Behandeling met SSRI’s bij vrouwen met PMS
Uitgangsvraag
Wat is de waarde van behandeling met SSRI’s bij vrouwen met PMS?
Aanbeveling
Geef SSRI’s, bij voorkeur, in de luteale (symptomatische) fase. Een continue dosering wordt gezien als alternatief.
Schrijf, bij behandeling van ernstige PMS, SSRI’s in een lage dosering voor en verhoog deze alleen bij uitblijvend of onvoldoende effect. Maak samen met de patiënte een afweging tussen verwacht effect en mogelijke bijwerkingen.
Stel, zo nodig, de medicatie bij, afgestemd op de individuele patiënt, op basis van voorkeur, klachtenreductie en bijwerkingen.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
De keuze voor een SSRI kan gemaakt worden op basis van kosten, ervaring, voorkeur van de patiënte en eventuele bijwerkingen. Bij voorkeur wordt met een zo laag mogelijke dosering gestart (bijvoorbeeld fluoxetine 10 mg per dag). Bij onvoldoende effect op vermindering van de klachten kan overwogen worden de dosering te verhogen. Alle onderzochte SSRI’s lijken effectief te zijn. Het voorschrijven van deze medicatie moet beperkt blijven tot medici met expertise op dit gebied. De minimum voorwaarden voor de anamnese worden vermeld in het hoofdstuk diagnose.
Vrouwen met PMS die behandeld worden met een SSRI dienen gewezen te worden op het mogelijk optreden van bijwerkingen, zoals misselijkheid, asthenie, vermoeidheid, somnolentie, verminderde zin in seks en transpireren. Deze bijwerkingen zijn dosisafhankelijk. Bij gebruik in de luteale fase treden klachten van een verminderd seksueel verlangen minder op ten opzichte van continu gebruik.
De lichamelijke bijwerkingen kunnen in belangrijke mate verklaard worden door de aanwezigheid van perifere serotoninereceptoren zoals in het gastro-intestinale stelsel.
Gastro-intestinale klachten, hoofdpijn, angst, duizeligheid, paresthesie, slaapstoornissen, vermoeidheid, afname van seksuele interesse, griepachtige symptomen en zweten zijn de meest voorkomende kenmerken van het abrupt staken van een SSRI of een duidelijke vermindering van de dosis.
Bij langdurig continu gebruik moet langzaam worden afgebouwd. Vanwege de mogelijke bijwerkingen en overige specifieke risico’s van SSRI-gebruik is het raadzaam dat het middel wordt voorgeschreven door een zorgverlener die hier ervaring mee heeft.
Wanneer een SSRI de klachten niet voldoende laat afnemen of wanneer de bijwerkingen van
de SSRI onvoldoende verdwijnen, kan een ander SSRI geprobeerd worden (Freeman, Jabara, Sondheimer, & Auletto, 2002). Ernstige PMS verbeterde significant met escitalopram tijdens de luteale fase of volgens een symptoomonset dosering (= starten op het moment dat de symptomen beginnen). Escitalopram wordt bij vrouwen met PMS vaak getolereerd.
Vrouwen met ernstige PMS reageren mogelijk beter wanneer een luteale fase dosering van 14 dagen wordt gegeven in vergelijking met een symptoomonset dosering (Freeman, Sondheimer, Sammel, Ferdousi, & Lin, 2005). Het voorschrijven van SSRI’s voor gebruik in de luteale fase geeft minder kans op bijwerkingen.
Ofschoon SSRI’s op basis van meta-analyses (Brown et al., 2009; Dimmock, Wyatt, Jones, & O'Brien, 2000) steeds meer gezien worden als middel van eerste keus bij de behandeling van PMS en PMDD (Freeman, Sammel, Lin, Rickels, & Sondheimer, 2011) blijkt in de praktijk dat ongeveer 40% van de vrouwen met PMS of PMDD vanwege onbekende oorzaken onvoldoende reageren op de behandeling met SSRI’s (Mitwaly, Kahn, & Halbreich, 2002).
Het is van belang bewust te zijn van de bijwerkingen van SSRI’s en het is aan te bevelen nauw samen te werken met huisartsen met ervaring met het gebruik van SSRI’s.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Het voornaamste doel van de behandeling is vermindering/wegnemen van PMS klachten. Indien effectief gebleken bij een patiënte, ervaart zij hier voordeel van. Mogelijke nadelen zijn de bijwerkingen ervan en deze dienen dan ook uitvoerig met patiënte besproken te worden. Daarnaast kan het een nadeel zijn dat patiënten dagelijks in de luteale fase, dan wel continu de medicatie moeten innemen. In de praktijk zijn redenen voor continu in plaats van luteaal gebruik; een irregulaire cyclus en daardoor onvoldoende duidelijkheid over het startmoment of een individuele voorkeur voor continu gebruik of onvoldoende effect van gebruik in de luteale fase.
Kosten (middelenbeslag)
Het voornaamste doel van de behandeling is vermindering/wegnemen van PMS klachten. Indien effectief gebleken bij een patiënte, ervaart zij hier voordeel van. Mogelijke nadelen zijn de bijwerkingen ervan en deze dienen dan ook uitvoerig met patiënte besproken te worden. Daarnaast kan het een nadeel zijn dat patiënten dagelijks in de luteale fase, dan wel continu, de medicatie moeten innemen.
