Start ziektemodulerende middelen
Uitgangsvraag
Bij welke patiënten met relapsing remitting MS is het starten van ziektemodulerende middelen geïndiceerd?
Aanbeveling
Adviseer aan alle patiënten met actieve relapsing remitting MS in het afgelopen jaar te starten met een ziektemodulerend middel.
Overweeg initieel een afwachtend beleid* bij:
- alle patiënten die het afgelopen jaar geen actieve relapsing remitting MS hebben
- bij patiënten met gunstige prognostische factoren
*Bij afwachtend beleid monitor de patiënt klinisch en radiologisch met MRI hersenen bij 3-6 en 12 maanden.
Betrek bij de indicatiestelling voor behandeling met een ziektemodulerend middel de volgende factoren:
- Klinisch prognostische factoren:
- ernst van relapse
- mate van herstel
- symptomen herleidbaar tot lokalisaties infratentorieel of myelum
- Klinisch gunstige factoren:
- 1 of geen relapse in de eerste 5 jaar ziekte
- EDSS-score <=2 na 5 jaar follow-up
- Diagnostisch ongunstig prognostische factoren:
- op MRI scan hersenen een groot aantal (10 of meer) of een groot volume van T2- laesies op voor MS karakteristieke locaties
- gadolinium aankleurende laesies
- laesies infratentorieel en/of myelum
- unieke oligoclonale banden in liquor
- Vroeg starten met een ziektemodulerend middel is een gunstig prognostische factor voor ziektebeloop
- Zwangerschapswens op korte termijn (binnen een jaar).
Bespreek met de patiënt diens overwegingen voor het wel of niet starten van medicatie en kom samen tot een besluit.
Overwegingen
Waarden en voorkeuren
Er is volgens de richtlijncommissie grote variatie in de voorkeuren van patiënten wat behandelen versus niet behandelen betreft. Shared-decision making is dan ook aangewezen. Shared-decision making is een overlegmodel waarin de behandelend arts patiënten helpt te bepalen wat de beste therapie is die hun waarden en voorkeuren belichaamt (Berger et al., 2018).
Ter info: in het onderzoek van Kremer et al. (2018), een Nederlandse studie, onderzocht men welke aspecten belangrijk zijn in de keuze van ziektemodulerende middelen. Hiervoor liet men neurologen, verpleegkundigen en patiënten dezelfde 27 aspecten scoren. De top 3 waren: effect op ziekteprogressie, effect op kwaliteit van leven en effect op relapse. Voor het aspect veiligheid verschilde de score: veiligheid scoorde op de vierde plaats bij neurologen en MS-verpleegkundigen en plaats acht bij patiënten. De gerapporteerde informatie had betrekking op de gemiddelde patiënt.
Professioneel perspectief
De aanbevelingen in dit hoofdstuk zijn gebaseerd op de klinische en niet-klinische parameters zoals deze in de geadapteerde richtlijn van de Europese werkgroep zijn meegenomen. Hersenatrofie gemeten met MRI wordt in verschillende klinische trials als eindpunt meegenomen. Hoewel deze parameter op groepsniveau verschillen tussen behandelarmen laat zien, heeft deze een grote interindividuele variatie en is derhalve (nog) niet bruikbaar als parameter in de dagelijkse klinische praktijk (Tur et al., 2018). Derhalve komt deze uitkomstmaat door de werkgroep in deze richtlijn niet aan bod. In de Europese richtlijn worden belangrijke uitkomstmaten als kwaliteit van leven en effect op cognitieve beperking niet gerapporteerd.
Verschillende fase 2 en 3 studies onderzochten cognitief functioneren met de Paced Auditory Serial Addition Test (PASAT, een onderdeel binnen de MSFC) en de Brief Repeatable Battery of Neuropsychological Tests (BRB-N). Deze studies (Kalb et al., 2018) laten gemengde resultaten zien, wat waarschijnlijk aan een korte follow-up duur en methodologische problemen te wijten is. Een recent MS Society expert panel adviseerde de Symbol Digit Modalities Test (SDMT) in trial designs mee te nemen om hier extra licht op te werpen (Kalb et al., 2018).
Tur et al. (2018) inventariseerden in een recente review de MS specifieke kwaliteit van leven meetinstrumenten die gebruikt zijn in klinische trials zoals de 29-items MS impact scale (MSIS-29), Patient-reported indices in MS (PRIMUS), en MS Quality of Life-54 (MSQoL-54). De MSIS-29 is niet als uitkomstmaat gebruikt in vergelijkende studies tussen ziektemodulerende therapie en placebo in relapsing remitting MS. Kappos et al. (2006) rapporteren zonder data te tonen dat in de BENEFIT studie de MSQoL-54 in essentie niet veranderde tijdens follow-up van mensen met een klinisch geïsoleerd syndroom en geen verschillen liet zien tussen interferon-b-1b en placebogroep. Calabresi et al. (2014) rapporteerden in de Freedoms II studie geen verschil in PRIMUS of MSQoL-54 tussen fingolimod en placebo-behandelde mensen met relapsing remitting MS. Hoewel meer studies naar niet MS-specifieke kwaliteit van leven gekeken hebben, bieden deze data volgens de werkgroep onvoldoende ondersteuning voor uitspraken over het effect van ziektemodulerende therapieën over MS-specifieke kwaliteit van leven.
Starten of watchful waiting:
Er zijn in verschillende onbehandelde MS cohorten subgroepen geïdentificeerd met een gunstig beloop van hun MS. De gerapporteerde proporties relapse-onset patiënten met een EDSS <3-≤4 na 10-15 jaar follow-up varieert van 26-37.6% met een uitschieter naar 80% (Reynders et al., 2017). Deze subgroepen werden vooral gekarakteriseerd door jonge vrouwen met 1 relapse in de eerste 5 jaar van hun ziekte en een EDSS-score < 2 na 5 jaar vervolg. Omgekeerd lijken dus vooral patiënten met actieve ziekte en progressie at-risk voor accumulatie van beperking. Dit wordt ondersteund door longitudinale cohortstudies, waarin de volgende voorspellers van blijvende beperking geïdentificeerd werden: aantal cerebrale T2 laesies bij begin van ziekte, aanwezigheid van gadolinium aankleurende laesies op moment van diagnose, laesies die infratentorieel en in het myelum gelokaliseerd zijn, piramidale verschijnselen. Het gebruik van ziektemodulerende therapie was in deze cohorten geassocieerd met een lagere kans op blijvende invaliditeit (Barkhof et al., 1997; Jokubaitis et al, 2015; Wattjes et al., 2015; Thompson et al., 2017; Tintoré et al., 2015). Hiermee kan behandeling van actieve ziekte in MS de kans op progressieve beperking en progressieve ziekte wellicht verminderen, al is dit nog nooit aangetoond in fase 3 registratie studies. Enige ondersteuning kan gevonden worden in studies naar het natuurlijk beloop van MS. In niet met ziektemodulerende middelen behandelde relapse-onset cohorten wordt een life-time risico van 50% op secundair progressieve MS beschreven (Tedeholm et al., 2015; Tremlett et al., 2008; Eriksson et al., 2003; Debouverie et al., 2009). In behandelde cohorten ligt deze puntschatting lager: mogelijk een life-time risico van 15-30% (Thompson et al., 2018; Lorscheider et al., 2016; Cree et al., 2016; Trojano et al., 2007). De interpretatie van deze cijfers wordt echter bemoeilijkt door de verschillende definities van secundair progressieve MS.
De werkgroep vindt dat, indien er geen aanwijzingen zijn voor actieve ziekte in het afgelopen jaar, initieel een afwachtend beleid tot de behandelmogelijkheid hoort. Behalve waarden en voorkeuren van de patiënt (zie hierboven), evenals mogelijke nadelige effecten van behandeling (zie hieronder) kunnen ook praktische overwegingen zoals een zwangerschapswens <1 jaar een rol spelen. Indien ervoor gekozen wordt om niet te starten met een ziektemodulerend middel dan is het advies om klinisch en radiologisch de patiënt te monitoren, op aanwijzingen voor actieve ziekte bij anamnese en lichamelijk onderzoek, door o.a. na 3-6 en 12 maanden na diagnose een MRI hersenen (in principe zonder contrast) te herhalen en op basis van de nieuw verkregen gegevens nogmaals het gekozen beleid te evalueren (Wattjes et al. 2015).
Vroeg of laat starten
Indien er wel aanwijzingen voor actieve ziekte zijn, hebben verschillende studies onderzocht of het behandelen versus wachten met therapie van invloed op eerder genoemde eindpunten is.
Extensiestudies waren beschikbaar voor 4 trials met interferon-b. Deze studies lieten zien dat vroeg starten ten opzichte van laat starten leidde tot een lagere annualized relapse rate, minder nieuwe of groter wordende T2 laesies bij een follow-up duur van 2 en 4 jaar (Kieseier et al., 2015; PRISMS-4, 2001), en geringer aantal studiedeelnemers met verslechterde invaliditeit na een follow-up duur van 2 en 8 jaar (Kieseier et al., 2015; Kappos et al., 2006). Na een follow-up duur van 16 jaar was er weinig verschil tussen de verschillende behandel groepen in de mate van invalditeit, het aantal studiedeelnemers die een EDSS-score van 6 haalden, of het aantal studiedeelnemers die converteerden naar secundair progressieve MS (Ebers et al., 2010).
De enige extensiestudie over glatirameeracetaat met ongeveer 6 jaar follow-up suggereert voordeel van voortdurende behandeling, omdat de deelnemers die in de studie bleven (ca 80%) persisterend een lage annualized relapse rate hielden en ongeveer 70% geen toename in EDSS score (Johnson et al., 2000).
In de enige extensiestudie over teriflunomide was er na een follow-up duur van 9 jaar weinig verschil tussen vroeg en later starten wat betreft de annualized relapse rate, terwijl het percentage studiedeelnemers met verslechterde invaliditeit lager was in de groep die vroeg startte (O’Connor et al., 2016).
Ten aanzien van dimethylfumaraat rapporteerde één extensiestudie na een follow-up duur van 5 jaar dat er weinig verschil was tussen de groepen die vroeg en later startten wat betreft de annualized relapse rate, terwijl het percentage studiedeelnemers met verslechterde invaliditeit lager was in de groep die vroeg startte (Gold et al., 2016).
