Nierfunctievervangende therapie: dosering
Uitgangsvraag
Wat is de optimale dosering van nierfunctievervangende therapie bij patiënten met septische acute nierschade?
Aanbeveling
Streef bij continue venoveneuze hemodiafiltratie naar een gerealiseerde dosis (effluent) van 20 tot 25 ml/kg/u bij patiënten met sepsis.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
RRT kan intermitterend (IHD) of continu (CRRT) worden toegepast. CRRT wordt alleen op de IC uitgevoerd. Het is onduidelijk wat de optimale dosis RRT is bij septische AKI. Er is daarom literatuuronderzoek verricht naar de optimale dosering.
De vergelijking van hogere en lagere dosering van RRT werd in een aantal studies beschreven bij patiënten met sepsis. In de diverse RCT’s werden uiteenlopende waarden gehanteerd. Ook waren er verschillen in pre- en postdilutie, en in substitutievloeistoffen (tabel 7.11). Als hoge dosis werd in 4 van de 5 studies een waarde aangehouden tussen de 65 en 85 mL/kg/u, en als standaard of lage dosis 35-50 mL/kg/u (Boussekey, 2008; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012). Bellomo (2008) vergeleek lagere doseringen: 40 en 25 mL/kg/u. Op basis van de cruciale uitkomstmaten mortaliteit en herstel van nierfunctie komt er geen duidelijke voorkeur voor hoge of lage dosis naar voren bij patiënten met sepsis. Door de zeer lage bewijskracht is het onduidelijk of er op 28, 60 en 90 dagen een verschil is in mortaliteit tussen hoge en lage dosering. Daarnaast lijkt de dosis van RRT niet of nauwelijks effect te hebben op de duur van RRT. Bij een hogere dosis lijkt er echter sprake te zijn van een langere verblijfsduur op de IC en in het ziekenhuis. Op basis van deze uitkomstmaten zou de voorkeur uitgaan naar een lagere dosis. In de geïncludeerde studies is dosering alleen onderzocht in continue RRT, er kunnen derhalve geen conclusies worden getrokken voor intermitterende RRT. De overall bewijskracht, de laagste bewijskracht voor een van de cruciale uitkomstmaten, is zeer laag. Hier ligt een kennislacune.
In de KDIGO guidelines (Khwaja, 2012) wordt de dosis van CRRT summier behandeld. Men bevestigt dat er geen onderzoeken zijn die een hogere dosis RRT ondersteunen en men adviseert de dosis van 20 tot 25 ml/kg/u. Wel benadrukt men dat deze dosis vaak niet gehaald wordt, en dat het daarom verstandig is om een iets hogere dosis in te stellen. De European Renal Best Practice position statement adviseert een dosis van 20 tot 25 ml/kg/u wanneer postdilutie wordt gegeven, en een hogere dosis wanneer ook predilutie wordt gegeven (Jörres, 2013). Er zijn geen studies bekend waarbij een lagere dosis dan 20 ml/kg/u is onderzocht.
In de studies wordt niet gespecificeerd welk lichaamsgewicht gehanteerd wordt. Vermoedelijk gaat het dan ook over TBW. Echter, omdat de lichaamssamenstelling uiteraard zeer kan verschillen onder IC-patiënten, is het de vraag of de geleverde dosis voor iedereen vergelijkbaar is. Het lijkt logisch om een andere parameter zoals lean body weight of ideaal lichaamsgewicht te nemen, of het lichaamsgewicht te maximeren tot 120 kg. Hier is echter geen onderzoek naar gedaan. In de praktijk zal veelal worden gekeken naar de individuele patiënt, waarbij de dosis op individuele basis kan worden bijgesteld.
Samenvattend levert de literatuur geen onderbouwing om voor hoge of lage dosis RRT te kiezen. Daarentegen zijn er wel mogelijke nadelen van hoge dosis RRT op de IC. Zo blijkt uit de literatuur dat hoge dosis RRT mogelijk een langere opnameduur op IC en in het ziekenhuis tot gevolg heeft. Verder heeft hoge dosis RRT een hogere klaring van sommige medicamenten tot gevolg (Ulldemolins, 2014) hetgeen kan leiden tot lagere, subtherapeutische spiegels. Ook kan de hoge bloedflow die nodig is voor hoge dosis RRT soms leiden tot problemen met katheter (aanvoerproblemen) en/of filter (hoge TMP). Verder zijn bij hoge dosis RRT meer metabole complicaties beschreven (desequilibrium syndroom, hypofosfatemie, te snelle correctie van hyponatriëmie, (Bellomo, 2008; Park, 2006)). Als laatste leidt hoge dosis RRT tot een toename in de kosten als gevolg van de grotere volumina substitutievloeistof en filtervervanging (Bellomo, 2008). Deze mogelijke nadelen, alsmede de mogelijk langere verblijfsduur op IC en op de afdeling, vormen voor de werkgroep een reden om een voorkeur uit te spreken voor lage of standaard dosis RRT.
Waarden en voorkeuren van patiënten (en eventueel hun verzorgers/familie)
De voorkeur van de patiënt voor deze interventies is niet onderzocht en onbekend. Goede voorlichting en communicatie over behandeldoelen, voor- en nadelen, risico’s en verwacht resultaat is belangrijk om een goede afweging te maken voor patiënten en naasten. Een hogere dosis van RRT heeft waarschijnlijk geen voordelen. Wel zijn er potentiële nadelen zoals metabole ontregeling, aanvoerproblemen met de katheter en mogelijk een langere verblijfsduur op de IC en op de afdeling. Vanuit patiëntperspectief is er dus mogelijk een voorkeur voor lage/standaarddosering (20 tot 25 ml/kg/u).
Kosten (middelenbeslag)
Een hogere dosering van CRRT leidt waarschijnlijk tot hogere kosten (Bellomo, 2008). Dit wordt veroorzaakt door meer verbruiksmiddelen (filters en vloeistoffen) maar mogelijk ook door een langere opnameduur. Tegelijk heeft hoge dosering CRRT waarschijnlijk geen positief effect op het ziektebeloop. Vanuit kostenperspectief is er daarom een voorkeur voor lage/standaarddosering CRRT.
Aanvaardbaarheid, haalbaarheid en implementatie
Hoge dosis CRRT heeft als gevolg dat er meer verbruik is van dialysevloeistoffen; de vloeistofzakken zullen vaker gewisseld moeten worden hetgeen een extra werkbelasting vormt. Hoge dosering CRRT leidt frequent tot katheter(aanvoer)problemen met als gevolg veel alarmen, downtime, en een verhoogd risico op stolling in het filter. Het filter moet dan vervangen worden hetgeen een negatief effect heeft op de behandeldosis en op de werkbelasting. Vanuit dit perspectief is er een voorkeur voor lage/standaarddosering CCRT.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Uit de gebruikte literatuur blijkt geen evidence om voor lage of hoge dosering CRRT (40 tot 85 l/kg/u) te kiezen, maar zijn er wel mogelijke nadelen van hoge dosering CRRT, zowel voor de patiënt als op het gebied van kosten en implementatie. De werkgroep spreekt daarom een voorkeur uit voor lage/standaard dosering CRRT (20 tot 25 ml/kg/u). Zij doet tevens de aanbeveling om de dosis aan te passen naar gelang er sprake is van pre- en/of postdilutie.