Aanvaardbaarheid, haalbaarheid en implementatie
Ook dit aspect wordt niet specifiek in de artikelen genoemd. Het voorschrijven van SSRI’s lijkt een haalbare interventie te zijn, aangezien er behalve het voorschrijven van de medicatie geen extra interventies of aanpassingen gedaan hoeven te worden (zoals scholing, aanschaf apparatuur, hogere opnamecapaciteit, etc.). Wel dient het door of in samenwerking met medici met expertise op dit gebied voorgeschreven te worden.
Onderbouwing
De huidige richtlijn beveelt SSRI’s (Selective serotonin reuptake inhibitor) aan in de symptomatische, luteale fase. Er wordt geadviseerd te starten in een lage dosering en alleen bij uitblijven van effect op te hogen. Hoewel enkele studies in het verleden hebben aangetoond dat SSRI's een goed effect hebben op premenstrueel syndroom (PMS) klachten, lijkt in de praktijk een groot aantal vrouwen met PMS onvoldoende baat te hebben bij de behandeling.
Conclusions
Marjoribanks (2013) concluded that SSRIs have been shown to be effective in reducing PMS symptoms, both when used in the luteal phase and continuously. Side effects are common, especially nausea and asthenia and are dose dependent. The level of evidence for the various studies included in this systematic review is low to moderate, mainly due to poor documentation of methods. There is also moderate heterogeneity in one of the primary analyses.
Yonkers (2013) concluded that fluoxetine has a clear therapeutic effect on PMS.
|
Low GRADE |
The evidence suggests that SSRI’s reduce PMS complaints when compared with placebo in a patient population of PMS.
Sources: Marjoribanks, 2013; Yonkers, 2013 |
Description of studies
The Cochrane review by Marjoribanks (2013) included 31 RCTs which compared SSRIs with placebo in a total of 4372 women who were clinically diagnosed with PMS. Five different SSRIs were analyzed in the studies (fluoxetine, paroxetine, sertraline, escitalopram and citalopram) and compared with placebo. The different dosages ('low,' 'medium' or 'high') and dosage forms ('luteal' or 'continuous') were also examined. The effect of SSRIs compared to placebo was examined for the following primary outcome measures: overall PMS symptoms and adverse events. In addition, the following secondary outcome measures were examined: psychological complaints, physical complaints, functional complaints and irritability.
Yonkers (2013) investigated in a RCT, in which 39 women with PMS were included, the effect of daily use of fluoxetine compared to calcium carbonate and placebo on PMS complaints over a period of four menstrual cycles. The primary outcome was improvement in symptoms, which were assessed using various scoring lists (The Inventory of Depressive Symptomatology, Premenstrual Tension Scale, Clinical Global Impression-Severity and -Improvement scales, and Daily Record of Severity of Problems).
Results
The results of the Cochrane review by Marjoribanks (2013) showed that SSRIs reduced overall self-rated symptoms significantly more effectively than placebo. This applied to low dose SSRIs (SMD - 0.67, 95% CI -0.29 to -1.05, two studies, 301 women; I2 = 59%), moderate dose SSRIs (SMD -0.65, 95% CI -0.46 to -0.84, nine studies, 1276 women; I2 = 58%) and high dose SSRIs (SMD -0.95, 95% CI - 0.58 to -1.31, one study, 134 women). The effect size was small when studies reporting change scores were pooled (for moderate dose SSRIs: SMD -0.36, 95% CI -0.20 to -0.51, four studies, 657 women; low heterogeneity (I2=29%), moderate quality evidence). (Figure 1) Eleven studies reported psychological symptom scores, either as end scores (five studies) or as change scores (four studies). When psychological symptoms were assessed with end scores, SSRIs reduced symptoms significantly more effectively than placebo. Low dose SSRIs were associated with a small effect size (SMD -0.38, 95% CI -20.0 to -0.57, three studies, 470 women; I2 = 0%) and moderate dose SSRIs with a moderate effect size (SMD -0.51, 95% CI -0.37 to -0.65, five studies, 795 women; I2 = 0%). This applied to both luteal and continuous administration. Heterogeneity was absent. Nine studies reported physical symptom scores, either as end scores (five studies) or as change scores (four studies). When physical symptoms were assessed with end scores, there were no data for low dose SSRIs. Moderate dose SSRIs reduced physical symptoms significantly more than placebo, with a small effect size and moderate heterogeneity (SMD -0.43, 95% CI -0.21 to -0.65, five studies, 781 women; I2 = 50%). The heterogeneity was attributable to differences in type of administration. In the single study of luteal administration there was no significant difference between moderate dose SSRIs and placebo for this outcome (OR -0.13, 95% CI -0.40 to 0.13, one study, 219 women), while in the studies of continuous administration there was a significant benefit for the SSRI group, of moderate effect size (OR -0.52, 95% CI -0.69 to -0.3, four studies, 562 women; I2 = 0%). High dose SSRIs were associated with a moderate effect size (SMD -0.56, 95% CI -0.26 to -0.86, one study, 179 women). In the SSRI group, significantly more dropout of participants was seen due to adverse events (OR 2.55, 95% CI 1.84 to 3.53). The most common side effects seen at an average dose were: nausea, asthenia, fatigue, somnolence, decreased sexual desire and sweating. The side effects appeared to be dose dependent. No significant difference was seen between luteal use and continuous use in side effects, except for loss of sexual interest, which was reported more frequently with continuous use.
