Lachgas versus placebo/ medicamenteus
Uitgangsvraag
Wat is de plaats van lachgas vergeleken met placebo/ andersoortige medicamenteuze pijnbehandeling bij zwangere vrouwen met het verzoek tot behandelen van de baringspijn?
Aanbeveling
Overweeg lachgas als optie voor zwangeren met verzoek tot behandelen van de baringspijn als één van de vormen van pijnbehandeling, en/of aan de zwangere die de bevalling in de eerste lijn wenst te voltooien.
Maak op regionaal niveau transparant waar lachgas tot de opties behoort voor het behandelen van pijn tijdens de bevalling.
Overwegingen
De onderstaande overwegingen gelden in principe voor de gehele patiëntenpopulatie zoals geformuleerd in de uitgangsvraag.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Lachgas lijkt een grotere mate van pijnstilling te geven vergeleken met placebo, waarbij een gemiddeld verschil van 2 punten op de VAS (10-puntsschaal) werd gezien. De duur van de ontsluiting lijkt korter te zijn bij zwangeren die lachgas gebruiken vergeleken met zwangeren die een placebo krijgen. Wel is er mogelijk sprake van een verhoogd risico op braken bij gebruik van lachgas vergeleken met placebo. Het risico op een sectio caesarea lijkt niet te verschillen tussen beide groepen. Overige cruciale en belangrijke uitkomstmaten, zoals tevredenheid ten aanzien van pijnstilling , vaginale kunstverlossing en Apgarscore < 7 na 5 minuten zijn niet in de literatuur gerapporteerd. De overall bewijskracht is laag.
Lachgas lijkt snellere pijnstilling te bieden met een significant lagere pijnscore gemeten na 30 minuten vergeleken met pethidine, waarbij het effect op 60 minuten na start gebruik van lachgas of pethidine niet meer significant lijkt te verschillen. Gemeten over de gehele duur van zowel de ontsluitings- als uitdrijvingsfase toont één studie een grotere mate van pijnstilling bij gebruik van lachgas vergeleken met pethidine (Mobaraki, 2016). Overige cruciale en belangrijke uitkomstmaten, zoals wijze van bevallen, misselijkheid/braken en Apgarscore < 7 bij 5 minuten zijn niet in de literatuur gerapporteerd. De overall bewijskracht is laag.
Er is slechts beperkt literatuur beschikbaar over de vergelijking lachgas versus remifentanil. Eén kleine RCT levert onvoldoende bewijs om met voldoende zekerheid conclusies te kunnen trekken over de verschillen in effectiviteit (en veiligheid) tussen deze strategieën. De overall bewijskracht is zeer laag. Er zijn geen aanwijzingen dat het gebruik van lachgas onveilig is. De bijwerkingen voor de neonaat zijn slechts beperkt onderzocht. De meest voorkomende bijwerkingen van lachgas zijn duizeligheid, licht in het hoofd, euforie, misselijkheid en braken (https://www.lareb.nl/).
Waarden en voorkeuren van patiënten
Beweegredenen voor de zwangere om voor pijnbehandeling met lachgas te kiezen kunnen zijn zelfcontrole, de niet-invasieve opzet en snelle werking en uitwerking van het middel. Als in een geboortehuis of op verlosafdelingen in ziekenhuizen de mogelijkheid bestaat te bevallen met deze vorm van pijnbehandeling, kan de zwangere onder begeleiding blijven van de eigen eerstelijns verloskundige, omdat het gebruik van lachgas geen indicatie vormt voor overdracht naar de tweede lijn voor continue foetale bewaking. Mogelijke nadelige bijwerkingen kunnen zijn: licht in het hoofd, euforie, misselijkheid en braken (https://www.lareb.nl/). Daarnaast is er sprake van beperkte bewegingsvrijheid voor de zwangere gedurende de periode dat zij het masker draagt. De zwangere kan het masker niet afzetten tijdens toediening en tijdens de eerste 20 tot 30 minuten na stoppen. Dit in verband met de veiligheid van de omringende hulpverleners. Zwangere vrouwen willen al tijdens de zwangerschap informatie over de verschillende vormen van pijnbehandeling, de voor- en de nadelen en wat dit betekent in hun situatie en willen hierover samen beslissen. Uit de meldactie blijkt dat dit nog onvoldoende in de praktijk gebeurt en vastgelegd wordt. Zie voor wijze van counselen en de vergelijking van de verschillende opties de module ‘Counseling: Samen beslissen‘.
Kosten (middelenbeslag)
Sinds een aantal jaren wordt lachgas vaker aangeboden in ziekenhuizen en geboortecentra (Openbare indicatoren Zwangerschap en bevalling, verslagjaar 2017). Echter, het beschikbaar maken van lachgas vergt logistieke aanpassingen, aanschaf van benodigde apparatuur en scholing van personeel. Dit vraagt een grote financiële investering van ziekenhuizen en/of geboortehuizen.
Aanvaardbaarheid voor de overige relevante stakeholders
Veiligheidsmaatregelen en strikte handhaving van protocollen ter bescherming van werknemers die beroepsmatig blootstaan aan lachgas om piekblootstelling te voorkomen is een randvoorwaarde voor de toepassing van lachgas. In verband met de mogelijke reprotoxische werking voor personeel blootgesteld aan inhalatieanesthetica, wordt gestreefd naar een zo laag mogelijke blootstelling. Voor de toepassing van lachgas gelden (bedrijfs)grenswaarden, waaraan men zich in elk geval moet houden. Lachgas kan alleen worden toegepast in geboortehuizen en ziekenhuizen die over de aangewezen faciliteiten beschikken (KNOV, 2011).
Haalbaarheid en implementatie
Nu duidelijk is op welke manier lachgas zonder risico voor omstanders kan worden toegediend, wordt het op grotere schaal in Nederland aangeboden als pijnbehandeling tijdens de bevalling. Een brede beschikbaarheid zou de keuzevrijheid van de zwangeren ten goede komen. Kanttekening is wel dat er voor deze veilige toediening vele maatregelen genomen moeten worden. Lachgas is alleen beschikbaar in geboortecentra en ziekenhuizen die voldoen aan de normen voor de aanvoer en afzuiging van het gas. Verloskundige zorgverleners kunnen lachgas voorschrijven en gebruiken bij hun cliënten, na adequate training, mits zij alle organisatorische en veiligheidsvoorschriften volgen voor het gebruik van lachgas. Er is geaccrediteerde scholing voor verloskundige zorgverleners. Daarnaast is er nog scholing voor verpleegkundigen en kraamverzorgenden vereist om de zwangeren die gebruik maken van lachgas adequaat te kunnen begeleiden en te bewaken.
Aanbeveling-1
Rationale/ balans tussen voor- en nadelen van de interventie
Lachgas kan voor zwangere vrouwen een aantrekkelijke vorm van pijnbehandeling zijn. Het lijkt effectiever tegen de pijn dan placebo of pethidine (de eerste 30 minuten), werkt snel en kan in diverse settingen ingezet worden, mits aan de veiligheidsvoorwaarden is voldaan. Er zijn geen aanwijzingen dat het gebruik van lachgas onveilig is. De gevolgen voor de neonaat zijn slechts beperkt onderzocht.
Aanbeveling-2
Rationale / balans tussen voor- en nadelen van de interventie
De kosten voor het gebruik van lachgas zijn hoog, vooral de aanlegkosten. Lachgas is daarom niet breed beschikbaar. Daarom zou het ten minste transparant moeten zijn voor de zwangere waar deze vorm van pijnbehandeling wel en niet tot de opties behoort.
Onderbouwing
Lachgas (distikstofmonoxide (N2O)) is een gas dat geïnhaleerd kan worden en gebruikt wordt voor analgesie tijdens de bevalling. In de obstetrie wordt meestal een mengel van 50% distikstofoxide (N2O) en 50% zuurstof (O2) (=entonox) gebruikt. De barende vrouw kan zichzelf lachgas toedienen tijdens een wee door inhalatie via een mond- of gezichtsmasker. Lachgas is een lichte vorm van sedatie, voor aanbevelingen ten aanzien van sedatie zie de NVA richtlijn sedatie. In deze uitgangsvraag wordt de effectiviteit van lachgas ten opzichte van placebo maar ook andere medicamenteuze vormen van pijnbehandeling beschreven.
Lachgas versus placebo
|
Laag GRADE |
Lachgas tijdens de baring lijkt een grotere mate van pijnstilling te geven in vergelijking tot behandeling met placebo.
