Ziektemodulerende middelen
Uitgangsvraag
Met welke ziektemodulerende middelen kan worden gestart bij patiënten met relapsing remitting MS met een indicatie voor behandeling?
Aanbeveling
Overweeg bij patiënten met een actieve relapsing remitting MS in het afgelopen jaar en afwezigheid van meerdere prognostisch ongunstige factoren, zie hieronder, te starten met een over het algemeen veiliger maar minder effectief eerstelijns ziektemodulerende middel. Eerstelijns middelen zijn interferon-β, glatirameer acetaat, fumaraatzuuresters, teriflunomide en de S1P modulatoren.
Overweeg bij patiënten met een actieve relapsing remitting MS in het afgelopen jaar en aanwezigheid van meerdere prognostisch ongunstige factoren, zie hieronder, of patiënten met een zeer actieve relapsing remitting MS direct te starten met een meer effectief maar meer risicovol tweedelijns ziektemodulerend middel. Tweedelijns middelen zijn cladribine, anti-CD20 monoclonalen, natalizumab en alemtuzumab.
Bespreek met patiënt de keuzeopties voor een ziektemodulerend middel. In de afwegingen dienen de volgende factoren meegenomen te worden:
- patiënt voorkeuren en behandeldoelen
- prognostische factoren:
- klinisch prognostische factoren zijn:
- aantal relapses in het afgelopen jaar
- ernst van relapse
- mate van herstel
- symptomen herleidbaar tot lokalisaties infratentorieel of myelum
- radiologisch prognostische factoren zijn:
- op MRI scan het aantal en grootte van T2 laesies op voor MS karakteristieke lokalisatie en aantal lokalisaties.
- prognostisch ongunstig zijn:
- gadolinium aankleurende laesies
- laesies infratentorieel en/of myelum
- unieke oligoclonale banden in liquor.
- prognostisch ongunstig zijn:
- op MRI scan het aantal en grootte van T2 laesies op voor MS karakteristieke lokalisatie en aantal lokalisaties.
- klinisch prognostische factoren zijn:
- comorbiditeit
- huidige of toekomstige zwangerschapswens
- veiligheid van een behandeling
Weeg bij middelen uit een vergelijkbare klasse (interferonen-β, fumaraatzuuresters, anti-CD20-monoclonalen en S1P modulatoren) en verschillende toedieningsvormen (natalizumab i.v. of s.c.) voor de individuele patiënt effectiviteit, neveneffecten en gebruikgemak af tegen de (maatschappelijke) kosten van deze middelen.
Overweeg na gedurende 6 maanden behandeling met natalizumab i.v. met 4 wekelijkse intervallen, de behandeling naar 6 wekelijkse intervallen uit te breiden.
Wees terughoudend met het opstarten van patiënten op natalizumab s.c.. Weeg de voordelen van s.c. toedienen tegenover mogelijke nadelen af. De kans op ontwikkelen van anti-natalizumab antilichamen is bij nataliuzmab s.c. nog niet bekend. Er zijn aanwijzingen dat bij switchen van natalizumab i.v. naar s.c. de spiegels van natalizumab in het bloed dalen
Behandel patiënten met alemtuzumab alleen in een centrum met de juiste IC faciliteiten en neurologen met uitgebreide expertise in MS behandeling en goed op de hoogte zijn van potentiële ernstige bijwerkingen.
Behandel MS patiënten niet met mitoxantron tenzij het verwachte resultaat opweegt tegen de potentiële ernstige risico’s en er geen alternatieve behandeling beschikbaar is.
Overwegingen
Waarden en voorkeuren
Er is volgens de richtlijnwerkgroep grote variatie in de voorkeuren van patiënten wat betreft keuze uit verschillende behandelingen. Shared-decision making is dan ook aangewezen. Shared-decision making is een overlegmodel waarin de behandelend arts patiënten helpt te bepalen wat de beste therapie is die hun waarden en voorkeuren belichaamt (Berger & Markowitz, 2018).
Kwalitatief onderzoek (Schlegel et al., 2018) suggereert dat patiënten een rol in de keuze van medicament op prijs stellen, maar er wel een grote rol van de neuroloog in het omkaderen van afweging van met name doeltreffendheid en veiligheid is. De werkgroep vindt dat er een grote rol voor de clinicus is weggelegd voor het in kaart brengen van prognostische factoren en aan de hand hiervan een triage van mogelijke ziektemodulerende middelen uit te voeren (Rotstein & Montalban, 2019). Patiëntgebonden voorkeuren wat betreft veiligheid en doeltreffendheid zullen hierin meegewogen worden, evenals voorkeuren t.a.v. toedieningswijze, doseringsfrequentie en intensiteit van monitoring (Rotstein & Montalban, 2019). Een geïnformeerde keuze kan wellicht tot een betere therapietrouw leiden.
Professioneel perspectief
Eerste- en tweedelijns ziektemodulerende middelen, toedieningsweg en ziekteprogressie
De relatieve plaatsbepaling van ziektemodulerende therapieën in de behandeling van MS is een uitdagend onderwerp. Voor deze richtlijn is gekozen voor twee benaderingen: de traditionele directe meta-analyse (head-to-head vergelijkingen) met beoordeling van kwaliteit van bewijs binnen het raamwerk van de GRADE-classificatie, en de netwerk meta analyse met beoordeling van kwaliteit van bewijs binnen het raamwerk van de GRADE-classificatie. Aan deze benaderingen zitten beperkingen die uitgebreid toegelicht zijn bij het onderdeel ‘beoordeling van de bewijskracht’.
Over het algemeen geldt dat meer doeltreffende ziektemodulerende middelen ook een relatief groter risico op complicaties laten zien (Merkel et al. 2017). Derhalve vindt de werkgroep dat bij gunstige prognostische factoren en niet-zeer actieve MS, behandeling met een veiliger middel dat wat minder effectief is, een doeltreffende behandeling is.
Prognostische factoren voor het beloop van MS zijn recent gereviewed door Rotstein & Montalban (2019).
Hoewel er variatie tussen individuele studies is, zijn met een ernstiger ziektebeloop geassocieerd:
- oudere leeftijd bij eerste klachten
- mannelijk geslacht
- roken
- lage circulerende 25-hydroxyvitamine D waarden
- niet-Europese afkomst
- veel comorbiditeit
- hoge relapse frequentie
- kort interval tussen eerste en tweede relapse
- hersenstam of ruggenmergsyndroom
- slecht herstel na eerste relapse
- hoge EDSS score bij diagnose
- poly-symptomatisch begin
- vroege cognitieve klachten
- veel T2 laesies op MRI scan
- hoog T2 laesie volume
- gadolinium aankleurende laesies
- infratentoriële en ruggenmerglaesies
- atrofie van grijze stof/gehele hersenen
- aanwezigheid van oligoclonale banden in liquor
Hierbij is de mate van ziekteactiviteit radiologisch en klinisch de belangrijkste voorspeller van prognose. Verder zijn er een aantal nieuwe biomarkers beschreven die een ongunstig beloop voorspellen, zoals lichte keten neurofilament en chitinase, en atrofie van de retinal nerve fiber layer bij optical coherence tomography (OCT). Deze nieuwe biomarkers hebben hun weg naar de klinische praktijk nog niet geheel gevonden (Rotstein & Montalban, 2019).
Eerste- en tweedelijnstherapie
Bij de stratificatie van ziektemodulerende middelen wordt van oudsher in de dagelijkse klinische praktijk de onderverdeling eerste en tweedelijnstherapie gemaakt, ofwel middelen die gemiddeld genomen minder effectief zijn met lager risicoprofiel of doeltreffender ziektemodulerende middelen met een relatief groter risico profiel (Merkel et al. 2017) (zie onderstaande tabel “Indeling van ziektemodulernde MS therapieën”). Deze keuzes zijn gebaseerd op de resultaten uit de literatuuranalyse met betrekking tot effectiviteit en veiligheid (traditionele meta-analyse en netwerk meta-analyse): annualised relapse rate, EDSS en studieuitval vanwege bijwerkingen. Met de eerstelijns middelen (in de internationale literatuur ook wel platformtherapie genoemd) worden de interferonen-b, glatirameeracetaat, teriflunomide en de fumaraatzuuresters bedoeld. De S1P modulatoren worden op basis van effectiviteit en veiligheid tegenwoordig ook als eerstelijns therapie gezien. Als tweedelijns therapieën (in de internationale literatuur ook wel hoog-effectieve therapie genoemd) worden cladribine, anti-CD20 monoclonalen en natalizumab genoemd (Merkel et al., 2017 et al., 2019; Rotstein & Montalban, 2019). Na herbeoordeling door EMA (zie onder) werd alemtuzumab tijdelijk als derdelijns therapie gezien ofwel zoals tegenwoordig geformuleerd: alemtuzumab is bedoeld voor patiënten met zeer actieve MS ondanks adequate behandeling met andere tweedelijns ziektemodulerende therapieën.
Tabel “Indeling van ziektemodulerende MS therapieën”
Middelen die gemiddeld genomen minder effectief zijn met lager risicoprofiel (eerstelijn) of doeltreffender ziektemodulerende middelen met een relatief groter risico profiel (tweedelijn)
|
Eerstelijn |
Tweedelijn |
|
Interferon-β |
|
|
Glatirameeracetaat |
Cladibrine |
|
Teriflunomide |
Anti-CD20 monoclonalen |
|
Fumaraatzuuresters |
Natalizumab |
|
S1P modulatoren (vroege tweedelijn) |
Alemtuzumab* |
*altemtuzumab is bedoeld voor patiënten met zeer actieve MS ondanks adequate behandeling met andere tweedelijns ziektemodulerende therapieën
De onderverdeling van MS therapieën in lijnen is vooral een overblijfsel van de financieringsstructuur rondom de introductie van nieuwe hoog-effectieve MS therapieën. In het huidige behandellandschap wordt de toewijzing van patiënten veel meer maatwerk, gericht op de bovengenoemde prognostische factoren en ziektebeloop. Bij patiënten met actieve ziekte en weinig prognostisch ongunstige risicofactoren dient met een eerstelijns ziektemodulerende therapie gestart te worden, bij aanwezigheid van meerdere ongunstige risicofactoren kan (direct) behandeling met een tweedelijns therapie overwogen worden. Persisterende inflammatoire ziekteactiviteit van MS onder adequate behandeling met een eerstelijns MS therapie wordt gezien als een zwaar wegende ongunstige prognostische factor. In dit raamwerk is de onderverdeling in eerstelijns en tweedelijns therapieën nog bruikbaar om de gelaagdheid in behandelstrategieën weer te geven, maar dient een adequate behandeling van MS bij veel ongunstige prognostische factoren niet ontraden te worden. Anderzijds wordt de keuze voor ziektemodulerende therapieën niet alleen door deze factoren gestuurd, maar ook door patiëntvoorkeuren, zwangerschapswens, etc.
Bij een zeer actieve MS (zie bijlage “Definities en begrippen”) kan overwogen worden om direct met een tweedelijns (hoog effectief) middel te starten.
Alemzutumab
In 2019 heeft alemtuzumab een herbeoordeling van de EMA ondergaan na meldingen van ernstige cardiovasculaire reacties, auto-immuun hepatitis en hemofagocytaire lymfohistiocytose (HLH). De EMA adviseert nu om alemtuzumab alleen voor te schrijven aan mensen met zeer actieve relapsing-remitting MS ondanks gebruik van een ziektemodulerend middel. Alemtuzumab mag niet worden voorgeschreven aan mensen met aandoeningen van het hart, bloedcirculatie, stollingsstoornissen of andere autoimmuunaandoeningen. Alemtuzumab mag alleen voorgeschreven worden in een ziekenhuis met de juiste IC faciliteiten en artsen die goed op de hoogte zijn van potentiële ernstige bijwerkingen en werd daarom in de praktijk gezien als een derdelijns middel.[17] Gezien de gunstige data uit trials en registries, lijkt een plaatsbepaling als tweedelijns therapie waarbij specifieke voorzorgen in acht genomen worden meer op zijn plaats.
[17] https://www.ema.europa.eu/en/medicines/human/referrals/lemtrada
Mitoxantron
De indicatie voor mitoxantron wordt door de EMA gedefinieerd als “Mitoxantrone is indicated for treatment of patients with highly active RRMS associated with rapidly evolving disability where no alternative therapeutic options exist”. De reden hiervoor zijn de ernstige bijwerkingen van mitoxantron, zoals de behandelings-geassocieerde acute lymfatische leukemie en cardiotoxiciteit. De werkgroep is derhalve van mening dat patiënten met MS gezien veelvuldige alternatieven niet met mitoxantron behandeld dienen te worden.[18]
[18] https://www.ema.europa.eu/en/documents/referral/novantrone-article-30-referral-annex-iii_en.pdf.Actueel : 31 juli 2019.
Vaccinatiegereedheid
Bij de keuze voor een ziektemodulerende therapie, ligt sinds de start van de COVID-19 pandemie een grotere nadruk op de invloed die deze therapieën op het beloop van een COVID-19 infectie hebben, maar ook op vaccinatierespons van patiënten onder behandeling. Aan het begin van de COVID-19 epidemie werd aangetoond dat patiënten behandeld met de anti-CD20-monoclonalen ocrelizumab en rituximab een groter risico liepen om met een symptomatische COVID-19 infectie op de intensive care te belanden (Simpson-Yap et al., 2021). Ook werd voor de anti-CD20-monoclonaal ocrelizumab en S1P receptor modulator fingolimod aangetoond dat er een verminderde humorale response op vaccinatie tegen COVID-19 is (Apostolidis et al., 2021; Milo et al., 2022; ). Hierbij werd bij mensen behandeld met ocrelizumab een intacte T cel respons tegen COVID-19 gezien, maar niet bij patiënten behandeld met fingolimod. De werkgroep is van mening dat bij keuze van ziektemodulerende therapie risicofactoren voor een ernstig verlopende COVID-19 infectie in de keuze van ziektemodulerende therapie meegenomen dienen te worden. Dit is geen specifieke overweging voor COVID-19, maar ook algemeen risico op ernstige complicaties van infecties dienen meegenomen te worden, alsmede respons op (reizigers-) vaccinaties bij patiënten die hier een indicatie voor hebben.
Vergelijkbare middelen uit dezelfde klasse
Er zijn voor de behandeling van relapsing MS inmiddels meerdere middelen uit vergelijkbare klassen geregistreerd, welke met name verschillen in gebruik of moleculaire samenstelling. Voorbeelden zijn Interferon-β (interferon-β en peginterferon-β), fumaraatzuur esters (dimethylfumaraat en diroximelfumaraat), S1P receptor modulatoren (fingolimod, ozanimod, ponesimod) en CD20 monoclonalen (ocrelizumab en ofatumumab). Hiernaast worden middelen ook in andere toedieningsvormen aangeboden zoals natalizumab intraveneus en subcutaan. Hierbij zijn de voordelen van individuele middelen spaarzaam direct met elkaar vergeleken.
Eén studie rapporteerde over de vergelijking dimethylfumaraat vs diroximelfumaraat. Er werden gastrointestinale klachten beschreven bij 34.8% van de patiënten in de diroximelfumaraat vs 49.0% van de patiënten in de dimethylfumaraat groep, wat een absoluut verschil gaf van 14.2%. Er stopten minder patiënten in de diroximelfumaraat groep vanwege bijwerkingen of gastrointestinale bijwerkingen (1.6% wn 0.8%, respectievelijk) vergeleken met de dimethylfumaraat groep (5.6% en 4.8%, respectievelijk). Een directe vergelijking van effectiviteit werd niet gerapporteerd. In de dagelijkse praktijk wordt vaak langzamer opgebouwd dan dat in bovenstaande studie gedaan werd, juist om het optreden van bijwerkingen te beperken. Of het verschil in optreden van bijwerkingen bij langzamer opbouwen overeind blijft is onbekend. Eén fase één studie rapporteerde over de directe vergelijking tussen interferon-β en peginterferon-β bij gezonde individuen (Hu et al., 2016).
De vergelijking van natalizumab elke 4 weken i.v. of subcutaan werd door één studie onderzocht (Trojano et al., 2021). Er werd een vergelijkbaar aantal ‘combined unique active lesions’ en aanvallen gerapporteerd in de i.v. en s.c groepen (0.23 vs 0.02 en 7.8% vs. 9.1%, respectievelijk). Behandeling iedere 12 weken was geassocieerd met een hoog risico op aanvallen. Omdat deelnemers aan deze studie patiënten waren die voorafgaand aan de studie onder behandeling met natalizumab i.v. geen anti-natalizumab neutraliserende antilichamen maakten (anti drug antibodies), is het risico op anti-natalizumab antilichamen niet bekend. In deze studie testten geen patiënten positief voor anti-natalizumab antilichamen in de behandelarm na vier weken behandeling. De mate van optreden van anti-natalizumab antilichamen zal in de praktijk nog moeten blijken, en noopt tot terughoudendheid met het opstarten van patiënten met natalizumab s.c. in plaats van i.v. Er zijn aanwijzingen dat bij switchen van natalizumab i.v. naar s.c. de spiegels van natalizumab in het bloed dalen (Toorop et al., 2023). Ook deze onbekendheid met natalizumab s.c. draagt er toe bij dat er op dit moment zorgvuldig de voor- en nadelen van nataliuzmab s.c. versus i.v. afgewogen dienen te worden.
Voor de klinische praktijk werd t.a.v. natalizumab doseringsschema de NOVA-studie gepubliceerd, welke een doseerschema van iedere 4 versus iedere 6 weken onderzocht (Foley et al., 2022). Hoewel hier methodologische kanttekeningen door de auteurs bij werden geplaatst, was het aantal nieuwe of groter-wordende T2 laesies groter in de 6 vergeleken met de 4 weken groep. Er werden verder geen signalen van toegenomen ziekteactiviteit gezien. Momenteel worden nog wetenschappelijke studies naar dosering van natalizumab op basis van spiegels in bloed gedaan. De werkgroep is van mening dat verlenging van de doseringsinterval van natalizumab naar 1x per 6 weken op basis van deze data te verantwoorden is, maar op individueel niveau moet worden afgewogen.
Er zijn geen studies gepubliceerd waarin individuele anti-CD20 monoclonalen of S1P modulatoren direct met elkaar vergeleken werden. Hierbij kunnen verschillen in de netwerk meta-analyse tussen verschillende middelen uit dezelfde klasse berusten op verschillen in effectiviteit of op verschillen tussen de onderzochte studiepopulaties. Hoewel er biologische verschillen zijn tussen de S1P receptoren waarop individuele middelen ingrijpen, en het exacte CD-20 antigeen waar CD-20 monoclonalen tegen gericht zijn, is het onduidelijk of dit voor individuele middelen resulteert in een verschil in effectiviteit of veiligheid ten opzichte van elkaar. De werkgroep is hierdoor van mening dat deze middelen onderverdeeld dienen te worden in dezelfde lijn. Hierbij dient benadrukt te worden dat bij start van therapie, vooral de mate van ziekteactiviteit, prognostische factoren, en kinderwens bepaalt met welke lijn en middel gestart dient te worden. Dit betekent dat een tweedelijns therapie zeker niet ecxlusief voorbehouden is aan mensen die falen op eerstelijns behandeling. Mogelijke verschillen in klinische en veiligheids-karakteristieken (risico op rebound na stoppen bij verschillende S1P modulatoren, vaccinatie-responsen bij S1P modulatoren en anti-CD20 monoclonalen) zijn volop in onderzoek en kunnen invloed hebben op de plaatsbepaling in het veld.
Tabel 1: Overzicht van ziektemodulerende middelen met contra-indicaties, risico’s en adviezen voor monitoring (update: tabel 5.1)
Deze tabel is voornamelijk gebaseerd op:
- De tabel die oorspronkelijk in de richtlijn was opgenomen en SPC teksten,
- Lokale werkafspraken van Amsterdam UMC
|
Middel |
Contra-indicaties |
Voorzichtigheid bij |
Te bespreken bijwerkingen |
|
Alemtuzumab |
- Cardiovasculaire voorgeschiedenis1 of veranderde bloedstolling2 - Ernstige (actieve) infectie3 - Immuungecompromiteerde status4 - IC behoeftige patiënt met agressieve vorm van MS
|
- Dragerschap HPV bij vrouwen - Bestaande en/of actieve maligniteit - Schildklieraandoeningen - Afwijkingen in bloedbeeld, lever- of nierfunctie - Andere auto-immuunziekten (naast MS) - Actieve maligniteit |
- Auto-immuunziekten (30-40% schildklierziekten, 1-4% immuun trombocytopenische purpura, 0,5% auto-immuun nefropathieën, hemofagocytaire lymfohistiocytose, auto-immuunhepatitis, EBV hepatitis, trombotische trombocytopenische purpura, hemofilie A) - Frequent (>90%): meestal geringe tot matig ernstige infusie gerelateerde reacties - Allergische reactie - Voornamelijk in de 1e maand na kuur: bovenste luchtweginfecties, HSV, VZV, oppervlakkige schimmels, een algemeen verhoogde vatbaarheid voor infecties (waaronder Listeria meningitis/encefalitis) - Cytopenie - (cardio)vasculaire risico’s: herseninfarcten, myocardinfarcten, longbloedingen, dissectie cervicale arteriën |
|
Cladribine |
- Ernstige (actieve) infectie3 - Immuungecompromiteerde status4 |
- Matig-ernstige leverfunctiestoornissen
|
- Lymfopenie - Infecties: orale herpes, herpes zoster, TBC, PML (bij oncologische patiënten waargenomen, nog niet bij MS patiënten) - Maligniteiten - Huiduitslag, alopecia |
|
Fumaraatzuuresters (Dimethylfumaraat en diroxymelfumaraat) |
- Ernstige (actieve) infectie3 - Immuungecompromiteerde status4 - Bekende maligniteiten (muv cutaan basaalcelcarcinoom na complete resectie) - Ernstige nier-/leverfunctiestoornissen
|
|
- Gastro-intestinale klachten - Flushing, opvliegers, jeuk, huiduitslag - Leverenzymstijging, proteïnurie, leukopenie/lymfopenie - PML (zeer zeldzaam) |
|
S1P1-modulatoren (Fingolimod, ozanimod en ponesimod) |
- Ernstige (actieve) infectie3 - Immuungecompromitteerde status4 - Bekende maligniteiten - Ernstige leverfunctiestoornissen - Cardiovasculaire voorgeschiedenis5 of geleidingsstoornissen6 - Specifieke contra-indicaties siponimod7
|
- Specifieke waarschuwingen tav fingolimod8 - Interacties met andere medicatie (vooraf checken) |
- Hoofdpijn - Bradycardie bij eerste inname - Leverfunctiestoornissen, waaronder zeldzaam acuut leverfalen waarvoor levertransplantatie (fingolimod) - Macula oedeem - Respiratoire effecten (oa afname FEV1 en DLCO) - Lymfopenie - Infecties: luchtweginfecties, urineweginfecties, HSV en VZV reactivatie, zeldzaam andere opportunistische infecties (waaronder cryptococcen meningitis of PML bij fingolimod) - Huidtumoren waaronder basaalcelcarcinoom en melanoom - Rebound MS ziekteactiviteit na staken |
|
Glatirameer acetaat |
|
- Hartafwijkingen - Nierfunctiestoornissen |
- Vasodilatatie/irritatie tpv injectieplaats - IPIR: Pijn op de borst, dyspnoe, palpitaties, tachycardie |
|
Interferon beta (1a, 1b en peg-interferon beta 1a) |
- Ernstige depressie of suïcidaliteit - Gedecompenseerde leverziekte - Overgevoeligheid voor interferon of hulpstoffen (humaan albumine, mannitol) |
- (ernstige) nierschade / nefrotisch syndroom - Hartziekten - Ernstige myelosuppressie |
- Griepachtige verschijnselen - Lokale huidreactie tpv injectie - Leukopenie en stijging leverenzymen - Schildklierafwijkingen, trombotische micro-angiopathie; kan ook bij langer gebruik optreden |
|
Natalizumab |
- Ernstige (actieve) infectie3 - PML in de voorgeschiedenis - Immuungecompromitteerde status4 - Actieve maligniteit (muv cutaan basaalcelcarcinoom) - Ernstige leverfunctiestoornissen
|
- JC virus positiviteit - Actieve herpesinfectie (koortslip); indien ten tijde van eerste infuus niet geven (latere infusen geen bezwaar)
|
- Frequent: urineweginfecties, nasofaryngitis, hoofdpijn, misselijkheid/braken - Allergische reactie (grootste risico na 4 giften maar ook na enkele jaren beschreven) - Optreden van PML - HSV en/of VZV reactivatie zoals cutane herpesinfectie en gordelroos, echter ook encefalitits/meningitis op basis van HSV of VZV en acute retinale necrose op basis van VZV - Trombocytopenie, incl. immuungemedieerde trombocytopenische purpura - Levertoxiciteit - Rebound MS ziekteactiviteit na staken |
|
Anti-CD20 monoclonalen (Ocrelizumab en ofatumumab) |
- Ernstige (actieve) infectie3 - Immuungecompromitteerde status4 - Bekende maligniteit |
- Chronische/recidiverende infecties |
- Infusie gerelateerde reacties (variërend van hoofdpijn, koorts, hypertensie, misselijkheid, etc. tot anafylaxie of ARDS) - Zeer vaak (>10%): griep achtige verschijnselen, bovenste luchtweginfecties of urineweginfecties - Vaak (1-10%): HZV en VZV reactivatie, infecties elders, PML zeer zeldzaam |
|
Teriflunomide |
- Ernstige (actieve) infectie3 - Immuungecompromitteerde status4 - Ernstige leverfunctiestoornis of nierfunctiestoornis met nierdialyse |
Gelijktijdig gebruik van inductoren CYP en transportereiwitten |
- Verminderde haardichtheid - Gastro-intestinale klachten - ALAT stijging - Leukopenie, neutropenie - Milde infecties - Hypertensie |
Legenda:
- O.a. ongecontroleerde hypertensie, cervicocefale dissectie, beroerte, anginapectoris of myocardinfarct
- Stollingsstoornissen, gebruik van trombocytenaggregatieremmers of anticoagulantia;
- O.a. hepatitis B/C, tuberculose, HIV
- Immuungecompromitteerde status door immunosuppressieve-, immunomodulerende- of antineoplastische behandeling (m.u.v. corticosteroiden), HIV infectie, bekend immunodeficientiesyndroom, lymfopenie
- Myocardinfarct, instabiele angina pectoris, beroerte, TIA, gedecompenseerd hartfalen, NYHA klasse III/IV in afgelopen 6 maanden.
- 2e graads AV-blok type II, 3e graads AV-blok, sick-sinussyndroom (tenzij functionerende pacemaker). Specifiek voor fingolimod: aritmieen behandeld met klasse Ia of III anti-aritmica, QTc-interval >500 msec.
- Ernstige OSAS, overgevoeligheid voor pinda of soja, homozygoot voor CYP2C9*3
- Bradyaritmie of negatief-chronotrope medicatie, ernstige astma, ontregelde diabetes mellitus
NB1: Voor alle middelen geldt dat overgevoeligheid voor de werkzame stof of een hulpstof een contra-indicatie is.
NB2: Voor adviezen omtrent zwangerschap: zie de desbetreffende module.
Afkortingen:
PML= progressive multifocale leukoencefalopathie, HSV = herpes simplex virus, VZV = varicella zoster virus, EBV = epstein-barr virus, ARDS = acute respiratory distress syndrome, HPV = human papillomavirus
Balans van gewenste en ongewenste effecten
Vanwege veiligheidsaspecten van tweedelijnsmiddelen is starten met een eerstelijnsmiddel de eerste optie.
Bij een zeer actieve MS (zie bijlage “Definities en begrippen”) kan overwogen worden om direct met een hoog-effectieve ziektemodulerende therapie te starten. Eerdere behandeling met een effectiever (maar minder veilig) middel kan leiden tot een betere controle van relapse activiteit.
Kosten en middelen
Er zijn bij de werkgroep geen voor Nederland toepasbare kosteneffectiviteitsstudies bekend waarin alle besproken ziektemodulerende middelen een plaats hebben. De werkgroep is van mening dat bij de keuze voor een individueel middel uit dezelfde klasse (interferon-β/peginterferon-β, fumaraatzuur esters, S1P modulatoren, anti-CD20 monoclonalen, natalizumab i.v./ s.c.) naast individuele patiëntkarakteristieken, voorkeuren, therapietrouw, en gebruiksgemak ook maatschappelijke kosten van een therapie afgewogen moeten worden. Hierbij dient bij identiek gebruikgemak, veiligheid, en patiëntvoorkeur de prioriteit gegeven te worden aan de economisch meest voordelige middelen uit deze klassen. In het geval van de S1P modulatoren heeft bijvoorbeeld ponesimod op dit moment de laagste kostprijs, en kan meestal buiten de dagbehandeling opgestart worden. Een modelleringsstudie van opeenvolgende behandelingen met ziektemodulerende therapie vergeleek effectiviteit en kosteneffectiviteit van verschillende combinaties van therapieen met elkaar. In dit model is er een grote overlap tussen effectiviteit en kosten van verschillende combinaties van ziektemodulerende middelen. Hierbij zijn combinaties met cladribine en fingolimod goedkoper dan combinaties met natalizumab en ocrelizumab, maar leveren ook minder qualy’s op (Versteegh et al., 2022).