The study by Yonkers (2013) showed that fluoxetine had a greater effect on symptom reduction than the use of calcium and placebo. There was a significant difference in the Daily Record of Severity of Problems (β -0.28; 95% CI -0.53 to -0.04; p=0.02) and the Clinical Global Impression-Severity and Improvement (β -1.03 95% CI -1.70 to -0.35; p=0.04). There were no severe adverse events. Events were uncommon and occurred in all 3 groups with a numerically higher number in the calcium group.
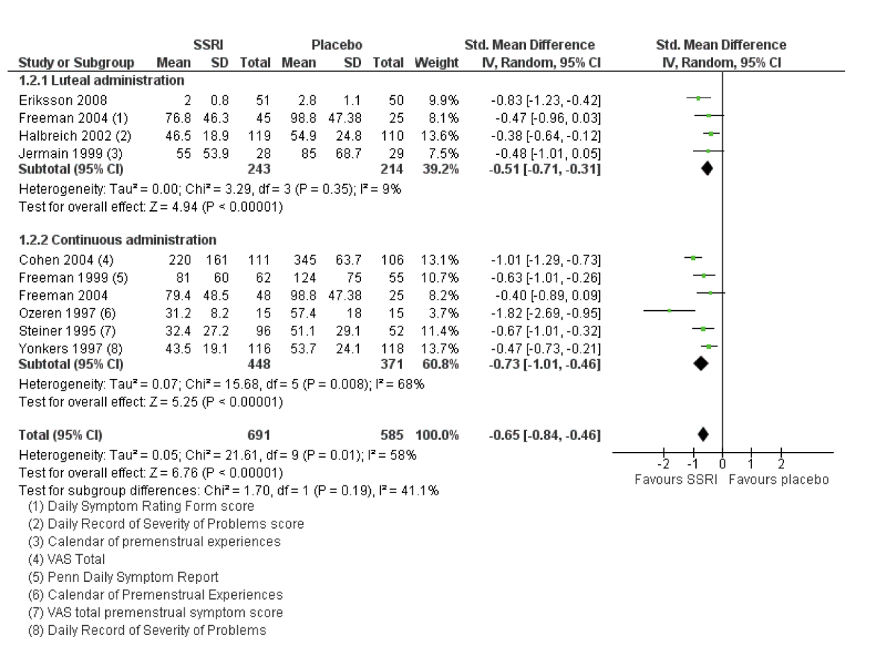
Figure 1. Forest plot of comparison: 1 SSRIs versus placebo - all symptoms (end scores), outcome: 1.2 Moderate dose SSRI. (Marjoribanks, 2013)
Level of evidence of the literature
The level of evidence (GRADE method) is determined per comparison and outcome measure and is based on results from RCTs and therefore starts at level “high”. Subsequently, the level of evidence was downgraded if there were relevant shortcomings in one of the several GRADE domains: risk of bias, inconsistency, indirectness, imprecision, and publication bias.
The level of evidence regarding the outcome measure PMS complaints was downgraded by two levels to a low GRADE because of study limitations (risk of bias: no adequate description of methods of randomization and allocation concealment; and high risk of attrition bias) and inconsistency (substantial overall heterogeneity).
A systematic review of the literature was performed to answer the following question:
What is the effectiveness of SSRIs compared to placebo in women with PMS?
P: Women with PMS.
I: SSRIs.
C: Placebo.
O: PMS complaints: abdominal pain, mastalgia, edema, bloating, depressive symptoms.
Relevant outcome measures
The guideline development group considered no outcome measure to be crucial for decision-making; and PMS complaints as important outcome measures for decision making.
A priori, the working group did not define the outcome measures listed above but used the definitions used in the studies.
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms on 21-03-2021. The detailed search strategy is depicted under the tab Methods. The systematic literature search resulted in 176 hits. Studies were selected based on the following criteria: 1) The study population consisted of women with PMS, 2) The study compared SSRIs with placebo.
13 studies were initially selected based on title and abstract screening. After reading the full text, 11 studies were excluded (see the table with reasons for exclusion under the tab Methods), and two studies were included.
Results
Two studies (Marjoribanks, 2013; Yonkers, 2013) were included in the literature analysis. Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
- Brown J, O' Brien PM, Marjoribanks J, Wyatt K. Selective serotonin reuptake inhibitors for premenstrual syndrome. Cochrane Database Syst Rev. 2009 Apr 15;(2):CD001396. Update in: Cochrane Database Syst Rev. 2013;6:CD001396.
- Dimmock PW, Wyatt KM, Jones PW, O'Brien PM. Efficacy of selective serotonin-reuptake inhibitors in premenstrual syndrome: a systematic review. Lancet. 2000 Sep 30;356(9236):1131-6.