Bronnen: (Attar, 2016; Parsa, 2017 en Talebi, 2009) |
|
Laag GRADE |
Zwangeren behandeld met lachgas tijdens de baring lijken een hoger risico op braken te hebben in vergelijking tot behandeling met placebo.
Bronnen: (Attar, 2016; Parsa, 2017 en Talebi, 2009, Zhang, 2001) |
|
Laag GRADE |
Er lijkt geen verschil in het risico op misselijkheid tussen zwangeren behandeld met lachgas tijdens de baring in vergelijking tot behandeling met placebo.
Bronnen: (Attar, 2016; Parsa, 2017 en Talebi, 2009) |
|
Laag GRADE |
Er lijkt geen verschil in het risico op een sectio caesarea tussen zwangeren behandeld met lachgas tijdens de baring in vergelijking tot behandeling met placebo.
Bronnen: (Rezaeipour, 2008; Zhang, 2001) |
|
Redelijk GRADE |
De duur van de ontsluitingsfase is waarschijnlijk korter bij zwangeren behandeld met lachgas vergeleken met placebo.
Bronnen: (Attar, 2016; Parsa, 2017) |
|
Laag GRADE |
Er lijkt geen verschil in de duur van de uitdrijving tussen zwangeren behandeld met lachgas tijdens de baring in vergelijking tot behandeling met placebo.
Bronnen: (Attar, 2016; Parsa, 2017) |
|
Laag GRADE |
Er lijkt geen verschil in het risico op sufheid (als maat voor sedatie) tussen zwangeren behandeld met lachgas tijdens de baring in vergelijking tot behandeling met placebo.
Bronnen: (Attar, 2016; Nasrollahi, 2017; Talebi, 2009) |
Lachgas versus pethidine
|
Laag GRADE |
Lachgas tijdens de baring lijkt na 30 minuten een grotere mate van pijnstilling te bieden dan pethidine. Na 60 minuten lijkt de mate van pijnstilling tussen behandeling met lachgas en pethidine vergelijkbaar.
Bronnen: (Mobaraki, 2016) |
|
Laag GRADE |
Er lijkt geen verschil in bevallingsduur tussen zwangere vrouwen behandeld met lachgas in vergelijking tot behandeling met pethidine.
Bronnen: (Mobaraki, 2016) |
Lachgas versus remifentanil
|
Zeer laag GRADE |
Het is onzeker of er een verschil is in mate van pijnstilling tussen zwangere vrouwen behandeld met lachgas in vergelijking tot remifentanil.
Bronnen: (Volmanen, 2005) |
|
Zeer laag GRADE |
Het is onzeker of er een verschil is in jeuk tussen zwangere vrouwen behandeld met lachgas in vergelijking tot remifentanil.
Bronnen: (Volmanen, 2005) |
|
Zeer laag GRADE |
Het is onzeker of er een verschil is in misselijkheid tussen zwangere vrouwen behandeld met lachgas in vergelijking tot remifentanil.
Bronnen: (Volmanen, 2005) |
|
Zeer laag GRADE |
Het is onzeker of er een verschil is in foetale hartslagafwijkingen tussen zwangere vrouwen behandeld met lachgas in vergelijking tot remifentanil.
Bronnen: (Volmanen, 2005) |
|
Zeer laag GRADE |
Het is onzeker of er een verschil is in de mate van sedatie tussen zwangere vrouwen behandeld met lachgas in vergelijking tot remifentanil.
Bronnen: (Volmanen, 2005) |
Beschrijving studies
De meta-analyse van Klomp (2012) includeerde vijf RCT’s of cross-over studies waarin pijnbehandeling tijdens de baring vergeleken werd met placebo (Carstoniu, 1994; Cheng, 2001; Rezaeipour, 2008; Talebi, 2009; Zhang, 2001). Deze meta-analyse werd geüpdatet met drie recente RCT’s (Attar, 2016; Parsa, 2017; Nasrollahi, 2017). In deze studies werd lachgas in doseringen variërend van 30-50% vergeleken met toegediende zuurstof of gecomprimeerde lucht. Lachgas werd in alle studies gegeven in de eerste fase van de bevalling (tot het moment van volledige ontsluiting en start van persweeën).
Daarnaast werden twee RCT’s geïncludeerd die lachgas vergelijken met andere pijnbehandelingen zoals pethidine (Mobaraki, 2016) en remifentanil (Volmanen, 2005). De cross-over studie van Volmanen (2005), onderzocht de effecten van lachgas (50%) in vergelijking met remifentanil. In totaal werden 20 patiënten gerandomiseerd, waarbij groep I startte met lachgas (50%) en vervolgens werd behandeld met remifentanil (bolus van 0,4 µg/kg met één minuut lockout tijd) en groep II startte met remifentanil en vervolgens overging naar lachgas. De cross-over tijd van beide interventies was 20 minuten en werd gevolgd door een neutralisatie periode van 20 minuten. Zwangeren met een ongecompliceerde zwangerschap, in de ontsluitingsfase (< 7 cm ontsluiting), met een kind in hoofdligging en met het verzoek tot behandelen van de baringspijn werden geïncludeerd. De RCT van Mobaraki (2016) onderzocht het effect van lachgas in vergelijking tot pethidine. In deze studie werd lachgas in een concentratie van 50% gebruikt en kregen zwangeren in de pethidine-groep 0,5 mg/kg pethidine intramusculair geïnjecteerd. De studies van Attar (2016) en Carstoniu (1994) includeerden zowel primipara als multipara zwangeren. De studies van Cheng (2001), Mobaraki (2016), Parsa (2017), Rezaeipour (2008) en Volmanen (2005) includeerden alleen primipara. De studies van Talebi (2009) en Zhang (2001) rapporteerden niet over pariteit. Tabel 1 geeft een overzicht van de belangrijkste studiekarakteristieken van de geïncludeerde studies.
Tabel 1 Geïncludeerde studies: lachgas versus placebo of andere medicamenteuze pijnbehandeling
|
Studie |
N |
Design |
Interventie |
Controle |
|
1. lachgas versus placebo |
||||
|
Attar, 2016 |
400 |
RCT |
N2O (50%) + O2 (50%) |
O2 |
|
Carstoniu, 1994 |
26 |
Cross-over RCT |
N2O (50%) + O2 (50%) |
Compressie lucht |
|
Cheng, 2001 |
50 |
RCT |
N2O (30-50%) + O2 |
Compressie lucht |
|
Nasrollahi, 2017 |
178 |
RCT |
N2O (50%) + O2 (50%) |
O2 |
|
Parsa, 2017 |
120 |
RCT |
N2O (50%) + O2 (50%) |
O2 |
|
Rezaeipour, 2008 |
155 |
RCT |
N2O (50%) + O2 (50%) |
O2 |
|
Talebi, 2009 |
534 |
RCT |
N2O (50%) + O2 (50%) |
O2 |
|
Zhang, 2001 |
110 |
RCT |
N2O (30-50%) + O2 (5 L∙min-1) |
O2 |
|
2. lachgas versus pethidine |
||||
|
Mobaraki, 2016 |
100 |
RCT |
N2O (50%) + O2 (50%) |
Pethidine 0,5 mg∙kg-1 |
|
3. lachgas versus remifentanil |
||||
|
Volmanen, 2005 |
20 |
Cross-over RCT |
N2O (50%) + O2 (50%) |
PCA 0,4 mcg/kg lockout 1 min |
Resultaten
Lachgas versus placebo
Pijnintensiteit (cruciale uitkomstmaat)
Pijnintensiteit werd in drie van de zeven RCT’s onderzocht (Attar, 2016; Parsa, 2017; Talebi, 2009). Alle studies maakten gebruik van een VAS-schaal of NRS-schaal welke één uur na start van de eerste pijnbehandeling werd afgenomen. De schalen variëren van 0= geen pijn tot 10= meest erge pijn. Het verschil in gemiddelde VAS-score één uur na start van de pijnbehandeling was -2,23 punt (95%BI (-4,10; -0,37)), statistisch significant in het voordeel van de lachgas-groep vergeleken met de placebogroep (p<0,02; n= 1029 zwangeren). Er is sprake van hoge statistische heterogeniteit waardoor de meta-analyse kritisch moet worden beschouwd (I2= 99%).
