1.50).
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until 2nd of June 2021. The detailed search strategy is depicted under the tab Methods. The systematic literature search resulted in 264 hits. Studies were selected based on the following criteria:
- Describing adult patients at the ICU in an analgosedation strategy.
- Comparing remifentanil with long-acting opioids (morphine, fentanyl, sufentanil or alfentanil);
- Study design: RCT or systematic reviews;
- Articles published in English or Dutch;
- Articles published between January 2000 and June 2021;
- Describing at least one of the outcome measures.
A total of 10 studies were initially selected based on title and abstract screening. After reading the full text, six studies were excluded (see the table with reasons for exclusion under the tab Methods), and four studies were included.
Results
Four studies (one systematic review and three clinical trials) were included in the analysis of the literature. Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
Referenties
- Cevik F, Celik M, Clark PM, Macit C. Sedation and analgesia in intensive care: a comparison of
fentanyl and remifentanil. Pain Res Treat. 2011;2011:650320. doi: 10.1155/2011/650320. Epub 2011 Jul 2. PMID: 22110929; PMCID: PMC3197257. - Liu D, Lyu J, Zhao H, An Y. The influence of analgesic-based sedation protocols on delirium
and outcomes in critically ill patients: A randomized controlled trial. PLoS One. 2017 Sep 14;12(9):e0184310. doi: 10.1371/journal.pone.0184310. PMID: 28910303; PMCID: PMC5598969. - Spies C, Macguill M, Heymann A, Ganea C, Krahne D, Assman A, Kosiek HR, Scholtz K,
Wernecke KD, Martin J. A prospective, randomized, double-blind, multicenter study comparing remifentanil with fentanyl in mechanically ventilated patients. Intensive Care Med. 2011 Mar;37(3):469-76. doi: 10.1007/s00134-010-2100-5. Epub 2010 Dec 17. PMID: 21165734. - Tan JA, Ho KM. Use of remifentanil as a sedative agent in critically ill adult patients: a meta
analysis. Anaesthesia. 2009 Dec;64(12):1342-52. doi: 10.1111/j.1365-2044.2009.06129.x. Epub 2009 Oct 22. PMID: 19849681.
Evidence tabellen
Evidence table for systematic reviews of RCTs and observational studies (intervention studies)
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Tan, 2009 |
SR and meta-analysis of RCTs
Literature search up to August 2009
A: Baillard, 2005 B: Dahaba, 2004 C: Breen, 2005 D: Muellejans, 2006 E: Muellejans, 2004
Study design: A: RCT B: RCT C: RCT D: RCT E: RCT
Setting and Country: A: France B: Austria C: 15 sites in 10 countries (UK + others). D: Germany E: 21 sites in 5 countries.
Source of funding and conflicts of interest: A n.r. B: GlaxoSmithKline C: GlaxoSmithKline D: GlaxoSmithKline E: GlaxoSmithKline
GlaxoSmithKline was the manufacturer of remifentayl.
The study was solely funded by Department of Intensive Care Medicine, Royal Perth Hospital. The funding source had no role in the collection, analysis, and interpretation of the data or in the decision to submit the manuscript for publication.
|
Inclusion criteria SR: Only randomised controlled trials comparing remifentanil with another opioid (sufentanil, alfentanil, fentanyl or morphine) or hypnotic or sedative agent (propofol or midazolam) in critically ill adult (> 18 years old) patients were included.
Exclusion criteria SR: n.r.
11 studies included
Important patient characteristics at baseline:
N, mean age A 41 patients, 58-59 yrs B: 40 patients, 54-58 yrs C: 105 patients, 52-57 yrs
Sex: A: n.r. in review B: 60% Men C: 67.6% Men D: 73.6% Men E: 70.4% Men
Groups comparable at baseline? Yes |
A: Remifentanil group (n = 21): 10 lg.kg) 1.h )1 and titrated. B: Remifentanil group (n = 20): 9–12 lg.kg)1.h)1 C: Remifentanil group (n = 57): 6–18 ug.kg)1.h)1 D: Remifentanil group (n = 39): 6–60 lg.kg)1.h)1. E: Remifentanil group (n = 77): 9–12 lg.kg)1.h)1.
|
A: Sufentanil group (n = 20): 0.125 lg.kg)1.h)1 and titrated C: Midazolam ± fentanyl ⁄ morphine group (n = 48): D: Midazolam with fentanyl group (n = 33): midazolam: 0.02– 0.04 mg.kg)1.h)1, fentanyl: 1–2 lg.kg)1.h)1 E: Fentanyl group (n = 75): 1–2 lg.kg)1.h)1
|
Endpoint of follow-up:
A: n.r. in review B: until IC discharge C: post-treatment or 10 days after administration, or death. D: after drug administration E: 24 hours after discontinuation of study drug
For how many participants were no complete outcome data available? (intervention/control) A: n.r. in review B: 0/0 C: 0/0 D: 1/7 E: 0/0
|
ICU length of stay Defined as the number of days spent at the ICU.