Onderbouwing
Achtergrond
Acute nierschade (acute kidney injury, AKI) is de moderne term voor een plotseling verslechterende nierfunctie en wordt geclassificeerd volgens de KDIGO (voorheen RIFLE en AKIN) consensus classificatie. Bij septische AKI treedt de acute verslechtering van de nierfunctie op als gevolg van sepsis. Logischerwijs beschrijft septische AKI een syndroom waarbij voldaan wordt aan de consensus definities van zowel sepsis als AKI (Bellomo, 2017). Een deel van de patiënten met septische AKI behoeft nierfunctievervangende therapie (renal replacement therapy, RRT), hetgeen geassocieerd is met een hoge morbiditeit, mortaliteit en zorgkosten. Het is momenteel onduidelijk of RRT dosering van invloed is op de klinische uitkomst voor patiënten met sepsis.
Conclusies
|
Zeer laag GRADE |
Het is onduidelijk of hoge dosering van continue nierfunctievervangende therapie (35 tot 85 ml/kg/u) invloed heeft op mortaliteit ten opzichte van standaard/lage dosering (20 tot 25 ml/kg/u) bij patiënten met septische AKI.
Bronnen: (Bellomo, 2008; Boussekey, 2008; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) |
|
Laag GRADE |
Hoge dosering van continue nierfunctievervangende therapie (35 tot 85 ml/kg/u) lijkt herstel van nierfunctie niet te verbeteren ten opzichte van standaard/lage dosering (20 tot 25 ml/kg/u) bij patiënten met septische AKI.
Bronnen: (Boussekey, 2008; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) |
|
Laag GRADE |
Hoge dosering van continue nierfunctievervangende therapie (35 tot 85 ml/kg/u) lijkt intensive-careverblijfsduur te verlengen ten opzichte van lage/standaard dosering (20 tot-25 ml/kg/u) bij patiënten met septische AKI.
Bronnen: (Boussekey, 2008; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) |
|
Laag GRADE |
Hoge dosering van continue nierfunctievervangende therapie (35 tot 85 ml/kg/u) lijkt ziekenhuisverblijfsduur te verlengen ten opzichte van lage/standaard dosering (20 tot 25 ml/kg/u) bij patiënten met septische AKI.
Bronnen: (Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) |
|
Laag GRADE |
De dosering lijkt geen of nauwelijks effect te hebben op de duur van continue nierfunctievervangende therapie bij patiënten met septische AKI.
Bronnen: (Boussekey, 2008; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) |
Samenvatting literatuur
Om RRT dosis te kunnen kwantificeren is een eenheid nodig. Het is gebruikelijk om hiervoor de klaring van kleine moleculen te gebruiken. Tijdens CRRT is de klaring van kleine moleculen gelijk aan het totale effluent volume (= totale filtraat en/of dialysaat volume) uitgedrukt per tijdseenheid per kilogram lichaamsgewicht. Dit geldt voor postdilutie CRRT. Tijdens predilutie CRRT zal door hemodilutie de klaring minder effectief zijn. Hiervoor dient gecorrigeerd te worden.
De verschillende studies hadden een eigen definitie van hoge en lage/standaard dosis van de behandeling. Alle studies beschreven continue RRT; pre- en postdilutie was in alle studies echter verschillend en dit kan een invloed hebben op de geleverde klaring (8 tot 14%). Ook waren er verschillen in het voorschrijven van dialysaat (zie tabel 7.5). In geen van de studies wordt het gewicht nader gespecificeerd, zodat uitgegaan moet worden van total body water (TBW).
Bellomo (2009) beschreef in de RENAL RCT mortaliteit in ernstig zieke patiënten met AKI, en onderscheidde vooraf gedefinieerde subgroepen, waaronder sepsis (49% van de patiënten). Mortaliteit op dag 90 werd beschreven bij 722 volwassen patiënten met sepsis die continue venoveneuze hemofiltratie kregen met hoge dosis (40 mL/kg/u) of lage dosis (25 mL/kg/u). Deze studie uit Australië en Nieuw-Zeeland had een redelijk risico op bias.
Boussekey (2008) vergeleek in een prospectieve gerandomiseerde pilotstudie hoge dosis hemofiltratie (65 ml/kg/u) met lage dosis hemofiltratie (35 ml/kg/u) bij 19 patiënten met septische shock en acuut nierfalen in Frankrijk. De studie had een redelijk risico op bias. Er was wel een effect op de bloeddruk bij hoge dosis CRRT, maar dit resulteerde niet in een betere overleving.
In de IVOIRE-studie, een multicenter RCT uit Frankrijk, België en Nederland, beschreven door Joannes-Boyau (2013), werden 140 patiënten geïncludeerd met septische shock en AKI. Mortaliteit, verblijfsduur en herstel van nierfunctie werden vergeleken tussen hoog- (70 mL/kg/u) en standaard dosis (35 mL/kg/u) continue hemofiltratie. De studie werd voortijdig beëindigd wegens trage inclusie. De studie had een laag risico op bias.
Park (2016) vergeleek hoge (80 mL/kg/u) en conventionele (40 mL/kg/u) dosis van continue venoveneuze hemodiafiltratie bij 212 volwassen patiënten met septische AKI op de IC in 2 ziekenhuizen in Seoul, Zuid-Korea. Mortaliteit, verblijfsduur en herstel van nierfunctie werden beschreven in deze prospectieve, gerandomiseerde studie. De studie had een redelijk risico op bias.
Zhang (2012) randomiseerde 280 patiënten met ernstige sepsis en acuut nierfalen op de IC in een Chinees onderzoek. Mortaliteit, verblijfsduur en herstel van nierfunctie werden beschreven bij behandeling met 85 of 50 mL/kg/u continue venoveneuze hemofiltratie. De studie had een hoog risico op bias.
Tabel 11 RRT behandeldosis per studie
|
|
Hoge dosis (mL/kg/u) |
Standaard/lage dosis (mL/kg/u) |
Predilutie |
Postdilutie |
Dialysaat |
Gewicht |
|
Bellomo, 2009 |
40 |
25 |
0% |
100% |
Ja |
TBW |
|
Boussekey, 2008 |
65 |
35 |
1/3 |
2/3 |
Nee |
TBW |
|
Joannes-Boyau, 2013 |
70 |
35 |
1/3 |
2/3 |
Nee |
TBW |
|
Park, 2016 |
80 |
40 |
100% |
0% |
Ja |
TBW |
|
Zhang, 2012* |
85 |
50 |
2/3 |
1/3 |
Nee |
TBW |
*online-productie van substitutievloeistof; TBW: total body water
Resultaten
Mortaliteit
Mortaliteit werd beschreven op verschillende tijdspunten, zoals weergegeven in figuur 7.12. Op 28 dagen werd in 4 studies (Boussekey, 2008; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) met in totaal 648 patiënten geen verschil gevonden tussen hoge en lage dosering, met een relatief risico (RR) van 0,99 en een 95% betrouwbaarheidsinterval (BI) van 0,86 tot 1,12. Op 60 dagen werd door 2 studies (Joannes-Boyau, 2013; Zhang, 2012) bij in totaal 417 patiënten een RR van 0,97 (95% BI van 0,82 tot 1,14) gevonden, een klinisch relevant verschil in het voordeel van laag volume. Op 90 dagen werd tot slot in 4 studies (Bellomo, 2009; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) met in totaal 1351 patiënten geen klinisch relevant verschil gevonden met een RR van 0,98 (95% BI van 0,90 tot 1,07).