Aanvaardbaarheid en haalbaarheid
Volgens de werkgroep zijn de aanbevelingen haalbaar en aanvaardbaar voor de belangrijkste betrokkenen omdat zij grotendeels aansluiten bij de huidige klinische praktijk.
Fingolimod wordt niet vergoed als deze direct als eerste middel bij zeer actieve ziekte worden ingezet. Fingolimod wordt alleen vergoed als eerst een ander middel niet effectief is gebleken. Er zijn andere S1P modulatoren (ozanimod, ponesimod) welke wel direct als eerstelijns therapie ingezet kunnen worden. De werkgroep is van mening dat alle eerste- en tweedelijns middelen direct inzetbaar moeten kunnen zijn bij patiënten met zeer actieve MS.
Bij de plaatsbepaling van ziektemodulerende therapieën met het oog op vergoeding, is ervoor gekozen om S1P modulatoren en anti-CD20 monoclonalen in verschillende lijnen van therapie in te schalen. Hierbij worden ozanimod en ponesimod als eerstelijns vergoed, en fingolimod als tweedelijns. Ocrelizumab wordt als eerstelijns vergoed, ofatumumab als tweedelijns. Gezien de overlappende werkingsmechanismen en veiligheidsprofielen, is de werkgroep van mening dat deze middelen best allemaal in dezelfde lijn geplaatst kunnen worden.
Rationale
De centrale vraag is hoe patiënten met MS moeten worden behandeld op basis van bovenstaande gegevens. Samengevat zijn er niet voor alle middelen direct vergelijkende studies gepubliceerd, zijn niet alle middelen in iedere denkbare studiepopulatie onderzocht (i.e. actieve vs. zeer actieve MS), daarom werd gebruik gemaakt van een netwerk meta-analyse. De netwerk meta-analyse liet zien dat alemtuzumab, natalizumab, cladribine, ofatumumab, ozanimod_1.0, ocrelizumab, fingolimod en dimethylfumaraat waarschijnlijk een klinisch relevante reductie van de annualized relapse rate geven (tabel A). Misschien geldt dit ook voor mitoxantron. Waarschijnlijk geven alemtuzumab, natalizumab, ocrelizumab en fingolimod een klinisch relevante toename van ‘vrij zijn van relapse’ (tabel B). Waarschijnlijk geven alemtuzumab, natalizumab en ocrelizumab een klinisch relevante afname van ‘toenemende invaliditeit’ (tabel C).
Waarschijnlijk geeft dimethylfumaraat geen klinisch relevante toename van stoppen vanwege bijwerkingen (tabel D). Ponesimod en mitoxantron geven misschien een klinisch relevante toename van stoppen vanwege bijwerkingen (tabel D). Bij de keuze voor een behandeling wordt een afweging gemaakt van de effectiviteit en verwachte bijwerkingen en overige factoren die in de aanbeveling worden beschreven waarbij de eerdere indeling in eerstelijns (interferon-β/peginterferon-β, glatirameeracetaat, teriflunomide en fumaraatzuuresters)- en tweedelijns middelen (natalizumab, fingolimod, alemtuzumab, ocrelizumab en cladribine) wordt gebruikt.
Onderbouwing
Achtergrond
In de module Bij welke patiënten met relapsing remitting MS is het starten van ziektemodulerende middelen geïndiceerd? zijn de effectiviteit en veiligheid van ziektemodulerende middelen in vergelijking met placebo bij relapsing remitting MS besproken. In deze paragraaf worden onderlinge verschillen qua effectiveit (e.g. netto-baten) van ziektemodulerende middelen geëvalueerd, indien er een indicatie is om te starten met een ziektemodulerend middel.
Conclusies / Summary of Findings
Conclusies Deel 1: samenvatting literatuur traditionele meta-analyse: directe vergelijking ziektemodulerende middelen
Conclusies Review 1. Interferon-β versus glatirameeracetaat
1.1 Annualized Relapse Rate bij een follow-up duur van 96-104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 96-104 weken voor de annualized relapse rate -gemiddeld voor beide doseringen en toedieningsroutes- 0.05 relapse minder (95% BI: 0.21 minder tot 0.11 meer) zien per patiënt per jaar in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Calabrese et al., 2012 |
1.2 Proportie vrij van relapse bij een follow-up duur van 96-104 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 96-104 weken voor de proportie patiënten vrij van relapse een vermindering van het relatieve effect zien met 2% (RR: 0.98; 95% BI: 0.90-1.06 en een risicoverschil van 12 patiënten vrij van relapse minder per 1000 (95% BI: 61 minder tot 36 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Cadavid et al., 2009 ; Mikol et al., 2008; O’Connor et al., 2009 |
1.3 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor toenemende invaliditeit een toename van het relatieve risico zien met 4% (RR: 1.04; 95% BI: 0.83-1.31) en een risicoverschil van 8 patiënten meer met toenemende invaliditeit (95% BI: 34 minder tot 62 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: O’Connor et al., 2009 |
1.4 Aantal patiënten zonder nieuwe T2 laesies of groter wordende T2 laesies bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal patiënten zonder nieuwe of groter wordende T2 laesies een toename van het relatieve effect zien met 8% (RR: 1.08; 95% BI: 0.86-1.36) en een risicoverschil van 30 patiënten meer zonder laesies per 1000 (95% BI: 52 minder tot 135 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Mikol et al., 2008 |
1.5 Aantal patiënten zonder gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal patiënten zonder gadolinium aankleurende laesies een toename van het relatieve effect zien met 21% (RR: 1.21; 95% BI: 1.08-1.35) en een risicoverschil van 141 patiënten meer zonder laesies per 1000 (95% BI: 54 meer tot 234 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Mikol et al., 2008 |
1.6 Aantal nieuwe gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal nieuw gadolinium aankleurende laesies een vermindering met gemiddeld 0.15 laesies zien (95% BI: 0.48 minder tot 0.17 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: O’Connor et al., 2009 |
1.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48-104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-104 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 15% (RR: 1.15; 95% BI: 0.75-1.77) en een risicoverschil van 5 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 9 minder tot 28 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bronnen: Cadavid et al., 2009; Calabrese et al., 2012; Mikol et al., 2008; O’Connor et al., 2009 |
1.7 Studieuitval om willekeurige reden bij een follow-up duur van 48-104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-104 weken voor studieuitval om een willekeurige reden een toename van het relatieve risico zien met 30% (RR: 1.30; 95% BI: 0.68-2.47) en een risicoverschil van 29 patiënten meer met studieuitval om een willekeurige reden per 1000 (95% BI: 31 minder tot 142 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bronnen: Cadavid et al., 2009; Calabrese et al., 2012; Mikol et al., 2008; O’Connor et al., 2009 |
1.8 Studieuitval vanwege bijwerkingen bij een follow-up duur van 208 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 208 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 78% (RR: 1.78; 95% BI: 0.62-5.16) en een risicoverschil van 56 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 27 minder tot 297 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: O’Connor et al., 2009 |
1.9 Studieuitval om willekeurige reden bij een follow-up duur van 208 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 208 weken voor studieuitval om een willekeurige reden een toename van het relatieve risico zien met 2% (RR: 1.02; 95% BI: 0.55-1.88) en een risicoverschil van 4 patiënten meer met studieuitval om een willekeurige reden per 1000 (95% BI: 96 minder tot 189 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: O’Connor et al., 2009 |
1.10 Mortaliteit bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor mortaliteit geen verschil zien (RR: 1.0; 95% BI: 1.0-1.01) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: O’Connor et al., 2009 |
Conclusies Review 2. Teriflunomide versus interferon
2.1 Annualized Relapse Rate bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor de annualized relapse rate 0.04 relapse meer (95% BI: 0.17 minder tot 0.25 meer) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.2 Proportie vrij van relapse bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor de proportie patiënten vrij van relapse een vermindering van het relatieve effect zien met 32% (RR: 0.68; 95% BI: 0.57-0.83 en een risicoverschil van 271 patiënten vrij van relapse minder per 1000 (95% BI: 152 minder tot 364 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.3 MRI uitkomsten
|
---------
|
Er werden geen MRI-uitkomsten gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
|
2.4 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor studieuitval vanwege bijwerkingen een vermindering van het relatieve risico zien met 49% (RR: 0.51; 95% BI: 0.27-0.98 en een risicoverschil van 104 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 4 minder tot 154 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.5 Studieuitval om willekeurige reden bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor studieuitval om willekeurige reden een vermindering van het relatieve risico zien met 31% (RR: 0.69; 95% BI: 0.42-1.11 en een risicoverschil van 89 patiënten minder met studieuitval om willekeurige reden per 1000 (95% BI: 167 minder tot 32 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.6 Infecties bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor infecties een toename van het relatieve risico zien met 8% (RR: 1.08; 95% BI: 0.81-1.43) en een risicoverschil van 36 patiënten meer met infecties per 1000 (95% BI: 86 minder tot 194 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
Conclusies Review 3. Fingolimod versus interferon-b
3.1 Annualized Relapse Rate bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor de annualized relapse rate 0.17 relapse minder (95% BI: 0.26 minder tot 0.08 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.2 Proportie vrij van relapse bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 19% (RR: 1.19; 95% BI: 1.11-1.29 en een risicoverschil van 131 patiënten vrij van relapse meer per 1000 (95% BI: 76 meer tot 201 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.3 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 26% (RR: 0.74; 95% BI: 0.45-1.22) en een risicoverschil van 21 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 43 minder tot 17 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Cohen et al., 2010 |
3.4 Aantal patiënten zonder nieuwe T2 laesies of groter wordende T2 laesies bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor het aantal patiënten zonder nieuwe of groter wordende T2 laesies een toename van het relatieve effect zien met 20% (RR: 1.20; 95% BI: 1.04-1.39) en een risicoverschil van 91 patiënten meer zonder laesies per 1000 (95% BI: 18 meer tot 178 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.5 Aantal patiënten zonder gadolinium aankleurende laesies bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor het aantal patiënten zonder gadolinium aankleurende laesies een toename van het relatieve effect zien met 12% (RR: 1.12; 95% BI: 1.05-1.19) en een risicoverschil van 97 patiënten meer zonder laesies per 1000 (95% BI: 40 meer tot 154 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.6 Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor het gemiddeld aantal gadolinium aankleurende laesies een vermindering met gemiddeld 0.28 laesies zien (95% BI: 0.50 minder tot 0.06 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Cohen et al., 2010 |
3.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 41% (RR: 1.41; 95% BI: 0.92-2.18) en een risicoverschil van 30 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 6 minder tot 88 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Cohen et al., 2010 |
3.8 Studieuitval om willekeurige reden bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor studieuitval om een willekeurige reden een vermindering van het relatieve risico zien met 31% (RR: 0.69; 95% BI: 0.45-1.07) en een risicoverschil van 32 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 57 minder tot 7 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: Cohen et al., 2010 |
3.9 Infecties bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor infecties geen verschil zien (RR: 1.0; 95% BI: 0.86-1.17) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: Cohen et al., 2010 |
3.10 Maligniteit bij een follow-up duur van 52 weken
|
Zeer laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor maligniteit geen verschil zien (RR: niet berekend vanwege niet optreden van een maligniteit) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect verschilt waarschijnlijk aanzienlijk verschillen het geschatte effect).
Bron: Cohen et al., 2010 |
Conclusies Review 4. Alemtuzumab versus interferon-b
4.1 Annualized Relapse Rate (follow-up 104-156 weken)
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor de annualized relapse rate 0.25 relapse minder (95% BI: 0.33 minder tot 0.18 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012 |
4.1 Annualized Relapse Rate (follow-up 260 weken)
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 260 weken voor de annualized relapse rate 0.23 relapse minder (95% BI: 0.30 minder tot 0.16 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Camms223, 2008 |
4.2 Proportie vrij van relapse bij een follow-up duur van 104-156 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 38% (RR: 1.38; 95% BI: 1.26-1.51) en een risicoverschil van 198 patiënten vrij van relapse meer per 1000 (95% BI: 136 meer tot 266 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.3 Proportie vrij van relapse bij een follow-up duur van 260 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 260 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 67% (RR: 1.67; 95% BI: 1.29-2.17 en een risicoverschil van 272 patiënten vrij van relapse meer per 1000 (95% BI: 118 meer tot 474 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.4 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 104-156, 260 weken)
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 41% (RR: 0.59; 95% BI: 0.40-0.86) respectievelijk 57% (RR: 0.43; 95% BI: 0.24-0.78) en een risicoverschil van 8 patiënten minder (95% BI: 34 minder tot 62 minder) respectievelijk 154 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 59 minder tot 205 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008* *Dit is de enige studie die ook voor een follow-up van 260 weken rapporteerde. |
4.5 Aantal patiënten met nieuwe T2 laesies of groter wordende T2 laesies bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal patiënten met nieuwe of groter wordende T2 laesies een vermindering van het relatieve risico zien met 23% (RR: 0.77; 95% BI: 0.60-1.00) en een risicoverschil van 139 patiënten minder met laesies per 1000 (95% BI: 242 minder tot 0 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Cohen et al., 2012; Coles et al., 2012 |
4.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 104-156, 260 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor studieuitval vanwege bijwerkingen een vermindering van het relatieve risico zien met 69% (RR: 0.31; 95% BI: 0.17-0.55) respectievelijk 65% (RR:0.35; 95% BI: 0.13-0.95) en een risicoverschil van 54 patiënten minder (95% BI: 35 minder tot 65 minder) respectievelijk 85 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 7 minder tot 114 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect). Dit is echter een artefact omdat alemtuzumab in een veel lagere frequentie wordt toegediend dan interferon-b.
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008* *Dit is de enige studie die ook voor een follow-up van 260 weken rapporteerde. |
4.7 Studieuitval om willekeurige reden bij een follow-up duur van 104-156 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor studieuitval om een willekeurige reden een vermindering van het relatieve risico zien met 64% (RR: 0.36; 95% BI: 0.25-0.52) en een risicoverschil van 178 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 133 minder tot 208 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect). Dit is echter een artefact omdat alemtuzumab in een veel lagere frequentie wordt toegediend dan interferon-b.
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.8 Infecties bij een follow-up duur van 104-156, 260 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor infecties een toename van het relatieve risico zien met 32% (RR: 1.32; 95% BI: 1.10-1.58) respectievelijk 41% (RR:1.41; 95% BI: 1.13-1.76) en een risicoverschil van 174 patiënten meer (95% BI: 53 meer tot 315 meer) respectievelijk 207 patiënten meer infecties per 1000 (95% BI: 66 meer tot 384 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.9 Mortaliteit bij een follow-up duur van 104-156, 260 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor mortaliteit een risicoverschil zien van 4 overleden patiënten meer per 1000 (95% BI: 11 minder tot 4 meer) en bij een follow-up duur van 260 weken geen verschil (RR 1; 95% BI: 0.97-1.03) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.10 Maligniteit bij een follow-up duur van 260 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 260 weken voor maligniteit een risicoverschil van 2 patiënten met een maligniteit minder per 1000 zien in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.11 Auto-immuunziekten (immuun-gemedieerde trombocytopenische purpura)** bij een follow-up duur van 104-156, 260 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor auto-immuunziekten een toename van het relatieve risico zien met 168% (RR: 2.68; 95% BI: 0.56-12.90) respectievelijk 98% (RR:1.98; 95% BI: 0.18-21.53) en een risicoverschil van 3 patiënten meer (95% BI: 1 minder tot 24 meer) respectievelijk 9 patiënten meer per 1000 (95% BI: 8 minder tot 192 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
Conclusies Review 5. Ocrelizumab versus interferon-b
5.1 Annualized Relapse Rate bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor de annualized relapse rate 0.13 relapse minder (95% BI: 0.18 minder tot 0.08 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.2 Proportie vrij van relapse bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 19% (RR: 1.19; 95% BI: 1.12-1.26 en een risicoverschil van 128 patiënten vrij van relapse meer per 1000 (95% BI: 81 meer tot 175 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Havrdová et al., 2018 |
5.3 Verbetering invaliditeit bevestigd na 12 en 24 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat voor verbetering invaliditeit een toename van het relatieve effect zien met 35% (RR: 1.35; 95% BI: 1.02-1.79) en een risicoverschil van 40 patiënten meer met verbetering invaliditeit (95% BI: 2 meer tot 91 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.4 Toenemende invaliditeit bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 40% (RR: 0.60; 95% BI: 0.46-0.80) en een risicoverschil van 67 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 90 minder tot 33 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.5 en 5.6 Studieuitval vanwege bijwerkingen en studieuitval om willekeurige reden bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor studieuitval vanwege bijwerkingen en om willekeurige reden een vermindering van het relatieve risico zien met 54% (RR: 0.46; 95% BI: 0.30-0.70) respectievelijk 40% (RR: 0.60; 95% BI: 0.48-0.75) en een risicoverschil van 42 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 54 minder tot 23 minder) respectievelijk 80 patiënten minder (95% BI: 50 minder tot 104 minder) vanwege studieuitval om willekeurige reden in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.7 (Parasitaire) infecties bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor infecties een toename van het relatieve risico zien met 11% (RR: 1.11; 95% BI: 1.02-1.22) en een risicoverschil van 58 patiënten met infecties meer per 1000 (95% BI: 10 meer tot 115 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.8 Mortaliteit bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor mortaliteit een halvering van het relatieve risico zien (RR: 0.50; 95% BI: 0.05-5.51) en een risicoverschil van 1 overleden patiënt minder per 1000 (95% BI: 8 minder tot 5 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.9 Maligniteit bij een follow-up duur van 96 weken
|
Laag
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor maligniteit een verdubbeling van het relatieve risico zien (RR: 2.00; 95% BI: 0.37-10.90) en een risicoverschil van 2 patiënten meer met een maligniteit per 1000 (95% BI: 5 minder tot 10 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Hauser et al., 2017 |
Conclusies Review 6. Ozanimod versus interferon-β
Conclusies Ozanimod versus interferon-β
6.1 Annualized Relapse Rate bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet of nauwelijks in een verschil in de annualized relapse rate ten opzicht van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS vermindert waarschijnlijk de annualized relapse rate ten opzicht van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.4 Toenemende invaliditeit na 12 of 24 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet of nauwelijks in 24 weken-verminderde toename van invaliditeit ten opzicht van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert niet of nauwelijks in niet of nauwelijks in 24 weken-bevestigde toename van lichamelijke beperking ten opzicht van interferon-β.
Bron: Cohen et al., 2019 |
6.5a Nieuwe of groter wordende laesies bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert in minder nieuwe of groter wordende laesies ten opzicht van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert in minder nieuwe of groter wordende laesies ten opzicht van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.5b Gadolinium aankleurende laesies bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een verschil in gadolinium aankleurende laesies ten opzichte van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een verschil in gadolinium aankleurende laesies ten opzicht van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg of 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet of nauwelijks in minder studieuitval vanwege bijwerkingen dan wanneer interferon-b werd gebruikt.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.7 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg of 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet of nauwelijks in minder studieuitval om willekeurige redenen dan wanneer interferon-b werd gebruikt.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.8 (Ernstige) infecties bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet in meer (ernstige) infecties vergeleken met interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert niet meer (ernstige) infecties vergeleken met interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.9 Mortaliteit bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet in een grotere of kleinere kans op mortaliteit dan gebruik van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert niet in een grotere of kleinere kans op mortaliteit dan gebruik van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.10 Neoplasmata/maligniteit bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een grotere of kleinere kans op neoplasmata/maligniteit dan gebruik van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een grotere of kleinere kans op neoplasmata/maligniteit dan gebruik van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
Conclusies Review 7. Ofatumumab versus teriflunomide
7.1 Annualized Relapse Rate bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS verlaagt waarschijnlijk de annualized relapse rate ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.2 Verbetering invaliditeit (uitgedrukt in EDSS)
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert niet in een verschil in verbetering van invaliditeit ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.3 Verminderde toename van invaliditeit (uitgedrukt in EDSS)
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert niet in een verminderde toename van invaliditeit ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.4 Nieuwe laesies bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert in minder nieuwe laesies ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.5 Gadolinium aankleurende laesies bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert in minder gadolinium aankleurende laesies ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder studieuitval ten gevolge van bijwerkingen ten opzichte van teriflunomide.
Bron: Hauser et al., 2020 |
7.7 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder studieuitval ten gevolge van willekeurige redenen ten opzichte van teriflunomide.
Bron: Hauser et al., 2020 |
7.8 (Ernstige) infecties
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert niet in meer infecties ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.9 Mortaliteit bij een follow-up duur van 48 weken
|
Laag
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS lijkt niet in meer mortaliteit te resulteren ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.10 Neoplasmata/maligniteit bij een follow-up duur van 96 weken
|
Laag
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS lijkt niet in meer of minder neoplasmata/maligniteit te resulteren ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
Conclusies Review 8. Ponesimod vs teriflunomide
8.1 Annualized Relapse Rate bij een follow-up duur van 108 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert niet of nauwelijks in een klinisch relevant verschil in de annualized relapse rate ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.4 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 12 of 24 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert niet of nauwelijks in een klinisch relevant verschil niet of nauwelijks in 24 weken-bevestigde toename van lichamelijke beperking ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.5 Combined unique active lesions
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder unieke nieuwe laesies ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.6 Gadolinium aankleurende laesies
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder gadolinium aankleurende laesies ten opzichte van teriflunomide.
Bron: Kappos et al., 2021 |
8.7 No evidence of disease activity
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder no evidence of disease activity (NEDA) ten opzichte van teriflunomide.
Bron: Kappos et al., 2021 |
8.8 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in meer studieuitval vanwege bijwerkingen ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.9 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in meer studieuitval om willekeurige redenen ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.10 Infecties bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in meer infecties ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.11 Mortaliteit bij een follow-up duur van 24-48 weken
|
Laag
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS lijkt niet te resulteren in meer mortaliteit ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.12 Neoplasmata/maligniteit bij een follow-up duur van 24-48 weken
|
Laag
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS lijkt niet te resulteren in meer neoplasmata /maligniteit ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
Conclusies Deel 2: netwerkmeta-analyse
|
‘Annualized relapse rate’
Waarschijnlijk geven alemtuzumab, natalizumab, cladribine, ofatumumab, ozanimod_1.0, ocrelizumab, fingolimod en dimethylfumaraat een klinisch relevante reductie van de annualized relapse rate (tabel A). Misschien geldt dit ook voor mitoxantron. De overige middelen hebben misschien geen (overtuigend) klinisch relevant effect (tabel A).
|
||||
|
Tabel A. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘annualized relapse rate’. |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
SMD: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant†† |
Alemtuzumab§ |
-0.940 (-1.219; -0.660) |
0.97 |
|
|
|
Natalizumab |
-0.620 (-0.759; -0.482) |
0.86 |
|
|
|
Cladribine |
-0.489 (-0.624; -0.354) |
0.73 |
|
|
|
Ofatumumab |
-0.468 (-0.599; -0.336) |
0.70 |
|
|
|
Ozanimod_1.0 |
-0.417 [-0.521; -0.314] |
0.61 |
|
|
|
Ocrelizumab |
-0.406 (-0.522; -0.290) |
0.58 |
|
|
|
Fingolimod |
-0.391 (-0.474; -0.307) |
0.55 |
|
|
|
Dimethylfumaraat |
-0.310 (-0.396; -0.224) |
0.37 |
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
- |
- |
- |
|
|
|
|
|
|
|
Zeer laag/laag |
Klinisch relevant |
Mitoxantron |
-0.822 (-1.396; -0.247) |
0.89 |
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
Ponesimod Ozanimod_0.5 Glatirameeracetaat Interferon-β Teriflunomide |
-0.339 (-0.489; -0.188) -0.301 [-0.405; -0.199] -0.184 (-0.250; -0.119) -0.168 (-0.232; -0.104) -0.132 (-0.226, -0.037) |
0.44 0.36 0.18 0.15 0.10 |
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
* SMD: standardized mean difference. Een groter verschil wijst op een grotere vermindering van de ‘annualized relapse rate’ bij gebruik van een ziektemodulerend middel. † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Bovengrens van 95% BI van SMD < -0.2 wijst op (mogelijk) klinisch relevant effect. Een SMD < -0.2 wordt gezien als een klein maar belangrijk effect (Guyatt et al., 2013). §alemtuzumab is het enige middel dat superieur is aan diverse andere klinisch relevante middelen (dimethylfumaraat, fingolimod, ocrelizumab, ozanomod_1.0): bovengrens van 95% BI van SMD < -0.2 . Zie league table (bijlage 3a). Opmerking: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
|
‘Vrij van relapse’
Waarschijnlijk geven alemtuzumab, natalizumab, ocrelizumab en fingolimod een klinisch relevante toename van ‘vrij zijn van relapse’ (tabel B). Waarschijnlijk heeft cladribine geen (overtuigend) klinisch relevant effect. De overige middelen hebben misschien geen (overtuigend) klinisch relevant effect (tabel B).
|
||||
|
Tabel B. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘vrij van relapse’. |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
RR: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant†† |
Alemtuzumab Natalizumab Fingolimod Ocrelizumab |
1.626 [1.447;1.826] 1.593 [1.402;1.810] 1.422 [1.327;1.524] 1.400 [1.274;1.538] |
0.95 0.92 0.74 0.69 |
|
|
||||
|
Niet (overtuigend) klinisch relevant |
Cladribine |
1.306 [1.203;1.418] |
0.51 |
|
|
|
|
|
|
|
|
Zeer laag/laag |
Klinisch relevant |
- |
|
|
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
Dimethylfumaraat Teriflunomide Glatirameeracetaat Interferon-β |
1.277 [1.183;1.378] 1.253 [1.155;1.359] 1.184 [1.120;1.251] 1.178 [1.096;1.265] |
0.44 0.38 0.20 0.18 |
|
|
|
|||
|
|
|
|||
|
|
||||
|
*RR> 1 wijst op een grotere mate van vrij zijn van relapses bij gebruik van een ziektemodulerend middel. † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Ondergrens van 95% BI van RR > 1.25 wijst op (mogelijk) klinisch relevant effect. Opmerkingen: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
|
‘Toenemende invaliditeit’
Waarschijnlijk geven alemtuzumab, natalizumab en ocrelizumab een klinisch relevante afname van ‘toenemende invaliditeit’ (tabel C). De overige middelen hebben misschien geen (overtuigend) klinisch relevant effect (tabel C).
|
||||
|
Tabel C. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘toenemende invaliditeit’ |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
RR: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant†† |
Ocrelizumab Alemtuzumab Natalizumab |
0.492 [0.346; 0.699] 0.495 [0.346; 0.708] 0.585 [0.458; 0.748] |
0.82 0.82 0.68 |
|
|
|
|||
|
|
|
|||
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
- |
|
|
|
|
||||
|
Zeer laag/laag |
Klinisch relevant |
- |
|
|
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
Mitoxantron Ofatumumab Ponesimod Dimethylfumaraat Fingolimod Teriflunomide Glatirameeracetaat Interferon-β Ozanimod_0.5 Ozanimod_1.0 |
0.198 [0.047; 0.826] 0.540 [0.379; 0.770] 0.626 [0.410; 0.956] 0.651 [0.525; 0.808] 0.689 [0.555; 0.854] 0.763 [0.624; 0.934] 0.811 [0.656; 1.003] 0.814 [0.655; 1.011] 0.902 [0.580; 1.403] 1.173 [0.762; 1.807] |
0.95 0.75 0.59 0.56 0.50 0.36 0.30 0.29 0.23 0.04 |
|
|
||||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
*RR<1 wijst op een geringere toename van invaliditeit bij gebruik van een ziektemodulerend middel. † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Bovengrens van 95% BI van RR < 0.75 wijst op (mogelijk) klinisch relevant effect. Opmerking: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
|
‘Stoppen vanwege bijwerkingen’
Waarschijnlijk geeft interferon-β een klinisch relevante toename van stoppen vanwege bijwerkingen. Waarschijnlijk geeft dimethylfumaraat geen klinisch relevante toename van stoppen vanwege bijwerkingen (tabel D).
Ponesimod en mitoxantron geven misschien een klinisch relevante toename van stoppen vanwege bijwerkingen (tabel D).
Voor de overige middelen geldt dat ze misschien geen (overtuigend) klinisch relevante toename van stoppen vanwege bijwerkingen geven (tabel D).
|
||||
|
Tabel D. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘studieuitval vanwege bijwerkingen’. |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
RR: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant ongunstig effect†† |
Interferon-β |
1.942 [1.371;2.750] |
0.74 |
|
|
Geen (overtuigend) klinisch relevant ongunstig effect |
Dimethylfumaraat |
0.969 [0.775;1.212] |
0.23 |
|
|
||||
|
|
||||
|
Zeer laag/laag |
Klinisch relevant ongunstig effect |
Ponesimod Teriflunomide |
2.670 [1.570;4.539] 1.849 [1.340;2.552] |
0.90 0.69 |
|
|
||||
|
|
|
|||
|
|
|
|||
|
|
Geen (overtuigend) klinisch relevant ongunstig effect |
Mitoxantron Ofatumumab Glatirameeracetaat Ozanimod_1.0 Fingolimod Natalizumab Ozanimod_0.5 Cladribine** Ocrelizumab** Alemtuzumab** |
4.440 [0.560;35.41] 2.016 [1.229;3.308] 1.864 [1.168;2.974] 1.484 [0.858;2.569] 1.413 [1.111;1.799] 1.256 [0.492;3.206] 1.214 [0.692;2.129] 1.130 [0.433;2.945] 0.769 [0.446;1.328] 0.610 [0.320;1.164] |
0.85 0.75 0.70 0.54 0.49 0.43 0.38 0.37 0.12 0.05 |
|
|
|
|||
|
|
|
|||
|
|
||||
|
|
||||
|
|
|
|||
|
|
|
|||
|
|
||||
|
* RR>1 wijst op toename van stoppen vanwege bijwerkingen bij gebruik van een ziektemodulerend middel † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Ondergrens van 95% BI van RR > 1.25 wijst op (mogelijk) klinisch relevant ongunstig effect. ** artefact: Het relatief geringe risico op stoppen vanwege bijwerkingen is een artefact omdat deze middelen in een veel lagere frequentie worden toegediend dan de andere middelen. Opmerking: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
Samenvatting literatuur
Deel 1: samenvatting literatuur traditionele meta-analyse: directe vergelijking ziektemodulerende middelen
Dit betreft een samenvatting van de studies waarin twee ziektemodulerende middelen direct met elkaar worden vergeleken, een beschrijving van de netwerk meta-analyse volgt in deel 2. De conclusies zijn per uitkomstmaat gegradeerd. Voor de volgende middelen is volgens de Europese richtlijn direct bewijs voorhanden: glatirameeracetaat en interferon; teriflunomide en interferon; fingolimod en interferon; alemtuzumab en interferon en ocrelizumab en interferon.