- Freeman EW, Jabara S, Sondheimer SJ, Auletto R. Citalopram in PMS patients with prior SSRI treatment failure: a preliminary study. J Womens Health Gend Based Med. 2002 Jun;11(5):459-64.
- Freeman EW, Sondheimer SJ, Sammel MD, Ferdousi T, Lin H. A preliminary study of luteal phase versus symptom-onset dosing with escitalopram for premenstrual dysphoric disorder. J Clin Psychiatry. 2005 Jun;66(6):769-73.
- Freeman EW, Sammel MD, Lin H, Rickels K, Sondheimer SJ. Clinical subtypes of premenstrual syndrome and responses to sertraline treatment. Obstet Gynecol. 2011 Dec;118(6):1293-1300.
- Mitwally MF, Kahn LS, Halbreich U. Pharmacotherapy of premenstrual syndromes and premenstrual dysphoric disorder: current practices. Expert Opin Pharmacother. 2002 Nov;3(11):1577-90.
- Marjoribanks, Brown, O' Brien, & Wyatt. Selective serotonin reuptake inhibitors for premenstrual syndrome. Cochrane Database of Systematic Reviews 2013, Issue 6. Art. No.: CD001396.
- Yonkers, Pearlstein, Gotman. A pilot study to compare fluoxetine, calcium and placebo in the treatment of premenstrual syndrome. Journal of clinical psychopharmacology 2013, 33:614-620.
Research question: What is the effectiveness of SSRIs compared to placebo in women with PMS?
Systematic review
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Marjoribanks , 2013
individual study characteristics deduced from [1st author, year of publication ]]
PS., study characteristics and results are extracted from the SR (unless stated otherwise) |
SR and meta-analysis of RCTs
Literature search up to February 2013. To be found in the article.
Study design: RCT (parallel-group design/ crossover)
Setting and Country: June 2013 New Zealand
Source of funding and conflicts of interest: No source of funding provided, Not specified.
One of the authors has been funded over many years from pharmaceutical companies.
At least 21 out of 31 of the studies received funding from pharmaceutical companies. |
Inclusion criteria SR: Published and unpublished RCTs Studies of women of any age who met the medically defined diagnostic criteria for PMS or PMDD or late luteal phase dysphoric disorder (LPDD)
Exclusion criteria SR: current or recent major or Axis 1 psychiatric diagnosis (other than PMDD); other clinically significant disease; recent hormonal contraceptive use; current or planned pregnancy; use of concurrent medication (including psychotropic drugs) non-randomised studies
31 studies included
|
Describe intervention:
SSRIs, at any dose and in any dosing regimen for any duration longer than one menstrual cycle
• Sertraline 50 to 150 mg (Arrendondo 1997; Freeman 1999; Freeman 2004; Halbreich 1997; Halbreich 2002; Jermain 1999; Kornstein 2006; Yonkers 1997; Young 1998); • Fluoxetine 10 to 20 mg (Cohen 2002; Crnobaric 1998; Menkes 1992; Miner 2002; Ozeren 1997; Pearlstein 1997; Steiner 1995; Stone 1991; Su 1997; Wood 1992); • paroxetine 5 to 25 mg (Cohen 2004; Eriksson 1995; Glaxo 1996; Glaxo 1996a; Glaxo 2001; Landen 2007; Pearlstein 2005; Steiner 2005; Steiner 2008); • escitalopram 10 to 20 mg (Eriksson 2008; Freeman 2010);c • italopram 10 to 30 mg (Wikander 1998).
|
Describe control: Placebo
|
End-point of follow-up: Not reported.
For how many participants were no complete outcome data available? (intervention/control)
14 studies analysed all or most women by intention to treat, and were rated as at low risk of attrition bias (Cohen 2002; Crnobaric 1998; Eriksson 2008; Glaxo 2001; Landen 2007; Miner 2002; Pearlstein 1997; Pearlstein 2005; Steiner 2005; Steiner 2008; Stone 1991; Su 1997; Wood 1992; Yonkers 1997). Four studies had data missing for over 20% of participants and were rated as at high risk of attrition bias (Halbreich 1997; Jermain 1999; Kornstein 2006; Young 1998). The other 13 studies were at unclear risk of attrition bias due to unclear reporting of numbers randomised or numbers analysed, or failure to include 10% to 20% of participants in the analysis. A number of these studies also imputed a high proportion of data.
|
Overall self-rated symptoms: low dose SSRIs: SMD - 0.67, 95% CI -0.29-1.05
moderate dose SSRIs: SMD -0.65, 95% CI -0.46 -0.84
high dose SSRIs: SMD -0.95, 95% CI - 0.58 -1.31
Psychological symptom scores: low dose SSRIs: SMD -0.38, 95% CI -20.0 -0.57
moderate dose SSRIs: SMD -0.51, 95% CI -0.37-0.65
Physical symptom scores: moderate dose SSRIs: SMD -0.43, 95% CI -0.21-0.65
high dose SSRIs: SMD -0.56, 95% CI -0.26 -0.86
|
Author’s conclusion: SSRIs are effective in reducing the symptoms of PMS, whether taken in the luteal phase only or continuously. Adverse effects are relatively frequent, the most common being nausea and asthenia. Adverse effects are dose-dependent.