Figuur 12 Mortaliteit bij hoge versus lage dosis continue RRT
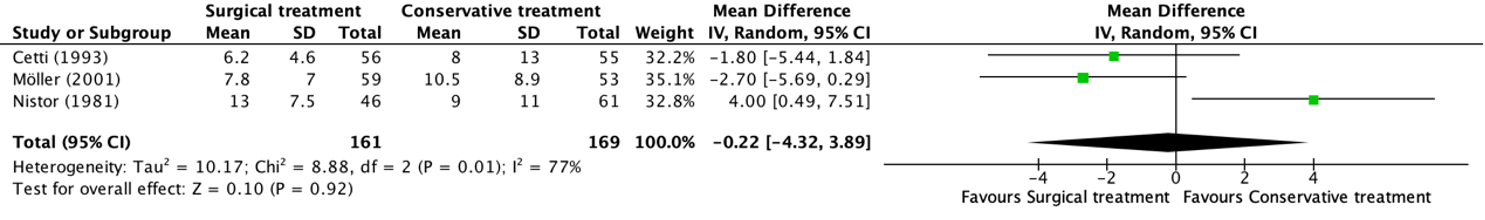
Mortaliteit (relatief risico) bij patiënten met sepsis. Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat mortaliteit is gebaseerd op gerandomiseerde trials en start daarom op hoog. Er is met 3 niveaus afgewaardeerd tot zeer laag vanwege de beperkte populatieomvang, overschrijding van de grenzen van klinische relevantie (beide imprecisie) en beperkingen in de extrapoleerbaarheid (klinische heterogeniteit van de studies).
Herstel van nierfunctie
In 4 studies met in totaal 306 patiënten werd herstel van nierfunctie beschreven. In beide groepen was aan het eind van de follow-up periode slechts een enkele patiënt nog afhankelijk van RRT. Er werd geen verschil gevonden tussen de groepen met een relatief risico van 1,02 en een 95% BI van 0,98 tot 1,06 (figuur 7.13).
Figuur 13 Herstel van nierfunctie bij hoge versus lage dosis continue RRT
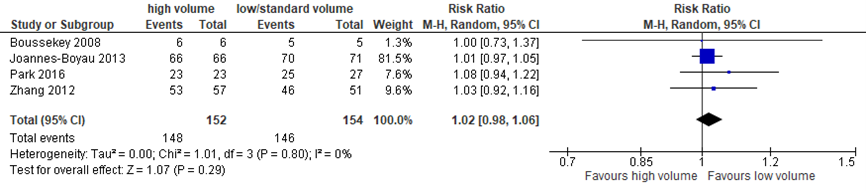
Herstel van nierfunctie (relatief risico) bij overlevende patiënten met sepsis. Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat herstel van nierfunctie is gebaseerd op gerandomiseerde trials en start daarom op hoog. Er is met 2 niveaus afgewaardeerd tot laag vanwege de beperkte populatieomvang (imprecisie) en beperkingen in de extrapoleerbaarheid (klinische heterogeniteit van de studies).
Intensive-careverblijfsduur
Verschillende studies beschreven verblijfsduur in het ziekenhuis en/of op de IC (figuur 7.14). In 4 studies (Boussekey, 2008; Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) met in totaal 648 patiënten werd een gemiddeld verschil in IC-verblijfsduur gevonden van 1,75 dagen (95% BI -3,77 tot 7,28) in het voordeel van lage dosis RRT. Dit verschil is klinisch relevant, maar niet statistisch significant.
Het verschil in ziekenhuisverblijfsduur werd beschreven in 3 studies (Joannes-Boyau, 2013; Park, 2006; Zhang, 2012) met in totaal 629 patiënten. Er werd een klinisch relevant verschil gevonden van 5,97 dagen in het voordeel van lage dosis met een zeer breed 95% betrouwbaarheidsinterval van -9,78 tot 21,71 (figuur 14).
Figuur 14 Ziekenhuis- en IC-verblijfsduur bij hoge versus lage dosis continue RRT
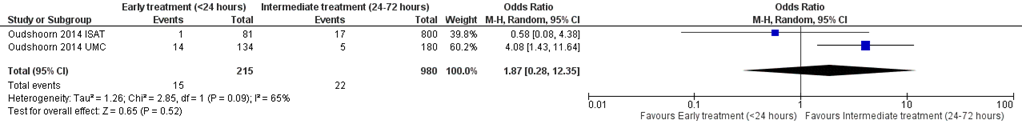
Verblijfsduur (gemiddeld verschil) van patiënten met sepsis in dagen. Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaten ziekenhuisverblijfsduur en IC-verblijfsduur is gebaseerd op gerandomiseerde trials en start daarom op hoog. Er is in beide gevallen met 3 niveaus afgewaardeerd tot zeer laag vanwege beperkingen in de extrapoleerbaarheid (klinische heterogeniteit van de studies), de beperkte populatieomvang en vanwege overschrijding van de grenzen van klinische relevantie (beide imprecisie).
Duur van RRT
Ten slotte werd de duur van RRT beschreven in 4 studies met in totaal 648 patiënten. Er werd geen klinisch relevant verschil gevonden tussen hoge en lage dosis, met een gemiddeld verschil van 0,19 en een 95% BI van -1,07 tot 1,45 (figuur 15).
Figuur 15 Duur van RRT bij hoge versus lage dosis continue RRT
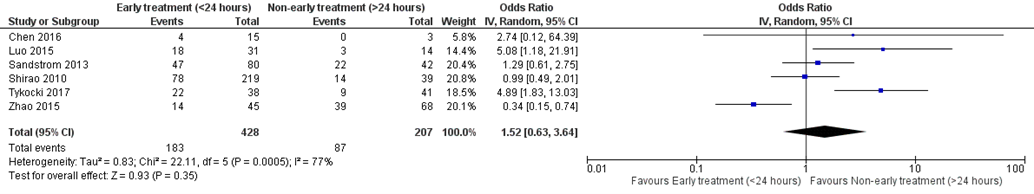
Duur van RRT (gemiddeld verschil) bij patiënten met sepsis in dagen. Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat duur van RRT is gebaseerd op gerandomiseerde trials en start daarom op hoog. Er is met 3 niveaus afgewaardeerd tot zeer laag vanwege beperkingen in de extrapoleerbaarheid (klinische heterogeniteit van de studies), de beperkte populatieomvang en vanwege overschrijding van de grenzen van klinische relevantie (beide imprecisie).
Zoeken en selecteren
Om deze vraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Deelvraag 3: Dosering
Wat is de optimale dosering van RRT bij patiënten met septische AKI?
P: volwassen patiënten met septische AKI;
I: hoge dosering RRT;
C: lage/standaard dosering RRT;
O: mortaliteit, herstel van nierfunctie, intensive-careverblijfsduur, ziekenhuisverblijfsduur, duur van RRT.
Relevante uitkomstmaten
De werkgroep achtte mortaliteit en herstel van nierfunctie voor de besluitvorming cruciale uitkomstmaten; en intensive-careverblijfsduur, ziekenhuisverblijfsduur en duur van RRT voor de besluitvorming belangrijke uitkomstmaten. Daarnaast werd voor de tweede deelvraag (timing) het wel of niet ontvangen van RRT als belangrijke uitkomstmaat gezien.
De werkgroep definieerde niet a priori de genoemde uitkomstmaten, maar hanteerde de in de studies gebruikte definities.