Beschrijving studies
Er werden 10 trials geïncludeerd in de analyse en bij de update in 2022 werden er 4 toegevoegd. De meeste trials waren multicenter trials en door de industrie gesponsord. De middelen glatirameeracetaat, teriflunomide, fingolimod, alemtuzumab en ocrelizumab werden vergeleken met interferon-β.
Studiepopulaties
De studiegrootte varieerde tussen de 75 en 1292. Het percentage vrouwen varieerde van 66% tot 70% in de studiepopulaties. De gemiddelde leeftijd van de studiedeelnemers varieerde van 32 tot 37 jaar. De gemiddelde initiële EDSS-score bij de start van het onderzoek varieerde van 1.9 tot 2.84. De gemiddelde follow-up duur in de studies varieerde van 52 tot 260 weken. Het percentage studiedeelnemers dat eerder een ziektemodulerend middel had gebruikt varieerde van 0 tot 74. De gemiddelde ziekteduur varieerde van 1.1 tot 7.3 jaar. Het gemiddelde aantal relapses in het voorgaande jaar varieerde van 1.2 tot 1.9.
De inclusie- en exclusiecriteria die voor de trials werden gehanteerd zijn samengevat in tabel 1.
Tabel 1. Inclusiecriteria gebruikt in 10 RCTs
|
|
Inclusie- en exclusiecriteria |
|
Cadavid et al., 2009 |
Inclusion criteria
Exclusion criteria Subjects were not enrolled in the study if they possessed any of the following conditions:
|
|
Calabrese et al., 2012 |
Inclusion criteria
Exclusion criteria
|
|
Mikol et al., 2008 |
Inclusion criteria
Exclusion criteria
|
|
O’Connor et al., 2009 |
Inclusion criteria
Exclusion criteria
|
|
Vermersch et al., 2014 |
Inclusion criteria
Exclusion criteria
|
|
Cohen et al., 2010 |
Inclusion criteria
Exclusion criteria
Previous recent therapy with either any type of interferon-b or glatiramer acetate was not a criterion for exclusion. |
|
Cohen et al., 2012 |
Inclusion criteria
Exclusion criteria
|
|
Coles et al., 2008 |
Inclusion criteria
Exclusion criteria
|
|
Coles et al., 2012 |
Inclusion criteria
Exclusion criteria
|
|
Hauser et al., 2017 (2 RCTs) |
Inclusion criteria
Exclusion criteria
|
|
Comi et al. 2019 SUNBEAM |
Inclusion Criteria
Exclusion Criteria
|
|
Cohen et al., 2019 RADIANCE |
Inclusion criteria
Exclusion Criteria
|
|
Hauser et al., 2020 ASCLEPIOS |
Inclusion criteria
Exclusion criteria
|
|
Kappos et al. 2021 OPTIMUM |
Inclusion criteria
Exclusion criteria
|
Voor de toegepaste dosering, toedieningsweg en follow-up duur in de hiervoor genoemde studies zie bijlage 2.
Review 1. Interferon-β versus glatirameeracetaat
- Klinische effectiviteit
1.1 “Annualized Relapse Rate” (ARR)
Eén RCT rapporteerde over de ARR (Calabrese et al., 2012; twee interferon-b-1a armen: subcutaan en intramusculair met een dosering van 44 µg [3x/week] respectievelijk 30 µg [1x/week]). Interferon-β verminderde -gemiddeld voor beide doseringen en toedieningsroutes- in vergelijking met glatirameeracetaat de ARR met 0.05 per persoon per jaar (95% BI: 0.21 minder tot 0.11 meer). De dosis van 30 µg gaf geen reductie, die van 44 µg gaf een reductie van 0.10 relapses per persoon per jaar.
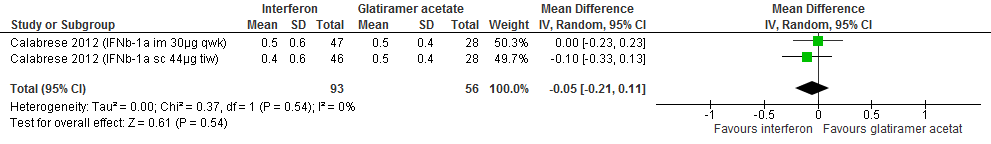
Figuur 1.1 Annualized relapse rate interferon-β verus glatirameeracetaat (104 weeks’follow-up)
1.2 Proportie vrij van relapse
Drie RCTs (Cadavid et al., 2009 ; Mikol et al., 2008; O’Connor et al., 2009) rapporteerden over de proportie patiënten die na 96-104 weken vrij van een relapse waren gebleven. Interferon-b was in deze studies iets minder effectief dan glatirameeracetaat: RR=0.98 (95% BI: 0.90-1.06), of in absolute termen: 12 minder per 1000 (95% BI: 61 minder tot 36 meer).
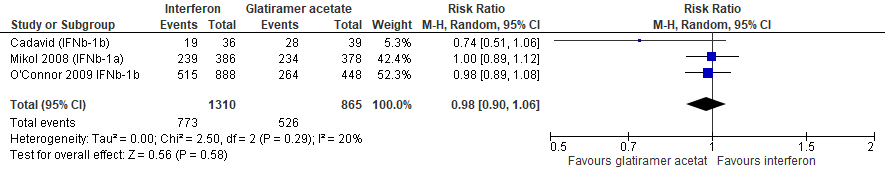
Figuur 1.2 Proportie vrij van relapse interferon-β verus glatirameeracetaat (104 weeks’ follow-up)
1.3 Toenemende invaliditeit uitgedrukt in EDSS
Eén RCT (O’Connor et al., 2009) rapporteerde over toenemende invaliditeit. Volgens deze studie vertraagde interferon-b toenemende invaliditeit iets minder dan glatirameeracetaat: RR=1.04 (95% BI: 0.83-1.31), of in absolute termen: 8 patiënten meer per 1000 met toenemende invaliditeit (95% BI: 34 minder tot 62 meer) bij gebruik van interferon-b vergeleken met glatirameeracetaat.
- Effectiviteit niet-klinisch
1.4 Aantal patiënten zonder nieuwe T2 laesies of groter wordende T2 laesies
Eén RCT (Mikol et al., 2008) rapporteerde over het aantal patiënten met nieuwe T2-laesies. Volgens deze studie ging gebruik van interferon-b met minder nieuwe laesies gepaard dan met glatirameeracetaat: RR=1.08 (95% BI: 0.86-1.36) of in absolute termen: 30 patiënten meer zonder nieuwe laesies per 1000 (95% BI: 52 minder tot 135 meer) met interferon-b dan met glatirameeracetaat.
1.5 Aantal patiënten zonder gadolinium aankleurende laesies
Eén RCT (Mikol et al., 2008) rapporteerde over het aantal patiënten zonder gadolinium aankleurende laesies. Volgens deze studie ging gebruik van interferon-b gepaard met een groter aantal patiënten zonder gadolinium aankleurende laesies dan gebruik van glatirameeracetaat: RR=1.21 (95% BI: 1.08-1.35), of in absolute termen: 141 patiënten meer per 1000 (95% BI: 54 tot 234 meer).
1.6 Aantal nieuwe gadolinium aankleurende laesies
Eén RCT (O’Connor et al., 2009) rapporteerde over het aantal nieuwe gadolinium aankleurende laesies. Volgens deze studie ging gebruik van interferon-b gepaard met minder nieuwe gadolinium aankleurende laesies dan glatirameeracetaat: gemiddeld 0.15 minder (95% BI: 0.48 minder tot 0.17 meer).
- No Evidence of Disease Activity (NEDA)
Er werden geen uitkomsten gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
- Tolereerbaarheid en veiligheid
1.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 96-104 weken
Vier RCTs (Cadavid et al., 2009; Calabrese et al., 2012; Mikol et al., 2008; O’Connor et al., 2009) rapporteerden over studieuitval vanwege bijwerkingen. Op grond van de gecombineerde uitkomsten van deze studies was er nauwelijks verschil tussen interferon-b en glatirameeracetaat: RR =1.15 (95% BI: 0.75-1.77), of in absolute termen: 5 patiënten meer per 1000 (95% BI: 9 minder tot 28 meer) die stopten bij gebruik van interferon-b in vergelijking met glatirameeracetaat.
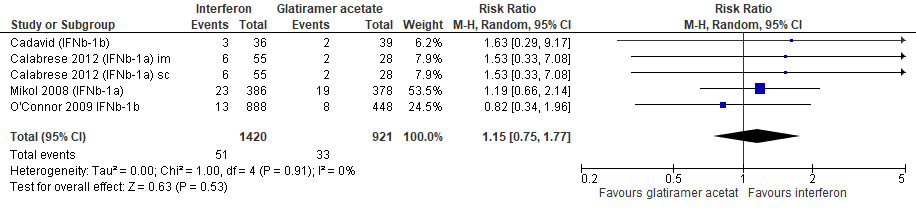
Figuur 1.4 Studieuitval vanwege bijwerkingen interferon-β versus glatirameeracetaat (96-104 weeks’ follow-up)
1.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 208 weken
Eén RCT (O’Connor et al., 2009) rapporteerde over studieuitval vanwege bijwerkingen na een follow-up duur van 208 weken. Volgens deze studie liet gebruik van interferon-b een groter risico op studieuitval zien dan glatirameeracetaat: RR=1.78 (95% BI: 0.62-5.16), of in absolute termen: 56 patiënten meer per 1000 (95% BI: 27 minder tot 297 meer) die stopten vanwege bijwerkingen.
1.8 Studieuitval om willekeurige reden bij een follow-up duur van 48-104 weken
Vier RCTs (Cadavid et al., 2009; Calabrese et al., 2012; Mikol et al., 2008; O’Connor et al., 2009) rapporteerden over studieuitval om een willekeurige reden. Op grond van de gecombineerde uitkomsten van deze studies had gebruik van interferon-b meer risico op studieuitval dan glatirameeracetaat: RR=1.30 (95% BI: 0.68-2.47), of in absolute termen: 29 patiënten meer per 1000 (95% BI: 31 minder tot 142 meer) die stopten.
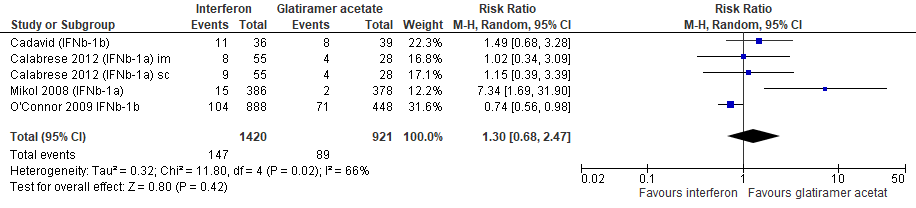
Figuur 1.5 Studieuitval om willekeurige reden interferon-β versus glatirameeracetaat
1.8 Studieuitval om willekeurige reden bij een follow-up duur van 208 weken)
Eén RCT (O’Connor et al., 2009) rapporteerde over studieuitval om een willekeurige reden na een follow-up duur van 208 weken. Volgens deze studie was er nauwelijks verschil tussen interferon-b en glatirameeracetaat: RR=1.02 (95% BI: 0.55-1.88), of in absolute termen: 4 patiënten meer per 1000 (95% BI: 96 minder tot 189 meer) die stopten bij gebruik van interferon.
1.9 (Ernstige) infecties
Hierover werd niet gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
1.10 Mortaliteit
Eén RCT (O’Connor et al., 2009) rapporteerde over mortaliteit. Volgens deze studie was er geen verschil tussen interferon-b en glatirameeracetaat: RR=1.0 (95% BI: (1.0-1.01).
1.11 Neoplasmata/maligniteit
Hierover werd niet gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
- Kwaliteit van bewijs
1.1 “Annualized Relapse Rate” (ARR)
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijke randomisatie en blindering, vertekening door veel ontbrekende uitkomsten) en met één niveau voor ernstige onnauwkeurigheid (<400 patiënten).
1.2 Proportie vrij van relapse
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijke randomisatie in alle studies en in sommige studies veel ontbrekende uitkomsten of onduidelijke blindering).
1.3 Toenemende invaliditeit
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijke randomisatie, ontbrekende uitkomsten, onduidelijke blindering).
1.4 Nieuwe T2 laesies of groter wordende T2 laesies
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor risk of bias (hoog risico op performance bias) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
1.5 Aantal patiënten zonder gadolinium aankleurende laesies
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor risk of bias (hoog risico op performance bias) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
1.6 Aantal nieuwe gadolinium aankleurende laesies
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijke randomisatie, onduidelijk risico op performance bias, veel ontbrekende data) en met één niveau voor ernstige onnauwkeurigheid (<400 patiënten).
1.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48-104 weken en stoppen om willekeurige reden bij een follow-up duur van 48-104 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd vanwege ernstige risk of bias (onduidelijke randomisatie in alle studies en in sommige studies veel ontbrekende uitkomsten of onduidelijke blindering) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
1.8 Studieuitval vanwege bijwerkingen en om willekeurige reden bij een follow-up duur van 208 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijke randomisatie, ontbrekende uitkomsten, onduidelijke blindering) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
1.10 Mortaliteit
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor risk of bias (hoog risico op performance bias) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
Conclusies
1.1 Annualized Relapse Rate bij een follow-up duur van 96-104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 96-104 weken voor de annualized relapse rate -gemiddeld voor beide doseringen en toedieningsroutes- 0.05 relapse minder (95% BI: 0.21 minder tot 0.11 meer) zien per patiënt per jaar in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Calabrese et al., 2012 |
1.2 Proportie vrij van relapse bij een follow-up duur van 96-104 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 96-104 weken voor de proportie patiënten vrij van relapse een vermindering van het relatieve effect zien met 2% (RR: 0.98; 95% BI: 0.90-1.06 en een risicoverschil van 12 patiënten vrij van relapse minder per 1000 (95% BI: 61 minder tot 36 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Cadavid et al., 2009 ; Mikol et al., 2008; O’Connor et al., 2009 |
1.3 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor toenemende invaliditeit een toename van het relatieve risico zien met 4% (RR: 1.04; 95% BI: 0.83-1.31) en een risicoverschil van 8 patiënten meer met toenemende invaliditeit (95% BI: 34 minder tot 62 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: O’Connor et al., 2009 |
1.4 Aantal patiënten zonder nieuwe T2 laesies of groter wordende T2 laesies bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal patiënten zonder nieuwe of groter wordende T2 laesies een toename van het relatieve effect zien met 8% (RR: 1.08; 95% BI: 0.86-1.36) en een risicoverschil van 30 patiënten meer zonder laesies per 1000 (95% BI: 52 minder tot 135 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Mikol et al., 2008 |
1.5 Aantal patiënten zonder gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal patiënten zonder gadolinium aankleurende laesies een toename van het relatieve effect zien met 21% (RR: 1.21; 95% BI: 1.08-1.35) en een risicoverschil van 141 patiënten meer zonder laesies per 1000 (95% BI: 54 meer tot 234 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Mikol et al., 2008 |
1.6 Aantal nieuwe gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal nieuw gadolinium aankleurende laesies een vermindering met gemiddeld 0.15 laesies zien (95% BI: 0.48 minder tot 0.17 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: O’Connor et al., 2009 |
1.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48-104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-104 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 15% (RR: 1.15; 95% BI: 0.75-1.77) en een risicoverschil van 5 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 9 minder tot 28 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bronnen: Cadavid et al., 2009; Calabrese et al., 2012; Mikol et al., 2008; O’Connor et al., 2009 |
1.7 Studieuitval om willekeurige reden bij een follow-up duur van 48-104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-104 weken voor studieuitval om een willekeurige reden een toename van het relatieve risico zien met 30% (RR: 1.30; 95% BI: 0.68-2.47) en een risicoverschil van 29 patiënten meer met studieuitval om een willekeurige reden per 1000 (95% BI: 31 minder tot 142 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bronnen: Cadavid et al., 2009; Calabrese et al., 2012; Mikol et al., 2008; O’Connor et al., 2009 |
1.8 Studieuitval vanwege bijwerkingen bij een follow-up duur van 208 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 208 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 78% (RR: 1.78; 95% BI: 0.62-5.16) en een risicoverschil van 56 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 27 minder tot 297 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: O’Connor et al., 2009 |
1.9 Studieuitval om willekeurige reden bij een follow-up duur van 208 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 208 weken voor studieuitval om een willekeurige reden een toename van het relatieve risico zien met 2% (RR: 1.02; 95% BI: 0.55-1.88) en een risicoverschil van 4 patiënten meer met studieuitval om een willekeurige reden per 1000 (95% BI: 96 minder tot 189 meer) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: O’Connor et al., 2009 |
1.10 Mortaliteit bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor mortaliteit geen verschil zien (RR: 1.0; 95% BI: 1.0-1.01) in vergelijking tot glatirameeracetaat (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: O’Connor et al., 2009 |
Review 2. Teriflunomide versus interferon
- Klinische effectiviteit
2.1 “Annualized Relapse Rate” (ARR)
Eén RCT (Vermersch et al., 2014) rapporteerde over de ARR. Teriflunomide verminderde in vergelijking met interferon-b de ARR niet, er was een verschil van + 0.04 relapses per persoon per jaar (95% BI: 0.17 minder tot 0.25 meer).
2.2 Proportie vrij van relapse
Eén RCT (Vermersch et al., 2014) rapporteerde over de proportie patiënten die na 48 weken vrij van een relapse waren gebleven. Teriflunomide was in deze studie minder effectief dan interferon-b: RR = 0.68 (95% BI: 0.57-0.82), of in absolute termen: 271 patiënten minder per 1000 (95% BI: 152 tot 364 minder).
Toenemende invaliditeit uitgedrukt in EDSS
Hierover werd niet gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
- Effectiviteit niet-klinisch
2.3 Over geen van de MRI-uitkomsten werd gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
- No Evidence of Disease Activity (NEDA)
Er werden geen uitkomsten gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
- Tolereerbaarheid en veiligheid
2.4 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48 weken
Eén RCT (Vermersch et al., 2014) rapporteerde over studieuitval vanwege bijwerkingen. In deze studie liet teriflunomide in vergelijking met interferon-b een kleiner risico op studieuitval vanwege bijwerkingen zien: RR=0.51 (95% BI: 0.27-0.98), of in absolute termen: 104 patiënten minder per 1000 (95% BI: 4 tot 154 minder).
2.5 Studieuitval om willekeurige reden bij een follow-up duur van 48 weken
Eén RCT (Vermersch et al., 2014) rapporteerde over studieuitval om een willekeurige reden na een follow-up duur van 48 weken. Volgens deze studie ging gebruik van teriflunomide in vergelijking met interferon-b gepaard met een kleiner risico op studieuitval: RR=0.69 (95% BI: 0.42-1.11), of in absolute termen: 89 patiënten minder per 1000 (95% BI: 167 minder tot 32 meer) die stopten om een willekeurige reden.
2.6 (Ernstige) infecties
Eén RCT (Vermersch et al., 2014) rapporteerde over infecties. Volgens deze studie ging gebruik van teriflunomide in vergelijking met interferon-b gepaard met een enigszins verhoogd risico op infecties: RR=1.08 (95% BI: 0.81-1.43), of in absolute termen: 36 patiënten meer per 1000 (95% BI: 86 minder tot 194 meer).
2.7 Mortaliteit
Hierover werd niet gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
2.8 Neoplasmata/maligniteit
Hierover werd niet gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
- Kwaliteit van bewijs
2.1 “Annualized Relapse Rate” (ARR)
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (interferon was open-label, verschil in percentage uitvallers tussen beide groepen, onduidelijke randomisatieprocedure) en met één niveau voor ernstige onnauwkeurigheid (<400 patiënten).
2.2 Proportie vrij van relapse
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (interferon-b was open-label, verschil in percentage uitvallers tussen beide groepen, onduidelijke randomisatieprocedure) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
2.4 Studieuitval vanwege bijwerkingen bij follow-up duur 48-104 weken en stoppen om willekeurige reden bij follow-up duur 48-104 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (interferon-b was open-label, verschil in percentage uitvallers tussen beide groepen, onduidelijke randomisatieprocedure) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
2.5 Studieuitval vanwege bijwerkingen en om willekeurige reden bij een follow-up duur van 208 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (interferon-b was open-label, verschil in percentage uitvallers tussen beide groepen, onduidelijke randomisatieprocedure) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
2.6 Infecties
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (interferon-b was open-label, verschil in percentage uitvaller tussen beide groepen, onduidelijke randomisatieprocedure) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
Conclusies
2.1 Annualized Relapse Rate bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor de annualized relapse rate 0.04 relapse meer (95% BI: 0.17 minder tot 0.25 meer) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.2 Proportie vrij van relapse bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor de proportie patiënten vrij van relapse een vermindering van het relatieve effect zien met 32% (RR: 0.68; 95% BI: 0.57-0.83 en een risicoverschil van 271 patiënten vrij van relapse minder per 1000 (95% BI: 152 minder tot 364 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.3 MRI uitkomsten
|
---------
|
Er werden geen MRI-uitkomsten gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
|
2.4 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor studieuitval vanwege bijwerkingen een vermindering van het relatieve risico zien met 49% (RR: 0.51; 95% BI: 0.27-0.98 en een risicoverschil van 104 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 4 minder tot 154 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.5 Studieuitval om willekeurige reden bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor studieuitval om willekeurige reden een vermindering van het relatieve risico zien met 31% (RR: 0.69; 95% BI: 0.42-1.11 en een risicoverschil van 89 patiënten minder met studieuitval om willekeurige reden per 1000 (95% BI: 167 minder tot 32 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
2.6 Infecties bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor infecties een toename van het relatieve risico zien met 8% (RR: 1.08; 95% BI: 0.81-1.43) en een risicoverschil van 36 patiënten meer met infecties per 1000 (95% BI: 86 minder tot 194 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Vermersch et al., 2014 |
Review 3. Fingolimod versus interferon-b
- Klinische effectiviteit
3.1 “Annualized Relapse Rate” (ARR)
Eén RCT (Cohen et al., 2010) rapporteerde over de ARR. Fingolimod verminderde in vergelijking met interferon-b de ARR met 0.17 relapses per persoon per jaar (95% BI: 0.26 tot 0.08 minder).
3.2 Proportie vrij van relapse
Eén RCT (Cohen et al., 2010) rapporteerde over de proportie patiënten die na 52 weken vrij van een relapse waren gebleven. Fingolimod was in deze studie effectiever dan interferon-b: RR=1.19 (95% BI: 1.11-1.29), of in absolute termen: 131 patiënten meer per 1000 (95% BI: 76 tot 201 meer) die vrij waren gebleven van een relapse.
3.3 Toenemende invaliditeit uitgedrukt in EDSS
Eén RCT (Cohen et al., 2010) rapporteerde over toenemende invaliditeit. Volgens deze studie vertraagde fingolimod toenemende invaliditeit iets meer dan interferon-b: RR=0.74 (95% BI: 0.45- 1.22), ofwel in absolute termen: 21 patiënten minder per 1000 (95% BI: 43 minder tot 17 meer) bij gebruik van fingolimod vergeleken met interferon-b.
- Effectiviteit niet-klinisch
3.4 Aantal patiënten zonder nieuwe T2 laesies of groter wordende T2 laesies
Eén RCT (Cohen et al., 2010) rapporteerde over het aantal patiënten met nieuwe of groter wordende T2-laesies. Volgens deze studie ging gebruik van fingolimod met minder nieuwe T2 laesies gepaard dan met interferon-b: RR=1.20 (95% BI: 1.04-1.39), ofwel in absolute termen: 91 patiënten meer per 1000 (95% BI: 18 tot 178 meer) met fingolimod dan met interferon-b.
3.5 Aantal patiënten zonder gadolinium aankleurende laesies laesies
Eén RCT (Cohen et al., 2010) rapporteerde over het aantal patiënten zonder gadolinium aankleurende laesies. Volgens deze studie ging gebruik van fingolimod gepaard met een groter aantal patiënten zonder gadolinium aankleurende laesies dan gebruik van interferon-b: RR=1.12 (95% BI: 1.05-1.19), ofwel in absolute termen 97 meer per 1000 (95% BI: 40 tot 154 meer).
3.6 Gemiddeld aantal gadolinium aankleurende laesies
Eén RCT (Cohen et al., 2010) rapporteerde over het gemiddelde aantal gadolinium aankleurende laesies. Volgens deze studie ging gebruik van fingolimod gepaard met een kleiner aantal gadolinium aankleurende laesies dan interferon-b: gemiddeld 0.28 minder (95% BI: 0.50 tot 0.06 minder).
- No Evidence of Disease Activity (NEDA)
Er werden geen uitkomsten gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
- Tolereerbaarheid en veiligheid
3.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 52 weken
Eén RCT (Cohen et al., 2010) rapporteerde over studieuitval vanwege bijwerkingen. Volgens deze studie ging gebruik van fingolimod in vergelijking met interferon-b gepaard met een groter risico op studieuitval vanwege bijwerkingen: RR=1.41 (95% BI: 0.92-2.18), ofwel in absolute termen 30 studie-uitvallers vanwege bijwerkingen meer per 1000 (95% BI: 6 minder tot 88 meer) met fingolimod dan met interferon-b.
3.8 Studieuitval om willekeurige reden bij een follow-up duur van 52 weken
Eén RCT (Cohen et al., 2010) rapporteerde over studieuitval om een willekeurige reden na een follow-up duur van 52 weken. Volgens deze studie liet gebruik van fingolimod een kleiner risico op studieuitval om willekeurige reden zien dan interferon-b: RR=0.69 (95% BI: 0.45-1.07) ofwel in absolute termen, 32 studie uitvallers om willekeurige reden minder per 1000 (95% BI: 57 minder tot 7 meer) met fingolimod dan met interferon-b.
3.9 (Ernstige) infecties bij een follow-up duur van 52 weken
Eén RCT (Cohen et al., 2010) rapporteerde over infecties. Volgens deze studie was er geen verschil in het optreden van infecties tussen fingolimod en interferon-b: RR=1.00 (95% BI: 0.86-1.17).
Mortaliteit bij een follow-up duur van 52 weken
Hierover werd niet gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
3.10 Neoplasmata/maligniteit bij een follow-up duur van 52 weken
Eén RCT (Cohen et al., 2010) rapporteerde over het optreden van een neoplasma. Noch in de fingolimod-groep noch in de interferon-b-groep werd het optreden van een neoplasma waargenomen.
- Kwaliteit van bewijs
3.1 “Annualized Relapse Rate” (ARR) bij follow-up duur van 52 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten).
3.2 Proportie vrij van relapse bij follow-up duur van 52 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten).
3.3 Toenemende invaliditeit bij follow-up duur van 52 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
3.4 Nieuwe T2 laesies of groter wordende T2 laesies bij follow-up duur van 52 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten).
3.5 Aantal patiënten zonder gadolinium aankleurende laesies bij follow-up duur van 52 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten).
3.6 Gemiddeld aantal gadolinium aankleurende laesies bij follow-up duur van 52 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten) en met één niveau vanwege ernstige onnauwkeurigheid (betrouwbaarheidsinterval sluit zowel verwaarloosbaar als aanmerkelijk effect in).
3.7 Studieuitval vanwege bijwerkingen bij follow-up duur van 52 weken en om willekeurige reden bij een follow-up duur van 52 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
3.9 Infecties
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
3.10 Neoplasmata/Maligniteit bij een follow-up duur van 52 weken
De kwaliteit van bewijs is zeer laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten), met één niveau vanwege ernstige onnauwkeurigheid (<300 events) en met één niveau voor indirect bewijs: de follow-up duur van 52 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk vindt voor een beoordeling van het risico op het optreden van maligniteiten.