Figuur 1 Forestplot met meta-analyse voor de uitkomstmaat pijnintensiteit
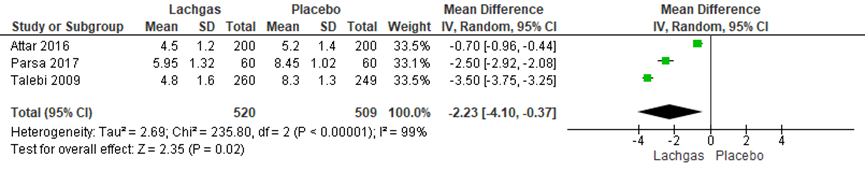
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2 statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat pijnintensiteit is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias) en met één niveau vanwege imprecisie (breed betrouwbaarheidsinterval). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Braken
Het aantal zwangeren dat braakte tijdens de pijnbehandeling werd in vier van de zeven RCT’s onderzocht (Attar, 2016; Parsa, 2017; Talebi, 2009; Zhang, 2001). Het percentage zwangeren dat braakte was 5,2% in de lachgasgroep (n= 30) versus 2,1% in de placebogroep (n= 12). Het gemiddelde relatieve risico op de bijwerking braken was hoger in de lachgasgroep vergeleken met de placebogroep (RR= 2,06; 95%BI (1,09;3,91); p= 0,03; n= 1139 zwangeren). Er is geen sprake van statistische heterogeniteit (I2= 0%).
Figuur 2 Forestplot met meta-analyse voor de uitkomstmaat braken
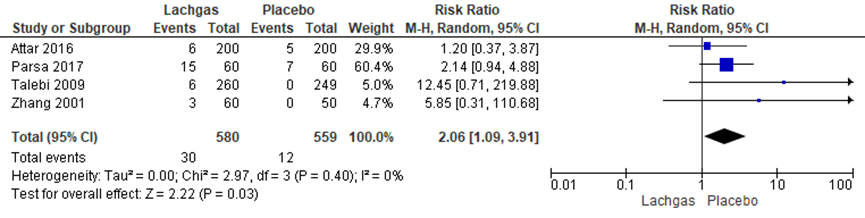
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2 statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat braken is met twee niveau’s verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias) en het geringe aantal events (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Misselijkheid
Misselijkheid werd in vier van de zeven RCT’s onderzocht (Attar, 2016; Parsa, 2017; Talebi, 2009; Nasrollahi, 2017). Het percentage zwangeren dat misselijk was betrof 12,2% in de lachgasgroep (n= 74) versus 3,7% in de placebogroep (n= 19). Het relatieve risico op misselijkheid was RR= 7,71 (95%BI= (0,48; 123,69); p= 0,15 ; n= 1207 zwangeren), geen statistisch significant verschil tussen beide groepen. Er was sprake van een hoge mate van heterogeniteit (I2= 91%). Vanwege de statistische heterogeniteit moet de meta-analyse kritisch worden beschouwd.
Figuur 3 Forestplot met meta-analyse voor de uitkomstmaat misselijkheid
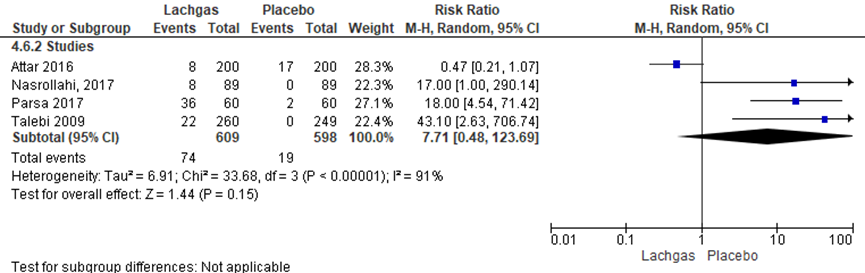
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2 statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat misselijkheid is met twee niveaus verlaagd gezien de beperkingen in onderzoeksopzet (risk of bias) en vanwege heterogeniteit tussen de puntschatters (inconsistentie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Sectio caesarea (wijze van bevallen)
Het aantal zwangeren bevallen middels een sectio caesarea werd in drie van de zeven RCT’s onderzocht (Rezaeipour, 2008; Zhang, 2001; Nasrollahi, 2017). Het percentage zwangeren met een sectio caesarea was 10,6% in de lachgasgroep (n= 24) en 13% in de placebogroep (n= 28). Het gemiddelde relatieve risico voor het aantal zwangeren met een sectio caesarea was RR= 0,80 (95%BI= (0,48; 1,33); p= 0,79; n= 443 zwangeren), geen statistisch significant verschil tussen de twee groepen. Er is geen sprake van statistische heterogeniteit (I2= 0%).
Figuur 4 Forestplot met meta-analyse voor de uitkomstmaat sectio caesarea
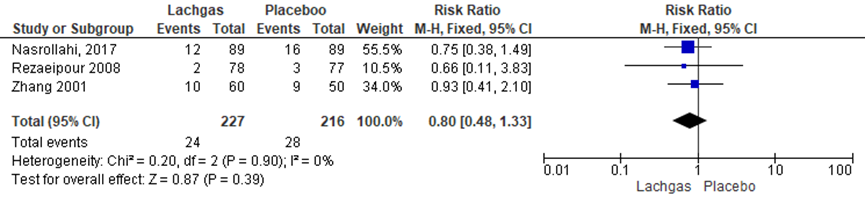
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2 statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat sectio caesarea is met twee niveaus verlaagd gezien het geringe aantal events en brede betrouwbaarheidsinterval (-2 graderingen imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Bevallingsduur - ontsluitingsfase
De bevallingsduur werd in twee RCT’s onderzocht (Attar, 2008; Parsa, 2017). De totale duur van de ontsluitingsfase was significant korter bij zwangeren in de lachgasgroep vergeleken met zwangeren in de placebogroep (gemiddeld verschil: -39,70 min; 95%BI= (-52,95; -26,46 min; p<0,001; n= 518). Er is sprake van een hoge mate van statistische heterogeniteit (I2 66%).
Figuur 5 Forestplot met meta-analyse voor de uitkomstmaat bevallingsduur ontsluitingsfase
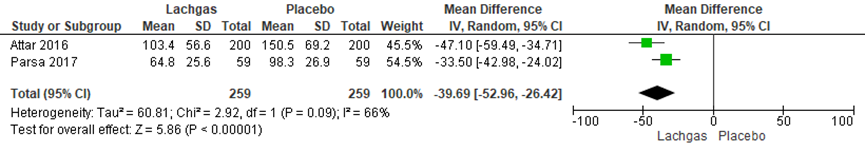
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2 statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat bevallingsduur is met één niveau verlaagd vanwege beperkingen in de onderzoeksopzet (risk of bias). Het niveau van bewijskracht wordt gegradeerd als ‘redelijk’.
Bevallingsduur - uitdrijvingsfase
De bevallingsduur van de uitdrijvingsfase werd in twee RCT’s onderzocht (Attar, 2016 Parsa, 2017). De totale uitdrijvingsduur verschilde niet significant tussen zwangeren in de lachgasgroep in vergelijking met zwangeren in de placebogroep (gemiddeld verschil: -10,93 min; 95%BI= (-27,45; 5,59 min); p<0,19; n= 518). Er is sprake van hoge statistische heterogeniteit (I2= 91%).
Figuur 6 Forestplot met meta-analyse voor de uitkomstmaat Bevallingsduur uitdrijvingsfase
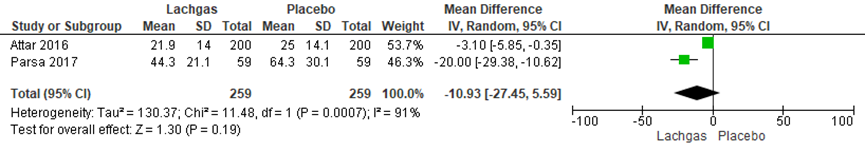
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2 statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat bevallingsduur is met twee niveaus verlaagd vanwege beperkingen in de onderzoeksopzet (risk of bias) en het brede betrouwbaarheidsinterval (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Sufheid (als maat voor sedatie)
Het aantal zwangeren met klachten van sufheid als maat voor sedatie tijdens de pijnbehandeling werd in drie van de zeven RCT’s onderzocht (Attar, 2016; Nasrollahi, 2017; Talebi, 2009). De studies rapporteerden deze uitkomstmaat als ‘drowsiness’ (niet nader gedefinieerd) volgens een dichotome schaal. Het percentage zwangeren met klachten van sufheid was 13,1% in de lachgasgroep (n= 72) versus 2,0% in de placebogroep (n= 11). Het gemiddelde relatieve risico voor het aantal zwangeren met klachten van sufheid was RR= 13,37 (95% BI 0,25 tot 722,55); p= 0,20; n= 1087 zwangeren), geen statistisch significant verschil tussen de twee groepen. Er is sprake van een hoge mate van statistische heterogeniteit (I2= 91%).