De werkgroep definieerde de grens van 3% als een klinisch (patiënt) relevant verschil voor mortaliteit (gebaseerd op de SDD-trial van de Smet, 2009), en 25% voor de overige dichotome uitkomstmaten. Voor verblijfsduur op de intensive care (IC), duur van RRT en RRT-vrije dagen werd 1 dag als klinisch relevant verschil gedefinieerd, voor ziekhuisverblijfsduur 2 dagen.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID) en Embase (via Embase.com) is op 28 augustus 2019 met relevante zoektermen gezocht naar literatuur voor deelvraag 1, 2 en 3. Omdat er weinig literatuur beschikbaar is over RRT, is de populatie uit de PICO uitgebreid met ernstig zieke IC-patiënten. Wanneer voldoende studies beschikbaar waren om een deelvraag te beantwoorden in een sepsispopulatie, dan werden IC-studies geëxcludeerd.
De literatuur-zoekactie leverde 521 treffers op. Op basis van titel en abstract werden voor deelvraag 3 in eerste instantie 15 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 10 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording) en 5 studies definitief geselecteerd.
Resultaten
In de literatuuranalyse van deelvraag 3 (RRT-dosering) zijn vijf studies meegenomen, die allen resultaten specifiek voor een sepsispopulatie beschreven. In eerste instantie werd de systematische review van Clark (2014) gebruikt voor het beschrijven van de cruciale uitkomstmaat mortaliteit bij patiënten met sepsis. Omdat Clark (2014) niet de cruciale uitkomstmaat “herstel van nierfunctie” beschreef, zijn de originele artikelen uit deze review gebruikt om alle uitkomstmaten te beschrijven. De review is daarom niet verder geanalyseerd. Aanvullend werd de RCT van Park (2016) geanalyseerd, evenals de RCT van Bellomo (2009), die mortaliteit rapporteerde in een vooraf gedefinieerde sepsis subgroep in de RENAL-trial. Posthoc-analyses van sepsis subgroepen die niet vooraf gedefinieerd waren, zijn niet meegenomen in de analyse. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk-of-biastabellen.
Referenties
- Bai, M., Zhou, M., He, L., Ma, F., Li, Y., Yu, Y., ... & Sun, S. (2015). Citrate versus heparin anticoagulation for continuous renal replacement therapy: an updated meta-analysis of RCTs. Intensive care medicine, 41(12), 2098-2110.
- Barbar SD, Clere-Jehl R, Bourredjem A, Hernu R, Montini F, Bruyère R, Lebert C, Bohé J, Badie J, Eraldi JP, Rigaud JP, Levy B, Siami S, Louis G, Bouadma L, Constantin JM, Mercier E, Klouche K, du Cheyron D, Piton G, Annane D, Jaber S, van der Linden T, Blasco G, Mira JP, Schwebel C, Chimot L, Guiot P, Nay MA, Meziani F, Helms J, Roger C, Louart B, Trusson R, Dargent A, Binquet C, Quenot JP; IDEAL-ICU Trial Investigators and the CRICS TRIGGERSEP Network. Timing of Renal-Replacement Therapy in Patients with Acute Kidney Injury and Sepsis. N Engl J Med. 2018 Oct 11;379(15):1431-1442. doi: 10.1056/NEJMoa1803213. PubMed PMID: 30304656.
- Bellomo R, Cass A, RENAL Replacement Therapy Study Investigators. Intensity of continuous renal-replacement therapy in critically ill patients. N Engl J Med. 2009;361(17):1627-1638. doi:10.1056/NEJMoa0902413.
- Boussekey N, Chiche A, Faure K, et al. A pilot randomized study comparing high and low volume hemofiltration on vasopressor use in septic shock. Intensive Care Med. 2008;34(9):1646‐1653. doi:10.1007/s00134-008-1127-3.
- de Smet AM, Kluytmans JA, Cooper BS, et al. Decontamination of the digestive tract and oropharynx in ICU patients. N Engl J Med. 2009;360(1):20‐31. doi:10.1056/NEJMoa0800394.
- Joannes-Boyau O, Honoré PM, Perez P, et al. High-volume versus standard-volume haemofiltration for septic shock patients with acute kidney injury (IVOIRE study): a multicentre randomized controlled trial. Intensive Care Med. 2013;39(9):1535‐1546. doi:10.1007/s00134-013-2967-z.
- Jörres A, John S, Lewington A, et al. A European Renal Best Practice (ERBP) position statement on the Kidney Disease Improving Global Outcomes (KDIGO) Clinical Practice Guidelines on Acute Kidney Injury: part 2: renal replacement therapy. Nephrol Dial Transplant. 2013;28(12):2940‐2945. doi:10.1093/ndt/gft297.
- Park JT, Lee H, Kee YK, et al. High-Dose Versus Conventional-Dose Continuous Venovenous Hemodiafiltration and Patient and Kidney Survival and Cytokine Removal in Sepsis-Associated Acute Kidney Injury: A Randomized Controlled Trial. Am J Kidney Dis. 2016;68(4):599‐608. doi:10.1053/j.ajkd.2016.02.049.
- Zhang P, Yang Y, Lv R, Zhang Y, Xie W, Chen J. Effect of the intensity of continuous renal replacement therapy in patients with sepsis and acute kidney injury: a single-center randomized clinical trial. Nephrol Dial Transplant. 2012;27(3):967‐973. doi:10.1093/ndt/gfr486.
Evidence tabellen
Evidence tables for intervention studies (randomized controlled trials and non-randomized observational studies (cohort studies, case-control studies, case series))1
This table is also suitable for diagnostic studies (screening studies) that compare the effectiveness of two or more tests. This only applies if the test is included as part of a test-and-treat strategy - otherwise the evidence table for studies of diagnostic test accuracy should be used.
Research question 3: What is the optimal dose of renal replacement therapy in patients with septic shock and acute renal failure?
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3 |
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Bellomo, 2009
RENAL trial |
Type of study: RCT
Setting and country: Multicenter trial in 35 ICUs in Australia and New Zealand
Funding and conflicts of interest: Supported by grants from the National Health and Medical Research Council of Australia (352550) and the Health Research Council of New Zealand (06-357). Several authors receive consulting fees, grant support and reimbursement from the industry. |
Inclusion criteria: Critically ill patients, 18 years of age or older, with acute kidney injury, deemed by the treating clinician to require RRT, and met at least one of the predefined criteria (e.g. oliguria, acidemia, organ edema).
Exclusion criteria: Patients who had received previous RRT during the same hospital admission or who were on maintenance dialysis for end-stage kidney disease.
N total at baseline: Intervention: 721 Control: 743
Age (mean±SD) Intervention: 64.7±14.5 Control: 64.4±15.3
Sex (%female) Intervention 34.3% Control: 36.5%
Mechanical ventilation: Intervention 73.5% Control: 74.2%
APACHE III score: Intervention 102.5±25.9 Control: 102.3±25.5
Groups were comparable at baseline.