Personal remarks on study quality, conclusions, and other issues (potentially) relevant to the research question
Level of evidence: GRADE:
The overall quality of the evidence was low to moderate, the main weakness in the included studies being poor reporting of methods.
SSRIs compared to placebo - all symptoms (end scores) for premenstrual syndrome: Moderate dose SSRI versus placebo Luteal or continuous Administration: Low GRADE
Moderate dose SSRI versus placebo Luteal administration: Moderate GRADE
Moderate dose SSRI versus placebo Continuous administration: Low GRADE
Heterogeneity: Heterogeneity was low or absent for most outcomes, though (as noted above) there was moderate heterogeneity for one of the primary analyses.
Overall self-rated symptoms (end scores): moderate heterogeneity (I2 = 58%)
Psychological symptoms: No heterogeneity (I2 =0%)
Physical symptom scores: moderate heterogeneity (I2 = 50%) |
Intervention study
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Yonkers, 2013 |
Type of study: parallel, double-blind randomized trial- pilot
Setting and country: 2001 2005 in Providence and 2001-2009 in New Haven. USA
Funding and conflicts of interest: This study was supported by a grant from Women’s Health Research at Yale and Grant MH62379 from the National Institute of Mental Health. Dr Yonkers discloses receipt of royalties from Up To Date. Dr Pearstein discloses receipt of a research grant from Pfizer. |
Inclusion criteria: (1) Women 18-48 years; (2) had regular menses; (3) met criteria for moderate to severe PMS; (4) retrospectively reported symptoms of moderate to severe PMS in at least 9 of 12 menstrual cycles during the year before screening; (5) in the investigators’ opinion were using an adequate method of contraception, including hormonal contraceptives, if treatment had endured a minimum of 6 months; and (6) had the capacity to provide verbal and written consent.
Exclusion criteria: (1) fulfilled DSM-IV criteria for another serious Axis I disorder (psychotic disorders, bipolar disorder, major depressive disorder, bulimia and anorexia, obsessive-compulsive disorder, panic disorder, or substance abuse disorder in the prior month); (2) posed a significant risk of suicide; (3) were taking an ongoing medication that, in the opinion of the investigator, could either treat her PMS symptoms (e.g., gonadotropin-releasing hormone agonist, calcium, or other psychotropic agent) or were contraindicated during fluoxetine or calcium therapy (digitalis, select antiarrhythmic drugs, select antihistamines); (5) had a history of hypersensitivity or adverse reaction to fluoxetine or calcium; (6) were lactating, pregnant, or imminently planning to become pregnant; or (7) had a clinically significant medical condition that could render participation unsafe (e.g., hepatic failure, renal disease, parathyroid disease, active peptic ulcer disease, or malabsorption).
N total at baseline: Randomized: 49 Intervention1: 13 Intervention2: 13 Control: 13
Important prognostic factors2: Mean age: Not provided. See Table 1 for age categories.
Groups comparable at baseline? No |
Describe intervention (treatment/procedure/test):
Fluoxetine 10 mg, twice daily
Calcium 600 mg twice daily
|
Describe control (treatment/procedure/test):
Placebo |
Length of follow-up: 4 months
Loss-to-follow-up: 10 (20.5%) Reason not provided.
Incomplete outcome data: Intervention: 0 (0%) Reasons (describe)
Control: 0 (0%) Reasons (describe)
|
Outcome measures and effect size (include 95%CI and p-value if available):
Change in Symptom Scores: IDS (Differences in changes over time between groups)
I1: B= -2.63 (95% CI: -5.51 to 0.24) P=0.07 Effect size*= 1.15
I2: B= -0.10 (95% CI: -2.85 to 2.65) P=0.94 Effect size*= 0.10
C: referent
DRSP (Differences in changes over time between groups)
I1: B= -0.28 (95% CI: -0.53 to -0.04) P=0.02 Effect size*= 2.08
I2: B= -0.06 (95% CI: -0.16 to 0.28) P=0.58 Effect size*= 0.18
C: referent
Response to Treatment DRSP LOCF** response I1: 8/10 (80%) I2: 5/12 (42%) C: 5/12 (42%) |
Criteria for moderate to severe PMS: as defined by the presence of at least 3 of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) symptoms problematic enough to warrant treatment, one of which is a functionally impairing affective symptom
*Effect size was calculated as change relative to placebo from baseline to Visit 5.
** The last observation carried forward (LOCF)
Authors conclusion: Fluoxetine had clear therapeutic benefit for premenstrual syndrome, whereas the effect of calcium was much smaller. Results of this pilot do not support the need for a larger study that would compare these compounds.
Limitations: The fluoxetine group had greater baseline severity on the DRSP. Second, the sample size for this pilot may not be sufficient to draw definitive conclusions about treatment efficacy. Higher attrition in the fluoxetine group possible overestimation of the fluoxetine effect |
Table of quality assessment
Systematic review
|
Study
First author, year |
Appropriate and clearly focused question? |
Comprehensive and systematic literature search? |
Description of included and excluded studies? |
Description of relevant characteristics of included studies? |
Appropriate adjustment for potential confounders in observational studies? |
Assessment of scientific quality of included studies? |
Enough similarities between studies to make combining them reasonable? |
Potential risk of publication bias taken into account? |
Potential conflicts of interest reported? |
|
Marjoribanks, 2013 |
Yes |
Yes |
Yes |
Yes |
Not applicable |
Yes |
Yes |
Yes |
Yes |
Intervention study
|
Study reference
(first author, publication year) |
Was the allocation sequence adequately generated?
