Biologicals als aanvullende behandeling bij ernstig astma
Uitgangsvraag
Wat is de toegevoegde waarde van immuunmodulerende therapie (biologicals) bij patiënten met ernstig astma?
De uitgangsvraag omvat de volgende deelvragen:
- Wat is de toegevoegde waarde van Omalizumab (anti-IgE) als behandeling bij ernstig astma?
- Wat is de toegevoegde waarde van Mepolizumab (anti-IL-5) als behandeling bij ernstig astma?
- Wat is de toegevoegde waarde van Reslizumab (anti-IL-5) als behandeling bij ernstig astma?
- Wat is de toegevoegde waarde van Benralizumab (anti-IL-5R) als behandeling bij ernstig astma?
- Wat is de toegevoegde waarde van Dupilumab (anti-IL-4R) als behandeling bij ernstig astma?
Aanbeveling
Bespreek de mogelijkheden en risico’s van gebruik biologicals met de ernstig astmapatiënt en kom gezamenlijk tot een besluit omtrent de toepassing hiervan. Leg dit besluit vast.
De indicatiestelling voor behandeling met biologicals vindt bij voorkeur plaats in of in nauw overleg met een kennis- of behandelcentrum voor patiënten met ernstig astma (zie ook module Organisatie van zorg).
Overweeg behandeling met omalizumab bij patiënten met ernstig, allergisch, IgE-gemedieerd astma die ondanks optimale medicamenteuze behandeling twee of meer astma-aanvallen per jaar hebben.
Overweeg behandeling met anti-IL-5 (mepolizumab, reslizumab) of anti-IL-5R (benralizumab) bij patiënten met ernstig astma met eosinofiele luchtweginflammatie, die ondanks optimale medicamenteuze behandeling twee of meer astma-aanvallen per jaar hebben.
Overweeg behandeling met anti-IL-5 (mepolizumab, reslizumab) of anti-IL-5R (benralizumab) bij patiënten met ernstig eosinofiel astma bij wie het astma slechts met onderhoudsbehandeling met orale corticosteroïden onder controle te houden is.
Overweeg behandeling met anti-IL-4R (dupilumab) bij patiënten met ernstig astma met type 2-inflammatie patroon, die ondanks optimale medicamenteuze behandeling twee of meer astma-aanvallen hebben per jaar.
Overweeg behandeling met anti-IL-4R (dupilumab) bij patiënten met ernstig type 2 astma bij wie het astma slechts met onderhoudsbehandeling met orale corticosteroïden onder controle te houden is.
Beoordeel na 4 tot 6 maanden het effect van de behandeling met biologicals en overweeg behandeling te switchen bij onvoldoende effect. Overleg met een kennis- of behandelcentrum over een alternatieve behandeling.
Overwegingen
Omalizumab
Behandeling met omalizumab bij ernstig allergisch astma is mogelijk effectief in het verbeteren van kwaliteit van leven en het reduceren van astma-aanvallen. Slechts bij langere follow-up (> 28 weken) is er een effect zichtbaar op astma-aanvallen, hoewel het verschil met de placebogroep vrij gering is. Het kan veilig subcutaan worden toegediend. Dosis en doseringsfrequentie worden berekend op basis van de hoogte van het totaal IgE in het bloed en het gewicht van de patiënt (EMA, 2012). De toedieningsfrequentie is 1x per 2 of 4 weken. De respons is hoger in patiënten met bloed eosinofielen > 250/mcl en ‘fraction exhaled nitric oxide’ (FeNO) > 20 ppb. Er is onvoldoende bewijs dat omalizumab ook werkzaam is buiten de doseringstabel. Hoewel er aanwijzingen zijn dat omalizumab ook werkzaam zou kunnen zijn bij niet-allergisch astma is het bewijs hiervoor vooralsnog te beperkt om hier enige aanbeveling over te kunnen doen (Pillai, 2016). Gezien de hoge kosten is het enkel geïndiceerd voor patiënten met ernstig IgE-gemedieerd allergisch astma.
Omalizumab lijkt een gunstig veiligheidsprofiel te hebben. Met betrekking tot eventuele bijwerkingen op langere termijn is er mogelijk een verhoogd risico op arteriële trombo-embolie, zoals TIA of myocardinfarct (Iribarren, 2017).
Op basis van twee studies (Nopp, 2010; Ledford, 2017) lijkt het mogelijk te zijn om bij astmapatiënten die meer dan 5 jaar omalizumab hebben gebruikt, te proberen de behandeling te stoppen. De kans op een astma-aanval is weliswaar hoger in de groep die stopt dan in de omalizumabgroep, maar iets meer dan de helft van de patiënten kreeg helemaal geen astma-aanval meer. Een subanalyse in de studie van Ledford (2017) liet zien dat de patiënten die ten tijde van het stoppen van de omalizumab hoge eosinofielen in het bloed hadden (> 300) een hogere kans hadden om een astma-aanval te krijgen. Bepalen van bloedeosinofielen zou kunnen helpen bij de selectie van patiënten die na 5 jaar kunnen stoppen.
Mepolizumab
Behandeling met mepolizumab bij ernstig eosinofiel astma is effectief, met name bij patiënten, bij wie ondanks behandeling met een combinatie hoge dosis ICS/LABA regelmatig astma-aanvallen optreden en eosinofile inflammatie persisteert. In een aantal studies van goede kwaliteit is een klinisch relevante reductie in astma-aanvallen en verbetering van kwaliteit van leven aangetoond. Daarnaast lijkt een afname in behoefte aan onderhoudsbehandeling met orale steroïden en verbetering in astmacontrole waarschijnlijk. In de studie van Bel (2014) worden de verbeteringen in ACQ, AQLQ, exacerbaties en longfunctie in de actieve behandelgroep waarschijnlijk zelfs nog onderschat. Mepolizumab is geregistreerd voor subcutane toediening (100 mg/ 4 weken).
Behandeling met mepolizumab dient voorbehouden te blijven voor patiënten met verhoogde perifere eosinofilie, waarbij als criterium wordt aanbevolen bloed eo’s > 300 of > 150 bij astmapatiënten die chronisch orale corticosteroïden gebruiken (Sectie Astma & Allergie NVALT 2016), of bij patiënten waarbij een verhoogde eosinofiele inflammatie in de luchtwegen wordt gevonden met sputum inductie en de eosinofile cellen > 2% zijn.
Mepolizumab lijkt een gunstig veiligheidsprofiel te hebben. De meest voorkomende bijwerkingen (incidentie ≥ 5%) zijn hoofdpijn, reactie op injectieplaats, rugpijn en moeheid. Bij mepolizumab kunnen daarnaast urineweginfecties optreden. Met betrekking tot eventuele bijwerkingen op langere termijn kan nog geen uitspraak worden gedaan. Evenmin kan op dit moment aangegeven worden hoe lang de behandeling gecontinueerd dient te worden. Gezien de hoge kosten is het enkel geïndiceerd voor patiënten met ernstig astma met aantoonbare eosinofiele inflammatie.
Reslizumab
Behandeling met reslizumab bij ernstig eosinofiel astma is effectief, met name bij patiënten bij wie ondanks behandeling met een combinatie hoge dosis ICS/LABA regelmatig astma-aanvallen optreden. In een aantal studies van goede kwaliteit is een klinisch relevante reductie in astma-aanvallen aangetoond. Behandeling met reslizumab dient voorbehouden te blijven voor patiënten met verhoogde perifere eosinofilie, waarbij als criterium wordt aanbevolen bloed eo’s > 400 of > 150 bij astmapatiënten die chronisch orale corticosteroïden gebruiken. Reslizumab is geregistreerd voor intraveneuze toediening (3mg/kg/ 4 weken) en is de enige anti-IL-5 waarbij de dosis afhankelijk is van het gewicht.
Reslizumab lijkt een gunstig veiligheidsprofiel te hebben. Een bijwerking die bij 1 tot 10% van de patiënten voorkomt, is stijging van het serumcreatinekinase. Deze bijwerking is tijdelijk en asymptomatisch (SmPC, 2019). Met betrekking tot eventuele bijwerkingen op langere termijn kan nog geen uitspraak worden gedaan. Evenmin kan op dit moment aangegeven worden hoe lang de behandeling gecontinueerd dient te worden.
Een nadeel is de intraveneuze toediening, wat overigens geen bezwaar hoeft te zijn voor een poliklinische setting. Gezien de hoge kosten is het enkel geïndiceerd voor patiënten met ernstig astma met aantoonbare eosinofiele inflammatie.
Benralizumab
Behandeling met benralizumab bij ernstig eosinofiel astma is effectief, met name bij patiënten, bij wie ondanks behandeling met een combinatie hoge dosis ICS/LABA regelmatig astma-aanvallen optreden. In een aantal studies van goede kwaliteit is een klinisch relevante reductie in astma-aanvallen aangetoond. Daarnaast lijkt een afname in behoefte aan onderhoudsbehandeling met orale corticosteroïden en verbetering in astmacontrole waarschijnlijk. In de studie van Nair (2017) worden de verbeteringen in ACQ, AQLQ, exacerbaties en longfunctie in de actieve behandelgroep waarschijnlijk zelfs nog onderschat. Behandeling met benralizumab dient voorbehouden te blijven voor patiënten met verhoogde perifere eosinofilie, waarbij als criterium wordt aanbevolen bloed eo’s > 300 of > 150 bij astmapatiënten die chronisch orale corticosteroïden gebruiken. Benralizumab is geregistreerd voor 30 mg subcutane toediening en kan na de eerste drie injecties (a 4 weken) 8-wekelijks worden toegediend.
Benralizumab lijkt een gunstig veiligheidsprofiel te hebben. De meest voorkomende bijwerkingen (incidentie > 1%) zijn faryngitis, overgevoeligheidsreacties, koorts en reacties op de injectieplaats. Met betrekking tot eventuele bijwerkingen op langere termijn kan nog geen uitspraak worden gedaan. Evenmin kan op dit moment aangegeven worden hoe lang de behandeling gecontinueerd dient te worden. Gezien de hoge kosten is het enkel geïndiceerd voor patiënten met ernstig astma met aantoonbare eosinofile inflammatie.
Dupilumab
Dupilumab heeft een aangetoonde effectiviteit op vrijwel alle uitkomstmaten. In een aantal studies van goede kwaliteit is een klinisch relevante reductie in astma-aanvallen en afname in behoefte aan onderhoudsbehandeling met orale corticosteroïden aangetoond. Daarnaast lijkt een verbetering van kwaliteit van leven, astmacontrole en longfunctie waarschijnlijk. In de studie van Rabe (2018) worden de verbeteringen in ACQ, exacerbaties en longfunctie in de actieve behandelgroep waarschijnlijk zelfs nog onderschat. Dupilumab is door de EMA geregistreerd voor de behandeling van matig tot ernstig astma met type 2-inflammatie patroon gekenmerkt door verhoogde bloed-eosinofielen en/of verhoogde FeNO. Het is in 2 dosis beschikbaar (200 en 300 mg) en wordt iedere 2 weken toegediend. De eerste dosis bestaat uit 2 injecties (400 resp 600 mg). De 300 mg/2 weken is geadviseerd voor astmapatiënten die systemische corticosteroïden gebruiken of bij wie er tevens sprake is van matig tot ernstig constiutioneel eczeem of neuspoliepen. Dupilumab is ook voor beide indicaties afzonderlijk geregistreerd. Wanneer dit niet het geval is, dan is 200 mg/2 weken voldoende. Gezien de hoge kosten is het enkel geïndiceerd voor patiënten met ernstig astma met een type 2-inflammatie patroon.
Dupilumab lijkt een gunstig veiligheidsprofiel te hebben. Bijwerkingen zijn vergelijkbaar met andere biologicals, met uitzondering van oogklachten hetgeen bij dupilumabgebruik kan voorkomen, met name wanneer er ook sprake is van atopisch eczeem. Dit betreft met name niet-ernstige oogklachten, zoals conjunctivitis of blefaroconjunctivitis, maar er zijn ook ernstigere oogklachten gemeld, waaronder limbale-stamceldeficiëntie. Incidenteel kan een tijdelijke toegenomen bloedeosinofilie worden gezien. Bij eo’s > 3000 per μL kan dit potentieel leiden tot eindorgaanschade en dient de behandeling te worden gestopt. Met betrekking tot eventuele bijwerkingen op langere termijn kan nog geen uitspraak worden gedaan. Evenmin kan op dit moment aangegeven worden hoe lang de behandeling gecontinueerd dient te worden.
Overkoepelende overwegingen - samenvatting en selectie van patiënten geschikt voor biological behandeling (Figuur 6 en 7)
Nu we de beschikking hebben gekregen over deze nieuwe biologicals voor de behandeling van ernstig astma, is er eindelijk hoop op een bijna normaal leven voor een deel van de patiënten met deze invaliderende ziekte. Als het veiligheidsprofiel van deze biologicals zo goed blijft als tot heden het geval is, zullen deze geneesmiddelen een belangrijke doorbraak betekenen in de behandeling van ernstig astma.
Ondanks dit veelbelovende nieuws zijn er nog steeds veel vragen, valkuilen en dilemma's in de behandeling van patiënten met ernstig astma in de dagelijkse praktijk.
Allereerst is het belangrijk bij patiënten met ernstig ongecontroleerd astma om te (her)checken of alle factoren aangepakt zijn en waar mogelijk geelimineerd die astmasymptomen kunnen verergeren, waaronder slechte therapietrouw, psychopathologie, inadequate inhalatietechniek, aanhoudende blootstelling aan sensibiliserende of niet-sensibiliserende triggers, comorbiditeiten, of gebruik van medicijnen zoals bètablokkers of NSAID's (Van der Meer, 2016).
Vervolgens is het essentieel om bij elke individuele patiënt te testen of de astmasymptomen gerelateerd zijn aan actieve type 2 inflammatie van de luchtwegen (zie figuur 1). Momenteel wordt dit het beste weerspiegeld door verhoogde surrogaatmarkers waaronder totaal IgE, bloed of sputum eosinofielen, of uitgeademde NO (Lim, 2018). Afkapwaarden van deze markers zijn nog steeds onderwerp van discussie, maar om praktische redenen zijn in het huidige algoritme afkapwaarden door de richtlijnwerkgroep voorgesteld. Een herhaalde meting van deze markers is nodig om type 2 inflammatie met zekerheid te bevestigen of uit te sluiten (Coumou, 2018). Hierbij kan het meten van type 2 markers ten tijde van een astma-aanval belangrijk zijn om het onderliggende mechanisme te begrijpen dat de astma-aanvallen veroorzaakt en type 2 inflammatie aan te tonen. Indien bloed of FeNo bij herhaling geen type 2 inflammatie tonen, maar wel type 2 inflammatie vermoed wordt, onder andere op grond van goed responderen op OCS, wordt aanbevolen om sputuminductie uit te voeren in een astma kennis- of behandelcentrum.
Het is belangrijk om te beseffen dat type 2 inflammatie onderdrukt kan worden bij patiënten die chronische OCS gebruiken. Bij deze patiënten kunnen type 2-markers weer verschijnen zodra de dosis OCS geleidelijk wordt verlaagd.
Bij patiënten met zeer hoge waarden van bloed eosinofielen (≥ 1500 cellen/ mL) of die hoge doses OCS nodig hebben (> 20 mg/ dag prednison-equivalent) om type 2 inflammatie te onderdrukken, is het raadzaam om andere oorzaken uit te sluiten (bijvoorbeeld parasitaire infecties, drug-geinduceerd, hypereosinofiel syndroom, eosinofiele granulomatose en polyangiitis (EGPA)).
Figuur 6 Selectie van astmapatiënten voor behandeling met biologicals
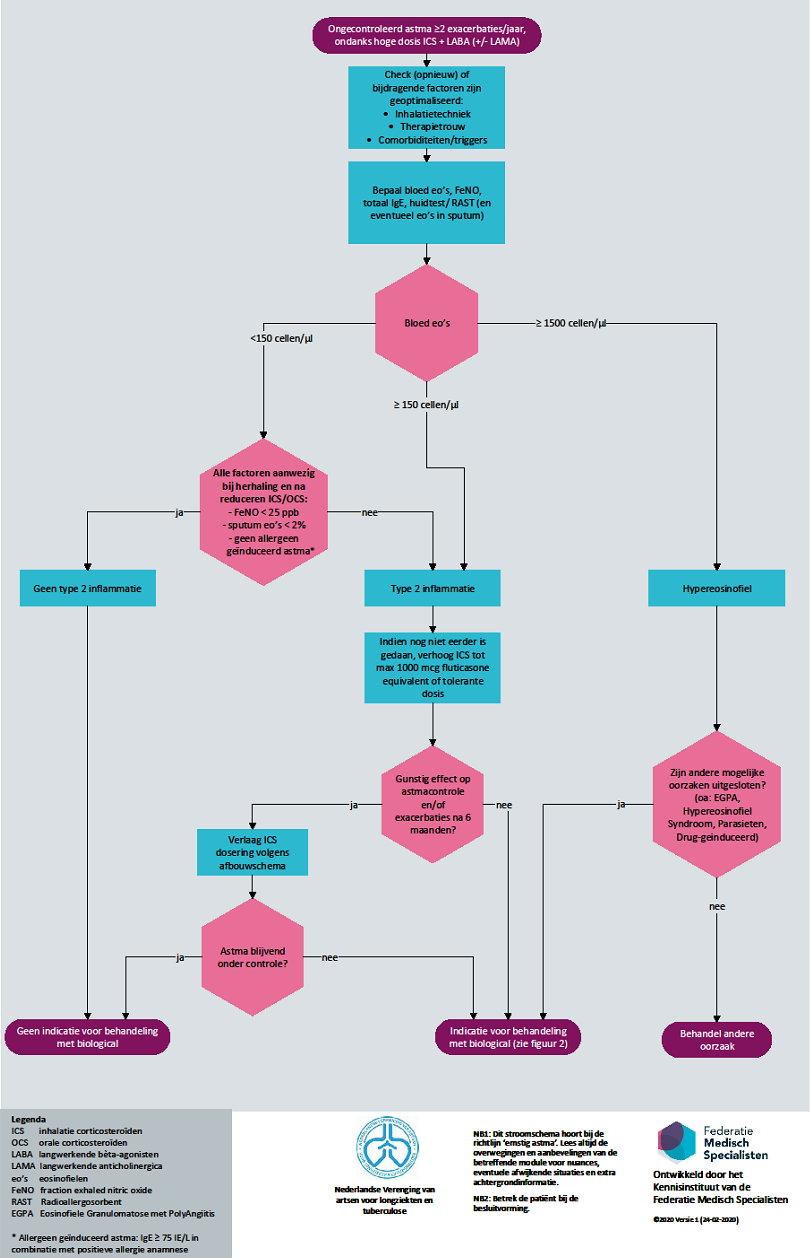
Bij patiënten met ernstig ongecontroleerd astma en een bewezen type 2 inflammatie kunnen biologicals worden overwogen. Niet al deze patiënten reageren echter even succesvol op biologicals. Sommigen reageren zeer goed op een anti-IL-5 of anti IL-5-receptor, anderen veel beter op anti-IgE of een anti-IL-4-receptor. Tevens is er het dilemma met betrekking tot de selectie tussen beschikbare anti-IL-5/ IL-5R-biologicals (mepolizumab, reslizumab of benralizumab). Hoewel er weinig gegevens beschikbaar zijn om te adviseren van welk anti-IL-5-geneesmiddel de patiënt waarschijnlijk het meeste baat zal hebben, leert de praktijk dat de drie middelen niet uitwisselbaar zijn. Het ontbreken van response op 1 anti-IL-5(R) middel sluit een goede respons op een ander anti-IL-5(R) middel zeker niet uit. Dit vraagt om het beschikbaar houden van de verschillende middelen zodat de mogelijkheid om een ander middel te proberen in voorkomende gevallen openblijft. Helaas kan met de momenteel beschikbare biomarkers de respons op een specifiek biological niet worden voorspeld bij de individuele patiënt met type 2-inflammatie (Carr, 2018). Dit vereist meer specifieke biomarkers geassocieerd met geactiveerde moleculaire routes. Op basis van de beschikbare onderzoeken en eigen klinische ervaring hebben we een suggestie gedaan voor een behandelalgoritme betreffende biologicals. Dit algoritme wordt aangeboden ter ondersteuning van de dagelijkse praktijk, waarbij duidelijk mag zijn dat de effectiviteit van dit algoritme niet is geëvalueerd.
