Opstellen validatie- of verificatieplan
Uitgangsvraag
Welke onderdelen moeten worden opgenomen in het validatie- of verificatieplan?
Aanbeveling
Validatie/verificatie moet geschieden volgens een vooraf opgesteld en geautoriseerd validatie/verificatieplan, dat ten minste de volgende onderdelen bevat:
1) beoogde toepassing van de onderzoeksprocedure
2) vastleggen van de measurand (geldt alleen bij validatie)
3) selectie van de relevante prestatiekarakteristieken
4) onderbouwde acceptatiecriteria in licht van beoogde toepassing
5) wijze van toetsing
6) identiteit van onderzoekers en bevoegde autorisator(en)
Overwegingen
Voor zowel validatie als verificatie geldt dat de prestatiekarakteristieken moeten worden getoetst tegen de acceptatiecriteria. Deze acceptatiecriteria worden vooraf vastgelegd en onderbouwd in respectievelijk het validatie- of verificatieplan. Naast de acceptatiecriteria moet in deze plannen ook worden vastgelegd wat de measurand is, wat de beoogde toepassing is, op welke wijze wordt getoetst en wie de onderzoekers en de autorisator/eindverantwoordelijke zijn.
Met name voor validatie van een (zelf ontwikkelde) onderzoeksprocedure is het noodzakelijk om duidelijk de measurand vast te leggen. Beschrijving van de measurand vereist kennis van de component of chemische entiteit (analyt) die gezocht wordt, de matrix en toestand waarin de analyt zich bevindt en de kenmerken van de gebruikte onderzoeksprocedure. Dit speelt bijvoorbeeld bij de bepaling van eiwitachtige componenten met immunochemische onderzoeksprocedures een zeer belangrijke rol.
Onderbouwing
Leidend is hetgeen hierover in de ISO15189 en in de EU wetgeving (Directive én Regulation) is voorgeschreven, zie de Inleiding. Tevens wordt uitgegaan van de vorige versie van deze NVKC Richtlijn uit 2016. Het is nodig om ook bij het opstellen van een plan onderscheid te maken tussen Verificatie en Validatie.
In de WHO guideline TGS-4 voor validatie van IVD’s (2017) wordt gewezen op de noodzaak te beginnen met een goede definitie van het doel van de beoogde test, dat wil zeggen de te meten stof in de gewenste matrix en met de geschikte methode. Feitelijk spreken we dan over de definitie van analyte en measurand. Vervolgens moeten de eisen worden vastgelegd van prestatiekarakteristieken en acceptatiecriteria.
Beschrijving van de measurand vereist kennis van de component of chemische entiteit (analyt) die gezocht wordt, de matrix en toestand waarin de analyt zich bevindt en de kenmerken van de gebruikte onderzoeksprocedure. Dit speelt bijvoorbeeld bij de bepaling van eiwitachtige componenten met immunochemische onderzoeksprocedures een zeer belangrijke rol.