Effect measure: , mean difference [95% CI]: A: 0.29 [-0.10 – 0.68] B: -0.88 [-1.05 - -0.71] C: -0.90 [-8.30 – 6.50] D: 0.05 [-0.51 – 061] E: -0.64 [-1.12 [-0.16]
Pooled effect (random effects model): -0.32 [95% CI -0.94 to 0.30] favoring remifentanil Heterogeneity (I2): 89%
Mortality Effect measure: risk ratio [95% CI]:
A: 0.95 [0.60 – 152] B: not estimatable 0/0 cases C: 1.18 [0.40 – 3.48] D: n.r. E: n.r.
Pooled effect (random effects model): 0.98 [95% CI 0.64 to 1.51] favoring remifentanil Heterogeneity (I2): 0%
Hemodynamic adverse events Defined as the incidence of hypotension.
Effect measure: risk ratio [95% CI]: A: n.r. B: 3.00 [0.13 – 69.52] C: 0.63 [0.15 – 2.68] D: 1.11 [0.45 – 2.73] E: 683 [0.36 – 128.02]
Pooled effect (random effects model): 1.24 [95% CI 0.62 to 2.47] favoring remifentanil Heterogeneity (I2): 0%
Delirium Effect measure: risk ratio [95% CI]:
A: n.r.
Pooled effect (random effects model): 0.76 [95% CI 0.31 to 1.84] favoring remifentanil Heterogeneity (I2): N.A.
Duration of mechanical ventilation Defined as the number of days on mechanical ventilation.
Effect measure: mean difference [95% CI]: A: n.r.
Pooled effect (random effects model): -0.84 [95% CI -2.47 to 0.78] favoring remifentanil Heterogeneity (I2): 57%
Selfextubations Effect measure: risk ratio [95% CI]:
A: n.r.
|
Author’s conclusion In conclusion, the current evidence does not support the routine use of remifentanil as a sedative agent in critically ill adult patients. A large randomised controlled trial, including daily interruption of sedative drug administration, is needed to assess whether remifentanil with or without another short acting hypnotic agent is cost effective in critically ill adult patients with multiple organ failure.
Sources of funding: |
Evidence table for RCTs
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3
|
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Liu, 2017 |
Type of study: prospective randomized controlled trial.
Setting and country: Single centre, Peking.
Funding and conflicts of interest: The authors have declared that no competing interests exist. |
Inclusion criteria: (1) signing a consent form by the patients' legal authorized principal; (2) admission to the surgical ICU; (3) requirement for mechanical ventilation with the time of mechanical ventilation anticipated to be greater than 24 hours; (4) requirement for midazolam sedation; and (5) age greater than 18 and less than 85 years.
Exclusion criteria: 1) intracranial lesions, neurosurgical intervention, mental disabilities or coma such that they were unable to cooperate; (2) alcohol abuse; (3) history of delirium or antipsychotic use at home described according to the medical history or family members; (4) allergy to the investigational drug or other contraindications; or (5) women who were pregnant or lactating.
N total at baseline: Intervention: 35 Control: 35
Important prognostic factors2: For example age (SD) I: 66.1 (11.9) C: 62 (10.0)
Sex: I: 60.0% M C: 48.6% M
Groups comparable at baseline? Yes |
Remifentanil 1μg/kg/hr and midazolam. Midazolam was administered with a loading dose of 0.05 mg/kg followed by 0.02±0.1 mg/kg/hr. The treatment administered until patients were weaned from the ventilator.
|
Fentanyl 1μg/ kg/hr and midazolam. Midazolam was administered with a loading dose of 0.05 mg/kg followed by 0.02±0.1 mg/kg/hr. The treatment administered until patients were weaned from the ventilator. |
Length of follow-up: Till die or the 28th day from admission to the ICU.
Loss-to-follow-up: Intervention: 0 (0%) Control: 0 (0%)
Incomplete outcome data: Intervention: 0 (0%) Control: 0 (0%)
|
ICU length of stay Defined as the number of days spent on the ICU
Effect measure: mean difference [95% CI]: 1.00 [-1.35 – 3.35] favoring remifentanil.