Indien verschillende opties voorhanden zijn, kan de keuze voor een bepaald middel medebepaald worden op grond van toedieningsvorm, gewichtsafhankelijke doseringsmogelijkheid, doseringsinterval of mogelijkheid van thuis/zelftoediening. Omalizumab wordt 2 tot 4-wekelijks subcutaan toegediend volgens doseerschema gebaseerd op totaal IgE en gewicht. Mepolizumab wordt toegediend als een vaste subcutane dosering elke 4 weken, en reslizumab als een 4-wekelijkse intraveneuze dosering welke gewichtsafhankelijk is. Daarnaast wordt benralizumab gegeven in een 8-wekelijkse vaste subcutane dosering (na opstartfase). Als laatste heeft dupilumab een 2-wekelijks vaste subcutane dosering. Zelftoediening is voor alle biologicals inmiddels beschikbaar. De werkgroep adviseert na aanvang van behandeling met biological eerst de OCS af te bouwen en zo mogelijk te stoppen, en vervolgens pas de andere add-on medicatie. Ten aanzien van ICS wordt geadviseerd na 3 tot 6 maanden waar mogelijk af te bouwen, maar wel minimaal een middelhoge dosis (zie module Diagnostiek, tabel 2) te handhaven. Verder adviseert de werkgroep alert te blijven ten aanzien van de mogelijkheid van bijnierschorsinsufficientie bij het afbouwen/staken van de OCS. De werkgroep beveelt aan om bij afbouw prednison ≤ 5 mg/dag standaard ochtendcortisol te bepalen en bij ochtendcortisolwaarden < 250 nmol/L ook een ACTH stimulatietest te laten verrichten.
Het effect van biological behandeling moet na 4 tot 6 maanden worden geëvalueerd. Het besluit om al dan niet de behandeling te continueren wordt gewoonlijk in samenspraak tussen patiënt en behandelaar genomen en gebaseerd op zowel objectieve metingen als ook subjectieve beoordeling, waarbij ruime ervaring bij de behandelaar uitermate behulpzaam kan zijn. Er is nog geen eenduidigheid hoe respons gedefinieerd dient te worden, maar er wordt sterk aanbevolen om gebruik te maken van objectieve gegevens, waarin het evalueren van OCS-dosering, astma-aanval frequentie, astmacontrole, kwaliteit van leven en longfunctie nuttig lijken te zijn. Als het effect niet bevredigend is en er andere biologicals beschikbaar zijn, kan worden overwogen om over te schakelen naar een ander type 2 biological, waarna het effect opnieuw na 4 tot 6 maanden dient te worden geëvalueerd. Als er geen alternatief biological beschikbaar is, kan de proefperiode worden verlengd tot maximaal 1 jaar voor een mogelijke later optredende respons.
In RCT’s, real-life studies en klinische ervaringen lijkt 60 tot 80% van de behandelde patiënten een goede respons te hebben op biological behandeling. Waarschijnlijk is er een klein percentage patiënten met type 2 inflammatie dat niet respondeert op alle momenteel 5 beschikbare biologicals. Voor deze patiënten kan OCS in onderhoudsdosis nodig blijken of een andere aanvullende behandeling worden overwogen te weten: bronchiale thermoplastiek, hooggebergtebehandeling, longrevalidatie, gewichtsvermindering of macroliden. Richtlijnen voor het veilig afbouwen van orale corticosteroïden zijn nog niet beschikbaar, maar men zou de taperingsschema's van de OCS-reductie trials met mepolizumab en benralizumab kunnen gebruiken (Bel, 2014; Nair, 2017).
Bij patiënten die geen type 2 inflammatie vertonen na uitgebreid testen (bloed-eosinofielen < 0,15 en sputum-eosinofielen < 2% en FeNO < 25 ppb en IgE < 75 IE L), zelfs na verlaging van de dosis OCS, is het toedienen van type 2 biologicals niet nuttig (Papi, 2018). Bij die patiënten moet men blijven zoeken naar andere factoren die astmasymptomen kunnen verergeren (Van der Meer, 2016). Afhankelijk van de bevindingen kan andere therapie geschikt zijn te weten: hooggebergtebehandeling, longrevalidatie, gewichtsvermindering, macroliden, bronchiale thermoplastiek.
In dit tijdperk van snelle ontwikkeling en een schat aan nieuwe behandelingen met een groot potentieel voor ernstige astmapatiënten, kan het voor artsen in de dagelijkse praktijk een uitdaging zijn om behandelingskeuzes te maken voor individuele patiënten. Hopelijk biedt het algoritme enige sturing en kan het bijdragen aan de personalized behandeling van ernstig astma (Chung, 2019).
Figuur 7 Stroomschema voor keuze* biological in type 2 ernstig astma
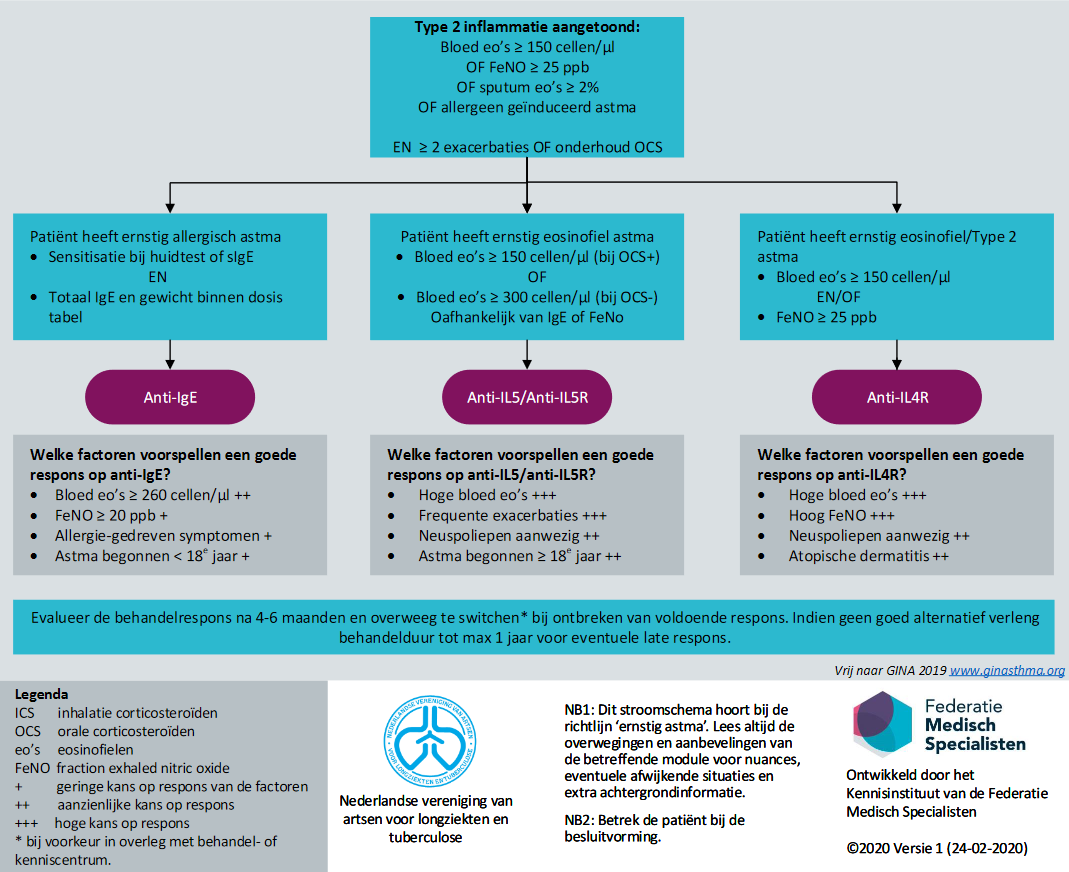
Indicatiestelling
Gezien het bovenstaande en mede omdat de behandeling kostbaar is, wordt geadviseerd de indicatiestelling voor behandeling met biologicals bij voorkeur plaats te laten vinden in overleg met een kennis- of behandelcentrum voor ernstig astma. Overleg met een kennis- en behandelcentra bij zwangerschap(swens).
De uiteindelijke keuze van de biological hangt af van het klinische plaatje, de aard van de inflammatie, de frequentie van toediening, de wens van de patiënt en de mogelijkheden voor zelftoediening. De verschillende productkenmerken van de biologicals staan beschreven in tabel 1, zodat mede aan de hand hiervan een optimale keus gemaakt kan worden.
Tabel 1 Productkenmerken biologicals volgens SMPC (samenvatting van productkenmerken)
|
|
Verpakking en toedieningsvorm |
Frequentie en wijze van toedienen |
Toediening toegestaan volgens SMPC door: |
|
omalizumab Xolair® |
poeder voor injectievloeistof 150 mg |
75–600 mg subcutaan per keer in 1 tot 4 injecties elke 2 of 4 weken. De dosis en de doseringsfrequentie worden bepaald door het lichaamsgewicht en door de serumconcentratie totaal IgE gemeten voor de start van de behandeling, zie hiervoor de SmPC van Xolair®; |
beroepsbeoefenaar in de gezondheidszorg |
|
wegwerpspuit 75 en 150 mg |
75–600 mg subcutaan per keer in 1 tot 4 injecties elke 2 of 4 weken. De dosis en de doseringsfrequentie worden bepaald door het lichaamsgewicht en door de serumconcentratie totaal IgE gemeten voor de start van de behandeling, zie hiervoor de SmPC van Xolair®; |
beroepsbeoefenaar in de gezondheidszorg patiënt* |
|
|
mepolizumab Nucala® |
poeder voor injectievloeistof 100 mg |
100 mg subcutaan eens per 4 weken |
beroepsbeoefenaar in de gezondheidszorg |
|
voorgevulde pen 100 mg |
100 mg subcutaan eens per 4 weken |
beroepsbeoefenaar in de gezondheidszorg patiënt* |
|
|
reslizumab Cinqaero® |
concentraat voor oplossing voor infusie 10 mg/ml. 25 en 100 mg injectieflacons |
3 mg/kg lichaamsgewicht intraveneus eens per 4 weken |
beroepsbeoefenaar in de gezondheidszorg |
|
benralizumab Fasenra® |
voorgevulde spuit 30 mg |
30 mg subcutaan elke 4 weken gedurende de eerste 3 giften, daarna 30 mg elke 8 weken |
beroepsbeoefenaar in de gezondheidszorg patiënt* |
|
voorgevulde pen 30 mg
|
30 mg subcutaan elke 4 weken gedurende de eerste 3 giften, daarna 30 mg elke 8 weken |
beroepsbeoefenaar in de gezondheidszorg patiënt* |
|
|
dupilumab Dupixent® |
wegwerpspuit 200 en 300 mg |
startdosis 400 mg en vervolgens 200 mg elke 2 weken subcutaan; bij gebruik van orale corticosteroïden of bij comorbide matig tot ernstig constitutioneel eczeem startdosis 600 mg en vervolgens 300 mg elke 2 weken subcutaan |
beroepsbeoefenaar in de gezondheidszorg patiënt* |
*mits de patiënt juist geïnstrueerd is en in staat is zelftoediening toe te passen
Onderbouwing
Astma is een complexe en heterogene aandoening waarbinnen verschillende subgroepen/fenotypen kunnen worden onderscheiden. Eosinofiele luchtweginflammatie wordt beschouwd als een van de belangrijkste, te beïnvloeden, kenmerken van astma. Aanvankelijk stond astma geassocieerd met eosinofiele luchtweginflammatie bekend als Th2-hoog astma vanwege de centrale rol van T-helper (Th) 2-cellen en bijbehorende cytokines (IL-4, IL-5 en IL-13) (Woodruff, 2009). In 2011 werd een andere belangrijke immuuncel geidentificeerd, de type 2 innate lymphoid cell (ILC2) (Mjösberg, 2011). Deze ILC2 cellen zijn in staat grote hoeveelheden IL-5 en IL-13 vrij te geven na blootstelling aan niet-specifieke stimuli zoals microben en verontreinigende stoffen (Godar, 2018). Sindsdien is de naam van 'Th2 astma' gewijzigd in 'type 2 astma' (Eger, 2019). Bij zowel het allergisch astma dat op kinderleeftijd ontstaat als ook het zogeheten late-onset eosinofiel, vaak niet-allergisch, astma speelt type 2 inflammatie een belangrijke rol. Op dit moment zijn er 5 biologicals beschikbaar voor de behandeling van ernstig astma met type 2 inflammatie, en zijn er nog verschillende monoclonale antilichaam en ‘small molecule’ behandelingen in ontwikkeling (Bel, 2017). In aanvulling op het reeds jaren beschikbare omalizumab (anti-IgE) betreft dit monoclonale antilichamen gericht tegen IL-5 (mepolizumab, reslizumab), IL-5 receptor alpha (benralizumab) en IL-4 receptor alpha (dupilumab).
Al deze biologicals richten zich tegen receptoren, cytokines of eiwitten die een belangrijke rol spelen in de “type 2” inflammatie: IL-4 stimuleert de productie van IgE door B-cellen en IgE activeert mestcellen, basofielen en dendritische cellen tot het vrijkomen van ontstekingsmediatoren, IL-5 is het belangrijkste cytokine voor het rekruteren en activeren van eosinofiele cellen en IL-13 stimuleert gladde spiercellen en mucus productie (Israel, 2017). IL-4 en IL-13 kunnen beide worden geremd door blokkade van de IL-4 receptor.
Voorheen waren systemische corticosteroïden de enige effectieve behandeling voor patiënten met ernstig astma en type 2 inflammatie. In aanvulling op de reeds jaren beschikbare omalizumab (anti-IgE) zijn er met de komst van nieuwe biologicals alternatieven bijgekomen voor deze groep van de ernstig astmapatiënten met veel minder bijwerkingen dan de orale corticosteroiden. In deze module wordt de effectiviteit van de vijf verschillende biologicals in kaart gebracht.
Omalizumab
Verbetering van astmacontrole
|
Laag GRADE |
Behandeling met omalizumab lijkt niet te resulteren in een verbetering van astmacontrole, gemeten met ACQ, in patiënten met ernstig allergisch astma.
Bronnen: (Garcia, 2013) |
Verbetering van kwaliteit van leven
|
Redelijk GRADE |
Behandeling met omalizumab resulteert mogelijk in een, niet-klinisch relevante, verbetering van kwaliteit van leven in patiënten met ernstig astma.
Bronnen: (Hanania, 2011; Humbert, 2005; Holgate, 2004) |
Astma-aanval frequentie
|
Laag GRADE |
Het is onduidelijk of omalizumab de astma-aanval frequentie doet afnemen. Op de korte termijn lijkt omalizumab geen effect te hebben, op langere termijn zou het mogelijk kunnen leiden tot minder astma-aanvallen.
Bronnen: (Hanania, 2011; Humbert, 2005; Holgate, 2004; Chanez, 2010; Garcia, 2013) |
Corticosteroïdreductie
|
- GRADE |
Er is onvoldoende onderzoek bij volwassenen met ernstig astma om de effecten van omalizumab op corticosteroïdreductie te kunnen beoordelen. |
Verbetering longfunctie
|
Laag GRADE |
Het is onduidelijk of omalizumab de longfunctie doet verbeteren.
Bronnen: (Humbert, 2005; Chanez, 2010; Garcia, 2013) |
Bijwerkingen
|
Redelijk GRADE |
Behandeling met omalizumab resulteert waarschijnlijk niet in het optreden van ernstige bijwerkingen tijdens de behandeling in patiënten met ernstig astma.
Bronnen: (Humbert, 2005; Holgate, 2004; Chanez, 2010; Garcia, 2013; Hanania, 2011) |
Mepolizumab
Verbetering van astmacontrole
|
Redelijk GRADE |
Behandeling met mepolizumab resulteert mogelijk in een, niet-klinisch relevante, verbetering van astmacontrole in patiënten met ernstig astma.
Bronnen: (Bel, 2014; Ortega, 2014; Chupp, 2017) |
Verbetering van kwaliteit van leven
|
Hoog GRADE |
Behandeling met mepolizumab geeft een klinisch relevante verbetering van kwaliteit van leven in patiënten met ernstig astma.
Bronnen: (Bel, 2014; Ortega, 2014; Chupp, 2017) |
Astma-aanval frequentie
|
Hoog GRADE |
Behandeling met mepolizumab geeft een klinisch relevante afname van de jaarlijkse astma-aanval frequentie in patiënten met ernstig astma.
Bronnen: (Bel, 2014; Ortega, 2014; Chupp, 2017) |
Corticosteroïdreductie
|
Redelijk GRADE |
Behandeling met mepolizumab lijkt een klinisch relevante reductie in corticosteroïdgebruik te geven in patiënten met ernstig astma.
Bronnen: (Bel, 2014) |
Verbetering longfunctie
|
Laag GRADE |
Behandeling met mepolizumab lijkt mogelijk een niet klinisch relevante verbetering van de longfunctie (FEV1) te geven.
Bronnen: (Bel, 2014; Ortega, 2014; Chupp, 2017) |
Bijwerkingen
|
Redelijk GRADE |
Behandeling met mepolizumab resulteert waarschijnlijk niet in het optreden van ernstige bijwerkingen tijdens de behandeling van patiënten met ernstig astma.
Bronnen: (Bel, 2014; Ortega, 2014; Chupp, 2017) |
Reslizumab
Verbetering van astmacontrole
|
Redelijk GRADE |
Behandeling met reslizumab resulteert mogelijk in een, niet-klinisch relevante, verbetering van astmacontrole in patiënten met ernstig astma.
Bronnen: (Li, 2017) |
Verbetering van kwaliteit van leven
|
Redelijk GRADE |
Behandeling met reslizumab resulteert mogelijk in een, niet-klinisch relevante, verbetering van kwaliteit van leven in patiënten met ernstig astma.