Definitely yes Probably yes Probably no Definitely no |
Was the allocation adequately concealed?
Definitely yes Probably yes Probably no Definitely no |
Blinding: Was knowledge of the allocated interventions adequately prevented?
Were patients blinded?
Were healthcare providers blinded?
Were data collectors blinded?
Were outcome assessors blinded?
Were data analysts blinded?
Definitely yes Probably yes Probably no Definitely no |
Was loss to follow-up (missing outcome data) infrequent?
Definitely yes Probably yes Probably no Definitely no |
Are reports of the study free of selective outcome reporting?
Definitely yes Probably yes Probably no Definitely no |
Was the study apparently free of other problems that could put it at a risk of bias?
Definitely yes Probably yes Probably no Definitely no |
Overall risk of bias If applicable/necessary, per outcome measure
Low Some concerns High
|
|
Yonkers, 2013
|
Definitely yes;
Reason: Block randomization |
Probably yes;
Reason: Allocation was concealed from the research staff and patients using sequentially numbered containers. |
Probably yes;
Reason: Patients, health care providers and data collectors blinded (blinding of outcome assessors and analysts not reported) |
Probably no;
Reason: Loss to follow-up was frequent in intervention in last visit. Adequate imputation methods were not used. Reason for loss to follow up not provided. |
Probably yes;
Reason: All predefined outcomes were reported. No protocol available |
Probably no;
Reason: Attrition and recall bias. |
Some concerns |
Table of excluded studies
|
Author and year |
Reason for exclusion |
|
Freeman, 2010 |
Does not comply with PICO (narrative review) |
|
Freeman, 2011 |
Does not comply with PICO (wrong outcome, included studies are included in Cochrane review by Marjoribanks et al. ) |
|
Gnanasambanthan, 2019 |
Does not comply with PICO (narrative review) |
|
Ismaili, 2013 |
Does not comply with PICO (ISPMD consensus about auditable standards) |
|
Kleinstäuber, 2012 |
Included studies in the meta-analysis overlap with Cochrane review by Marjoribanks et al. |
|
Nazari, 2013 |
Does not comply with PICO (wrong study design, no placebo) |
|
Shehata, 2020 |
Does not comply with PICO (wrong Intervention: combined oral contraceptives+ SSRI ) |
|
Steiner, 2013 |
Does not comply with PICO (narrative review+ wrong population: PMDD) |
|
Rapkin, 2013 |
Does not comply with PICO (narrative review+ wrong population: PMDD) |
|
Panay, 2011 |
Does not comply with PICO (narrative review) |
|
Yonkers, 2015 |
Does not comply with PICO (wrong population: PMDD) |
Beoordelingsdatum en geldigheid
Publicatiedatum : 13-11-2023
Beoordeeld op geldigheid : 09-11-2023
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodules.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodules zijn in 2020 per module schrijvers en meelezers benoemd. Deze personen werden aangewezen als vertegenwoordigers door de relevante beroepsgroepen die betrokken zijn bij de in de module beschreven zorg (zie hiervoor de Samenstelling van de werkgroep). Alle schrijvers van modules vallend onder één richtlijn vormden samen een schrijfgroep. Alle meelezers van modules vallend onder één richtlijn vormden samen een clusterwerkgroep. In totaal resulteerde dit dus in zes werkgroep en zes clusterwerkgroepen.
Voorzitter project (technisch voorzitter)
Timmermans A. (Anne), gynaecoloog, AmsterdamUMC, NVOG
Werkgroep richtlijn Premenstrueel Syndroom
Dijkstra J.R. (Jeroen), gynaecoloog, Isala Ziekenhuis te Zwolle, NVOG
Traas-Hofmans M.A.F. (Maaike), gynaecoloog, Gelre Ziekenhuizen Apeldoorn en Zutphen, NVOG
Uijt de Haag J.A.M. (Jenna), ANIOS gynaecologie en obstetrie, Wilhelmina KinderZiekenhuis (WKZ), NVOG
van Leeuwen J. (Jeanette), gynaecoloog, UMCU te Utrecht, NVOG
Versluis M.A.C. (Marco), gynaecoloog, UMCG te Groningen, NVOG
Clusterwerkgroep richtlijn Premenstrueel syndroom
Bosch M. (Marlies), patiëntvertegenwoordiger, Bekkenbodem4All
de Boer M.K. (Marrit), psychiater, Synaeda Psycho Medisch Centrum te Leeuwarden, NVvP
de Voogd I. (Ingrid), huisarts, NHG
Labots-Vogelesang S.M. (Marijke), huisarts, NHG
Vlaardingerbroek H. (Hester), kinderarts-endocrinoloog, LUMC te Leiden, NVK
Ondersteuning project
Abdollahi M. (Mohammadreza), adviseur Kennisinstituut van de Federatie van Medisch Specialisten
Labeur Y.J. (Yvonne), adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Sussenbach A.E. (Annelotte), junior adviseur Kennisinstituut van de Federatie van Medisch Specialisten
Verhoeven M. (Maxime), adviseur, Kennisinstituut van de Federatie van Medisch Specialisten
Projectleiding