Conclusies
3.1 Annualized Relapse Rate bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor de annualized relapse rate 0.17 relapse minder (95% BI: 0.26 minder tot 0.08 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.2 Proportie vrij van relapse bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 19% (RR: 1.19; 95% BI: 1.11-1.29 en een risicoverschil van 131 patiënten vrij van relapse meer per 1000 (95% BI: 76 meer tot 201 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.3 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 26% (RR: 0.74; 95% BI: 0.45-1.22) en een risicoverschil van 21 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 43 minder tot 17 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Cohen et al., 2010 |
3.4 Aantal patiënten zonder nieuwe T2 laesies of groter wordende T2 laesies bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor het aantal patiënten zonder nieuwe of groter wordende T2 laesies een toename van het relatieve effect zien met 20% (RR: 1.20; 95% BI: 1.04-1.39) en een risicoverschil van 91 patiënten meer zonder laesies per 1000 (95% BI: 18 meer tot 178 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.5 Aantal patiënten zonder gadolinium aankleurende laesies bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor het aantal patiënten zonder gadolinium aankleurende laesies een toename van het relatieve effect zien met 12% (RR: 1.12; 95% BI: 1.05-1.19) en een risicoverschil van 97 patiënten meer zonder laesies per 1000 (95% BI: 40 meer tot 154 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Cohen et al., 2010 |
3.6 Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor het gemiddeld aantal gadolinium aankleurende laesies een vermindering met gemiddeld 0.28 laesies zien (95% BI: 0.50 minder tot 0.06 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Cohen et al., 2010 |
3.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 41% (RR: 1.41; 95% BI: 0.92-2.18) en een risicoverschil van 30 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 6 minder tot 88 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Cohen et al., 2010 |
3.8 Studieuitval om willekeurige reden bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor studieuitval om een willekeurige reden een vermindering van het relatieve risico zien met 31% (RR: 0.69; 95% BI: 0.45-1.07) en een risicoverschil van 32 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 57 minder tot 7 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: Cohen et al., 2010 |
3.9 Infecties bij een follow-up duur van 52 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor infecties geen verschil zien (RR: 1.0; 95% BI: 0.86-1.17) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bron: Cohen et al., 2010 |
3.10 Maligniteit bij een follow-up duur van 52 weken
|
Zeer laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor maligniteit geen verschil zien (RR: niet berekend vanwege niet optreden van een maligniteit) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect verschilt waarschijnlijk aanzienlijk verschillen het geschatte effect).
Bron: Cohen et al., 2010 |
Review 4. Alemtuzumab versus interferon-b
- Klinische effectiviteit
4.1 “Annualized Relapse Rate” (ARR) bij een follow-up duur (104-156, 260 weken)
Twee RCTs (Cohen et al., 2012; Coles et al., 2012) rapporteerden over de ARR bij een follow-up duur van 104-156 weken. Eén RCT (Camms223, 2008) rapporteerde ARR bij een follow-up duur van 260 weken. Alemtuzumab verminderde in vergelijking met interferon-b de ARR met 0.25 (95% BI: 0.33 tot 0.18 minder) relapses per persoon per jaar bij een follow-up duur van 104-156 weken. Bij een follow-up duur van 260 weken bleef dit verschil gehandhaafd: 0.23 (95% BI: 0.3 tot 0.16 minder) relapses per persoon per jaar minder met alemtuzumab in vergelijking met interferon-b.
4.2 Proportie vrij van relapse bij een follow-up duur (104-156 weken, 260 weken)
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over de proportie patiënten die na 104-156 weken vrij van een relapse waren gebleven. Alemtuzumab was in deze studies effectiever dan interferon-b: RR=1.38 (95% BI: 1.26-1.51), of in absolute termen, 198 patiënten meer per 1000 (95% BI: 136 tot 266 meer) die vrij waren gebleven van een relapse. Bij een follow-up duur van 260 weken (Camms223, 2008) was dit verschil nog uitgesprokener: RR= 1.67 (95% BI: 1.29-2.17), ofwel in absolute termen, 272 patiënten meer per 1000 (95% BI: 118 tot 474 meer) die vrij van een relapse bleven met alemtuzumab in vergelijking met interferon-b.
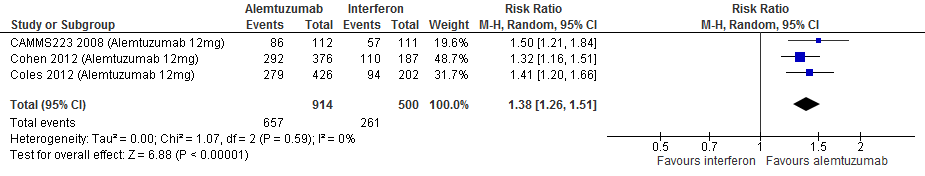
Figuur 4.1 Proportie vrij van relapse alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
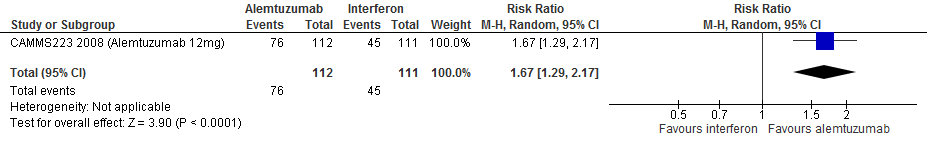
Figuur 4.2 Proportie vrij van relapse alemtuzumab versus interferon-β (260 weeks’ follow-up)
4.3 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur (104-156, 260 weken)
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over toenemende invaliditeit na 104-156 weken. Alemtuzumab was in deze studies effectiever dan interferon-b: RR=0.59 (95% BI: 0.4-0.86) ofwel in absolute termen, 69 patiënten minder per 1000 (95% BI: 24 tot 101 minder) bij wie sprake was van toenemende invaliditeit met alemtuzumab in vergelijking met interferon-b.
Bij een follow-up duur van 260 weken (Camms223, 2008) was dit verschil nog uitgesprokener: RR=0.43 (95% BI: 0.24-0.78), ofwel in absolute termen, 154 patiënten minder per 1000 (95% BI: 59 tot 205 minder) bij wie sprake was van toenemende invaliditeit met alemtuzumab in vergelijking met interferon-b.
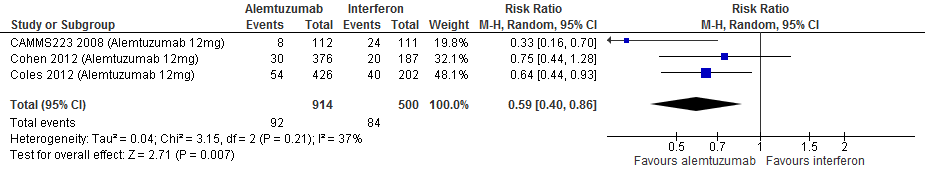
Figuur 4.3 Toenemende invaliditeit alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
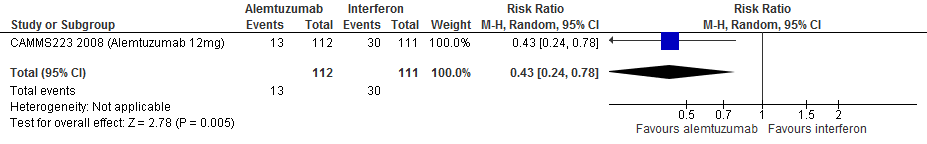
Figuur 4.4 Toenemende invaliditeit alemtuzumab versus interferon-β (260 weeks’ follow-up)
- Effectiviteit niet-klinisch
4.4 Aantal patiënten met nieuwe T2 laesies of groter wordende T2 laesies
Twee RCTs (Cohen et al., 2012; Coles et al., 2012) rapporteerden over het aantal patiënten met nieuwe of groter wordende T2-laesies bij een follow-up duur van 104 weken. Volgens deze studies ging gebruik van alemtuzumab met minder nieuwe of groter wordende T2 laesies gepaard dan met interferon-b: RR=0.77 (95% BI: 0.60-1.00), ofwel in absolute termen, 139 patiënten per 1000 (95% BI: 242 minder tot 0 minder) bij wie sprake was van nieuwe of groter wordende T2 laesies bij gebruik van alemtuzumab in vergelijking met interferon-b.
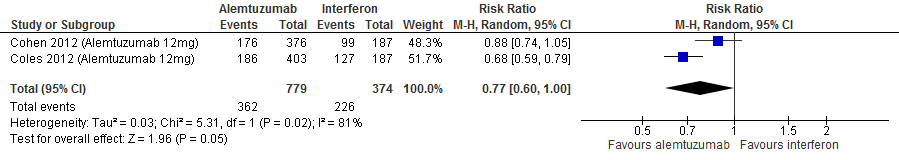
Figuur 4.5 Aantal patiënten met nieuwe T2 laesies of groter wordende T2 laesies alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
Overige MRI-uitkomsten (aantal patiënten zonder gadolinium aankleurende laesies, gemiddeld aantal gadolinium aankleurende laesies) werden niet gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
- No Evidence of Disease Activity (NEDA)
Er werden geen uitkomsten gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
- Tolereerbaarheid en veiligheid
4.5 Studieuitval vanwege bijwerkingen bij een follow-up duur van 104-156, 260 weken
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over studieuitval vanwege bijwerkingen na 104-156 weken. Volgens deze studies kwam studieuitval vanwege bijwerkingen minder vaak voor onder patiënten bij gebruik van alemtuzumab in vergelijking met interferon-b: RR=0.31 (95% BI: 0.17-0.55), ofwel in absolute termen, 54 studie uitvallers minder per 1000 (95% BI: 35 tot 65 minder) wanneer alemtuzumab in plaats van interferon-b werd gebruikt. Bij een follow-up duur van 260 weken (Camms223, 2008) bleef dit verschil gehandhaafd: RR=0.35 (95% BI: 0.13-0.95), ofwel in absolute termen, 85 patiënten minder per 1000 (95% BI: 7 tot 114 minder) wanneer alemtuzumab in plaats van interferon-b werd gebruikt.
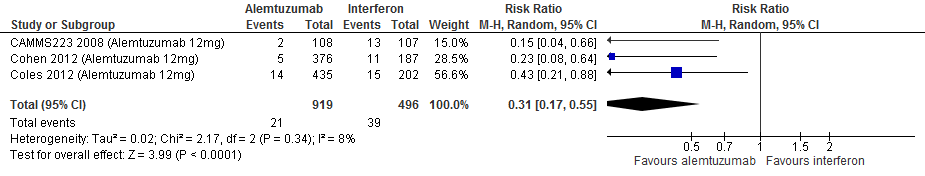
Figuur 4.6 Studieuitval vanwege bijwerkingen alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
4.6 Studieuitval om willekeurige reden bij een follow-up duur van 104-156 weken
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over studieuitval om willekeurige reden na 104-156 weken. Volgens deze studies verminderde alemtuzumab in vergelijking met interferon-b het risico op studieuitval om willekeurige reden: RR=0.36 (95% BI: 0.25-0.52), ofwel in absolute termen, 178 studie uitvallers minder per 1000 (95% BI: 133 tot 208 minder) wanneer zij alemtuzumab gebruikten in plaats van interferon-b.
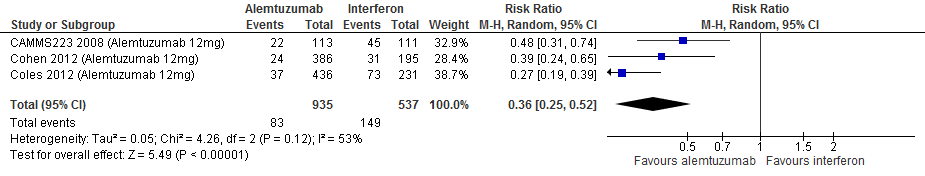
Figuur 4.7 Studieuitval om willekeurige redenen alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
4.7 (Ernstige) infecties bij een follow-up duur van 104-156, 260 weken
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over infecties. Volgens deze studies hadden bij een follow-up duur van 104-156 weken meer patiënten een infectie met alemtuzumab in vergelijking met interferon-b: RR=1.32 (95% BI: 1.10-1.58), ofwel in absolute termen, 174 patiënten meer per 1000 (95% BI: 54 tot 315 meer) met een infectie bij gebruik van alemtuzumab in vergelijking met interferon-b. Bij een follow-up duur van 260 weken (Camms223, 2008) bleef dit verschil gehandhaafd: RR=1.41 (95% BI: 1.13-1.76), ofwel 207 patiënten meer per 1000 (95% BI: 66 tot 384 meer) met een infectie bij gebruik van alemtuzumab in vergelijking met interferon-b.
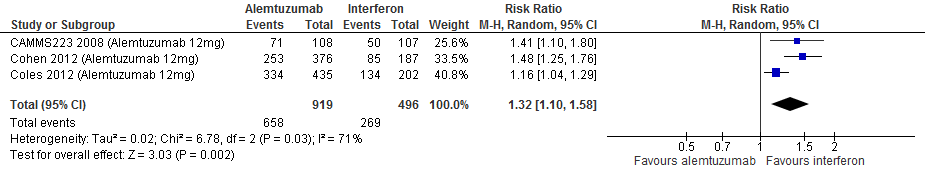
Figuur 4.8 Risico op infectie alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
4.8 Mortaliteit bij een follow-up duur van 104-156, 260 weken
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over mortaliteit bij een follow-up duur van 104-156 weken. Volgens deze studies kwamen er vier sterfgevallen op 919 patiënten voor die alemtuzumab gebruikten en geen sterfgevallen in de interferon-b-groep (n=496). In de RCT die over 260 weken rapporteerde (Camms223, 2008) kwam in zowel de alemtuzumab-groep (n=108) als de interferon-b-groep (n-107) één sterfgeval voor.
[7] In de besproken studies werden geen suïcides gezien.
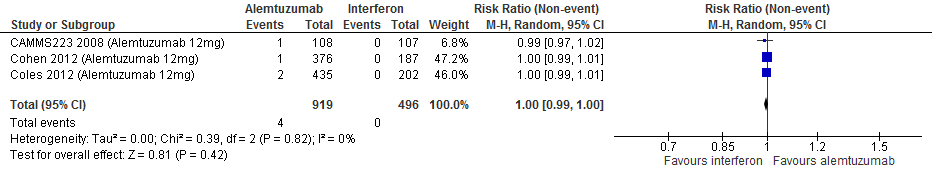
Figuur 4.9 Mortaliteit alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
4.9 Neoplasmata/maligniteit bij een follow-up duur van 104-156 weken
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over maligniteit bij een follow-up duur van 104-156 weken. In de alemtuzumab-groep was de incidentie van een maligniteit 0.4%, in de interferon-groep 0.6%, hetgeen suggereert dat er geen verschil is in risico op een maligniteit tussen alemtuzumab en interferon-b.[8]
[8] De Europese werkgroep berekende geen relatief risico of risicoverschil en verwijst naar een commentaar dat echter niet te vinden is.
4.10 Auto-immuunziekten (immuun-gemedieerde trombocytopenische purpura [ITP]) (104-156, 260 weken)
Drie RCTs (Coles et al., 2012; Cohen et al., 2012; Camms223, 2008) rapporteerden over immuungemedieerde trombocytopenische purpura (ITP) bij een follow-up duur van 104-156 weken. Alemtuzumab verhoogt in vergelijking met interferon-b het risico op ITP enigszins: RR=2.68 (95% BI: 0.56-12.9), ofwel in absolute termen, 3 patiënten meer per 1000 (95% BI: 1 minder tot 24 mee) met ITP wanneer alemtuzumab in vergelijking met interferon-b werd gebruikt. Bij een follow-up duur van 260 weken (Camms223, 2008) bleef dit verschil gehandhaafd: 9 patiënten meer per 1000 (95% BI: 8 minder tot 192 meer) met ITP bij gebruik van alemtuzumab in vergelijking met interferon-b. Dit is de enige auto-immuungemedieerde bijwerking die in de Europese richtlijn gerapporteerd wordt. Zie verder in paragraaf Professioneel perspectief van dit hoofdstuk waar ook andere auto-immuungemedieerde bijwerkingen in relatie tot alemtuzumab worden genoemd.
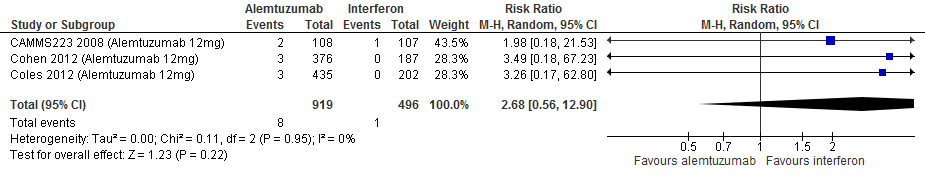
Figuur 4.10 Auto-immuunziekten alemtuzumab versus interferon-β (104-156 weeks’ follow-up)
- Kwaliteit van bewijs
4.1 “Annualized Relapse Rate” (ARR) bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (alle studies waren open label; uitkomstenbeoordelaars waren in twee studies niet geblindeerd).
4.1 “Annualized Relapse Rate” (ARR) bij een follow-up duur van 260 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (open label studie) en met één niveau voor ernstige onnauwkeurigheid (<400 patiënten).
4.2 Proportie vrij van relapse bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (alle studies waren open label; uitkomstenbeoordelaars waren in twee studies niet geblindeerd).
4.2 Proportie vrij van relapse bij een follow-up duur van 260 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (open label studie) en met één niveau voor ernstige onnauwkeurigheid (<300 events).
4.3 Toenemende invaliditeit bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (alle studies waren open label; uitkomstenbeoordelaars waren in twee studies niet geblindeerd) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
4.3 Toenemende invaliditeit bij een follow-up duur van 260 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (open label studie) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
4.4 Nieuwe T2 laesies of groter wordende T2 laesies bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten).
4.5a Studieuitval vanwege bijwerkingen bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (alle studies waren open label; uitkomstenbeoordelaars waren in twee studies niet geblindeerd) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
4.5b Studieuitval vanwege bijwerkingen bij een follow-up duur van 260 weken
De kwaliteit van bewijs is laag. De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (open label studie) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
4.6 Studieuitval om willekeurige reden bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (alle studies waren open label; uitkomstenbeoordelaars waren in twee studies niet geblindeerd) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
4.7a Infecties bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (onduidelijk of blindering van uitkomstenbeoordelaar heeft plaatsgevonden, groot risico op selectieve rapportage van uitkomsten) en met één niveau vanwege ernstige inconsistentie (een van de studies verschilt sterk in grootte effect van de twee andere studies; I2=71%; p-waarde chi2-toets op heterogeniteit: 0.03).
4.7b Infecties bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd voor ernstige risk of bias (alle studies waren open label; uitkomstenbeoordelaars waren in twee studies niet geblindeerd) en met één niveau vanwege ernstige onnauwkeurigheid (<300 events).
4.8 Mortaliteit bij een follow-up duur van 104-156 weken, 260 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events).
4.9 Maligniteit bij een follow-up duur van 104-156 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events).
4.10 Auto-immuunziekten (immuun-gemedieerde trombocytopenische purpura [ITP]) bij een follow-up duur van 104-156, 260 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events).
Conclusies
4.1 Annualized Relapse Rate (follow-up 104-156 weken)
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor de annualized relapse rate 0.25 relapse minder (95% BI: 0.33 minder tot 0.18 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012 |
4.1 Annualized Relapse Rate (follow-up 260 weken)
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 260 weken voor de annualized relapse rate 0.23 relapse minder (95% BI: 0.30 minder tot 0.16 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Camms223, 2008 |
4.2 Proportie vrij van relapse bij een follow-up duur van 104-156 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 38% (RR: 1.38; 95% BI: 1.26-1.51) en een risicoverschil van 198 patiënten vrij van relapse meer per 1000 (95% BI: 136 meer tot 266 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.3 Proportie vrij van relapse bij een follow-up duur van 260 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 260 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 67% (RR: 1.67; 95% BI: 1.29-2.17 en een risicoverschil van 272 patiënten vrij van relapse meer per 1000 (95% BI: 118 meer tot 474 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.4 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 104-156, 260 weken)
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 41% (RR: 0.59; 95% BI: 0.40-0.86) respectievelijk 57% (RR: 0.43; 95% BI: 0.24-0.78) en een risicoverschil van 8 patiënten minder (95% BI: 34 minder tot 62 minder) respectievelijk 154 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 59 minder tot 205 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008* *Dit is de enige studie die ook voor een follow-up van 260 weken rapporteerde. |
4.5 Aantal patiënten met nieuwe T2 laesies of groter wordende T2 laesies bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal patiënten met nieuwe of groter wordende T2 laesies een vermindering van het relatieve risico zien met 23% (RR: 0.77; 95% BI: 0.60-1.00) en een risicoverschil van 139 patiënten minder met laesies per 1000 (95% BI: 242 minder tot 0 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Cohen et al., 2012; Coles et al., 2012 |
4.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 104-156, 260 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor studieuitval vanwege bijwerkingen een vermindering van het relatieve risico zien met 69% (RR: 0.31; 95% BI: 0.17-0.55) respectievelijk 65% (RR:0.35; 95% BI: 0.13-0.95) en een risicoverschil van 54 patiënten minder (95% BI: 35 minder tot 65 minder) respectievelijk 85 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 7 minder tot 114 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect). Dit is echter een artefact omdat alemtuzumab in een veel lagere frequentie wordt toegediend dan interferon-b.
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008* *Dit is de enige studie die ook voor een follow-up van 260 weken rapporteerde. |
4.7 Studieuitval om willekeurige reden bij een follow-up duur van 104-156 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor studieuitval om een willekeurige reden een vermindering van het relatieve risico zien met 64% (RR: 0.36; 95% BI: 0.25-0.52) en een risicoverschil van 178 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 133 minder tot 208 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect). Dit is echter een artefact omdat alemtuzumab in een veel lagere frequentie wordt toegediend dan interferon-b.
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.8 Infecties bij een follow-up duur van 104-156, 260 weken
|
Laag
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor infecties een toename van het relatieve risico zien met 32% (RR: 1.32; 95% BI: 1.10-1.58) respectievelijk 41% (RR:1.41; 95% BI: 1.13-1.76) en een risicoverschil van 174 patiënten meer (95% BI: 53 meer tot 315 meer) respectievelijk 207 patiënten meer infecties per 1000 (95% BI: 66 meer tot 384 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.9 Mortaliteit bij een follow-up duur van 104-156, 260 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 weken voor mortaliteit een risicoverschil zien van 4 overleden patiënten meer per 1000 (95% BI: 11 minder tot 4 meer) en bij een follow-up duur van 260 weken geen verschil (RR 1; 95% BI: 0.97-1.03) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.10 Maligniteit bij een follow-up duur van 260 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 260 weken voor maligniteit een risicoverschil van 2 patiënten met een maligniteit minder per 1000 zien in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
4.11 Auto-immuunziekten (immuun-gemedieerde trombocytopenische purpura)** bij een follow-up duur van 104-156, 260 weken
|
Redelijk
GRADE |
Gebruik van alemtuzumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-156 en 260 weken voor auto-immuunziekten een toename van het relatieve risico zien met 168% (RR: 2.68; 95% BI: 0.56-12.90) respectievelijk 98% (RR:1.98; 95% BI: 0.18-21.53) en een risicoverschil van 3 patiënten meer (95% BI: 1 minder tot 24 meer) respectievelijk 9 patiënten meer per 1000 (95% BI: 8 minder tot 192 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Coles et al., 2012; Cohen et al., 2012; Camms223, 2008 |
Review 5. Ocrelizumab versus interferon-b
- Klinische effectiviteit
5.1 “Annualized Relapse Rate” (ARR)
Twee RCTs (beide gerapporteerd in Hauser et al., 2017) rapporteerden over de ARR na een follow-up van 96 weken. Ocrelizumab verminderde in vergelijking met interferon-b de ARR met 0.13 (95% BI: 0.18 tot 0.08 minder) relapses per persoon per jaar.
5.2 Proportie vrij van relapse
Hierover werd niet gerapporteerd in de Europese richtlijntekst en bijbehorende bijlagen. De werkgroep droeg vanwege het belang van deze uitkomstmaat voor de netwerkmeta-analyse een studie aan die na de publicatie van de Europese richtlijn verscheen en waarin wel data over de proportie vrij van relapse werden gepubliceerd (Havrdová et al., 2018). Deze data zijn gebaseerd op de OPERA I and OPERA II studies. De follow-up duur hiervan was 96 weken.
Ocrelizumab was in deze studies effectiever dan interferon-b: RR=1.19 ((95% BI: 1.12–1.26), of in absolute termen, 128 patiënten meer per 1000 (95% BI: 81 meer tot 175 meer) die vrij waren gebleven van een relapse.
5.3 Verbetering invaliditeit (uitgedrukt in EDSS) bevestigd na 12 en na 24 weken
Eén RCT (Hauser et al., 2017) rapporteerde over verbetering van invaliditeit.
Voor patiënten met een initiële EDSS score van ≥2.0 en ≤5.5, werd verbetering gedefinieerd als een reductie van de EDSS score met ≥1.0 punt in vergelijking met de initiële EDSS score. Voor patiënten met een initiële EDSS score van >5.5, werd verbetering gedefinieerd als een reductie van de EDSS score met ≥0.5 punt.
Ocrelizumab was in deze RCT effectiever dan interferon-b: RR=1.32 (95% BI: 1.04-1.68), ofwel in absolute termen, 50 patiënten meer per 1000 (95% BI: 6 tot 106 meer) met verbeterde invaliditeit, bevestigd na 12 weken. Voor zover het verbeterde invaliditeit na 24 weken bevestigd betrof, was ocrelizumab eveneens effectiever dan interferon-b: RR=1.35 (95% BI: 1.02-1.79), ofwel in absolute termen, 40 patiënten meer per 1000 (95% BI: 2 tot 91 meer) met verbeterde invaliditeit.
5.4 Toenemende invaliditeit uitgedrukt in EDSS
Twee RCTs (beide gerapporteerd in Hauser et al., 2017) rapporteerden over toenemende invaliditeit na 96 weken. Toenemende invaliditeit werd gedefinieerd als een toename van de EDSS score met >0.5 punt na 96 weken ten opzichte van de initiële EDSS score.
Ocrelizumab was in deze studies effectiever dan interferon-b: RR=0.6 (95% BI: 0.46 -0.8), ofwel in absolute termen, 67 patiënten minder per 1000 (95% BI: 90 tot 33 minder) bij wie sprake was van toenemende invaliditeit met ocrelizumab in vergelijking met interferon-b.
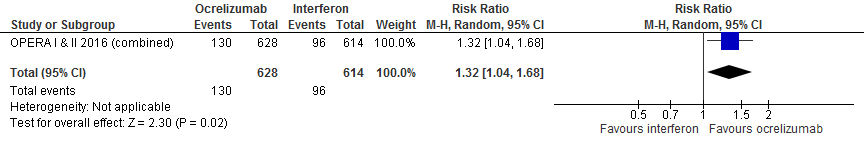
Figuur 5.2 Toenemende invaliditeit (bevestigd na 12 weken) ocrelizumab versus interferon-β (52-96 weeks’ follow-up)
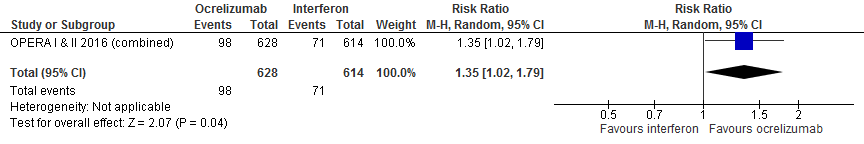
Figuur 5.3 Toenemende invaliditeit (bevestigd na 24 weken) ocrelizumab versus interferon-β (52-96 weeks’ follow-up)
- Effectiviteit niet-klinisch
MRI-uitkomsten (o.a. aantal patiënten zonder gadolinium aankleurende laesies, gemiddeld aantal gadolinium aankleurende laesies, aantal nieuwe of groter wordende T2 laesies) werden niet gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen
- No Evidence of Disease Activity (NEDA)
Er werden geen uitkomsten gerapporteerd in de Europese richtlijn en daarbij behorende bijlagen.
- Tolereerbaarheid en veiligheid
5.5 Studieuitval vanwege bijwerkingen bij een follow-up duur van 96 weken
Twee RCTs (beide gerapporteerd in Hauser et al., 2017) rapporteerden over studieuitval vanwege bijwerkingen na 96 weken. Volgens deze studies is bij patiënten die ocrelizumab gebruiken minder vaak sprake van studieuitval vanwege bijwerkingen dan onder patiënten die interferon-b gebruiken: RR=0.46 (95% BI: 0.3-0.7), ofwel in absolute termen, 42 patiënten minder per 1000 (95% BI: 54 tot 23 minder) wanneer ocrelizumab in plaats van interferon-b werd gebruikt.
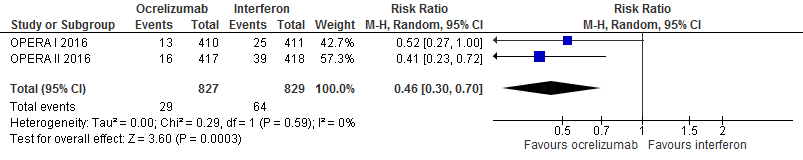
Figuur 5.4 Studieuitval vanwege bijwerkingen ocrelizumab versus interferon-β (96 weeks’ follow-up)
5.6 Studieuitval om willekeurige reden bij een follow-up duur van 96 weken
Twee RCTs (beide gerapporteerd in Hauser et al., 2017) rapporteerden over studieuitval om willekeurige reden na 96 weken. Volgens deze studies verminderde ocrelizumab in vergelijking met interferon-b het risico op studieuitval om willekeurige reden: RR=0.60 (95% BI: 0.48-0.75), ofwel in absolute termen, 80 studie uitvallers om willekeurige reden minder per 1000 (95% BI: 104 tot 50 minder) wanneer zij ocrelizumab gebruikten in plaats van interferon-b.