Bronnen: (Castro, 2015; Bjermer, 2016) |
Astma-aanval frequentie
|
Hoog GRADE |
Behandeling met reslizumab geeft een klinisch relevante afname van de astma-aanval frequentie in patiënten met matig tot ernstig astma.
Bronnen: (Li, 2017) |
Corticosteroïdreductie
|
GRADE |
Er is onvoldoende onderzoek bij volwassenen met ernstig astma om de effecten van reslizumab op corticosteroïdreductie te kunnen beoordelen. |
Verbetering longfunctie
|
Redelijk GRADE |
Behandeling met reslizumab geeft waarschijnlijk geen klinische relevante verbetering van de longfunctie (FEV1) in patiënten met matig tot ernstig astma.
Bronnen: (Li, 2017) |
Bijwerkingen
|
Redelijk GRADE |
Behandeling met reslizumab resulteert waarschijnlijk niet in het optreden van ernstige bijwerkingen tijdens de behandeling van patiënten met ernstig astma.
Bronnen: (Li, 2017) |
Benralizumab
Verbetering van astmacontrole
Verbetering van kwaliteit van leven
|
Redelijk GRADE |
Behandeling met benralizumab geeft mogelijk een, niet-klinisch relevante verbetering van kwaliteit van leven in patiënten met ernstig astma.
Bronnen: (Bleecker, 2016; FitzGerald, 2016; Nair, 2017) |
Astma-aanval frequentie
|
Hoog GRADE |
Behandeling met benralizumab geeft een klinisch relevante afname van de astma-aanval frequentie in patiënten met ernstig astma.
Bronnen: (Bleecker, 2016; FitzGerald, 2016; Nair, 2017) |
Corticosteroïdreductie
|
Redelijk GRADE |
Behandeling met benralizumab lijkt een klinische relevante reductie in corticosteroïd gebruik te geven in patiënten met ernstig astma.
Bronnen: (Nair, 2017) |
Verbetering longfunctie
|
Redelijk GRADE |
Behandeling met benralizumab resulteert waarschijnlijk in een niet klinisch relevante verbetering van de longfunctie in patiënten met ernstig astma.
Bronnen: (Bleecker, 2016; FitzGerald, 2016; Nair, 2017) |
Bijwerkingen
|
Redelijk GRADE |
Behandeling met benralizumab resulteert waarschijnlijk niet in het optreden van ernstige bijwerkingen in patiënten met ernstig astma.
Bronnen: (Bleecker, 2016; FitzGerald, 2016; Nair, 2017) |
Dupilumab
Verbetering van astmacontrole
|
Redelijk GRADE |
Behandeling met dupilumab resulteert waarschijnlijk in een verbetering van astmacontrole in patiënten met matig tot ernstig astma.
Bronnen: (Wenzel, 2016; Castro, 2018; Rabe, 2018) |
Verbetering van kwaliteit van leven
|
Redelijk GRADE |
Behandeling met dupilumab resulteert waarschijnlijk in een verbetering van kwaliteit van leven in patiënten met matig tot ernstig astma en ≥300 eosinofielen per μL.
Bronnen: (Wenzel, 2016; Castro, 2018) |
Astma-aanval frequentie
|
Hoog GRADE |
Behandeling met dupilumab leidt tot een afname van de astma-aanval frequentie in patiënten met matig tot ernstig astma.
Bronnen: (Wenzel, 2016; Castro, 2018; Rabe, 2018) |
Corticosteroïdreductie
|
Redelijk GRADE |
Behandeling met dupilumab resulteert waarschijnlijk in een reductie van corticosteroïdgebruik in patiënten met ernstig astma.
Bronnen: (Rabe, 2018) |
Verbetering longfunctie
|
Redelijk GRADE |
Behandeling met dupilumab resulteert waarschijnlijk in een verbetering van de longfunctie in patiënten met matig tot ernstig astma.
Bronnen: (Wenzel, 2016; Castro, 2018; Rabe, 2018) |
Bijwerkingen
|
Redelijk GRADE |
Behandeling met dupilumab resulteert waarschijnlijk niet in het optreden van ernstige bijwerkingen tijdens de behandeling in patiënten met matig tot ernstig astma.
Bronnen: (Wenzel, 2016; Castro, 2018; Rabe, 2018) |
Omalizumab (anti-IgE)
Beschrijving studies
Er is één systematische review (Normansell, 2014) geïncludeerd die vijf RCT’s beschrijft (Chanez, 2010; Hanania, 2011; Humbert, 2005; Garcia, 2013; Holgate, 2004), die de effecten van omalizumab onderzochten in volwassen en adolescente patiënten met ernstig of ongecontroleerd astma, ondanks behandeling met medium-hoge dosis ICS plus LABA’s
In de RCT van Chanez (2010) kregen 31 patiënten (≥ 18 jaar) met ongecontroleerde ernstig allergisch astma, gedurende 16 weken ofwel omalizumab (n=20) of placebo (n=11).
De dubbelblinde RCT van Hanania (2011) includeerde 850 patiënten (leeftijd 12 tot 75 jaar) met inadequaat gecontroleerd ernstig allergisch astma. Patiënten ontvingen omalizumab (n=427) of een placebo (n=423) gedurende 48 weken.
In de dubbelblinde gerandomiseerde INNOVATE-studie (Humbert, 2005) werden 482 patiënten, van wie 419 patiënten geïncludeerd werden in de efficacy analyses (leeftijd 12 tot 75 jaar) met inadequaat gecontroleerd astma gerandomiseerd tot behandeling met omalizumab (n=209) of placebo (n=210) gedurende 28 weken.
Garcia (2013) onderzocht in zijn dubbelblinde RCT de werking van omalizumab in 41 patiënten (18 tot 70 jaar) met ernstig, persisterend, niet-atopisch astma. Patiënten werden gerandomiseerd tot het ontvangen van omalizumab (n=20) of placebo (n=21) gedurende 16 weken.
De dubbelblinde RCT van (Holgate, 2004) onderzocht de werking van omalizumab in 246 patiënten (12 tot 75 jaar) met ernstig allergisch astma door ofwel te behandelen met omalizumab (n=126) of met placebo (n=120) gedurende 16 weken.
In alle studies werden patiënten behandeld met een dosis subcutaan omalizumab afhankelijk van van lichaamsgewicht en serum IgE concentratie, maar tenminste met 0,016 mg/kg per IU/mL omalizumab.
Resultaten
Meta-analyses waren niet mogelijk door de verschillende manieren van data rapporteren of het ontbreken van data.
Verbetering van astmacontrole (cruciale uitkomstmaat)
Eén studie rapporteerde astmacontrole, gemeten met de ACQ-vragenlijst, als uitkomstmaat in de behandeling met omalizumab. Garcia (2013) vond geen verschil in verbetering in ACQ-score tussen de omalizumab-groep en de placebogroep: gemiddelde verschil (SD) omalizumab -0,5 (0,98) versus placebo -0,5 (1,43) p=0,744.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van astmacontrole is met twee niveaus verlaagd gezien het kleine aantal studies (imprecisie).
Verbetering van kwaliteit van leven (AQLQ) (cruciale uitkomstmaat)
Drie studies rapporteerden kwaliteit van leven, gemeten met de AQLQ-vragenlijst, als uitkomstmaat in de behandeling met omalizumab. Deze studies vonden een significante maar niet klinisch relevante verbetering in kwaliteit van leven (Hanania, 2011: MD 0,29 (95% BI 0,15 tot 0,43); (Holgate, 2004): 58% van de patiënten in de omalizumab-groep had ≥ 0,5 punten verbetering versus 39% in de placebogroep, p<0.01; Humbert, 2005: 60,8% van de patiënten in de omalizumab-groep had ≥ 0,5 punten verbetering versus 47,8% in de placebogroep, p=0,008).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van kwaliteit van leven is met één niveau verlaagd gezien het effect te klein is om klinische relevantie te behalen (imprecisie).
Astma-aanval frequentie (belangrijke uitkomstmaat)
Vijf studies rapporteerden astma-aanvallen als uitkomstmaat in de behandeling met omalizumab. Twee langlopende (> 28 weken) studies vonden een significante maar niet klinisch relevante afname in astma-aanvallen in de omalizumab-groep ten opzichte van de placebogroep (Hanania, 2011: rate ratio 0,75 (0,61 tot 0,92); Humbert, 2005: rate ratio 0,74 (0,55 tot 1,00)).
Drie kortlopende (16 weken) studies vonden geen verschil in astma-aanval frequentie tussen beide groepen (Holgate, 2004; Chanez, 2010: Garcia, 2013).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat astma-aanval frequentie is met twee niveaus verlaagd gezien het effect te klein is om klinische relevantie te behalen (imprecisie) en de grote heterogeniteit in resultaten (inconsistentie).
Corticosteroïdreductie (belangrijke uitkomstmaat)
De uitkomstmaat steroïdreductie is niet gerapporteerd als uitkomstmaat in de studies naar de plaats van omalizumab als aanvullende behandeling bij ernstig astma.
Verbetering longfunctie (FEV1) (belangrijke uitkomstmaat)
Vier studies rapporteerden longfunctie (FEV1) als uitkomstmaat in de behandeling met omalizumab.
Eén studie, Garcia (2013), vond een significante en klinisch relevante verbetering in longfunctie in de omalizumab-groep ten opzichte van de placebogroep (+250 ml, p=0,032; +10%, p=0,029).
Een studie vonden een significante maar niet klinisch relevante verbetering in longfunctie (Humbert, 2005: verbetering in FEV1 % voorspelde waarde in omalizumab ten opzichte van placebo: 2,8%, p=0,043).
Chanez (2010) vond geen significante verbetering in longfunctie in de omalizumab-groep ten opzichte van de placebogroep (mediane verandering in FEV1: 2,6% in omalizumab-groep (-10 tot 60), versus 1,7% in de placebogroep (-19 tot 7), p=0,312).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering longfunctie is met twee niveaus verlaagd gezien het effect te klein is om klinische relevantie te behalen (imprecisie) en de grote heterogeniteit in resultaten (inconsistentie).
Bijwerkingen
Vijf studies rapporteerden bijwerkingen als uitkomstmaat in de behandeling met omalizumab.
Vier studies vonden dat het optreden van bijwerkingen vergelijkbaar was in de omalizumab- en placebogroep (Chanez, 2010; Hanania, 2011; Humbert, 2005; Garcia, 2013).
Holgate (2004) daarentegen vond dat de incidentie van ernstige bijwerkingen lager was in de omalizumab-groep (6,3% (8/126) versus 18,3% (22/120) in de placebogroep). De meest voorkomende bijwerkingen waren infecties van de onderste luchtwegen en nasofaryngitis (Humbert, 2005).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat bijwerkingen is met één niveau verlaagd aangezien deze op heterogene wijze werd gerapporteerd in de verschillende studies.
Mepolizumab
Beschrijving studies
Er zijn drie RCT’s geïncludeerd die de effecten van mepolizumab 100 mg subcutaan (s.c.) onderzochten in patiënten met ernstig astma. De orale corticosteroïd-sparende studie van Bel (2014) includeerde 135 patiënten (leeftijd 16 tot 74 jaar) met ernstig eosinofiel astma (≥150 eosinofielen /µl bij aanvang van de studie, of ≥ 300 eosinofielen/µl in het jaar voor aanvang van de studie), ondanks onderhoudsbehandeling met orale corticosteroïden, naast hoge dosis inhalatiesteroïden (ICS) plus additionele controller. De interventiegroep (n=69) werd behandeld met mepolizumab 100 mg s.c., en de controlegroep (n=66) met een placebo, elke 4 weken gedurende 20 weken.
De studie van Ortega (2014) onderzocht 576 patiënten (leeftijd 12 tot 84 jaar) met ernstig eosinofiel astma (≥ 150 eosinofielen /µl bij aanvang van de studie, of ≥ 300 eosinofielen/µl in het jaar voor aanvang van de studie, FEV1 van < 80% van de voorspelde waarde), ondanks behandeling met tenminste 880 μg fluticason of equivalent en additionele controller. De interventiegroep ontving mepolizumab intraveneus (i.v.) 75 mg (n=191) of s.c. 100 mg (n=194), en de controlecontrolegroep (n=191) een placebo, elke 4 weken gedurende 32 weken. De interventiegroep die mepolizumab intraveneus ontving is niet meegenomen in de literatuuranalyse.
De RCT van Chupp (2017) onderzocht 551 patiënten met ernstige eosinofiel astma (≥ 150 eosinofielen /µl bij aanvang van de studie, of ≥300 eosinofielen/µl in het jaar voor aanvang van de studie, FEV1 van < 80% van de voorspeld waarde), ondanks behandeling met hoge dosis ICS plus additionele controller. Patiënten werden gerandomiseerd tot behandeling met mepolizumab 100 mg s.c. (n=274) of met placebo (n=277) elke 4 weken, gedurende 24 weken.
Resultaten
Meta-analyses waren niet mogelijk door de verschillende manieren van data rapporteren of het ontbreken van data.
Verbetering van astmacontrole (ACQ) (cruciale uitkomstmaat)
Drie studies rapporteerden astmacontrole, gemeten met de ACQ-5 vragenlijst, als uitkomstmaat in de behandeling met mepolizumab. De drie studies vonden significante verbeteringen in ACQ-5-score in de mepolizumab-groep ten opzichte van de placebogroep (Bel, 2014: MD -0,52 punten (-0,87 tot -0,17); Ortega, 2014: -0,44 (-0,63 tot -0,25); Chupp, 2017: -0,4 punten (-0,6 tot -0,2)). Alleen de resultaten van Bel (2014) waren als klinisch relevant te waarderen.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van astmacontrole is met één niveau verlaagd gezien het effect in 2 van de 3 studies te klein is om klinische relevantie te behalen (imprecisie).
Verbetering van kwaliteit van leven (SGRQ) (cruciale uitkomstmaat)
Drie studies rapporteerden kwaliteit van leven als uitkomstmaat in de behandeling met mepolizumab, aan de hand van de St. George’s Respiratory Questionnaire (SGRQ)-vragenlijst, waarbij een verschil van 4 punten klinisch relevant is. De drie studies vonden een significante en klinisch relevante verbetering in kwaliteit van leven in de interventiegroep in vergelijking met de controlegroep (Bel, 2014: MD -5,8 punten (-10,6 tot -1.0); Ortega, 2014: -7,0 (-10,2 tot -3,8); Chupp, 2017: -7,7 (-10,5 tot -4,9)).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat kwaliteit van leven is niet verlaagd.
Astma-aanval frequentie (belangrijke uitkomstmaat)
Drie studies, met een follow-up van >20 weken, rapporteerden astma-aanvallen als uitkomstmaat in de behandeling met mepolizumab. Een astma-aanval was gedefinieerd als een verergering van astma leidend tot de verdubbeling (of meer) van de bestaande onderhoudsdosis van orale corticosteroïden gedurende 3 of meer dagen, of ziekenhuisopname of een bezoek aan de spoedeisende hulp voor behandeling van astma. De drie studies vonden een significante en klinisch relevante reductie in jaarlijkse astma-aanvallen in de mepolizumab-groep in vergelijking met de placebogroep (Bel, 2014: 32% reductie, RR 0,68 (0,47 tot 0,99); Ortega, 2014: 53% reductie, RR 0,47; Chupp, 2017: 58% reductie, RR 0,42 (0,31 tot 0,56)).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat astma-aanval frequentie is niet verlaagd.
Corticosteroïdreductie (belangrijke uitkomstmaat)
Eén studie rapporteerde orale corticosteroïdreductie als uitkomstmaat in de behandeling met mepolizumab. Bel (2014) vond dat de kans op een verlaging van de orale corticosteroïd-dosis 2,39 keer groter was in de mepolizumab (100 mg) groep dan in de placebogroep (95% BI 1,25 tot 4,56). Na 6 maanden behandeling was de mediane procentuele reductie vanaf baseline in corticosteroïden 50% in de mepolizumab-groep, vergeleken met geen verlaging van de placebogroep (P = 0,007). Dit verschil is klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat corticosteroïdreductie is met een niveau verlaagd gezien het kleine aantal studies (imprecisie).
Verbetering longfunctie (belangrijke uitkomstmaat)
Drie studies rapporteerden longfunctie als uitkomstmaat in de behandeling met mepolizumab. Twee studies vonden een significante maar niet klinisch relevante verbetering in prebronchodilatator FEV1 in de mepolizumab-groep ten opzichte van de placebogroep (Ortega, 2014: MD 98 ml (11 tot 184); Chupp, 2017: 120 ml (47 tot 192)). Bel (2014) vond geen verschillen in pre- noch postbronchodilatator FEV1 tussen behandeling met mepolizumab en placebo.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering longfunctie is met twee niveaus verlaagd gezien het effect te klein is om klinische relevantie te behalen (imprecisie) en de heterogeniteit in resultaten (inconsistentie).
Bijwerkingen
Drie studies rapporteerden bijwerkingen ten gevolge van behandeling met mepolizumab. In de studie van Bel (2014) was de incidentie van niet-astma-gerelateerde bijwerkingen 83% in de mepolizumab-groep en 91% in de placebogroep. Astma-aanvallen waarvoor hospitalisatie nodig was en pneumonie waren de meest voorkomende ernstige bijwerkingen, en kwamen alleen voor in de placebogroep. Ortega (2014) en Chupp (2017) vonden een vergelijkbaar veiligheidsprofiel tussen mepolizumab en placebo. De incidentie van ernstige bijwerkingen (inclusief astma-gerelateerde gebeurtenissen) was 8% in de mepolizumab-groep en 14% in de placebogroep (Ortega, 2014). De drie studies rapporteerden hoofdpijn en nasofaryngitis als meest frequent gemelde bijwerkingen in zowel de mepolizumab-groep als de placebogroep.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat bijwerkingen is met één niveau verlaagd aangezien deze op heterogene wijze werd gerapporteerd in de verschillende studies.
Reslizumab
Beschrijving studies
De systematische review van Li (2017) includeerde de onderstaande vier publicaties die de effecten van reslizumab onderzochten in patiënten met ernstig of ongecontroleerd astma. Eén van deze studies beschrijft de resultaten van twee RCT’s, waardoor er uiteindelijk vijf RCT’s geïncludeerd zijn.
De RCT van Castro (2011) includeerde 106 patiënten (leeftijd 18 tot 75 jaar) met ernstig eosinofiel astma, hetgeen slecht gecontroleerd was bij hoge dosis ICS en LABA’s. De interventiegroep (n=53) werd gedurende 12 weken behandeld met intraveneus (i.v.) reslizumab, in een dosis van 3,0 mg/kg, en de controlegroep (n=53) met een placebo.