Figuur 7 Forestplot met meta-analyse voor de uitkomstmaat sufheid (als maat voor sedatie) uitdrijvingsfase
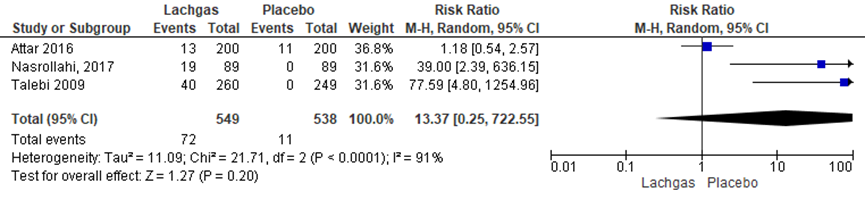
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2 statistische heterogeniteit; CI: betrouwbaarheidsinterval
Overige uitkomstmaten
Geen van de studies naar het effect van lachgas versus placebo rapporteerde de uitkomstmaten: borstvoeding, vaginale kunstverlossing, ademhalingsdepressie, hypotensie, jeuk, opname NICU, foetale harstslag afwijkingen, Apgarscore < 7 bij 5 min, pH a. umbilicalis, mortaliteit, negatieve uitkomstmaten voor de baby op lange termijn of kosten. De uitkomstmaat tevredenheid werd in de studie van Attar (2016) niet bij de zwangere zelf gemeten, maar onderzoekers deden een aanname van hoge tevredenheid indien klachten als misselijkheid/braken niet voorkwamen. Deze meting voldoet niet aan de schaal waarmee de werkgroep deze uitkomst beoogde te meten en is om die reden niet gerapporteerd.
Lachgas versus pethidine
Pijnintensiteit - gemeten 30 en 60 minuten na start analgesie
De pijnintensiteit op 30 en 60 minuten na de start van de analgesie is in één studie onderzocht (Mobaraki, 2016, n= 100) met de VAS-schaal. Mobaraki (2016) rapporteerde een significant lagere VAS-score 30 minuten na start van de pijnbehandeling bij zwangeren behandeld met lachgas vergeleken met pethidine (resp. 3,94 ± 1,4 versus 5,6 ± 1,1) met een gemiddeld verschil van -1,66 cm (95% BI -2,15 tot -1,17; P<0,001). Echter, 60 minuten na start pijnbehandeling werd geen statistisch significant verschil meer waargenomen tussen de lachgas-groep en de pethidine groep (respectievelijk 5,06 ± 1,4 versus 4,7 ± 1,1) met een gemiddeld verschil van 0,36 punt (95% BI -0,13 tot 0,85; P= 0,15).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat pijnintensiteit is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias) en het geringe aantal patiënten (1 RCT; imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Bevallingsduur - ontsluitingsfase
De duur van de ontsluitingsfase werd gemeten in de studie van Mobaraki (2016) (n= 100). De duur van de ontsluitingsfase verschilde niet significant tussen zwangeren behandeld met lachgas vergeleken met pethidine (respectievelijk 3,15 ± 1,65 uur versus 3,65 ± 1,76 uur) met een gemiddeld verschil van een half uur (MD -0,50 uur (95% BI -1,17 tot 0,17 uur; P=0,14). Ook de duur van de uitdrijvingsfase verschilde niet significant tussen zwangeren behandeld met lachgas vergeleken met pethidine (33,7 ± 13,2 versus 31,6 ± 7,5 minuten) met een gemiddeld verschil van 2,10 minuten (95% BI 2,11 tot 6,31 minuten; P=0,33).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat bevallingsduur is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias), het geringe aantal patiënten (1 RCT; imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Bevallingsduur - uitdrijvingsfase
De duur van de uitdrijvingsfase werd gemeten in de studie van Mobaraki (2016) (n= 100). De duur van de uitdrijvingsfase verschilde niet significant tussen zwangeren behandeld met lachgas vergeleken met pethidine (respectievelijk 33,7 ± 13,2 minuten versus 31,6 ± 7,5 minuten) met een gemiddeld verschil van een half uur (MD -2,1 minuten (95% BI 2,11 tot 6,31 minuten; P=0,33).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat duur uitdrijvingsfase is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias), het geringe aantal patiënten (1 RCT; imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Overige uitkomstmaten
Geen van de studies naar het effect van lachgas versus pethidine rapporteerde de uitkomstmaten: cross-over naar aanvullende pijnbehandeling, borstvoeding, vaginale kunstverlossing, ademhalingsdepressie, hypotensie, jeuk, opname NICU, foetale harstslag afwijkingen, Apgarscore < 7 bij 5 min, pH a. umbilicalis, mortaliteit, negatieve uitkomstmaten voor de baby op lange termijn, kosten of tevredenheid
Lachgas versus remifentanil
Pijnintensiteit
In één kleine cross-over RCT (Volmanen, 2005; n= 15) werd de pijnintensiteit gemeten met een VAS-schaal 0, 10, 20, 30 en 40 minuten na de start van de pijnmedicatie. De mediane verschilscore op de VAS-schaal was 1,5 punt (25ste - 75ste percentiel 1,0 tot 3,0) in de remifentanil-groep en 0,5 punt (25ste - 75ste percentiel -0,5 tot 1,0) in de lachgas-groep, significant in het voordeel van de remifentanil groep (P= 0,01).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat pijnintensiteit is met drie niveaus verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias), het zeer geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘zeer laag’.
Jeuk
In één kleine cross-over RCT (Volmanen, 2005; n=15) werd de klacht jeuk onderzocht. Er werd geen significant verschil waargenomen in het risico op jeuk bij zwangeren behandeld met lachgas vergeleken met remifentanil (RR= 4,44; 95%BI= (0,23; 85,8); p= 0,32, n=15).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat jeuk is met drie niveau’s verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias) en het zeer geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘zeer laag’.
Misselijkheid
In één kleine cross-over RCT (Volmanen, 2005; n=15) werd de klacht misselijkheid onderzocht. Er werd geen significant verschil waargenomen in het risico op misselijkheid bij zwangeren behandeld met lachgas vergeleken met remifentanil (RR= 0,88; 95%BI= (0,32; 2,42); p= 0,80, n= 15).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat misselijkheid is met drie niveau’s verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias) en het zeer geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘zeer laag’.
Foetale hartslagafwijkingen
Foetale hartslagafwijkingen werd onderzocht in één kleine cross-over RCT (Volmanen, 2005; n=15). Foetale hartslagafwijkingen werden bij drie foetussen geconstateerd (alle drie verlaagde hartslag variabiliteit) en bij drie foetussen tijdens de behandeling met lachgas (één met vroege hartslag vertragingen en twee met verlaagde hartslag variabiliteit), geen statistisch significant verschil tussen beide behandelmodaliteiten (RR= 1,0; 95%BI= (0,23; 4,31); p= 1,00, n=15).
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat foetale hartslagafwijkingen is met drie niveau’s verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias) en het zeer geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘zeer laag’.
Sedatie
In één kleine cross-over RCT (Volmanen, 2005; n=15) werd de mate van sedatie onderzocht met een 4-punts NRS-score (3=ernstig; 2=gemiddeld; 1=mild; 0=geen). De gemiddelde NRS-score voor sedatie was 2,0 (1,5-2,5) in de groep zwangeren behandeld met remifentanil en 0,5 in de groep zwangeren behandeld met lachgas (MD= 1,50; 95%BI= (0,31; 2,68); p=0,001), een statistisch significant hogere NRS-score in de groep zwangeren behandeld met remifentanil.
Bewijskracht van de literatuur
De bewijskracht van de uitkomstmaat sedatie is met drie niveaus verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias) en het zeer geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘zeer laag’.
Overige uitkomstmaten
Geen van de studies naar het effect van lachgas versus remifentanil rapporteerden de uitkomstmaten: tevredenheid ten aanzien van pijnstilling, cross-over naar aanvullende pijnbehandeling, borstvoeding, sectio caesarea, vaginale kunstverlossing, ademhalingsdepressie, hypotensie, jeuk, opname NICU, foetale harstslag afwijkingen, Apgarscore < 7 bij 5 min, pH a. umbilicalis, mortaliteit, negatieve uitkomstmaten voor de baby op lange termijn of kosten.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat zijn de (on)gunstige effecten van geïnhaleerde analgesie vergeleken met placebo/ andersoortige medicamenteuze pijnbehandeling bij zwangere vrouwen met het verzoek tot behandelen van de baringspijn?