N sepsis subgroup: Intervention: 359 Control: 363 |
High volume CRRT, 40 mL/kg/h |
Low volume CRRT, 25 mL/kg/h
(Replacement fluid was delivered into the extracorporeal circuit after the filter (i.e., postdilution), with a ratio of dialysate to replacement fluid of 1:1. Fluid was removed by decreasing the flow of the replacement fluid and of the dialysate in equal proportion, so that effluent exceeded them both by any amount prescribed by the clinician. Filters with the AN69 membrane (Gambro) were used. Hemosol BO fluid (Gambro) was used as the dialysate and replacement fluid.) |
Length of follow-up: 90 days
Loss-to-follow-up: Intervention: 1 Control: 0
Incomplete outcome data: N/A |
In critically ill population
Mortality (90 days, primary outcome) High volume: 322/721 (44.7%) Low volume: 332/743 (44.7%) P=0.99
Mortality (28 days) High volume: 278/722 (38.5%) Low volume: 274/743 (36.9%) P=0.52
RRT dependence among survivors (after 90 days) High volume: 27/399 (6.8%) Low volume: 18/411 (4.4%) P=0.14
RRT duration (days) High volume: 13.0±20.8 Low volume: 11.5±18.0 P=0.14
Hospital length of stay (days) High volume: 26±25.8 Low volume: 25.7±24.7 P=0.79
ICU length of stay (days) High volume: 11.8±14.1 Low volume: 11.8±14.2 P=0.95
In sepsis subgroup: Mortality (90 days) High volume: 168/359 (46.8%) Low volume: 186/363 (51.2%)
OR (95% confidence interval) 0.84 (0.62–1.12) |
“Operational characteristics such as frequent filter clotting could have influenced solute clearance. The difference between the prescribed dose and the delivered dose highlights the risk of overestimating the effective delivery of therapy and the need to improve operational measures in continuous renal-replacement therapy. Specifically, basing the delivered dose on effluent volume most likely overestimates true solute clearance.” |
|
Boussekey, 2008 |
Type of study: Pilot RCT
Setting and country: Single center, France
Funding and conflicts of interest: Tourcoing hospital provided the financial support for this study. The authors have no financial involvement with any organization or entity with a financial interest in the subject discussed in the manuscript. |
Inclusion criteria: Patients with septic shock and ARF with at least one of the following criteria requiring RRT: urine output <200 ml/12 h or anuria>12 h, serum urea>30 mmol/l, creatinine >500 µmol/l or doubling of base creatinine for patients with chronic renal failure.
Exclusion criteria: Patients were excluded if they suffered from obstructive or prerenal renal failure, severe chronic renal failure, were included in another study, had severe immunosuppression or were moribund.
N total, median age, sex: High volume: N=9, age 68, 78% male Low volume: N=10, age 72.5, 80% male
APACHE II score (median (IQR)) High volume: 31 (26–33) Low volume: 33.5 (28–37) |
High volume hemofiltration, 65 mL/kg/h |
Low volume hemofiltration, 35 mL/kg/h
(All patients were treated with the Prismaflex machine (Hospal, Lyon, France) using a 1.4 m2 polyethersulfone filter (HF 1400, Hospal, Lyon, France) with a cut-off point of 20 kDa. Vascular access was obtained with dual-lumen catheters (Prismaccess, 13F, 20 cm long) inserted in the femoral or jugular vein. Blood flow was set between 180 and 250 ml/min in the LVHF group, and between 200 and 300 ml/min in the HVHF group, to reach a filtration fraction below 20%. Ultrafiltrate flow was delivered prefilter in one-third and postfilter in two-thirds of the patients. Bicarbonate-buffered replacement fluids were used for all RRT procedures.) |
Length of follow-up: 28 days
Loss-to-follow-up: Intervention: 0 Control: 0
Incomplete outcome data: N/A |
The primary end point was the decrease of vasopressor dose along with a stable MAP>65 mmHg.
Mortality (28 days) High volume: 3/9 (33%) Low volume: 6/10 (60%) P=0.65
RRT duration (median (IQR) High volume: 6 (2-14) Low volume: 7 (2-17)
ICU length of stay (median (IQR) days) High volume: 18 (15–23) Low volume: 14.5 (7–29)
Recovery of renal function High volume: 6/6 Low volume: 4/4 |
“Study was not powered before enrolment because it was monocentric and we wanted to perform our study in a limited period (<2 years) to limit the bias related to treatment variations that could have occurred in a longer duration of time.” |
|
Joannes-Boyau, 2013
IVOIRE trial |
Type of study: RCT
Setting and country: Multicenter trial in 18 ICUs in France, Belgium and the Netherlands
Funding and conflicts of interest: The study was supported by a grant from the French Health Ministry (Hospital Clinical Research Program—PHRC). Dr. Bagshaw is supported by a Canada Research Chair in Critical Care Nephrology and Clinical Investigator Award from Alberta Innovates—Health Solutions. Other authors do not have any conflict of interest. |
Inclusion criteria: (1) patient age C18 years, (2) admission to ICU, (3) presence of septic shock, defined according to the consensus definition of the American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference Committee, for a period of no more than 24 h, and (4) presence of AKI fulfilling the INJURY category or greater according to the (RIFLE) classification scheme, by either the serum creatinine and/or urine output criteria
Exclusion criteria: (1) age ≥80 years, (2) estimated life expectancy of <3 months, (3) metastatic cancer, (4) decompensated cirrhosis, (5) acute necrotizing pancreatitis, (6) prior diagnosis of ESKD, (7) confirmed pregnancy, (8) severe coagulopathy (defined as international normalized ratio (INR) >3 and/or platelet count <20,000 cells/mL), and (9) lack of commitment to full medical support.
N total at baseline Intervention: N=66; Control: N=71
Age, median (IQR) Intervention: 68 (58–77) Control: 70 (58–75)
Sex (%male) Intervention: 68 Control: 54
RIFLE injury/failure Intervention: 70/30% Control: 80/20%
Groups were comparable at baseline. |
High volume haemofiltration, 70 mL/kg/h
CVVH was prescribed at a dose of 70 mL/kg/h for 96 h. The blood flow rate was modified in order to maintain a filtration fraction ≤25 %, with average blood flow rates ranging between 200 and 320 mL/min.
In both groups, target transmembrane pressure was maintained between 100 and 300 mmHg. |
Standard volume haemofiltration, 35 mL/kg/h
For all patients, haemo-filtration was performed with an Aquarius haemofiltration circuit (Edwards Life Sciences) equipped with a 1.9 m2 Aquamax polyethersulfone filter (Edwards Life Sciences; molecular weight cut-off approximately 35,000 Da; low adsorption capacity). The same circuit and haemofilter were applied for all the participants to avoid bias. Every haemofiltration was delivered in the continuous veno-venous haemofiltration (CVVH) mode.
In addition to the allocated haemofiltration therapy, all the patients received therapy for septic shock consistent with best contemporary practice and consensus guideline recommendations (Surviving Sepsis Campaign, recommendations for septic shock patients from the French Intensive Care Society (SFAR + SRLF)) |
Length of follow-up: 90 days
Loss-to-follow-up: Intervention: n=0 Control: n=0
Incomplete outcome data: N/A |
Mortality (28 days), primary outcome High volume: 25/66 (38%) standard: 29/71 (41%) P=0.94
Mortality (60 days) High volume: 33/66 (50%) standard: 35/71 (49%) P=0.93
Mortality (90 days) High volume: 37/66 (56%) standard: 36/71 (51%) P=0.53
RRT-free days (median (IQR) at day 90) High volume: 84 (78–86) standard: 83 (74–85) P=0.22
ICU-free days (median (IQR) days) at day 90) High volume: 75 (59–86) standard: 74 (59–83) P=0.46
hospital-free days (median (IQR) days) at day 90) High volume: 61 (34–86) standard: 60 (26–81) P=0.51
RRT dependency in survivors at day 90 High volume: 0/29 standard: 1/35 P=0.95 |
To detect this 15 % absolute reduction in 28-day mortality with an 85 % power, a sample size of 230 patients per group (a total 460) was required. Unfortunately, because of slow participant accrual, the trial was prematurely terminated and the decision was taken to keep only one final analysis, despite the possible lower power than expected and the higher risk of β error. |
|
Park, 2016
HICORES trial |
Type of study: RCT
Setting and country: Dual center in medical ICUs of 2 large academic Hospitals in Korea
Funding and conflicts of interest: This work was supported by Gambro Korea Ltd, Seoul, Korea. The supporter did not have any role in study design; collection, analysis, and interpretation of data; writing the report; or the decision to submit the report for publication. The authors declare that they have no other relevant financial interests. |
Inclusion criteria: Critically ill adults 20 years or older who had AKI due to sepsis and required CRRT; patients with AKI at a level greater than the injury stage according to the RIFLE (risk, injury, failure, loss, end-stage renal disease) criteria, which was consistent with urine output , 0.5 mL/kg/h over 12 hours or a more than 2-fold increase in serum creatinine level compared with baseline.