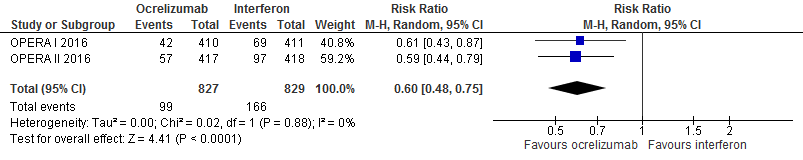
Figuur 5.5 Studieuitval om willekeurige redenen ocrelizumab versus interferon-β (96 weeks’ follow-up)
5.7 (Ernstige) infecties
Eén RCT (Hauser et al., 2017) rapporteerde over infecties en binnengedrongen parasieten (‘infestations’). Volgens deze studie hadden bij een follow-up duur van 96 weken meer patiënten infecties en parasieten met ocrelizumab in vergelijking met interferon-b: RR=1.11 (95% BI: 1.02-1.22), ofwel in absolute termen, 58 patiënten meer per 1000 (95% BI: 10 tot 115 meer) met een (parasitaire) infectie bij gebruik van ocrelizumab in vergelijking met interferon-b.
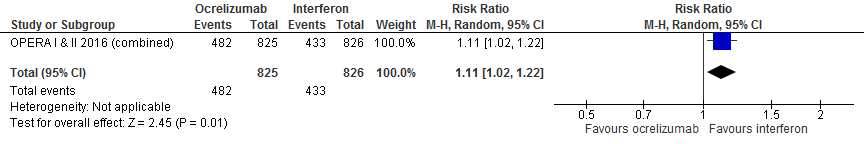
Figuur 5.6 Risico op infecties ocrelizumab versus interferon-β (96 weeks’ follow-up)
5.8 Mortaliteit
Eén RCT (Hauser et al., 2017) rapporteerde over mortaliteit bij een follow-up duur van 96 weken. Volgens deze studie kwam er één sterfgeval op 825 patiënten voor die ocrelizumab gebruikten en twee sterfgevallen in de interferon-b-groep (n=826) hetgeen een relatief risico geeft van 0.5 (95% BI: 0.05-5.51), ofwel in absolute termen, 1 patiënt minder per 1000 (95% BI: 8 minder tot 5 meer) die overleden na gebruik van ocrelizumab in vergelijking met interferon-b.[9]
[9] De Europese werkgroep vermeldt in bijlage een RR van 1.0. Dit is evident onjuist. RR en risicoverschil zijn door de werkgroep opnieuw berekend.
Figuur 5.5 Mortaliteit ocrelizumab versus interferon-β (96 weeks’ follow-up)
5.9 Neoplasmata/maligniteit bij een follow-up duur van 96 weken
Eén RCT (Hauser et al., 2017) rapporteerde over maligniteiten bij een follow-up duur van 96 weken. In de ocrelizumab-groep was de incidentie van een maligniteit 0.5%, in de interferon-b-groep 0.2%, hetgeen een relatief risico geeft van 2.0 (95% BI: 0.37-10.90), ofwel in absolute termen, 2 patiënten meer per 1000 (95% BI: 5 minder tot 10 meer) met een maligniteit bij gebruik van ocrelizumab ten opzichte van interferon-b.[10]
[10] De Europese werkgroep vermeldt in bijlage een RR van 1.0. Dit is evident onjuist. RR en risicoverschil zijn door de werkgroep opnieuw berekend.
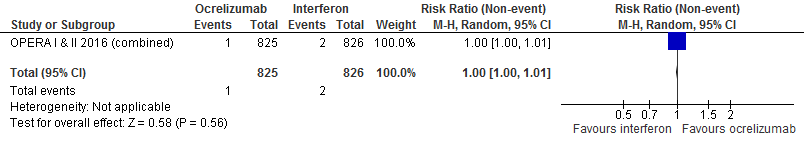
Figuur 5.6 Neoplasmata/maligniteiten versus interferon-β (96 weeks’ follow-up)
- Kwaliteit van bewijs
5.1 “Annualized Relapse Rate” (ARR)
De kwaliteit van bewijs is hoog.
5.2 Proportie vrij van relapse
De kwaliteit van bewijs is hoog.
5.3 Verbetering invaliditeit bevestigd na 12 en 24 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige onnauwkeurigheid (<300 events).
5.4 Toenemende invaliditeit
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige onnauwkeurigheid (<300 events).
5.5 en 5.6 Studieuitval vanwege bijwerkingen en om willekeurige reden
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events).
5.7 (Parasitaire) infecties
De kwaliteit van bewijs is hoog.
5.8 Mortaliteit
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events).
5.9 Maligniteit bij een follow-up duur van 96 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events) en met één niveau voor indirect bewijs: de follow-up duur van 96 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk vindt voor een beoordeling van het risico op het optreden van maligniteiten.
Conclusies
5.1 Annualized Relapse Rate bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor de annualized relapse rate 0.13 relapse minder (95% BI: 0.18 minder tot 0.08 minder) zien per patiënt per jaar in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.2 Proportie vrij van relapse bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 19% (RR: 1.19; 95% BI: 1.12-1.26 en een risicoverschil van 128 patiënten vrij van relapse meer per 1000 (95% BI: 81 meer tot 175 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Havrdová et al., 2018 |
5.3 Verbetering invaliditeit bevestigd na 12 en 24 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat voor verbetering invaliditeit een toename van het relatieve effect zien met 35% (RR: 1.35; 95% BI: 1.02-1.79) en een risicoverschil van 40 patiënten meer met verbetering invaliditeit (95% BI: 2 meer tot 91 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.4 Toenemende invaliditeit bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 40% (RR: 0.60; 95% BI: 0.46-0.80) en een risicoverschil van 67 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 90 minder tot 33 minder) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.5 en 5.6 Studieuitval vanwege bijwerkingen en studieuitval om willekeurige reden bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor studieuitval vanwege bijwerkingen en om willekeurige reden een vermindering van het relatieve risico zien met 54% (RR: 0.46; 95% BI: 0.30-0.70) respectievelijk 40% (RR: 0.60; 95% BI: 0.48-0.75) en een risicoverschil van 42 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 54 minder tot 23 minder) respectievelijk 80 patiënten minder (95% BI: 50 minder tot 104 minder) vanwege studieuitval om willekeurige reden in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.7 (Parasitaire) infecties bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor infecties een toename van het relatieve risico zien met 11% (RR: 1.11; 95% BI: 1.02-1.22) en een risicoverschil van 58 patiënten met infecties meer per 1000 (95% BI: 10 meer tot 115 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.8 Mortaliteit bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor mortaliteit een halvering van het relatieve risico zien (RR: 0.50; 95% BI: 0.05-5.51) en een risicoverschil van 1 overleden patiënt minder per 1000 (95% BI: 8 minder tot 5 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Hauser et al., 2017 |
5.9 Maligniteit bij een follow-up duur van 96 weken
|
Laag
GRADE |
Gebruik van ocrelizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor maligniteit een verdubbeling van het relatieve risico zien (RR: 2.00; 95% BI: 0.37-10.90) en een risicoverschil van 2 patiënten meer met een maligniteit per 1000 (95% BI: 5 minder tot 10 meer) in vergelijking tot interferon-β (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Hauser et al., 2017 |
Review 6. Ozanimod versus interferon-β
- Klinische effectiviteit
6.1 “Annualized Relapse Rate” (ARR)
Twee RCTs (Comi et al., 2019, Cohen et al., 2019) rapporteerden over de ARR na een follow-up van 24 tot 48 weken (zie figuur 6.1). Ozanimod 0.5mg/d (894 patiënten) verminderde in vergelijking met interferon-b (889 patiënten) over 24-48 weken de ARR met 0.07 (95% BI: 0,02 tot 0,12 minder) relapses per persoon per jaar. Ozanimod 1.0 mg/d (881 patiënten) verminderde in vergelijking met interferon-b over 24-48 weken de ARR met 0.13 (95% BI: 0,07 tot 0,18 minder) relapses per persoon per jaar.
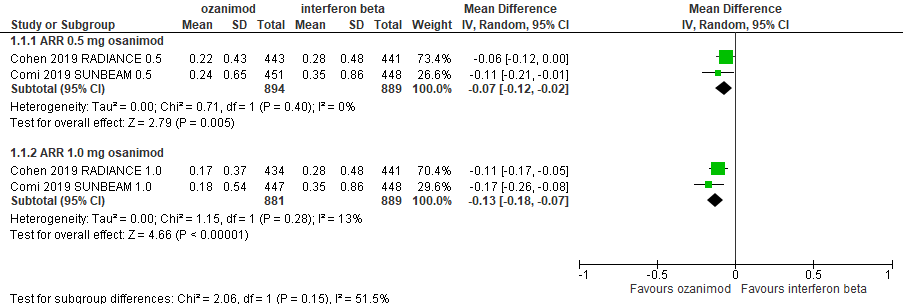
Figuur 6.1: Annualized relapse rate ozanimod 0.5 mg versus interferon-β en ozanimod 1.0 mg versus interferon-β
6.2 Proportie vrij van relapse
Hierover werd niet gerapporteerd in de RCTs.
6.3 Verbetering invaliditeit (uitgedrukt in EDSS) bevestigd na 12 en na 24 weken
Hierover werd niet gerapporteerd in de RCTs
6.4 Toenemende invaliditeit uitgedrukt in EDSS
Hierover werd in één RCT gerapporteerd (Cohen et al., 2019). Het risico op 12 weken-bevestigde toename van lichamelijke beperking weergegeven in de EDSS-score was 50/441 (11.3%) in de interferon-b groep, 41/439 (9.3%) in de ozanimod 0.5mg/d groep (RR 0.82 (95% BI 0.56; 1.22), en 54/443 (12.5%) in de ozanimod 1.0mg/d groep (1.08 (95% BI 0.75; 1.54). Deze proporties waren niet significant verschillend (p=0.283 en p=0.822 resp). Het risico op 24 weken-bevestigde toename van lichamelijke beperking weergegeven in de EDSS-score was 29/441 (6.6%) in de interferon-b groep, 32/439 (7.3%) in de ozanimod 0.5mg/d groep (RR 1,11 (95% BI 0.68; 1.80), en 42/443 (9.7%) in de ozanimod 1.0mg/d groep (RR 1,44 (95% BI 0.92; 2.27). Deze proporties waren niet significant verschillend (p=0.715 en p=0.135 resp).
- Effectiviteit niet-klinisch
6.5a Nieuwe of groter wordende laesies
Als secundaire MRI-uitkomsten werden gerapporteerd in twee RCTs (Comi et al., 2019, Cohen et al., 2019) het gemiddeld aantal nieuwe of groter-wordende laesies in 12 en 24 maanden, en het aantal gadolinium-aankleurende laesies. Ozanimod 0.5mg/d verminderde in vergelijking met interferon-b het gemiddeld aantal nieuwe of groter-wordende laesies in 12-24 maanden met 0.89 (95% BI: 0.39 tot 1.38 minder) per persoon per jaar. Ozanimod 1.0 mg/d verminderde in vergelijking met interferon-b het gemiddeld aantal nieuwe of groter-wordende laesies in 12-24 maanden met 1.36 (95% BI: 0.95 tot 1.77 minder) per persoon per jaar.
6.5b Gadolineum aankleurende laesies
Ozanimod 0.5 mg/d verminderde in vergelijking met interferon-b het gemiddeld aantal gadolinium-aankleurende laesies in 12-24 maanden met 0.16 (95% BI: 0,02 tot 0,30 minder) per persoon per jaar. Ozanimod 1.0 mg/d verminderde in vergelijking met interferon-b het gemiddeld aantal gadolinium-aankleurende laesies in 12-24 maanden met 0.22 (95% BI: 0.10 tot 0.34 minder) per persoon per jaar.
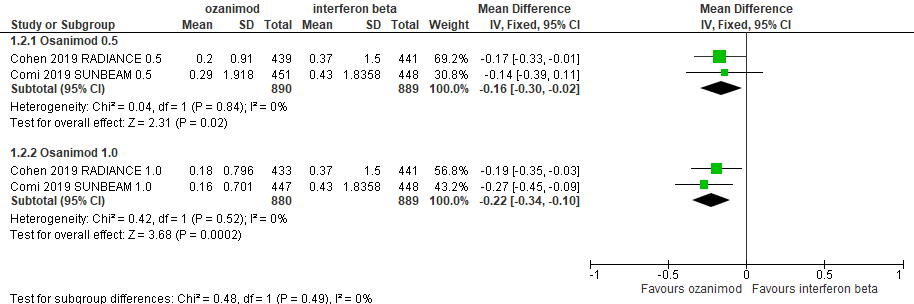
Figuur 6.2 Nieuwe of groter wordende laesies ozanimod 0.5 mg versus interferon-β en ozanimod 1.0 mg versus interferon-β
Figuur 6.3 Gadolinium aankleurende laesies ozanimod 0.5 mg versus interferon-β en tussen ozanimod 1.0 mg versus interferon-β
- No Evidence of Disease Activity (NEDA)
Er werden geen uitkomsten gerapporteerd in de RCTs.
- Tolereerbaarheid en veiligheid
6.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 12 tot 24 weken
Twee RCTs (Comi et al., 2019, Cohen et al., 2019) rapporteerden over studieuitval vanwege bijwerkingen na 24 tot 48 weken. Volgens deze studies was bij patiënten die ozanimod 0.5 mg of 1.0 mg gebruikten studieuitval vanwege bijwerkingen vergelijkbaar met patiënten die interferon-b gebruiken: ozanimod 0.5 mg RR=0.62 (95% BI: 0.35; 1.09); ozanimod 1.0 mg RR=0.77 (95% BI 0.46; 1.27), ofwel in absolute termen15 patiënten minder per 1000 voor ozanimod 0.5 mg en 9 per 1000 minder voor ozanimod 1.0 mg, wanneer ozanimod in plaats van interferon-b werd gebruikt.
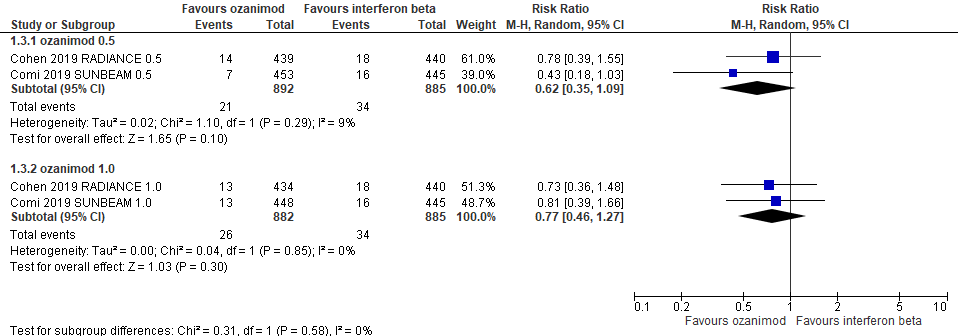
Figuur 6.4 Studieuitval vanwege bijwerkingen ozanimod 0.5 mg versus interferon-β en ozanimod 1.0 mg versus interferon-β
6.7 Studieuitval om willekeurige reden bij een follow-up duur van 24-48 weken
Twee RCTs (Comi et al., 2019, Cohen et al., 2019) rapporteerden over studieuitval om willekeurige redenen na 24 tot 48 weken. Volgens deze studies was bij patiënten die ozanimod 0.5 mg of 1.0 mg gebruikten studieuitval vanwege bijwerkingen vergelijkbaar met patiënten die interferon-b gebruiken: ozanimod 0.5 mg RR=0.89 (95% BI: 0.64; 1.23); ozanimod 1.0 mg RR=0.74 (95% BI 0.55; 0.98), ofwel in absolute termen 102 patiënten minder per 1000 voor ozanimod 0.5 mg en 84 per 1000 minder voor ozanimod 1.0 mg, wanneer ozanimod in plaats van interferon-b werd gebruikt.
6.8 (Ernstige) infecties
Twee RCTs (Comi et al., 2019, Cohen et al., 2019) rapporteerde over infecties. Volgens deze studie hadden bij een follow-up duur van 24-48 weken evenveel patiënten infecties met ozanimod in vergelijking met interferon-b. De incidentie van herpes infecties was over 48 weken in de ozanimod 1.0mg/d-groep 0.9% (4 casus), In de ozanimod 0.5 mg/d-groep 0.5% (2 casus), en in de interferon-b-groep 0.2% (1 casus). Over 96 weken was dit in de ozanimod 1.0mg/d-groep 0.9% (4 casus), in de ozanimod 0.5 mg/d-groep 0.7% (3 casus), en in de interferon-b-groep 1.1% (5 casus).
6.9 Mortaliteit
Één RCT (Comi et al., 2017) rapporteerde over mortaliteit bij een follow-up duur van 48 weken. Volgens deze studie kwamen er geen sterfgevallen voor bij alle deelnemers aan de studie (n=1346). Een andere RCT (Cohen et al., 2019) rapporteerde over mortaliteit bij een follow-up duur van 96 weken. Hierbij waren er geen sterfgevallen in de interferon-b en ozanimod 1.0mg/d groepen, in de ozanimod 0.5mg/d groep overleed 1 deelnemer tijdens de studie (0.2%) door verdrinking bij een ongeval.
6.10 Neoplasmata/maligniteit bij een follow-up duur van 48 weken
Één RCT (Comi et al., 2017) rapporteerde over maligniteiten bij een follow-up duur van 48 weken. In de ozanimod 1.0mg/d-groep was de incidentie van een maligniteit 0.2% (1 casus), In de ozanimod 0.5 mg/d-groep 0.4% (2 casus), en in de interferon-b-groep werden geen maligniteiten gedetecteerd.
- Kwaliteit van bewijs
6.1 “Annualized Relapse Rate” (ARR)
De kwaliteit van bewijs voor ARR is redelijk. De studies waren kwalitatief goed uitgevoerd.
Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
6.4 Toenemende invaliditeit uitgedrukt in EDSS
De kwaliteit van bewijs voor toenemende invaliditeit 6.4 is redelijk. De studies waren kwalitatief goed uitgevoerd.
Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
6.5a Nieuwe of groter wordende laesies
De kwaliteit van bewijs voor nieuwe of groter wordende lasies 6.5a is hoog. De studies waren kwalitatief goed uitgevoerd en er waren geen redenen om af te waarderen.
6.5b Gadolineum aankleurende laesies
De kwaliteit van bewijs voor gadolinium aankleurende laesies 6.5b is hoog. De studies waren kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor kinische besluitvorming (-0,10).
6.6 Studieuitval vanwege bijwerkingen
De kwaliteit van bewijs voor studieuitval vanwege bijwerkingen is redelijk. De studies waren kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie en geen effect
6.7 Studieuitval om willekeurige reden
De kwaliteit van bewijs voor studieuitval om willekeurige redenen is redelijk. De studies waren kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
6.8 (Ernstige) infecties
De kwaliteit van bewijs voor (ernstige) infecties is hoog. De studies waren kwalitatief goed uitgevoerd.
6.9 Mortaliteit
De kwaliteit van bewijs voor mortaliteit is hoog. De studies waren kwalitatief goed uitgevoerd.
6.10 Neoplasmata/maligniteit bij een follow-up duur van 48 weken
De kwaliteit van bewijs voor neoplasmata/maligniteit is redelijk. De studies waren kwalitatief goed uitgevoerd, maar gezien het geringe aantal events werd afgewaardeerd voor imprecisie.
Conclusies Ozanimod versus interferon-β
6.1 Annualized Relapse Rate bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet of nauwelijks in een verschil in de annualized relapse rate ten opzicht van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS vermindert waarschijnlijk de annualized relapse rate ten opzicht van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.4 Toenemende invaliditeit na 12 of 24 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet of nauwelijks in 24 weken-verminderde toename van invaliditeit ten opzicht van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert niet of nauwelijks in niet of nauwelijks in 24 weken-bevestigde toename van lichamelijke beperking ten opzicht van interferon-β.
Bron: Cohen et al., 2019 |
6.5a Nieuwe of groter wordende laesies bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert in minder nieuwe of groter wordende laesies ten opzicht van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert in minder nieuwe of groter wordende laesies ten opzicht van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.5b Gadolinium aankleurende laesies bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een verschil in gadolinium aankleurende laesies ten opzichte van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een verschil in gadolinium aankleurende laesies ten opzicht van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg of 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet of nauwelijks in minder studieuitval vanwege bijwerkingen dan wanneer interferon-b werd gebruikt.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.7 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg of 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet of nauwelijks in minder studieuitval om willekeurige redenen dan wanneer interferon-b werd gebruikt.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.8 (Ernstige) infecties bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet in meer (ernstige) infecties vergeleken met interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert niet meer (ernstige) infecties vergeleken met interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.9 Mortaliteit bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert niet in een grotere of kleinere kans op mortaliteit dan gebruik van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert niet in een grotere of kleinere kans op mortaliteit dan gebruik van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
6.10 Neoplasmata/maligniteit bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ozanimod 0,5 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een grotere of kleinere kans op neoplasmata/maligniteit dan gebruik van interferon-β.
Het gebruik van ozanimod 1,0 mg door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in een grotere of kleinere kans op neoplasmata/maligniteit dan gebruik van interferon-β.
Bron: Comi et al., 2019, Cohen et al., 2019 |
Review 7. Ofatumumab versus teriflunomide
- Klinische effectiviteit
7.1 “Annualized Relapse Rate” (ARR)
Twee RCTs (beiden besproken in Hauser et al., 2020) rapporteerden over de ARR na een follow-up van ten minste 30 maanden. Ofatumumab verminderde in vergelijking met teriflunomide de ARR met 0.13 (95% BI: 0.09 tot 0.16 minder) relapses per persoon per jaar.
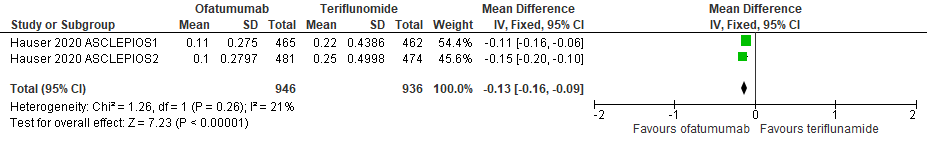
Figuur 7.1: Annualised relapse rate ofatumumab versus teriflunomide
Proportie vrij van relapse
Hierover werd niet gerapporteerd in de RCTs.
7.2 Verbetering invaliditeit (uitgedrukt in EDSS) bevestigd na 24 weken
Verbetering van beperking uitgedrukt in EDSS score en bevestigd na 24 weken werd in twee RCTs onderzocht (beiden besproken in Hauser et al., 2020). In de ofatumumab groep verbeterde de EDSS-score in 74/749 deelnemers (11%) en in de teriflunomide groep in 53/723 deelnemers (8.1%), wat resulteert in een verschil van 2.9% (95% BI 0% tot 5% meer).
7.3 Toenemende invaliditeit uitgedrukt in EDSS
Toename van beperking uitgedrukt in EDSS score en bevestigd na 24 weken werd in twee RCTs onderzocht (beiden besproken in Hauser et al., 2020). Het risico op 24 weken-bevestigde toename van lichamelijke beperking weergegeven in de EDSS score was 71/944 (8.1%) in de ofatumumab groep, en 99/931 (12.0%) in de teriflunomide groep, wat resulteert in een verschil van 3% (95% BI 1% tot 6%).
- Effectiviteit niet-klinisch
7.4 Secundaire MRI-uitkomsten werden gerapporteerd in 2 RCTs (beiden besproken in Hauser et al., 2020) het gemiddeld aantal nieuwe of groter-wordende laesies in op het einde van de studie (minstens 30 maanden), en het aantal gadolinium-aankleurende laesies gedurende de studie. Ofatumumab verminderde in vergelijking met teriflunomide het gemiddeld aantal nieuwe of groter-wordende laesies gedurende de studie met 3.40 (95% BI: 2.98 tot 3.82 minder), zie figuur 7.2. Ofatumumab verminderde in vergelijking met teriflunomide het gemiddeld aantal gadolinium-aankleurende laesies gedurende de studie met 0.46 (95% BI: 0.37 tot 0.54 minder) per persoon per jaar, zie figuur 7.3).
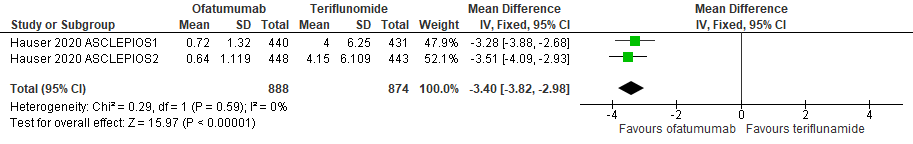
Figuur 7.2: Nieuwe laesies ofatumumab versus teriflunomide
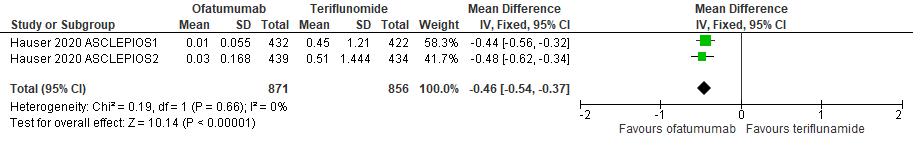
Figuur 7.3: Gadolinium aankleurende laesies ofatumumab versus teriflunomide
- No Evidence of Disease Activity (NEDA)
Er werden geen uitkomsten gerapporteerd in de RCTs.
- 7.6 Tolereerbaarheid en veiligheid
Studieuitval vanwege bijwerkingen bij een follow-up duur van 48 weken
Twee RCTs (beiden besproken in Hauser et al., 2020) rapporteerden over studieuitval vanwege bijwerkingen na 48 weken. Volgens deze studies was studieuitval vanwege bijwerkingen dan onder patiënten die ofatumumab 54/946 (5,7%) gebruikten vergelijkbaar met teriflunomide 49/938 (5,1%): RR=1.09 (95% BI: 0.75-1.59), ofwel in absolute en, 5 patiënten meer per 1000 wanneer ofatumumab in plaats van teriflunomide werd gebruikt, zie figuur 7.4.
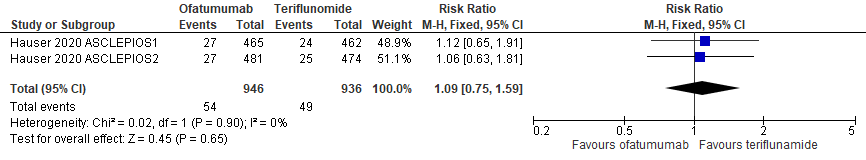
Figuur 7.4 Studieuitval vanwege bijwerkingen ofatumumab versus teriflunomide
7.7 Studieuitval om willekeurige reden bij een follow-up duur van 48 weken
Twee RCTs (beiden besproken in Hauser et al., 2020) rapporteerden over studieuitval om willekeurige reden. Volgens deze studies was er geen verschil tussen ofatumumab in vergelijking met teriflunomide voor het risico op studieuitval om willekeurige reden: RR=0.98 (95% BI: 0.73-1.31), ofwel in absolute termen, 2 studie uitvallers minder om willekeurige reden minder per 1000 wanneer zij ofatumumab gebruikten in plaats van teriflunomide.
7.8 (Ernstige) infecties
Twee RCTs (beiden besproken in Hauser et al., 2020) rapporteerde over ernstige infecties. Volgens deze studies hadden bij een follow-up duur van 48 weken evenveel patiënten infecties met ofatumumab in vergelijking met teriflunomide. De incidentie van ernstige infecties was over 48 weken in de ofatumumabgroep 2.5% (24 casus), en in de teriflunomide-groep 1.8% (17 casus), wat overeenkomt met een RR van 1.40 (95% BI 0.76; 2.58).
7.9 Mortaliteit
Twee RCTs (beiden besproken in Hauser et al., 2020) rapporteerden over mortaliteit bij een follow-up duur van 48 weken. Volgens deze studie kwamen er geen sterfgevallen voor in de ofatumumab groepen en één in de teriflunomidegroep, door aorta dissectie.
7.10 Neoplasmata/maligniteit bij een follow-up duur van 48 weken
Twee RCTs (beiden besproken in Hauser et al., 2020) rapporteerden over maligniteiten bij een follow-up duur van 48 weken. In de ofatumumabgroep was de incidentie van een maligniteit 0.5% (5 casus), in de teriflunomidegroep was de incidentie van een maligniteit 0.4% (4 casus).
- Kwaliteit van bewijs
7.1 Annualized Relapse Rate bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor ARR is redelijk. De studies waren kwalitatief goed uitgevoerd.
Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
7.2 Verbetering invaliditeit (uitgedrukt in EDSS)
De kwaliteit van bewijs voor verminderde toename van invaliditeit is hoog. De studies waren kwalitatief goed uitgevoerd.
7.3 Verminderde toename van invaliditeit (uitgedrukt in EDSS)
De kwaliteit van bewijs voor verminderde toename van invaliditeit is hoog. De studies waren kwalitatief goed uitgevoerd.
7.4 Nieuwe laesies bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor nieuwe laesies is hoog. De studies waren kwalitatief goed uitgevoerd.
7.5 Gadolinium aankleurende laesies bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor gadolinium aankleurenden laesies is hoog. De studies waren kwalitatief goed uitgevoerd.