Bij het opstellen van een validatie- of verificatieplan en de toetsing en verslaglegging hoort ook een flowschema over het overall proces (zie Flowschema in Aanverwante items):
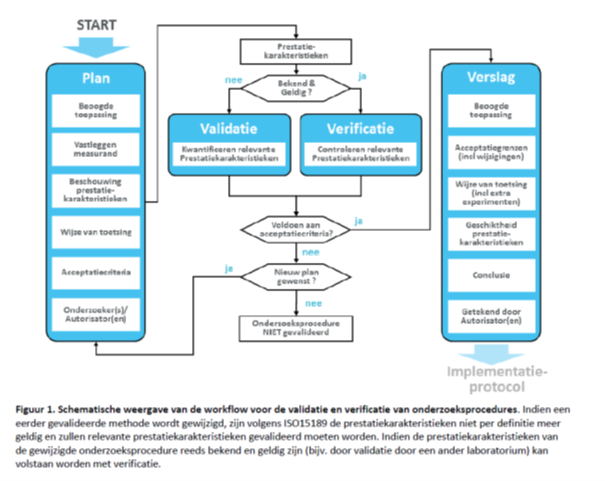
Ter kennisgeving: dit schema wordt opgenomen bij de Aanverwante items
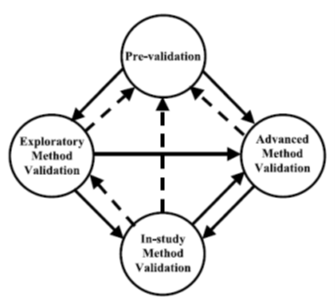
Voor het opstellen van een validatieplan is door Mulder et al (2018) en door Ravisankar (2015) gewezen op de noodzakelijke relevante voorbereiding. Die omvat onder meer: chemische kennis verzamelen over de te meten analyt, nagaan welke meetprincipe of methode potentieel geschikt is, welke monstermatrix gebruikt kan worden, hoe het monster bewerkt en geconserveerd kan worden, etc. De wijze waarop het analyt gemeten wordt en het gedetailleerde meetprincipe dat daarbij gebruikt wordt, bepaalt uiteindelijk de measurand. Bij validatie zien we vaak een duidelijke pendelbeweging tussen het ontwikkelen van de onderzoeksprocedure en validatie onderzoek. Dit kan explorerende methode validatie worden genoemd. Lee et al (2006) onderscheiden vier verschillende soorten validatieprocessen, die met elkaar verweven en iteratief zijn (zie bijgaande figuur). Bij het ontwikkelen van een methode is een basis (explorerende) validatie in een vroege fase nodig, om te beginnen met betrekking tot de stabiliteit van het monster en de gebruikte reagentia en standaarden. Vervolgens komen initiële onderzoeken naar lineariteit, reproduceerbaarheid en bereik van de beoogde methode aan de orde, enzovoort. Ook Lee noemt method feasability studies als mogelijkheid om in een vroege fase van de methode ontwikkeling een schatting te kunnen maken van de uiteindelijke bruikbaarheid.
De selectie van de relevante prestatiekarakteristieken wordt in hoge mate bepaald door enerzijds het beoogde doel en anderzijds de beschikbaarheid van relevante en betrouwbare gegevens uit ander onderzoek. Indien relevante en betrouwbare gegevens beschikbaar zijn, dan moet voor kritische en mogelijk laboratoriumafhankelijke prestatiekarakteristieken feitelijk een verificatie worden uitgevoerd. In alle overige gevallen een validatie.
Mulder et al, Lee en Ravisankar geven nuttige overzichten van prestatie karakteristieken. Een opsomming van de te gebruiken prestatiekarakteristieken volgens de IVD Regulation (performance characteristics ‘where applicable’) wordt gegeven in Annex I, paragraaf 9.1 en in paragraaf 20.4 (over de informatie die de fabrikant aan de gebruiker moet aanleveren). In ISO15189 wordt in paragraaf 5.5.1.3 onder het kopje ‘Opmerking’ een vergelijkbare set prestatiekarakteristieken voorgesteld.
Een verdere uitwerking voor de noodzakelijke acceptatie criteria en wijze van toetsing is besproken in hoofdstuk 3. Het is denkbaar dat bij een validatie of verificatie de vooraf opgestelde acceptatiecriteria niet gehaald worden. In dat geval kan overwogen worden om de acceptatie criteria nogmaals kritisch te bezien ten opzichte van het beoogde doel. Hierbij moet ook de gekozen afkappunten en de daarbij acceptabele meetonzekerheid getoetst worden bij relevante medische richtlijnen en medische specialisten.
Voor de bij validatie of verificatie betrokken personen geldt dat deze de juiste bevoegdheden moeten bezitten om de resultaten te beoordelen en een verslag op te maken van de beoordeling. ISO15189 par 5.5.1.2 en 5.5.1.3. In het algemeen geldt dat alle betrokken werkzaamheden verricht moeten worden door personen die goed opgeleid en competent zijn paragraaf 5.1.6 t/m 5.1.8. Volgens ISO 15189:2012 5.5.1.1 moet de identiteit van de personen die in onderzoeksprocessen handelingen verrichten worden vastgelegd.
- Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, Keller S, Weinryb I, Green M, Duan L, Rogers JA, Millham R, O’Brien PJ, Sailstad J, Khan M, Ray C, Wagner JA. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res 2006; 23: 312-328.
- Mulder L, van der Molen R, Koelman C, van Leeuwen E, Roos A, Damoiseaux J. Validation conform ISO-15189 of assays in the field of autoimmunity: Joint efforts in The Netherlands. Autoimmun Rev 2018; 17: 513-517.
- Ravisankar P, Naga Navya Ch, Pravallika D, Navya Sri D. A review on step-by-step analytical method validation. IOSR Journal Of Pharmacy 2015; 5: 07-19.
Beoordelingsdatum en geldigheid
Publicatiedatum : 28-12-2022
Beoordeeld op geldigheid : 28-12-2022
Algemene gegevens
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.kennisinstituut.nl) en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS).
Doel en doelgroep
Doel
Validatie en verificatie van onderzoeksprocedures in medische laboratoria betekent het toetsen of de beoogde onderzoeksprocedures geschikt zijn voor het beoogde doel, namelijk het leveren van betrouwbare informatie voor de aanvrager ten behoeve van diagnose, prognose, behandeling of vervolgen van therapie.
De voorliggende richtlijn beschrijft de vereiste analytische en toetsingsprocedures bij validatie of verificatie – conform ISO 15189 – met inachtneming van de daarbij benodigde prestatiekarakteristieken en acceptatiecriteria. Na beschouwing van additionele wetenschappelijke literatuur (indien beschikbaar) en op basis van consensus zijn de aanbevelingen opgesteld.
Doelgroep
Hoewel deze richtlijn met name bedoeld is voor toepassing in de klinische chemie, kan zij ook nuttig zijn voor andere disciplines binnen de medische laboratoria, zoals klinische farmacie, medische microbiologie en medische immunologie.
Samenstelling werkgroep
Voor het (door)ontwikkelen van de richtlijn is in 2017 een werkgroep ingesteld. De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
- Dr. drs. W.P. (Wytze) Oosterhuis, arts klinische chemie, Zuyderland Medisch Centrum, Heerlen/Sittard-Geleen (voorzitter)
- Dr. W.P.H.G. (Wilhelmine) Verboeket-van de Venne, wetenschappelijk onderzoeker klinische chemie, Zuyderland Medisch Centrum, Heerlen (secretaris)
- Dr. ir. A.J. (Arjan) van Adrichem, AIOS klinische chemie, Zuyderland Medisch Centrum, Heerlen/Sittard-Geleen
- Dr. ir. R.J.A.C. (Roseri) Roelofsen-de Beer, laboratoriumspecialist klinische chemie, Ziekenhuis Rivierenland Tiel, Tiel
- Dr. ir. J.P.M. (Jos) Wielders, gepensioneerd laboratoriumspecialist klinische chemie, Amersfoort
Met ondersteuning van:
- Drs. E.E. (Eva) Volmeijer, adviseur Kennisinstituut van de Federatie Medisch Specialisten (tot juli 2018)
- Dr. J. (Janneke) Hoogervorst-Schilp, adviseur Kennisinstituut van de Federatie Medisch Specialisten (vanaf juli 2018)
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben. Een overzicht van de belangen van werkgroepleden is op verzoek beschikbaar; er zijn geen restricties geconstateerd m.b.t. deelname aan de werkgroep.
Inbreng patiëntenperspectief
Gezien de technische aard van de richtlijn is – in afstemming met Patiëntenfederatie Nederland – het patiëntenperspectief niet meegenomen. De Patiëntenfederatie is wel uitgenodigd voor de commentaar- en autorisatiefase.
Methode ontwikkeling
Evidence based
Implementatie
Tijdens de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn (module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is opgenomen bij de aanverwante producten.