M.b.t. fingolimod lieten twee extensiestudies zien dat de groep die vroeg startte een lagere annualized relapse rate had, een groter percentage studiedeelnemers bij wie de invaliditeit niet verslechterde, minder nieuwe T2 laesies, gadolinium aankleurende laesies en percentage veranderingen in het hersenvolume na een follow-up duur van 4.5 jaar (Kappos et al., 2015).
De enige extensie studie over cladribine laat geen duidelijke verschil zien tussen de verschillende behandelgroepen (cladribine gevolgd door cladribine, cladribine gevolgd door placebo, placebo gevolgd door cladribine waarvan 2 jaar in fase drie studieverband gevolgd door 2 jaar extensie fase) ten aanzien van aantal studiedeelnemers die relapse vrij waren of kans op bevestigde ziekteprogressie uitgedrukt als bevestigde EDSS progressie (Giovannoni et al. 2018).
Deze extensiestudies tonen vaak nadelen van uitstel van behandeling met ziektemodulerende therapie bij actieve relapsing remitting MS. Hiernaast wordt in prospectieve cohort studies ook een voordeel van vroege behandeling met ziektemodulerende therapie op het risico op nieuwe aanvallen en progressie van EDSS-score gerapporteerd (Tintore et al., 2015) . Derhalve is de werkgroep van mening dat het uitstellen van therapie bij patiënten met actieve ziekte nadelige gevolgen kan hebben en er dus een indicatie is tot het behandelen van relapsing remitting MS patiënten met actieve ziekte.
Therapietrouw en keuzehulp
Voor een doeltreffende behandeling met een ziektemodulerende therapie is therapietrouw van belang. Burks et al. (2017) rapporteerden in een Amerikaanse studie zowel bij orale als geïnjecteerde ziektemodulerende behandelingen een therapietrouw van 73%, waarbij therapietrouw geassocieerd was met een 42% lagere kans op relapses, en 53% lagere kans op ziekenhuisopnames. In een Spaans cohort (Morillo Verdugo et al., 2019) werd een vergelijkbare therapietrouw van 71% gerapporteerd. Hierbij moet onvoldoende doeltreffendheid als reden van therapieontrouw in de interpretatie van deze data meegenomen worden. Deze orde van grootte sluit aan bij cijfers van de WHO (Sabate, 2003), waarbij een gemiddelde therapietrouw van 80% voor chronische medicatie en 20% voor leefstijladviezen wordt gerapporteerd. Behandelaars van mensen met MS schatten in studies bijwerkingen in als de voornaamste reden om medicatie niet in te nemen (86%), terwijl mensen met MS in verschillende real-world studies een scala aan redenen rapporteren, waaronder vergeten van inname, bijwerkingen, ‘behandelmoeheid’, praktische bezwaren m.b.t. injectie, de perceptie van onvoldoende doeltreffendheid, en niet-realistische verwachtingen m.b.t. positieve effecten op symptomen van MS. Een positief effect van een goede counseling vooraf op therapietrouw lijkt daarom aannemelijk. In een recent review paper concluderen Ben-Zacharia et al. (2018) dat er redelijk bewijs is uit vragenlijsten en cohortstudies dat shared decision making therapietrouw in MS bevordert. Gecontroleerde studies laten wisselende resultaten zien, maar worden in betrouwbaarheid beperkt door gebrek aan standaardisatie in definities en meetinstrumenten. Door mensen met MS wordt shared decision making hoog gewaardeerd. In een Amerikaanse studie (Kasper et al., 2008) geeft 90% van de respondenten met MS de voorkeur aan een shared decision making, terwijl 10% een beslissing exclusief door de behandelaar hoger waardeert. De doeltreffendheid van een keuzehulp in dit proces wordt ondersteund door een Cochrane review (Stacey et al., 2011) waarin 86 gecontroleerde studies naar keuzehulpen vergeleken werden. Door gebruik van een keuzehulp hadden deelnemers meer kennis en een betere risicoperceptie, waren meer betrokken bij het overlegproces, en maakten keuzes die beter bij de individuele voorkeuren pasten. Er was een trend zichtbaar naar een gunstig effect op therapietrouw, maar die was niet significant. Gemiddeld genomen duurde een consult bij het gebruik van de keuzehulp 2.5 minuut langer. De werkgroep vindt hierom een gezamenlijk proces van besluitvorming uitgevoerd door een zorgverlener met goede kennis van zaken aan te bevelen, waarbij de keuzehulp zoals de consultkaart een potentieel doeltreffende toevoeging is.
Balans van gewenste en ongewenste effecten
De kwaliteit van bewijs is voor de meeste uitkomstmaten wat betreft tolereerbaarheid en veiligheid (zeer) laag. Dit geldt in wat mindere mate voor de met effectiviteit samenhangende uitkomstmaten. Dit impliceert dat de netto-baten (gewenste versus ongewenste effecten) van de besproken middelen onzeker zijn.
Samenvattend is MS een chronische ziekte waarbij het moeilijk is om voor de individuele patiënt te voorspellen hoe de ziekte zal gaan verlopen. Alle geregistreerde ziektemodulerende medicijnen voor MS hebben een remmend effect op de ziekte, ofschoon de mate van zekerheid hierover varieert van groot tot gering. Ze kunnen allemaal bijwerkingen hebben. Het effect en de bijwerkingen van de medicatie zijn voor de individuele patiënt moeilijk van tevoren te voorspellen. Per patiënt zal op basis van prognostische factoren, comorbiditeit en de voorkeur van de patiënt een beslissing genomen moeten worden over wel/niet starten met een ziektemodulerend middel.
Kosten en middelen
Nederlandse kosteneffectiviteitsstudies voor starten met medicatie versus afwachten zijn de richtlijnwerkgroep niet bekend. In een recente modelleringsstudie gebaseerd op de Nederlandse situatie (Huygens et al., 2021) werd gerapporteerd dat het behandelen van MS met ziektemodulerende therapieën kosten met zich meebrengt, maar dat er hiermee ook een winst in quality-adjusted life years (QALY’s) voor mensen met MS gerealiseerd wordt (Versteegh et al., 2022). Deze modellen zijn vooral gebaseerd op resultaten van klinische trials, waarin vooral patiënten geïncludeerd zijn met klinisch en/of radiologisch actieve ziekte en daarom ook enkel op deze groep van toepassing.
Aanvaardbaarheid en haalbaarheid
Volgens de werkgroep zijn de aanbevelingen haalbaar en aanvaardbaar voor de belangrijkste betrokkenen omdat zij grotendeels aansluiten bij de huidige klinische praktijk.
Rationale
Deze aanbevelingen leggen de nadruk op het zo veel mogelijk voorkomen van onderbehandeling van relevante ziekteactiviteit, maar ook op het voorkomen van overbehandeling bij afwezige ziekteactiviteit. De rol van shared decision making in deze afwegingen en hieruit volgende behandelbeslissingen wordt benadrukt.
Onderbouwing
Achtergrond
Ziektemodulerende therapie kan worden ingezet om het risico op relapses en ziekteprogressie te verminderen. In onderstaande literatuursamenvatting worden de voordelen en nadelen van de verschillende ziektemodulerende middelen ten opzichte van placebo besproken. In de volgende module (2.4.2) worden de relatieve voordelen en nadelen van deze middelen ten opzichte van elkaar besproken.
Voor de definitie van relapsing remitting MS zie ‘Definities en begrippen’.
Samenvatting literatuur
Beschrijving studies
De meeste trials waren multicenter trials en werden door de industrie gesponsord. De middelen interferon-b/peginterferon-β, glatirameeracetaat, teriflunomide, dimethylfumaraat, fingolimod, natalizumab en cladribine werden vergeleken met een placebo.
Studiepopulaties
De studiegrootte varieerde tussen de 301 en 1430. In 70% van de trials was het aantal deelnemers meer dan 1000. Meer vrouwen dan mannen werden geïncludeerd (% vrouwen varieerde van 68% tot 85%). De gemiddelde leeftijd van de studiedeelnemers varieerde van 35 tot 41 jaar. De gemiddelde initiële EDSS-score bij de start van het onderzoek varieerde van 2.4 tot 2.9. De gemiddelde follow-up duur in de studies varieerde van 48 tot 156 weken. Het percentage studiedeelnemers dat eerder een ziektemodulerend middel had gebruikt varieerde van 7.6 tot 75. De gemiddelde ziekteduur varieerde van 1.3 tot 10.6 jaar. In de helft van de studies was dit circa 5 jaar.[1] De inclusie- en exclusiecriteria die voor de trials werden gehanteerd zijn samengevat in tabel 1.
[1]De cijfers kunnen afwijken van de cijfers die in de Europese richtlijntekst worden vermeld. De reden is dat de Europese richtlijntekst geen onderscheid heeft gemaakt tussen placebo gecontroleerde en head-to-head vergelijkingen.
Tabel 1. Inclusiecriteria gebruikt in 16 RCTs
|
|
Inclusie- en exclusiecriteria |
|
Vollmer et al., 2014
BRAVO Study |
Inclusion criteria -age 18–55 years -diagnosis of RRMS (revised McDonald criteria), and EDSS scores of 0–5.5 -patients must have had at least one relapse in the previous 12 months, two relapses in the previous 24 months, or one relapse in the previous 12–24 months plus one GdE lesion in the previous 12 months.
Exclusion criteria -progressive forms of MS -corticosteroid use for relapses in the previous 30 days -use of experimental drugs, investigational drugs, or immunosuppressive therapy (including mitoxantrone) in the previous 6 months -use of glatiramer acetate in the previous 2 months; and prior use of natalizumab, laquinimod, cladribine, or any IFNb at any time. |
|
Confavreux et al., 2014
TOWER study |
Inclusion criteria -age 18 to 55 years -relapsing multiple sclerosis (RRMS) meeting 2005 McDonald criteria, with or without underlying progression -expanded Disability Status Scale (EDSS) score of 5.5 points or less -at least one relapse in the previous year or at least two relapses in the previous 2 years, and no relapse in the 30 days before randomization.