De studie van Castro (2015) beschrijft de resultaten van twee verschillende RCT’s naar de effecten van reslizumab in patiënten met ongecontroleerd matig tot ernstig astma, met verhoogde eosinofielen waarden (> 400 eosinofielen/µl), ondanks middelhoge dosis ICS en LABA’s. Eén studie includeerde 489 patiënten in de leeftijd van 12 tot 75 jaar, de andere studie 464 patiënten in de leeftijd van 12 tot 75 jaar. In beide studies werd de interventiegroep (n=477 (245 in studie 1 en 232 in studie 2)) behandeld met reslizumab i.v., 3,0 mg/kg, en de controlegroep (n=476 (244 en 232)) met een placebo, voor de duur van 52 weken.
Bjermer (2016) onderzocht de werking van reslizumab in 315 patiënten (leeftijd 12 tot 75 jaar) met inadequaat gecontroleerd astma met tenminste een behandeling met middelhoge dosis van ICS en LABA’s en een bloed eosinofielen-gehalte van ≥ 400 cellen/ml. Patiënten werden gerandomiseerd (1:1:1) over drie studiegroepen: een groep (n=106) ontving reslizumab i.v. 3,0 mg/kg, een groep (n=104) ontving reslizumab i.v. 0,3 mg/kg, en de derde groep (n=105) ontving een placebo, elke vier weken gedurende 16 weken (een totaal van vier doses). De interventiegroep die 0,3 mg/kg reslizumab ontving is niet meegenomen in de voor de richtlijn uitgevoerde meta-analyse.
Corren (2016) onderzocht de werking en veiligheid van reslizumab in patiënten met slecht gecontroleerd astma (leeftijd 18 tot 65 jaar), bij behandeling met een middelhoge dosis ICS en LABA’s, en analyseerde het effect in de subgroepen patiënten met < 400 eosinofielen/ml en ≥400 eosinofielen/ml. Patiënten werden gerandomiseerd tot het ontvangen van reslizumab 3,0 mg/kg i.v. (n=398) of een placebo (n=98) elke vier weken gedurende 16 weken. In de meta-analyse zijn alleen de resultaten voor patiënten met ≥400 eosinofielen/ml meegenomen.
Resultaten
Verbetering van astmacontrole (ACQ) (cruciale uitkomstmaat)
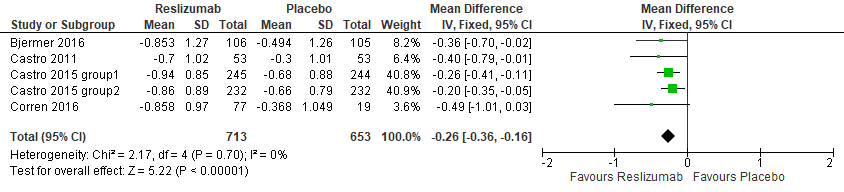
De review van Li (2017) rapporteerde de resultaten van vijf RCT’s die astmacontrole onderzochten, middels de Asthma Control Questionnaire (ACQ). De studies vonden een grotere verbetering van astmacontrole in de reslizumab groep dan in de placebogroep, zie figuur 1 (MD -0,26 (-0,36 tot -0,16)). Het verschil tussen de groepen in verbetering op de ACQ van 0,26 punten is echter niet klinisch relevant.
Figuur 1 Meta-analyse astmacontrole, reslizumab versus placebo (Li, 2017)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van astmacontrole is met één niveau verlaagd aangezien het effect te gering is om klinische relevantie te behalen (imprecisie).
Verbetering van kwaliteit van leven (cruciale uitkomstmaat)
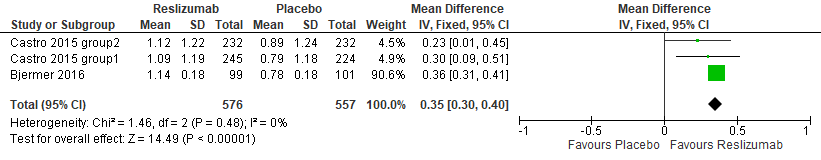
De review van Li (2017) rapporteerde niet de uitkomstmaat kwaliteit van leven, gebaseerd op de Asthma Quality of Life Questionnaire (AQLQ). De twee RCT’s van Castro (2015) en de RCT van Bjermer (2016) deden dit wel. De voor de richtlijn uitgevoerde meta-analyse van de resultaten van behandeling met reslizumab in de dosis 3,0 mg/kg laat zien dat de AQLQ-score significant hoger was in de reslizumab groep dan in de placebogroep, zie figuur 2 (MD 0,35 (95% BI 0,30 tot 0,40)). Het effect haalde niet de klinisch relevante grens van 0,5. Het grote verschil in behandelingsduur (52 weken versus 16 weken) leek geen invloed te hebben op de resultaten.
Figuur 2 Meta-analyse Kwaliteit van leven, reslizumab versus placebo
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van kwaliteit van leven is met één niveau verlaagd aangezien het effect te gering is om klinische relevantie te behalen (imprecisie).
Astma-aanval frequentie (belangrijke uitkomstmaat)
De review van Li (2017) beschreef drie studies die het risico op astma-aanvallen rapporteerden. Deze studies vonden dat het risico op astma-aanvallen significant lager was in de reslizumab groep dan in de placebogroep, zie figuur 3 (RR 0,46 (95% BI 0,35 tot 0,59)). De gerapporteerde afname in risico op astma-aanvallen van 54% is klinisch relevant. De studie van Castro (2011) had een follow-up duur van 12 maanden, en beide studies van Castro (2015) hadden een follow-up van 52 weken.
Figuur 3 Meta-analyse astma exacerbaties, reslizumab versus placebo (Li, 2017)
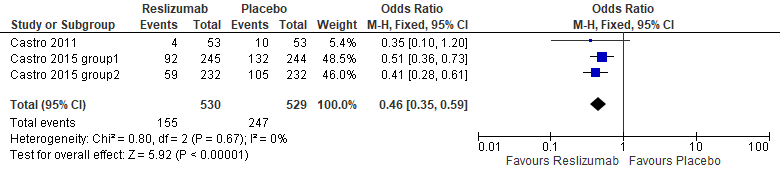
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat astma-aanval frequentie is niet verlaagd.
Corticosteroïdreductie (belangrijke uitkomstmaat)
De uitkomstmaat corticosteroïdreductie is niet gerapporteerd als uitkomstmaat in de studies naar de plaats van reslizumab als aanvullende behandeling bij ernstig astma.
Verbetering longfunctie (belangrijke uitkomstmaat)
De review van Li (2017) rapporteerde vijf studies die de longfunctie (FEV1) onderzochten. De studies vonden dat de longfunctie significant meer verbeterde in de reslizumab groep dan in de placebogroep, zie figuur 4 (MD 0,16 (0,09 tot 0,23)). Dit verschil is echter niet klinisch relevant.
Figuur 4 Meta-analyse verbetering longfunctie, reslizumab versus placebo (Li, 2017)
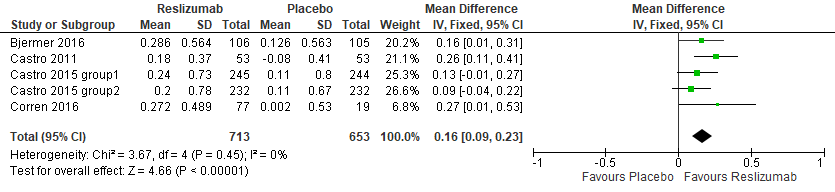
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat kwaliteit van leven is met één niveau verlaagd gezien het effect niet groot genoeg was om klinische relevantie te behalen (imprecisie).
Bijwerkingen
Li (2017) includeerde vijf RCT’s die het voorkomen van bijwerkingen als gevolg van de behandeling met reslizumab rapporteerde.
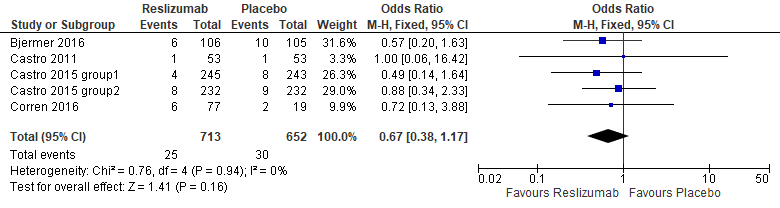
De studies vonden dat behandeling met reslizumab niet resulteerde in een grotere studie-uitval door het optreden van bijwerkingen in vergelijking met placebo (OR 0,86 (0,68 tot 1,10)), zie figuur 5a. Tevens vonden de vijf studies dat er geen verschil was tussen behandeling met reslizumab en placebo in het risico op optreden van bijwerkingen in de bovenste luchtwegen, zoals bovenste luchtweginfecties, nasopharyngitis en sinusitis, (OR 0,67 (0,38 tot 1,17)), zie figuur 5b. Bij minimaal 1% van de patiënten met reslizumab stijgt de serumcreatinekinasewaarde (SmPC).
Figuur 5 Meta-analyse optreden van bijwerkingen, reslizumab versus placebo (Li, 2017)
(a)
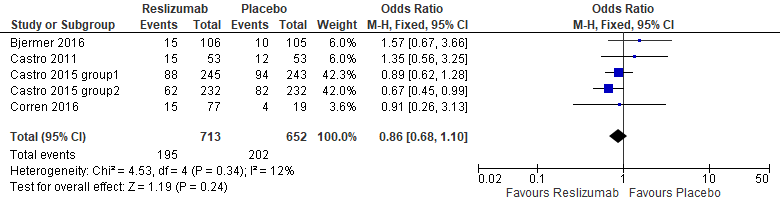
(b)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat kwaliteit van leven is met één niveaus verlaagd gezien het verschil in effect tussen de studies, en het grote verschil in studie grootte (inconsistentie).
Benralizumab
Beschrijving studies
Er zijn drie RCT’s geïncludeerd die de effecten van benralizumab in de dosis 30 mg subcutaan (s.c.) als aanvullende behandeling onderzochten in patiënten met ernstig astma.
In de studie van Bleecker (2016) werden 1205 patiënten onderzocht (leeftijd 12 tot75 jaar) met ernstig, ongecontroleerd astma ondanks behandeling met hoge dosis ICS en LABA’s. De interventiegroep (n=798) ontving 30 mg benralizumab s.c. elke 4 weken (n=400) of elke 8 weken (n=398) met de eerste drie doses om de 4 weken, en de controlegroep (n=407) ontving een placebo, voor de duur van 48 weken. Patiënten werden gestratificeerd (2:1) op baseline bloed-eosinofielen waarden, respectievelijk ≥ 300 of < 300 cellen/μL.
De RCT van FitzGerald (2016) includeerde 1306 patiënten (leeftijd 12 tot 75 jaar) met ernstig, ongecontroleerd astma, ondanks behandeling met medium tot hoge dosis ICS en LABA’s. Patiënten werden gerandomiseerd tot benralizumab 30 mg s.c. elke 4 weken (n=425) of elke 8 weken (n=441) met de eerste drie doses om de 4 weken, of tot placebo (n=440), gedurende 56 weken. Patiënten werden gestratificeerd (2:1) op baseline bloed-eosinofielen waarden, respectievelijk ≥ 300 of < 300 cellen/μL.
De studie van Nair (2017) onderzocht de werking van benralizumab in volwassenen met ernstig astma en een eosinofiel gehalte van ≥ 150 cellen/μL ondanks onderhoudsbehandeling met orale corticosteroïden (OCS) (n=220). Patiënten werden gerandomiseerd tot behandeling met benralizumab 30 mg s.c., elke 4 weken (n=72) of 30 mg elke 8 weken (n=73) met de eerste drie doses om de 4 weken, of tot behandeling met een placebo elke vier weken (n=75), gedurende de interventieperiode van 28 weken.
De interventiegroepen die benralizumab elke 4 weken ontvingen zijn niet meegenomen in de literatuuranalyse. De literatuuranalyse beschrijft de effecten van benralizumab in de dosis 30 mg per 8 weken, met de eerste drie doses om de 4 weken.
Resultaten
Verbetering van astmacontrole (cruciale uitkomstmaat)
Alle drie studies rapporteerden de uitkomstmaat verbetering van astmacontrole, gemeten met de ACQ-6-vragenlijst. Behandeling met benralizumab leidt in patiënten met ≥ 300 eosinofielen/μL tot een significante, maar niet klinisch relevante, verbetering in ACQ-6-score in vergelijking met placebo (Bleecker, 2016: -0,29 (-0,48 tot -0,10), FitzGerald, 2016: -0,23 (-0,43 tot -0,04)).
In patiënten met OCS afhankelijke astma leidt behandeling met benralizumab tot een klinisch relevante verbetering in ACQ-6-score in vergelijking met placebo (Nair, 2017: -0,55 (-0,23 tot -0,86)).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van astmacontrole is met één niveau verlaagd gezien het geringe klinisch relevante effect (imprecisie).
Verbetering van kwaliteit van leven (cruciale uitkomstmaat)
De drie studies rapporteren de uitkomst verbetering van kwaliteit van leven, gebaseerd op de Asthma Quality of Life Questionnaire (AQLQ). Behandeling met benralizumab leidt in patiënten met ≥ 300 eosinofielen/μL tot een significante, maar niet klinisch relevante verbetering in AQLQ-score in vergelijking met placebo (Bleecker, 2016: 0,30 (0,10 tot 0,50), FitzGerald, 2016: 0,24 (0,04 tot 0,45)). Ook in patiënten met OCS afhankelijke astma leidt behandeling met benralizumab tot een niet klinisch relevante verbetering in AQLQ-score in vergelijking met placebo (Nair, 2017: 0,45 (0,14 tot 0,76)).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van kwaliteit van leven is met één niveau verlaagd aangezien het effect te gering is om klinische relevantie te behalen (imprecisie).
Astma-aanval frequentie (belangrijke uitkomstmaat)
De drie studies rapporteren de uitkomstmaat astma-aanvallen. Deze studies, met een follow-up > 28 weken, vonden een klinisch relevante verlaging van de astma-aanval frequentie.
De studies in patiënten met ≥ 300 eosinofielen/μL laten zien dat behandeling met benralizumab resulteerde in significante en klinisch relevante reducties in jaarlijkse frequentie van astma-aanvallen, in vergelijking met placebo (Bleecker, 2016: rate ratio 0,49 (95% BI 0,37 tot 0,64), FitzGerald, 2016: rate ratio 0,72 (0,54 tot 0,95)). Tevens was de tijd tot de eerste astma-aanval verhoogd in vergelijking met placebo (Bleecker, 2016: HR 0,60 (0,46 tot 0,78)), FitzGerald, 2016: 0,73 (0,55 tot 0,95)).
De orale corticosteroïd-sparende studie van Nair (2017) in OCS-afhankelijke patiënten laat een jaarlijkse astma-aanval frequentie zien die 70% lager was dan met placebo (rate ratio 0,30 (95% BI 0,17 tot 0,53)). Behandeling met benralizumab was geassocieerd met een lagere kans op het hebben van ten minste één astma-aanval dan placebo (OR 0,28 (0,14 tot 0,56)) en resulteerde in een langere tijd tot de eerste astma-aanval dan placebo (Nair, 2017).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat astma-aanval frequentie is niet verlaagd.
Corticosteroïdreductie (belangrijke uitkomstmaat)
Eén studie rapporteerde de uitkomstmaat corticosteroïdreductie.
Nair (2017) onderzocht de gevolgen van behandeling met benralizumab op corticosteroïdreductie. De studie vond dat benralizumab resulteerde in een significante afname in orale corticosteroïden dosis ten opzichte van placebo; een 75% vermindering in corticosteroïden dosis ten opzichte van een 25% vermindering bij behandeling met placebo (p<0,001). De kans op afname in corticosteroïden dosis was hoger bij behandeling met benralizumab: OR 4,12 (2,22 tot 7,63)). Dit verschil is klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat corticosteroïdreductie is met een niveau verlaagd vanwege het kleine aantal studies (imprecisie).
Verbetering longfunctie (belangrijke uitkomstmaat)
De drie studies rapporteren de uitkomstmaat longfunctie, gemeten in prebronchodilatator FEV1 (L).
Twee studies vonden dat behandeling met benralizumab in patiënten met ≥ 300 eosinofielen/μL leidt tot een niet klinisch relevante verbetering in longfunctie in vergelijking met placebo (Bleecker, 2016: 0,16 L (0,07 tot 0,25), FitzGerald, 2016: 0,12 L (0,03 tot 0,20)).
Nair (2017) vond in OCS afhankelijke patiënten geen verbetering in longfunctie in vergelijking met placebo.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van longfunctie is met één niveau verlaagd aangezien er geen klinisch relevant effect is behaald (imprecisie).
Bijwerkingen
De drie studies rapporteerden over het optreden van bijwerkingen als gevolg van behandeling met benralizumab.
Bleecker (2016) vond vergelijkbare percentages van patiënten die bijwerkingen rapporteerden (72% in de benralizumab-groep versus 76% in de placebogroep). De meest voorkomende bijwerkingen bij patiënten in de benralizumab-groep waren verslechtering van astma (13%), nasopharyngitis (12%) en bovenste luchtwegen infectie (10%). Bijwerkingen gerelateerd aan astma werden gemeld door, naar verhouding, minder patiënten in de benralizumab-groep dan in de placebogroep. Ernstige bijwerkingen werden gerapporteerd door vergelijkbare percentages van patiënten in de benralizumab-groep (99 (12%)) en placebo-groep (55 (14%)).
Fitzgerald (2016) vond dat minder patiënten behandeld met benralizumab (642 (74%) van 866) versus placebo (342 (78%) van 440) bijwerkingen ervaarden. De meest voorkomende bijwerking was nasopharyngitis (20%).
Nair (2017) vond dat 75% van de patiënten in de benralizumab-groep, en 83% van de patiënten in de placebogroep minstens één bijwerking hadden. De meest voorkomende bijwerkingen waren nasopharyngitis (in 17% van de patiënten), verergering van astma (in 13%), en bronchitis (in 10%).
Bewijskracht van de literatuur
De bewijskracht voor bijwerkingen werd verlaagd aangezien deze op heterogene wijze werden gerapporteerd in de verschillende studies.
Dupilumab
Beschrijving studies
Drie studies deden onderzoek naar de werking van dupilumab in volwassenen met matig tot ernstig astma.
Wenzel (2016) includeerde in haar RCT 776 patiënten (>18 jaar) met een ongecontroleerd, persisterend matig tot ernstig astma, bij behandeling met middelhoge-tot-hoge dosis ICS en LABA’s. Patiënten werden gerandomiseerd (1:1:1:1:1) in vijf groepen: dupilumab s.c 200 mg elke vier weken, oplaaddosis 400 mg (N=154), 300 mg elke vier weken, oplaaddosis 600 mg (n=157), 200 mg elke twee weken, oplaaddosis 400 mg (n=150), 300 mg elke twee weken, oplaaddosis 600 mg (n=157), of placebo (n=158), gedurende een periode van 24 weken. De werking van dupilumab werd onderzocht in de totale interventiegroep, en per subgroep gebaseerd op eosinofiel-gehalte: ≥300 eosinofielen per μl (n=325) en <300 eosinofielen per μl (n=451). Voor de literatuuranalyse in deze richtlijn zijn alleen de resultaten van de groepen 200 mg/2 weken 300 mg/2 weken meegenomen.