Exclusion criteria: Patients older than 80 years; with life expectancy less than 3 months, terminal cancer, Child-Pugh class C liver cirrhosis, or history of dialysis; and those who were pregnant or lactating prior to randomization.
N total: Intervention: 109 Conventional: 110
Age, mean±SD Intervention: 62.3±13.4 Conventional: 61.9±12.8
Sex (% male) Intervention: 59 Conventional: 71
APACHE II score Intervention: 28.6 ± 6.6 Conventional: 28.9 ± 7.9
Groups were comparable at baseline. |
High volume continuous venovenous hemodiafiltration, 80 mL/kg/h |
Conventional volume continuous venovenous hemodiafiltration, 40 mL/kg/h
CVVHDF therapies were delivered by the Gambro Prisma or Prisma Flex machines using ST100 (surface area, 1.0 m2) filter sets, which contain a polyacrylonitrile AN 69 membrane (Gambro). For cases that required flow rates .>2,000 mL/h, the Prisma Flex RRT machine as preferred; in other cases, either the Prisma or Prisma Flex RRT machine was used. Vascular access for CVVHDF was obtained by the insertion of a 14F double-lumen catheter into the femoral or internal jugular vein. Blood flow rate was initiated at 100 mL/min and gradually increased to 150 mL/min. The replacement and dialysate volumes were set using the 1:1 balanced-predilution method. Half the calculated total effluent volume was given as replacement Hemosol (Gambro), and the other half was administrated as dialysate. Only the Hemosol replacement fluid was administered intravenously through the predialyzer replacement pump. The dialysate remained outside the dialyzer membrane and was not given intravenously. |
Length of follow-up: 90 days
Loss-to-follow-up: Intervention: N=4 Reasons: transferred to other hospital (3) and discharged against medical advice (1)
Control: N=3 Reasons: transferred to other hospital (3)
Incomplete outcome data: N/A |
Mortality (28 days), primary outcome High volume: 69/105 (66%) standard: 69/107 (65%) P=0.5
Mortality (90 days) High volume: 82/105 (78%) standard: 80/107 (75%) P=0.6
RRT duration (mean days (95% CI)) High volume: 6.2 (4.9, 7.6) standard: 5.4 (4.1, 6.8) P=0.4
ICU length of stay (in days, mean±SD) High volume: 18.2±16.6 standard: 11.5±10.9 P=0.05
hospital length of stay (in days, mean±SD) High volume: 59.3±45.2 standard: 38.8±32.1 P=0.08
RRT dependency* in survivors at day 28 High volume: 13/36 (36%) standard: 13/38 (34%) P=0.9
RRT dependency* in survivors at day 90 High volume: 0/23 (0%) standard: 2/27 (7.4%) P=0.1
*table describes “non-dialysis-dependent” but the values are more likely to indicate “dialysis dependent” |
“The predilution method used in these patients could have lowered the clearance rate. In addition, the study could have been underpowered for the clinical outcomes due to the relatively small number of participants.” |
|
Zhang, 2012 |
Type of study: RCT
Setting and country: Single center, China
Funding and conflicts of interest: This study was supported by the Science and Technology of Zhejiang Province office (grant no. 2007C13055). Conflict of interest statement: None declared |
Inclusion criteria: >18 years, suffered from severe sepsis and AKI in the ICU, required CRRT and met at least one of the following criteria: oliguria (urine output <100 mL in a 6-h period and unresponsive to fluid resuscitation), serum potassium concentration >6.5 mmol/L, severe acidemia (pH < 7.2), serum creatinine (SCr) >250 lmol/L or presence of severe organ edema (e.g. pulmonary edema).
Exclusion criteria: Presence of a malignant tumor, chronic renal insufficiency (SCr > 133 lmol/L) or receiving any kind of renal replacement therapy before randomization.
N total at baseline: Intervention: 141 Control: 139
Age, mean±SD Intervention: 56.62±16.38 Control: 59.96±18.81
Sex (% male) Intervention: 59 Control: 64
Septic shock (%) Intervention: 51 Control: 50
APACHE II score Intervention: 21.97±8.25 Control: 22.60±7.59
Groups were comparable at baseline. |
Very high volume continuous venous hemofiltration, 85 mL/kg/h.
The actual flow rate of effluent was 87.54 mL/kg/h. |
High volume continuous venous hemofiltration, 50 mL/kg/h.
The actual flow rate of effluent was 49.99 mL/kg/h.
Replacement fluid was delivered into the extracorporeal circuit at a pre-dilution/post-dilution ratio of 2/1. Central venous access was used and the catheters of 11.5 or 13.5 FR316 cm, 11.5 or 13.5 FR 3 19.5 cm (Kendall, Tyco healthcare Group LP, Mansfield, MA) were utilized. Blood flow was kept >250 mL/min. Polysulfone filters (AV600; Fresenius Medical Care, Bad Homburg, Germany) were used for all patients and the filter was changed when the transmembrane pressure of the filter was >250 mmHg.. AK-ultra 200 (Gambro, Lund, Sweden) online replacements (bicarbonate solution) were used as the replacement fluid and were infused into the CRRT circuit instantly whenever it was produced. |
Length of follow-up: 90 days
Loss-to-follow-up: Intervention: 0 Control: 0
Incomplete outcome data: N/A |
Mortality (90 days), primary outcome Very high volume: 84/141 (60%) High volume: 88/139 (63%) P=NS
Mortality (28 days) Very high volume: 81/141 (57%) High volume: 81/139 (58%) P=NS
Mortality (60 days) Very high volume: 84/141 (60%) High volume: 87/139 (63%) P=NS
Cessation of RRT Very high volume: 53/57 (93%) High volume: 46/51 (90%) P=NS
ICU length of stay (days) Very high volume: 21.91 ± 28.04 High volume: 25.92 ± 40.36 P=NS
Hospital length of stay (days) Very high volume: 35.53 ± 61.40 High volume: 38.47 ± 54.24 P=NS
Duration of RRT (days) Very high volume: 9.38 ± 12. 06 High volume: 8.88 ± 10.79 P=NS |
“Our study has several limitations. Firstly, the time of renal support initiation was relatively late which may have had influenced the effectiveness of the CRRT treatment leading to the high mortality rate seen and is one of the major issues. Secondly, we did not include a standard dose group (20–35 mL/kg/h) in our study. Finally, we did not assess all possible side effects of EHVHF.” |
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures.