Exclusion criteria -other relevant diseases -pregnant, breastfeeding, or planned to conceive or father a child during the study -previously or concomitantly received cytokine therapy, interferon-b, or glatiramer acetate within 3 months of randomization, or use of natalizumab or other immunosuppressive agents. |
|
Calabresi et al., 2014a
ADVANCE study |
Inclusion criteria -diagnosis of RRMS as defined by the McDonald criteria -age 18–65 years -a score of 0–5 on the Expanded Disability Status Scale (which ranges from 0 to 10, with higher scores indicating greater disability), and at least two clinically documented relapses in the previous 3 years, with at least one having occurred within the past 12 months.
Exclusion criteria -progressive forms of MS -pre-specified laboratory abnormalities, and -previous treatment with interferon for MS for more than 4 weeks or discontinuation less than 6 months before baseline. |
|
Calabresi et al., 2014b
FREEDOMS II study |
Inclusion criteria -age 18–55 years -diagnosed with RRMS according to the 2005 revised McDonald criteria -one or more confirmed relapses during the preceding year (or two or more confirmed relapses during the previous 2 years) -expanded Disability Status Scale (EDSS) score of 0–5.5, and no relapse or steroid treatment within 30 days before randomization -both treatment naive and previously treated patients were included in the study -previously treated patients if interferon-b or glatiramer acetate therapy was stopped at least 3 months before randomization and natalizumab treatment at least 6 months before randomization.
Exclusion criteria -clinically significant systemic disease or immune suppression (drug-induced or disease-induced) -active infection or macular oedema, diabetes mellitus, or a history of malignancy (apart from successfully treated basal or squamous-cell skin carcinoma), and specific cardiac, pulmonary, or hepatic disorders. |
|
Khan et al., 2013
GALA study |
Inclusion criteria -age 18 to 55 years -a confirmed RRMS diagnosis (according to the revised McDonald criteria16) -expanded Disability Status Scale (EDSS) score of ≤5.5, and were relapse-free for ≥30 days -≥ 1 documented relapse in the 12 months prior to screening, ≥ 2 documented relapses in the 24 months prior to screening, or 1 documented relapse between 12 and 24 months prior to screening with at least 1 documented T1 gadolinium (Gd)-enhancing lesion in an MRI performed within 12 months of screening. Women of childbearing potential were required to practice an acceptable method of birth control.
Exclusion criteria -progressive forms of MS and previous treatment with GA or any other glatiramoid -treatment with immunomodulators including interferon-b and intravenous immunoglobulin, within 2 months of screening -use of immunosuppressive agents, including mitoxantrone and fingolimod, cytotoxic agents, or chronic (>30 days) systemic corticosteroid treatment within 6 months of screening -treatment with cladribine, natalizumab, or any other monoclonal antibody treatment within 2 years of screening -known sensitivity to Gd or mannitol -inability to successfully undergo MRI scanning. |
|
Gold et al., 2012
DEFINE study |
Inclusion criteria -age of 18 to 55 years -a diagnosis of RRMS as defined according to theMcDonald criteria -a baseline score of 0 to 5.0 on the Expanded Disability Status Scale and disease activity as evidenced by at least one clinically documented relapse within 12 months before randomization or a brain magnetic resonance imaging (MRI) scan, obtained within 6 weeks before randomization, that showed at least one gadolinium-enhancing lesion.
Exclusion criteria -progressive forms of MS -another major disease that would preclude participation in a clinical trial -abnormal results on prespecified laboratory tests, or -recent exposure to contraindicated medications. |
|
Fox et al., 2012
CONFIRM trial |
Inclusion criteria -age 18 to 55 years -diagnosis of RRMS (McDonald criteria) -a score of 0 to 5 on the Expanded Disability Status Scale and at least one clinically documented relapse in the previous 12 months or at least one gadolinium-enhancing lesion 0 to 6 weeks before randomization
Exclusion criteria -progressive forms of MS -other clinically significant illness -prespecified laboratory abnormalities, and prior exposure to glatiramer acetate or contraindicated medications. |
|
O’Connor et al., 2011
TEMSO study |
Inclusion criteria -age 18 to 55 years -diagnosis of MS (McDonald criteria), and a relapsing clinical course, with or without progression -a score of 5.5 or lower on the Expanded Disability Status Scale -at least two clinical relapses in the previous 2 years or one relapse during the preceding year, but no relapses in the 60 days before randomization.
Exclusion criteria -other systemic diseases, were pregnant -planned to conceive during the trial period. |
|
Giovannoni et al., 2010
CLARITY study |
Inclusion criteria -a diagnosis of RRMS (according to the McDonald criteria) -lesions consistent with MS on magnetic resonance imaging (MRI) (according to the Fazekas criteria) -at least one relapse within 12 months before study entry -a score of no more than 5.5 on the Kurtzke Expanded Disability Status.
Exclusion criteria -two or more previous disease-modifying therapies failed or immunosuppressive therapy at any time before study entry or cytokine based therapy, intravenous immunoglobulin therapy, or plasmapheresis within 3 months before study entry -abnormal results on hematologic testing (a platelet or neutrophil count below the lower limit of the normal range or a leukocyte count of half the lower limit of the normal range) within 28 days before study entry -a disorder that could compromise immune function (including systemic disease or infection with the human immunodeficiency virus or human T-cell lymphotropic virus), or a relapse within 28 days before study entry. |
|
Kappos et al., 2010
FREEDOMS study |
Inclusion criteria -age of 18 to 55 years -a diagnosis of MS, according to the revised McDonald criteria -a relapsing–remitting course -one or more documented relapses in the previous year or two or more in the previous 2 years, and a score of 0 to 5.5 on the Expanded Disability Status Scale -interferon-b or glatiramer acetate therapy had to have been stopped 3 or more months before randomization.
Exclusion criteria -relapse or corticosteroid treatment within 30 days before randomization -active infection, macular edema, diabetes mellitus, immune suppression (drug- or disease-induced), or clinically significant systemic disease. |
|
Polman et al., 2006
AFFIRM study |
Inclusion criteria -age of 18 to 50 years -diagnosis of RRMS; -a score of 0 to 5.0 on the Expanded Disability Status Scale (EDSS), -undergone magnetic resonance imaging (MRI) showing lesions consistent with MS; and -at least one medically documented relapse within the 12 months before the study began.
Exclusion criteria -primary progressive, secondary progressive, or progressive RRMS -a relapse within 50 days before the administration of the first dose of the study drug, -treatment with cyclophosphamide or mitoxantrone within the previous year, or treatment with interferon-b, glatiramer acetate, cyclosporine, azathioprine, methotrexate, or intravenous immunoglobulin within the previous 6 months, -treatment with interferon-b, glatiramer acetate, or both for more than six months. |
|
PRISMS 1998 |
Inclusion criteria -at least two relapses in the preceding 2 years and had Kurtzke EDSS scores of 0–5.0
Exclusion criteria -any previous systemic treatment with interferons, lymphoid irradiation, or cyclophospamide, or with other immunomodulatory or immunosuppressive treatments in the preceding 12 months. |
|
Jacobs et al., 1996
MSCRG study |
Inclusion criteria -definite MS for at least 1 year, -baseline Expanded Disability Status Score of 1.0 to 3.5 inclusive, -at least 2 documented exacerbations in the prior 3 years, -no exacerbations for at least 2 months at study entry, and age 18 to 55 years Our definition of RRMS included patients with complete remissions (returned to baseline pre-exacerbation disability status) and patients with incomplete remissions (did not return to their baseline pre-exacerbation disability status because of new residua).
Exclusion criteria -prior immunosuppressant or interferon therapy, adrenocorticotropic hormone or corticosteroid treatment within 2 months of study entry, -pregnancy or nursing, -an unwillingness to practice contraception, -the presence of chronic-progressive MS, or any disease other than MS compromising organ function. |
|
Johnson et al., 1995
DEFINE trial |
Inclusion criteria -all patients met the criteria of clinically definite MS or laboratory supported definite MS -male and female patients between the ages of 18 and 45 years were eligible -they were all ambulatory with an EDSS score of 0 through 5.0 -a history of at least two clearly identified and documented relapses in the 2 years prior to entry, -onset of the first relapse at least 1 year before randomization, and a period of neurologic stability and freedom from corticosteroid therapy of at least 30 days prior to entry.
Exclusion criteria -ever received copolymer 1 or previous immunosuppressive therapy with cytotoxic chemotherapy (azathioprine, cyclophosphamide, or cyclosporine) or lymphoid irradiation. -pregnancy or lactation, -insulin-dependent diabetes mellitus, -positive HIV or HTLV-I serology, -evidence of Lyme disease, or -required use of aspirin or chronic nonsteroidal anti-inflammatory drugs during the course of the trial. |
|
IFNB MS Group, 1993
|
Inclusion criteria - ages of 18 and 50 years, - ambulatory with Kurtzke Expanded Disability Status Scale (EDSS) scores of 5.5 or less, and - at least two acute exacerbations during the previous 2 years. - clinically stable for at least 30 days before entry and received no ACTH or prednisone during this period.
Exclusion criteria -Prior treatment with azathioprine or cyclophosphamide. |
*Een studie (Fox et al., 2012) had naast placebo twee behandelarmen met verschillende middelen (dimethylfumaraat en glatirameeracetaat) en telt als 2 RCTs mee.
Interventies
Deze zijn samengevat in tabel 2.