P: zwangere vrouwen met het verzoek tot behandelen van de baringspijn;
I: lachgas;
C: placebo/ andere medicamenteuze pijnbehandeling (epidurale analgesie, remifentanil, tramadol, pethidine, morfine);
O: zie onderstaand:
- Pijnintensiteit (VAS/ NRS-schaal of een ander gevalideerd instrument).
- Cross-over naar andere/ aanvullende pijnmedicatie.
- Tevredenheid ten aanzien van pijnstilling (rapportcijfer/ VAS/ NRS-schaal of een ander gevalideerd instrument).
- Borstvoeding.
- Modus partus (vaginale kunstverlossing, sectio caesarea).
- Maternale uitkomsten: ademhalingsdepressie, bevallingsduur, hypotensie, misselijkheid/braken, jeuk, sedatie (volgens gevalideerde schalen bijvoorbeeld NRS).
- Neonatale complicaties: opname NICU, Apgarscore < 7 bij 5 min, foetale hartslag afwijkingen, temperatuur, pH a. umbilicalis, negatieve uitkomsten voor de baby op lange termijn.
- Maternale/ neonatale sterfte.
- Kosten.
Relevante uitkomstmaten
De werkgroep achtte pijnintensiteit, tevredenheid en modus partus (vaginale kunstverlossing, sectio caesarea) voor de besluitvorming kritieke uitkomstmaten; en misselijkheid/braken en Apgarscore < 7 bij 5 min voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde de uitkomstmaten als volgt: pijnintensiteit (VAS/ NRS-schaal of een ander gevalideerd instrument) en tevredenheid (rapportcijfer/ VAS/ NRS-schaal of een ander gevalideerd instrument), waarbij scores door de patiënt zelf gedurende of direct na de bevalling gerapporteerd werden. Voor neonatale Apgarscore wordt de definitiescore van ≤ 7 bij 5 minuten aangehouden. Voor de overige uitkomstmaten definieerde de werkgroep niet a priori de genoemde uitkomstmaten, maar hanteerde de in de studies gebruikte definities.
Naast significantie wordt de klinische besluitvorming vooral bepaald door de klinische relevantie van de waargenomen verschillen tussen behandelopties. Voor dichotome uitkomstmaten definieerde de werkgroep een minimaal klinisch (patiënt) relevant verschil volgens de grenzen van de GRADE-working group, namelijk een verschil in relatief risico van 25%. Voor de continue kritieke uitkomstmaten definieerde de werkgroep een verschil van 10% op pijnintensiteit of tevredenheid als een klinisch (patiënt) relevant verschil.
Zoeken en selecteren (Methode)
In de databases Medline (OVID), Embase (via Embase.com) en de Cochrane Library (via Wiley) is op 26 februari 2018 met relevante zoektermen gezocht naar studies die pijnbehandeling middels lachgas vergeleken met placebo of andere medicamenteuze vormen van pijnbehandeling (epidurale analgesie, remifentanil, tramadol, pethidine, morfine) bij zwangere vrouwen met baringspijn. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 329 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: voldoet aan de PICO-criteria, één van volgende studiedesigns: RCT of systematische reviews en beschrijft minimaal één van de bovengenoemde uitkomstmaten. Op basis van titel en abstract werden in eerste instantie 38 studies voorgeselecteerd, waaronder een systematische Cochrane review van Klomp (2012). Na raadpleging van de volledige tekst, werd de meta-analyse van Klomp (2012) als uitgangspunt gebruikt en aangevuld met vijf recente RCT’s (Attar, 2016; Mobaraki, 2016; Nasrollahi, 2017; Parsa, 2017; Volmanen, 2005). Er werden 32 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), waaronder de individuele studies die al in de meta-analyse van Klomp (2012) zijn geïncludeerd. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk of bias tabellen.
- Klomp T, van Poppel M, Jones L, Lazet J, Di Nisio M, Lagro-Janssen AL. Inhaled analgesia for pain management in labour. Cochrane Database Syst Rev. 2012 Sep 12;(9):CD009351. doi: 10.1002/14651858.CD009351.pub2. Review. PubMed PMID: 22972140.
- Attar AS, Feizabadi AS, Jarahi L, Feizabadi LS, Sheybani S. Effect of Entonox on reducing the need for Pethidine and the Relevant Fetal and Maternal Complications for Painless Labor. Electron Physician. 2016 Dec 25;8(12):3325-3332. doi: 10.19082/3325. eCollection 2016 Dec. PubMed PMID:28163844; PubMed Central PMCID: PMC5279962.
- Parsa P, Saeedzadeh N, Roshanaei G, Shobeiri F, Hakemzadeh F. The Effect of Entonox on Labour Pain Relief among Nulliparous Women: A Randomized Controlled Trial. J Clin Diagn Res. 2017 Mar;11(3):QC08-QC11. doi: 10.7860/JCDR/2017/21611.9362. Epub 2017 Mar 1. PubMed PMID: 28511452. Central PMCID: PMC5427378.
- Nasrollahi, S., Otogara, M., Jahan-ara, S., & Shayan, A. (2017). An investigation into the side effects of entonox on primiparas in painless labor. Indian Journal of Forensic Medicine & Toxicology, 11(2), 232-236.
- Mobaraki N, Yousefian M, Seifi S, Sakaki M. A Randomized Controlled Trial Comparing Use of Enthonox With Pethidine for Pain Relief in Primigravid Women During the Active Phase of Labor. Anesth Pain Med. 2016 Jul 24;6(4):e37420. eCollection 2016 Aug. PubMed PMID: 27843776; PubMed Central PMCID: PMC5100341.
- Volmanen P, Akural E, Raudaskoski T, Ohtonen P, Alahuhta S. Comparison of remifentanil and nitrous oxide in labour analgesia. Acta Anaesthesiol Scand. 2005 Apr;49(4):453-8. PubMed PMID: 15777291.
- KNOV, 2011 Randvoorwaarden voor het gebruik van Relivopan in eerstelijns geboortecentra:
- http://www.lachgasbevalt.nl/assets/files/files/Randvoorwaarden%20voor%20het%20gebruik%20van%20Relivopan%20in%20eerstelijns%20geboortecentra.pdf
Evidence table for systematic review of RCTs and observational studies (intervention studies)
Research question: Wat is de effectiviteit van Lachgas vergeleken met placebo/ andersoortige medicamenteuze pijnbehandeling bij zwangere vrouwen met met verzoek tot behandelen van de baringspijn?
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Klomp, 2012
Study characteristics and results are extracted from the SR (unless stated otherwise) |
SR and meta-analysis of (RCTs)
A: Carstoniu, 1994; B: Cheng, 2001; C: Rezaeipour, 2008; D: Talebi, 2009; E: Zhang, 2001 F: Volmanen, 2005
Study design: RCT
|
Inclusion criteria SR:
All published randomised controlled trials comparing combined Entonox with placebo during labour. Women in labour including women in high-risk groups, e.g. preterm labour or following induction of labour. Randomised controlled trials (RCTs) and studies with a cross-over design were included. We did not include quasi-RCTs.
8 studies included
Important patient characteristics at baseline:
A: n=26 participants, 14 in the experimental group and 12 controls. Exclusion criteria: age < 18 years, maternal cardiorespiratory disease, fetal distress, any condition affecting the accuracy of pulse oximetry or the use of opioids or regional anaesthesia. B: n=75 participants, 25 in each group. Inclusion criteria: healthy full term 22-30 years old singleton vertex presentation primipara. C: n=155 participants, 78 in the experiment group and 77 in the control group Inclusion criteria: primipara, 18-35 years of age, not have used any anaesthesia, not for inducing labour. D: n=534 ASA I and II parturients, 260 in experimental group and 249 in control group Inclusion criteria: scheduled for elective labour, term (38-42 weeks) in active stage of labour (dilation more then 4 cm). E: n=20 participants, 10 in the experimental group (Entonox) en 10 in the Remifentanil group.