- Provide data per treatment group on the most important prognostic factors ((potential) confounders).
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls.
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders.
Risk of bias table for intervention studies (randomized controlled trials)
Research question 3: What is the optimal dose for renal replacement therapy in patients with septic shock and acute renal failure?
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome accessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Bellomo, 2009 |
Not reported |
unclear |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
|
Boussekey, 2008 |
Not reported. “out of every four patients, two were allocated to LVHF, and two to HVHF treatment at random.” |
unclear |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
|
Joannes-Boyau, 2013 |
The allocation sequence was computer-generated by the clinical trials unit statistician. The randomization ratio was 1:1. Randomization was stratified by centre, using blocks of four for centres with expected enrolment below 30, and random blocks of four or six for centres with an expected enrolment greater than 30. The allocation process was centralized and the allocation group was concealed until it was implemented. |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
|
Park, 2016” |
“Patients eligible for inclusion were “randomly assigned in a 1:1 ratio to 1 of the 2 treatment groups by means of a centralized computer-generated adaptive randomization scheme at the time of CRRT initiation.” |
unclear |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unclear |
|
Zhang, 2012 |
“Patient randomization was generated from a computer-generated sequence of random numbers” |
unclear |
unlikely |
unlikely |
unlikely |
unclear |
unclear |
unclear |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules..
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear.
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Verantwoording
Autorisatiedatum en geldigheid
Laatst beoordeeld : 01-09-2022
Laatst geautoriseerd : 05-10-2022
Geplande herbeoordeling :
Algemene gegevens
De ontwikkeling van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2017 een multidisciplinaire werkgroep ingesteld voor sepsis fase 1, de samenstelling van de werkgroep is gewijzigd in 2019 voor de ontwikkeling van sepsis fase II en III, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de zorg voor patiënten met sepsis in het ziekenhuis.
Werkgroep
- Prof. Dr. A.R.H. van Zanten, internist-intensivist, Ziekenhuis Gelderse Vallei te Ede, NIV (voorzitter)
- Dr. C.H.S.B. van den Berg, intensivist-infectioloog, UMC Groningen, NVIC
- D. Bolman, patiëntvertegenwoordiger, IC Connect en FCIC
- V. Bon, verpleegkundig Specialist spoedeisende hulp, Onze Lieve Vrouwe Gasthuis, te Amsterdam en Ambulanceverpleegkundige bij Ambulance Amsterdam, V&VN-VS
- Dr. C.S.C. Bouman, internist-intensivist, Amsterdam UMC, locatie AMC, NIV
- Dr. L.P.G Derde, internist-intensivist, Universitair Medisch Centrum Utrecht te Utrecht, NVIC
- Drs. M. Hoogendoorn, manager Vakgroep Anesthesiologie & Intensive Care, Isala te Zwolle, V&VN-IC
- Dr. W.G. Ista, universitair hoofddocent Sector Verplegingswetenschap afdeling Interne Geneeskunde en universitair hoofddocent Kinder IC, Erasmus MC te Rotterdam, V&VN-IC
- Dr. R.W.M.M. Jansen, klinisch geriater, Noordwest ziekenhuisgroep te Alkmaar, NVKG
- Dr. H. P. Krepel, nefroloog, Bravis ziekenhuis te Roosendaal en Bergen op Zoom, NIV
- Dr. M.C.G. van de Poll, chirurg-intensivist, Maastricht UMC, NVIC
- Dr. B. P.C. Ramakers, internist-intensivist, RadboudUMC te Nijmegen, NVIC
- Dr. M.J.A. de Regt, internist-infectioloog/ internist-acute geneeskunde, Onze Lieve Vrouwe Gasthuis te Amsterdam, NIV
- Dr. S.U.C. Sankatsing, internist-infectioloog/ internist-acute geneeskunde, Diakonessenhuis te Utrecht, NIV
- Drs. R. Schellaars, intensivist, Ziekenhuis Gelderse Vallei te Ede, NVA
- Drs. R.M. Wilting, chirurg-intensivist, Elisabeth-TweeSteden ziekenhuis te Tilburg, NVvH
- Dr. J. Sommers, fysiotherapeut en onderzoeker, Amsterdam UMC, locatie AMC, KNGF
Met ondersteuning van
- Dr. F. Willeboordse, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Dr. R. Zwarts - van de Putte, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Dr. M.S. Ruiter, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
A. van Zanten |
lnternist-intensivist, afdeling lntensive Care Ziekenhuis Gelderse Vallei, Ede Medisch Directeur, Ziekenhuis Gelderse Vallei, Ede |
Onbetaald: Voorzitter Sectie lC NIV Lid Concilium Interne Geneeskunde Lid GIC Lid platform kwaliteit NIV Lid ESPEN richtlijn commissie Voeding volwassen lC patiënten Lid diverse congrescommissies (Nationale Voedingscongres, internationaal Sepsis Symposium Netherlands, Mythen Missers en Maatwerk infectieuze bedreigingen, MasterclasslC Schiermonnikoog)
Betaald: Spreker voor voedingsfirma's (niet sepsis gerelateerd): Danone-Nutricia, Abbott, BBraun, Baxter, Fresenius-Kabi, Lyric,Mermaid, Nestle-Novartis.
initiator Masterclass Voeding en lC |
Participatie als onderzoeker in lC sepsis trials (b.v. KPA-ART 123 trombomoduline & adrenomedulline trial), opbrengsten vloeien naar Stichting lC research en worden niet uitgekeerd aan onderzoekers.
Echtgenote heeft een Congres- en Organisatiebureau interactie dat voor vele wetenschappelijke verenigingen congressen organiseert (onder andere sepsis congres).
|
Belangen zijn besproken, voorzitterschap is akkoord |
|
C. van den Berg |
intensivist-infectioloog, UMC Groningen |
Geen |
Geen |
Geen restricties |
|
D. Bolman |
vertegenwoordiger FCIC en IC Connect, ervaringsdeskundige |
vrijwilliger FCIC en IC Connect (onbetaald) vrijwilliger Nederlandse adoptievoorziening (onbetaald) |
Geen |
Geen restricties |
|
V. Bon |
Verpleegkundig Specialist SEH OLVG Ambulanceverpleegkundige bij Ambulance Amsterdam |
instructeur bij opleidingsinstituut spoedeisende geneeskunde in Houten (betaald). |
Ik heb meegewerkt aan de Phantasi trial, het prehospitaal toedienen van AB bij sepsis (https://classic.clinicaltrials.gov/ct2/show/NCT01988428). |
Geen restricties |
|
C. Bouman |
Internist-intensivist Care Volwassenen Amsterdam UMC locatie AMC |
Geen |
Geen |
Geen restricties |
|
L. Derde |
Intensivist UMC Utrecht (0,8 fte) Co-lead WP5 PREPARE (0,2 fte) |
EU coordinating investigator, voorzitter EU Regionale Management Commissie en lid Internationale Trial Stuur Commissie REMAP-CAP, onder andere gefinancierd via PREPARE (FP7) en RECOVER (H2020) grants
Lid NVVM influenza richtlijn
Lid ESICM (European Society of Intensive Care Medicine) commissie voor ernstige CAP (community acquired pneumonia)
Voorzitter van de Clinical Training Committee (CTC) van ESICM.