Tabel 2 Interventies
|
Eerste auteur, jaar van publicatie (follow-up duur) |
Geneesmiddel |
Toedieningsweg |
Dosering |
Controle |
|
Vollmer et al., 2014 (follow-up: 152 wk) |
Interferon b-1a |
Intramusculair |
30 µg 1x/week |
placebo |
|
PRISMS 1998 (follow-up: 104 wk) |
Interferon b-1a |
Subcutaan |
44µg 3x/week |
placebo |
|
Jacobs et al., 1996; (follow-up: 52 wk) |
Interferon b-1a |
Intramusculair |
30 µg 1x/week |
placebo |
|
IFNB MS Group, 1993 (follow-up: 156 wk) |
Interferon b-1b |
Subcutaan |
1.6 milj. IE om de dag |
placebo |
|
Calabresi et al., 2014a (follow-up: 48 wk) |
Peginterferon b-1a |
Subcutaan |
125 µg om de 2 weken |
placebo |
|
Khan et al., 2013 (follow-up: 52 wk) |
Glatirameeracetaat |
Subcutaan |
40 mg 3x/week |
placebo |
|
Fox et al., 2012 (follow-up: 104 wk) |
Glatirameeracetaat |
Subcutaan |
20 mg/dag |
placebo |
|
Johnson et al., 1995; (follow-up: 104 wk) |
Glatirameeracetaat |
Subcutaan |
20 mg/dag |
placebo |
|
Confavreux et al., 2014; (follow-up: 104 wk) |
Teriflunomide |
Oraal |
14mg/dag |
placebo |
|
O'Connor et al., 2011 (follow-up: 104 wk) |
Teriflunomide |
oraal |
14mg/dag |
placebo |
|
Fox et al., 2012 (follow-up: 104 wk) |
Dimethylfumaraat |
Oraal |
240 mg/2x daags
|
placebo |
|
Gold et al., 2012 (follow-up: 104 wk) |
Dimethylfumaraat |
Oraal |
240 mg/2x daags
|
placebo |
|
Calabresi et al., 2014b (follow-up: 104 wk) |
Fingolimod |
Oraal |
0.5 mg/dag |
placebo |
|
Kappos et al., 2010 (follow-up: 104 wk) |
Fingolimod |
Oraal |
0.5 mg/dag |
placebo |
|
Polman et al., 2006 (follow-up: 104 wk) |
Natalizumab |
intraveneus |
300 mg om de 4 weken |
placebo |
|
Giovannoni et al, 2010 (follow-up: 96 wk) |
Cladribine |
oraal |
1. Cladribine tabletten (2 kuren van 3.5 mg per kg lichaamsgewicht gevolgd door 2 kuren met placebo in de eerste 48 weken; in de tweede periode van 48 weken 2 kuren) 2. Cladribine (4 kuren van 5.25 mg per kg lichaamsgewicht in de eerste 48 weken; in de tweede periode van 48 weken 2 kuren) |
placebo |
Review 1: Interferon versus placebo[2]
[2] Conform de Europese richtlijn zijn alle soorten interferon samengenomen.
Klinische effectiviteit
1.1 “Annualized Relapse Rate”
Twee RCTs (Calabresi et al., 2014; Vollmer et al.2014) rapporteerden over de annualized relapse rate (ARR). (Peg)Interferon-β verminderde in vergelijking met een placebo de ARR met 0.1 (95% BI: 0.04 tot 0.16 minder).
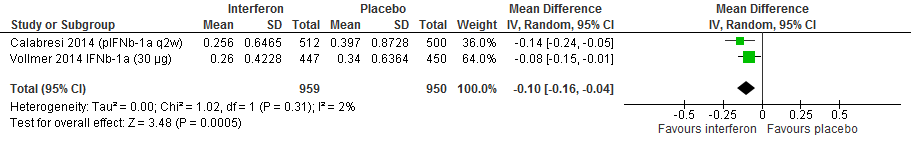
Figuur 1.1 Annualised relapse rate interferon versus placebo (48-104 weeks’ follow-up)
1.2 Proportie vrij van een relapse
Vijf RCTs onderzochten deze uitkomstmaat. Drie RCTs (PRISMS, 1998; Jacobs et al., 1996; INFB MS group, 1993) rapporteerden over de proportie patiënten die na 104 weken vrij van een relapse waren gebleven. Calabresi (2014a) rapporteerde over de proportie patiënten die na 48 weken vrij van een relapse waren gebleven, Vollmer (2014) rapporteerde over de proportie patiënten die na 156 weken vrij van een relapse waren gebleven. De resultaten zijn in tabel 1 samengevat. Gebruik van interferon-β zorgde er in deze studies voor dat meer patiënten vrij bleven van een relapse: ca. 8-13% meer dan de patiënten die een placebo kregen in een tijdsbestek van 48 tot 156 weken.
Tabel 1. Proportie patiënten vrij van een relapse; interferon-β versus placebo
|
Follow-up duur |
Proportie vrij van een relapse |
Relatieve toename proportie vrij van een relapse interferon-β vs. Placebo |
|
|
Interferon-β |
Placebo |
||
|
48 weken |
82.4% |
71.6% |
RR: 1.15 (95% BI: 1.08-1.23) |
|
104 weken |
31.1% |
18.3% |
RR: 1.73 (95% BI: 1.35-2.21) |
|
156 weken |
21.8% |
13.8% |
RR: 1.58 (95% BI: 0.91-2.74) |
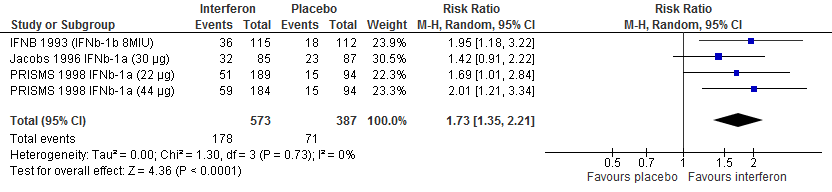
Figuur 1.2 Proportie vrij van een relapse interferon versus placebo (48-104 weeks’ follow-up)
1.3 Toenemende invaliditeit uitgedrukt in Expanded Disability Status Scale (EDSS)
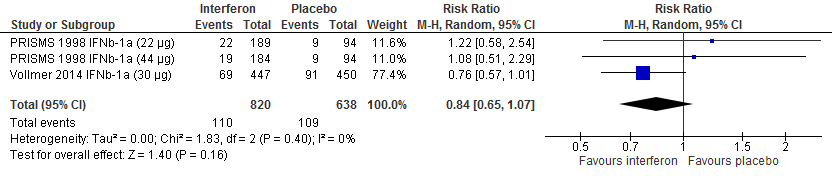
Drie RCTs onderzochten deze uitkomstmaat. Twee RCTs met een follow-up duur van 104 weken (Vollmer et al., 2014; Jacobs et al., 1996) rapporteerden over de proportie patiënten met toenemende invaliditeit die na 6 maanden werd bevestigd. Calabresi (2014a) rapporteerde over de proportie patiënten met toenemende invaliditeit die na 3 maanden werd bevestigd met een follow-up duur van 48 weken, Vollmer (2014) rapporteerde over de proportie patiënten met toenemende invaliditeit met een follow-up duur van 156 weken. De resultaten zijn in tabel 2 samengevat. Gebruik van interferon-β zorgde er in deze studies voor dat bij minder patiënten de invaliditeit toenam: ca. 4-12% minder dan bij patiënten die een placebo kregen.
Tabel 2. Proportie patiënten met toenemende invaliditeit; interferon-β versus placebo
|
Bevestigd na hoeveel maanden |
Proportie patiënten met toenemende invaliditeit |
Reductie risico op toenemende invaliditeit interferon-β vs. Placebo |
|
|
|
Interferon-β |
placebo |
|
|
Bevestigd na 3 maanden |
6.1% |
10% |
RR: 0.61 (95% BI: 0.39-0.93) |
|
Bevestigd na 6 maanden |
10% |
14% |
RR: 0.71 (95% BI: 0.51-0.98) |
|
Niet bevestigd |
26.6% |
39% |
RR: 0.68 (95% BI: 0.47-0.98) |
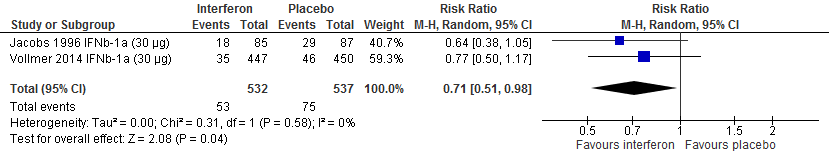
Figuur 1.3 Toenemende invaliditeit uitgedrukt in EDSS interferon versus placebo (48-104 weeks’ follow-up)
Effectiviteit-niet klinisch
1.4 Nieuwe T2-laesies of groter wordende T2-laesies
Twee RCTs onderzochten deze uitkomstmaten. Calabresi (2014a) rapporteerde over het aantal nieuwe T2-laesies of groter wordende T2-laesies. Patiënten die interferon-β kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 7.3 laesies minder (95% BI: 5.75-8.85). PRISMS (1998) rapporteerde over het aantal patiënten die vrij waren van nieuwe of groter wordende T2 laesies. 24.8% van de patiënten die interferon-β kregen, waren vrij van nieuwe of groter wordende T2 laesies. Bij patiënten die een placebo kregen, was dit percentage 8.7. In relatieve termen: RR=2.8 (95% BI: 1.69-4.63).
1.5 Gadolinium aankleurende laesies (gemiddeld aantal; aantal patiënten zonder deze laesies; aantal nieuwe gadolinium aankleurende laesies; cumulatieve aantal gadolinium aankleurende laesies)
Een RCT (Vollmer et al., 2014) rapporteerde over het cumulatieve aantal gadolinium aankleurende laesies. Bij patiënten die interferon-β kregen was het cumulatieve aantal gadolinium aankleurende laesies na 12 en 24 maanden 1.44 minder (95% BI: 0.91-1.97) dan bij patiënten die een placebo kregen.
Tolereerbaarheid en veiligheid
1.6 Studieuitval vanwege bijwerkingen of om willekeurige reden (zoals gerapporteerd in Europese richtlijn)
Drie RCTs (Vollmer et al., 2014; PRISMS, 1998; Jacobs et al., 1996) rapporteerden over studieuitval vanwege bijwerkingen na een follow-up duur van 104 weken, zie figuur 1.4. Twee RCTs (Vollmer et al., 2014; PRISMS, 1998) rapporteerden over studieuitval om willekeurige redenen na een follow-up duur van 104 weken vanwege bijwerkingen, zie figuur 1.5. Een RCT (Calabresi et al., 2014a) rapporteerde over studieuitval vanwege bijwerkingen en om een willekeurige reden na een follow-up duur van 48 weken. Een RCT (IFNB MS group, 1993) rapporteerde over het studieuitval vanwege bijwerkingen en om een willekeurige reden na een follow-up duur van 156 weken. De resultaten zijn in tabel 3 samengevat. Gebruik van interferon-β zorgde er in deze studies voor dat bijna 2 tot 5 keer zo veel patiënten die interferon-β gebruikten stopten met deelname aan de studie vanwege bijwerkingen. Op stoppen om een willekeurige reden lijkt gebruik van interferon-β minder effect te hebben.