De gerandomiseerde dubbelblinde studie van Castro (2018) onderzocht de werking van dupilumab in 1902 patiënten (> 12 jaar) met matig tot ernstig ongecontroleerd astma, ondanks behandeling met middelhoge-tot-hoge dosis ICS en LABA’s. Patiënten werden gerandomiseerd in vier groepen: dupilumab s.c. 200 mg, oplaaddosis 400 mg (n=631), dupilumab s.c. 300 mg, oplaaddosis 600 mg (n=633), of placebo in vergelijkbare volumes, respectievelijk 1,14 ml (n=317) of 2,00 ml (n=321) elke twee weken voor 52 weken.
Rabe (2018) includeerde in zijn gerandomiseerde dubbelblinde orale corticosteroïd-sparende studie 210 patiënten (> 12 jaar) met ernstig astma en behandeling met hoge dosis ICS en LABA’s. Patiënten ontvingen subcutaan dupilumab in een dosis van 300 mg, oplaadosis 600 mg (n=103) of een placebo (n=107), elke twee weken gedurende 24 weken.
Resultaten
Verbetering van astmacontrole (cruciale uitkomstmaat)
Drie studies rapporteerden de uitkomstmaat verbetering van astmacontrole, gemeten met ACQ-5, als gevolg van behandeling met dupilumab.
Dosis 200 mg/2 weken
Wenzel (2016) vond een significante verbetering in de totaalscore van ACQ-5 in de dupilumab-groep vergeleken met de placebogroep zowel in de totale interventiegroep (MD -0,35 (-0,57 tot -0,14)), als in de subgroep ≥ 300 eosinofielen per (MD -0,42 (-0,76 tot -0,07)), Als ook in de subgroep < 300 eosinofielen per μL (MD -0,33 (-0,61 tot -0,05)). Ook Castro (2018) vond een significante verbetering in ACQ-5 score bij 200 mg/2 weken dupilumab (-0,39 (-0,53 tot -0,25)). Er zijn geen klinisch relevante verschillen behaald.
Dosis 300 mg/2 weken
Wenzel (2016) vond een significante verbetering in de totaalscore van ACQ-5 in de dupillumab-groep vergeleken met de placebogroep zowel in de totale interventiegroep (MD -0,31 (-0,52 tot -0,09)), als in de subgroep ≥ 300 eosinofielen per μL (MD -0,55 (-0,90 tot -0,20), dit verschil is klinisch relevant), maar niet in de subgroep <300 eosinofielen per μL. Ook Castro (2018) vond een significante maar niet klinisch relevante verbetering in ACQ-5 score (-0,22 (-0,36 tot -0,08)) in de dupilumab-groep. Rabe (2018) vond een klinisch relevante verbetering in ACQ-5 score in de dupilumab-groep vergeleken met de placebogroep (MD -0,47 (-0,76 tot -0,18)).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van astmacontrole is met één niveau verlaagd gezien het geringe klinische relevante effect (imprecisie).
Verbetering van kwaliteit van leven (cruciale uitkomstmaat)
Twee studies rapporteerden de uitkomstmaat verbetering van kwaliteit van leven, gemeten met de AQLQ-vragenlijst ten gevolge van behandeling met dupilumab.
Dosis 200 mg/2 weken
In de RCT van Wenzel (2016) waren in de totale interventiegroep de globale AQLQ-scores in week 24 ten opzichte van baseline significant hoger bij patiënten in de dupilumab-groep ten opzichte van de placebogroep (MD 0,31 (0,08 tot 0,55)). In de subgroep ≥ 300 eosinofielen per μL, waren de AQLQ-scores significant en klinisch relevant hoger in vergelijking met placebo (MD 0,67 (0,31 tot 1,0)). In de subgroep < 300 eosinofielen per μL waren geen significante verbeteringen gevonden in de dupilumab-groepen ten opzichte van de placebogroep. Resultaten van de studie van Castro (2018) laten een significante maar niet klinisch relevante verbetering in kwaliteit van leven zien (MD 0,29 (0,15 tot 0,44)).
Dosis 300 mg/2 weken
Wenzel (2016) vond een significant maar niet klinisch relevant verschil in AQLQ-scores in de totale interventiegroep in vergelijking met placebo (MD 0,36 (0,12 tot 0,59)). In de subgroep ≥ 300 eosinofielen per μL, waren de AQLQ-scores in vergelijking met placebo significant en klinisch relevant hoger (MD 0,78 (0,42 tot 1,15)). In de subgroep < 300 eosinofielen per μL waren geen significante verbeteringen gevonden ten opzichte van de placebogroep. Resultaten van de studie van Castro (2018) laten een significante maar niet klinisch relevante verbetering in kwaliteit van leven zien (MD 0,26 (0,12 tot 0,40)).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verbetering van kwaliteit van leven is met één niveau verlaagd wegens het kleine aantal studies (imprecisie).
Astma-aanval frequentie (belangrijke uitkomstmaat)
Drie studies rapporteerden de uitkomstmaat astma-aanval frequentie ten gevolge van behandeling met dupilumab.
Dosis 200 mg/2 weken
De resultaten van Wenzel (2016) laten zien dat toediening van dupilumab in alle subgroepen resulteert in een klinisch relevante verlaging van de jaarlijkse incidentie van het aantal ernstige astma-aanvallen in vergelijking met toediening van een placebo (totale interventiegroep: risicoreductie 70,0% (43,5 tot 84,1); ≥ 300 eosinofielen per μL: risicoreductie 71,2% (24,3 tot 89,1); < 300 eosinofielen per μL: risicoreductie 67,6% (24,4 tot 85,9)).
Castro (2018) vond een klinisch relevante lagere jaarlijkse frequentie van ernstige astma-aanvallen in patiënten in de dupilumab-groep versus de placebogroep (RR 0,52 (0,41 tot 0,66)) in de placebogroep. In de subgroep ≥ 300 eosinofielen/ml was het relatieve risico op astma-aanvallen in de dupilumab-groep versus de placebogroep 0,34 (0,24 tot 0,48).
In de subgroep 150 tot 300 eosinofielen/ml was het relatieve risico versus de placebogroep 0,64 (0,41 tot 1,02). In de subgroep < 150 eosinofielen/ml was de astma-aanval frequentie in de dupilumab vergelijkbaar met die in de placebogroep.
Dosis 300 mg/2 weken
Wenzel (2016) vond een klinisch relevante verlaging van de jaarlijkse incidentie van het aantal ernstige astma-aanvallen in de dupilumab-groep in vergelijking met de placebogroep (totale interventiegroep: risicoreductie 70,5% (45,4 tot 84,1); ≥ 300 eosinofielen per μL: risicoreductie 80,7% (44,1 tot 93,3); < 300 eosinofielen per μL: risicoreductie 59,9% (16,1 tot 80,8)). Rabe (2018) vond dat behandeling met dupilumab resulteerde in een klinisch relevant lager percentage van 59% (95% BI 37 tot 74) van astma-aanvallen dan bij behandeling met placebo. De afname in astma-aanvallen was meer uitgesproken in patiënten met eosinofielenwaarden ≥ 300 cellen/ml (71% (40 tot 86)). Ook Castro (2018) vond een klinisch relevante lagere jaarlijkse frequentie van ernstige astma-aanvallen in patiënten in de dupilumab-groep versus de placebogroep (RR 0,54 (0,43 tot 0,68)) in de placebogroep. In de subgroep ≥300 eosinofielen/ml was het relatieve risico op astma-aanvallen 0,33 (0,23 tot 0,45) ten opzichte van de placebogroep. In de subgroep 150-300 eosinofielen/ml was het relatieve risico versus de placebogroep 0,56 (0,35 tot 0,89). In de subgroep < 150 eosinofielen/ml was de astma-aanval frequentie in de dupilumab vergelijkbaar met die in de placebogroep.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat astma-aanval frequentieis niet verlaagd.
Corticosteroïdreductie (belangrijke uitkomstmaat)
Eén studie rapporteerde de uitkomstmaat corticosteroïdreductie ten gevolge van behandeling met dupilumab. Rabe (2018) vond een afname in corticosteroïden dosis van 70% in de dupilumab-groep (300 mg/2 weken) versus een afname van 42% in de placebogroep (p <0,001). 80% versus 50% van de patiënten had een dosisverlaging van minstens 50%, 69% versus 33% had een dosisverlaging tot minder dan 5 mg per dag. In totaal gebruikte 52% van de patiënten in de dupilumab-groep geen orale corticosteroïden meer in week 24, vergeleken met 29% van patiënten in de placebogroep. De afname was het grootst bij patiënten met >300 eosinofielen/ml (OR voor een afname van ten minste 50%: 6,59 (2,13 tot 20,42)) in vergelijking met patiënten met < 300 eosinofielen/ml (OR 2,91 (1,28 tot 6,63)). De verschillen zijn klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat corticosteroïdreductie is met één niveau verlaagd gezien het kleine aantal studies (imprecisie).
Verbetering longfunctie (belangrijke uitkomstmaat)
Drie studies rapporteerden de uitkomstmaat verbetering longfunctie ten gevolge van behandeling met dupilumab.
Dosis 200 mg/2 weken
Wenzel (2016) vond een significante stijging in percentage FEV1 ten opzichte van de placebogroep in de totale interventiegroep (MD 9,60% (4,47 tot 14,74)), in de subgroep met ≥300 eosinofielen per μL (MD 10,1% (1,23 tot 18,90)), en in de subgroep < 300 eosinofielen per μL (8,75% (2,70 tot 14,81)). Castro (2018) vond een grotere verbetering in FEV1 (%) in de dupilumab-groep dan in de placebogroep (MD 9,2% (5,5 tot 12,9)). Alleen in de totale interventiegroep was het verschil klinisch relevant.
Dosis 300 mg/2 weken
Wenzel (2016) vond een significante en klinisch relevante stijging in percentage FEV1 ten opzichte van de placebogroep in de totale interventiegroep (MD 10,33% (5,26 tot 15,40)), in de subgroep met ≥ 300 eosinofielen per μL (MD 12,09% (3,20 tot 20,97)), en in de subgroep < 300 eosinofielen per μL (7,90% (1,98 tot 13,81)), echter is deze niet klinisch relevant. Ook Castro (2018) vond een grotere verbetering in FEV1 (%) in de dupilumab-groep dan in de placebogroep (MD 9,4% (5,7 tot 13,1)), maar niet klinisch relevant. Rabe (2018) vond hogere FEV1 waarden in de dupilumab-groep dan in de placebogroep (0,22 liter (0,09 tot 0,34)). In de groep patiënten met > 300 eosinofielen/ml was dit verschil nog groter en klinisch relevant (0,32 liter (0,10 tot 0,54)).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat verbetering van longfunctie is met één niveau verlaagd vanwege de geringe klinische relevantie.
Bijwerkingen
Drie studies rapporteerden bijwerkingen als uitkomstmaat in hun onderzoek naar de effecten van dupilumab.
Er werd een vergelijkbare incidentie van bijwerkingen gevonden in de dupilumab-groep en de placebogroep (Wenzel, 2016; Rabe, 2018; Castro, 2018). De meest voorkomende bijwerkingen waren infecties van de bovenste luchtwegen en reacties op de injectieplaats (Wenzel, 2016; Rabe, 2018).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat bijwerkingen is met één niveau verlaagd aangezien deze op heterogene wijze werd gerapporteerd in de verschillende studies.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat is de effectiviteit van immuunmodulerende therapie (omalizumab, mepolizumab, reslizumab, benralizumab, dupilumab) als aanvullende behandeling bij ernstig astma?
P (Patiënten) patiënten met ernstig astma ondanks behandeling met medium- hoge dosis ICS plus LABA’s;
I (Interventie) omalizumab, mepolizumab, reslizumab, benralizumab, dupilumab;
C (Comparison) placebo, usual care;
O (Outcomes) astmacontrole (ACQ), kwaliteit van leven (AQLQ en SGRQ), astma-aanval frequentie, corticosteroïdreductie, verbetering longfunctie (FEV), bijwerkingen.
Relevante uitkomstmaten
De werkgroep achtte ‘verbetering van astmacontrole’, en ‘verbetering van kwaliteit van leven’ voor de besluitvorming cruciale uitkomstmaten; en ‘astma-aanval frequentie’, ‘corticosteroïdreductie’, ‘verbetering van de longfunctie’, voor de besluitvorming belangrijke uitkomstmaten.
Verbetering van astmacontrole
De werkgroep definieerde ≥ 0,5 punt verbetering op Asthma Control Questionnaire (ACQ) of >3 punten op de Asthma Control Test (ACT) als een klinisch (patiënt) relevant verschil.
Verbetering van kwaliteit van leven
De werkgroep definieerde ≥ 0,5 punt verbetering op Asthma Quality of Life Questionnaire (AQLQ) en SGRQ (4 punten verschil = klinisch relevant) als een klinisch (patiënt) relevant verschil.
Astma-aanval frequentie
De werkgroep definieerde ≥ 30% afname van aantal astma-aanvallen als een klinisch (patiënt) relevant verschil.
Corticosteroïdreductie
De werkgroep definieerde ≥ 2,5 mg/dag dosisafname prednison of equivalent van ander systemisch steroïd als een klinisch (patiënt) relevant verschil.
Verbetering longfunctie
De werkgroep definieerde een toename FEV1 ≥ 10% van voorspelde waarde als een klinisch (patiënt) relevant verschil.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID), en Embase (via Embase.com) is op 21 april 2018 met relevante zoektermen gezocht naar reviews en vergelijkende studies, gepubliceerd vanaf 2000, die de effectiviteit van immuunmodulerende therapie (biologicals) als aanvullende behandeling bij ernstig astma, ondanks behandeling met medium-hoge dosis ICS plus LABA’s, onderzochten. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 1341 treffers. Vanwege het hoge aantal treffers is er besloten eerst de systematische reviews te beoordelen. Studies werden geselecteerd op grond van de volgende selectiecriteria: relevantie, matig-tot ernstig astma ondanks behandeling met medium-hoge dosis ICS plus LABA’s, en leeftijd van de patiënt. Op basis van titel en abstract werden in eerste instantie 22 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 20 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), en twee systematische reviews geselecteerd en opgenomen in de literatuuranalyse. Vervolgens zijn de RCT’s uit de literatuurzoekactie die gepubliceerd zijn na de zoekdatum van de systematische reviews (vanaf 2013 tot 21 april 2018) beoordeeld. Op basis van titel en abstract werden in eerste instantie 17 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 5 studies geëxcludeerd op basis van studiedesign, relevantie, mate van ernst van astma, leeftijd van de patiënt, en gebruikte dosis. Tenslotte zijn er bij de overgebleven 12 geïncludeerde RCT’s nog twee RCT’s toegevoegd die zijn verschenen na de zoekdatum. In totaal zijn er 14 RCT’s naast de systematische reviews opgenomen in de literatuuranalyse. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk of bias tabellen.
- 1 - Bel EH, Ten Brinke A. New Anti-Eosinophil Drugs for Asthma and COPD: Targeting the Trait! Chest. 2017 Dec;152(6):1276-1282.
- 2 - Bel EH, Wenzel SE, Thompson PJ, Prazma CM, Keene ON, Yancey SW, Ortega HG, Pavord ID; SIRIUS Investigators. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014 Sep 25;371(13):1189-97. doi: 10.1056/NEJMoa1403291. Epub 2014 Sep 8. PubMed PMID: 25199060.
- 3 - Bjermer L, Lemiere C, Maspero J, Weiss S, Zangrilli J, Germinaro M. Reslizumab for Inadequately Controlled Asthma With Elevated Blood Eosinophil Levels: A Randomized Phase 3 Study. Chest. 2016 Oct;150(4):789-798. doi: 10.1016/j.chest.2016.03.032. Epub 2016 Apr 4. PubMed PMID: 27056586.
- 4 - Bleecker ER, FitzGerald JM, Chanez P, Papi A, Weinstein SF, Barker P, Sproule S, Gilmartin G, Aurivillius M, Werkström V, Goldman M; SIROCCO study investigators. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β(2)-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet. 2016 Oct 29;388(10056):2115-2127. doi: 10.1016/S0140-6736(16)31324-1. Epub 2016 Sep 5. PubMed PMID: 27609408.
- 5 - Carr TF, Zeki AA, Kraft M. Eosinophilic and Noneosinophilic Asthma. Am J Respir Crit Care Med. 2018 Jan 1;197(1):22-37Coumou H, Westerhof GA, de Nijs SB, Amelink M, Bel EH. Diagnosing persistent blood eosinophilia in asthma with single blood eosinophil or exhaled nitric oxide level. Respir Med. 2018 Aug;141:81-86.
- 6 - Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, Busse WW, Ford L, Sher L, FitzGerald JM, Katelaris C, Tohda Y, Zhang B, Staudinger H, Pirozzi G, Amin N, Ruddy M, Akinlade B, Khan A, Chao J, Martincova R, Graham NMH, Hamilton JD, Swanson BN, Stahl N, Yancopoulos GD, Teper A. Dupilumab Efficacy and Safety in Moderate-to-Severe Uncontrolled Asthma. N Engl J Med. 2018 Jun 28;378(26):2486-2496. doi: 10.1056/NEJMoa1804092. Epub 2018 May 21. PubMed PMID: 29782217.
- 7 - Castro M, Mathur S, Hargreave F, Boulet LP, Xie F, Young J, Wilkins HJ, Henkel T, Nair P; Res-5-0010 Study Group. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011 Nov 15;184(10):1125-32. doi: 10.1164/rccm.201103-0396OC. Epub 2011 Aug 18. PubMed PMID: 21852542.
- 8 - Castro M, Zangrilli J, Wechsler ME, Bateman ED, Brusselle GG, Bardin P, Murphy K, Maspero JF, O'Brien C, Korn S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015 May;3(5):355-66. doi: 10.1016/S2213-2600(15)00042-9. Epub 2015 Feb 23. Erratum in: Lancet Respir Med. 2015 Apr;3(4):e15. Lancet Respir Med. 2016 Oct;4(10 ):e50. PubMed PMID: 25736990.
- 9 - Chanez P, Contin-Bordes C, Garcia G, Verkindre C, Didier A, De Blay F, de Lara MT, Blanco P, Moreau JF, Robinson P, Bourdeix I, Trunet P, Le Gros V, Humbert M, Molimard M. Omalizumab-induced decrease of FcξRI expression in patients with severe allergic asthma. Respir Med. 2010 Nov;104(11):1608-17. doi: 10.1016/j.rmed.2010.07.011. Epub 2010 Aug 30. PubMed PMID: 20801010.
- 10 - Chung KF, Adcock IM. Precision Medicine for the discovery of treatable mechanisms in severe Asthma. Allergy. 2019 Mar 13. (Epub ahead of print) Review.
- 11 - Chupp GL, Bradford ES, Albers FC, Bratton DJ, Wang-Jairaj J, Nelsen LM, Trevor JL, Magnan A, Ten Brinke A. Efficacy of mepolizumab add-on therapy on health-related quality of life and markers of asthma control in severe eosinophilic asthma (MUSCA): a randomised, double-blind, placebo-controlled, parallel-group, multicentre, phase 3b trial. Lancet Respir Med. 2017 May;5(5):390-400. doi: 10.1016/S2213-2600(17)30125-X. Epub 2017 Apr 5. PubMed PMID: 28395936.