Groups comparable at baseline? YES |
Describe intervention:
A: Experimental group received self-administered 50% nitrous oxide and oxygen for 5 consecutive contractions; B: Experimental group received nitrous oxide 30% to 50% and oxygen; C: Experiment group received Entonox; D: Experimental group received self-administration of pre-prepared mixture of 50% nitrous oxide and oxygen started as early as the onset of pain with each contraction; E: Experiment group received 30% to 50% nitrous oxide and oxygen 5L/min; F: Experimental group received 50% nitrous oxide (Entonox). |
Describe control:
A: Control group received 5 contractions compressed air; B: Control group received air; C: Control group inhaled oxygen; D: Control group received self-administration of 50% oxygen as early as the onset of pain with each contraction; E: Controls received only oxygen 5L/min; F: Control group received remifentanil hydrochloride, Glaxo Operations UK Ltd, Barnard Castle, Durham, UK; diluted with saline and given as a solution of 25 mg/ml). |
End-point of follow-up: na
For how many participants were no complete outcome data available? (intervention/control) A: Entonox (n=3); Control (-) B: Entonox (-); Control (-) C: Entonox (n=2); Control (n=3) D: Entonox (-); Control (-) E: Entonox (-); Control (-) F: Entonox (n=4); Remifentanil (n=1)
|
Entonox versus Placebo
Outcome measure Pain intensity. Effect measure: mean difference (95% CI): D: -0,70 (-0,96; -0,44) F: -2,50 (-2,92; -2,08) Pooled effect: -2,17 (95% CI -2,34 to -2,01) favoring Entonox Heterogeneity (I2): 99%
Outcome measure Vomiting. Effect measure: Risk Ratio (95% CI):
D: 12,45 (0,71; 219,88) E: 5,85 (0,31; 110,68) F: 1,20 (0,37; 3,87) G: 2,14 (0,94; 4,88) Pooled effect: 2,34 (95% CI 1,26 to 4,36) favoring Control Heterogeneity (I2): 0%
Outcome measure Nausea. Effect measure: Risk Ratio (95% CI): D: 43,10 (2,63; 706,74) F: 0,47 (0,21; 1,07) G: 18,00 (4,54; 71,42) H: 17,00 (1,00; 290,14) Pooled effect: 3,72 (95% CI 2,29 to 6,04) favoring Control Heterogeneity (I2): 91%
Outcome measure Sectio Caesarea. Effect measure: Risk Ratio (95% CI): C: 0,66 (0,11; 3,83) E: 0,93 (0,41; 2,10); H: 0,75 (0,38; 1,49); Pooled effect: 0,80 (95% CI 0,48 to 1,33) No difference. Heterogeneity (I2): 0%
Outcome measure Duration labour 1st stage. Effect measure: Mean difference (95% CI): F: -47,10 (-59,49; 34,71) G: -33,53 (-43,00; -24,06) Pooled effect: -39,70 (95% CI -52,95 to -26,46) favoring Control Heterogeneity (I2): 66%
Outcome measure Duration labour 2nd stage. Effect measure: Mean difference (95% CI): F: -3,10 (-5,85; 0,35) G: -19,99 (-29,38; -10,60) Pooled effect: -10,92 (95% CI -27,43 to -5,58) No difference. Heterogeneity (I2): 91%
Entonox versus Pethidine
Outcome measure Pain intensity 30 minutes. Effect measure: mean difference (95% CI): I: -1,66 (-2,15; -1,77) favouring Entonox
Outcome measure Pain intensity 60 minutes. Effect measure: mean difference (95% CI): I: 0,36 (-0,13; 0,85) no difference
Outcome measure Duration labour 1st stage. Effect measure: Mean difference (95% CI): I: -0,50 (-1,17; 0,17) no difference
Outcome measure Duration labour 2nd stage. Effect measure: Mean difference (95% CI): I: 2,10 (-2,11; 6,31) no difference.
Entonox versus Remifentanil Outcome measure Pain intensity Effect measure: mean difference (95% CI): K: 0,50 (-0,50 (25th); 1,00 (75th)) favouring remifentanil
Outcome measure Itching Effect measure: Relative Risk (95% CI): K: 4,44 (0,23; 85,8) no difference
Outcome measure Nausea Effect measure: Relative Risk (95% CI): K: 0,88 (0,32; 2,42) no difference |
Facultative:
Brief description of author’s conclusion:
‘Inhaled analgesia appears to be effective in reducing pain intensity and in giving pain relief in labour. However, substantial heterogeneity was detected for pain intensity. When inhaled analgesia is compared with no treatment or placebo, nitrous oxide appears to result in even more side effects such as nausea, vomiting, dizziness and drowsiness.’ |
|
Attar, 2016;
|
Design: Double-blind clinical trial |
Insclusion Nulliparous women aged 18-40 years, term pregnancy (37-42 weeks), singleton, normal fetus, no medical and midwifery complications of mother (heart problems or other internal diseases), no use of other anesthesia or analgesia drugs, cephalic presentation, no restrictions for Entonox gas inhalation (respiratory diseases, pneumothorax, history of head trauma), having a dilatation of 4 cm at the beginning of the active phase of delivery when entering into the study, and normal fetal heart pattern were included in this study, and patients with any complication during childbirth, lack of consent to enter the project, and those who required cesarean delivery were excluded from the study. |
Entonox The Entonox group received inhaled gas containing 50% oxygen and 50% nitric oxide and the Oxygen group received 4-6 lit/min oxygen only |
Control: The control group inhaled oxygen |
Na. |
Outcome measures Pain intensity Entonox: 4,5 ± 1,2 O2: 5,2 ± 1,4 95%BI= (-0,96 tot -0,44)
Outcome measures Vommiting Entonox: 6/200 O2: 5/200 95%BI= NS
Outcome measures Nausea Entonox: 8/200 O2: 17/200 95%BI= (0,21 tot 1,07)
Outcome measures Time delivery phase I Entonox: 103,4 (56,6) O2: 150,5 (69,2) 95%BI= (-59,5 tot -34,7)
Outcome measures Time delivery phase II Entonox: 21,9 (14) O2: 25 (14,1) 95%BI= (-5,85 tot -0,35)
Outcome measures Drowsiness Entonox: 6/200 O2: 5/200 95%BI= NS
|
‘In our study, Entonox significantly reduced pain during delivery without significant increase in maternal and neonatal complications.’ |
|
Parsa, 2017 |
Design: Clinical controlled trial study |
Insclusion Inclusion criteria were, nulliparous, spontaneous active phase (cervical dilatation 3-4 cm and effacement of 40-50%). The exclusion criteria were: medical problems in foetus or mother, administration of oxytocin, narcotics and sedatives. Informed consent forms were obtained. A basic demonstration was presented to mothers for the use of masks during uterine contractions. |
Entonox ENTONOX® cylinders made by BOC health care factory were used. They were supplied to the following specification: oxygen 50.0%±2.0%; nitrous oxide 50.0%±2.0%. |
Control For the control group in labour room, only oxygen gas inhalation was initiated at the onset of pain with each contraction. |
NA. |
Outcome measures Pain intensity Entonox: 5,95 ± 1,32 O2: 8,45 ± 1,02 95%BI= (-2,92 tot -2,08)
Outcome measures Vommiting Entonox: 15/60 O2: 7/60 95%BI= NS
Outcome measures Nausea Entonox: 36/60 O2: 2/60 95%BI= (4,54 tot 70,42)
Outcome measures Time delivery phase I Entonox: 64,8 (25,6) O2: 98,3 (26,9) 95%BI= (-42,98 tot -24,02)
Outcome measures Time delivery phase II Entonox: 44,3 (21,1) O2: 64,3 (30,1) 95%BI= (-29,38 tot -10,62) |
|
|
Nasrollahi, 2017 |
Design: Randomized controlled trial |
Inclusion: Inclusion criteria were being primipara, gestational age ranging between 37 and 42 weeks, being in the active labor phase (3-4 cm dilation), absence of medical problems and lack of obstetric complications in the mother and fetus |
Entonox After entering the active phase of labor, the patients could inhale 2-6 times in the face mask connected to Entonox cylinder, 30 seconds before pain started and they could breathe room air in the pain intervals |
Contol The control group used oxygen cylinders after entering the active labor phase. |
NA |
Outcome measures Nausea Entonox: 8/89 O2: 0/89 95%BI= (1,00 tot 290,14)
Outcome measures Sectio C Entonox: 12/89 O2: 16/86 p-value= NS Outcome measures Drowsiness Entonox: 19/89 O2: 0/89 95%BI= (2,39 tot 636,15) |
|
|
Mobaraki, 2012 |
Design: Randomized controlled trial |
Inclusion: The inclusion criteria for this study were the commencement of spontaneous labor pain along with appropriate maternal and fetal indications for vaginal delivery.
|
Entonox Patients were taught to use an Entonox facemaskat the beginning of uterine contractionsandto continue deep inspirations at times when there was pain and cramps. Use of Entonox could be started or cut at any moment during labor according to the needs and preferences of the woman. |
Contol The pethidine group received an intramuscular injection of 0.5 mg/kg of pethidine. If a patient’s pain rated higher than 5 VAS, 0.25 mg/kg of pethidine was injected. |
NA |
Outcome measure: Pain intensity Entonox: 3,94 ± 1,4 Pethidine: 5,6 ± 1,1 95% BI= (-2,15 tot -1,17)
Outcome measure: Delivery time Entnox: 3,15 ± 1,65 uur Pethidine: 3,65 ± 1,76 uur 95% BI = (-1,17 tot 0,17 uur)
|
|
Table of quality assessment for systematic reviews of RCTs and observational studies
Based on AMSTAR checklist (Shea, 2007; BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher, 2009; PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
Research question: Wat is de effectiviteit van CSE vergeleken met epidurale analgesie bij zwangere vrouwen met het verzoek tot behandelen van de baringspijn?