Lid SSC/ESICM/SCCM COVID-19 guideline committee
Voorzitter NVIC taskforce COVID-19 |
Betrokken bij onderzoek dat door EU wordt gefinancierd:
1. EU coordinating investigator REMAP-CAP: doel is zoeken naar optimale behandeling voor patiënten met community acquired pneumonie op de IC. REMAP-CAP wordt in andere regio's van de wereld ondersteund door andere funding (investigator driven). Diverse medicamenten (tocilizumab, sarilumab, interferon, lopinavir/ritonavir, anakinra) zijn (deels) door de fabrikanten geleverd voor de studie. In de Verenigde Staten wordt één van de domeinen (eritoran, apremilast) ondersteund door de fabrikanten (Eisai, Amgen). De funders, sponsors en fabrikanten hadden geen rol in de opzet van de studie, de analyses, publicaties, of beslissingen die in de trial zijn genomen. |
Geen restricties |
|
M. Hoogendoorn |
Vakgroep manager Anesthesiologie & Intensive Care, Isala, Zwolle |
Managing Director Athena Care B.V. - Research organisatie Vakgroep Anesthesiologie & Intensive Care (betaald) Bestuurslid V&VN-IC (onbetaald) Bestuurslid NICE (onbetaald)" |
Geen |
Geen restricties |
|
E. Ista |
Universitair docent/senior onderzoeker, IC Kinderen, Erasmus MC-Sophia Kinderziekenhuis, Rotterdam |
Universitair Hoofddocent/Senior onderzoeker, Verplegingswetenschap, Interne Geneeskunde, Erasmus MC, Rotterdam
Lid Bestuurscommissie Richtlijnen V&VN, Utrecht (onbetaald) tot 2018
Lid CCMO, discipline verplegingswetenschap (betaald) |
Geen |
Geen restricties |
|
R. Jansen |
Klinisch geriater, Noordwest ziekenhuisgroep, locatie Alkmaar |
Onderwijs aan Amstelacademie voor verschillende verpleegkundigen opleidingen (betaald).
Onbetaald medeorganisator ouderengeneeskunde congres Maastricht 2.0 in 2018, en 2019.
Onbetaald medeorganisator van nieuw congres Cardiovasculaire aandoeningen bij ouderen (CarVascGer) 2020 te Utrecht. |
Honoraria van Bayer Nederland, Boehringer-Ingelheim, Daiichi-Sankyo en BMS-Pfizer voor houden lezingen, webTV, et cetera over atriumfibrilleren en gebruik NOAC's en schrijven van bijdrage aan zakboekjes. |
Geen restricties |
|
H. Krepel |
Nefroloog, Bravis Ziekenhuis Roosendaal/Bergen op Zoom |
Geen |
Domestico studie (thuisdialyse) |
Geen actie |
|
M. van de Poll |
Intensivist MUMC+ |
Lid van richtlijncommissie NVIC Voeding |
Investigator-initiated grants van Fresenius Kabi en Nutricia, ZON-MW, KCE |
Restricties ten aanzien van het opstellen van aanbevelingen over voeding. |
|
B. Ramakers |
Intensivist in het Radboudumc |
Voorzitter stichting Venticare (kennisplatform voor medewerkers in de acute zorg): onbetaald |
Geen |
Geen restricties |
|
M. de Regt |
Internist Acute Geneeskunde/infectioloog Onze Lieve Vrouwe Gasthuis, Amsterdam |
Geen |
Geen |
Geen restricties |
|
S. Sankatsing |
Internist-infectioloog/ internist-acute geneeskunde, Diakonessenhuis Utrecht |
Bestuurslid Nederlandse Vereniging van lnternist-lnfectiologen (NVll), onbetaald
Lid Commissie Richtlijnen Nederlandse lnternisten Vereniging (NlV), waarvoor vacatiegelden
Lid Expertise team behandeling Covid-19 van de Federatie Medisch Specialisten (FMS), onbetaald |
Geen |
Geen restricties |
|
R. Schellaars |
Intensivist, Ziekenhuis Gelderse Vallei, Ede |
Voormalig bestuurslid SIC-NVA |
Geen |
Geen restricties |
|
R. Wilting |
Chirurg-intensivist, afdeling IC Elisabeth-TweeSteden ziekenhuis Tilburg |
Lid GIC (NVvH) |
Geen |
Geen restricties |
|
J. Sommers |
Fysiotherapeut en onderzoeker, Amsterdam UMC, locatie AMC |
Cursuscoördinator en docent NPI, scholing IC fysiotherapie (betaald) |
Geen |
Geen restricties |
|
M. Ruiter |
Adviseur Kennisinstituut van de Federatie Medisch Specialisten |
Geen |
Geen |
Geen restricties |
|
R. Zwarts - van de Putte |
Adviseur Kennisinstituut van de Federatie Medisch Specialisten |
Geen |
Geen |
Geen restricties |
|
F. Willeboordse |
Adviseur Kennisinstituut van de Federatie Medisch Specialisten |
Geen |
Geen |
Geen restricties |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een patiëntenfocusgroep en afvaardiging van een patiëntvertegenwoordiger in de werkgroepen. Het verslag van de focusgroep (zie bijlage) is besproken in de werkgroep. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen. De conceptrichtlijn is voor commentaar voorgelegd aan Stichting Family and patient Centered Intensive Care (Stichting FCIC), aan IC Connect, de patiëntenorganisatie voor (voormalig) IC-patiënten en naasten en aan de Patiëntenfederatie Nederland en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Patiëntenparticipatie bij deze richtlijn werd medegefinancierd uit de Kwaliteitsgelden Patiënten Consumenten (SKPC) binnen het programma KIDZ.
Methode ontwikkeling
Evidence based
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep de knelpunten in de zorg voor patiënten met sepsis. Tevens zijn er knelpunten aangedragen door vertegenwoordigers van Ambulancezorg Nederland, Inspectie Gezondheidszorg en Jeugd, Nederlandse Internisten Vereniging, Nederlandse Vereniging van Artsen voor Longziekten en Tuberculose, Nederlandse Vereniging voor Klinische Chemie en Laboratoriumgeneeskunde, Nederlandse Vereniging van Ziekenhuizen, Nederlandse Vereniging van Ziekenhuisapothekers, Family and Patient Centered Intensive Care en IC Connect, Stichting Werkgroep Antibiotica Beleid, Vereniging Innovatieve Geneesmiddelen en Dutch Acute Medicine via een Invitational conference. Een verslag hiervan is opgenomen onder aanverwante producten.
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
Definitie |
|
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello, 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE-methodiek.
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE-gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij fase II/III van de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst. Uit de kwalitatieve raming blijkt dat er geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
|
Module |
Uitkomst kwalitatieve raming |
Toelichting |
|
Vasopressoren |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Inotropica |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Nierfunctievervangende therapie: modaliteit |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Nierfunctievervangende therapie: timing |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Nierfunctievervangende therapie: dosering |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Nierfunctievervangende therapie: antistolling |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Sedatiemiddel |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Sedatie: continu vs. interruptie |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Medicamenteuze behandeling van delier |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Niet-medicamenteuze behandeling van delier |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Transfusiebeleid |
n.v.t. |
Verwijzing naar bestaande module. |
|
Mobilisatie |
geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Voeding |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbevelingen niet breed toepasbaar zijn (<5.000 patiënten) en zullen daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
|
Voorlichting langetermijngevolgen |
geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Randvoorwaarden |
geen substantiële financiële gevolgen |
In deze module wordt vooral verwezen naar andere relevante documenten verwezen. Er worden daarom geen financiële gevolgen verwacht. |
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html.
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbo.