Zuurstof
Uitgangsvraag
Wat is de plaats van aanvullende zuurstof toediening bij patiënten die procedurele sedatie en/of analgesie (PSA) ondergaan?
Aanbeveling
Op plaatsen waar lichte PSA uitgevoerd wordt is aanbevolen zuurstof direct beschikbaar te hebben om hypoxemie te kunnen behandelen.
Dien laagdrempelig additionele zuurstof toe voorafgaande en tijdens matige of diepe PSA om desaturatie en hypoxemie te voorkomen of om hypoxemie te behandelen.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Er is literatuuronderzoek verricht naar de effecten van routinematige toediening van zuurstof ten opzichte van geen toediening van zuurstof voorafgaande en tijdens procedurele sedatie en/of analgesie (PSA).
Complicaties waren gedefinieerd als cruciale uitkomstmaat. Op basis van de literatuur kan geconcludeerd worden dat desaturatie mogelijk minder vaak voorkomt wanneer er voorafgaande en tijdens PSA aanvullende zuurstof wordt toegediend. Voor overige complicaties was de bewijskracht zeer laag en kan op basis van de literatuur geen uitspraak worden gedaan. De overall bewijskracht van de cruciale uitkomstmaat is hierdoor beoordeeld als zeer laag. Voor de belangrijke uitkomstmaten aanvullende interventies en veiligheid is bewijskracht voor deze uitkomsten zeer laag, waardoor deze geen richting kunnen geven aan de besluitvorming.
Routinematige toediening van zuurstof is een controversieel punt, omdat deels de mening bestaat dat het toedienen van zuurstof een respiratoire insufficiëntie/apneu zou kunnen maskeren. Echter dient het ademhalingspatroon altijd goed geobserveerd te worden en is op veel plekken waar matige tot diepe sedatie wordt toegepast capnografie ter beschikking waarmee de ademexcursies en ademfrequenties beter kunnen worden bewaakt, en dus het boven beschreven potentiële nadeel van extra zuurstof toediening komt te vervallen.
Pathofysiologisch resulteert een verhoogd aanbod zuurstof in het compenseren van potentiële risico’s (bijv. lagere functionele residuale capaciteit (FRC) bij de liggende patiënt, kleinere gasuitwisselingsoppervlakte, perfusie/ventilatie (P/Q) mismatch i.v.m. voorgeschiedenis, kleinere diffusiecapaciteit) en het verhogen van de zuurstofreserve in het geval van een respiratoire depressie of apneu. Bij het optreden van deze complicaties zou de patiënt zonder interventies, die mogelijk ook weer complicaties kunnen bevorderen, vanzelf kunnen herstellen.
Omdat het risico op desaturatie en hypoxemie door de toediening van zuurstof voor- en tijdens PSA waarschijnlijk lager is en de indicaties voor potentieel gevaarlijke luchtwegmanoeuvres minder zou worden, beveelt de commissie bij matig tot diepe sedatie een zo goed mogelijke en adequate (pre)oxygenatie aan. Bij lichte sedatie is het aanbevolen zuurstof binnen korte tijd beschikbaar te hebben.
Omdat toediening van zuurstof een respiratoire depressie of apneu kan maskeren dient de ademhaling van de patiënt nauwkeurig geobserveerd te worden. Dit kan visueel of met behulp van capnografie gebeuren.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
De patiënt heeft baat bij het veilig uitvoeren van PSA. Het toepassen van adequate (pre)oxygenatie en zuurstoftoediening tijdens PSA verhoogt de zuurstofreserve en reduceert daarmee de risico’s van hypoxemie voor de patiënt.
Kosten (middelenbeslag)
Zuurstof is in de Nederlandse zetting op locaties waar PSA toegepast wordt meestal laagdrempelig beschikbaar (centraal/decentraal). Voor de infrastructuur zijn waarschijnlijk geen extra kosten noodzakelijk. De prijs voor zuurstof zelf fluctueert enorm afhankelijk van de infrastructuur (centraal vs. decentraal) en de hoeveelheid zuurstof die door een zorginstelling verbruikt wordt. Exemplarisch voor één Nederlands ziekenhuis zijn de kosten voor zuurstof 0,00013 cent/liter (centraal) vs. 0,012 cent/liter (decentraal = zuurstoffles). Per minuut zijn de kosten bij een flow van bijvoorbeeld 15 l/min via non-rebreather masker dan 0,002 cent vs. 0,18 cent. Met betrekking tot de kosten(effectiviteit) is de interventie zuurstoftoediening verantwoordbaar en niet met hoge extra kosten verbonden.
Aanvaardbaarheid, haalbaarheid en implementatie
Op locaties in Nederland waar PSA wordt toegepast is zuurstof meestal laagdrempelig beschikbaar (zowel centraal als decentraal). De werkgroep voorziet daarom geen problemen op het gebied van de aanvaardbaarheid en haalbaarheid.
Rationale van aanbeveling 1: weging van argumenten voor en tegen de interventies
De meest voorkomende bijwerkingen en complicaties tijdens PSA betreffen de functie ademhaling met als risico desaturatie en hypoxemie. Een basis voor de preventie en de behandeling van luchtwegproblemen vormt de toediening van zuurstof.
Rationale van aanbeveling 2: weging van argumenten voor en tegen de interventies
Pathofysiologisch resulteert een verhoogd aanbod zuurstof in het compenseren van potentiële risico’s (bijv. lagere FRC bij de liggende patiënt, kleinere gasuitwisselingsoppervlakte, P/Q mismatch i.v.m. voorgeschiedenis, kleinere diffusiecapaciteit) en het verhogen van de zuurstofreserve in het geval van een respiratoire depressie of apneu. Bij het optreden van deze complicaties zou de patiënt zonder interventies (indicaties voor potentieel gevaarlijke luchtwegmanoeuvres), die mogelijk ook weer complicaties kunnen bevorderen, kunnen herstellen van een lagere zuurstofsaturatie.
Omdat het risico van desaturatie en hypoxemie door de toediening van zuurstof voor en tijdens PSA waarschijnlijk lager is en de indicaties voor potentieel gevaarlijke luchtwegmanoeuvres potentieel minder zou worden, beveelt de commissie een zo goed mogelijke en adequate (pre)oxygenatie aan.
Onderbouwing
Zuurstof toediening voorafgaande (pre-oxygenatie) en tijdens procedurele sedatie en/of analgesie (PSA) kan de zuurstofreserve vergroten, waardoor de incidentie van hypoxemie lager zou kunnen worden en de noodzaak voor luchtweg interventies, die ook weer extra risico’s zouden kunnen vormen, zou kunnen afnemen.
In de oude CBO-richtlijn staat beschreven dat routinematige toediening van zuurstof voorafgaande en tijdens PSA niet noodzakelijk is, maar in ieder geval dient plaats te vinden bij eerste aanwijzingen van hypoxemie (perifere zuurstofsaturatie <92% of een daling van > 5 procentpunten van de uitgangswaarde). De sedatierichtlijn van de American Society of Anesthesiologists (ASA) van 2018 beveelt zuurstof toediening aan, voor en tijdens matige tot diepe sedatie, indien er geen specifieke contra-indicaties bestaan. In de ESA-richtlijn van 2018 wordt aanbevolen dat zuurstof beschikbaar moet staan bij het starten van PSA en gegeven kan worden om hypoxemie te vermijden, met name bij lange procedures en als een hypoxemie verwacht kan worden (good consensus, evidence B: strong).
(De)saturation
|
Low GRADE |
Supplemental oxygen possibly results in less desaturation and a lower incidence of hypoxia compared to no supplemental oxygen in patients undergoing PSA.
Sources: Arawaka, 2013; Deitch, 2007; Deitch, 2008; Deitch, 2011; Rozario, 2008 |
Respiratory depression
|
Very low GRADE |
The evidence is very uncertain about the effect of supplemental oxygen on respiratory depression during PSA.
Sources: Deitch, 2007; Deitch, 2008; Deitch, 2011 |
Adverse events
|
Very low GRADE |
The evidence is very uncertain about the effect of supplemental oxygen on cardiac arrhythmia, hypotension, bradycardia or vomiting during PSA.
Sources: Arawaka, 2013; Deitch, 2007; Deitch, 2008; Deitch, 2011 |
Interventions
|
Very low GRADE |
The evidence is very uncertain about the effect of supplemental oxygen on airway maneuvers, respiratory support or the need for antidotes during PSA.
Sources: Arawaka, 2013, Deitch, 2007; Deitch, 2008; Deitch, 2011 |
Safety - Physician’s identification of respiratory depression
|
Very low GRADE |
The evidence is very uncertain about the effect of supplemental oxygen on the identification of respiratory depression during PSA.
Sources: Deitch, 2007; Deitch, 2008 |
Summary of literature
Description of studies
Arawaka (2013) performed a RCT to examine the masking effects of oxygen supplementation in SpO2. The study included 70 adult patients who underwent sedated diagnostic colonoscopy. Patients were randomly divided into two groups: the intervention group received supplemental oxygen prior to and during the procedure at a rate of 2 L/min by nasal cannula (n = 35), the control group breathed room air (n = 35). The mean age of the patients in the intervention group was 54.4y (± 10.8) and in the control group 59.2y (± 12.1). In all patients, the underlying disease in the ASA class II were hypertension and diabetes mellitus. Sedation and analgesia (using 0.3 mg flunitrazepam and 35 mg meperidine) were administered through an intravenous catheter. A supplemental dose of flunitrazepam for abdominal pain or discomfort was given to three patients in the intervention group and two patients in the control group, but no supplemental meperidine was administered to any patient in either group. One patient in the intervention group and two patients in the control group did not complete the colonoscopy due to abdominal discomfort and pain. Measurements included SpO2 and etCO2 and were taken during the procedure using a pulse oximetry probe and a non-intubated O2/CO2 oral-nasal cannula, which was connected to a capnography monitor. The method of randomisation and concealment of allocation were not described, which could influence the risk of bias.