7.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor studieuitval vanwege bijwerkingen is redelijk. De studies waren kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grenzen voor klinische relevantie.
7.7 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor studieuitval om willekeurige reden is redelijk. De studies waren kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grenzen voor klinische relevantie.
7.8 (Ernstige infecties)
De kwaliteit van bewijs voor (ernstige) infecties is hoog. De studies waren kwalitatief goed uitgevoerd.
7.9 Mortaliteit
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events) en met één niveau voor indirect bewijs: de follow-up duur van 48 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk vindt voor een beoordeling van het risico op het optreden van maligniteiten.
hoog. De studies waren kwalitatief goed uitgevoerd.
7.10 Neoplasmata/maligniteit bij een follow-up duur van 48 weken
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events) en met één niveau voor indirect bewijs: de follow-up duur van 48 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk vindt voor een beoordeling van het risico op het optreden van maligniteiten. De studies waren kwalitatief goed uitgevoerd
Conclusies
7.1 Annualized Relapse Rate bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS verlaagt waarschijnlijk de annualized relapse rate ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.2 Verbetering invaliditeit (uitgedrukt in EDSS)
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert niet in een verschil in verbetering van invaliditeit ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.3 Verminderde toename van invaliditeit (uitgedrukt in EDSS)
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert niet in een verminderde toename van invaliditeit ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.4 Nieuwe laesies bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert in minder nieuwe laesies ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.5 Gadolinium aankleurende laesies bij een follow-up duur van 24-48 weken
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert in minder gadolinium aankleurende laesies ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder studieuitval ten gevolge van bijwerkingen ten opzichte van teriflunomide.
Bron: Hauser et al., 2020 |
7.7 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder studieuitval ten gevolge van willekeurige redenen ten opzichte van teriflunomide.
Bron: Hauser et al., 2020 |
7.8 (Ernstige) infecties
|
Hoog
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS resulteert niet in meer infecties ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.9 Mortaliteit bij een follow-up duur van 48 weken
|
Laag
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS lijkt niet in meer mortaliteit te resulteren ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
7.10 Neoplasmata/maligniteit bij een follow-up duur van 96 weken
|
Laag
GRADE |
Het gebruik van ofatumumab door patiënten met relapsing remitting MS lijkt niet in meer of minder neoplasmata/maligniteit te resulteren ten opzicht van teriflunomide.
Bron: Hauser et al., 2020 |
Review 8. Ponesimod vs teriflunomide
8.1 “Annualized Relapse Rate” (ARR)
Eén RCT (besproken in Kappos et al., 2021) rapporteerde over de ARR na een follow-up van 108 weken. Ponesimod verminderde in vergelijking met teriflunomide over 108 weken de ARR met 0.09 (95% BI: 0.04 tot 0.14 minder, 0.202 vs 0.290, resp) relapses per persoon per jaar.
8.2 Proportie vrij van relapse;
Hierover werd niet gerapporteerd in de RCT.
8.3 Verbetering invaliditeit (uitgedrukt in EDSS) bevestigd na 12 en na 24 weken
Hierover werd niet gerapporteerd in de RCT.
8.4 Toenemende invaliditeit uitgedrukt in EDSS
Toename van invaliditeit uitgedrukt in EDSS score en bevestigd na 12 weken werd in één RCT onderzocht (besproken in Kappos et al., 2021). Het risico op 12 weken-bevestigde toename van lichamelijke beperking weergegeven in de EDSS score was 57 van de 567 patiënten (10.1%) in de ponesimod groep, en 70 van de 566 patiënten (12.4%) in de teriflunomide groep. Dit resulteert in een RR van 0.81 (95% BI -0.58 tot 1.13 minder). Toename van beperking uitgedrukt in EDSS score en bevestigd na 24 weken werd in één RCT onderzocht (besproken in Kappos et al., 2021). Het risico op 24 weken-bevestigde toename van lichamelijke beperking weergegeven in de EDSS score was 46 van de 567 patiënten (8.1%) in de ponesimod groep, en 56 van de 566 patiënten (9.9%) in de teriflunomide groep, wat resulteert in een RR van 0.82 (95% BI 0.57 tot 1.19 minder).
- 8.5 Effectiviteit niet-klinisch
Als secundaire MRI-uitkomsten werden gerapporteerd in één RCT (besproken in Kappos et al., 2021) het gemiddeld aantal nieuwe, groter-wordende of aankleurende laesies (Combined Unique Active lesions, CUA) per jaar, en het aantal gadolinium-aankleurende laesies per scan. Ponesimod verminderde in vergelijking met teriflunomide het gemiddeld aantal CUA per jaar met 1.76 (95% BI: 1,26 tot 2,25 minder, 1.405 vs. 3.164 rep). Ponesimod verminderde in vergelijking met teriflunomide het gemiddeld aantal gadolinium-aankleurende laesies per scan met 0.25 (95% BI: 0,12 tot 0,38 minder, 0.18 vs. 0.43).
- 8.7 No Evidence of Disease Activity (NEDA)
Één RCT (besproken in Kappos et al., 2021) rapporteerde over NEDA-3 na een follow-up van 108 weken. Ponesimod vermeerdere in vergelijking met teriflunomide de NEDA met 8.6% (95% BI: 3.66% tot 13.54% minder, 25.0% vs 16.4,% resp).
- Tolereerbaarheid en veiligheid
8.8 Studieuitval vanwege bijwerkingen bij een follow-up duur van 108 weken
Één RCT (Kappos et al., 2021) rapporteerde over studieuitval vanwege bijwerkingen na 108 weken. Volgens deze studies is bij patiënten die ponesimod gebruiken 49 van de 565 patiënten minder vaak sprake van studieuitval vanwege bijwerkingen dan onder patiënten die teriflunomide gebruiken 34 van de 566 : RR=1.44 (95% BI: 0.95-2.20).
8.9 Studieuitval om willekeurige reden bij een follow-up duur van 108 weken
Eén RCT (Kappos et al., 2021) rapporteerde over studieuitval om willekeurige reden. Volgens deze studies verminderde ponesimiod in vergelijking met teriflunomide het risico op studieuitval om willekeurige reden: 94 van de 567 patiënten RR=1.01 (95% BI: 0.78; 1.32), ofwel in absolute termen, 2 studieuitvallers om willekeurige reden meer per 1000 wanneer zij ponesimod gebruikten in plaats van teriflunomide.
8.10 Infecties
Eén RCT (Kappos et al., 2021) rapporteerde over infecties. Volgens deze studie hadden bij een follow-up duur van 108 weken iets meer patiënten met ponesimod (9 casus) een infectie in vergelijking met teriflunomide (5). Over 108 weken was dit in de ponesimod groep 1.6% (9 casus), in de teriflunomide groep 0.9% (5 casus). In beide groepen werden 27 herpes infecties (4.8%) gezien.
8.11 Mortaliteit
Eén RCT (Kappos et al., 2021) rapporteerde over mortaliteit bij een follow-up duur van 108 weken. Volgens deze studie waren er geen sterfgevallen in de ponesimodgroep en twee sterfgevallen in de teriflunomidegroep, één geval van hartfalen (coronary artery insufficiency) en één door MS. Volgens de onderzoekers waren deze sterftegevallen niet gerelateerd aan de behandeling.
8.12 Neoplasmata/maligniteit bij een follow-up duur van 108 weken
Eén RCT (Kappos et al., 2021) rapporteerde over nieuwvormingen, zowel benigne als maligne bij een follow-up duur van 108 weken. In de ponesimodgroep was de incidentie hiervan 1.1% (6 casussen), in de teriflunomidegroep was de incidentie 0,5% (3 casussen).
- Kwaliteit van bewijs
8.1 Annualized Relapse Rate bij een follow-up duur van 108 weken
De kwaliteit van bewijs voor ARR is redelijk. De studies waren kwalitatief goed uitgevoerd.
Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
8.4 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 12 of 24 weken
De kwaliteit van bewijs voor toenemende invaliditeit is redelijk. De studie was kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
8.5 Combined unique active lesions en 8.6 Gadolinium aankleurende laesies
De kwaliteit van bewijs voor nieuwe laesies en gadolinium aankleurende laesies is redelijk. De studie was kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
8.7 No evidence of disease activity
De kwaliteit van bewijs voor NEDA is redelijk. De studie was kwalitatief goed uitgevoerd. Er werd afgewaardeerd voor imprecisie wegens overlap met de grens voor klinische relevantie.
8.8 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor studieuitval vanwege bijwerkingen is redelijk. De studies waren kwalitatief goed uitgevoerd. Er werd afgewaardeerd wegens imprecies vanwege overlap met de grenzen voor klinische relevantie
8.9 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor studieuitval om willekeurige reden is gemiddeld. De studies waren kwalitatief goed uitgevoerd. Er werd afgewaardeerd wegens impreciesie vanwege overlap met de grenzen voor klinische relevantie.
8.10 Infecties bij een follow-up duur
De kwaliteit van bewijs is laag. Er werd met één niveau afgewaardeerd vanwege imprecisie (<300 events) en met één niveau voor indirect bewijs: de follow-up duur van 48 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk vindt voor een beoordeling van het risico op het optreden van maligniteiten. De studies waren kwalitatief goed uitgevoerd uitgevoerd.
8.11 Mortaliteit bij een follow-up duur van 24-48 weken
De kwaliteit van bewijs voor mortaliteit is laag. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events) en met één niveau voor indirect bewijs: de follow-up duur van 48 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk vindt voor een beoordeling van het risico op het optreden van maligniteiten.De studies waren kwalitatief goed uitgevoerd.
8.12 Neoplasmata/maligniteit bij een follow-up duur van 108 weken
De kwaliteit van bewijs voor neoplasmata/maligniteit is redelijk. De studies waren kwalitatief goed uitgevoerd, maar gezien het geringe aantal events werd afgewaardeerd voor imprecisie.
Conclusie
8.1 Annualized Relapse Rate bij een follow-up duur van 108 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert niet of nauwelijks in een klinisch relevant verschil in de annualized relapse rate ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.4 Toenemende invaliditeit uitgedrukt in EDSS bij een follow-up duur van 12 of 24 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert niet of nauwelijks in een klinisch relevant verschil niet of nauwelijks in 24 weken-bevestigde toename van lichamelijke beperking ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.5 Combined unique active lesions
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder unieke nieuwe laesies ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.6 Gadolinium aankleurende laesies
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder gadolinium aankleurende laesies ten opzichte van teriflunomide.
Bron: Kappos et al., 2021 |
8.7 No evidence of disease activity
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in minder no evidence of disease activity (NEDA) ten opzichte van teriflunomide.
Bron: Kappos et al., 2021 |
8.8 Studieuitval vanwege bijwerkingen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in meer studieuitval vanwege bijwerkingen ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.9 Studieuitval om willekeurige redenen bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in meer studieuitval om willekeurige redenen ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.10 Infecties bij een follow-up duur van 24-48 weken
|
Redelijk
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS resulteert waarschijnlijk niet in meer infecties ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.11 Mortaliteit bij een follow-up duur van 24-48 weken
|
Laag
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS lijkt niet te resulteren in meer mortaliteit ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
8.12 Neoplasmata/maligniteit bij een follow-up duur van 24-48 weken
|
Laag
GRADE |
Het gebruik van ponesimod door patiënten met relapsing remitting MS lijkt niet te resulteren in meer neoplasmata /maligniteit ten opzicht van teriflunomide.
Bron: Kappos et al., 2021 |
Deel 2: netwerkmeta-analyse
Inleiding
Ziektemodulerende therapieën kunnen worden ingezet om het risico op relapses en ziekteprogressie te verminderen. In de module 2.4 werden de relatieve voordelen en nadelen van deze middelen ten opzichte placebo (2.4.1) en ten opzicht van elkaar besproken in een traditionele meta-analyse (2.4.2). In onderstaande literatuursamenvatting worden de voor- en nadelen van de verschillende ziektemodulerende middelen ten opzichte van elkaar besproken en worden ze onderling vergeleken in een netwerk meta-analyse.
Zoeken en selecteren
Om de uitgangsvraag te kunnen beantwoorden heeft de Nederlandse richtlijnwerkgroep gebruik gemaakt van dezelfde literatuursearch als bij module 2.4, zie deze module voor alle details.
Methode
De netwerkmeta-analyse (NMA) werd uitgevoerd met de netmeta en meta packages van R versie 4.0.2 (RStudio, Boston, MA) en NMAstudio voor de netwerk plots en forest plots.[11]
[11] G. Rücker, U. Krahn, J. König, O. Efthimiou, A. Davies, T. Papakonstantinou & G. Schwarzer. netmeta: Network Meta-Analysis using Frequentist Methods, 2021. Metelli S, Chaimani A. NMAstudio: a fully interactive web-application for producing and visualising network meta-analyses. SRSM Annual Meeting 2021, Bern, Switzerland.
Deze netwerkmeta-analyse is gebaseerd op de studies en uitkomsten zoals vermeld in de Europese richtlijn. Een uitzondering is het middel mitoxantron, waarvoor de oorspronkelijke studie is geraadpleegd (Millefiorini et al., 1997). In de update van 2022 zijn de volgende RCTs toegevoegd: Cohen et al., 2019; Comi et al., 2019; Hauser et al., 2020; Kappos et al., 2021.
Het beoordelen van de kwaliteit van bewijs van netwerkmeta-analyses met de GRADE-benadering is beschreven in Puhan et al. (2014). Kort samengevat:
- presenteer directe en indirecte schattingen van het effect van behandelingen voor iedere vergelijking in het netwerk;
- beoordeel de kwaliteit van bewijs van iedere directe en indirecte effectschatting;
- presenteer de NMA effectschatting voor iedere vergelijking in het netwerk;
- beoordeel de kwaliteit van bewijs van iedere NMA effectschatting.
Voor een indirecte vergelijking (bijvoorbeeld: A vs. B) geldt dat het laagste niveau van de kwaliteit van bewijs van twee directe vergelijkingen (bijvoorbeeld A vs. Placebo, B vs. placebo) de kwaliteit van bewijs van een indirecte vergelijking bepaalt voor een bepaalde uitkomstmaat.
Bij een NMA gelden de gebruikelijke GRADE-domeinen en één additioneel domein: intransiviteit. Intransiviteit is een beoordeling of het placebo-effect verandert door de jaren heen; met andere woorden of de placebo groepen van de verschillende studies vergelijkbaar zijn. Als de trials die de basis zijn voor een indirecte effectschatting in belangrijke mate van elkaar verschillen (zoals trials van A vs. C en van B vs. C) kan er sprake zijn van intransiviteit, en kan de kwaliteit van bewijs afgewaardeerd worden. Dit op voorwaarde dat intransitiviteit geen globale inconsistentie van het netwerk veroorzaakt (zie ook de leeswijzer).
Wanneer alleen direct of indirect bewijs voor een bepaalde vergelijking beschikbaar is, wordt de kwaliteit van bewijs voor de NMA-effectschatting gebaseerd op de ene of de andere beschikbare effectschatting.
Wanneer zowel direct als indirect bewijs beschikbaar is, bepaalt het hoogte niveau van de kwaliteit van bewijs van een van beide de kwaliteit van bewijs van de NMA-effectschatting. De rationale hiervan is:
- als de directe en indirecte effectschattingen coherent zijn (‘gelijk’ zijn), dan kan de lagere kwaliteit van bewijs van de ene effectschatting de hogere kwaliteit van bewijs van de andere effectschatting alleen maar ondersteunen;
- de effectschatting met de hogere kwaliteit van bewijs is in het algemeen de nauwkeuriger schatting, die het geheel van de evidence dan ook domineert.
Het is mogelijk dat directe en indirect effectschattingen incoherent zijn (‘ongelijk’ zijn). In dat geval zijn er twee mogelijke opties: (1) keuze van de effectschatting met de hoogste kwaliteit van bewijs beschouwen als de beste effectschatting, en dus afzien van een NMA-effectschatting, (2) de NMA-effectschatting gebruiken maar de kwaliteit van bewijs ervan afwaarderen vanwege incoherentie.
De mate van coherentie kan naast het vergelijken van de schattingen van het directe en indirecte effect worden beoordeeld door na te gaan in welke mate de betrouwbaarheidsintervallen overlappen en een statistische test van de ratio van directe en indirecte effectschatting.
Of er van ernstige onnauwkeurigheid van een NMA-effectschatting sprake is, daarvoor wordt de volgende regel gehanteerd:
- voor dichotome uitkomstmaten: sluit het betrouwbaarheidsinterval een relatief risico in van 0.75 of van 1.25 (c.q. een afname respectievelijk toename van het relatieve effect met 25%) in, dan wordt afgewaardeerd voor onnauwkeurigheid. Een relatief risico van 0.75 of 1.25 wordt als grenswaarde gehanteerd voor een klinisch relevant effect. Valt een waarde van 0.75 of 1.25 in het betrouwbaarheidsinterval, dan zou zowel een klinisch relevant als een klinisch niet-relevant effect nog tot de mogelijkheden behoren, hetgeen de onzekerheid over de effectschatting vergroot;
- voor continue uitkomstmaten: sluit het betrouwbaarheidsinterval een standardized mean difference (SMD) van -0.2 of 0.2 dan wordt afgewaardeerd voor onnauwkeurigheid. Een SMD van -0.2 of 0.2 wordt als grenswaarde gehanteerd voor een klinisch relevant effect (Guyatt et al., 2013).
Onlangs zijn enkele modificaties voorgesteld (Brignardello-Petersen et al., 2018):
- het is niet nodig om de GRADE-factor ernstige onnauwkeurigheid toe te passen bij het beoordelen van de kwaliteit van bewijs van directe en indirecte effectschattingen. Alleen de nauwkeurigheid van de NMA-effectschatting hoeft te worden beoordeeld. Dit impliceert dat voor de directe en indirecte effectschatting vier GRADE-factoren (risk of bias; inconsistentie; indirect bewijs en publicatiebias) worden beoordeeld;
- het is niet nodig om de kwaliteit van indirect bewijs te beoordelen wanneer de kwaliteit van direct bewijs hoog is en het meeste bijdraagt aan de NMA-effectschatting. De effectschatting met het smalste betrouwbaarheidsinterval draagt het meeste bij aan de NMA-effectschatting;
- een statistische test van globale incoherentie van het netwerk is niet betrouwbaar voor het uitsluiten van incoherentie van directe en indirecte effectschattingen voor afzonderlijke vergelijkingen in het netwerk. Ook dient de mate van overeenkomst tussen indirecte en directe effectschatting na te worden nagegaan.
In schema:

Figuur 1. Process for rating the certainty of the network estimate for each pairwise comparison in an NMA (Brignardello-Petersen et al., 2018)
De GRADE Working Group (Brignardello-Petersen et al., 2020) adviseert de volgende aanpak voor het trekken van conclusies ten aanzien van een netwerkmeta-analyse:
- stel vast welke ‘comparator’ (veelal placebo) het meest frequent voorkomt in een netwerk van vergelijkingen van interventies, en gebruik deze als referentie;
- stel vast wat een acceptabele effectgrootte is voor een klinisch relevant effect of ‘minimal important difference’. In deze richtlijn wordt een toename of afname van het relatief effect van 25% (voor dichotome uitkomstmaten) en 0.2 voor continue uitkomstmaten gehanteerd;
- classificeer de behandelingen met de meest frequente ‘comparator’ als referentie naar gelang het wel of geen klinisch relevant effect betreft. In deze richtlijn betekent dit dat een gunstig klinisch relevant effect (bijvoorbeeld een vermindering van het risico op progressie van invaliditeit) van een bepaald middel een bovengrens van het 95% BI heeft dat kleiner is dan 0.75. Dit levert meestal 2 categorieën op: behandelingen die niet (overtuigend) klinisch relevant zijn [categorie 0] en behandelingen die klinisch relevant zijn [categorie 1];
- probeer te differentiëren binnen categorie 1 op basis van (a) effectgrootte en betrouwbaarheidsinterval en (b) hoge/redelijke kwaliteit van bewijs. Als een behandeling beter is dan de andere én er is hoge/ redelijke kwaliteit van bewijs, dan komt de behandeling in een volgende categorie [categorie 2] terecht. In de praktijk zal dit weinig gebeuren. In deze richtlijn zou dit betekenen dat het effect van middel A vergeleken met middel B voor bijvoorbeeld vermindering van progressie van invaliditeit een bovengrens van het 95% BI heeft van 0.75. Voor veel middelen die als klinisch relevant zijn geclassificeerd zal niet gelden dat een van die middelen meer dan 25% effectiever is dan een ander middel;
- verdeel categorieën 0 en 1 in twee subcategorieën: hoge/redelijke kwaliteit van bewijs versus lage/zeer lage kwaliteit van bewijs
- presenteer in dalende volgorde van effectiviteit, waarvoor p-scores kunnen worden gebruikt, de uitkomsten van elke behandeling per categorie kwaliteit van bewijs [hoog/redelijk versus laag/zeer laag], en per categorie kwaliteit van bewijs naar de mate van klinische relevantie.
Resultaten
- Klinische effectiviteit
Annualized Relapse Rate bij een follow-up duur van 48-129 weken
Transitiviteit in het globale netwerk voor deze uitkomstmaat werd nagegaan door analyse van een eventuele tijdtrend in de placebogroepen en statistische test van globale consistentie van het netwerk. Geen van beide leverde aanwijzingen voor intransitiviteit op (bijlage 3a).
De meest frequente ‘comparator’ is placebo (figuur 2).[12] Alle ziektemodulerende middelen zullen in de netwerkmeta-analyse daarom worden vergeleken met placebo.[13] Het resultaat van de netwerkmeta-analyse voor de uitkomstmaat annualized relapse rate wordt gepresenteerd in figuur 3. Deze forest plot geeft niet meer dan een eerste indruk van de onderlinge verschillen in effectiviteit; er wordt namelijk geen rekening gehouden met de kwaliteit van bewijs noch met de klinisch relevantie van het effect.
Met name alemtuzumab, mitoxantron en natalizumab lijken tot de meest effectieve middelen te behoren, terwijl glatirameeracetaat, teriflunomide en interferon-β tot de minst effectieve middelen lijken te behoren. Zie verder onder conclusies.
[12] Elke interventie, inclusief de comparator, wordt afgebeeld als een cirkel. Interventies die direct zijn vergeleken in de studies zijn met een lijn met elkaar verbonden. De breedte van een lijn is evenredig met het aantal studies die de twee interventies hebben vergeleken. De grootte van de cirkel is evenredig aan het aantal armen in de studies waarin de interventie voorkomt. In deze plot komen placebo en verschillende typen interferon, gevolgd door glatirameeracetaat, dus het meest voor.
[13] Mitoxantron is meegenomen ter wille van een vergelijking met de uitkomsten van eerdere netwerk-meta-analyse (Tramecere et al., 2015).
Figuur 2. Netwerk-plot voor uitkomstmaat annualized relapse rate bij een follow-up duur van 48-129 weken. De omvang van de knooppunten weerspiegelt het aantal gerandomiseerde patiënten, de dikte van de lijnen het aantal studies waarin een vergelijking van interventies werd onderzocht.
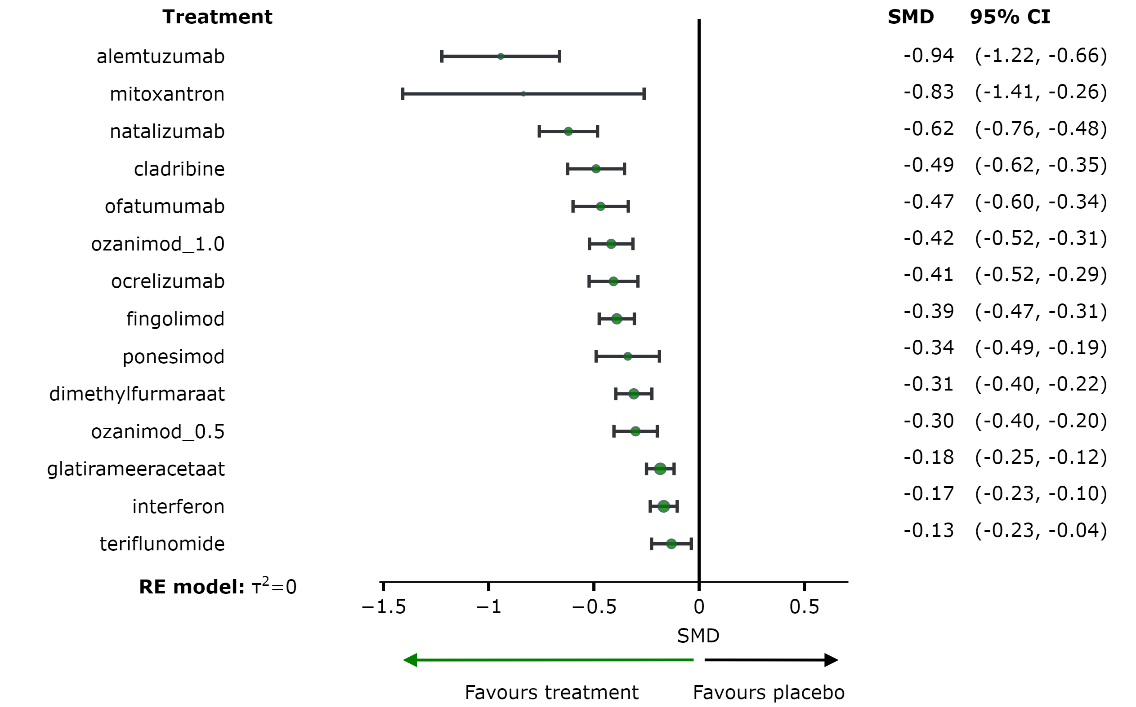
Figuur 3. Effect van ziektemodulerende middelen op de annualized relapse rate bij een follow-up duur van 48-129 weken[14]
[14] Tau2 geeft aan in welke mate de aanname dat alle effectgroottes in alle interventies dezelfde “between-study variation” hebben, correct is. Tau2 = 0 geeft aan dat deze aanname correct is
Proportie vrij van relapse bij een follow-up duur van 48-156 weken[15]
Transitiviteit in het globale netwerk voor deze uitkomstmaat werd nagegaan door analyse van een tijdtrend in de placebogroepen en statistische test van globale consistentie van het netwerk. Op grond hiervan zijn er sterke aanwijzingen voor intransitiviteit. Dit probleem werd geadresseerd door exclusie van vijf studies (waaronder de mitoxantron studie); twee studies werden als indirect bewijs geclassificeerd (bijlage 3b).
De meest frequente ‘comparator’ is placebo (figuur 4). Alle ziektemodulerende middelen zullen in de netwerkmeta-analyse daarom worden vergeleken met placebo. Het resultaat van de netwerkmeta-analyse voor de uitkomstmaat vrij van relapse wordt gepresenteerd in figuur 5. Deze forest plot geeft niet meer dan een eerste indruk van de onderlinge verschillen in effectiviteit; er wordt namelijk geen rekening gehouden met de kwaliteit van bewijs noch met de klinisch relevantie van het effect.
Met name alemtuzumab en natalizumab lijken tot de meest effectieve middelen te behoren, terwijl interferon-β en glatirameeracetaat tot de minst effectieve middelen lijken te behoren. Zie verder onder conclusies.
[15] Voor de update van 2022 werden geen studies gevonden die hierover rapporteerden. De tekst is op grond van overwegingen inzake transitiviteit aangepast.
Figuur 4. Netwerk-plot voor uitkomstmaat ‘vrij zijn van relapse bij een follow-up duur van 48-156 weken. De omvang van de knooppunten weerspiegelt het aantal gerandomiseerde patiënten, de dikte van de lijnen het aantal studies waarin een vergelijking van interventies werd onderzocht.

Figuur 5. Effect van ziektemodulerende middelen op ‘vrij zijn van relapse’ bij een follow-up duur van 48-156 weken. Deze uitkomstmaat werd niet gerapporteerd voor ponesimod, ozanimod en ofatumumab.
Toegenomen invaliditeit bij een follow-up duur van 48-156 weken
Transitiviteit in het globale netwerk voor deze uitkomstmaat werd nagegaan door analyse van een eventuele tijdtrend in de placebogroepen en statistische test van globale consistentie van het netwerk. Op grond hiervan zijn er aanwijzingen voor intransitiviteit. Dit probleem werd geadresseerd door twee studies als indirect bewijs te classificeren (bijlage 3c).
De meest frequente ‘comparator’ is placebo naast interferon-β (figuur 6). Alle ziektemodulerende middelen zullen in de netwerkmeta-analyse daarom worden vergeleken met placebo. Het resultaat van de netwerkmeta-analyse voor de uitkomstmaat toegenomen invaliditeit wordt gepresenteerd in figuur 7. Deze forest plot geeft niet meer dan een eerste indruk van de onderlinge verschillen in effectiviteit; er wordt namelijk geen rekening gehouden met de kwaliteit van bewijs noch met de klinisch relevantie van het effect.
Figuur 6. Netwerk plot voor uitkomstmaat toegenomen invaliditeit bij follow-up duur van 48-156 weken. De omvang van de knooppunten weerspiegelt het aantal gerandomiseerde patiënten, de dikte van de lijnen het aantal studies waarin een vergelijking van interventies werd onderzocht.

Figuur 7. Effect van ziektemodulerende middelen op toegenomen invaliditeit bij follow-up duur van 48-156 weken
Nota bene: Cladribine ontbreekt omdat Giovannoni et al. (2010) anders dan de studies over andere middelen, als effectmaat een hazard ratio gebruikt in plaats van een risk ratio of relatief risico. Een hazard ratio kan niet worden geconverteerd naar een risk ratio.