Mortality Effect measure: risk ratio [95% CI]: 1.00 [0.85 – 1.18] favoring long-acting opioids.
Hemodynamic adverse events Defined as the incidence of hypotension. n.r.
Delirium Effect measure: risk ratio [95% CI]:
1.29 [1.04 – 1.37] favoring remifentanil.
Duration of mechanical ventilation Defined as the number of days on mechanical ventilation.
Effect measure: mean difference [95% CI]: 1.00 [-0.33 – 2.33] favoring remifentanil.
Selfextubations Effect measure: risk ratio [95% CI]: n.r.
|
Author’s conclusion Patients who received benzodiazepines have a relatively greater risk of delirium; analgesics can reduce the amount of sedatives required and can further reduce the occurrence of delirium and improve the prognosis. Remifentanil may have an advantage over fentanyl in reducing delirium. |
|
Spies, 2011 |
Type of study: a registered clinical trial
Setting and Country: 2-Center, Germany.
Funding and conflicts of interest: This study was fully supported by GlaxoSmithKline GmbH & Co (manufacturer of remifentanil) |
Inclusion criteria: All patients requiring intensive care therapy were considered for enrolment. Additional inclusion criteria were expected ventilation for more than 24 h, ventilation for less than 48 h at the time of enrolment, age 18 years or older, and written informed consent.
Exclusion criteria: included known allergy to any of the study medications; pregnancy; World Health Organization chronic pain grade 3 or above, i.e., regular use of potent opioids such as morphine, recent opioid analgesia via a spinal catheter, epidural or any other regional technique; American Society of Anesthesiologists (ASA) classification 5 patients (moribund); participation in a clinical study within the previous 30 days; and neurotrauma or organic brain pathology.
N total at baseline: Intervention: 28 Control: 32
Important prognostic factors2:
age (SD): I: 64 (15) C: 63 (12)
Sex: I: 71.4% M C: 84.4% M
Groups comparable at baseline? Yes |
Remifentanil was infused at 0.1–0.4 lg/kg ideal body weight/min. The intravenous infusion of both study agents was titrated up to the analgesia target.
The study protocol did not allow any bolus application of either fentanyl or remifentanil. If the patient was awake, we used the VAS and if the patient was sedated we used the BPS. The target scores were a VAS B3 and/or a BPS B6 at rest. Sedation was delivered by using propofol (to a maximum of 4 mg/kg ideal body weight/h), and midazolam (0.01–0.18 mg/kg ideal body weight/h) could be added if required. Sedation was titrated according to the Richmond Agitation Sedation Scale (RASS). The target RASS was 0 to -1. There were a few exceptions where deeper sedation (up to RASS of -4) was allowed (e.g., patients requiring prone positioning). Unless contraindicated, all patients could receive the following adjuvant analgesics: metamizole (1 g four times daily enterally or intravenously), and/or paracetamol (1 g four times daily enterally or intravenously), and/or clonidine (0.32–1.3 lg/ kg/h intravenously).
Patients in the remifentanil group received a morphine bolus (0.1 mg/kg) 30 min before ending the study medication.
Rescue pain therapy with morphine boli was allowed at all time points for all patients. Delirium diagnosis was performed by using the Confusion Assessment Method for the ICU (CAM-ICU) in all patients with a RASS[-3] |
Fentanyl was infused at 0.02–0.08 lg/kg ideal body weight/min. The intravenous infusion of both study agents was titrated up to the analgesia target.
The study protocol did not allow any bolus application of either fentanyl or remifentanil. If the patient was awake, we used the VAS and if the patient was sedated we used the BPS. The target scores were a VAS B3 and/or a BPS B6 at rest. Sedation was delivered by using propofol (to a maximum of 4 mg/kg ideal body weight/h), and midazolam (0.01–0.18 mg/kg ideal body weight/h) could be added if required. Sedation was titrated according to the Richmond Agitation Sedation Scale (RASS). The target RASS was 0 to -1. There were a few exceptions where deeper sedation (up to RASS of -4) was allowed (e.g., patients requiring prone positioning). Unless contraindicated, all patients could receive the following adjuvant analgesics: metamizole (1 g four times daily enterally or intravenously), and/or paracetamol (1 g four times daily enterally or intravenously), and/or clonidine (0.32–1.3 lg/ kg/h intravenously).
Patients in the fentanyl group received a placebo bolus instead of morphine 30 minutes before ending the study medication.
Rescue pain therapy with morphine boli was allowed at all time points for all patients. Delirium diagnosis was performed by using the Confusion Assessment Method for the ICU (CAM-ICU) in all patients with a RASS[-3] |
Length of follow-up: 30 after the start of the study drug.