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Klomp, 2012 |
YES |
YES |
Yes |
YES |
Unclear |
YES |
Unclear |
YES |
YES |
- Research question (PICO) and inclusion criteria should be appropriate and predefined.
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched.
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons.
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported.
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs).
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table et cetera).
Risk of bias table for intervention studies (randomized controlled trials)
Research question: Wat is de effectiviteit van CSE vergeleken met epidurale analgesie bij zwangere vrouwen met het verzoek tot behandelen van de baringspijn?
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Attar, 2016 |
By using random number tables, the mothers were randomly allocated to one of the two groups |
Unlikely |
Likely |
Likely |
Unclear |
Unlikely |
Unlikely |
Unlikely |
|
Parsa, 2017 |
The allocation sequence was determined by one of the members of the research team, not involved in the sample selection, using a four-block randomized design. |
Unlikely
|
Unlikely |
Likely |
Unclear |
Unlikely |
Unclear |
Unclear |
|
Nasrollahi, 2017 |
After obtaining the informed consent, the women meeting inclusion criteria were randomly assigned into experimental (89 women inhaling Entonox) and control (89 women inhaling oxygen) groups |
Unclear |
Likely |
Likely |
Unclear |
Unlikely |
Unlikely |
Unlikely |
|
Mobakari, 2012 |
By using random numbers, the subjects were randomly allocated into two groups |
Unclear |
Likely |
Unlikely |
Unclear |
Unclear |
Unlikely |
Unlikely |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear.
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Pinyan, 2017 |
Studiedesign voldoet niet (geen vergelijkende studie) |
|
Weibel, 2017 |
Bevat geen vergelijkende studies volgens PICO |
|
Madden, 2016 |
Voldoet niet aan PICO (gaat over hypnose) |
|
Ji, 2002 |
Artikel in het chinees (Cochrane review van Klomp, 2012 beschrijft enkel de vergelijking met ‘geen behandeling’ niet de vergelijking met CSE). |
|
Devabhakthuni, 2013 |
Review. Echter, Volmanen, 2005 is al beschreven in literatuuranalyse. |
|
Likis, 2014 |
Review. Relevante studies worden al beschreven in de literatuuranalyse. |
|
Likis, 2012 |
Review. Relevante studies worden al beschreven in de literatuuranalyse. |
|
Rosen, 2002 |
Review. Relevante studies worden al beschreven in de literatuuranalyse. |
|
Zhonghua, 2002 |
Artikel in het chinees. |
|
Shao, 2000 |
Artikel in het chinees. |
|
Othman, 2012 |
Bevat geen vergelijkende studies volgens PICO |
|
Rooks, 2011 |
Narrative review |
|
Ullman, 2010 |
Bevat geen vergelijkende studies volgens PICO |
|
Varposhti, 2013 |
Vergelijking voldoet niet aan PICO |
|
Pita, 2012 |
Studiedesign voldoet niet (geen vergelijkende studie) |
|
Zare, 2010 |
Artikel in het Arabisch. |
|
Griffin, 1995 |
Vergelijking voldoet niet aan PICO |
|
Arfeen, 1994 |
Geen gerandomiseerde studie |
|
Griffin, 1994 |
Geen gerandomiseerde studie |
|
Ranta, 1994 |
Geen gerandomiseerde studie |
|
Harrison, 1987 |
Vergelijking voldoet niet aan PICO |
|
Stefani, 1982 |
Vergelijking voldoet niet aan PICO |
|
MRC, 1970 |
Vergelijking voldoet niet aan PICO |
|
Richardson, 2017 |
Studiedesign voldoet niet (survey studie) |
|
Feng, 2016 |
Studie design voldoet niet (retrospectieve studie) |
|
Najafi, 2016 |
Studiedesign voldoet niet (geen vergelijkende studie) |
|
Lindholm, 2015 |
Studiedesign voldoet niet (survey studie) |
|
Dammer, 2014 |
Studiedesign voldoet niet (geen vergelijkende studie) |
|
Rosenstein, 2014 |
Betreft een conference abstract |
|
Teimoori, 2011 |
Vergelijking voldoet niet aan PICO combinatie met prometazine |
|
Holdcroft, 1974 |
Vergelijking voldoet niet aan PICO |
|
Zanardo, 2017 |
Observationele studie, maar geen multivariate analyse |
Beoordelingsdatum en geldigheid
Publicatiedatum : 09-07-2020
Beoordeeld op geldigheid : 03-07-2020
Bij het opstellen van de modules heeft de werkgroep een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijnmodules komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De andere aan deze richtlijnmodule deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijnmodule delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
|
Module[1] |
Regiehouder(s)[2] |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn[3] |
Frequentie van beoordeling op actualiteit[4] |
Wie houdt er toezicht op actualiteit[5] |
Relevante factoren voor wijzigingen in aanbeveling[6] |
|
Lachgas versus placebo/medicamenteus |
NVA |
2019 |
2024 |
5-jaarlijks |
NVA |
Nieuwe evidence |
[1] Naam van de module
[2] Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders)
[3] Maximaal na vijf jaar
[4] (half)Jaarlijks, eens in twee jaar, eens in vijf jaar
[5] regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) werkgroep die in stand blijft
[6] Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen
Algemene gegevens
Deze richtlijn is ontwikkeld in samenwerking met:
- Patiëntenfederatie Nederland
De ontwikkeling van de richtlijnmodules werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodules.
Samenstelling werkgroep
- Dr. W.L.M.C.M. Schellekens, extern voorzitter
- Drs. I.C.M. Beenakkers, anesthesioloog, werkzaam in het UMC Utrecht, NVA
- Drs. F.A. Klerk, anesthesioloog, werkzaam in het Diakonessenhuis Utrecht, NVA
- Drs. C.E. Kam-Endtz, anesthesioloog, werkzaam in het Haaglanden Medisch Centrum, NVA
- Dr. F.T.H. Lim, gynaecoloog, werkzaam in het IJssellandziekenhuis, NVOG
- Dr. L.M. Freeman, gynaecoloog, werkzaam in het Ikazia Ziekenhuis Rotterdam, NVOG
- Dr. J.M. Middeldorp, gynaecoloog, werkzaam in het Leids Universitair Medisch Centrum, NVOG
- Drs. A.G. Kaspers, kinderarts, werkzaam in het Medisch Spectrum Twente, NVK
- Drs. L.A.M. Moll, klinisch verloskundige, werkzaam in het St. Antonius Ziekenhuis Nieuwegein, KNOV
- Dr. J. de Boer, beleidsmedewerker bij KNOV
- Drs. S. Ratsma-Wesselius, Obstetrisch verpleegkundige, werkzaam bij het Amsterdam UMC, Locatie AMC, V&VN
- Dr. J.E. Nagtegaal, Ziekenhuisapotheker, werkzaam in het Meander Medisch Centrum, NVZA
- Dr. A.M.D.E. Timmerman, Klinisch Fysicus, werkzaam in het UMC Utrecht, NVKF
- Drs. J.C. Mooij, adviseur patiëntenbelang, Patiëntenfederatie Nederland.