Deitch (2007) performed a RCT to examine whether supplemental oxygen reduces the incidence of hypoxia in patients who underwent emergency department procedural sedation and analgesia. The study included 80 patients who were randomized into two groups. The intervention group received supplemental oxygen (n = 44) and the control group received room air (n = 36), both at 2 L per minute by nasal cannula. The mean age of the patients in the intervention group was 36y (range 2 to 77) and in the control group 32y (range 13 to 68). Procedural sedation and analgesia was performed according to standard emergency department protocol. Intravenous midazolam and fentanyl were titrated to the desired level of sedation and analgesia, and the procedure was performed. Patients in the intervention group received a median dose of Fentanyl of 150.0 (range 25 to 400) µg and Midazolam of 5.0 (range 0.5 to 10.0) mg. Patients in the control group received a median (range) dose of Fentanyl of 100.0 (range 50 to 400) µg and Midazolam of 4.0 (range 1.0 to 8.0) mg. Patients were monitored during the procedure until they were back to their baseline alertness. Outcomes included oxygen saturation, etCO2 levels and blood pressure. This study had a low risk of bias.
Deitch (2008) performed a RCT to examine whether supplemental oxygen reduces the incidence of hypoxia in adult study patients who underwent emergency department procedural sedation. The study included 110 adults who were randomized into two groups. The intervention group received supplemental oxygen (n = 56) and the control group received room air (n = 54), both at 3 L per minute by nasal cannula. The mean age of the patients in the intervention group was 37y (range 19 to 86) and in the control group 37y (range 18 to 75). Procedural sedation with propofol was performed according to standard emergency department protocol. The initial dose of intravenous propofol was 1 to 1.5 mg/kg (ideal body weight). Subsequent doses of 0.5 mg/kg were administered until the desired level of sedation was achieved. Patients in the intervention group received a mean initial propofol dose of 1.19 (± 0.32) mg/kg and a total propofol dose of 1.71 (± 0.66) mg/kg. Patients in the control group received a mean initial propofol dose of 1.33 (± 0.32) mg/kg and a total propofol dose of 1.99 (± 0.79) mg/kg. Patients were monitored during the procedure until they were back to their baseline alertness. Outcomes included oxygen saturation, etCO2 levels and blood pressure. This study had a low risk of bias.
Deitch (2011) performed a RCT to examine whether high-flow oxygen reduces the incidence of hypoxia in adults who underwent emergency department sedation. The study included 117 adults who were randomized into two groups. The intervention group received high-flow oxygen (n = 59) and the control group received room air (n = 58), both administered at 15 L per minute by a non-rebreather mask. The mean age of the patients in the intervention group was 37y (range 27 to 55) and in the control group 32y (range 21.5 to 45.5). Procedural sedation with propofol was performed in accordance with standard practice. Patients wore a capnography nasal cannula under their mask and received 5 minutes of oxygen or room air administration before an initial propofol dose of 1 mg/kg according to ideal body weight. An additional dose of 0.5 mg/kg propofol was administered to achieve and maintain the desired level of sedation. In both groups, the median initial propofol dose was 1.0 (IQR 1.0 to 1.0) mg/kg. Patients in the intervention group received a median total propofol dose of 1.6 (IQR 1.1 to 2.8) mg/kg. The control group received a median total propofol dose of 1.5 (IQR 1.0 to 2.0) mg/kg. Patients were monitored during the procedure using electronic monitoring. Outcomes included oxygen saturation, etCO2, and blood pressure. In this study, 15 patients were excluded after enrolment, but it was not described to which treatment group these patients were allocated. It is unclear how this could have influenced the results.
Rozario (2008) performed a non-blinded RCT to examine whether supplemental oxygen administered prior to and during moderate sedation decreases episodes of oxygen desaturation in adults undergoing endoscopic procedures. The study included 389 patients who were randomized into two groups. The intervention group received oxygen (2 L/min) prior to moderate sedation (n = 194) and the control group received room air instead of oxygen prior to the procedure (n = 195). The mean age of the patients in the intervention group was 56y (95% CI 54 to 58) and in the control group 56y (95% CI 55 to 58). Moderate sedation was administered via an intravenous line and consisted of fentanyl and midazolam or fentanyl and diazepam. It was not reported which and how many patients received midazolam or diazepam and in which dosage. The outcome was oxygen saturation, which was measured during the procedure with monitors. The study was non-blinded and the concealment of allocation was not described. If a patient in the experimental group had an episode of desaturation, oxygen flow was increased, and further interventions were implemented as needed. If desaturation (oxygen saturation <95%) occurred in a patient in the control group, low-flow oxygen at 2 L/min was administered via nasal cannula for the remaining duration of the procedure. It was not described how many and at what moment the additional oxygen flow was administered.
Results
Complications
(De)saturation
All included studies measured oxygen saturation; one study analyzed oxygen saturation continuously and the other four studies defined hypoxia a priori by applying a cut-off value for oxygen desaturation. Different definitions for hypoxia were used.
Arawaka (2013) measured oxygen saturation continuously and reported that at peak etCO2, SpO2 was higher in the oxygen supplementation group (98.6% ± 1.4%) when compared with the room air breathing group (93.1% ± 2.9%). The mean difference is 5.50% (95% CI 4.43 to 6.57). This is not a clinically relevant difference in this patient group (colonoscopy, lateral decubitus, free airway).
Deitch (2007) defined hypoxia as oxygen saturation <90%. This study reported an incidence of hypoxia of 14% in both the intervention (6/44) and the control group (5/36). The RR was 0.98 (95% CI 0.33 to 2.96), in favor of the intervention group. This is not a clinically relevant difference.
Two studies defined hypoxia as oxygen saturation <93% (Deitch, 2008; Deitch, 2011), of which one study specified that this should be >15 seconds (Deitch, 2011).
Deitch (2008) found a lower incidence of hypoxia in the intervention group compared to the control group. In the intervention group, hypoxia was reported for 10/56 (18%) patients, compared to 15/54 (28%) patients in the control group. The RR was 0.64 (95% CI 0.32 to 1.30), in favor of the intervention group.
Deitch (2011) found a lower incidence of hypoxia in the intervention group compared to the control group. In the intervention group, hypoxia was reported for 11/59 (19%) patients, compared to 24/58 (41%) patients in the control group. The RR was 0.45 (95% CI 0.24 to 0.83), in favor of the intervention group. The pooled RR for these two studies is 0.53 (95% CI 0.33 to 0.84) (Figure 1). This is a clinically relevant difference.
Rozario (2008) defined desaturation as oxygen saturation <95% and also made a distinction between mild (90% - 94.9%) and severe (<90%) desaturation. This study reported that in the intervention group, hypoxia was found in 24/194 (12%) patients, compared to 138/195 (71%) patients in the control group. The RR was 0.17 (95% CI 0.12 to 0.26), in favor of the intervention group. This is a clinically relevant difference.
Figure 1. Incidence of hypoxia for supplemental oxygen versus no supplemental oxygen
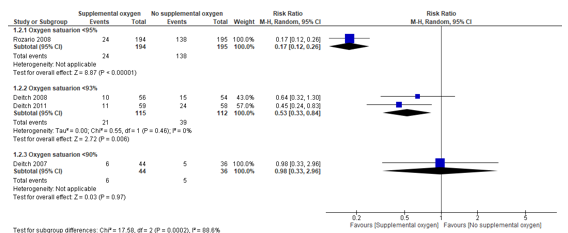
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Additionally, Rozario (2008) made a distinction between mild (oxygen saturation 90% - 94.9%) and severe hypoxia (oxygen saturation <90%). In the intervention group, 21/194 (11%) patients developed mild hypoxia compared to 132/195 (68%) in the control group. The RR is 0.16 (95% CI 0.11 to 0.24), in favor of the intervention group. This is a clinically relevant difference.
Severe hypoxia was found in 3/194 (2%) patients in the intervention group compared to 6/195 (3%) in the control group. The RR is 0.50 (0.13 to 1.98), in favor of the intervention group. This is a clinically relevant difference.
Level of evidence of the literature
The level of evidence regarding (de)saturation came from RCTs and started as high. The level of evidence was downgraded by two levels because of risk of bias (concealment of allocation unclear, lack of intention to treat analysis; -1) and imprecision (wide 95% confidence intervals crossing border for clinical relevance; -1) resulting in a low level of evidence.
Respiratory depression
Three studies examined respiratory depression. If patients met at least one of the following criteria, they were considered to experience respiratory depression: 1) etCO2 ≥ 50 mm Hg; 2) an absolute etCO2 change from baseline ≥ 10 mm Hg; 3) loss of the etCO2 waveform; 4) oxygen saturation < 90% (Deitch, 2007) or < 93% (Deitch, 2008). One study defined respiratory depression as meeting at least one of the following criteria: 1) etCO2 > 50 mm Hg; 2) an absolute increase or decrease from baseline ≥ 10%; 3) a loss of waveform for ≥ 15 seconds (Deitch, 2011).