Loss-to-follow-up: 0%
Incomplete outcome data: n.r. |
ICU length of stay Defined as the number of days spent on the ICU
Effect measure: mean difference [95% CI]: n.r.
Mortality Effect measure: risk ratio [95% CI]: n.r.
Hemodynamic adverse events Defined as the incidence of hypotension. n.r.
Delirium Effect measure: risk ratio [95% CI]:
1.26 [1.04 – 1.54] favoring remifentanil
Duration of mechanical ventilation Defined as the number of days on mechanical ventilation.
Effect measure: mean difference [95% CI]: n.r.
Selfextubations Effect measure: risk ratio [95% CI]: n.r.
|
Author’s conclusion The use of a remifentanil-based analgesia regime in critically ill patients was not superior regarding the achievement and maintenance of sufficient analgesia compared with a fentanyl-based analgesia.
Limitation In the remifentanil group there were 16 violations to the protocol versus 11 in the fentanyl group (p = 0.34).
|
|
Cevik, 2011 |
Type of study: prospective, randomized, open-label, and controlled trial
Setting and Country: Turkey
Funding and conflicts of interest: n.r. |
Inclusion criteria: Age ≥ 18; patient requiring mechanical ventilation and sedation.
Exclusion criteria: Age ≤ 18; patients who have neuromuscular disease; patients who are receiving neuromuscular blockers; patients with a known or suspected allergy or intolerance to midazolam, fentanyl, or remifentanil; patients who die during the study period; patients who use toxic substances; alcoholic patients; patients suspected of being pregnant; patients who are moribund, that is, who are classified as ASA grade V according to the American Society of Anesthesiologists (not expected to live > 24 hours).
N total at baseline: Intervention: 16 Control: 16
Important prognostic factors2:
age (SD): I: 50.6 (25.2) C: 51.9 (120.8)
Sex: I: 43.8% M C: 62.5% M
Groups comparable at baseline? Yes |
The R group received remifentanil at an initial dose of 0.05 μg/kg/min and midazolam infusion at an initial dose of 0.03mg/kg/h.
No adjuvant analgesics such as paracetamol or morphine were administered, and no rescue pain therapy was provided. Bolus doses of remifentanil, fentanyl, or midazolam were not allowed in the protocol and therefore were not administered. |
the F group received fentanyl at an initial dose of 0.015 μg/kg/min and midazolam infusion at an initial dose of 0.03mg/kg/h.
No adjuvant analgesics such as paracetamol or morphine were administered, and no rescue pain therapy was provided. Bolus doses of remifentanil, fentanyl, or midazolam were not allowed in the protocol and therefore were not administered. |
Length of follow-up: n.r.
Loss-to-follow-up: Intervention: 1/17 Control 1/17
Reason: Died before weaning
Incomplete outcome data: n.r. |
ICU length of stay Defined as the number of days spent on the ICU
Effect measure: mean difference [95% CI]: 1.18 [ -4.69 – 7.05] favoring remifentanil.
Mortality Effect measure: risk ratio [95% CI]: n.r.
Hemodynamic adverse events Defined as the incidence of hypotension. 1.00 [0.63 – 1.60] favoring remifentanil.
Delirium Effect measure: risk ratio [95% CI]: n.r.
Duration of mechanical ventilation Defined as the number of days on mechanical ventilation.
Effect measure: mean difference [95% CI]: 0.00 [-1.80 – 1.80].
Selfextubations Effect measure: risk ratio [95% CI]: n.r.
|
Author’s conclusion In conclusion, this study demonstrates that there were a small number of significant differences between remifentanil and fentanyl statistically with respect to hemodynamic parameters, analgesic and sedative requirements, sedation time, mechanical ventilation time, ICU discharge time, organ dysfunction, or adverse effects. Compared to fentanyl, remifentanil provided significantly more potent and rapid analgesia based on BPS measurements and a statistically non significantly shorter time to discharge. On the other hand, remifentanil caused a significantly sharper fall in heart rate within the first six hours of treatment. Differences between groups in terms of BUN and ALT were also recorded. Although both fentanyl-midazolam and remifentanil-midazolam can safely provide sedation and analgesia, fine differences between the fentanyl and remifentanil should be considered when selecting a suitable regimen for individual patients. |
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors [(potential) confounders]
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
Risk-of-bias tables
Table of quality assessment for systematic reviews of RCTs and observational studies
Based on AMSTAR checklist (Shea et al.; 2007, BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher et al 2009, PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Tan, 2009 |
Yes |
Yes |
Yes |
Yes |
Not applicable. |
No |
Yes |
Yes |
Yes |
- Research question (PICO) and inclusion criteria should be appropriate and predefined
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs)
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table etc.)