Augustus 2022- nu Mostovaya I.M. (Irina) (projectleider), senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten
April 2020 tot augustus 2021: Bijlsma-Rutte A. (Anne), adviseur, Kennisinstituut van de Federatie van Medisch Specialisten
September 2021 tot januari 2022: Venhorst K. (Kristie), adviseur, Kennisinstituut van de Federatie van Medisch Specialisten
Februari 2022 tot juni 2022: Göthlin M. (Mattias), adviseur, Kennisinstituut van de Federatie van Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoek financiering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Timmermans (technisch voorzitter van het project) |
Gynaecoloog, Amsterdam UMC (0.5 fte) |
Commissie kwaliteitsdocumenten NVOG (onbetaald); projectgroep Gynae Goes Green NVOG (onbetaald) |
Geen |
Geen actie |
|
Premenstrueel syndroom - werkgroep |
||||
|
Dijkstra |
Gynaecoloog, fulltime, Isala Zwolle |
(Plaatsvervangend) opleider gynaecologie, onbetaald |
Geen |
Louter betrokken bij besluitvorming rondom modules PMS. Niet betrokken bij de besluitvorming rondom HMB, myomectomieën, endometriumablatie etc. |
|
Traas-Hofmans |
Gynaecoloog - subspecialist voortplantingsgeneeskunde |
Secretaris NVOG-werkgroep Psychosomatische Obstetrie en Gynaecologie (onbetaald), tot 2021 |
Geen |
Geen actie |
|
Uijt de Haag |
ANIOS gynaecologie en obstetrie, Wilhelmina KinderZiekenhuis (WKZ) |
Geen |
Geen |
Geen actie |
|
Van Leeuwen |
Gynaecoloog UMC Utrecht/WKZ 0,8fte aandacht benigne gynaecologie & kindergynaecologie |
Geen |
Geen |
Geen actie |
|
Versluis |
- Gynaecoloog, vakgroep gynaecologie, Universitair Medisch Centrum Groningen (0.9) fte |
Geen |
Geen |
Geen actie |
|
Premenstrueel syndroom - clusterwerkgroep |
||||
|
Bosch |
Stichting Bekkenbodem4All. PR | Belangenbehartiging |
Fotograaf Bisdom Groningen-Leeuwarden. Deels betaald, deels vrijwilligerswerk |
Functie Belangenbehartiging Patiënten organisatie |
Geen |
|
De Boer |
Psychiater/ eerste geneeskundige, Synaeda Psycho Medisch Centrum, Leeuwarden. |
Copromotor, Universitair Centrum Psychiatrie, UMC Groningen. Onderwerp onderzoeksproject: hormonen en depressie. Onbetaald. Afgerond in 2021. |
Geen |
Geen |
|
De Voogd |
Praktijk-houdend huisarts Kaderhuisarts Urogynaecologie |
Geen |
Geen betaling voor kaderhuisarts functies |
Geen |
|
Labots |
Huisarts niet-praktiserend |
Onderzoeker Radboudumc, afdeling Eerstelijnsgeneeskunde/ Vrouwenstudie Medische Wetenschappen . Onbezoldigd |
Praktiserend huisarts ruim 30 jaar |
Geen actie |
|
Vlaardingerbroek |
Kinderarts-endocrinoloog, LUMC Leiden |
Geen |
Geen |
Geen actie |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door het uitnodigen van Patiëntenfederatie Nederland en Stichting Bekkenbodem4All voor de schriftelijke knelpunteninventarisatie en voor deelname aan de clusterwerkgroepen. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen. De richtlijn is voor commentaar voorgelegd aan Patiëntenfederatie Nederland en Stichting Bekkenbodem4All en de eventueel aangeleverde commentaren worden bekeken en verwerkt.
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld volgens de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerden de werkgroep de knelpunten in de zorg en de actualiteit van de aanbevelingen beschreven in de te reviseren modules. Tevens zijn er knelpunten aangedragen door de Nederlandse Vereniging voor Obstetrie en Gynaecologie (NVOG), de Nederlandse Vereniging van Maag-Darm-Leverartsen (NVMDL), Vereniging Klinische Genetica Nederland (VKGN), Inspectie Gezondheidszorg en Jeugd (IGJ), Koninklijke Nederlandse Organisatie van Verloskundigen (KNOV), Nederlands Huisartsen Genootschap (NHG), Nederlandse Vereniging voor Bekkenfysiotherapie (NVFB) / Koninklijk Nederlands Genootschap voor Fysiotherapie (KNGF), Nederlandse Vereniging van Ziekenhuizen (NVZ), Patiëntenfederatie Nederland (PFN), Zorginstituut Nederland (ZiNL), Zelfstandige Klinieken Nederland (ZKN) en Zorgverzekeraars Nederland (ZN) via een schriftelijke knelpunteninventarisatie.
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Ook definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Hultcrantz, 2017; Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nul effect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE-methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Volgens de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren worden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren wordt de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule wordt aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en akkoord.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwaliteit.