Tabel 3. Uitkomsten met betrekking tot studieuitval vanwege bijwerkingen of anderszins (interferon-β vs. placebo)
|
Uitkomstmaten |
Risicoverschil (interferon-β vs. placebo) |
Relatieve risico (RR; 95% BI) |
|
Studieuitval vanwege bijwerkingen |
3.7% |
4.69 (1.8-12.19) |
|
Studieuitval vanwege bijwerkingen |
2.1% |
1.72 (1.04-2.86) |
|
Studieuitval vanwege bijwerkingen |
6.5% |
4.96 (1.11-22.17) |
|
Studieuitval om willekeurige reden |
5.7% |
1.64 (1.15-2.34) |
|
Studieuitval om willekeurige reden |
-3.7% |
0.84 (0.65-1.07) |
|
Studieuitval om willekeurige reden |
-1.0% |
0.95 (0.57-1.59) |
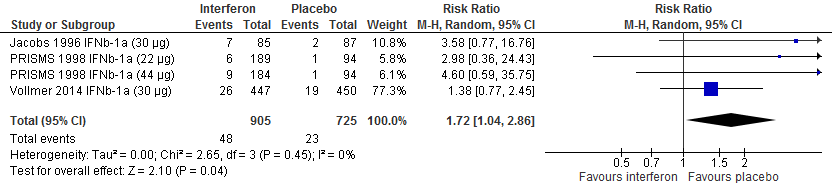
Figuur 1.4 Studieuitval vanwege bijwerkingen interferon versus placebo (104 weeks’ follow-up)
Figuur 1.5 Studieuitval vanwege willekeurige reden interferon versus placebo (104 weeks’ follow-up)
Infecties, mortaliteit, neoplasmata (alle), maligniteiten
Hierover werd niet gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
Kwaliteit van bewijs
In deze studies waren nog niet de relapsing remitting MS patiënten conform 2017 Mc Donald criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. Inclusie van deze patiënten zou de uitkomsten annualized relapse rate, progressie van invaliditeit en vrij van relapses in deze studies wellicht hebben beïnvloed. De werkgroep meent dat het effect op deze uitkomsten niet dusdanig groot is dat er sprake is van een ernstige mate van indirect bewijs. De werkgroep heeft om die reden dan ook niet afgewaardeerd.
1.1 Annualized relapse rate bij een follow-up duur van 48-104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (detectiebias en performance bias).
1.2a Proportie vrij van relapse bij een follow-up duur van 48 weken
De kwaliteit van bewijs is redelijk. Er is afgewaardeerd met één niveau vanwege ernstige risk of bias (detectiebias).
1.2b Proportie vrij van relapse bij een follow-up duur van 104 en 156 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau vanwege ernstige risk of bias (onduidelijk randomisatieprocedure en blindering van randomisatie, en mogelijk selectieve rapportage van uitkomsten).
1.3 Toenemende invaliditeit bij een follow-up duur van 48, 104 en 156 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau vanwege ernstige risk of bias (onduidelijk randomisatieprocedure en blindering van randomisatie, en mogelijk selectieve rapportage van uitkomsten).
1.4a Nieuwe of groter wordende T2 laesies bij een follow-up duur van 48 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (detectiebias).
1.4b Vrij van nieuwe of groter wordende T2 laesies bij een follow-up duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (ernstig risico op selectieve rapportage van uitkomsten) en met één niveau vanwege ernstige onnauwkeurigheid (gering aantal events).
1.5 Cumulatieve gadolinium aankleurende laesie bij een follow-up duur van 12 en 24 maanden
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (performance bias).
1.6a Studieuitval vanwege bijwerkingen bij een follow-up duur van 48 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’).
1.6b Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 en 156 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau vanwege ernstige risk of bias (onduidelijk randomisatieprocedure en blindering van randomisatie, en mogelijk selectieve rapportage van uitkomsten).
1.6c Studieuitval om willekeurige reden bij een follow-up duur van 48 en 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’).
1.6d Studieuitval om willekeurige reden bij een follow-up duur van 156 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau vanwege ernstige risk of bias (onduidelijk randomisatieprocedure en blindering van randomisatie, en mogelijk selectieve rapportage van uitkomsten).
Conclusies interferon versus placebo bij patiënten met relapsing remitting MS
1.1 Annualized relapse rate bij een follow-up duur van 48-104 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-104 weken voor de annualized relapse rate gemiddeld 0.1 relapse (95% BI: 0.04 tot 0.16 minder) per patiënt per jaar minder zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014; Vollmer et al.2014 |
1.2a Proportie vrij van relapse bij een follow-up duur van 48 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 15% (RR: 1.15; 95% BI: 1.08-1.23) en een risicoverschil van 107 patiënten vrij van relapse meer per 1000 (95% BI: 57 meer tot 165 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014 |
1.2b Proportie vrij van relapse bij een follow-up duur van 104 en 156 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 en 156 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 73% (RR: 1.73; 95% BI: 1.35-2.21) respectievelijk 58% (RR: 1.58; 95% BI: 0.91-2.74), en een risicoverschil van 134 patiënten vrij van relapse meer per 1000 (95% BI: 64 meer tot 222 meer) respectievelijk 80 meer per 1000 (95% BI: 12 minder tot 240 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: PRISMS, 1998; Jacobs et al., 1996; INFB MS group, 1993; Vollmer et al., 2014 |
1.3 Toenemende invaliditeit bij een follow-up duur van 48, 104 en 156 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48, 104 en 156 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 39% (RR: 0.61; 95% BI: 0.39-0.93), 29% (RR: 0.71; 95% BI: 0.51-0.98) en 32% (RR: 0.68; 95% BI: 0.47-0.98), en een risicoverschil van 39 patiënten met toenemende invaliditeit minder (95% BI: 7 minder tot 61 minder), 41 patiënten met toenemende invaliditeit minder (95% BI: 3 minder tot 68 minder) en 125 patiënten met toenemende invaliditeit minder per 1000 (95% BI: 8 minder tot 207 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Vollmer et al., 2014; Jacobs et al., 1996; Calabresi et al., 2014 |
1.4a Nieuwe of groter wordende T2 laesies bij een follow-up duur van 48 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor nieuwe of groter wordende T2 laesies gemiddeld 7.3 laesies minder (95% BI: 5.75-8.85) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Calabresi et al., 2014a |
1.4b Vrij van nieuwe of groter wordende T2 laesies bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor de proportie vrij van nieuwe of groter wordende T2 laesies een toename van het relatief effect zien met 180% (RR: 2.8; 95% BI: 1.69-4.63) en een risicoverschil van 157 patiënten vrij van nieuwe of groter wordende T2 laesies meer per 1000 (95% BI: 60 meer tot 316 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: PRISMS, 1998 |
1.5 Cumulatieve aantal gadolinium aankleurende laesies bij een follow-up duur van 12 en 24 maanden
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 12-24 maanden voor het cumulatieve aantal gadolinium aankleurende laesies 1.44 laesies minder (95% BI: -1.97;-0.91) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Vollmer et al., 2014 |
1.6a Studieuitval vanwege bijwerkingen bij een follow-up duur van 48 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor studieuitval vanwege bijwerkingen een bijna vijfvoudige toename van het relatieve risco zien (RR: 4.69; 95% BI: 1.8-12.19) en een risicoverschil van 37 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 8 meer tot 112 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Calabresi et al., 2014a |
1.6b Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 en 156 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 en 156 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 72% (RR: 1.72; 95% BI: 1.04-2.86) respectievelijk een bijna vijfvoudige toename van het relatieve risco (RR: 4.96; 95% BI: 1.11-22.17) en een risicoverschil van 23 patiënten meer met studieuitval vanwege bijwerkingen (95% BI: 1 meer tot 59 meer) respectievelijk 64 patiënten meer per 1000 (95% BI: 2 meer tot 344 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Vollmer et al., 2014; PRISMS, 1998; Jacobs et al., 1996; IFNB MS group, 1993 |
1.6c Studieuitval om willekeurige reden bij een follow-up duur van 48 en 104 weken
|
Redelijk
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 en 104 weken voor studieuitval om willekeurige reden een toename van het relatieve risico zien met 64% (RR:1.64; 95% BI: 1.15-2.34) respectievelijk een afname van het relatieve risico met 16% (RR: 0.84; 95% BI: 0.65-1.07) en een risicoverschil van 56 patiënten meer met studieuitval om willekeurige reden (95% BI: 13 meer tot 118 meer) respectievelijk 27 patiënten minder per 1000 (95% BI: 60 minder tot 12 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Vollmer et al., 2014; PRISMS, 1998; Jacobs et al., 1996; IFNB MS group, 1993 |
1.6d Studieuitval om willekeurige reden bij een follow-up duur van 156 weken
|
Laag
GRADE |
Gebruik van interferon-β door patiënten met relapsing remitting MS laat bij een follow-up duur van 156 weken voor studieuitval om willekeurige reden een afname van het relatieve risico zien met 5% (RR: 0.95; 95% BI: 0.57-1.59) en een risicoverschil van 10 patiënten minder met studieuitval om willekeurige reden per 1000 (95% BI: 84 minder tot 115 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: IFNB MS group, 1993 |
Review 2: Glatirameeracetaat versus placebo
Klinische effectiviteit
2.1“Annualized Relapse Rate”
Twee RCTs (Fox et al., 2012; Khan et al., 2013) rapporteerden over de annualized relapse rate (ARR). Glatirameeracetaat verminderde in vergelijking met een placebo de ARR met 0.14 (95% BI: -0.21;-0.06), zie figuur 2.1.
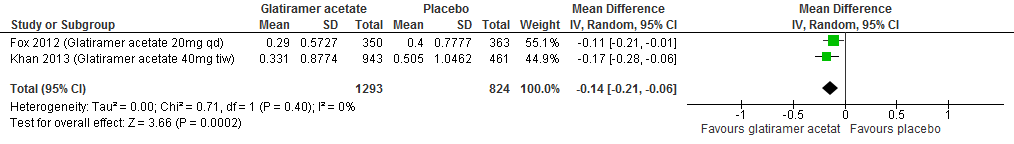
Figuur 2.1 Annualised relapse rate glatirameer versus placebo (52-96 weeks’ follow-up)
2.2 Proportie vrij van een relapse
Drie RCTs (Johnson et al., 1995; Fox et al., 2012; Khan et al., 2013) rapporteerden over de proportie patiënten die na 52-104 weken vrij van een relapse waren gebleven. Patiënten die naar glatirameeracetaat waren gerandomiseerd bleven in deze studies in een tijdsbestek van 52 tot 104 weken meer vrij van een relapse: de relatieve toename was 17% (RR: 1.17; 95% BI: 1.10-1.24), het risicoverschil 9.8% (95% BI: 5.8 meer tot 13.9 meer) vergeleken met patiënten met een placebo, zie figuur 2.2.