- Clinical and statistical heterogeneity should be assessed; clinical: enough similarities in patient characteristics, intervention and definition of outcome measure to allow pooling? For pooled data: assessment of statistical heterogeneity using appropriate statistical tests (e.g. Chi-square, I2)?
- An assessment of publication bias should include a combination of graphical aids (e.g., funnel plot, other available tests) and/or statistical tests (e.g., Egger regression test, Hedges-Olken). Note: If no test values or funnel plot included, score “no”. Score “yes” if mentions that publication bias could not be assessed because there were fewer than 10 included studies.
- Sources of support (including commercial co-authorship) should be reported in both the systematic review and the included studies. Note: To get a “yes,” source of funding or support must be indicated for the systematic review AND for each of the included studies.
Table of quality assessment for RCTs
|
Study reference
(first author, publication year) |
Was the allocation sequence adequately generated? a
Definitely yes Probably yes Probably no Definitely no |
Was the allocation adequately concealed?b
Definitely yes Probably yes Probably no Definitely no |
Blinding: Was knowledge of the allocated interventions adequately prevented?c
Were patients blinded?
Were healthcare providers blinded?
Were data collectors blinded?
Were outcome assessors blinded?
Were data analysts blinded?
Definitely yes Probably yes Probably no Definitely no |
Was loss to follow-up (missing outcome data) infrequent?d
Definitely yes Probably yes Probably no Definitely no |
Are reports of the study free of selective outcome reporting?e
Definitely yes Probably yes Probably no Definitely no |
Was the study apparently free of other problems that could put it at a risk of bias?f
Definitely yes Probably yes Probably no Definitely no |
Overall risk of bias If applicable/necessary, per outcome measure
LOW Some concerns HIGH
|
|
Liu, 2017 |
Definitely yes;
Reason: Different treatments were offered to patients in identical vials and boxes. Each box was also labelled with a numerical code, unique to treatment allocation and again blinded from both the investigator and study participant, as an additional measure to allow review of the correct treatment allocation by the study nurse. |
Definitely yes;
Reason: Randomization was performed by the sealed envelope system, in which the study nurse randomly opened a preformed envelope containing the allocated treatment regimen. |
No information;
Reason: No information was provided about the blinding of patients, healthcare providers, data collectors, outcome assessors and data analysts (except from the study nurses). |
Definitely yes;
Reason: Missing data / loss to follow-up was infrequent (n=0) |
Definitely yes;
Reason: All predefined outcome measures were reported. |
Definitely yes;
Reason: No other problems reported. |
LOW |
|
Spies, 2011 |
Definitely yes;
Reason: Patients who met enrolment criteria were randomly allocated in a 1:1 ratio to the fentanyl or remifentanil group. The randomization list was created by using computer based block randomization by a biostatistician with no patient contact. |
Definitely yes;
Reason: Allocation concealment was ensured by pharmacy-controlled randomization: on the basis of the randomization list, the pharmacies in the respective centres distributed patient-specific syringe pumps and bolus syringes (morphine in the remifentanil group and saline in the fentanyl group) that were identical in size, weight, and appearance. |
Definitely yes;
Reason: All study investigators, staff members, and patients were blinded to the study medication |
Probably yes;
Reason: Loss to follow-up was infrequent (five patients were excluded) |
Definitely yes;
Reason: All predefined outcome measures were reported. |
Probably no;
Reason: The trial was stopped early due to futility.
Even though median RASS values as well as median duration of propofol infusion did not differ significantly between groups we cannot exclude that ‘‘oversedation’’ might have masked a possible outcome benefit. |
LOW |
|
Cevik, 2011 |
No information;
Reason: No information was provided about the generation of the allocation sequence. |
No information;
Reason: No information was proved about the allocation concealment. |
No information;
Reason: No information was provided about the blinding of patients, healthcare providers, data collectors, outcome assessors and data analysts. |
Probably yes: patients (one patient in the fentanyl/midazolam group and one patient in the remifentanil/midazolam group) had to be excluded because they died before weaning during the study. |
Definitely yes;
Reason: All relevant outcomes were reported. |
Definitely yes;
Reason: No other problems reported. |
Some concerns |
- Randomization: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomization process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomization (performed at a site remote from trial location). Inadequate procedures are all procedures based on inadequate randomization procedures or open allocation schedules..