- Tolereerbaarheid en veiligheid
Stoppen vanwege bijwerkingen
Transitiviteit in het globale netwerk voor deze uitkomstmaat werd nagegaan door middel van een statistische test van globale consistentie van het netwerk. Deze leverde geen aanwijzingen voor intransitiviteit op (bijlage 3d).[16]
[16] Inusah et al. (2010), Nicholas et al. (2011), Steinvorth et al. (2013) en Röver et al. (2015) verschaffen geen aanwijzingen dat placebogroepen wat stoppen vanwege bijwerkingen betreft in de loop van de tijd veranderd zouden zijn. Daarom is inzake transitiviteit alleen een globale test voor inconsistentie van het netwerk uitgevoerd (bijlage 3d).
De meest frequente ‘comparators’ zijn placebo en interferon-β (figuur 8). Vanwege de eerdere keuzes voor placebo zal ook voor deze uitkomstmaat placebo als ‘comparator’ worden gebruikt. Het resultaat van de netwerkmeta-analyse voor de uitkomstmaat stoppen vanwege bijwerkingen wordt gepresenteerd in figuur 9. Deze forest plot geeft niet meer dan een eerste indruk van de onderlinge verschillen in effectiviteit; er wordt namelijk geen rekening gehouden met de kwaliteit van bewijs noch met de klinisch relevantie van het effect.
Bij middelen die minder frequent gedoseerd worden, ligt a priori de kans om te stoppen met een middel lager. Daarom is ‘stoppen vanwege bijwerkingen’ beperkt bruikbaar als uitkomstmaat voor vergelijking van veiligheid tussen middelen met een verschillende doseringsfrequentie. Met name alemtuzumab en ocrelizumab lijken het minste te leiden tot stoppen vanwege bijwerkingen. Deze middelen worden in een lagere frequentie toegediend dan de meeste andere middelen waarmee in het netwerk vergeleken wordt.
Figuur 8. Netwerk plot voor stoppen vanwege bijwerkingen bij een follow-up duur van 48-129 weken. De omvang van de knooppunten weerspiegelt het aantal gerandomiseerde patiënten, de dikte van de lijnen het aantal studies waarin een vergelijking van interventies werd onderzocht.
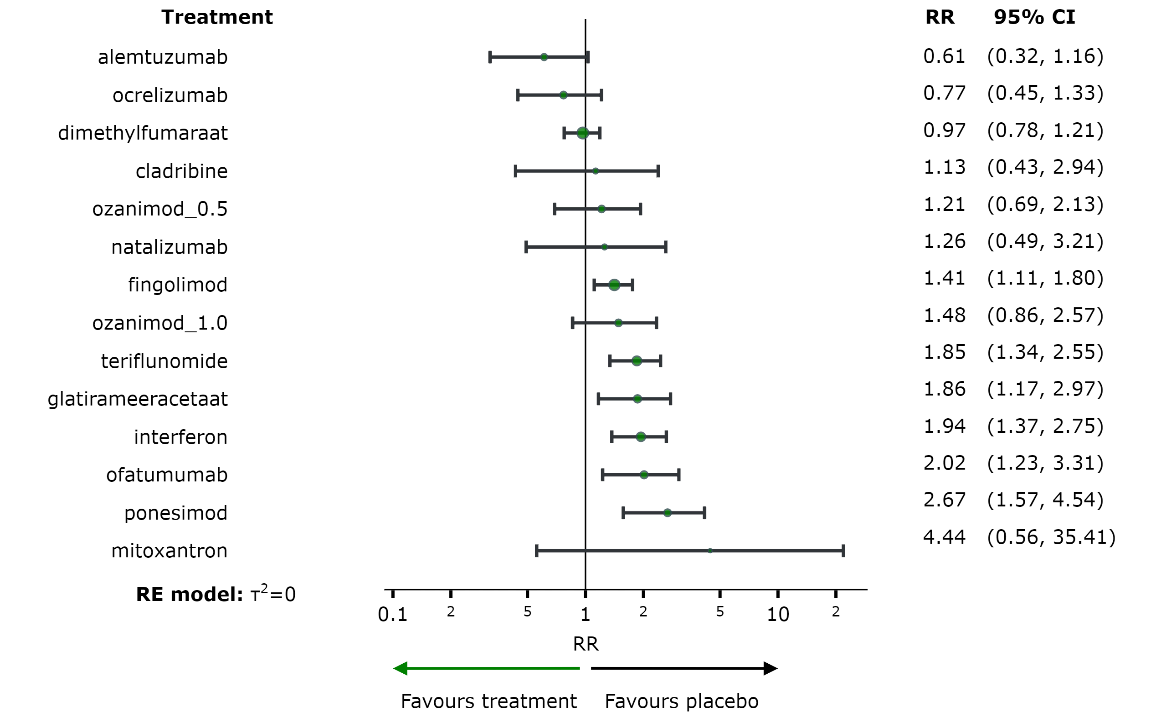
Figuur 9. Effect van ziektemodulerende middelen op stoppen vanwege bijwerkingen bij follow-up duur van 48-129 weken
- Kwaliteit van bewijs
Bij een netwerkmeta-analysen kunnen de volgende kanttekeningen worden gemaakt:
- een netwerkmeta-analyse heeft altijd betrekking op een individuele uitkomstmaat;
- het gebruik van netwerkmeta-analyse voor informatie over klinische besluitvorming berust op de relatieve effecten van ieder paar interventies én de kwaliteit van bewijs van ieder relatief effect;
- noch de grootte van de relatieve effecten noch de kwaliteit van bewijs is op zichzelf voldoende om conclusies te trekken. Immers de suggestie dat de ene behandeling beter is dan de anders kan berusten op zowel hoge als lage kwaliteit van bewijs. Hoge kwaliteit van bewijs kan er ook zijn voor het ontbreken van verschil in effectiviteit van een behandeling;
- zelden zal met een netwerkmeta-analyse vastgesteld kunnen worden dat één behandeling duidelijk superieur is aan de andere behandelingen.
Met andere woorden, er kunnen categorieën van interventies worden beschreven zoals interventies met een klinisch relevant effect en interventies zonder een (overtuigend) klinisch relevant effect. Het categoriseren berust op zowel de relatieve effecten ten opzichte van een drempelwaarde die geldt als klinisch relevant als op de kwaliteit van bewijs.
Hierbij is uitgegaan van de beoordeling van de kwaliteit van bewijs door de Europese richtlijnwerkgroep voor de GRADE-factoren risk of bias, inconsistentie, indirect bewijs en publicatiebias. Echter, de Europese richtlijnwerkgroep heeft geen rekening gehouden met het gegeven dat in de loop der tijd de placebogroepen veranderd zijn, wat consequenties kan hebben voor de beoordeling van de GRADE-factor ‘indirectness’. Zie bijlage 3 voor veranderingen in de placebogroepen en de eerder gemaakte opmerkingen over transitiviteit. Voor een netwerkmeta-analyse is het van belang zich hiervan rekenschap te geven. Om deze reden kan de kwaliteit van bewijs van directe effectschattingen (d.w.z. hier in vergelijking met placebo) soms afwijken van de kwaliteit van bewijs zoals vermeld in 5.1. Daarnaast is aanvullende ‘guidance’ gebruikt, zoals vermeld in de methode sectie.
Annualized relapse rate
Voor alle ziektemodulerende middelen behalve voor mitoxantron, teriflunomide, interferon-β, ozanimod_0.5, ponesimod en glatirameeracetaat is er redelijke tot hoge kwaliteit van bewijs wat het effect op de annualized relapse rate betreft (tabel 2).
Tabel 2. Resultaten netwerkmeta-analyse en kwaliteit van bewijs voor ‘annualized relapse rate’ (aantal relapses per patiënt per jaar)
|
Interventie versus placebo |
K |
Prop |
Direct standardized mean difference [95%-BI]* |
Kwaliteit van bewijs |
Indirect standardized mean difference [95%-BI]* |
Kwaliteit van bewijs |
NMA standardized mean difference [95%-BI] |
Kwaliteit van bewijs |
|
Alemtuzumab |
0 |
0 |
|
|
-0.940 [-1.219; -0.660] |
Redelijk |
Redelijk (s-RoB) |
|
|
Cladribine |
1 |
1 |
-0.489 |
Hoog |
|
Hoog |
||
|
Dimethylfumaraat |
2 |
0.71 |
-0.307 |
Redelijk (s-RoB) |
-0.318 |
Redelijk |
Redelijk |
|
|
Fingolimod |
2 |
0.78 |
-0.377 |
Redelijk |
-0.421 |
Redelijk |
Redelijk |
|
|
Glatirameeracetaat |
2 |
0.4 |
-0.180 |
Redelijk |
-0.190 |
Redelijk |
Laag |
|
|
Interferon-β |
2 |
0.56 |
-0.167 |
Redelijk |
-0.169 |
Redelijk |
Laag |
|
|
Mitoxantron† |
1 |
1 |
-0.822 [-1.396; -0.247] |
Redelijk |
|
|
Zeer laag (s-Rob; vs-imprec.) |
|
|
Natalizumab |
1 |
1 |
-0.620 [-0.759; -0.482] |
Redelijk |
|
Redelijk (s-indirect.) |
||
|
Ocrelizumab |
0 |
0 |
|
|
-0.406 |
Redelijk |
Redelijk |
|
|
Ofatumumab |
0 |
0 |
|
|
-0.468 |
Redelijk (s-RoB) |
Redelijk (s-RoB) |
|
|
Ozanimod_0.5 |
0 |
0 |
|
|
-0.301 |
Redelijk (s-RoB) |
Laag (s-RoB; s-imprec.) |
|
|
Ozanimod_1.0 |
0 |
0 |
|
|
-0.417 |
Redelijk |
Redelijk |
|
|
Ponesimod |
0 |
0 |
|
|
-0.339 |
Redelijk |
Laag |
|
|
Teriflunomide |
2 |
0.9 |
-0.156 |
Redelijk |
0.052 |
Redelijk |
Laag (s-RoB; s-imprec.) |
†Niet opgenomen in Europese richtlijntekst; wel vermeld als excluded studie; omwille van volledigheid overgenomen, evenals is gedaan door Tramacere et al. (2015).
K: aantal studies met directe evidence; prop: fractie directe evidence dat bijdraagt aan NMA effect.
s-RoB: serious risk of bias; s-indirect.: serious indirectness; s-incons.: serious inconsistency (I2>50% of variabele puntschatters); s-incoher.: serious incoherence; vs-incoher.: very serious incoherence; s-imprec.: serious imprecision (95% BI doorkruist SMD=-0.2 of SMD=0.2); vs-imprec.: very serious imprecision (95% BI doorkruist SMD=-0.2 én SMD=0.2 of optimal information size <<400).
Global test for incoherence: χ2 statistic: 2.055; dF=4, P= 0.726; geen lokale incoherentie (alle p-waarden van z-waarden voor ratio van direct en indirect effect>0.10).
*imprecision niet beoordeeld.
‘Vrij zijn van relapses’
Voor cladribine, alemtuzumab, natalizumab, fingolimod en ocrelizumab is er redelijke tot hoge kwaliteit van bewijs wat het effect op ‘vrij zijn van relapses’ betreft (tabel 3).
Tabel 3. Resultaten netwerkmeta-analyse en kwaliteit van bewijs voor ‘vrij zijn van relapse’ (relatieve effecten)†
|
Interventie versus placebo |
K |
Prop |
Direct RR [95%-BI] |
Kwaliteit van bewijs* |
Indirect RR [95%-BI] |
Kwaliteit van bewijs* |
NMA RR [95%-BI] |
Kwaliteit van bewijs |
|
Alemtuzumab |
0 |
0 |
|
|
1.626 [1.447;1.826] |
Redelijk |
1.626 [1.447;1.826] |
Redelijk |
|
Cladribine |
2 |
1 |
1.306 [1.203;1.418] |
Hoog |
|
|
1.306 [1.203;1.418] |
Redelijk |
|
Dimethylfumaraat |
2 |
1 |
1.277 [1.183;1.378] |
Redelijk (s-RoB) |
|
1.277 [1.183;1.378] |
Laag |
|
|
Fingolimod |
2 |
0.67 |
1.443 [1.326;1.571] |
Redelijk (s-RoB)
|
1.381 [1.226;1.557] |
Redelijk (s-RoB) |
1.422 [1.327;1.524] |
Redelijk |
|
Glatirameeracetaat |
3 |
0.82 |
1.17 [1.101;1.245] |
Redelijk (s-RoB) |
1.247 [1.093;1.422] |
Redelijk (s-RoB) |
1.184 [1.120;1.251] |
Laag (s-RoB; s-imprec.) |
|
Interferon-β |
1 |
0.03 |
1.424 [0.913;2.221] |
Laag (s-RoB; s-indirect.)
|
1.172 [1.090;1.260] |
Redelijk (s-RoB) |
1.178 [1.096;1.265] |
Laag (s-RoB; s-imprec.) |
|
Natalizumab |
1 |
1 |
1.593 [1.402;1.810] |
Hoog |
|
1.593 [1.402;1.810] |
Hoog |
|
|
Ocrelizumab |
0 |
0 |
|
|
1.400 [1.274;1.538] |
Redelijk |
1.400 [1.274;1.538] |
Redelijk (s-RoB) |
|
Teriflunomide |
2 |
1 |
1.253 [1.155;1.359] |
Redelijk (s-RoB) |
|
|
1.253 [1.155;1.359] |
Laag |
† gedefinieerd als percentage ‘vrij van relapse’ in ziektemodulerend middel-groep gedeeld door percentage ‘vrij van relapse’ in placebogroep.
K: aantal studies met directe evidence; prop: fractie directe evidence dat bijdraagt aan NMA effect.
s-RoB: serious risk of bias; s-indirect.: serious indirectness; s-incons.: serious inconsistency; s-incoher.: serious incoherence; s-imprec.: serious imprecision (95% BI doorkruist 1.250).
*imprecision niet beoordeeld.
Voor Ofatumumab, Ozanimod en Ponesimod waren geen data beschikbaar.
Global test for incoherence: χ2 statistic: 1.216, dF= 2, p= 0.544. Geen lokale incoherentie: alle p-waarden van z-waarden voor ratio van direct en indirect effect>0.30.
Opmerking: de twee studies (Johnson1995 en Jacobs1996; glatirameeracetaat respectievelijk interferon-β vs. placebo) die als indirect bewijs werden geclassificeerd dragen nauwelijks bij aan de schatting van directe en indirecte effecten (bijlage 3b. Per studie bijdragen). Daarom is afwaarderen voor indirect bewijs voor vergelijking van een middel met placebo niet nodig. De studie over mitoxantron werd geëxcludeerd (bijlage 3b).
‘Toenemende invaliditeit’
Voor alemtuzumab, ocrelizumab en natalizumab is er redelijke tot hoge kwaliteit van bewijs voor het effect op ‘toenemende invaliditeit’ (tabel 4).
Tabel 4. Resultaten netwerkmeta-analyse en kwaliteit van bewijs voor ‘toenemende invaliditeit’ (relatieve effecten)†
|
Interventie versus placebo |
K |
Prop |
Direct RR [95%-BI] |
Kwaliteit van bewijs* |
Indirect RR [95%-BI] |
Kwaliteit van bewijs* |
NMA RR [95%-BI] |
Kwaliteit van bewijs |
|
Alemtuzumab |
0 |
0 |
|
|
0.495 [0.346; 0.708] |
Redelijk |
0.495 [0.346; 0.708] |
Redelijk |
|
Cladribine |
|
|
Geen data |
|
Geen data |
|
- |
|
|
Dimethylfumaraat |
2 |
1.00 |
0.651 [0.525; 0.808] |
Redelijk |
|
|
0.651 [0.525; 0.808] |
Laag |
|
Fingolimod |
2 |
0.85 |
0.710 [0.561; 0.897] |
Redelijk (s-RoB) |
0.583 [0.336; 1.013] |
Redelijk |
0.689 [0.555; 0.854] |
Laag |
|
Glatirameeracetaat |
2 |
0.65 |
0.855 [0.657; 1.114] |
Redelijk |
0.736 [0.514; 1.052] |
Redelijk |
0.811 [0.656; 1.003] |
Laag |
|
Interferon-β |
2 |
0.45 |
0.710 [0.514; 0.981] |
Redelijk |
0.911 [0.679; 1.221] |
Redelijk |
0.814 [0.655; 1.011] |
Laag |
|
Mitoxantron |
1 |
1.00 |
0.198 [0.047; 0.826] |
Laag |
|
|
0.198 [0.047; 0.826] |
Zeer laag |
|
Natalizumab |
1 |
1.00 |
0.585 [0.458; 0.748] |
Hoog |
|
|
0.585 [0.458; 0.748] |
Hoog |
|
Ocrelizumab |
0 |
0 |
|
|
0.492 [0.346; 0.699] |
Redelijk |
0.492 [0.346; 0.699] |
Redelijk |
|
Ofatumumab |
0 |
0 |
|
|
0.540 [0.379; 0.770] |
Redelijk |
0.540 [0.379; 0.770] |
Laag |
|
Ozanimod_0.5 |
0 |
0 |
|
|
0.902 |
Redelijk |
0.902 |
Zeer laag (s-RoB; vs-imprec.) |
|
Ozanimod_1.0 |
0 |
0 |
|
|
1.173 |
Redelijk |
1.173 |
Zeer laag |
|
Ponesimod |
0 |
0 |
|
|
0.626 [0.410; 0.956] |
Redelijk |
0.626 [0.410; 0.956] |
Laag |
|
Teriflunomide |
2 |
1.00 |
0.763 [0.624; 0.934] |
Redelijk |
|
|
0.763 [0.624; 0.934] |
Laag |
†gedefinieerd als percentage ‘toenemende invaliditeit’ in ziektemodulerend middel-groep gedeeld door percentage ‘toenemende invaliditeit’ in placebogroep.
K: aantal studies met directe evidence; prop: fractie directe evidence dat bijdraagt aan NMA effect.
s-RoB: serious risk of bias; vs-RoB: very serious risk of bias; s-indirect.: serious indirectness; s-incons.: serious inconsistency (I2>50% of variabele puntschatters); s-incoher.: serious incoherence; vs-incoher.: very serious incoherence; s-imprec.: serious imprecision (95% BI doorkruist RR=0.75 of RR=1.25)., vs-imprec.: very serious imprecision (95% BI doorkruist RR=0.75 én RR=1.25 of substantieel minder dan 300 events).
Global test for incoherence: χ2 statistic: 1.304; dF=2, P= 0.521; Geen lokale incoherentie (alle p-waarden>0.25 van z-waarden voor ratio van direct en indirect effect).
*imprecision niet beoordeeld.
Stoppen vanwege bijwerkingen
Voor alemtuzumab, dimethylfumaraat, en interferon-β is er redelijke tot hoge kwaliteit van bewijs wat het effect op ‘stoppen vanwege bijwerkingen’ betreft (tabel 5).
Tabel 5. Resultaten netwerkmeta-analyse en kwaliteit van bewijs voor ‘stoppen vanwege bijwerkingen’ (relatieve effecten)†
|
Interventie versus placebo |
K |
Prop |
Direct RR [95%-BI] |
Kwaliteit van bewijs* |
Indirect RR [95%-BI] |
Kwaliteit van bewijs* |
NMA RR [95%-BI] |
Kwaliteit van bewijs |
|
Alemtuzumab |
0 |
0 |
|
|
0.61 [0.320;1.164] |
Redelijk |
0.61 [0.320;1.164] |
Redelijk |
|
Cladribine |
2 |
1 |
1.13 [0.433;2.945] |
Hoog |
|
|
1.13 [0.433;2.945] |
Laag (vs-imprec.) |
|
Dimethylfumaraat |
2 |
1 |
0.969 [0.775;1.212] |
Redelijk (s-RoB) |
|
|
0.969 [ 0.775;1.212] |
Redelijk |
|
Fingolimod |
2 |
0.87 |
1.408 [1.087; 1.823] |
Laag |
1.451 [0.744;2.828] |
Redelijk |
1.413 [1.111;1.799] |
Laag |
|
Glatirameeracetaat |
2 |
0.34 |
2.634 [1.175; 5.903] |
Redelijk (s-RoB) |
1.566 [0.883;2.777] |
Redelijk |
1.864 [1.168;2.974] |
Laag |
|
Interferon-β |
4 |
0.47 |
1.72 [1.036; 2.856] |
Redelijk |
2.163 [1.340;3.492] |
Laag (s-RoB; s-incons.) |
1.942 [1.371;2.750] |
Redelijk |
|
Mitoxantron |
1 |
1 |
4.44 [0.56; 35.51] |
Laag (s-RoB) |
|
|
4.44 [0.56; 35.51] |
Zeer laag (s-RoB; vs-imprec.) |
|
Natalizumab |
1 |
1 |
1.256 [0.492; 3.206] |
Hoog |
|
|
1.256 [0.492;3.206] |
Laag |
|
Ocrelizumab |
0 |
0 |
|
0.769 [0.446;1.328] |
Redelijk |
0.769 [0.446;1.328] |
Zeer laag |
|
|
Ofatumumab |
0 |
0 |
|
|
2.016 [1.229;3.308] |
Laag (s-RoB; s-incons.) |
2.016 [1.229;3.308] |
Zeer laag (s-RoB; s-incons.; s-imprec.) |
|
Ozanimod_0.5 |
0 |
0 |
|
|
1.214 [0.692;2.129] |
Redelijk |
1.214 [0.692;2.129] |
Zeer laag |
|
Ozanimod_1.0 |
0 |
0 |
|
|
1.484 |
Redelijk |
1.484 |
Laag |
|
Ponesimod |
0 |
0 |
|
|
2.670 [1.570;4.539] |
Laag |
2.670 [1.570;4.539] |
Zeer laag |
|
Teriflunomide |
2 |
1 |
1.849 [1.340; 2.552] |
Laag s-incons.) |
|
1.849 [1.340;2.552] |
Laag |
† gedefinieerd als percentage ‘stoppen vanwege bijwerkingen’ in ziektemodulerende-groep gedeeld door percentage ‘stoppen vanwege bijwerkingen’ in placebogroep.
K: aantal studies met directe evidence; prop: fractie directe evidence dat bijdraagt aan NMA effect
s-RoB: serious risk of bias; s-indirect.: serious indirectness; s-incons.: serious inconsistency (I2>50% of variabele puntschatters); s-incoher.: serious incoherence; vs-incoher.: very serious incoherence; s-imprec.: serious imprecision (95% BI doorkruist RR=0.75 én RR=1.25); vs-imprec.: very serious imprecision (95% BI doorkruist RR=0.75 én RR=1.25).
Global test for incoherence: χ2 statistic: 1.130; dF=2, P= 0.568; Geen lokale incoherentie (alle p-waarden van z-waarden voor ratio van direct en indirect effect>0.30).
*imprecision niet beoordeeld
Conclusies
|
‘Annualized relapse rate’
Waarschijnlijk geven alemtuzumab, natalizumab, cladribine, ofatumumab, ozanimod_1.0, ocrelizumab, fingolimod en dimethylfumaraat een klinisch relevante reductie van de annualized relapse rate (tabel A). Misschien geldt dit ook voor mitoxantron. De overige middelen hebben misschien geen (overtuigend) klinisch relevant effect (tabel A).
|
||||
|
Tabel A. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘annualized relapse rate’. |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
SMD: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant†† |
Alemtuzumab§ |
-0.940 (-1.219; -0.660) |
0.97 |
|
|
|
Natalizumab |
-0.620 (-0.759; -0.482) |
0.86 |
|
|
|
Cladribine |
-0.489 (-0.624; -0.354) |
0.73 |
|
|
|
Ofatumumab |
-0.468 (-0.599; -0.336) |
0.70 |
|
|
|
Ozanimod_1.0 |
-0.417 [-0.521; -0.314] |
0.61 |
|
|
|
Ocrelizumab |
-0.406 (-0.522; -0.290) |
0.58 |
|
|
|
Fingolimod |
-0.391 (-0.474; -0.307) |
0.55 |
|
|
|
Dimethylfumaraat |
-0.310 (-0.396; -0.224) |
0.37 |
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
- |
- |
- |
|
|
|
|
|
|
|
Zeer laag/laag |
Klinisch relevant |
Mitoxantron |
-0.822 (-1.396; -0.247) |
0.89 |
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
Ponesimod Ozanimod_0.5 Glatirameeracetaat Interferon-β Teriflunomide |
-0.339 (-0.489; -0.188) -0.301 [-0.405; -0.199] -0.184 (-0.250; -0.119) -0.168 (-0.232; -0.104) -0.132 (-0.226, -0.037) |
0.44 0.36 0.18 0.15 0.10 |
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
* SMD: standardized mean difference. Een groter verschil wijst op een grotere vermindering van de ‘annualized relapse rate’ bij gebruik van een ziektemodulerend middel. † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Bovengrens van 95% BI van SMD < -0.2 wijst op (mogelijk) klinisch relevant effect. Een SMD < -0.2 wordt gezien als een klein maar belangrijk effect (Guyatt et al., 2013). §alemtuzumab is het enige middel dat superieur is aan diverse andere klinisch relevante middelen (dimethylfumaraat, fingolimod, ocrelizumab, ozanomod_1.0): bovengrens van 95% BI van SMD < -0.2 . Zie league table (bijlage 3a). Opmerking: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
|
‘Vrij van relapse’
Waarschijnlijk geven alemtuzumab, natalizumab, ocrelizumab en fingolimod een klinisch relevante toename van ‘vrij zijn van relapse’ (tabel B). Waarschijnlijk heeft cladribine geen (overtuigend) klinisch relevant effect. De overige middelen hebben misschien geen (overtuigend) klinisch relevant effect (tabel B).
|
||||
|
Tabel B. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘vrij van relapse’. |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
RR: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant†† |
Alemtuzumab Natalizumab Fingolimod Ocrelizumab |
1.626 [1.447;1.826] 1.593 [1.402;1.810] 1.422 [1.327;1.524] 1.400 [1.274;1.538] |
0.95 0.92 0.74 0.69 |
|
|
||||
|
Niet (overtuigend) klinisch relevant |
Cladribine |
1.306 [1.203;1.418] |
0.51 |
|
|
|
|
|
|
|
|
Zeer laag/laag |
Klinisch relevant |
- |
|
|
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
Dimethylfumaraat Teriflunomide Glatirameeracetaat Interferon-β |
1.277 [1.183;1.378] 1.253 [1.155;1.359] 1.184 [1.120;1.251] 1.178 [1.096;1.265] |
0.44 0.38 0.20 0.18 |
|
|
|
|||
|
|
|
|||
|
|
||||
|
*RR> 1 wijst op een grotere mate van vrij zijn van relapses bij gebruik van een ziektemodulerend middel. † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Ondergrens van 95% BI van RR > 1.25 wijst op (mogelijk) klinisch relevant effect. Opmerkingen: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
|
‘Toenemende invaliditeit’
Waarschijnlijk geven alemtuzumab, natalizumab en ocrelizumab een klinisch relevante afname van ‘toenemende invaliditeit’ (tabel C). De overige middelen hebben misschien geen (overtuigend) klinisch relevant effect (tabel C).
|
||||
|
Tabel C. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘toenemende invaliditeit’ |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
RR: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant†† |
Ocrelizumab Alemtuzumab Natalizumab |
0.492 [0.346; 0.699] 0.495 [0.346; 0.708] 0.585 [0.458; 0.748] |
0.82 0.82 0.68 |
|
|
|
|||
|
|
|
|||
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
- |
|
|
|
|
||||
|
Zeer laag/laag |
Klinisch relevant |
- |
|
|
|
|
|
|
|
|
|
|
Niet (overtuigend) klinisch relevant |
Mitoxantron Ofatumumab Ponesimod Dimethylfumaraat Fingolimod Teriflunomide Glatirameeracetaat Interferon-β Ozanimod_0.5 Ozanimod_1.0 |
0.198 [0.047; 0.826] 0.540 [0.379; 0.770] 0.626 [0.410; 0.956] 0.651 [0.525; 0.808] 0.689 [0.555; 0.854] 0.763 [0.624; 0.934] 0.811 [0.656; 1.003] 0.814 [0.655; 1.011] 0.902 [0.580; 1.403] 1.173 [0.762; 1.807] |
0.95 0.75 0.59 0.56 0.50 0.36 0.30 0.29 0.23 0.04 |
|
|
||||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
|
|
|||
|
*RR<1 wijst op een geringere toename van invaliditeit bij gebruik van een ziektemodulerend middel. † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Bovengrens van 95% BI van RR < 0.75 wijst op (mogelijk) klinisch relevant effect. Opmerking: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
|
‘Stoppen vanwege bijwerkingen’
Waarschijnlijk geeft interferon-β een klinisch relevante toename van stoppen vanwege bijwerkingen. Waarschijnlijk geeft dimethylfumaraat geen klinisch relevante toename van stoppen vanwege bijwerkingen (tabel D).
Ponesimod en mitoxantron geven misschien een klinisch relevante toename van stoppen vanwege bijwerkingen (tabel D).