Met ondersteuning van
- Dr. E.M.E. den Breejen, senior adviseur Kennisinstituut van de Federatie Medisch Specialisten
- Dr. W.J. Harmsen, adviseur Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Achternaam werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Schellekens |
gepensioneerd, ZZP: strategisch adviseur |
Lid RvT Diakonessenhuis, Utrecht |
Geen |
Geen |
|
Klerk |
Staf Anesthesiologie Diakonessenhuis Utrecht. |
bestuurslid Obst. Anesth. onbetaald |
Geen |
Geen |
|
Ratsma Wesselius |
Senior verpleegkundige Verloscentrum AMC |
Gastdocent VU Amstel Academie verpleegkundige vervolgopleidingen Obstetrie, betaald |
Geen |
Geen |
|
Beenakkers |
Anesthesioloog WKZ/UMCU |
Voorzitter sectie obstetrische anesthesie van de NVA. Onbetaald |
Echtgenoot werkzaam bij GSK |
Geen |
|
Nagtegaal |
Ziekenhuisapotheker Meander Medisch Centrum |
Beroepenveldcommissie Farmakunde Hogeschool Utrecht, onbetaald |
Geen |
Geen |
|
Timmerman |
Staffunctionaris Klinische Fysica & Patiëntveiligheid |
Lid NIVEL expertgroep infuustechnologie - advies maken kennistoets voor verpleegkundigen – onbetaald Lid ondernemingsraad UMC Utrecht - onbetaald |
EMRP Researcher Grant Metrology for Drug Delivery HLT07- REG1 €120,422.88 USPTO Applicaton #: #20160106909 Apparatus for simultaneous multiple medicament administration |
Geen |
|
Kam-Endtz |
Anesthesioloog Haaglanden MC |
Geen |
Geen |
Geen |
|
Middeldorp |
Gynaecoloog-perinatoloog |
Geen |
Geen |
Geen |
|
Moll |
Klinisch verloskundige/research verloskundige in het St. Antoniusziekenhuis in Nieuwegein |
Geen |
Geen |
Geen |
|
Freeman |
Gynaecoloog |
voorzitter multidiciplinaire werkgroep obstetrische anesthesie
|
Mijn promotieonderzoek naar epidurale analgesie en remifentanil is gesubsidieerd door ZonMw. Dit onderzoek is afgerond maar de resultaten zullen gebruikt worden in deze richtlijn |
Geen |
|
Mooij |
Beleidsmedewerker Patiëntenvereniging Nederland |
Vrijwilligerswerk (onbetaald) patiëntenorganisatie CCUVN |
Geen |
Geen |
|
Kaspers |
Kinderarts-neonatoloog, MST Enschede |
Geen |
Geen |
Geen |
|
De Boer |
Beleidsmedewerker richtlijnontwikkeling |
Geen nevenwerkzaamheden |
Geen |
Geen |
|
Lim |
gynaecoloog |
Geen |
Geen |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde van de Patientenfederatie Nederland in de werkgroep te laten deelnemen. De conceptmodule is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland. Daarnaast is door de Patiëntenfederatie Nederland een achterbanraadpleging verricht, waarvan de uitkomsten zo veel mogelijk meegenomen zijn in de overwegingen van de modules.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van het ontwikkelproces is rekening gehouden met de implementatie van de richtlijnmodule en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de module in de praktijk kunnen bevorderen of belemmeren. De implementatietabel is te vinden bij de aanverwante producten.
Werkwijze
AGREE
Deze modules zijn opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, (2010)), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based module tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse
Uit inventarisatie van de knelpunten door de commissie van de NVA bleek dat er een noodzaak was voor revisie van deze richtlijnmodules. Tevens zijn tijdens een fysieke knelpunteninventarisatie knelpunten aangedragen door aanpalende stakeholders inclusief patiëntenorganisaties. Een verslag hiervan is opgenomen onder aanverwante producten.
Uitgangsvraag en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroepleden en de adviseur uitgangsvragen opgesteld. Vervolgens inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet kritiek) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Aan de hand van specifieke zoektermen werd gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De geselecteerde databases waarin is gezocht en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie is opvraagbaar bij de Richtlijnendatabase, zie het tabblad ‘Zoekverantwoording’ voor verdere details.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: QUADAS II - voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt, kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
Bij de ontwikkeling van de modules is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag, randvoorwaarden die van invloed zijn op de implementatie van de aanbeveling zijn opgenomen in de implementatietabel.
Indicatorontwikkeling
Indicatoren over zwangerschap en geboorte zijn reeds onderdeel van de vervaardigde indicatoren bij de zorgstandaard integrale geboortezorg. Derhalve zijn er bij deze modules geen indicatoren ontwikkeld.
Kennislacunes
Tijdens de ontwikkeling van deze modules is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvraag. Er is nagegaan of (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Mocht dit bij deze module het geval zijn, dan is er een aanbeveling voor het doen van onderzoek opgenomen in de Kennislacunes. Deze zijn te vinden onder de aanverwante producten.
Commentaar- en autorisatiefase
De conceptmodules werden aan de betrokken (wetenschappelijke) verenigingen, instanties en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werden de conceptmodules aangepast en definitief vastgesteld door de werkgroep. De definitieve modules werden aan de deelnemende (wetenschappelijke) verenigingen en de Patiëntenfederatie Nederland voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd. De commentaartabel is op te vragen bij het Kennisinstituut via secretariaat@kennisinstituut.nl
Literatuur
Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, et al. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008;336(7654). doi: 10.1136/bmj.a139. PubMed PMID: 18483053.
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van de Federatie Medisch Specialisten.
Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
Zoekverantwoording
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
(1946-2018)
|
1 exp Labor, Obstetric/ or exp Delivery, obstetric/ or exp Labor pain/ or exp Analgesia, obstetric/ or (labor or labour or childbirth or birth* or parturient*).ti,ab. 431733 2 exp Analgesia, obstetric/ or exp Labor pain/ or analges*.ti,ab. or pain.ti,ab. 571908 3 (Entonox or nitronox or nitralgin or eutonal or relivopan).ti,ab. or (exp Nitrous Oxide/ or (nitrous oxide or n2o or laughing gas).ti,ab.) or ((inhalation or inhaled) adj3 analges*).ti,ab. 22893 4 1 and 2 and 3 338 5 limit 4 to (dutch or english) 286 6 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) 331939 7 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) 1697963 8 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ 2867692 9 5 and 6 14 10 5 and (7 not 6) 88 11 5 and (8 not (6 or 7)) 24 |
(na ontdubbeling) 329 |
|
Embase (Elsevier) |
#8 AND #6 NOT (#5 OR #4) 40 #10 #8 AND #5 NOT #4 179 #9 #8 AND #4 34 #8 #1 AND #2 AND #3 AND ((dutch)/lim OR (english)/lim) 720 #7 #1 AND #2 AND #3 826 #6 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR ('prospective study'/de NOT 'randomized controlled trial'/de) OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (case:ab,ti AND ((control NEAR/1 (study OR studies)):ab,ti)) OR (follow:ab,ti AND ((up NEAR/1 (study OR studies)):ab,ti)) OR ((observational NEAR/1 (study OR studies)):ab,ti) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti) 1,895,346 #5 ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it 2,071,721 #4 ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) NOT 'conference abstract':it 303,184 #3 'nitrous oxide plus oxygen'/exp OR entonox:ab,ti OR nitronox:ab,ti OR nitralgin:ab,ti OR eutonal:ab,ti OR relivopan:ab,ti OR 'nitrous oxide'/exp OR 'nitrous oxide':ab,ti OR n2o:ab,ti OR 'laughing gas':ab,ti OR (((inhalation OR inhaled) NEAR/3 analges*):ab,ti) 39,681 #2 'obstetric analgesia'/exp OR 'labor pain'/exp OR analges*:ab,ti OR pain:ab,ti 837,126 #1 'labor'/exp OR 'obstetric delivery'/exp OR 'labor pain'/exp OR 'obstetric analgesia'/exp OR labor:ab,ti OR labour:ab,ti OR childbirth:ab,ti OR birth*:ab,ti OR parturient*:ab,ti 576,154 |
|
|
Cochrane library (Wiley) |
(labor:ab,ti OR labour:ab,ti OR childbirth:ab,ti OR birth*:ab,ti OR parturient*:ab,ti) and (analges*:ab,ti OR pain:ab,ti) and (entonox:ab,ti OR nitronox:ab,ti OR nitralgin:ab,ti OR eutonal:ab,ti OR relivopan:ab,ti OR 'nitrous oxide':ab,ti OR n2o:ab,ti OR 'laughing gas':ab,ti OR ((inhalation OR inhaled) NEAR/3 analges*):ab,ti) 98 |