- 12 - Corren J, Weinstein S, Janka L, Zangrilli J, Garin M. Phase 3 Study of Reslizumab in Patients With Poorly Controlled Asthma: Effects Across a Broad Range of Eosinophil Counts. Chest. 2016 Oct;150(4):799-810. doi: 10.1016/j.chest.2016.03.018. Epub 2016 Mar 25. PubMed PMID: 27018175.
- 13 - Eger KA, Bel EH. The emergence of new biologics for severe asthma. Curr Opin Pharmacol. 2019 Jun;46:108-115.
- 14 - European Medicines Agency. Xolair: Summary of Product Characteristics. 2012
- 15 - FitzGerald JM, Bleecker ER, Nair P, Korn S, Ohta K, Lommatzsch M, Ferguson GT, Busse WW, Barker P, Sproule S, Gilmartin G, Werkström V, Aurivillius M, Goldman M; CALIMA study investigators. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2016 Oct 29;388(10056):2128-2141. doi: 10.1016/S0140-6736(16)31322-8. Epub 2016 Sep 5. PubMed PMID: 27609406.
- 16 - Garcia G, Magnan A, Chiron R, Contin-Bordes C, Berger P, Taillé C, Devouassoux G, de Blay F, Couderc LJ, Didier A, O'Callaghan DS, Girodet PO, Bourdeix I, Le Gros V, Humbert M. A proof-of-concept, randomized, controlled trial of omalizumab in patients with severe, difficult-to-control, nonatopic asthma. Chest. 2013 Aug;144(2):411-419. doi: 10.1378/chest.12-1961. PubMed PMID: 23579324.
- 17 - Godar M, Blanchetot C, de Haard H, Lambrecht BN, Brusselle G. Personalized medicine with biologics for severe type 2 asthma: current status and future prospects. MAbs. 2018 Jan;10(1):34-45.
- 18 - Hanania NA, Alpan O, Hamilos DL, Condemi JJ, Reyes-Rivera I, Zhu J, Rosen KE, Eisner MD, Wong DA, Busse W. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med. 2011 May 3;154(9):573-82. doi: 10.7326/0003-4819-154-9-201105030-00002. PubMed PMID: 21536936.
- 19 - Holgate ST, Chuchalin AG, Hébert J, Lötvall J, Persson GB, Chung KF, Bousquet J, Kerstjens HA, Fox H, Thirlwell J, Cioppa GD; Omalizumab 011 International Study Group. Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clin Exp Allergy. 2004 Apr;34(4):632-8. PubMed PMID: 15080818.
- 20 - Humbert M, Beasley R, Ayres J, Slavin R, Hébert J, Bousquet J, Beeh KM, Ramos S, Canonica GW, Hedgecock S, Fox H, Blogg M, Surrey K. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy. 2005 Mar;60(3):309-16. PubMed PMID: 15679715.
- 21 - Iribarren C, Rahmaoui A, Long AA, Szefler SJ, Bradley MS, Carrigan G, Eisner MD, Chen H, Omachi TA, Farkouh ME, Rothman KJ. Cardiovascular and cerebrovascular events among patients receiving omalizumab: Results from EXCELS, a prospective cohort study in moderate to severe asthma. J Allergy Clin Immunol. 2017 May;139(5):1489-1495.e5.
- 22 - Israel E, Reddel HK. Severe and Difficult-to-Treat Asthma in Adults. N Engl J Med. 2017 Sep 7;377(10):965-976.Lim HF, Nair P. Airway Inflammation and Inflammatory Biomarkers. Semin Respir Crit Care Med. 2018 Feb;39(1):56-63.
- 23 - Ledford D, Busse W, Trzaskoma B, Omachi TA, Rosén K, Chipps BE, Luskin AT, Solari PG. A randomized multicenter study evaluating Xolair persistence of response after long-term therapy. J Allergy Clin Immunol. 2017 Jul;140(1):162-169.e2. doi: 10.1016/j.jaci.2016.08.054.
- 24 - Li J, Wang F, Lin C, Du J, Xiao B, Du C, Sun J. The efficacy and safety of reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: A systematic review and meta-analysis. J Asthma. 2017 Apr;54(3):300-307. doi: 10.1080/02770903.2016.1212371. Epub 2016 Jul 19. Review. PubMed PMID: 27435534.
- 25 - Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, Spits H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011 Sep 11;12(11):1055-62
- 26 - Nair P, Wenzel S, Rabe KF, Bourdin A, Lugogo NL, Kuna P, Barker P, Sproule S, Ponnarambil S, Goldman M; ZONDA Trial Investigators. Oral Glucocorticoid-Sparing Effect of Benralizumab in Severe Asthma. N Engl J Med. 2017 Jun 22;376(25):2448-2458. doi: 10.1056/NEJMoa1703501. Epub 2017 May 22. PubMed PMID: 28530840.
- 27 - Nopp A, Johansson SG, Adédoyin J, Ankerst J, Palmqvist M, Oman H. After 6 years with Xolair; a 3-year withdrawal follow-up. Allergy. 2010 Jan;65(1):56-60. doi: 10.1111/j.1398-9995.2009.02144.x.
- 28 - Normansell R, Walker S, Milan SJ, Walters EH, Nair P. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev. 2014 Jan 13;(1):CD003559. doi:10.1002/1 4651858.CD003559.pub4. Review. PubMed PMID: 24414989.
- 29 - Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, Humbert M, Katz LE, Keene ON, Yancey SW, Chanez P; MENSA Investigators. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014 Sep 25;371(13):1198-207. doi: 10.1056/NEJMoa1403290. Epub 2014 Sep 8. Erratum in: N Engl J Med. 2015 Apr 30;372(18):1777. PubMed PMID: 25199059.
- 30 - Papi A, Beghé B, Fabbri LM. We Have to Learn to Do without Knowing Enough: Antieosinophilic Treatments for Severe Asthma. Am J Respir Crit Care Med. 2018 Jan 1;197(1):1-2.
- 31 - Pillai P, Chan YC, Wu SY, Ohm-Laursen L, Thomas C et al. Omalizumab reduces bronchial mucosal IgE and improves lung function in non-atopic asthma. Eur Respir J 2016; 48: 1593–1601
- 32 - Rabe KF, Nair P, Brusselle G, Maspero JF, Castro M, Sher L, Zhu H, Hamilton JD, Swanson BN, Khan A, Chao J, Staudinger H, Pirozzi G, Antoni C, Amin N, Ruddy M, Akinlade B, Graham NMH, Stahl N, Yancopoulos GD, Teper A. Efficacy and Safety of Dupilumab in Glucocorticoid-Dependent Severe Asthma. N Engl J Med. 2018 Jun 28;378(26):2475-2485. doi: 10.1056/NEJMoa1804093. Epub 2018 May 21. PubMed PMID: 29782224.
- 33 - van der Meer AN, Pasma H, Kempenaar-Okkema W, Pelinck JA, Schutten M, Storm H, Ten Brinke A. A 1-day visit in a severe asthma centre: effect on asthma control, quality of life and healthcare use. Eur Respir J. 2016 Sep;48(3):726-33.
- 34 - Wenzel S, Castro M, Corren J, Maspero J, Wang L, Zhang B, Pirozzi G, Sutherland ER, Evans RR, Joish VN, Eckert L, Graham NM, Stahl N, Yancopoulos GD, Louis-Tisserand M, Teper A. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet. 2016 Jul 2;388(10039):31-44. doi: 10.1016/S0140-6736(16)30307-5. Epub 2016 Apr 27. PubMed PMID: 27130691.
- 35 - Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009 Sep 1;180(5):388-95. doi:10.1164/rccm.200903-0392OC. Epub 2009 May 29. Erratum in: Am J Respir Crit Care Med. 2009 Oct 15;180(8):796.
Research question: What is the added value of immune modulating therapy (biological drugs) as an add-on treatment for severe asthma?
Table Evidence table for systematic reviews - Biologicals
Table - Quality assessment for systematic reviews of RCTs and observational studies – biologicals
Based on AMSTAR checklist (Shea, 2007; BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher, 2009; PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
|
Study
First author, year |
Appropriate and clearly focused question?
Yes/no/unclear |
Comprehensive and systematic literature search?
Yes/no/unclear |
Description of included and excluded studies?
Yes/no/unclear |
Description of relevant characteristics of included studies?
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?
Yes/no/unclear/no tapplicable |
Assessment of scientific quality of included studies?
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?
Yes/no/unclear |
Potential risk of publication bias taken into account?
Yes/no/unclear |
Potential conflicts of interest reported?
Yes/no/unclear |
|
Normansell, 2014 |
Yes |
Yes |
Yes |
Yes |
N/A |
Yes |
Yes |
Yes |
Yes |
|
Li, 2017 |
Yes |
Yes |
Yes |
Yes |
N/A |
Yes |
Yes |
Yes |
No |
Table - Evidence table for intervention studies - Biologicals
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Wenzel, 2016
Dupilumab |
Type of study: randomised, double-blind, placebo-controlled, parallel-group, pivotal phase 2b clinical trial
Setting: Multi-center (174)
Country: Multinational
Source of funding: Sanofi and Regeneron Pharmaceuticals |
Inclusion criteria: - Adults (aged ≥18 years) with asthma diagnosis for >12 months - have existing treatment with medium-to high- dose inhaled corticosteroids plus a long-acting β2 agonist (fluticasone propionate ≥250 μg, or equivalent inhaled corticosteroids, twice daily); - a pre-bronchodilator forced expiratory volume in 1s (FEV1) of 40–80% predicted at screening and at baseline; - a ACQ-5 score of 1·5 or higher at screening and at baseline; - reversibility of >12% and 200 mL in FEV1 after 200−400 μg of salbutamol at screening. - Patients were also required for study inclusion to have had any of the following within 1 year before screening: at least one systemic (oral or parenteral) corticosteroid burst therapy, or a hospital admission or an emergency or urgent medical care visit that required treatment with systemic steroids for worsening asthma.
N total at baseline: Intervention: 618 Control: 158
Important prognostic factors2: Not specified by intervention versus control group, but by eosinophilic count
|
Subcutaneous dupilumab 200 mg every 4 weeks (n=154), 300 mg every 4 weeks (n=157), 200 mg every 2 weeks (n=150), 300 mg every 2 weeks (n=157) (= 4 intervention groups).
|
Placebo
|
Length of follow-up: 24-week treatment and 16-week post-treatment follow-up
Loss-to-follow-up: Intervention: 55 (9%) Reasons: adverse events other reasons
Control: 12 (8%) Reasons: lack of efficacy adverse events poor compliance
|
Outcome measures and effect size (include 95%CI and p-value if available):
Change in FEV1 (L) subgroup ≥ 300 eosinophils: 300 mg/4wks, p=0·0212; 200 mg/2wks, p=0·0008; 300 mg/2wks, p=0·0063).
overall population: 200 mg/2wks, p<0·0001; 300 mg/2wks, p=0·0002;
< 300 eosinophils per μL: 200/2wks, p=0·0057; 300 mg/2wks, p=0·0262).
Asthma exacerbations Total population: 200 mg/2wks, p<0,001, risk reduction 70%; 300 mg/2wks, p<0,001, risk reduction 71%;
≥300 eosinofielen per μL: 200 mg/2wks, p=0,001, risk reduction 71%; 300 mg/2wks, p=0,005, risk reduction 81%;
<300 eosinofielen per μL: 200 mg/2wks, p=0,009, risk reduction 68%; 300 mg/2wks, p=0,013, risk reduction 60%.
ACQ-5 Total population: 200 mg/2wks MD –0,35, 95%BI –0,57 tot –0,14), P=0,002; 300 mg/2 wks MD –0,31, 95% BI –0,52 tot –0,09, P=0,0049
subgroup ≥ 300 eosinophils 200 mg/2wks MD –0,42, 95% BI –0,76 tot –0,07, P= 0,017; 300 mg/2wks MD –0,55, 95% BI –0,90 tot –0,20, P=0,002.
< 300 eosinophils per μL: Only with 200mg/2wks significant difference: MD -0,33, 95% BI -0,61 tot -0,05.
AQLQ Total population: 200 mg/4wks MD 0,23, 95% BI –0,00 tot 0,47, P=0,053; 300 mg/4wks MD 0,30, 95% BI 0,07 tot 0,53, P=0,012; 200 mg/2wks MD 0,31, 95% BI 0,08 tot 0,55, P=0,009; 300 mg/2wks MD 0,36, 95% BI 0,12 tot 0,59, P= 0,003
subgroup ≥ 300 eosinophils 200 mg/4wks MD 0,53, 95% BI 0,16 tot 0,90, P=0,005; 300 mg/4wks MD 0,43, 95% BI 0,07 tot 0,79, P=0,018; 200 mg/2wks MD 0,67, 95% BI 0,31 tot 1,0, P=<0,001; 300 mg/2wks MD 0,78, 95% BI 0,42 tot 1,15; P=<0,001
< 300 eosinophils per μL: No differences. |
|
|
Dupilumab |
Type of study: randomized, double-blind, placebo-controlled, parallel-group trial
Setting: Multi-center
Country: Multinational
Source of funding: Sanofi and Regeneron Pharmaceuticals |
Inclusion criteria: - Patients> 12 years - physician diagnosed persistent asthma for >12 months - current treatment with a medium-to-high-dose inhaled glucocorticoid (fluticasone propionate at a total daily dose of ≥500 μg) plus up to two additional controllers - a FEV1 before bronchodilator use of 80% or less of the predicted normal value - FEV1 reversibility of at least 12% and 200 ml; - ACQ-5 score on of 1.5 or higher - a worsening of asthma in the previous year that led to hospitalization, emergency medical care, or treatment with systemic glucocorticoids for 3 days or more.
N total at baseline: Intervention1 (200mg): 631 Intervention2 (300mg): 633 Control (1.14mL): 317 Control (2.0mL): 321 Totaal=1902
Important prognostic factors2: age ± SD: I1: 47.9±15.3 I2: 47.7±15.6 C1: 48.2±15.6 C2: 48.2±14.7
Sex (females, %): I1: 387 (61.3%) I2: 394 (62.2%) C1: 198 (62.5%) C2: 218 (67.9%)
ACQ5 score (at baseline) I1: 2.76±0.80 I2: 2.77±0.76 C1: 2.71±0.73 C2: 2.77±0.77
FEV1 % predicted I1: 58.38±13.52 I2: 58.51±13.52 C1: 58.43±13.22 C2: 58.35±13.87
Groups are comparable at baseline
|
Dupilumab, SC, 200mg or 300mg, every 2 weeks for 52 weeks |
Placebo, 1.14 or 2.00 mL, every 2 weeks for 52 weeks
|
Length of follow-up: 64 weeks
Loss-to-follow-up: Intervention 200 mg: 70 (14%) Reasons: - adverse event: 21 (30%) - lack of efficacy: 4 (6%) - non-compliance: 3 (4%) - other: 42 (60%)
Intervention 300 mg: 85 (18%) Reasons: - adverse event: 46 (54%) - lack of efficacy: 3 (4%) - non-compliance: 1 (1%) - other: 36 (42%)
Control 1.14 ml: 38 (17%) Reasons: - adverse event: 19 (50%) - lack of efficacy: 3 (8%) - non-compliance: 4 (10%) - other: 12 (32%)
Control 2 ml: 35 (14%) Reasons: - adverse event: 10 (29%) - lack of efficacy: 5 (14%) - non-compliance: 3 (9%) - other: 17 (49%)
|
Asthma exacerbations Annualized rate of severe asthma exacerbations: 200mg: 0.46 (95% confidence interval (CI), 0.39 to 0.53) versus 0.87 (0.72 to 1.05) placebo (47.7% lower rate with dupilumab than with placebo, P<0.001), 300mg: 0.52 (95% CI, 0.45 to 0.61) versus 0.97 (0.81 to 1.16) placebo (46.0% lower rate with dupilumab than with placebo, P<0.001)
subgroup >300 eosinophils: 200mg: 0.37 (95% CI, 0.29 to 0.48) versus 1.08 (95% CI, 0.85 to 1.38) placebo (65.8% lower rate with dupilumab than with placebo; 95% CI, 52.0 to 75.6), 300mg: 0.40 (95% CI, 0.32 to 0.51) versus 1.24 (95% CI, 0.97 to 1.57) placebo (67.4% lower rate with dupilumab than with placebo, P<0.001).
subgroup 150-300 eos: 200mg: 0.56 (95% CI, 0.42 to 0.75) versus 0.87 (95% CI, 0.59 to 1.27) placebo (35.6% lower rate), 300mg: 0.47 (95% CI, 0.35 to 0.64) versus 0.84 (95% CI, 0.58 to 1.23) placebo (44.3% lower rate).
subgroup <150 eos, the exacerbation rate was similar with dupilumab and with placebo.
Severe exacerbation events resulting in hospitalization or an emergency department visit: combined dupilumab 0.035 (95% CI, 0.025 to 0.048) versus combined placebo 0.065 (95% CI, 0.047 to 0.090).
Quality of life (AQLQ) dupilumab showed benefits over matched placebo in AQLQ score: (200 mg: 0,29 (0,15 to 0,44), 300 mg: 0,26 (0,12 to 0,40)).
Change in FEV1 difference versus. matched placebo at 52 weeks, 200 mg: 0.20 liters (95% CI, 0.14 to 0.25) 300 mg: 0.13 liters (0.08 to 0.19).
The percentage change from baseline to week 12 in the FEV1 before bronchodilator: 200 mg: 9.2 % (95% CI, 5.5 to 12.9) 300 mg: 9.4 % (95% CI, 5.7 to 13.1)
Change in ACQ score ACQ-5 scores were lower (indicating better asthma control) with dupilumab than with placebo as early as week 2, and the effect was sustained over the 52-week intervention period (200 mg: -0,39 (-0,53 tot -0,25), 300 mg: -0,22 (-0,36 tot -0,08))
Adverse events The incidence of adverse events was similar across intervention groups (81.0% in the combined dupilumab groups and 83.1% in the combined placebo groups) in the safety population
Steroïd reduction - |
|
|
Rabe, 2018
Dupilumab |
Type of study: randomized, double-blind, placebo-controlled, phase 3 trial
Setting: Multi-center
Country: Multinational
Source of funding: Sanofi and Regeneron Pharmaceuticals |
Inclusion criteria: - >12 years - physician-diagnosed asthma for 1 year or more according to the Global Initiative for Asthma 2014 guidelines - receiving treatment with regular systemic glucocorticoids in the previous 6 months (5 to 35 mg per day of prednisone or prednisolone or equivalent) - During the 4 weeks before screening, treatment had to also include a high-dose inhaled glucocorticoid (fluticasone propionate at a total daily dose of >500 μg or equipotent equivalent) in combination with up to two controllers (i.e., a long-acting β2-agonist or leukotriene-receptor antagonist) for at least 3 months. - FEV1 before bronchodilator use of 80% or less of the predicted normal value (or ≤90% of the predicted normal value in adolescents) - FEV1 reversibility of at least 12% and 200 ml, or airway hyperresponsiveness documented in the 12 months before screening visit 1.