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments, but this should not affect the risk of bias judgement. Blinding of those assessing and collecting outcomes prevents that the knowledge of patient assignment influences the process of outcome assessment or data collection (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is usually not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary. Finally, data analysts should be blinded to patient assignment to prevents that knowledge of patient assignment influences data analysis.
- If the percentage of patients lost to follow-up or the percentage of missing outcome data is large, or differs between treatment groups, or the reasons for loss to follow-up or missing outcome data differ between treatment groups, bias is likely unless the proportion of missing outcomes compared with observed event risk is not enough to have an important impact on the intervention effect estimate or appropriate imputation methods have been used.
- Results of all predefined outcome measures should be reported; if the protocol is available (in publication or trial registry), then outcomes in the protocol and published report can be compared; if not, outcomes listed in the methods section of an article can be compared with those whose results are reported.
- Problems may include: a potential source of bias related to the specific study design used (e.g. lead-time bias or survivor bias); trial stopped early due to some data-dependent process (including formal stopping rules); relevant baseline imbalance between intervention groups; claims of fraudulent behavior; deviations from intention-to-treat (ITT) analysis; (the role of the) funding body. Note: The principles of an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
- Overall judgement of risk of bias per study and per outcome measure, including predicted direction of bias (e.g. favors experimental, or favors comparator). Note: the decision to downgrade the certainty of the evidence for a particular outcome measure is taken based on the body of evidence, i.e. considering potential bias and its impact on the certainty of the evidence in all included studies reporting on the outcome.
Exclusietabel
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Chinachoti, 2002 |
Was al geïncludeerd in de review van Tan (2009) |
|
Dahaba, 2004 |
Was al geïncludeerd in de review van Tan (2009) |
|
Devabhakthuni, 2021 |
Bevat geen meta-analyse. |
|
Kunisawa, 2014 |
Verkeerde uitkomstmaat meegenomen (general dexmedetomidine (DEX) concentration required for sedation) |
|
Muellejans, 2004 |
Was al geïncludeerd in de review van Tan (2009) |
|
Muellejans, 2006 |
Was al geïncludeerd in de review van Tan (2009) |
Verantwoording
Autorisatiedatum en geldigheid
Laatst beoordeeld : 16-12-2022
Laatst geautoriseerd : 16-12-2022
Geplande herbeoordeling :
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Doel en doelgroep
Het doel is herziening van bestaande trias van Sedatie en Analgesie richtlijnen, waar sedatie en analgesie op de IC een onderdeel van is. De Sedatie en Analgesie richtlijnen hebben betrekking op alle patiënten die sedatie krijgen buiten de OK. De richtlijn zal blijven bestaan uit drie delen – volwassenen; kinderen; en IC. Deze patiëntencategorieën kennen specifieke risico’s waardoor specifieke aanbevelingen nodig zijn.
Sedatie en analgesie van de patiënten die een IC-behandeling krijgen verschilt steeds meer van procedurele sedatie en analgesie. Daarom staat deze IC-richtlijn in de nieuwe sedatie en alagesie-richtlijn meer los van de twee sedatie en analgesie richtlijnen (volwassenen en kinderen). Voorliggende richtlijn beschrijft het comfortabel krijgen en houden van patiënten die een IC-behandeling ondergaan, het tegengaan van pijn, agitatie en stress. Het beschrijft de medicamenteuze en niet medicamenteuze opties om de patiënt met zo min mogelijk complicaties in zo kort mogelijke tijd naar zo hoog mogelijk functioneel herstelniveau te krijgen, zowel fysiek als mentaal.
De richtlijn heeft betrekking op intensivisten en IC-verpleegkundigen. Daarnaast kan de richtlijn gebruikt worden door zorgverleners die betrokken zijn bij de behandeling op of na de Intensive Care zoals de mee behandelende medisch specialisten, paramedici, verpleegkundig specialisten en apothekers.
Er is afstemming met de twee andere onderdelen van de richtlijn Sedatie en Analgesie, namelijk die voor volwassenen en voor kinderen en mag samen met deze onderdelen als geheel worden gezien.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij sedatie en analgesie van patiënten op de IC. Inbreng patiënten- en naastenperspectief via Stichting FCIC en patiëntenorganisatie IC Connect.