Voor de overige middelen geldt dat ze misschien geen (overtuigend) klinisch relevante toename van stoppen vanwege bijwerkingen geven (tabel D).
|
||||
|
Tabel D. Ziektemodulerende middelen gerangschikt naar kwaliteit van bewijs, klinische relevant effect en effectgrootte voor de uitkomst ‘studieuitval vanwege bijwerkingen’. |
||||
|
Kwaliteit van bewijs |
Hoe is het verschil vs. placebo? |
Ziektemodulerend middel |
RR: Middel vs. placebo |
P-score† |
|
Redelijk/hoog |
Klinisch relevant ongunstig effect†† |
Interferon-β |
1.942 [1.371;2.750] |
0.74 |
|
|
Geen (overtuigend) klinisch relevant ongunstig effect |
Dimethylfumaraat |
0.969 [0.775;1.212] |
0.23 |
|
|
||||
|
|
||||
|
Zeer laag/laag |
Klinisch relevant ongunstig effect |
Ponesimod Teriflunomide |
2.670 [1.570;4.539] 1.849 [1.340;2.552] |
0.90 0.69 |
|
|
||||
|
|
|
|||
|
|
|
|||
|
|
Geen (overtuigend) klinisch relevant ongunstig effect |
Mitoxantron Ofatumumab Glatirameeracetaat Ozanimod_1.0 Fingolimod Natalizumab Ozanimod_0.5 Cladribine** Ocrelizumab** Alemtuzumab** |
4.440 [0.560;35.41] 2.016 [1.229;3.308] 1.864 [1.168;2.974] 1.484 [0.858;2.569] 1.413 [1.111;1.799] 1.256 [0.492;3.206] 1.214 [0.692;2.129] 1.130 [0.433;2.945] 0.769 [0.446;1.328] 0.610 [0.320;1.164] |
0.85 0.75 0.70 0.54 0.49 0.43 0.38 0.37 0.12 0.05 |
|
|
|
|||
|
|
|
|||
|
|
||||
|
|
||||
|
|
|
|||
|
|
|
|||
|
|
||||
|
* RR>1 wijst op toename van stoppen vanwege bijwerkingen bij gebruik van een ziektemodulerend middel † P-score: varieert van 0 tot 1 en kan worden geïnterpreteerd als de gemiddelde mate van zekerheid dat een interventie beter is dan de andere interventies in het netwerk. Wordt in GRADE gebruikt bij wijze van consistentiecontrole: d.w.z. om per niveau van de kwaliteit van bewijs na te gaan of de klinisch relevante effecten ook hogere p-scores hebben dan de klinisch niet-relevante effecten. ††Ondergrens van 95% BI van RR > 1.25 wijst op (mogelijk) klinisch relevant ongunstig effect. ** artefact: Het relatief geringe risico op stoppen vanwege bijwerkingen is een artefact omdat deze middelen in een veel lagere frequentie worden toegediend dan de andere middelen. Opmerking: Zie voor details van de beoordeling van de kwaliteit van bewijs de paragraaf ‘kwaliteit van bewijs’. |
||||
Zoeken en selecteren
Om de uitgangsvraag te kunnen beantwoorden is een systematische literatuuranalyse verricht met de volgende PICO-vraagstelling:
Wat zijn bij patiënten met relapsing remitting MS en met een indicatie voor ziektemodulerende behandeling, de relatieve netto-baten van de verschillende ziektemodulerende middelen?
-
- interferon-β (interferon-β/peginterferon-β)
- glatirameeracetaat
- teriflunomide
- fumaraatzuur esters (dimethylfumaraat/diroximel fumaraat)
- S1P receptor modulatoren (fingolimod/ozanimod/ponesimod)
- natalizumab (intraveneus of subcutaan)
- anti-CD20 monoclonalen (ocrelizumab/ofatumumab)
- cladribine
- alemtuzumab
Selectie- en exclusiecriteria:
|
Type studies |
|
|
Type patiënten |
|
|
Interventie |
|
|
Controle |
|
|
Type uitkomstmaten |
1. Effectiviteit - klinisch
2. Effectiviteit – niet klinisch
3. Tolereerbaarheid en veiligheid
|
|
Type setting |
|
|
Exclusiecriteria |
|
Deze search betreft een update van de moduletekst van 2021 (met search datum april 2018). Hierbij werd uitgegaan van de systematische literatuuranalyse in de Europese richtlijn. In de Europese richtlijn is gezocht in verschillende databases: Medline (OVID), Embase en Cochrane. De zoekverantwoording is weergegeven in de bijlage.
De Europese werkgroep vond vier RCTs (Cadavid et al., 2009; Calabrese et al., 2012; Mikol et al., 2008; O’Connor et al., 2009) waarin glatirameeracetaat en interferon-β werden vergeleken, twee RCTs (Vermersch et al., 2014; Cohen et al., 2010) waarin teriflunomide en fingolimod met interferon-β werden vergeleken, drie RCTs (Cohen et al., 2012; Coles et al., 2008; Coles et al., 2012) waarin alemtuzumab met interferon-β werd vergeleken en twee RCTs (beide gerapporteerd in Hauser et al., 2017) waarin ocrelizumab met interferon-β werd vergeleken.
De Nederlandse werkgroep verrichtte een netwerk meta-analyse met behulp van de in de Europese richtlijn samengevatte studies en de in de literatuursearch gevonden studies. In de netwerk meta-analyse worden de gegevens uit alle studies samen gebruikt om de effectiviteit ten opzicht van elkaar te vergelijken. Zie bijlage 1 voor een toelichting op het verschil tussen een traditionele en een netwerk meta-analyse.
Referenties
- Apostolidis SA, Kakara M, Painter MM, Goel RR, Mathew D, Lenzi K, Rezk A, Patterson KR, Espinoza DA, Kadri JC, Markowitz DM, E Markowitz C, Mexhitaj I, Jacobs D, Babb A, Betts MR, Prak ETL, Weiskopf D, Grifoni A, Lundgreen KA, Gouma S, Sette A, Bates P, Hensley SE, Greenplate AR, Wherry EJ, Li R, Bar-Or A. Cellular and humoral immune responses following SARS-CoV-2 mRNA vaccination in patients with multiple sclerosis on anti-CD20 therapy. Nat Med. 2021 Nov;27(11):1990-2001.
- Berger, J. R., & Markowitz, C. (2018). Deciding on the Best Multiple Sclerosis Therapy: Tough Choices. JAMA neurology, 75(12), 14611462.
- Brignardello-Petersen R, Bonner A, Alexander PE, Siemieniuk RA, Furukawa TA, Rochwerg B, Hazlewood GS, Alhazzani W, Mustafa RA, Murad MH, Puhan MA, Schünemann HJ, Guyatt GH; GRADE Working Group. Advances in the GRADE approach to rate the certainty in estimates from a network meta-analysis. J Clin Epidemiol. 2018 Jan;93:36-44.
- Cadavid, D., Wolansky, L. J., Skurnick, J., Lincoln, J., Cheriyan, J., Szczepanowski, K., Kamin, S. S., Pachner, A. R., Halper, J., & Cook, S. D. (2009). Efficacy of treatment of MS with IFNbeta-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology, 72(23), 1976-1983
- Calabrese M, Bernardi V, Atzori M, et al. Effect of disease-modifying drugs on cortical lesions and atrophy in relapsingremitting multiple sclerosis. Mult Scler 2012; 18: 418424.
- Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010; 362: 402415.
- Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing_remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 2012; 380: 18191828.
- Cohen, J. A., Comi, G., Selmaj, K. W., Bar-Or, A., Arnold, D. L., Steinman, L., et al. & RADIANCE Trial Investigators (2019). Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24-month, phase 3 trial. The Lancet. Neurology, 18(11), 1021-1033.
- Coles AJ, Compston DA, Selmaj KW, et al. (Camms223) Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med 2008; 359: 17861801.
- Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 2012; 380: 18291839.
- Comi, G., Kappos, L., Selmaj, K. W., Bar-Or, A., Arnold, D. L., Steinman, L., Hartung, H. P., Montalban, X., Kubala Havrdová, E., Cree, B. A. C., Sheffield, J. K., Minton, N., Raghupathi, K., Ding, N., Cohen, J. A., & SUNBEAM Study Investigators (2019). Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. The Lancet. Neurology, 18(11), 1009-1020.
- Guyatt GH, Thorlund K, Oxman AD, Walter SD, Patrick D, Furukawa TA, Johnston BC, Karanicolas P, Akl EA, Vist G, Kunz R, Brozek J, Kupper LL, Martin SL, Meerpohl JJ, Alonso-Coello P, Christensen R, Schunemann HJ. GRADE guidelines: 13. Preparing summary of findings tables and evidence profiles-continuous outcomes. J Clin Epidemiol. 2013 Feb;66(2):173-83.
- Havrdová E, Arnold DL, Bar-Or A, Comi G, Hartung HP, Kappos L, Lublin F, Selmaj K, Traboulsee A, Belachew S, Bennett I, Buffels R, Garren H, Han J, Julian L, Napieralski J, Hauser SL, Giovannoni G. No evidence of disease activity (NEDA) analysis by epochs in patients with relapsing multiple sclerosis treated with ocrelizumab vs interferon beta-1a. Mult Scler J Exp Transl Clin. 2018 Mar 12;4(1):2055217318760642.
- Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017; 376: 221-234.
- Hauser, S. L., Bar-Or, A., Cohen, J. A., Comi, G., Correale, J., Coyle, P. K., Cross, A. H., de Seze, J., Leppert, D., Montalban, X., Selmaj, K., Wiendl, H., Kerloeguen, C., Willi, R., Li, B., Kakarieka, A., Tomic, D., Goodyear, A., Pingili, R., Häring, D. A., ASCLEPIOS I and ASCLEPIOS II Trial Groups (2020). Ofatumumab versus Teriflunomide in Multiple Sclerosis. The New England journal of medicine, 383(6), 546557.
- Hu X, Shang S, Nestorov I, Hasan J, Seddighzadeh A, Dawson K, Sperling B, Werneburg B. COMPARE: Pharmacokinetic profiles of subcutaneous peginterferon beta-1a and subcutaneous interferon beta-1a over 2 weeks in healthy subjects. Br J Clin Pharmacol. 2016 Aug;82(2):380-8.
- Kappos, L., Fox, R. J., Burcklen, M., Freedman, M. S., Havrdová, E. K., Hennessy, B., Hohlfeld, R., Lublin, F., Montalban, X., Pozzilli, C., Scherz, T., D'Ambrosio, D., Linscheid, P., Vaclavkova, A., Pirozek-Lawniczek, M., Kracker, H., & Sprenger, T. (2021). Ponesimod Compared With Teriflunomide in Patients With Relapsing Multiple Sclerosis in the Active-Comparator Phase 3 OPTIMUM Study: A Randomized Clinical Trial. JAMA neurology, 78(5), 558-567.
- Matthijs M Versteegh, Simone A Huygens, Beatrijs W H Wokke, Joost Smolders. Effectiveness and Cost-Effectiveness of 360 Disease-Modifying Treatment Escalation Sequences in Multiple Sclerosis. Value Health 2022 Jun;25(6):984-991
- Merkel, B., Butzkueven, H., Traboulsee, A. L., Havrdova, E., & Kalincik, T. (2017). Timing of high-efficacy therapy in relapsing-remitting multiple sclerosis: A systematic review. Autoimmunity reviews, 16(6), 658-665.Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol 2008; 7: 903-914.
- Millefiorini, E., Gasperini, C., Pozzilli, C., D'Andrea, F., Bastianello, S., Trojano, M., Morino, S., Morra, V. B., Bozzao, A., Calo', A., Bernini, M. L., Gambi, D., & Prencipe, M. (1997). Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. Journal of neurology, 244(3), 153-159.
- Milo R, Staun-Ram E, Karussis D, Karni A, Hellmann MA, Bar-Haim E, Miller A; Israeli Neuroimmunology Study Group on COVID-19 Vaccination in Multiple Sclerosis. Humoral and Cellular Immune Responses to SARS-CoV-2 mRNA Vaccination in Patients with Multiple Sclerosis: An Israeli Multi-Center Experience Following 3 Vaccine Doses. Front Immunol. 2022 Apr 1;13:868915.
- Naismith et al 2021
- O'Connor P, Filippi M, Arnason B, et al. 250 lg or 500 lg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol 2009; 8: 889-897.
- Puhan MA, Schünemann HJ, Murad MH, Li T, Brignardello-Petersen R, Singh JA, Kessels AG, Guyatt GH; GRADE Working Group. A GRADE Working Group approach for rating the quality of treatment effect estimates from network meta-analysis. BMJ. 2014 Sep 24;349:g5630.
- Rotstein D, Montalban X. Reaching an evidence-based prognosis for personalized treatment of multiple sclerosis. Nat Rev Neurol. 2019.
- Schlegel V, Leray E. From Medical Prescription to Patient Compliance: A Qualitative Insight into the Neurologist-Patient Relationship in Multiple Sclerosis. Int J MS Care. 2018 Nov-Dec;20(6):279-286.
- Simpson-Yap S, De Brouwer E, Kalincik T, Rijke N, Hillert JA, Walton C, Edan G, Moreau Y, Spelman T, Geys L, Parciak T, Gautrais C, Lazovski N, Pirmani A, Ardeshirdavanai A, Forsberg L, Glaser A, McBurney R, Schmidt H, Bergmann AB, Braune S, Stahmann A, Middleton R, Salter A, Fox RJ, van der Walt A, Butzkueven H, Alroughani R, Ozakbas S, Rojas JI, van der Mei I, Nag N, Ivanov R, Sciascia do Olival G, Dias AE, Magyari M, Brum D, Mendes MF, Alonso RN, Nicholas RS, Bauer J, Chertcoff AS, Zabalza A, Arrambide G, Fidao A, Comi G, Peeters L. Associations of Disease-Modifying Therapies With COVID-19 Severity in Multiple Sclerosis. Neurology. 2021 Nov 9;97(19):e1870-e1885.
- Toorop, A. A., van Kempen, Z. L. E., Steenhuis, M., Nielsen, J., Sinnige, L. G. F., van Dijk, G., Roosendaal, C. M., Arnoldus, E. P. J., Hoitsma, E., Lissenberg-Witte, B. I., de Jong, B. A., Oosten, B. W. V., Strijbis, E. M. M., Uitdehaag, B. M. J., Rispens, T., Killestein, J., & NEXT-MS study group (2023). Decrease of natalizumab drug levels after switching from intravenous to subcutaneous administration in patients with multiple sclerosis. Journal of neurology, neurosurgery, and psychiatry, jnnp-2022-330467. Advance online publication.
- Trojano M, Ramió-Torrentà L, Grimaldi LM, Lubetzki C, Schippling S, Evans KC, Ren Z, Muralidharan KK, Licata S, Gafson AR. A randomized study of natalizumab dosing regimens for relapsing-remitting multiple sclerosis. Mult Scler. 2021 Dec;27(14):2240-2253.
- Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler J 2014; 20: 705716.
Verantwoording
Beoordelingsdatum en geldigheid
Laatst beoordeeld : 11-12-2023
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de modules voor het addendum is in 2021 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de indicatiestelling en monitoring van ziektemodulerende therapie bij mensen met MS (zie hiervoor de samenstelling van de werkgroep).
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname. De werkgroep werkte gedurende 2 jaar aan de totstandkoming van de richtlijn. De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Werkgroep
- Mevr. dr. B.A. de Jong, neuroloog, NVN (voorzitter)
- Mevr. B. Bakker, patiëntvertegenwoordiger, MS-patiënten Vereniging Nederland
- Dhr. drs. R. Haenen, revalidatiearts, VRA
- Mevr. K. Harrison, verpleegkundig specialist, V&VN
- Dhr. drs. B.J.A. van Hoeve, neuroloog, NVN
- Mevr. dr. E. Hoitsma, neuroloog, NVN
- Dhr. prof. dr. J. Killestein, neuroloog, NVN
- Dhr. drs. J.B. Masselink, ziekenhuisapotheker, NVZA
- Dhr. dr. B. Moraal, radioloog, NVVR
- Dhr. dr. J. Mostert, neuroloog, NVN
- Dhr. dr. J. Murk, arts-microbioloog, NVMM
- Mevr. M. Pippel, patiëntvertegenwoordiger, MS-patiënten Vereniging Nederland
- Dhr. dr. J. Smolders, neuroloog, NVN
- Dhr. dr. M.T. Wijburg, AIOS neurologie, NVN
- Mevr. Dr. A. Coumans, Gynaecoloog, NVOG
Met dank aan:
- Dhr. Dr. Ir. J.J.A. de Beer, zelfstandig richtlijnmethodoloog
- Mevr. dr. S. Huygens, gezondheidseconoom, Huygensandversteegh.com
- Dhr. dr. M. Versteegh, gezondheidseconoom, Huygensandversteegh.com
Met ondersteuning van:
- Mevr. dr. C.T.J. Michels, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Mevr. dr. M.L. Molag, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Achternaam werkgroeplid |
Hoofdfunctie |
Nevenwerkzaamheden |
Gemelde belangen |
BelandenCie |
|
Bakker |
MS Patiënt vertegenwoordiger MSVN |
Functionel tester software Thunderbyte AI |
geen |
Geen restrictie |
|
Coumans |
Gynaecoloog |
Geen |
geen |
Geen restrictie |
|
De Jong (voorzitter) |
Functie: neuroloog |
Voorzitter commissie Addendum MS richtlijn van NVN (onbetaald). Voorzitter commissie MS registratie van NVN (onbetaald). Lid van wetenschappelijk adviesraad van Prinses Beatrixfonds (onbetaald) Lid van het PROMS initiative, onderdeel van Global MSIF Movement (onbetaald) |
MS Centrum Amsterdam, Amsterdam UMC, locatie VUmc ontvangt financiële ondersteuning van: Bayer AG, Biogen, Roche, GlaxoSmithKline, Merck, Celgene BMS, Sanofi Genzyme, Novartis, TEVA, Stichting MS Research, NWO, ZonMw, Hersenstichting, Nationaal MS Fonds, Zorgverzekeraars Nederland (ZN) en Health~Holland (TKI*).
*TKI: Dit is een studie gesponsord door Sanofi, het gaat om een Bruton’s tyrosine kinase inhibitor: tolebrutinib (komt niet voor in richtlijn).
Dit zijn deels wel en niet persoonsgebonden subsidies (= deels contractonderzoek). Subsidies van instanties zoals MS research, ZonMw, hersenstichting zijn allemaal persoonsgebonden maar hierbij ik ben hierbij geen PI. Ik ben wel PI bij: - subsidie van ZN om onderzoek te doen met de landelijke MS registratie. - fase IV studie waarbij observationeel de kwaliteit en veiligheid van alemtuzumab (Sanofi Genzyme) gemonitord wordt (zit niet in adviesraad, krijg er geen geld voor). Studie gaat over post-surveillance en houdt geen relatie met indicatiestelling. - fase III studie naar tyrosine kinase inhibitors bij MS (dit middel staat niet in de richtlijn en komt naar verwachting niet op korte termijn op de markt).
Ontvangen onderzoekssubsidie van MS Vereniging Nederland in 2022: “Prevalentie van MS in Nederland” met een bedrag €60.167 Ontvangen onderzoekssubsidie van Stichting MS Research in 2016: "Improving cognitieve functioning among patients with multiple sclerosis by cognitive rehabilitation therapy or mindfulness based cognitive therapy: a dual-center, investigator-blinded, parallel group randomized controlled trial" met een bedrag van € 250.000.
|
Restrictie als TKIs ter sprake komt in een module. |
|
Haenen |
Functie: revalidatiearts |
Geen |
Geen |
Geen restrictie. |
|
Harrisson |
Functie: Verpleegkundig Specialist Werkgever: Tergooi ziekenhuis |
Richtlijn Revalidatie bij MS, vergoeding van gemaakte uren. |
Geen |
Geen restrictie. |
|
Hoitsma |
Functie: neuroloog |
Voorzitter landelijke MS werkgroep (onbetaald). |
Lokale onderzoeker bij (geen PI over gehele studies):
Betreft bij allen betaling onderzoeksactiviteiten. |
Geen restrictie. |
|
Killestein |
Functie: neuroloog (hoogleraar MS) |
Adviseur Springer Healthcare - Mednet Neurologie (betaald). Hoofdredacteur Tijdschrift voor Neurologie en Neurochirurgie (TNN), tot februari 2021, (betaald).
Voorzitter Horizonscan geneesmiddelen Zorginstituut Nederland. Werkgroep Neurologische aandoeningen inclusief gedrag (onbetaald). Lid Wetenschappelijke Adviesraad Stichting MS Research (onbetaald). Lid editorial board Neurology (onbetaald). Lid editorial board MS Journal (onbetaald).
|
MS Centrum Amsterdam, Amsterdam UMC, locatie VUmc ontvangt financiële ondersteuning van: Bayer AG, Biogen, Roche, GlaxoSmithKline, Merck, Celgene BMS, Sanofi Genzyme, Novartis, TEVA, Immunic Therapeutics, Stichting MS Research, NWO, ZonMw, Hersenstichting, Nationaal MS Fonds, Zorgverzekeraars Nederland (ZN) en Health~Holland (TKI)
1. Principle investigator NEXT-MS -natalizumab extended dosing bij MS -NCT04225312. Sponsor: Hersenstichting en Stichting MS Research. 2. Principle investigator DOT-MS - discontinuation of therapy in MS, NCT04260711- Sponsor: ZonMW en Stichting MS research 3. Principle investigator BLOOMS trial: efficacy, safety and cost-effectiveness of B cell tailored ocrelizumab versus standard ocrelizumab in RRMS - Sponsor: ZonMW en Treatmeds, moet nog starten. 4. Principle investigator Apps-MS studie - Sponsor: Stichting MS-Research en Health~Holland TKI. 5. Principle investigator Blood platelet-based liquid biopsies for optimization of diagnosis and treatment of patients with multiple sclerosis - Sponsor Merck. à Merck maakt medicatie die gebruikt wordt bij MS, Rebif en Mavencla. Betrfet onderzoek m.b.t. diagnostische biomarker (wel/geen MS). Geen enkele bemoeienis van Merck met de inhoud. 6. Deelname RELAMS - onderzoek naar relapses en pseudorelapses bij MS. 7. Deelname RAM-MS trial: Randomized Autologous heMatopoietic stem cell transplantation versus alemtuzumab, cladribine or ocrelizumab for patients with RRMS. EudraCT Number: 2017-001362-25 Sponsor: Stichting MS Research en Nationaal MS fonds. 8. Lid Steering Committee Ensemble trial Roche, vergoeding naar VUmc. 9. Lid Steering Committee Liberto trial Roche, vergoeding naar VUmc. 10. Lid Steering Committee NOVA trial Biogen, vergoeding naar VUmc. 11. Lid Adjudication Committee Immunic Calliper Phase II trial, vergoeding naar VUmc |
Geen restrictie. |
|
Masselink |
Functie: ziekenhuisapotheker Werkgever: Medisch Spectrum Twente |
Lid werkgroep neurologische aandoeningen (incl. gedrag) Horizonscan Geneesmiddelen, |
Geen |
Geen restrictie. |
|
Mostert |
Functie: neuroloog |
Lid hoofdredactieraad Tijdschrift voor Neurologie en Neurochirurgie (onbetaald).
Tot mei 2021: 1.5 jaar voorzitter en daarvoor 2 jaar secretaris van de landelijke MS werkgroep (onbetaald). |
Lokale onderzoeker bij (geen PI over gehele studies):
De financiering op contractbasis (per verrichting vergoeding). |
Bij wijzigingen, graag contact opnemen met belangencie (n.a.v. non-inferiority study) |
|
Murk |
Functie: arts-microbioloog Werkgever: Elisabeth-Tweesteden ziekenhuis Tilburg |
Redactielid tijdschrift infectieziekten (onbetaald)
Sectielid infectieziektenserologie SKML, coördinator rondzending H. pylori serologie (Nederlandse organisatie die externe kwaliteitsrondzendingen voor laboratoria organiseert) (onbetaald)
|
Ik werk als arts-microbioloog in vrije vestiging. Het verrichten van laboratoriumdiagnostiek is een onderdeel van de werkzaamheden waardoor een vrijgevestigde arts-microbioloog gefinancierd wordt. |
Geen restrictie. |
|
Pippel |
Functie: Gemeentesecretaris Werkgever: Gemeente Zandvoort |
Lid Adviesplatform MS vereniging |
Geen |
Geen restrictie. |
|
Smolders |
Neuroloog, 0.8 FTE afdeling Neurologie, Erasmus Medisch Centrum Rotterdam |
Bestuurslid MS werkgroep (onbetaald).
Kosteneffectiviteitsonderzoek MS in samenwerking met het bedrijf Huygens & Versteegh |
Onderzoek wordt gefinancierd door de MS research stichting (Voorschoten), het Nationaal MS fonds (Rotterdam) en de Erasmus Foundation (Rotterdam) Van de collectebusfondsen-studies ben ik PI. De bedrijven betreft onderzoeksprojecten met het centrum, waarbij ik niet als PI fungeer. Onderzoeksprojecten binnen MS centrum ErasMS worden gefinancierd door Merck (BTK inhibitor), Idorsia (preklinisch onderzoek compound), GSK (preklinisch onderzoek compound) en Roche (federated learning project).
In verleden:
|
Geen restrictie. |
|
Van Hoeve |
Functie: neuroloog Werkgever: ZorgSaam Zeeuws-Vlaanderen, Terneuzen |
Lid van MS zorg Zuid Zuid West, regionaal samenwerkingsverband tussen neurologen uit verschillende ziekenhuis, met MS als aandachtsgebied (onbetaald). |
April 2023: Binnenkort deelnemend centrum aan SPIN-P studie (gericht op o.a. etiologische/ prognostische factoren m.b.t. primair progressieve MS). Deelname Ectrims congres (oktober 2023) op uitnodiging Merck |
Geen restrictie. |
|
Wijburg |
Functie: AIOS Neurologie, Werkgever: Amsterdam UMC |
Geen |
Geen |
Geen restrictie.
|
|
Huygens (extern adviseur) |
Eigenaar Huygens & Versteegh B.V. |
Tijdelijk senior adviseur ziektemodellen Zorginstituut Nederland (betaald) |
Het ErasmuMC/iMTA MS model is ontwikkeld door Simone Huygens & Matthijs Versteegh met een onderzoeksgrant van ErasmusMC tijdens hun dienstverband bij iMTA Erasmus Universiteit Rotterdam. In hun Huygens & Versteegh B.V. maken zij melding van vergoedingen door IQVIA (niet MS gerelateerd), Beigene (niet MS gerelateerd), Optimax (niet MS gerelateerd), ErasmusMC (niet MS gerelateerd), ICER-US (MS gerelateerd, onafhankelijk expert advies of ICER-US rapport kosteneffectiviteit MS geneesmiddelen), EuroQoL (niet MS gerelateerd), iMTA (niet MS gerelateerd), Takeda (niet MS gerelateerd), Chiesi (niet MS gerelateerd) en Merck KGgA (MS gerelateerd, niet gerelateerd aan geneesmiddelen opgenomen in de richtlijn of het model (nieuwe moleculaire entiteit)). De werkzaamheden voor de richtlijn met het ErasmusMC/iMTA model zijn onbezoldigd uitgevoerd. |
Geen restrictie.
|
|
Versteegh (extern adviseur) |
Eigenaar Huygens & Versteegh B.V. |
Tijdelijk senior adviseur ziektemodellen Zorginstituut Nederland (betaald) Editorial board member Medical Decision Making (onbetaald) Lid EuroQoL research foundation (onbetaald maar met vergoeding reiskosten en congresbezoek) Columnist NTvG (onbetaald) |
Het ErasmuMC/iMTA MS model is ontwikkeld door Simone Huygens & Matthijs Versteegh met een onderzoeksgrant van ErasmusMC tijdens hun dienstverband bij iMTA Erasmus Universiteit Rotterdam. In hun Huygens & Versteegh B.V. maken zij melding van vergoedingen door IQVIA (niet MS gerelateerd), Beigene (niet MS gerelateerd), Optimax (niet MS gerelateerd), ErasmusMC (niet MS gerelateerd), ICER-US (MS gerelateerd, onafhankelijk expert advies of ICER-US rapport kosteneffectiviteit MS geneesmiddelen), EuroQoL (niet MS gerelateerd), iMTA (niet MS gerelateerd), Takeda (niet MS gerelateerd), Chiesi (niet MS gerelateerd) en Merck KGgA (MS gerelateerd, niet gerelateerd aan geneesmiddelen opgenomen in de richtlijn of het model (nieuwe moleculaire entiteit)). De werkzaamheden voor de richtlijn met het ErasmusMC/iMTA model zijn onbezoldigd uitgevoerd. |
Geen restrictie.
|
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door in de voorbereidende fase de MS-patiënten Vereniging Nederland te vragen om schriftelijke input omtrent knelpunten en aandachtspunten. Patiënten werden tevens in de werkgroep vertegenwoordigd door twee afgevaardigden van de MS-patiënten Vereniging Nederland. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen. De conceptrichtlijn is tevens voor commentaar voorgelegd aan Patiëntenfederatie Nederland en MS-patiënten Vereniging Nederland. De eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep de knelpunten in de zorg voor patiënten met MS. De werkgroep beoordeelde de aanbeveling(en) uit de eerdere richtlijnmodule (NVN, 2012; NVN, 2020) op noodzaak tot revisie. Tevens zijn er knelpunten aangedragen via een schriftelijke knelpunteninventarisatie. Een rapportage hiervan kan worden aangevraagd via de Richtlijnendatabase. Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.