Werkwijze
ISO 15189
De richtlijn is gebaseerd op de officiële Nederlandse vertaling van de Europese norm EN ISO 15189:2012 (formeel aangeduid als NEN-EN-ISO 15189+C11:2015nl); afgekort aangeduid als ISO 15189. De vereiste analytische en toetsingsprocedures bij validatie of verificatie, met inachtneming van de daarbij benodigde prestatiekarakteristieken en acceptatiecriteria, worden beschreven en – indien van toepassing – nader toegelicht.
Knelpuntenanalyse
Tijdens de voorbereidende fase werden de knelpunten geïnventariseerd. De richtlijn uit 2016 was niet modulair opgebouwd en niet opgenomen in de Richtlijnendatabase. Een modulair opgebouwde richtlijn vergemakkelijkt de integratie tussen (onderdelen van) richtlijnen en maakt onderlinge verwijzing mogelijk. Een ander voordeel is dat verouderde onderdelen van een richtlijn efficiënter herzien kunnen worden, met als gevolg dat richtlijnaanbevelingen sneller (en beter) aansluiten bij de dagelijkse praktijk.
Daarnaast is bij de richtlijn uit 2016 uitsluitend gebruik gemaakt van normatieve verwijzingen (zoals NEN-EN-ISO 15189, ISO 22870 en European Commission Directive 98/79/EC) en Europese/internationale standaarden. In de doorontwikkeling is gestreefd om zoveel mogelijk (aanvullende) wetenschappelijke c.q. consensus-based onderbouwing te geven voor de aanbevelingen in de richtlijn.
Uitgangsvragen
Het uitgangspunt was de in 2016 geautoriseerde Richtlijn Validatie en verificatie van onderzoeksprocedures in medische laboratoria. De werkgroep heeft vervolgens een aantal uitgangsvragen opgesteld. Tevens werd een additionele uitgangsvraag geformuleerd, gezien de overgang van het huidige besluit voor In Vitro Medical Devices (Directive 98/79/EC ofwel IVDD) naar de toekomstige verordening, Regulation (EU) 2017/745 (IVDR) die in mei 2022 in de gehele EU van kracht is.
Strategie voor zoeken en selecteren van literatuur
Er is oriënterend gezocht naar bestaande (inter)nationale richtlijnen en wetenschappelijke publicaties over validatie en verificatie van onderzoeksprocedures. Tevens is aanvullend gezocht naar publicaties aan de hand van de literatuurlijsten van de eerder gevonden documentatie.
Samenvatten van de literatuur
De belangrijkste bevindingen uit de normatieve en wetenschappelijke literatuur zijn beschreven in de samenvatting van de literatuur. Gezien de aard en (beperkte) beschikbaarheid van de literatuur kon geen gebruik gemaakt worden van de GRADE- of vergelijkbare methodologie.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast normatieve en wetenschappelijke publicaties ook andere aspecten belangrijk om te worden meegewogen, zoals de expertise van de werkgroepleden, kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op (de invulling c.q. toepassing van normelementen uit) ISO 15189, het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen. Normaliter is aan elke aanbeveling een implementatietermijn gekoppeld. Gezien er geen inhoudelijke veranderingen zijn t.o.v. de vigerende richtlijn zijn deze implementatietermijnen nog steeds geldig.
Randvoorwaarden (organisatie van zorg)
Bij de (door)ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie rondom validatie en verificatie van onderzoeksprocedures: alle aspecten die randvoorwaardelijk zijn voor het inrichten van het proces (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag.
Kennislacunes
Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is. Een overzicht van de aanbevelingen voor nader/vervolgonderzoek is opgenomen in de bijlage Kennislacunes (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptversie van de richtlijn is aan de leden van de NVKC en aan Patiëntenfederatie Nederland voorgelegd ter commentaar. De commentaren zijn verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren is de richtlijn aangepast en definitief vastgesteld door de werkgroep. De autorisatieversie van de richtlijn is ter stemming gebracht tijdens de algemene ledenvergadering van de NVKC, en voorgelegd aan Patiëntenfederatie Nederland ter autorisatie c.q. instemming.