Werkgroep
Drs. N.C. (Niels) Gritters van de Oever, anesthesioloog-intensivist, Treant zorggroep, NVIC
Dr. K.S. (Koen) Simons, internist-intensivist, Jeroen Bosch Ziekenhuis, NVIC
Dr. M. (Marissa) Vrolijk, anesthesioloog-intensivist-, LangeLand Ziekenhuis Zoetermeer, NVIC
Dr. L. (Lena) Koers, anesthesioloog-kinderintensivist, Leiden UMC, NVA
Dr. H. (Rik) Endeman, internist-intensivist, Erasmus MC, NIV
Dr. M. (Mark) van den Boogaard, IC verpleegkundige, senior onderzoeker, Radboud UMC, V&VN
Drs. R. (Roel) van Oorsouw, fysiotherapeut, Radboud UMC, KNGF
Dr. N.G.M. (Nicole) Hunfeld, ziekenhuisapotheker, Erasmus MC, NVZA
Drs. W.P. (Wai-Ping) Manubulu-Choo, ziekenhuisapotheker, Martini Ziekenhuis, NVZA
Drs. M. (Marianne) Brackel, patiëntvertegenwoordiger, Stichting FCIC en patiëntenorganisatie IC Connect, Jeugdarts knmg niet praktiserend
Met methodologische ondersteuning van
• Drs. Florien Ham, junior adviseur, Kennisinstituut van de Federatie Medisch Specialisten
• Drs. Toon Lamberts, senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten
• Dr. Mirre den Ouden - Vierwind, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
• Drs. Ingeborg van Dusseldorp, senior informatiespecialist, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Drs. N.C. (Niels) Gritters van de Oever |
Anesthesioloog-intensivist Treant zorggroep. |
Onbezoldigd en t/m juni 2021: NVIC bestuur ( NVIC accreditatie commissie NVIC luchtwegcommissie, congrescommissie) Chief Medical Officer van de LCPS (dagvergoeding) t/m maart 2022 voorzitter farmacotherapiecommissie NVIC per juli 2022 lid stafbestuur Treant ziekenhuisgroep per aug 2022 (vacatievergoeding) lid scientific board covidpredict per 2020 (onbezoldigd), commissie Acute Tekorten Geneesmiddelen per 2020 (vergadervergoeding) |
Geen. |
Geen actie. |
|
Dr. K.S. (Koen) Simons |
Internist-intensivist Jeroen Bosch Ziekenhuis. |
FCCS-instructeur bij NVIC (ca. 2 dagen/jaar, betaald). |
Geen. |
Geen actie. |
|
Dr. M. (Marissa) Vrolijk |
Anesthesioloog- intensivist LangeLand Ziekenhuis. |
Medisch beoordelaar adoptie bij Adoptie Stichting Meiling. |
Geen. |
Geen actie. |
|
Dr. L. (Lena) Koers |
Anesthesioloog-kinderintensivist LUMC |
Geen. |
Geen. |
Geen actie. |
|
Dr. H. (Rik) Endeman |
Internist-intensivist, Erasmus MC. |
Voorzitter van de gemeenschappelijke intensivisten commissie (GIC) (onbetaald). |
TravelGrant van Getinge om te spreken op een lunchsymposium in Kenya voor het jaarcongres (2018) van de Kenyan Society of Anaesthesiology and Intensive Care Medicine. Dit congres wordt ook veelbezocht door anaesthesisten en intensivisten uit omliggende landen. Het onderwerp was 'Hemodynamic monitoring in low resource and high resource environments', duurde een uur, en ging over wat voor meetinstrumenten je kan gebruiken om de hemodynamiek te bewaken indien je geen middelen hebt (low resource) en wel (high resource); duurde uiteindelijk 90 minuten. GETINGE verkoopt high-end HD-monitoring, deze kwamen in de presentatie voor, maar ook die van concurrenten. GETINGE had mij niets opgelegd m.b.t. de inhoud van de presentatie. PI van: Open Lung Concept 2.0 studie: Flow controlled ventilation (gesponsord door Ventinova). |
Geen actie. Het gesponsorde onderzoek is niet gerelateerd aan het onderwerp van de richtlijn. |
|
Dr. M. (Mark) van den Boogaard |
Senior onderzoeker, afdeling lntensive Care, Radboudumcc. |
Onbezoldigde functies: - Bestuurslid European Delirium Association. - Adviseur Network for lnvestigation of Delirium: Unifying Scientists (NIDUS). - Organisator IC-café regio Nijmegen & Omstreken. - Lid werkgroep Longterm Outcome and ICU Delirium van de European Society of lntensive Care Medicine. - Lid richtlijn Nazorg en revalidatie IC-patiënten. |
PI van onderstaande gesubsidieerde projecten: ZonMw subsidies: - programma GGG [2013]: Prevention of ICU delirium and delirium-related outcome with haloperidol; a multicentre randomized controlled trial. - programma DO [2015]: The impact of nUrsing DEliRium Preventive lnterventions in the lntensive Care Unit (UNDERPIN-ICU). Delirium komt terug in beide projecten en in de richtlijn, maar er zullen geen aanbevelingen naar vormen komen die belangenverstrengeling veroorzaken. Geen problemen worden voorzien, vanwege het niet betrokken zijn bij aanbevelingen over medicatie/verpleegkundige interventie obv door ZonMw gesubsidieerde projecten. ZIN subsidie: - programma Gebruiken van uitkomsteninformatie bij Samen beslissen [2018]: Samen beslissen op de IC: het gebruik van (patiëntgerapporteerde) uitkomst informatie bij gezamenIijke besluitvorming over IC-opname en behandelkeuzes op de IC. |