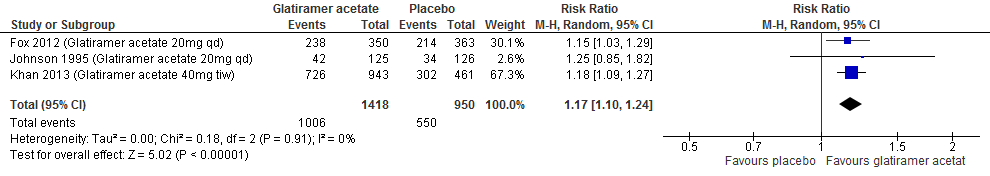
Figuur 2.2 Proportie vrij van een relapse glatirameer versus placebo (52-104 weeks’ follow-up)
2.3 Toenemende invaliditeit uitgedrukt in Expanded Disability Status Scale (EDSS)
Twee RCTs (Johnson et al., 1995; Fox et al., 2012) rapporteerden over de proportie patiënten met toenemende invaliditeit na een follow-up duur van 96-104 weken, zie figuur 2.3. Johnson (1995) rapporteerde over de proportie patiënten met toenemende invaliditeit na een follow-up duur van 128 weken. De resultaten zijn in tabel 4 samengevat. Glatirameeracetaat zorgde er in deze studies voor dat bij minder patiënten de invaliditeit toenam: ca. 3-6% minder dan bij patiënten die een placebo kregen.
Tabel 4. Proportie patiënten met toenemende invaliditeit; glatirameeracetaat versus placebo
|
Follow-up duur |
Proportie patiënten met toenemende invaliditeit |
Reductie risico op toenemende invaliditeit glatirameeracetaat vs. placebo |
|
|
Glatirameeracetaat |
placebo |
||
|
96-104 weken |
17.3% |
20.0% |
RR: 0.86 (95% BI: 0.66-1.11) |
|
128 weken |
23.2% |
29.4% |
RR: 0.79 (95% BI: 0.52-1.20) |
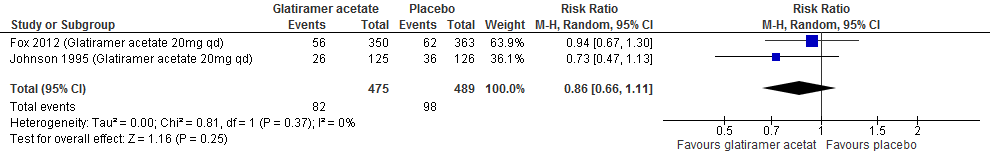
Figuur 2.3 Toenemende invaliditeit glatirameeracetaat versus placebo (96-104 weeks’ follow-up)
Effectiviteit-niet klinisch
2.4 Nieuwe T2-laesies of groter wordende T2-laesies
Een RCT (Fox et al., 2012) rapporteerde over het aantal nieuwe T2-laesies of groter wordende T2-laesies. Patiënten die glatirameeracetaat kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 9.4 laesies minder (95% BI: -14.26;- 4.54) bij een follow-up duur van 96 weken.
2.5 Gadolinium aankleurende laesies (gemiddeld aantal; aantal patiënten zonder deze laesies; aantal nieuwe gadolinium aankleurende laesies; cumulatieve aantal gadolinium aankleurende laesies)
Een RCT (Fox et al., 2012) rapporteerde over het gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 96 weken. Patiënten die glatirameeracetaat kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 1.3 gadolinium aankleurende laesies minder (95% BI: -2.26; -0.34).
Een RCT (Khan et al., 2013) rapporteerde over het cumulatieve aantal gadolinium aankleurende laesies bij een follow-up duur van 52 weken. Patiënten die glatirameeracetaat kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 0.73 laesies minder (95% BI: -1.15;-0.31). Over het aantal patiënten zonder gadolinium aankleurende laesies en het aantal nieuwe gadolinium aankleurende laesies werd niet gerapporteerd in de Europese richtlijn en bijlagen.
Tolereerbaarheid en veiligheid
2.6 Studieuitval vanwege bijwerkingen of om willekeurige reden (zoals gerapporteerd in Europese richtlijn)
Drie RCTs onderzochten deze uitkomstmaten. Twee RCTs (Fox et al., 2012; Johnson et al., 1995) rapporteerden over studieuitval vanwege bijwerkingen en over studieuitval om een willekeurige reden na een follow-up duur van 96-104 weken. Kahn (2013) rapporteerde over studieuitval vanwege bijwerkingen of om een willekeurige reden na een follow-up duur van 52 weken. De resultaten zijn in tabel 5 samengevat. Een eenduidig beeld levert dit niet op. De studies die 2 jaar duurden laten wel zien dat glatirameeracetaat het risico op stoppen met een factor 2 à 3 verhoogt.
Tabel 5. Uitkomsten met betrekking tot studieuitval vanwege bijwerkingen of anderszins (glatirameeracetaat vs. placebo)
|
Uitkomstmaten |
Risicoverschil |
Relatieve risico (RR; 95% BI) |
|
Studieuitval vanwege bijwerkingen |
2.0% |
2.63 (1.17-5.90) |
|
Studieuitval vanwege bijwerkingen |
-0.2% |
0.92 (0.39-2.13) |
|
Studieuitval om willekeurige reden |
-3.0% |
0.86 (0.66-1.11) |
|
Studieuitval om willekeurige reden |
2.2% |
1.32 (0.89-1.97) |
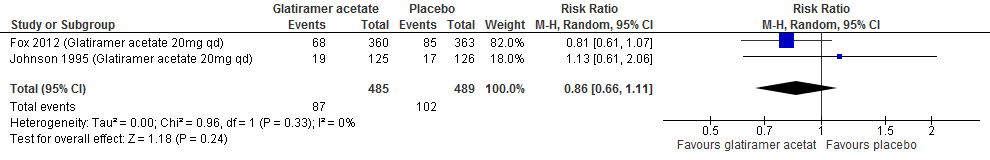
Figuur 2.4 Studieuitval vanwege willekeurige redenen glatirameeracetaat versus placebo (96-104 weeks’ follow-up)
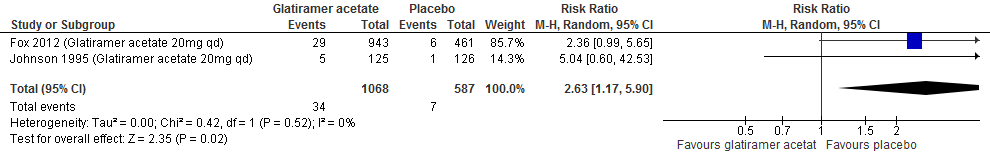
Figuur 2.5 Studieuitval vanwege bijwerkingen glatirameeracetaat versus placebo (96-104 weeks’ follow-up)
2.7 Infecties, mortaliteit, neoplasmata (alle), maligniteiten
Hierover werd niet gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen. Voor aanvullende infromatie zie SmPC van betreffende medicijn en tabel in hoofdstuk 2.3.2.
Kwaliteit van bewijs
In deze studies waren nog niet de relapsing remitting MS patiënten conform 2017 Mc Donald criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. Inclusie van deze patiënten zou de uitkomsten annualized relapse rate, progressie van invaliditeit en vrij van relapses in deze studies wellicht hebben beïnvloed. De werkgroep meent dat het effect op deze uitkomsten niet dusdanig groot is dat er sprake is van een ernstige mate van indirect bewijs. De werkgroep heeft om die reden dan ook niet afgewaardeerd.
2.1 Annualized relapse rate bij een follow-up duur van 52-96 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (performance bias, redenen voor uitval verschillen tussen behandel- en placebogroep).
2.2 Proportie vrij van relapse bij een follow-up duur van 52-96 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (performance bias, redenen voor uitval verschillen tussen behandel- en placebogroep, en selectieve rapportage van uitkomsten).
2.3 Toenemende invaliditeit bij een follow-up duur van 96-104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau vanwege ernstige risk of bias (performance bias, redenen voor uitval verschillen tussen behandel- en placebogroep, en selectieve rapportage van uitkomsten).
2.4 Nieuwe of groter wordende T2 laesies bij een follow-up duur van 96 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal events) en met één niveau vanwege ernstige risk of bias (performance bias, redenen voor uitval verschillen tussen behandel- en placebogroep).
2.5a Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 96 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal events) en met één niveau vanwege ernstige risk of bias (performance bias, redenen voor uitval verschillen tussen behandel- en placebogroep).
2.5b Cumulatieve aantal gadolinium aankleurende laesies bij een follow-up duur van 52 weken
De kwaliteit van bewijs is hoog.
2.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 52 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’).
2.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 96-104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau vanwege ernstige risk of bias (performance bias, redenen voor uitval verschillen tussen behandel- en placebogroep, en selectieve rapportage van uitkomsten).
2.8 Studieuitval om willekeurige reden bij een follow-up duur van 52 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’).
2.9 Studieuitval om willekeurige reden bij een follow-up duur van 96-104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau vanwege ernstige risk of bias (performance bias, redenen voor uitval verschillen tussen behandel- en placebogroep, en selectieve rapportage van uitkomsten).