Deitch (2007) reported a lower incidence of respiratory depression in the intervention group compared to the control group. In the intervention group, 20/44 (45%) patients experienced respiratory depression compared to 19/36 (52%) patients in the control group. The RR was 0.86 (95% CI 0.55 to 1.35), in favor of the intervention group. This is not a clinically relevant difference
Deitch (2008) reported that in the intervention group, 30/56 (53%) patients experienced respiratory depression compared to 22/54 (40%) patients in the control group. The RR was 1.31 (95% CI 0.88 to 1.97), in favor of the control group. This is a clinically relevant difference.
Deitch (2011) also reported a lower incidence of respiratory depression in the control group. In the intervention group, 30/59 (51%) patients experienced respiratory depression compared to 28/58 (48%) patients in the control group. The RR was 1.05 (95% CI 0.73 to 1.52), in favor of the control group. This is not a clinically relevant difference.
These results were pooled in a meta-analysis. The pooled RR from these three studies for incidence of respiratory depression was 1.07 (95% CI 0.85 to 1.35), in favor of the control group (Figure 2). This is not a clinically relevant difference.
Figure 2. Incidence of respiratory depression for supplemental oxygen versus no supplemental oxygen

Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Additionally, Deitch (2011) reported that in the intervention group, 10/59 (17%) patients had both respiratory depression and hypoxia, compared to 19/58 (33%) in the control group. The RR is 0.52 (95% CI 0.26 to 1.02), in favor of the intervention group. This is a clinically relevant difference. In the intervention group 20/59 (24%) experienced respiratory depression, but no hypoxia, compared to 9/58 (16%) in the control group. The RR is 2.18 (95%CI 1.09 to 4.39), in favor of the control group. This is a clinically relevant difference.
Level of evidence of the literature
The level of evidence regarding respiratory depression came from RCTs and started as high. The level of evidence was downgraded by three levels because of risk of bias (inadequate blinding, lack of intention to treat analysis; -1), inconsistency (conflicting results and inconsistency in definition of respiratory depression; -1) and imprecision (wide 95% confidence intervals crossing the border for clinical relevance; -1) resulting in very low.
Adverse events
Cardiac arrhythmia
Deitch (2008) reported that 1/56 (2%) patient in the intervention group experienced a transient arrhythmia compared to 0/54 (0%) in the control group. The RD is 0.02 (95% CI -0.03 to 0.07), in favor of the control group.
Deitch 2011 reported that 0/59 (0%) patients from the intervention group and 0/58 (0%) patients from the control group experiences an arrhythmia.
There were no clinically relevant differences for cardiac arrhythmia.
Other adverse events
Arawaka (2013) that reported 0/35 (0%) of patients in the intervention group and 0/35 (0%) patients in the control group experienced any cardiopulmonary or endoscopic complications that required medical intervention.
Three studies measured the incidence of hypotension, bradycardia and vomiting during the procedure.
Deitch (2007) reported that 0/44 (0%) patients in the intervention group and 0/36 (0%) patients in the control group experienced any adverse events. The RD is 0.00 (95% CI -0.05 to 0.05), indicating there is no difference between the two groups.
Deitch (2008) reported that 3/56 (5%) patients in the intervention group experienced mild hypotension, compared to 3/54 (6%) patients in the control group. The RD is 0.00 (95% CI -0.09 to 0.08), indicating there is no difference between the two groups.
Deitch (2011) reported adverse events in 2/59 (3%) patients in the intervention group experienced an adverse event (1 patient had bradycardia of 40 beats/min lasting for 5 minutes, 1 patient experienced hypotension 80/50 mmHg), compared to 1/58 (2%) patient in the control group (hypotension 85/40 mmHg). The RD is 0.02 (95%CI -0.04 to 0.07), in favor of the control group. This is not a clinically relevant difference.
Level of evidence of the literature
The level of evidence regarding adverse events came from RCTs and started as high. The level of evidence was downgraded by two levels because of risk of bias (concealment of allocation unclear, inadequate blinding, lack of intention to treat analysis; -2) and imprecision (low number of events; -1) resulting in a very low level of evidence.
Interventions
Airway maneuvers
Deitch (2007), Deitch (2008) and Deitch (2011) all reported that no patients in the intervention group (0%) or control group (0%) were intubated.
Respiratory support
Deitch (2007) reported that 0/44 (0%) patient in the intervention group and 0/36 (0%) in the control group required assisted ventilation.
Deitch (2008) reported that 1/56 (2%) patient in the intervention group received a period of assisted ventilation, compared to 0/54 (0%) in the control group. The RD is 0.02 (95% CI
-0.03 to 0.07), in favor of the control group.
Deitch (2011) reported that 0/59 (0%) patient in the intervention group received a period of assisted ventilation, compared to 1/58 (2%) in the control group (for 2 minutes). The RD is
-0.02 (95% CI -0.06 to 0.03), in favor of the intervention group.
There were no clinically relevant difference for respiratory support.
Other interventions - Fluids
Deitch (2011) reported that 1/59 (2%) patients of the intervention group received intravenous fluids, compared to 1/58 (2%) patient in the control group. The patient from the intervention group (80/50 mmHg) responded to these fluids in 10 minutes and the patient from the control group (85/40 mmHg) responded in 5 minutes. The RR for intravenous fluids is 0.98 (95% CI 0.06 to 15.35), in favor of the intervention group. This is not a clinically relevant difference.
Other interventions - Additional dose of flunitrazepam or meperidine
Arawaka (2013) reported that 3/35 (9%) patients from the intervention group and 2/35 patients (6%) from the control group received a supplemental dose of flunitrazepam for abdominal pain or discomfort. None of the patients received a supplemental dose of meperidine. The RR for a supplemental dose of flunitrazepam is 1.50 (95% CI 0.27 to 8.43), in favor of the control group (Figure 3). This is a clinically relevant difference.
Figure 3. An additional dose of flunitrazepam or meperidine for supplemental oxygen versus no supplemental oxygen
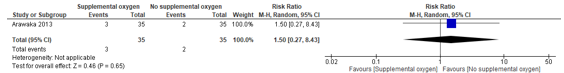
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Other interventions - Admission
Deitch (2007) reported that 0/44 (0%) patient in the intervention group and 0/36 (0%) patient in the control group required admission.
Deitch (2008) reported that 1/56 (2%) patient in the intervention group was admitted for observation, compared to 0/54 (0%) in the control group. The RD is 0.02 (95% CI -0.03 to 0.07), in favor of the control group.
Deitch (2011) reported that 0/59 (0%) patient in the intervention and 0/58 (0%) patient in the control group required admission.
There were no clinically relevant differences for admission.
Level of evidence of the literature
The level of evidence regarding interventions came from RCTs and started as high. The level of evidence was downgraded by three levels because of risk of bias (concealment of allocation unclear, inadequate blinding and lack of intention to treat analysis; -1) and imprecision (low number of events and crossing both borders for clinical relevance; -2) resulting in a very low level of evidence.
Safety
Physicians identification of respiratory depression
Two studies evaluated the ability of the physician to identify respiratory depression in patients that became hypoxic.
Deitch (2007) reported that physicians could identify respiratory depression in 3/6 (50%) patients that became hypoxic in the intervention group, compared to 5/5 patients (100%) in the control group. The RD is -0.50 (95% CI 0.25 to 1.19), in favor of the control group. Physicians failed to recognize respiratory depression in any patient who did not become hypoxic. In the intervention group, physicians could identify respiratory depression in 0/14 (0%) patients that did not become hypoxic in the intervention group, compared to 0/14 (0%) patients in the control group.
Deitch (2008) reported that physicians identified the presence of respiratory depression in 10/10 (100%) patients that became hypoxic in the intervention group, compared to 13/15 (87%) patients in the control group. The RD is 0.13 (95% CI -0.08 to 0.35), in favor of the intervention group. This is not a clinically relevant difference. In the 2 patients whose hypoxic events went unrecognized, the duration of hypoxia was brief (<60 seconds) and the severity was mild (lowest oxygen saturation was 88%).
Physicians recognized respiratory depression in 1/20 (5%) patients who did not become hypoxic in the intervention group, compared to 0/7 (0%) patients in the control group. The RD is 0.05 (95%CI -0.15 to 0.25), in favor of the intervention group. This is not a clinically relevant difference.
These results were pooled in a meta-analysis. The RD for the identification of respiratory depression in patients that became hypoxic is -0.16 (96%CI -0.80 to 0.49), in favor of the control group (Figure 4).
The RD for the identification of respiratory depression in patients that did not become hypoxic is 0.01 (95%CI -0.09 to 0.12), in favor of the intervention group (Figure 4).
In total, the pooled RD for physicians identification of respiratory depression either with or without hypoxia is -0.01 (95%CI -0.17 to 0.16) (Figure 4). This is not a clinically relevant difference.
Figure 4. Physicians identification of respiratory depression either with or without hypoxia for supplemental oxygen versus no supplemental oxygen
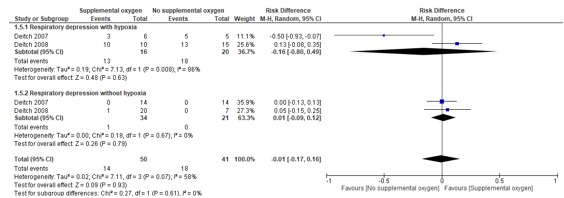
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Level of evidence of the literature
The level of evidence regarding safety came from RCTs and started as high. The level of evidence was downgraded by three levels because of risk of bias (inadequate blinding; -1) and imprecision (low number of events, wide 95% confidence interval crossing border for clinical relevance; -2) resulting in a very low level of evidence.