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013.
Schünemann HJ, Oxman AD, Brozek J, Glasziou P, Jaeschke R, Vist GE, Williams JW Jr, Kunz R, Craig J, Montori VM, Bossuyt P, Guyatt GH; GRADE Working Group. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008 May 17;336(7653):1106-10. Erratum in: BMJ. 2008 May 24;336(7654).
Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324.
Zoekverantwoording
Algemene informatie
|
Richtlijn: Premenstrueel syndroom (PMS) > herziening 7 modules in het ’24 modules project’ |
|
|
Uitgangsvraag: Wat is de waarde van behandeling met SSRI’s bij vrouwen met PMS? |
|
|
Database(s): Medline (OVID), Embase |
Datum: 21-03-2021 |
|
Periode: >2010 |
Talen: Engels, Nederlands |
|
Literatuurspecialist: Laura Boerboom |
|
|
BMI zoekblokken: voor verschillende opdrachten wordt (deels) gebruik gemaakt van de zoekblokken van BMI-Online https://blocks.bmi-online.nl/ Bij gebruikmaking van een volledig zoekblok zal naar de betreffende link op de website worden verwezen. |
|
|
Toelichting en opmerkingen: → Voor deze vraag is gezocht op de elementen PMS (in het blauw) en SSRI’s (in het groen). → De werkgroep had twee actuele sleutelpublicaties, en deze opgegeven artikelen van Marjoribanks (2013) en Yonkers (2013) komen beide uit onderstaande search. |
|
|
Te gebruiken voor richtlijnen tekst: In de databases Embase (via embase.com) en Medline (via OVID) is op 21-03-2021 met relevante zoektermen gezocht naar systematische reviews, RCT’s en observationele studies over de waarde van behandeling met SSRI’s bij vrouwen met PMS. De literatuurzoekactie leverde 176 unieke treffers op. |
|
Zoekopbrengst
|
|
EMBASE |
OVID/MEDLINE |
Ontdubbeld |
|
SRs |
60 |
18 |
59 |
|
RCTs |
79 |
28 |
83 |
|
Observationele studies |
28 |
17 |
34 |
|
Totaal |
167 |
63 |
176 |
Zoekstrategie
|
Database |
Zoektermen |
|||||||||||||||||||||||||||||||||
|
Embase
|
|
|||||||||||||||||||||||||||||||||
|
Medline (OVID)
|
1 exp Premenstrual Syndrome/ or exp Premenstrual Dysphoric Disorder/ or ((premenstrual or premenstruation or menstrual or pre-menstrual or pre-menstruation) adj3 (syndrome* or pain or tension* or dysphor* or distress or symptoms or stress or complaint* or disease* or dysfor* or disorder*)).ti,ab,kf. or 'late luteal'.ti,ab,kf. or 'luteal phase'.ti,ab,kf. or (luteal adj5 symptom*).ti,ab,kf. or pms.ti,ab,kf. or pmd.ti,ab,kf. or pmdd.ti,ab,kf. or llpdd.ti,ab,kf. or (dysphoric adj2 disorder*).ti,ab,kf. (26642) 2 exp Serotonin Uptake Inhibitors/ or ((serotonin adj5 inhibitor*) or amoxapine or citalopram or clomipramine or fenfluramine or fluoxetine or fluvoxamine or norfenfluramine or paroxetine or sertraline or trazodone or zimeldine or ssri* or ('5 hydroxytryptamine' adj5 inhibitor*) or ('5 ht' adj5 inhibitor*)).ti,ab,kf. (61421) 3 1 and 2 (542) 4 limit 3 to ((english or dutch) and yr="2010 -Current") (137) 5 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (2095906) 6 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ [Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies] (3669120) 7 (meta-analysis/ or meta-analysis as topic/ or (metaanaly* or meta-analy* or metanaly*).ti,ab,kf. or systematic review/ or cochrane.jw. or (prisma or prospero).ti,ab,kf. or ((systemati* or scoping or umbrella or "structured literature") adj3 (review* or overview*)).ti,ab,kf. or (systemic* adj1 review*).ti,ab,kf. or ((systemati* or literature or database* or data-base*) adj10 search*).ti,ab,kf. or ((structured or comprehensive* or systemic*) adj3 search*).ti,ab,kf. or ((literature adj3 review*) and (search* or database* or data-base*)).ti,ab,kf. or (("data extraction" or "data source*") and "study selection").ti,ab,kf. or ("search strategy" and "selection criteria").ti,ab,kf. or ("data source*" and "data synthesis").ti,ab,kf. or (medline or pubmed or embase or cochrane).ab. or ((critical or rapid) adj2 (review* or overview* or synthes*)).ti. or (((critical* or rapid*) adj3 (review* or overview* or synthes*)) and (search* or database* or data-base*)).ab. or (metasynthes* or meta-synthes*).ti,ab,kf.) not (comment/ or editorial/ or letter/ or ((exp animals/ or exp models, animal/) not humans/)) (485255) 8 4 and 7 (18) – SR’s 9 (4 and 5) not 8 (28) – RCT’s 10 (4 and 6) not 8 not 9 (17) – Observationele studies 11 8 or 9 or 10 (63) |