N total at baseline: Intervention: 103 Control: 107 Total: 210
Important prognostic factors2: age ± SD: I: 51.9±12.5 C: 50.7±12.8
Sex (males, %): I: 41 (40) C: 42 (39)
ACQ5 score (at baseline) I: 2.42±1.24 C: 2.58±1.09
FEV1 % predicted I: 51.64±15.28 C: 52.69±15.14
Groups are comparable at baseline |
subcutaneous dupilumab, 300 mg, every 2 weeks, for 24 weeks |
Placebo, every 2 weeks, for 24 weeks |
Length of follow-up: 36 weeks
Loss-to-follow-up: Two patients in the dupilumab group and one in the placebo group discontinued the trial. |
Asthma exacerbations 59% (95% CI, 37 to 74) lower rate of severe asthma exacerbations in dupilumab than placebo in the overall population.
Quality of life (AQLQ) -
Change in FEV1 0.22 liters (95% CI, 0.09 to 0.34)
Change in ACQ score MD −0.47 (95% CI, −0.76 to −0.18).
Adverse events the incidence of adverse events was similar in the dupilumab and placebo group (62% versus 64%). The most frequent adverse events were viral upper respiratory tract infection (9% dupilumab versus. 18% placebo group), bronchitis (7% versus. 6%), sinusitis (7% versus. 4%), influenza (3% versus. 6%), and the laboratory measure of eosinophilia (14% versus. 1%)
Steroïd reduction The percentage change in the glucocorticoid dose was 70.1% in the dupilumab group, as compared with −41.9% in the placebo group (P<0.001); 80% versus 50% of the patients had a dose reduction of at least 50%, 69% versus 33% had a dose reduction to less than 5 mg per day.
|
|
|
Nair, 2017
Benralizumab |
Type of study: randomized, double-blind, parallel-group, placebo-controlled trial
Setting: Multi-center
Country: Global
Source of funding: Funded by AstraZeneca |
Inclusion criteria: - blood eosinophil count of >150 cells - asthma that had been treated with medium-dose to high-dose inhaled glucocorticoid and LABA therapy for at least 12 months before enrollment -treated with high-dose inhaled glucocorticoid and LABA therapy for at least 6 months before enrollment. - Patients had been receiving oral glucocorticoid therapy for at least 6 continuous months directly before enrolment (equivalent to a prednisolone or prednisone dose of 7.5 to 40.0 mg per day).
N total at baseline: Intervention 1: 72 Control: 75
Important prognostic factors2: Age (yrs): I1: 50.2±12.0 I2: 52.9±10.1 C: 49.9±11.7
Sex (n, % females): I1: 40 (56) I2: 47 (64) C: 48 (64)
ACQ-6 score (at baseline) I1: 2.6±1.1 I2: 2.4±1.2 C: 2.7±1.0
FEV1 (% predicted) I1: 57.4±18.0 I2: 59.0±17.9 C: 62.0±16.5
Groups are comparable at baseline
|
Benralizumab (at a dose of 30 mg administered subcutaneously either every 4 weeks or every 8 weeks)
|
Placebo
|
Length of follow-up: 28 weeks
Loss-to-follow-up: A total of 11 patients did not complete the trial, including 5 who withdrew of their own decision. |
Asthma exacerbations Dose/4wks annual asthma exacerbation rate: 0.45; 95% CI, 0.27 to 0.76; P = 0.003). Dose/8wks: 0.30; 95% CI, 0.17 to 0.53; P<0.001). Odds ratio for dose/4wks versus. placebo: 0.32; 95% CI, 0.16 to 0.65; P = 0.001. Dose/8wks: 0.28; 95% CI, 0.14 to 0.56; P<0.001.
Quality of life (AQLQ) Dose/8wks: 0.45 points (95% CI, 0.14 to 0.76) higher than the increase with placebo (P = 0.004). Dose/4wks: 0.23; 95% CI, –0.08 to 0.53; P = 0.15).
Change in FEV1 FEV1 before bronchodilation dose/4wks: 256 ml (95% CI, 109 to 403). Dose/8wks: 222 ml (95% CI, 75 to 370).
Change in ACQ score Dose/8wks: 0.55 points (95% CI, 0.23 to 0.86) greater than the decrease with placebo (P = 0.001). Dose/4wks: –0.24 points; 95% CI, –0.55 to 0.08; P = 0.14).
Adverse events 75%) had at least one adverse event. The most frequently reported adverse events were nasopharyngitis (17%), worsening asthma (13%), and bronchitis (10%). Worsening asthma was the most common serious adverse event (in 7 patients).
Steroïd reduction the median reduction from baseline in the final oral glucocorticoid dose was 75% in both benralizumab groups, as compared with a reduction of 25% in placebo group (P<0.001). Dose/4wks: OR 4.09 times (95% confidence interval (CI), 2.22 to 7.57). Dose/8wks: 4.12 times (95% CI, 2.22 to 7.63). |
|
|
Bleecker, 2016
Benralizumab |
Type of study: randomised, double-blind, parallel-group, placebo-controlled phase 3 study
Setting: Multi-center
Country: Global
Source of funding: AstraZeneca and Kyowa Hakko Kirin |
Inclusion criteria: - 12–75 years, weight at least 40 kg - diagnosed by a physician with asthma needing treatment with medium-dosage or highdosage ICS plus LABA for at least 1 year before enrolment - at least two documented asthma exacerbations needing systemic corticosteroid treatment or a temporary increase in their usual maintenance dosages of oral corticosteroids within 1 year before enrolment. - treatment of ICS plus LABA with or without oral corticosteroids and additional asthma controllers for at least 3 months before enrolment - a prebronchodilator FEV1 of < 80% predicted at screening - a documented post-bronchodilator reversibility of at least 12% and at least 200 mL in FEV1 within 12 months before enrolment - an ACQ-6 score of at least 1,5 at enrolment.
N total at baseline: Intervention 1: 399 Control: 407
Important prognostic factors2: Age (yrs): I1: 50.1±13.4 I2: 47.6±14.5 C: 48.7±14.9
Sex (n, % males): I1: 124 (31%) I2: 146 (37%) C: 138 (34%)
ACQ-6 score (at baseline) I1: 2.8±1.0 I2: 2.8±0.9 C: 2.9±0.9
FEV1 (% predicted) I1: 57.4±14.1 I2: 56.1±14.6 C: 66.6±15.0
Groups are comparable at baseline |
Benralizumab; SC 30mg, every 4 or 8 weeks, for 48 weeks |
placebo; for 48 weeks |
Length of follow-up: 48 weeks
Loss-to-follow-up: I1 (4wks): 50 discontinued treatment 20 patient decision 9 adverse events 3 study-specific discontinuation criteria 4 lost to follow-up 5 severe non-compliance with the protocol 9 other reasons
I2 (8 wks): 40 discontinued treatment 16 patient decision 8 adverse events 2 study-specific discontinuation criteria 4 lost to follow-up 3 severe non-compliance with the protocol 7 other reasons
C: 45 discontinued treatment 20 patient decision 5 adverse events 3 study-specific discontinuation criteria 2 lost to follow-up 3 severe non-compliance with the protocol 11 other reasons 1 missing |
Asthma exacerbations RR 0.62 (0.50-0.77)
Quality of life (AQLQ) 4 weeks: MD 0,18 (–0,02 to 0,37) 8 weeks 0,30 (0,10–0,50)
Change in FEV1 MD 0.12 (0.05-0.19)
Change in ACQ score 8 weeks: –0.29 (95% CI –0.48 to –0.10); p=0·0028 4 weeks: –0,15 (–0,34 to 0,04); 0·1107)
Adverse events similar percentages of AE between benralizumab (574 (72%) of 797) and placebo (311 (76%) of 407). The most common adverse events in benralizumab group were worsening asthma (105 (13%)), nasopharyngitis (93 (12%)), and upper respiratory tract infection (76 (10%)); adverse events related to asthma were reported by proportionally less patients in the benralizumab-treated groups than in the placebo group. Serious adverse events were also reported by similar percentages of patients with benralizumab (99 (12%)) or placebo (55 (14%)). The most common serious adverse event in all patients was worsening asthma (31 (8%) in the placebo group versus 22 (5%) in the benralizumab Q4W group versus 24 (6%) in the benralizumab Q8W group); no other serious adverse event was experienced by more than 1% of patients.
Steroïd reduction - |
|
|
FitzGerald, 2016
Benralizumab |
Type of study: randomised, double-blind, parallel-group, placebo-controlled, phase 3 study (CALIMA)
Setting: Multi-center
Country: Global
Source of funding: AstraZeneca and Kyowa Hakko Kirin |
Inclusion criteria: - 12–75 years, weight at least 40 kg – physician diagnosed asthma requiring treatment with medium to high-dosage inhaled corticosteroids (>250 μg (medium) or ≥500 μg (high) fluticasone dry powder formulation or equivalent total daily dosage) plus LABA, for 12 months or more before enrolment - two or more asthma exacerbations in the 12 months before enrolment that required use of a systemic corticosteroid or temporary increase in the patient’s usual maintenance dosage of oral corticosteroids. - a pre bronchodilator FEV1 of <80% predicted (<90% predicted for patients aged 12–17 years) at screening; - ACQ-6 score ≥1,5 at enrolment; - post-bronchodilator reversibility in FEV1 of >12% and 200 mL or greater in FEV1 within 12 months before enrolment
N total at baseline: Intervention 1: 425 Control: 440
Important prognostic factors2: Age (yrs): I1: 50.0±13.6 I2: 49.0±14.3 C: 48.8±15.1
Sex (n, % males): I1: 155 (36%) I2: 168 (38%) C: 176 (40%)
ACQ-6 score (at baseline) I1: 2.7±0.9 I2: 2.8±0.9 C: 2.7±0.9
FEV1 (% predicted) I1: 58.9±14.8 I2: 57.9±14.9 C: 58.0±14.9
Groups are comparable at baseline |
Benralizumab; 14 SC doses of 30mg or 8 SC doses of 30mg, for 56 weeks |
placebo; for 56 weeks |
Length of follow-up: 56 weeks
Loss-to-follow-up: I1 (4wks): 41 discontinued treatment 16 patient decision 8 adverse events 5 study-specific discontinuation criteria 1 lost to follow-up 1 severe non-compliance with the protocol 10 other reasons
I (8wks): 59 discontinued treatment 28 patient decision 9 adverse events 5 study-specific discontinuation criteria 4 lost to follow-up 1 severe non-compliance with the protocol 12 other reasons
C: 49 discontinued treatment 19 patient decision 5 adverse events 9 study-specific discontinuation criteria 4 lost to follow-up 2 severe non-compliance with the protocol 10 other reasons |
Asthma exacerbations 4 weeks: RR 0,64 (0,49–0,85) 8 weeks: RR 0,72 (0,54–0,95)
Quality of life (AQLQ) 4 weeks: MD 0,16 (–0,04 to 0,37) 8 weeks: MD 0,24 (0,04 to 0,45)
Change in FEV1 4 weeks: MD 0,125 (0,037–0,213) 8 weeks: MD 0,116 (0,028–0,204)
Change in ACQ score 4 weeks: MD –0,19 (–0,38 to –0,01) 8 weeks: MD –0,25 (–0,44 to –0,07)
Adverse events Fewer patients treated with benralizumab (642 (74%) of 866) versus placebo (342 (78%) of 440) experienced adverse events. The most common adverse event was nasopharyngitis (20%). Adverse events were related to study treatment in 141 (11%) patients, 22 (2%) patients experienced adverse events that led to discontinuation of treatment, and four (<1%) patients died because of adverse events (table 4). Adverse events were mild in 317 (24%) patients, moderate in 551 (42%) patients, and severe in 116 (9%) patients, when classified according to their maximum intensity during the on-treatment period.
Steroïd reduction - |
|
|
Chupp, 2017
Mepolizumab |
Type of study: randomised, double-blind, placebo-controlled, parallel-group, phase 3b trial
Setting: Multi-center
Country: Global
Source of funding: GlaxoSmithKline |
Inclusion criteria: - patients > 12 years – severe eosinophilic asthma who had experienced at least two exacerbations requiring treatment with systemic corticosteroids in the previous 12 months (for patients on maintenance oral corticosteroids, two-fold or greater dose increases were required for inclusion), despite treatment with regular high-dose inhaled corticosteroids in the 12 months before screening, plus additional controller medication(s) for at least 3 months before screening. - airflow obstruction as indicated by a pre-bronchodilator FEV1 of < 80% predicted in those aged ≥18 years, or less than 90% predicted in those aged 12–17 years. - blood eosinophil count of ≥ 300 cells/μL within the 12 months before screening, or a blood eosinophil count of ≥150 cells/μL at screening
N total at baseline: Intervention: 274 Control: 277
Important prognostic factors2: Age (yrs): I: 49,8±14,0 C: 52,1±12,9
Sex (n, % males): I: 125 (46%) C: 101 (36%)
ACQ-5 score (at baseline) I: 2.2±1.1 C: 2.2±1.2
FEV1 (% predicted) I: 55.5±14.4 C: 55.2±14.6
Groups are comparable at baseline |
Subcutaneous injection of mepolizumab 100 mg, every 4 weeks for 24 weeks
|
Placebo, every 4 weeks for 24 weeks
|
Length of follow-up: 24 weeks
Loss-to-follow-up: Not described |
Asthma exacerbations Annualised rates of clinically significant exacerbations: 0·42 (0·31–0·56; p<0·0001). Exacerbations requiring an emergency room visit or admission to hospital: 0·32 (0·12–0·90; p=0·031).
Quality of life (SGRQ) Least squares mean (SE) change from baseline −15.6 (1.00) −7.9 (1.01), a treatment difference of −7,7 (95% CI −10,5 to −4,9; p<0·0001).
Change in FEV1 pre-bronchodilator FEV1 120 ml (47–192; p=0·001)
Change in ACQ score MD –0·4 (95%CI –0·6 to –0·2; p<0·0001)
Adverse events 192 (70%) of 273 patients who received mepolizumab and 207 (74%) of 278 patients who received placebo reported at least one on-treatment adverse event, and 31 (11%) and 25 (9%) patients reported at least one treatment-related adverse event in the mepolizumab and placebo groups, respectively (table 4). The most frequently reported adverse events in both the mepolizumab group and the placebo group were headache and nasopharyngitis.
Steroïd reduction - |
|
|
Ortega, 2014
Mepolizumab |
Type of study: randomized, double-blind, double-dummy, phase 3, placebo controlled trial
Setting: Multi-center
Country: international
Source of funding: GlaxoSmithKline |
Inclusion criteria: - age 12 - 82 years - clinical diagnosis of asthma by a physician - FEV1 < 80% of the predicted value (for adults) or an FEV1 of < 90% of the predicted value or a ratio of the FEV1 to FVC of <0.8 (for adolescents <18 years). - one or more of the following three test results: FEV1 reversibility of >12%, positive results on methacholine or mannitol challenge at visit 1 or 2 or during the previous year, and FEV1 variability (≥20%) between two clinic visits in the past 12 months. - at least two asthma exacerbations in the previous year that were treated with systemic glucocorticoids while they were receiving treatment with at least 880 μg of fluticasone propionate or the equivalent by inhalation per day and at least 3 months of treatment with an additional controller. - eosinophil count of at least 150 cells/µl in the peripheral blood at screening or at least 300 cells/µl at some time during the previous year.
N total at baseline: Intervention 1: 191 Control: 191
Important prognostic factors2: Age (range): I1: 50 (13–82) I2: 51 (12–81) C: 49 (12–76)
Sex (n, % females): I1: 106 (55%) I2: 116 (60%) C: 107 (56%)
ACQ-6 score (at baseline) I1: 2.12±1.13 I2: 2.26±1.27 C: 2.28±1.19
FEV1 (% predicted) I1: 61.4±18.3 I2: 59.3±17.5 C: 62.4±18.1
Groups are comparable at baseline |
Mepolizumab, 9 doses 75 IV or 100 mg SC, 32 weeks |
placebo; for 32 weeks |
Length of follow-up 40 weeks
Loss-to-follow-up: I1: 16 (8%) Discontinued study 9 Withdrew 1 Had lack of efficacy 2 Were lost to follow-up 3 Had protocol violation 1 Was withdrawn by physician
I2: 9 (5%) Discontinued study 4 Withdrew 1 Had adverse event 2 Had lack of efficacy 2 Were lost to follow-up
C: 12 (6%) Discontinued study 5 Withdrew 4 Had adverse event 1 Had lack of efficacy 2 Were withdrawn by physician |
Asthma exacerbations I1: MD 47 (28 to 60) I2: MD 53 (36 to 65)
Quality of life (SGRQ) I1: MD −6.4 (−9.7 to −3.2) I2: MD −7.0 (−10.2 to −3.8)
Change in FEV1 I1: MD 100 (13 to 187) I2: MD 98 (11 to 184)
Change in ACQ score I1: MD −0.42 (−0.61 to −0.23) I2: MD −0.44 (−0.63 to −0.25)
Adverse events The overall incidence of adverse events during treatment was similar in the three groups (84% in the intravenous-mepolizumab group, 78% in the subcutaneous-mepolizumab group, and 83% in the placebo group). The most frequently reported adverse events were nasopharyngitis and headache.
Steroïd reduction - |
|
|
Bel, 2014
Mepolizumab |
Type of study: randomized, double-blind trial
Setting: Multi-center
Country: international
Source of funding: GlaxoSmithKline |
Inclusion criteria: - at least a 6-month history of maintenance treatment with systemic glucocorticoids (5 to 35 mg per day of prednisone or its equivalent) before entering the study. - presence of eosinophilic inflammation, determined by a blood eosinophil level of either ≥300 cells/µl during the 12-month period before screening or ≥150 cells/µl during the optimization phase. - treatment with high-dose ICS + additional controller.
N total at baseline: Intervention: 69 Control: 99
Important prognostic factors2: age (range): I: 50 (16–74) C: 50 (28–70)
Sex (n, % females): I: 44 (64%) C: 30 (45%)
ACQ5 score (at baseline) I: 2.2±1.3 C: 2.0±1.2
FEV1 (% predicted) I: 59.6±17.0 C: 57.8±18.5
Groups are comparable at baseline |
Mepolizumab, 6 doses 100 mg SC, 20 weeks |
placebo; for 20 weeks |
Length of follow-up: 32 weeks
Loss-to-follow-up: I: 3 (4%) Discontinued study owing to adverse event
C: 4 (6%) Discontinued study 3 Had adverse event 1 Withdrew |
Asthma exacerbations RR 0.69 (0.50-0.95)
Quality of life (SGRQ) MD −5.8 points (95% CI, −10.6 to −1.0)
Change in FEV1 MD 0.14 (0.06-0.22)
Change in ACQ score MD: −0.52 points (95% CI, −0.87 to −0.17)
Adverse events The incidence of nonasthma-related adverse events was 83% in the mepolizumab group and 91% in the placebo group. The most frequently reported adverse events in the two study groups were headache and nasopharyngitis. Seven patients (4 in the mepolizumab group and 3 in the placebo group) had systemic reactions, and 6 patients (4 in the mepolizumab group and 2 in the placebo group) had local injection-site reactions.