Search and select
A systematic review of the literature was performed to answer the following question:
What is the effect of supplemental oxygen compared to no supplemental oxygen on complications, adverse events and airway maneuvers in patients undergoing Procedural Sedation and Analgesia (PSA)?
P: patients undergoing PSA
I: supplemental oxygen
C: no supplemental oxygen
O: complications, adverse events, interventions, safety
Relevant outcome measures
The guideline development group considered incidence of complications as a critical outcome measure for decision making; and adverse events and interventions as an important outcome measure for decision making.
A priori, the working group did not define the outcome measures listed above but used the definitions used in the studies.
The working group defined the GRADE standard limit of 25% difference for dichotomous outcomes (RR <0.8 or >1.25) and 10% for continuous outcomes as a minimal clinically (patient) important difference.
Search and select (Methods)
The databases [Medline (via OVID) and Embase (via Embase.com)] were searched with relevant search terms until 3rd of September 2020. The detailed search strategy is depicted under the tab Methods. The systematic literature search resulted in 543 hits. Studies were selected based on the following criteria: RCTs comparing outcomes of patients undergoing PSA with or without supplemental oxygen, published after 2005. A total of 35 studies were initially selected based on title and abstract screening. After reading the full text, 30 studies were excluded (see the table with reasons for exclusion under the tab Methods), and 5 studies were included.
Results
A total of five RCTs were included in this literature summary.
- Arakawa, H., Kaise, M., Sumiyama, K., Saito, S., Suzuki, T., & Tajiri, H. (2013). Does pulse oximetry accurately monitor a patient's ventilation during sedated endoscopy under oxygen supplementation. Singapore Med J, 54(4), 212-215.
- Deitch, K., Chudnofsky, C. R., & Dominici, P. (2007). The utility of supplemental oxygen during emergency department procedural sedation and analgesia with midazolam and fentanyl: a randomized, controlled trial. Annals of emergency medicine, 49(1), 1-8.
- Deitch, K., Chudnofsky, C. R., & Dominici, P. (2008). The utility of supplemental oxygen during emergency department procedural sedation with propofol: a randomized, controlled trial. Annals of emergency medicine, 52(1), 1-8.
- Deitch, K., Chudnofsky, C. R., Dominici, P., Latta, D., & Salamanca, Y. (2011). The utility of high-flow oxygen during emergency department procedural sedation and analgesia with propofol: a randomized, controlled trial. Annals of emergency medicine, 58(4), 360-364.
- 360-364.
- Rozario, L., Sloper, D., & Sheridan, M. J. (2008). Supplemental oxygen during moderate sedation and the occurrence of clinically significant desaturation during endoscopic procedures. Gastroenterology Nursing, 31(4), 281-285.
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3
|
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
|
Arakawa 2013 |
Type of study: RCT
Setting and country: Japan
Funding and conflicts of interest: Not described |
Patients who underwent sedated diagnostic colonoscopy
Inclusion criteria:
Exclusion criteria:
colonoscopy
drugs
N total at baseline: 70 Intervention: 35 Control: 35
Important prognostic factors2: age ± SD: I: 54.4y ± 10.8 C: 59.2y ± 12.1
Sex, male: I: 24/35 (69%) C: 22/35 (63%)
Baseline etCO2: I: 37.1 ± 2.6 mmHg C: 37.0 ± 2.6 mmHg
BMI (kg/m2) I: 24.1 ± 4.0 C: 22.7 ± 3.2
ASA status (no.) Class I I: 23/35 (67%) C: 24/35 (69%) Class II O: 12/35 (34%) C: 11/35 (31%)
Groups comparable at baseline.
|
Sedated diagnostic colonoscopy with oxygen supplementation
Administered routinely prior to and during the procedure at a rate of 2 L/min
Procedure time (min) I: 19.2 ± 7.8 Dosage Flunitrazepam (mg) I: 0.3 ± 0.1 Meperidine (mg) I: 35 OAA/S score I: 3.6 ± 0.6
|
Sedated diagnostic colonoscopy breathing room air
Room air was breathed during the procedure; oxygen supplementation was administered if SpO2 fell below 90% for more than 20-second intervals during the procedure
Procedure time (min) C: 22.3 ± 9.3 Dosage Flunitrazepam (mg) C: 0.3 ± 0.1 Meperidine (mg) C: 35 OAA/S score C: 3.7 ± 0.7
|
Length of follow-up: n.a.; measurement during the procedure
Incomplete outcome data: I: 1/35 (3%) C: 2/35 (6%) Reasons: All not completed colonoscopy due to abdominal discomfort and pain.
|
Mean±sd are reported for continues variables
Cardiopulmonary or endoscopic complications that required medical intervention I: 0/35 (0%) C: 0/35 (0%)
A supplemental dose flunitrazepam for abdominal pain or discomfort: I: 3/35 (9%) C: 2/35 (6%) Meperidine: I: 0/35 (0%) C:0/35 (0%)
etCO2 during procedure I: 44.9 ± 4.1 mmHg C: 44.1 ± 4.0 mmHg p = 0.4
Increase in etCO2 at its peak from baseline I: 7.7 ± 3.3 mmHg C: 7.0 ± 2.6 mmHg
SpO2 before oxygen administration I: 97.9% ± 1.4%, C: nearly comparable, data not shown (p = 0.21)
Baseline SpO2 value (after 3 min of oxygen suppl. in I-group) I: 99.2% ± 1.2% C: 97.3% ± 2.1% SpO2 after sedation No data reported; “SpO2 decreased as alveolar hypoventilation developed in each group” SpO2 at peak etCO2 I: 98.6% ± 1.4% C: 93.1% ± 2.9% p < 0.001 Reduction in SpO2 from baseline at peak etCO2 I: 0.5% ± 1.1% C: 4.1% ± 3.1% p < 0.001)
Respiratory rate; at baseline & time peak etCO2 No differences between groups
|
|
|
|
Deitch, 2007 |
Type of study: RCT
Setting and country: Oct 1, 2004 - March 1, 2005; Emergency department at Albert Einstein Medical Center, a Level I trauma center; USA
Funding and conflicts of interest: ‘The authors report this study did not receive any outside funding or support.’ |
Patients receiving midazolam and fentanyl for emergency department procedural sedation and analgesia
Inclusion criteria:
Exclusion criteria:
pulmonary disease
study drugs
N total at baseline: 80 Intervention: 44 Control: 36
Important prognostic factors2: Age median (range): I: 36y (Range 2–77) C: 32y (Range 13–68) Sex, male: I: 20/44 (45%) C: 12/36 (33%) Children by age: I:0–2y: 0 2–6y: 2 12–18y: 4 C: 0–2y: 0 2–6y: 0 12–18y: 6
Weight, kg, median (range) I: 77 (18–109) C: 80 (45–147) Abscess incision and drainage (%) I: 26/44 (59%) C: 24/36 (67%) Fracture/joint reduction (%) I: 17/44 (39%) C: 10/36 (28%) Other procedures (Tube thoracostomy, lumbar puncture, and haemorrhoid thrombectomy) (%) I: 1/44 (2%) C: 2/36 (5%)
Groups comparable at baseline. |
Emergency department PSA using midazolam and fentanyl with supplemental oxygen (delivered by nasal cannula at 2L/min)
Median (range) Fentanyl dose, µg 150.0 (25–400) Midazolam dose, mg 5.0 (0.5–10.0) Ramsey scores (90 s after the last dose of preprocedural medication) 4 (2–5)
|
Emergency department PSA using midazolam and fentanyl, breathing room air (delivered by nasal cannula at 2L/min)
Median (range) Fentanyl dose, µg 100.0 (50–400) Midazolam dose, mg 4.0 (1.0–8.0) Ramsey scores (90 s after the last dose of preprocedural medication) 4 (3–5) |
Length of follow-up: n.a.; outcomes during procedure
Loss-to-follow-up/ Incomplete outcome data: I: 0/44 (0%) C: 0/36 (0%) |
Hypoxia Oxygen saturation <90% I: 6/44 (14%) C: 5/36(14%) P=0.97; effect size 0%; 95% CI –15% to 15%
Respiratory depression N meeting 1 or more criteria* for RD I: 20/44 (45%) C: 19/36 (52%) N meeting both oxygen saturation and ETCO2 criteria* for RD I: I 5/44 (11%) C: 2/36 (6%) N meeting only oxygen saturation criteria* for RD I: 1/44 (2%) C: 3/36 (8%) N meeting only ETCO2 criteria* for RD I: 14/44 (32%)
Physicians identification of respiratory depression in patients that became hypoxic I: 3/6 (50%) C: 5/5 (100%)
Duration of hypoxia, seconds, median (range) Oxygen saturation decrease <90% I: 60s (1-120) = RD comparable Only ETCO2 criteria for respiratory depression I: 417s (3-600) C: 180s (60-540) = RD longer in the patients receiving supplemental oxygen
Adverse events bradycardia, or vomiting or required assisted ventilation, intubation, or admission I: 0/44 (0%) C: 0/36 (0%)
|
Respiratory depression defined as:
These criteria were considered present if they occurred any time during the procedure, regardless of their duration. When capnographic evidence of respiratory depression occurred, the patient’s vital signs, oxygen saturation, and ETCO2 level were recorded. |
|
Deitch, 2008 |
Type of study: RCT
Setting and country: Nov 2005 - Oct 2006: Emergency department at Albert Einstein Medical Center, a Level I trauma center; USA
Funding and conflicts of interest: the authors have stated no conflicts of interest were present and no financial relationships related to the subject of this article were declared |
Adult study patients receiving propofol for emergency department procedural sedation.