Geen actie. |
|
Drs. R. (Roel) van Oorsouw |
Fysiotherapeut/ PhD-kandidaat Radboudumc. |
Visiterend docent op de Hogeschool. Arnhem Nijmegen (10 uur per jaar) Congrescommissie NVZF (tot oktober 2021) Lid ethiekcommissie KNGF (sinds oktober 2021). |
Geen. |
Geen actie. |
|
Dr. N.G.M. (Nicole) Hunfeld |
Ziekenhuisapotheker ErasmusMC. |
Bestuurslid KNMP (functie: penningmeester, betaald). |
Boegbeeldfunctie, maar sedatie heeft geen relatie met openbare farmacie. |
Geen actie. |
|
Drs. W.P. (Wai-Ping) Manubulu-Choo |
Ziekenhuisapotheker, Martini Ziekenhuis. |
Geen. |
Geen. |
Geen actie. |
|
Drs. M. (Marianne) Brackel |
Patiëntvertegenwoordiger, Stichting FCIC en patiëntenorganisatie IC Connect, Jeugdarts knmg niet praktiserend. |
Geen. |
Geen. |
Geen actie. |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door het uitnodigen van Stichting Family and patient Centered Intensive Care (FCIC) en de patiëntenorganisatie IC Connect voor deelname aan de werkgroep. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de FCIC en IC Connect en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
Uit de kwalitatieve raming blijkt dat er geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
|
Module |
Uitkomst kwalitatieve raming |
Toelichting |
|
Module 1 Pijnprotocol op de IC |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module 2 Lichte sedatie op de IC |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module 3a Analgetica versus hypnotica |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat [het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module 3b Analgeticum bij analgosedatie |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module 4 Intraveneuze medicatie op de IC |
Geen substantiële financiële gevolgen. |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module 5 Inhalatieanesthetica op de IC |
Geen substantiële financiële gevolgen. |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module 6 Nonfarmacologische interventies |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module 7 Organisatie van zorg |
Geen substantiële financiële gevolgen. |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep de knelpunten in de zorg voor patiënten met sedatie en analgesie op de IC. Er is een knelpuntenanalyse gehouden samen met de andere Sedatie en analgesie richtlijnen. Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld. Ook beoordeelde de werkgroep de aanbeveling(en) uit de eerdere richtlijn Procedurele Sedatie en analgesie bij volwassenen op de Intensive Care (NVK/NVA, 2012) op noodzaak tot revisie. Tevens is er gekeken naar samenhang met andere bestaande en te ontwikkelen richtlijnen, o.a. de richtlijn Delirium op de Intensive Care (NVIC, 2010) en de richtlijn Nazorg en revalidatie van intensive care patiënten (VRA/NVIC) en aansluiting op de Europese richtlijn PADIS, 2018, die over hetzelfde onderwerp gaat. Ten tijde van de autorisatiefase van deze richtlijn verscheen de richtlijn sepsis (NIV), waarin onderdelen van de sedatie van de septische IC-patiënt worden besproken. Beide richtlijnen vullen elkaar aan.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
er is hoge zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; het is zeer onwaarschijnlijk dat de literatuurconclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Redelijk |
er is redelijke zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; het is mogelijk dat de conclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Laag |
er is lage zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; er is een reële kans dat de conclusie klinisch relevant verandert wanneer er resultaten van nieuw grootschalig onderzoek aan de literatuuranalyse worden toegevoegd. |
|
Zeer laag |
er is zeer lage zekerheid dat het ware effect van behandeling dichtbij het geschatte effect van behandeling ligt; de literatuurconclusie is zeer onzeker. |
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Zoekverantwoording
Zoekacties zijn opvraagbaar. Neem hiervoor contact op met de Richtlijnendatabase.