Steroïd reduction OR 2.39 (1.25–4.56) |
|
Table - Risk of bias for intervention studies (randomized controlled trials) - biologicals
|
Study reference
(first author, publication year) |
Describe method of randomisation |
Bias due to inadequate concealment of allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?
(unlikely/likely/unclear) |
Bias due to loss to follow-up?
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?
(unlikely/likely/unclear) |
|
Wenzel, 2016 |
Patients were randomised (1:1:1:1:1) by a centralised treatment allocation system |
Unlikely
Note: centralized system |
Unlikely
Note: treatment and placebo are in same form. Administers and patients unaware of difference. |
Unlikely
Note: treatment and placebo are in same form. Administers and patients unaware of difference. |
Unlikely
Note: investigators and site personnel masked to study treatment. |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unlikely
Note: percentages loss to follow-up are similar between intervention and control + reasons are reported. |
Unlikely
Note: The intention-to-treat population included all patients randomly assigned to treatment. |
|
Castro, 2018 |
Patients were randomly assigned in a 2:2:1:1 ratio, by a by means of interactive voice–Web response technology.
|
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Not described |
Note: Efficacy analyses were performed in the intention-to-treat population, which included all patients who underwent randomization. |
|
Rabe, 2018 |
Patients were randomly assigned in a 1:1 ratio, by means of interactive voice–Web response technology. |
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unlikely
Note: Only two patients in the dupilumab group and one in the placebo group discontinued the trial or had missing data. |
Unlikely
Note: Efficacy analyses were performed in the intention-to-treat population, which included all patients who underwent randomization. |
|
Nair, 2017 |
Patients underwent randomization in a 1:1:1 ratio, with the use of an interactive Web- or voiceresponse system |
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant and investigator |
Unlikely
Note: Masking of participant and investigator |
Unlikely
Note: Masking of participant and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unclear
Note: not described in article. |
Unlikely
Note: All patients who underwent randomization were included in the full analysis set. |
|
Chupp, 2017 |
Patients were randomly assigned in a 1:1 ratio, by means of a centralized computer system |
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unclear
Note: not described in article. |
Unlikely
Note: Efficacy analyses were performed in the intention-to-treat population. |
|
Ortega, 2014 |
Patients were randomly assigned in a 1:1 ratio, by means of a centralized computer system |
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unlikely
Note: percentages loss to follow-up are similar between intervention and control + reasons are reported. |
Unlikely
Note: Efficacy analyses were performed in the intention-to-treat population. |
|
Bel, 2014 |
Patients were randomly assigned in a 1:1 ratio, by means of a centralized computer system |
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unlikely
Note: percentages loss to follow-up are similar between intervention and control + reasons are reported. |
Unlikely
Note: Efficacy analyses were performed in the intention-to-treat population. |
|
Bleecker, 2016 |
Patients were randomly assigned in a 1:1:1 ratio, by means of a centralized computer system |
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unlikely
Note: percentages loss to follow-up are similar between intervention and control + reasons are reported. |
Unlikely
Note: Efficacy analyses were performed in the intention-to-treat population. |
|
FitzGerald, 2016 |
Patients were randomly assigned in a 1:1:1 ratio, by means of a centralized computer system |
Unlikely.
Note: centralized system |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: Masking of participant, care provider, and investigator |
Unlikely
Note: outcomes described in methods section are reported in the results. |
Unlikely
Note: percentages loss to follow-up are similar between intervention and control + reasons are reported. |
Unlikely
Note: Efficacy analyses were performed in the intention-to-treat population. |
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Reviews |
|
|
Bermejo, 2018 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Meng, 2018 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Nachef, 2018 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Wang, 2018 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Alhossan, 2017 |
Non-controlled real-life studies |
|
Cabon, 2017 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Cockle, 2017 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Farne, 2017 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Yancey, 2017 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Abraham, 2016 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Wang, 2016 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Lai, 2015 |
Resultaten kinderen en volwassenen niet los gerapporteerd |
|
Liu, 2013 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Norman, 2013 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Rodrigo, 2011 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Cox, 2007 |
Komt niet overeen met de PICO |
|
Hendeles, 2007 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Chipps, 2006 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Niebauer, 2006 |
Ander review met meer geïncludeerde studies beschikbaar |
|
Holgate, 2001 |
Ander review met meer geïncludeerde studies beschikbaar |
|
RCT’s |
|
|
Ferguson, 2017 |
Geen ernstig astma |
|
Iribarren, 2017 |
Prospective cohort study |
|
Ledford, 2017 |
Geen ernstig astma |
|
Castro, 2014 |
Er wordt niet de gangbare dosis gebruikt |
|
Pillai, 2016 |
Open-label studie |
|
Bousquet, 2011 |
Open-label studie |
|
Ayres, 2004 |
Open-label studie |
|
Wenzel, 2013 |
Onjuiste dosering |
Beoordelingsdatum en geldigheid
Publicatiedatum : 21-09-2020
Beoordeeld op geldigheid : 01-07-2020
Voor het beoordelen van de actualiteit van deze richtlijn is de werkgroep niet in stand gehouden. Uiterlijk in 2024 bepaalt het bestuur van de NVALT of de modules van deze richtlijn nog actueel zijn. Op modulair niveau is een onderhoudsplan beschreven. Bij het opstellen van de richtlijn heeft de werkgroep per module een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De NVALT is regiehouder van deze richtlijn en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
De voorliggende richtlijn betreft een herziening van de NVALT richtlijn Diagnostiek en behandeling van ernstig astma uit 2013. Alle modules zijn beoordeeld op actualiteit. Vervolgens is een prioritering aangebracht welke modules een daadwerkelijke update zouden moeten krijgen. Hieronder staan de modules genoemd met de wijzigingen. Tevens is per module een inschatting gemaakt voor de beoordeling voor herziening.
|
Uitgangsvraag/onderwerpen |
Wijzigingen richtlijn 2019 |
Uiterlijk jaar voor herziening |
|
Definities |
Opgenomen in Algemene inleiding |
2024 |
|
Diagnostiek van ernstig astma |
Minimale (tekstuele) aanpassingen |
2024 |
|
Behandelprincipes |
Herbevestigd |
2024 |
|
Antifungale therapie |
Minimale aanpassingen |
2024 |
|
Bariatrische chirurgie |
Nieuw ontwikkeld |
2024 |
|
Anti-IgE (omalizumab) |
Herzien in bredere module Biologicals |
2024 |
|
Bronchiale thermoplastiek |
Gereviseerd |
2024 |
|
Fysiotherapie |
Nieuw ontwikkeld |
2024 |
|
Hooggebergtebehandeling |
Wordt in Q2 2020 herzien in module Longrevalidatie en hooggebergtebehandeling |
2024 |
|
Leukotrieën |
Minimale aanpassingen |
2024 |
|
Longrevalidatie (inspanningstraining, educatie/zelfmanagement, ademhalingsoefeningen/yoga) |
Wordt opgenomen in module Longrevalidatie en hooggebergtebehandeling |
2024 |
|
Steroïdsparende behandeling |
Module wordt teruggetrokken. |
|
|
Systemische corticosteroïden |
Vervangen door nieuwe module |
2024 |
|
Theofylline |
Herbevestigd |
2024 |
|
Tiotropium Bromide |
Minimale aanpassingen |
2024 |
|
Optimaliseren en monitoren van zorg |
Gereviseerd |
2024 |
|
Organisatie van zorg |
Gereviseerd |
2024 |
|
Kwaliteitssystemen |
Opgenomen in Verantwoording en Implementatieplan |
2024 |
Algemene gegevens
De richtlijn is goedgekeurd door:
- Vereniging Nederland Davos
- Longfonds
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). Patiëntenparticipatie bij deze richtlijn werd medegefinancierd uit de Stichting Kwaliteitsgelden Patiënten Consumenten (SKPC) binnen het programma KIDZ.
Doel en doelgroep
Doel
Deze richtlijn geeft aanbevelingen ten aanzien van diagnostiek en behandeling van volwassen patiënten (leeftijd ≥ 18 jaar) met ernstig astma. Doel van deze herziening is om een richtlijn te verkrijgen waarin de meeste recente medische kennis omtrent de zorg voor patiënten met ernstig astma wordt meegenomen.
Doelgroep
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen die betrokken zijn bij de zorg voor patiënten met de ernstig astma en astma.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2018 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met ernstig astma te maken hebben.
Werkgroep
- Dr. E.J.M. Weersink, longarts, Amsterdam Universitaire medisch centrum, locatie AMC, NVALT
- Dr. G.J. Braunstahl, longarts, Franciscus Gasthuis en Vlietland, Rotterdam, NVALT
- Dr. A. ten Brinke, longarts, Medisch Centrum, Leeuwarden, NVALT
- Dr. H.P.A.A. van Veen, Longarts, Medisch Spectrum Twente, Enschede, NVALT
- Drs. L.H. Conemans, longarts, Maastricht UMC+, Maastricht, NVALT
- Dr. A. Aardenburg-van Huisstede, longarts in opleiding, Franciscus Gasthuis en Vlietland, Rotterdam, NVALT
- Dr. S.W. Zielhuis, Ziekenhuisapotheker, Medisch Centrum Leeuwarden, NVZA
- W. van Litsenburg, Verpleegkundig specialist, Catharina Ziekenhuis, Eindhoven, V&VN
- Dr. H.J. Hulzebos, Medisch fysioloog en fysiotherapeut, UMC Utrecht, Utrecht, KNGF
- M.A.P. Poulissen-Erinkveld, projectleider, Longfonds/ VND
- L.A.M. Frankemölle, Ervaringsdeskundige, Longfonds
- E.M. van der Roest, Senior kernmedeweker pre-analyse poli Wilhelmina Kinderziekenhuis, Ervaringsdeskundige, VND
Met ondersteuning van
- Dr. ir. N.L. van der Zwaluw, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Dr. A.J.G. Wirix, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- I. van Dusseldorp, literatuurspecialist, Kenniscentrum Medisch Centrum Leeuwarden
- S. Wouters, projectsecretaresse, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De KNMG-Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling” is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of ze in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatie management, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met evt. belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van Medisch Specialisten.
|
Werkgroep-lid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Dr. E.J.M. Weersink |
Longarts afdeling longziekten Academisch Universitair Medisch Centrum, Amsterdam. |
Geen |
In 2017 voor meerdere farmaceutische bedrijven een betaald adviseurschap (GSK, Novartis, TEVA, Chiesi, Boehringer). In 2018 nog wel adviseurschap niet meer tegen betaling.
|
Adviseurschappen zijn bij de start van de richtlijn gestaakt. Gekeken is naar een vice-voorzitter. Bij relevante onderwerpen zal de vice-voorzitter de vergadering leiden. |
|
Dr. G.J. Braunstahl |
Longarts, Franciscus Gasthuis full-time |
'- Nul-aanstelling Erasmus MC/promotiebegeleiding 4 uur/week (onbetaald) - Redactieraad Nederlands tijdschrift Allergie en Astma 2 uur/maand (onbetaald) - Secretaris Netherlands Respiratory Society (NRS) 4 uur/maand (onbetaald) - Vergoedingen voor advieswerk, richtlijncommissies, ontwikkeling studieprogramma's, presentaties e.d.: (incidenteel) betaald |
- Geen persoonlijke financiele belangen van structurele aard. - De stichting van onze onderzoeksafdeling heeft in het verleden unrestricted grants gekregen van Stichting astmabestrijding, stichting Coolsingel, stichting 't Trekpaert, stichting Bevordering Onderziek Fransciscus, GSK, Teva, Chiesi, Novartis, AstraZeneca, ALK Abello. |
Betaalde adviesfuncties zijn per 1-1-2018 neergelegd. Vergoedingen zijn overkoepelend aan de richtlijn. Geen directe acties nodig. |
|
Dr. A. ten Brinke |
Longarts, vrijgevestigd werkzaam in medisch centrum Leeuwarden |
Geen |
- Unrestricted research grants GSK, TEVA Research advisory boards GSK, Novartis, AstraZeneca, Boehringer Ingelheim, Chiesi, Sanofi. Honoraria lectures GSK, Novartis, Teva, Boehringer Ingelheim. - Actief in expertisecentrum ernstig astma in MCL |
Voor onderwerpen aangaande medicatie kan Ten Brinke, wanneer het zou komen tot stemming, niet mee mogen stemmen. Gezien haar expertise kan ze wel deelnemen aan de discussie over deze onderwerpen. |
|
H.P.A.A. van Veen |
Longarts Medisch spectrum Twente Enschede |
Adviesraden deelname: AstraZeneca, Teva, Novartis, Sanofi (alles betaald) |
Deelname aan wetenschappelijk onderzoek met AstraZeneca |
Geen trekker of meelezer bij modules aangaande medicatie. |
|
L.H. Conemans |
longarts Maastricht UMC |
Geen |
Geen |
Geen actie |
|
Dr. A. Aardenburg- van Huisstede |
Longarts in opleiding Franciscus Gasthuis Rotterdam |
Geen |
Promototieonderzoek verricht op deelgebied van richtlijn (relatie obesitas en astma) |
Expertise wordt juist ingezet op de module Bariatrie en astma. Geen directe actie. Bij onderwerpen waar mogelijke belangen van de voorzitter spelen, zal werkgroeplid optreden als onafhankelijk voorzitter. |
|
Dr. H.J. Hulzebos |
Medisch Fysioloog en (sport)fysiotherapeut in het Universitair Medisch Centrum Utrecht |
Secretaris VHVL (Vereniging Hart, Vaat en Longfysiotherapie) (onbetaald) |
Geen |
Geen actie |
|
W. van Litsenburg |
Verpleegkundig Specialist Astma en COPD Catharina ziekenhuis Eindhoven afdeling Longgeneeskunde 36 uur
Walter van Litsenburg Zorg, ZZP er thuiszorg 24 uur |
-HAN/VDO, opleiding voor praktijkondersteuners Astma/COPD module (betaald) -Workshops en gastlessen voor IMIS (inhalatietechniek), Fontys, UVT (betaald) -Kernteamlid van PICASSO een stichting die wetenschappelijk onderzoek ondersteunt. Sterke link met Boehringer Ingelheim (betaald) |
Geen |
Geen actie |
|
M.A.P. Poulissen-Erinkveld |
Projectleider Longfonds + Vereniging Nederland-Davos |
|
Geen |
Geen actie |
|
L.A.M. Frankemölle |
Ervarings-deskundige; Lid van de Longfonds ErvaringsDeskundigen groep |
- Task Force Group guidelines severe asthma op internationaal niveau. - Diverse werkgroepen zowel internationaal als nationaal, niet gerelateerd aan richtlijnen ernstig astma (onbetaald, reis- en verblijfskosten-vergoedingen). - Vrijwilligster voor het Longfonds en van daaruit ook voor ELF/ERS. - Lid van de werkgroep herziening NHG-standaard astma. |
Geen |
Geen actie |
|
E. van Golen-van der Roest |
Ervaring-sdeskundige; Pre-analyse medewerker UMC Utrecht 25 u/pw |
'- Redactielid, vrijwilliger VND (onbetaald) - bibliotheek, redactielid, vrijwilliger VONK (onbetaald) - Ledenadministratie Sleutelclub hamerik (onbetaald) |
Geen |
Geen actie |
|
Dr. S.W. Zielhuis |
Ziekenhuis-apotheker Medisch Centrum Leeuwarden |
Geen
|
In de afgelopen jaren aanwezig geweest bij eenmalige adviesraden voor ziekenhuisapothekers van een aantal bedrijven waaronder Sanofi, Novartis en Astra Zeneca. De bedrijven hebben producten binnen het indicatiegebied van deze richtlijn. Bij alle bovengenoemde firma's is geen sprake (geweest) van een dienstverband. Tot 2007 werkzaam geweest bij Teva Nederland, maar nu 11 jaar later op geen enkele wijze meer betrokken bij deze firma. |
Geen acties |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door afgevaardigden van de patiëntenverenigingen Longfonds en Vereniging Nederland Davos in de werkgroep en door deelname van de patiëntenvereniging aan de invitational conference. Een verslag hiervan (zie aanverwante producten) is besproken in de werkgroep en de belangrijkste knelpunten zijn verwerkt in de richtlijn. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de patiëntenverenigingen.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijnmodules en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden bij de aanverwante producten. De werkgroep heeft geen interne kwaliteitsindicatoren ontwikkeld.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt, wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. De werkgroep beoordeelde de aanbevelingen uit de eerdere richtlijn (NVALT Richtlijn Diagnostiek en behandeling van ernstig astma, 2013) op noodzaak tot revisie. Tevens zijn er knelpunten aangedragen door middel van een invitational conference. De werkgroep stelde vervolgens een lijst met knelpunten op en prioriteerde de knelpunten op basis van: (1) klinische relevantie, (2) de beschikbaarheid van (nieuwe) evidence, (3) en de te verwachten impact op de kwaliteit van zorg en patiëntveiligheid.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de voorzitter en de adviseur concept-uitgangsvragen opgesteld. Deze zijn met de werkgroep besproken waarna de werkgroep de definitieve uitgangsvragen heeft vastgesteld. Vervolgens inventariseerde de werkgroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als kritiek, belangrijk (maar niet kritiek) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de kritieke uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden (zie pagina 6 voor de uitkomstmaten en de bijbehorende klinisch relevante verbetering).
Strategie voor zoeken en selecteren van literatuur
Voor de afzonderlijke uitgangsvragen werd aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De gespecificeerde zoekstrategieën zijn op te vragen bij het Kennisinstituut.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR - voor systematische reviews; Cochrane - voor gerandomiseerd gecontroleerd onderzoek; ACROBAT-NRS - voor observationeel onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A) Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
*in 2017 heeft het Dutch GRADE Network bepaald dat de voorkeursformulering voor de op een na hoogste gradering ‘redelijk’ is in plaats van ‘matig’
B) Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008), en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (eindconclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een eindconclusie. In dat geval werden de conclusies uit de systematische literatuuranalyse samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Indicatorontwikkeling
Gelijktijdig met het ontwikkelen van de conceptrichtlijn heeft de werkgroep overwogen om interne kwaliteitsindicatoren te ontwikkelen om het toepassen van de richtlijn in de praktijk te volgen en te versterken. Meer informatie over de methode van indicatorontwikkeling is op te vragen bij het Kennisinstituut van de Federatie Medisch Specialisten. In samenwerkingen met de commissie kwaliteitsvisitatie NVALT wordt er een indicatorenset ontwikkeld die gebruikt kan worden tijdens de kwaliteitsvisitaties en die de astma- en ernstig astma zorg omvat. Er zijn derhalve geen specifieke indicatoren ontwikkeld behorende bij de voorliggende richtlijn.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site.html
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, et al. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008;336(7654). doi: 10.1136/bmj.a139. PubMed PMID: 18483053.
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van Medisch Specialist