Inclusion criteria:
facilitate a painful procedure
Exclusion criteria:
pulmonary disease
study drugs
N total at baseline: 110 Intervention: 56 Control: 54
Important prognostic factors2: Age, median (range) I: 37y (19-86) C: 37y (18-75) Sex, male: I: 23/56 (41%) C: 25/54 (46%) Weight, kg, median (range) I: 86 kg (49-145) C: 79 kg (43-134)
Abscess incision and drainage (%) I: 37/56 (66%) C: 32/54 (59%) Fracture reduction (%) I: 6/56 (10%) C: 9/54 (17%) Joint reduction (%) I: 13/56 (23%) C: 7/54 (13%)
Groups comparable at baseline.
|
Emergency department procedural sedation using propofol with supplemental oxygen (delivered at 3 L per minute by nasal cannula)
Initial propofol dose (mg/kg) Mean (SD) I: 1.19 (SD 0.32) Total propofol dose (mg/kg) I: 1.71 (SD 0.66) Ramsey scores 90 s after the last dose of preprocedure Medication, median (range) I: 5 (2-6) Time from first dose of medication to return to baseline alertness, min (range) I: 10 min (3-28)
|
Emergency department procedural sedation using propofol, breathing room air (delivered at 3 L per minute by nasal cannula)
Initial propofol dose (mg/kg) Mean (SD) C: 1.33 (SD 0.32) Total propofol dose (mg/kg) C: 1.99 (SD 0.79) Ramsey scores 90 s after the last dose of preprocedure Medication, median (range) C: 5 (2-6) Time from first dose of medication to return to baseline alertness, min (range) C: 12 min (4-35) |
Length of follow-up: n.a.; outcomes during procedure
Loss-to-follow-up/ Incomplete outcome data: I: 0/56 (0%) C: 0/54 (0%) |
Hypoxia Oxygen saturation <93% I: 10/56(18%) C: 15/54(28%) P=0.3; effect size 10%; 95% CI –24% to 7%)
Respiratory depression N meeting 1 or more criteria* for RD I: 30/56 (53%) C: 22/54 (40%) N meeting both oxygen saturation and ETCO2 criteria* for RD I: 3/56 (5%) C: 6/54 (11%) N meeting only oxygen saturation criteria* for RD I: 7/56 (12%) C: 9/54 (16%) N meeting only ETCO2 criteria* for RD I: 20/56 (36%)
Physicians identification of respiratory depression in patients that became hypoxic I: 10/10 (100%) C: 13/15 (87%)
ETCO2 Changes N patients (number patients that became hypoxic) ETCO2 >50 mm Hg I: 1 (0) C: 2 (1) ETCO2 >10 mm Hg above baseline I: 4 (0) C: 2 (2) ETCO2 >10 mm Hg below baseline I: 19 (3) C: 11 (4) Loss of the ETCO2 Waveform I: 1 (0) C: 1 (0)
Adverse events bradycardia, or vomiting or required assisted ventilation, intubation, or admission I: 4/56 (7%), 1 patient received a period of assisted ventilation, 3 patients developed mild hypotension C: 3/54 (6%), 3 patients developed mild hypotension
|
Respiratory depression defined as:
These criteria were considered present if they occurred any time during the procedure, regardless of their duration.
The mean initial and total propofol doses were slightly higher in the room air group, which may have contributed to the difference in the incidence of hypoxia observed between the 2 groups.
|
|
|
Deitch, 2011 |
Type of study: RCT, placebo-controlled
Setting and country: Jan 2009 -Nov 2010: Emergency department at Albert Einstein Medical Center, a Level I trauma center; USA
Funding and conflicts of interest: The authors report this study received funding from the Albert Einstein Society and the Pennsylvania Department of Health National Tobacco Settlement.
|
Adult patients receiving propofol procedural sedation
Inclusion criteria:
Exclusion criteria:
pulmonary disease
study drugs
N total at baseline: Intervention: 59 Control: 58
Important prognostic factors2: Age, median (IQR) I: 37y (27-55) C: 32y (21.5-45.5) Sex, male: I: 24/59 (41%) C: 30/58 (52%) Weight, kg, median (IQR) I: 77 kg (68-95) C: 86 kg (74-95)
Abscess incision and Drainage, % (95% CI) I: 42% (29-55) C: 51% (38-64) Fracture reduction, % (95%CI) I: 11% (3-19) C: 17% (7-27) Joint reduction, % (95%CI) I: 47% (34-60) C: 32% (20-44)
Groups comparable at baseline.
|
Procedural sedation using propofol with high-flow oxygen (administered at 15 L per minute by a nonrebreather mask)
Initial propofol dose (mg/kg) Median (IQR) I: 1.0 (1.0-1.0) Total propofol dose (mg/kg) I: 1.6 (1.1-2.8) Ramsey scores 90 s after the last dose of preprocedure Medication, median (IQR) I: 4 (2-5) Time from first dose of medication to return to baseline alertness, min (IQR) I: 14 min (10-19)
|
Procedural sedation using propofol with room air (administered at 15 L per minute by a nonrebreather mask
Initial propofol dose (mg/kg) Median (IQR) I: 1.0 (1.0-1.0) Total propofol dose (mg/kg) I: 1.5 (1.0-2.0) Ramsey scores 90 s after the last dose of preprocedure Medication, median (IQR) I: 4 (2-5) Time from first dose of medication to return to baseline alertness, min (IQR) I: 15 min (11-23)
|
Length of follow-up: n.a.; outcomes during procedure Loss-to-follow-up/ Incomplete outcome data: I: 0/59 (0%) C: 0/58 (0%)
|
Outcome measures and effect size (include 95%CI and p-value if available): Hypoxia C: 24/58 (41%)
Respiratory depression mm Hg, an absolute increase or decrease from baseline of >10%, or a loss of waveform for > 15 seconds I: 30/59 (51%) C: 28/58 (48%) I: 10/59 (17%) C: 19/58 (33%)
Respiratory depression without hypoxia I: 1/59 (2%) No hypoxia + physician intervention C: 5/58 Effect size 8%; 95% CI -4% to 20% I: 42/59 of hypotension to 80/50 mm Hg that responded to intravenous fluids in 10 minutes hypotension to 85/40 mm Hg that responded to intravenous fluids in 5 minutes. |
|
|
|
Rozario, 2008 |
Type of study: RCT
Setting and country: October 2005 - gastroenterology laboratory of the Inova Fairfax Hospital, Falls Church, Virginia, USA
Funding and conflicts of interest: Not reported |
Inclusion criteria:
procedure
cleared by primary physician to undergo endoscopy
Exclusion criteria:
oxygen therapy
rhythmias
N total at baseline: 389 Intervention: 194 Control: 195
Important prognostic factors2: Age, mean (95% CI): I: 56y (54-58) C: 56y (55-58) Sex, male: I: 79/194 (41%) C: 84/195 (43%) BMI, mean (95% CI) C: 25.9 (25.0-26.8) ASA-class I, % ASA-class III, % C: 123 (130-126) mmHg C: 69 (67-71) mmHg Pulse, mean (95%CI) Groups comparable at baseline, except for systolic and diastolic blood pressure – higher in intervention group. |
Moderate sedation for an endoscopic procedure in the gastroenterology laboratory with supplemental oxygen (2 L/min via nasal cannula).
|
Moderate sedation for an endoscopic procedure in the gastroenterology laboratory without supplemental oxygen. |
Length of follow-up: n.a.; outcomes during procedure
Loss-to-follow-up/ Incomplete outcome data: I: 0/194 (0%) C: 0/195 (0%)
|
Desaturation |
|
|
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors [(potential) confounders]
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
Risk of bias table
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Arakawa 2013 |
Not described
“Patientswere randomised into two study groups” |
Unclear |
Unlikely
Computerized recording of data not subjective |
Unlikely
Computerized recording of data not subjective
“the capnographic data were monitored by an independent physician at the bedside and were not accessible to the endoscopists.”
|
Unclear
Unclear whether knowledge about treatment could have influenced procedure. However, data recorded by computer linked to monitor
“the capnographic data were monitored by an independent physician at the bedside and were not accessible to the endoscopists.” |
Unclear
No trial register reported; outcomes described in methods are reported |
Unlikely
Unclear whether patients that did not complete the colonoscopy are included in results. Due to comparable drop-out rate (3 vs. 6%) no bias expected |
Unlikely
Unclear whether patients that did not complete the colonoscopy are included in results. Due to comparable drop-out rate (3 vs. 6%) no bias expected |
|
Deitch, 2007 |
Computerized
“Randomization was done with a computerized randomization table” |
Unlikely
Sequence not sealed, but unlikely that order of patients could be changed in emergency setting.
“Patients were assigned to their respective groups sequentially down a numbered list.” |
Unlikely
Patient not aware of study arm |
Unlikely
“Members of the clinical staff were blinded to the type of gas being administered and to the ETCO2 data, and they were unaware that their ability to recognize respiratory depression was being evaluated.” |
Unclear
“Research associate monitored and administrated outcomes. The research associates were not blinded to the purpose of this study, which could have resulted in bias during data collection. All research associates participating in this study were physicians who received training specifically directed at identifying a physician intervention for respiratory depression.” |
Unclear
No trial register reported; outcomes described in methods are reported |
Unlikely
All patient enrolled and randomized are included in the analysis |
Unlikely
No randomization reported for 3 patients that were enrolled but excluded; therefore all randomized patients seem to be included in the analysis per intention-to-treat protocol |
|
Deitch, 2008 |
Computerized
“Randomization was done with a computerized randomization table” |
Unlikely
Sequence not sealed, but unlikely that order of patients could be changed in emergency setting.
“Patients were assigned to their respective groups sequentially down a numbered list.” |
Unlikely
Patient not aware of study arm |
Unlikely
“To ensure that the treatment team was blinded to the type of gas being administered, the gases were delivered from one of 2 identical D-tanks marked “A” and “B”.” |
Unclear
“Research associate monitored and administrated outcomes. The research associates were not blinded to the purpose of this study, which could have resulted in bias during data collection. All research associates participating in this study were physicians who received training specifically directed at identifying a physician intervention for respiratory depression.” |
Unclear
No trial register reported; outcomes described in methods are reported |
Unlikely
All patient enrolled and randomized are included in the analysis |
Unlikely
No randomization reported for 2 patients that were enrolled but excluded; therefore all randomized patients seem to be included in the analysis per intention-to-treat protocol |
|
Deitch, 2011 |
Computerized
“We used a computer-generated, concealed randomization schedule to assign patients to receive either high-flow oxygen or room air,..” |
Unlikely
“We used a computer-generated, concealed randomization schedule” |
Unlikely
Patient not aware of study arm |
Unlikely
“The treatment team was blinded by delivering the gases with one of 2 identical-appearing D-tanks marked “A” and “B.” |
Unlikely
“Blinded, separate research physicians enrolled patients and recorded data. […] They had no clinical responsibilities and were instructed to observe and document but not interact with the clinical team or influence their actions.” |
Unclear
Trial not registered; reported that trial was conducted in accordance to protocol
“The trial was not preregistered but was conducted and analyzed in accordance with its original protocol.” |
Unclear
15 patients excluded after enrolment (n=4 protocol violation; n=11 ≥35% data loss, mostly due to patient movement or blood pressure cuff insufflations. Unclear whether treatment could influence these factors. Not described to which treatment group these patients were allocated |
Unclear
15 patients excluded, data not included in analysis and not imputed. However, no cross-over between treatment groups described |
|
Rozario, 2008 |
Computerized
“To assign subjects to groups, a computerized block randomization scheme was used, which ensured equal numbers of subjects in each group after every 10 allocations, up to 400 subjects.” |
Unclear
“The nurse researcher was randomly assigned to a procedure room and the patients scheduled in that room for the day were all potential study participants if they met the inclusion/exclusion criteria.” |
Unclear
Study was nonblinded
|
Unclear
Study was nonblinded
|
Unclear
|
Unclear
No trial register reported; if a patient experiences desaturation, oxygen flow was administered. It was not described how many patients actually received additional oxygen and it was not reported if any further intervention or admission was needed. |
Unlikely
All patient enrolled and randomized are included in the analysis |
Unlikely
All patient enrolled and randomized are included in the analysis |
Table of excluded studies
|
Author and year |
Reason for exclusion |
|
Abouzgheib 2019 |
C does not meet PICO |
|
Dobronte 2013 |
Not in Englisch or Dutch |
|
Douglas 2018 |
C does not meet PICO |
|
Dumonceau 2010 |
No RCT |
|
Eugene 2020 |
C does not meet PICO |
|
Ferreira 2016 |
I and C do not meet PICO |
|
Ghio 2007 |
n = 6 |
|
Homfray 2018 |
No RCT |
|
Hong 2015 |
I and C do not meet PICO |
|
Horiuchi 2009 |
No RCT |
|
Ishiwata 2018 |
I and C do not meet PICO |
|
Kalling 2007 |
No RCT |
|
Keidan 2008 |
P does not meet PICO; study in children |
|
Khiani 2009 |
No RCT |
|
Labaste 2016 |
No RCT |
|
Lin 2019 |
C does not meet PICO |
|
Mora 2014 |
Not in Englisch or Dutch |
|
Qin 2017 |
C does not meet PICO |
|
Restrepo 2014 |
Does not meet PICO |
|
Riccio 2019 |
C does not meet PICO |
|
Sago 2015 |
I and C do not meet PICO |
|
Schumann 2016 |
No RCT |
|
Siddiqui 2013 |
No RCT |
|
Sivilotti 2010 |
I and C do not meet PICO |
|
Smally 2007 |
Does not meet PICO |
|
Takakuwa 2018 |
No RCT |
|
Teng 2019 |
C does not meet PICO |
|
Thomson 2017 |
No RCT |
|
Tohda 2006 |
I and C do not meet PICO |
|
Vargo 2007 |
Does not meet PICO |
Beoordelingsdatum en geldigheid
Publicatiedatum : 23-05-2024
Beoordeeld op geldigheid : 23-05-2024
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij sedatie en/of analgesie bij volwassen patiënten.
Werkgroep
- Prof. dr. B. Preckel (voorzitter), anesthesioloog, Amsterdam UMC locatie AMC, NVA
- dr. C.R.M. Barends, anesthesioloog, UMCG, NVA
- L.R.M. Braam, BSc. Sedatie Praktijk Specialist, Catharina Ziekenhuis, NVAM
- drs. R. Brethouwer, abortusarts, Beahuis & Bloemenhovekliniek Heemstede, NGvA
- dr. J.M. van Dantzig, cardioloog, Catharina Ziekenhuis, NVVC
- drs. V.A.A. Heldens, anesthesioloog, Maxima MC, NVA
-
dr. C. Heringhaus, SEH-arts/anesthesioloog, LUMC (t/m 12-2022), Medisch manager Hyperbare Zuurstoftherapie Goes, MCHZ (vanaf 01-2023), NVSHA
- T. Jonkergouw, MA. Adviseur Patiëntbelang, Patiëntenfederatie Nederland (tot april 2023)
- Broere, M. Junior beleidsadviseur, Patiëntenfederatie Nederland (vanaf april 2023)
- dr. M. Klemt-Kropp, MDL-arts, Noordwest Ziekenhuisgroep, NVMDL
- drs. B.M.F. van der Leeuw, anesthesioloog, Albert Schweitzer ziekenhuis, NVA
- S. Reumkens, MSc. Physician Assistant, Diakonessenhuis, NAPA
Klankbordgroep
- drs. T.E.A. Geeraedts, radioloog, Erasmus MC, NVvR
- drs. J. Friederich, gynaecoloog, Noordwest Ziekenhuisgroep, NVOG
- dr. E.H.F.M. van der Heijden, longarts, Radboud UMC, NVALT
- drs. J. de Hoog, oogarts, Amsterdam UMC locatie AMC, NOG
- drs. A. Kanninga, arts voor verstandelijk gehandicapten, Cordaan, NVAVG
- drs. H.W.N. van der Pas, tandarts, UMC Utrecht, VMBZ
- dr. ir. C. van Pul, klinisch fysicus, Maxima MC, NVKF
- dr. R.J. Robijn, MDL-arts, Rijnstate, NVMDL
- drs. W.S. Segers, klinisch Geriater, Catharina Ziekenhuis, NVKG
- Prof. dr. A. Visser, hoogleraar geriatrische tandheelkunde, UMCG en Radboud UMC, KNMT
Met ondersteuning van:
- dr. L. Wesselman, adviseur, Kennisinstituut van Medisch Specialisten
- dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
- drs. I. van Dusseldorp, senior literatuurspecialist, Kennisinstituut van Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Preckel |
Anesthesioloog, hoogleraar anesthesiologie (in het bijzonder veiligheid in het perioperatieve proces) Amsterdam Universitair Medische Centra locatie AMC |
Onbetaalde nevenwerkzaamheden – commissie-werkzaamheden:
Lid Patient Safety and Quality Committee of the European Society of Anesthesiologists;
Lid Patient Safety Committee van de World Federation of Societies of Anesthesiologists;
Lid Raad Wetenschap en Innovatie van de Federatie Medisch Specialisten FMS;
Lid Commissie Wetenschap & Innovatie van de Nederlandse Vereniging voor Anesthesiologie NVA
Representative Council European Association of Cardiothoracic Anesthesiology and Intensive Care (EACTAIC)
Hoger leidinggevend personeel (penningmeester) van de “Stichting ter bevordering van de wetenschap en opleiding in de anesthesiologie”;
|
Research grants: European Society of Anesthesiology and Intensive Care ESAIC ZonMw NovoNordisk Netherland
Advisory board Sensium Healthcare United Kingdom
Geen van de gemelde belangen heeft relatie met het onderwerp van het advies/de richtlijn
|
Geen actie vereist |
|
Barends |
Anesthesioloog in het Universitair Medisch Centrum Groningen |
Geen |
Geen |
Geen actie vereist |
|
Braam |
Sedatiepraktijkspecialist Catharina Ziekenhuis Eindhoven |
Lid sedatie commissie NVAM (onbetaald) |
Geen |
Geen actie vereist |
|
Brethouwer |
Abortusarts te Beahuis & Bloemenhovekliniek (0,56 fte) en SAA (0,22 fte), Medisch coördinator Beahuis&Bloemenhovekliniek (0,33 fte) |
Penningmeester van het Nederlands Genootschap van Abortusartsen (onbetaald) Voorzitter landelijke werkgroep PSA van het NGvA (onbetaald) Bestuurslid van FIAPAC, een Europese abortus organisatie (onbetaald) |
Geen |
Geen actie vereist |
|
Broere |
Junior beleidsadviseur Patiëntenbelang - fulltime |
geen |
geen |
Geen actie vereist |
|
van Dantzig |
Cardioloog vrij gevestigd, Catharina Ziekenhuis 100% |
Lid Plenaire Visitatie Commissie NVVC (onbetaald) |
Op onze afdeling wordt extern gefinancierd onderzoek uitgevoerd maar niet op het gebied van de werkgroep. |
Geen actie vereist |
|
Heldens |
Anesthesioloog Maxima MC |
Geen |
Geen |
Geen actie vereist |
|
Heringhaus |
Vanaf 01-2023 Medisch manager Hyperbare Zuurstoftherapie Goes, MCHZ
t/m 12-2022 SEH-arts KNMG |
Trainingen voor verschillende onderwerpen gerelateerd aan acute zorg, hyperbare geneeskunde, PSA |
Geen |
Geen actie vereist |
|
Jonkergouw |
Junior beleidsadviseur - Patiëntenfederatie Nederland - 32 tot 36 uur per week |
Vrijwilliger activiteiten - Diabetes Vereniging Nederland - Zeer incidenteel |
Geen |
Geen actie vereist |
|
Klemt-Kropp |
MDL-arts, Noordwest Ziekenhuisgroep Alkmaar - Schagen - Den Helder (0.9 fte) |
Secretaris Concilium Gastroenterologicum, NVMDL tot 11 april 2022 (niet betaald) Voorzitter PSA commissie NVMDL (niet betaald) Docent Teach the Teacher AUMC en Noordwest Ziekenhuisgroep cursussen voor aios en medisch specialisten (betaalde functie, ongeveer 40 Std. per jaar)
Voorzitter Stichting MDL Holland-Noord (KvK 56261225) vanaf okt. 2012 t/m 31-12-2019. De stichting heeft in de laatste 3 jaar grants ontvangen van de farmaceutische industrie en van de farmaceutische industrie gesponsorde onderzoeken gefaciliteerd:
1. Ondersteuning optimalisering van zorg voor IBD-patiënten. Therapeutic drugmonitoring en PROMs bij patiënten met IBD. Zorgverbetertraject. PhD student, looptijd van 2015 tot op heden. Tot 2018 grant van Dr. Falk Pharma. vanaf 2018 grant van Janssen Cilag Geen relatie met sedatie 2.Retrieval of patients chronically infected with Hepatitis B or Hepatitis C in Northern Holland. Afgesloten 2018. Project gefinancierd met grant van Gilead. Geen relatie met sedatie 3. SIPI. Screening op Infectieuze aandoeningen in Penitentiaire Inrichtingen. Project gefinancierd met grants van AbbVie, MSD en Gilead. Project begin 2019 afgesloten. Geen relatie met sedatie 4. 3DUTCH trial. Een observationeel onderzoek naar de effectiviteit van een behandeling van chronische hepatitis C met een combinatie van de antivirale middelen ombitasvir - paritaprevir /ritonavir, ± dasabuvir, ± ribavirine. Sponsor AbbVie. Studie afgesloten Jan. 2018 Geen relatie met sedatie 5. Remsima switch IFX9501 - An open-label, multicenter, non- inferiority monitoring program to investigate the quality of life, efficacy and safety in subjects with Crohn’s Disease (CD), Ulcerative Colitis (UC) in stable remission after switching from Remicade® (infliximab) to Remsima® (infliximab biosimilar) L016-048. Sponsor: Munipharma. Afgelsoten augustus 2018. Geen relatie met sedatie 6. NASH - NN9931-4296 Investigation of efficacy and safety of three dose levels of subcutaneous semaglutide once daily versus placebo in subjects with non-alcoholic steatohepatitis L016-061. Sponsor NovoNordisk. Studie afgesloten Feb. 2020. Geen relatie met sedatie 7. Randomized, Double-blind, Placebo-controlled, Parallel-group Efficacy and Safety Study of SHP647 as Induction Therapy in Subjects with Moderate to Severe Crohn's Disease (CARMEN CD 305). SHD-647-305. Sponsor Shire. Studie loopt sinds 2019. Geen relatie met sedatie 8. Randomized, Double-blind, Placebo-controlled, Parallel-group Efficacy and Safety Study of SHP647 as Maintenance Therapy in Subjects with Moderate to Severe Crohn's Disease (CARMEN CD 307). SHD-647-307. Sponsor Shire. Studie loopt sinds 2019. Geen relatie met sedatie 9. Estimating the prevalence of advanced liver fibrosis in a population cohort in care in Northern-Holland with the use of the non-invasive FIB-4 index. Grant van Gilead. Onderzoek afgesloten sept. 2019. Geen relatie met sedatie |
Incidenteel deelname aan advisory boards van de farmaceutische industrie (Gilead, Janssen Cilag, AbbVie: (hepatologische onderwerpen, vooral hepatitis C) Incidenteel voordrachten tijdens symposia gesponsord van de farmaceutische industrie (Gilead, AbbVie)
|
Geen actie vereist; meeste studies afgerond; nr. 1,7,8 lopen. Sponsoren (Dr. Falk Pharma & Shire) hebben geen relatie met sedatie. |
|
Reumkens |
Physician Assistant Anesthesiologie Radboud UMC |
Voorzitter vakgroep PA Anesthesiologie NAPA |
Geen |
Geen actie vereist |
|
Van der Leeuw |
Anesthesioloog |
Geen |
Geen |
Geen actie vereist |
|
Klankbordgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Robijn |
Mdl arts rijnstate ziekenhuis Arnhem |
Geen |
Geen |
Geen actie vereist |
|
Van der Heijden |
Longarts |
Voormalig Secretaris Sectie Interventie longziekten NVALT (onbezoldigd)
Lid Board of National Delegates European Association of Bronchology and International Pulmonology (namens NL, onbezoldigd)
|
Buiten het veld van deze richtlijn heeft mijn afdeling de afgelopen 3 jaar vergoedingen ontvangen voor de volgende activiteiten: - unrestricted research grants: Pentax Medical Europe, Philips, Astra Zeneca, Johnson&Johnson. - adviseur / consultant: Pentax Medical, Philips IGT, Johnson&Johnson. - spreker: Pentax Medical. |
Geen actie vereist |
|
Van der Pas |
Tandarts, UMC Utrecht |
Commissielid Horace Wells van de KNMT, stimulatie van de intercollegiale samenwerking bij tandheelkundige behandeling van bijzondere zorggroepen met farmacologische ondersteuning. Onbetaald. Voormalig commissielid Bijzondere Zorggroepen van de KNMT, toegankelijkheid van mondzorg voor kwetsbare zorggroep. Betaald via vacatiegelden. Gastdocent opleiding mondzorgkunde HU. Lezing mondzorg aan mensen met een verstandelijke beperking. Betaald. Gastdocent opleiding verpleegkundig-specialist GGZ. Lezing mondzorg in de geestelijke gezondheidszorg Betaald. Cursusleider lichte sedatie in de mondzorg, BT Academy. Meerdaagse cursus voor tandartsen en mondhygienisten om zich te bekwamen in lichte sedatie, m.n. training in de inhalatiesedatie met lachgas-zuurstof mengsel middels titratietechniek. Betaald. |
Geen |
Geen actie vereist |
|
De Hoog |
Oogarts in Amsterdam UMC (0,2 fte.) en Retina Operatie Centrum Amstelveen (0,4 fte.). Medisch manager Retina Operatie Centrum (0,2 fte.) |
Voorzitter Werkgroep Vitreoretinale Chirurgie Nederland (onbetaald) Lid redactieraad vaktijdschrift 'De Oogarts', uitgave van BPM-medica (onbezoldigd) Medeorganisator Eilanddagen (bijscholing uveïtis voor oogartsen, onbetaald) |
Geen |
Geen actie vereist |
|
Geeraedts |
Interventieradioloog Afdeling Radiologie en Nucleaire geneeskunde Erasmus Medisch Centrum, Rotterdam |
Geen |
Geen |
Geen actie vereist |
|
Van Pul |
Klinisch fysicus in Maxima Medisch Centrum |
Universitair Hoofd Docent aan de Technische Universiteit van Eindhoven (0,2 fte). Daar supervisor van PhD studenten bij HTSM (NWO-TTW gesubsidieerd) project waaraan ook een industriële partner deelneemt (https://www.nwo.nl/projecten/15345-0).
|
Geen |
Geen actie vereist |
|
Friederich |
Gynaecoloog NWZ Den Helder, Algemeen gynaecoloog met als aandachtsgebieden benigne gynaecologie, minimaal invasieve chirurgie en bekkenbodemproblematiek |
Vicevoorzitter calamiteitencommissie NVZ lid klachtencommissie NWZ Den Helder |
Geen |
Geen actie vereist |
|
Segers |
Klinisch geriater, St. Jans Gasthuis, Weert
|
Klinisch farmacoloog in opleiding, Catharina ziekenhuis, Eindhoven Onbetaald |
Geen |
Geen actie vereist |
|
Kanninga |
Arts Verstandelijk Gehandicapten (arts VG) bij Cordaan Amsterdam Anesthesioloog niet praktiserend |
Geen |
Geen |
Geen actie vereist |
|
Visser |
Hoogleraar geriatrische tandheelkunde fulltime (1 fte) - Afdeling Gerodontologie, Centrum voor Tandheelkunde en Mondzorgkunde, Universitair Medisch Centrum Groningen, Rijksuniversiteit Groningen, Nederland - Afdeling Gerodontologie, Faculteit Tandheelkunde, Radboud UMC, Radboud Universiteit Nijmegen, Nederland |
Geen |
Geen |
Geen actie vereist |
Inbreng patiëntenperspectief
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door het uitnodigen van de Patiëntenfederatie Nederland voor de invitational conference. Het verslag hiervan (zie aanverwante producten) is besproken in de werkgroep. Aanvullend heeft een afgevaardigde van de Patiëntenfederatie Nederland deelgenomen in de werkgroep. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de richtlijn. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland en de aangeleverde commentaren zijn bekeken en verwerkt.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming gedaan of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie ook het hiervoor gebruikte stroomschema dat als uitgangspunt voor de beoordeling is gebruikt).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn. Een overzicht van uitkomsten van de kwalitatieve raming met bijbehorende toelichting vindt u in onderstaande tabel.
|
Module |
Uitkomst raming |
Toelichting |
|
Module Zuurstof |
geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (>40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet en het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. |
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep de knelpunten in de zorg voor patiënten die procedurele sedatie en/of analgesie ondergaan. Tevens zijn er knelpunten aangedragen door de Nederlandse Vereniging voor Anesthesiologie, de Nederlandse Vereniging voor Heelkunde, de Nederlandse Vereniging voor Obstetrie en Gynaecologie, Nederlandse Vereniging voor Cardiologie, de Nederlandse Vereniging van Maag-Darm-Leverartsen, de Vereniging voor Keel-Neus-Oorheelkunde en Heelkunde van het Hoofd-Halsgebied, de Nederlandse Vereniging voor Intensive Care, de Nederlandse Internisten Vereniging, de Nederlandse Vereniging van Spoedeisende Hulp Artsen, het Nederlands Genootschap van Abortusartsen, de Nederlandse Vereniging van Anesthesiemedewerkers, de Verpleegkundigen & Verzorgenden Nederland, de Nederlandse Vereniging voor Mondziekten, Kaak- en Aangezichtschirurgie, de Vereniging Mondzorg voor Bijzondere Zorggroepen, Stichting Kind & Ziekenhuis, Inspectie Gezondheidszorg en Jeugd en de Vereniging van Artsen voor Verstandelijk Gehandicapten via een invitational conference. Een verslag hiervan is opgenomen onder aanverwante producten. Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. Indien mogelijk werd de data uit verschillende studies gepoold in een random-effects model. Review Manager 5.4 werd gebruikt voor de statistische analyses. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nul effect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Zoekverantwoording
Literature search strategy
Algemene informatie
|
Richtlijn: PSA - volwassenen |
|
|
Uitgangsvraag: UV5 zuurstoftoediening |
|
|
Database(s): Ovid/Medline, Embase |
Datum: 25-8, 2-9, 3-9-2020 |
|
Periode: 2005 |
Talen: nvt |
|
Literatuurspecialist: Ingeborg van Dusseldorp |
|
|
Toelichting en opmerkingen: 3-9-2020 De term oxygenation is niet meegenomen in de zoekstrategie omdat hiermee voornamelijk artikelen werden gevonden over extracorporeal membrane oxygenation (ECMO). Het aantal referenties dat daarmee in totaal werd gevonden in 2 databases (nog niet ontdubbeld) bedroeg 1251, terwijl het aantal referenties zonder oxygenation in 2 databases (nog niet ontdubbeld) 768 bedroeg. NB. De term preoxygenation is wel meegenomen. Omdat over de periode vanaf 2005 is gezocht werd het artikel van Jurrell Effect of supplemental oxygen on cardiopulmonary changes during gastrointestinal endoscopy uit 1994 niet gevonden. De overige sleutelartikelen werden wel gevonden.
2-9-2020 Naar aanleiding van onderstaand zoekblok worden door de werkgroep de volgende termen aangeleverd: Supplemental oxygen Oxygen supply oxygen treatment oxygen administration oxygen therapy Oxygenation Preoxygenation Low flow oxygen high-flow oxygen low flow nasal cannula low flow oxygen therapy high flow nasal cannula High flow oxygen therapy Non rebreather valve Non rebreather mask / NRM
25-8-2020 Voor het zoekblok zuurstoftoediening heb ik voor COVID-19 uitgebreid gezocht met onderstaande strategie. Graag hoor ik of de terminologie die hierin wordt gebruikt ook van toepassing is op PSA. Ter informatie: in het zoekformulier worden de volgende termen voorgesteld: oxygen supply, Supplemental oxygen, high-flow oxygen
'oxygen therapy'/exp OR 'high flow nasal cannula'/exp OR 'high flow oxygen therapy'/exp OR 'non invasive positive pressure ventilation'/exp OR 'nasal intermittent positive pressure ventilation'/exp OR 'invasive mechanical ventilation'/exp OR 'o2 administration':ti,ab,kw OR 'o2 therapy':ti,ab,kw OR 'oxygen administration':ti,ab,kw OR 'oxygen inhalation therapy':ti,ab,kw OR 'oxygen insufflation':ti,ab,kw OR 'oxygen therapy':ti,ab,kw OR 'oxygen supply':ti,ab,kw OR 'supplemental oxygen':ti,ab,kw OR 'oxygen treatment':ti,ab,kw OR 'high flow nasal cannula*':ti,ab,kw OR hfnc:ti,ab,kw OR 'non-invasive ventilation':ti,ab,kw OR 'non-invasive positive pressure ventilation':ti,ab,kw OR 'nasal intermittent positive pressure ventilation':ti,ab,kw OR nippv:ti,ab,kw OR bipap:ti,ab,kw OR 'bilevel positive airway pressure':ti,ab,kw OR cpap:ti,ab,kw OR 'continuous positive airway pressure':ti,ab,kw OR 'non rebreathing valve'/exp OR 'non rebreather':ti,ab,kw OR 'non rebreathing':ti,ab,kw OR nrb:ti,ab,kw OR nrbm:ti,ab,kw OR 'low flow nasal cannula':ti,ab,kw OR 'invasive mechanical ventilation':ti,ab,kw OR optiflow:ti,ab,kw OR vapotherm:ti,ab,kw |
|
Zoekopbrengst
|
|
EMBASE |
OVID/MEDLINE |
Ontdubbeld |
|
SRs |
40 |
16 |
41 |
|
RCTs |
298 |
122 |
315 |
|
Observationele studies |
162 |
130 |
187 |
|
Overig |
|
|
|
|
Totaal |
|
|
543 |
Zoekverantwoording
Ovid/Medline
1 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or (systematic*or literature adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (300662)
2 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (2021557)
3 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ [Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies] (3511189)
4 exp Deep Sedation/ or exp Conscious Sedation/ or sedati*.ti,ab,kf. (59595)
5 exp Oxygen inhalation therapy/ or (oxygen* adj3 (suppl* or treatment* or administration or therap* or low flow or high flow)).ti,ab,kf. or preoxygenat*.ti,ab,kf. or (flow adj3 nasal cannul*).ti,ab,kf. or non rebreather valve*.ti,ab,kf. or non rebreather mask*.ti,ab,kf. (51720)
6 4 and 5 (814)
7 6 not ((exp animals/ or exp models, animal/) not humans/) not (letter/ or comment/ or editorial/) (731)
8 limit 7 to yr="2005 -Current" (449)
9 1 and 8 (16) SR
10 2 and 8 (133)
11 3 and 8 (201)
12 10 not 9 (122) RCT
13 11 not 10 not 9 (130) OBS
Embase
|
No. |
Query |
Results |
|
#18 |
#13 OR #16 OR #17 |
500 |
|
#17 |
#15 NOT #14 NOT #13 OBS |
162 |
|
#16 |
#14 NOT #13 RCT |
298 |
|
#15 |
#11 AND #12 |
588 |
|
#14 |
#10 AND #12 |
528 |
|
#13 |
#9 AND #12 SR |
40 |
|
#12 |
#3 AND [1-1-2005]/sd NOT ('conference abstract'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
1058 |
|
#11 |
'major clinical study'/de OR 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR 'prospective study'/de OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (('case control' NEAR/1 (study OR studies)):ab,ti) OR (('follow up' NEAR/1 (study OR studies)):ab,ti) OR (observational NEAR/1 (study OR studies)) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti) |
5395725 |
|
#10 |
('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it |
2340458 |
|
#9 |
('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
473856 |
|
#8 |
#3 AND #7 |
4 |
|
#7 |
#4 OR #5 OR #6 |
4 |
|
#6 |
effect AND of AND supplemental AND oxygen AND on AND cardiopulmonary AND changes AND during AND endoscopy. AND gastrointestinal AND endoscopy AND jurell |
1 |
|
#5 |
the AND utility AND of AND 'high flow' AND oxygen AND during AND emergency AND department AND procedural AND sedation AND analgesia AND with AND propofol |
2 |
|
#4 |
the AND utility AND of AND supplemental AND oxygen AND during AND emergency AND department AND procedural AND sedation AND analgesia AND with AND midazolam AND fentanyl |
1 |
|
#3 |
#1 AND #2 |
2109 |
|
#2 |
'non rebreathing valve'/exp OR 'high flow nasal cannula'/exp OR 'high flow oxygen therapy'/exp OR 'preoxygenation'/exp OR 'oxygen therapy'/exp OR 'oxygenator'/exp OR 'oxygen supply'/exp OR ((oxygen* NEAR/3 (suppl* OR treatment* OR administration OR therap* OR 'low flow' OR 'high flow')):ti,ab,kw) OR preoxygenat*:ti,ab,kw OR ((flow NEAR/3 'nasal cannul*'):ti,ab,kw) OR 'non rebreather valve*':ti,ab,kw OR 'non rebreather mask*':ti,ab,kw |
93585 |
|
#1 |
'procedural sedation and analgesia'/exp/mj OR 'procedural sedation'/exp OR 'sedation'/exp OR 'sedati*':ti,ab |
113121 |