Conclusies glatirameeracetaat versus placebo
2.1 Annualized relapse rate bij een follow-up duur van 52-96 weken
|
Redelijk
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 52-96 weken voor de annualized relapse rate gemiddeld 0.14 relapses minder (95% BI: -0.21;-0.06) per patiënt per jaar zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Fox et al., 2012; Khan et al., 2013 |
2.2 Proportie vrij van relapse bij een follow-up duur van 52-96 weken
|
Redelijk
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 52-96 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 17% (RR: 1.17; 95% BI: 1.10-1.24) en een risicoverschil van 98 patiënten vrij van relapse meer per 1000 (95% BI: 58 meer tot 139 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Johnson et al., 1995; Fox et al., 2012; Khan et al., 2013 |
2.3 Toenemende invaliditeit bij een follow-up duur van 96-104 en 128 weken
|
Laag
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 96-104 en 128 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 14% (RR: 0.86; 95% BI: 0.66-1.11) respectievelijk 21% (RR: 0.79; 95% BI: 0.52-1.20) en een risicoverschil van 28 patiënten minder (95% BI: 68 minder tot 22 meer) respectievelijk 62 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 141 minder tot 59 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Johnson et al., 1995; Fox et al., 2012 |
2.4 Nieuwe of groter wordende T2 laesies bij een follow-up duur van 96 weken
|
Laag
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor nieuwe of groter wordende T2 laesies gemiddeld 9.4 laesies minder (95% BI: -14.26;-4.54) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Fox et al., 2012 |
2.5a Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 96 weken
|
Laag
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor het gemiddeld aantal gadolinium aankleurende laesies gemiddeld 1.3 laesies minder (95% BI: -2.26; -0.34) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Fox et al., 2012 |
2.5b Cumulatieve aantal gadolinium aankleurende laesies bij een follow-up duur van 52 weken
|
Hoog
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken voor het cumulatieve aantal gadolinium aankleurende laesies gemiddeld 0.73 laesies minder (95% BI: -1.15; -0.31) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Khan et al., 2013 |
2.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken een vermindering van het relatieve risico zien met 8% (RR: 0.92; 95% BI: 0.39-2.13) en een risicoverschil van 2 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 18 minder tot 34 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Fox et al., 2012; Johnson et al., 1995 |
2.7 Studieuitval vanwege bijwerkingen bij een follow-up duur van 96-104 weken
|
Laag
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 96-104 weken bijna een verdrievouding van het relatieve risico zien (RR: 2.63; 95% BI: 1.17-5.90) en een risicoverschil van 19 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 2 meer tot 58 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Fox et al., 2012; Johnson et al., 1995 |
2.8 Studieuitval om een willekeurige reden bij een follow-up duur van 52 weken
|
Redelijk
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 weken een toename van het relatieve risico zien met 32% (RR: 1.32; 95% BI: 0.89-1.97) en een risicoverschil van 22 patiënten meer met studieuitval om een willekeurige reden per 1000 (95% BI: 7 minder tot 65 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Kahn et al., 2013 |
2.9 Studieuitval om een willekeurige reden bij een follow-up duur van 96-104 weken
|
Laag
GRADE |
Gebruik van glatirameeracetaat door patiënten met relapsing remitting MS laat bij een follow-up duur van 96-104 weken een vermindering van het relatieve risico zien met 14% (RR: 0.86; 95% BI: 0.66-1.11) en een risicoverschil van 28 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 68 minder tot 22 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Kahn et al., 2013 |
Review 3: Teriflunomide versus placebo
Klinische effectiviteit
3.1 “Annualized Relapse Rate”
Twee RCTs (Confavreux et al., 2014; O’Connor et al., 2011) rapporteerden over de annualized relapse rate (ARR). Teriflunomide verminderde in vergelijking met een placebo de ARR met 0.18 (95% BI: 0.11-0.24) ten opzichte van placebo bij een follow-up duur van 48 tot 108 weken, zie figuur 3.1.
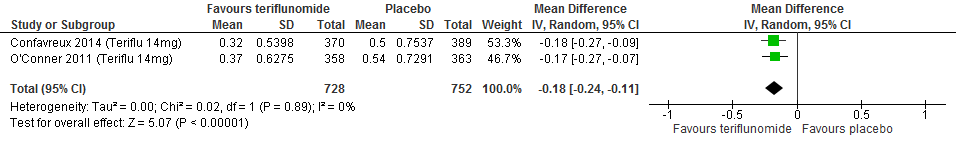
Figuur 3.1 Annualised relapse rate teriflunomide versus placebo (48-108 weeks’ follow-up)
3.2 Proportie vrij van een relaps
Twee RCTs (Confavreux et al., 2014; O’Connor et al., 2011) rapporteerden over de proportie patiënten die na 48-108 weken vrij van een relapse waren gebleven. Patiënten die naar teriflunomide waren gerandomiseerd bleven in deze studies in een tijdsbestek van 48 tot 108 weken meer vrij van een relapse: ca. 13% verschil met patiënten met een placebo (RR: 1.25; 95% BI: 1.16-1.36).
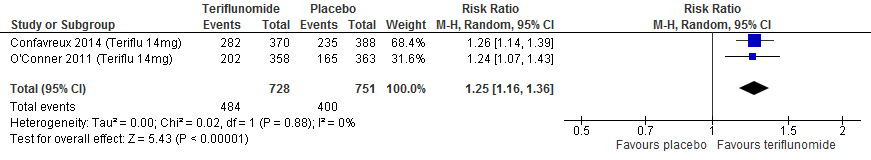
Figuur 3.2 Proportie vrij van een relapse teriflunomide versus placebo (48-108 weeks’ follow-up)
3.3 Toenemende invaliditeit uitgedrukt in Expanded Disability Status Scale (EDSS)
Twee RCTs (Confavreux et al., 2014; O’Connor et al., 2011) rapporteerden over de proportie patiënten met toenemende invaliditeit na een follow-up duur van 104-108 weken. Teriflunomide zorgde er in deze studies voor dat bij minder patiënten de invaliditeit toenam: ca. 5-6% minder dan bij patiënten die een placebo kregen (RR: 0.76; 95% BI: 0.62-0.93).
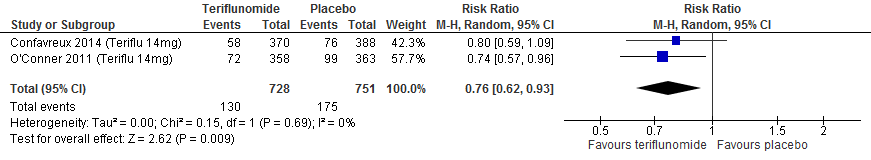
Figuur 3.3 Toenemende invaliditeit uitgedrukt in EDSS teriflunomide versus placebo (48-108 weeks’ follow-up)
Effectiviteit-niet klinisch
3.4 Nieuwe T2-laesies of groter wordende T2-laesies
Hierover werd niet gerapporteerd in de Europese richtlijntekst en bijlagen.
3.5 Gadolinium aankleurende laesies (gemiddeld aantal; aantal patiënten zonder deze laesies; aantal nieuwe gadolinium aankleurende laesies; cumulatieve aantal gadolinium aankleurende laesies)
Een RCT (O’Connor et al., 2011) rapporteerde over het gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 108 weken. Patiënten die teriflunomide kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 1.07 gadolinium aankleurende laesies minder (95% BI: 0.74-1.40) bij een follow-up duur van 108 weken. In de Europese richtlijntekst en bijlagen werd niet gerapporteerd over het aantal patiënten zonder gadolinium aankleurende laesies, aantal nieuwe gadolinium aankleurende laesies en het cumulatieve aantal gadolinium aankleurende laesies.
Tolereerbaarheid en veiligheid
3.6 Studieuitval vanwege bijwerkingen of om willekeurige reden (zoals gerapporteerd in Europese richtlijn)
Twee RCTs (Confavreux et al., 2014; O’Connor et al., 2011) rapporteerden over studieuitval vanwege bijwerkingen en over studieuitval om een willekeurige reden na een follow-up duur van 48-108 weken. De resultaten zijn in tabel 6 samengevat. Volgens deze studies verhoogt teriflunomide het risico op stoppen vanwege bijwerkingen met een factor 1.5 à 2, en heeft teriflunomide geen effect op stoppen om een willekeurige reden.
Tabel 6. Uitkomsten met betrekking tot studieuitval vanwege bijwerkingen of anderszins (teriflunomide vs. placebo)
|
Uitkomstmaten |
Risicoverschil |
Relatieve risico (RR; 95% BI) |
|
Studieuitval vanwege bijwerkingen |
5.9% |
1.77 (1.02-3.07) |
|
Studieuitval om willekeurige reden |
-0.2% |
1.0 (0.86-1.16) |
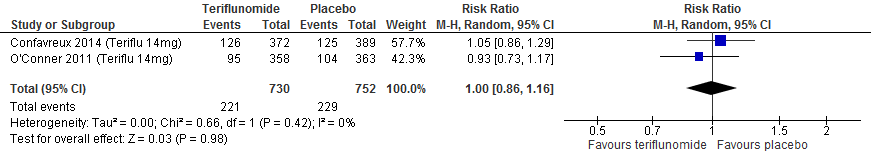
Figuur 3.4 Studieuitval om willekeurige redenen teriflunomide versus placebo (48-108 weeks’ follow-up)
3.7 Infecties
Twee RCTs (Confavreux et al., 2014; O’Connor et al., 2011) rapporteerden over infecties bij een follow-up duur van 48-108 weken. Patiënten die teriflunomide kregen hadden in vergelijking met placebo minder infecties: een risicoverschil van 5.8% (RR 0.85; 95% BI: 0.75-0.98).
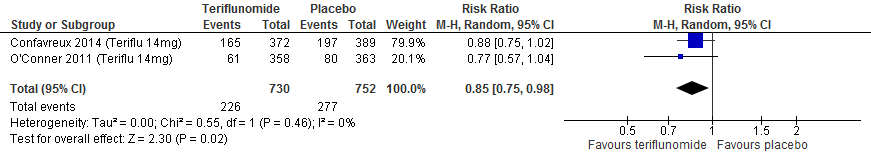
Figuur 3.5 Risico op infecties teriflunomide versus placebo (48-108 weeks’ follow-up)
3.8 Mortaliteit
Een RCT (Confavreux et al., 2014) rapporteerde over mortaliteit bij een follow-up duur van 48 weken. Teriflunomide had in vergelijking met placebo geen effect op de mortaliteit (RR 1.0; 95% BI: 0.99-1.01).
3.9 Neoplasmata (alle), maligniteiten
Twee RCTs (Confavreux et al., 2014; O’Connor et al., 2011) rapporteerden over neoplasmata bij een follow-up duur van 48-108 weken. Teriflunomide had in vergelijking met placebo geen effect op het optreden van neoplasmata volgens deze studies (RR 1.0; 95% BI: 0.99-1.01).
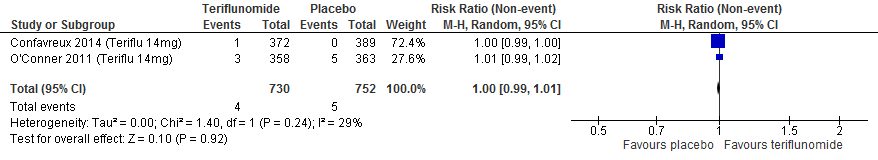
Figuur 3.6 Risico op kanker teriflunomide versus placebo (48-108 weeks’ follow-up)
In de Europese richtlijn en bijlagen wordt niet separaat over maligniteiten gerapporteerd.
Kwaliteit van bewijs
In deze studies waren nog niet de relapsing remitting MS patiënten conform huidige diagnostische criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. Inclusie van deze patiënten zou de uitkomsten annualized relapse rate, progressie van invaliditeit en vrij van relapses in deze studies wellicht hebben beïnvloed. De Nederlandse richtlijnwerkgroep meent dat het effect op deze uitkomsten niet dusdanig groot is dat er sprake is van een ernstige mate van indirect bewijs. De Nederlandse werkgroep heeft om die reden dan ook niet afgewaardeerd.
3.1 “Annualized Relapse Rate” bij een follow-up duur van 48-108 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
3.2 Proportie vrij van een relapse bij een follow-up duur van 48-108 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
3.3 Toenemende invaliditeit bij een follow-up duur van 104-108 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
3.4 Gadolinium aankleurende laesies (gemiddeld aantal) bij een follow-up duur van 104-108 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (onduidelijke blindering van randomisatie).
3.5 Studieuitval om een willekeurige reden bij een follow-up duur van 48-108 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
3.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48-108 weken
De kwaliteit van bewijs is zeer laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie), met één niveau vanwege inconsistente uitkomsten, en met één niveau vanwege ernstige onnauwkeurigheid (geringe aantal ‘events’).
3.7 Infecties bij een follow-up duur van 48-108 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
3.8 Mortaliteit bij een follow-up duur van 48-108 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd voor ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie) en met één niveau voor ernstige onnauwkeurigheid (gering aantal ‘events’).
3.9 Neoplasmata (alle) bij een follow-up duur van minimaal 60 maanden
De kwaliteit van bewijs is zeer laag. Er is met één niveau afgewaardeerd voor ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie), met één niveau voor ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau voor indirect bewijs: de follow-up duur van 48-108 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk acht voor een beoordeling van het risico op het optreden van maligniteiten.
Conclusies
3.1 Annualized relapse rate bij een follow-up duur van 48-108 weken
|
Redelijk
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-108 weken voor de annualized relapse rate gemiddeld 0.18 relapses minder (95% BI: -0.24;-0.11) per patiënt per jaar zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Confavreux et al., 2014; O’Connor et al., 2011 |
3.2 Proportie vrij van relapse bij een follow-up duur van 48-108 weken
|
Redelijk
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-104 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 25% (RR: 1.25; 95% BI: 1.16-1.36) en een risicoverschil van 133 patiënten vrij van relapse meer per 1000 (95% BI: 85 meer tot 192 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Confavreux et al., 2014; O’Connor et al., 2011 |
3.3 Toenemende invaliditeit bij een follow-up duur van 104-108 weken
|
Redelijk
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 104-108 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 24% (RR: 0.76; 95% BI: 0.62-0.93) en een risicoverschil van 56 patiënten minder met toenemende invaliditeit (95% BI: 16minder tot 89 minder) respectievelijk 56 patiënten minder met toenemende invaliditeit per 1000 (95% BI: 16 minder tot 89 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Confavreux et al., 2014; O’Connor et al., 2011 |
3.4 Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor het gemiddelde aantal gadolinium aankleurende laesies gemiddeld 1.07 laesies minder zien (95% BI: -1.40;-0.74) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: O’Connor et al., 2011 |
3.5 Studieuitval om een willekeurige reden bij een follow-up duur van 48-108 weken
|
Redelijk
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-108 weken voor studieuitval om een willekeurige reden geen verschil zien (RR: 1.0; 95% BI: 0.86-1.16) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Confavreux et al., 2014; O’Connor et al., 2011 |
3.6 Studieuitval vanwege bijwerkingen bij een follow-up duur van 48-108 weken
|
Zeer laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-108 weken voor studieuitval vanwege bijwerkingen bijna een verdubbeling van het relatieve risico zien (RR: 1.77; 95% BI: 1.02; 3.07) en een risicoverschil van 56 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 1 meer tot 151 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect verschilt waarschijnlijk aanzienlijk van het geschatte effect).
Bronnen: Confavreux et al., 2014; O’Connor et al., 2011 |
3.7 Infecties bij een follow-up duur van 48-108 weken
|
Redelijk
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-108 weken voor infecties een vermindering van het relatieve risico met 15% zien (RR: 0.85; 95% BI: 0.75;0.98) en een risicoverschil van 55 patiënten minder met infecties per 1000 (95% BI: 7 minder tot 92 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Confavreux et al., 2014; O’Connor et al., 2011 |
3.8 Mortaliteit bij een follow-up duur van 48 weken
|
Laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48 weken voor mortaliteit geen verschil zien (RR: 1.0; 95% BI: 0.99-1.01) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Confavreux et al., 2014 |
3.9 Neoplasmata bij een follow-up duur van 48-108 weken
|
Zeer laag
GRADE |
Gebruik van teriflunomide door patiënten met relapsing remitting MS laat bij een follow-up duur van 48-108 weken voor neoplasmata geen verschil zien (RR: 1.0; 95% BI: 0.99-1.01) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect verschilt waarschijnlijk aanzienlijk van het geschatte effect).
Bronnen: Confavreux et al., 2014; O’Connor et al., 2011 |
Review 4: dimethylfumaraat versus placebo
Klinische effectiviteit
4.1 “Annualized Relapse Rate”
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over de annualized relapse rate (ARR). Dimethylfumaraat verminderde in vergelijking met een placebo de ARR met 0.19 (95% BI: 0.13-0.25) ten opzichte van placebo bij een follow-up duur van 104 weken, zie figuur 4.1.
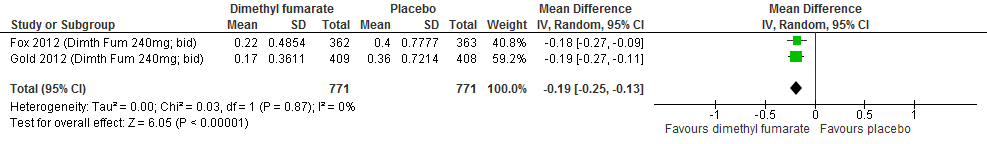
Figuur 4.1 Annualized relapse rate dimethylfumaraat versus placebo (104 weeks’ follow-up)
4.2 Proportie vrij van een relapse
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over de proportie patiënten die na 104 weken vrij van een relapse waren gebleven. Patiënten die naar dimethylfumaraat waren gerandomiseerd bleven in deze studies in een tijdsbestek van 104 weken meer vrij van een relapse: ca. 13.7% verschil met patiënten met een placebo (RR: 1.28; 95% BI: 1.14-1.43), zie figuur 4.2.
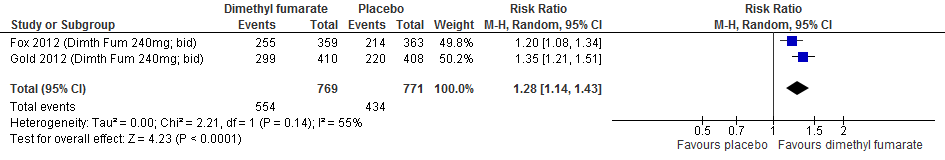
Figuur 4.2 Proportie vrij van relapse dimethylfumaraat versus placebo (104 weeks’ follow-up)
4.3 Toenemende invaliditeit uitgedrukt in Expanded Disability Status Scale (EDSS)
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over de proportie patiënten met toenemende invaliditeit na een follow-up duur van 104 weken. Dimethylfumaraat zorgde er in deze studies voor dat bij minder patiënten de invaliditeit toenam: ca. 7-8% minder dan bij patiënten die een placebo kregen (RR: 0.66; 95% BI: 0.51-0.85), zie figuur 4.3.
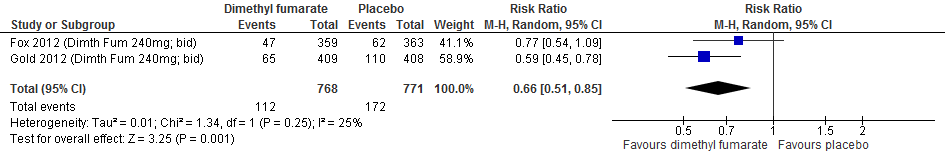
Figuur 4.3 Toenemende invaliditeit uitgedrukt in EDSS dimethylfumaraat versus placebo (104 weeks’ follow-up)
Effectiviteit-niet klinisch
4.4 Nieuwe T2-laesies of groter wordende T2-laesies
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over het gemiddelde aantal nieuwe of groter wordende T2 laesies bij een follow-up duur van 104 weken. Dimethylfumaraat verminderde dit aantal met 13.36 (95% BI: 10.09-16.63) ten opzichte van placebo.
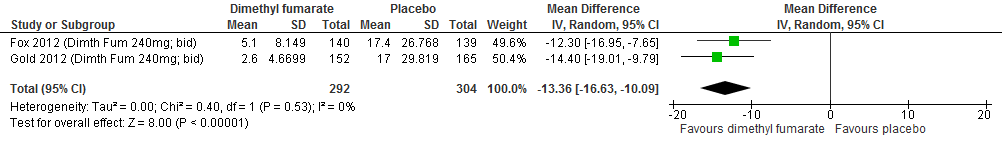
Figuur 4.4 Nieuwe T2 laesies of groter wordende T2 laesies dimethylfumaraat versus placebo (104 weeks’ follow-up)
4.5 Gadolinium aankleurende laesies (gemiddeld aantal; aantal patiënten zonder deze laesies; aantal nieuwe gadolinium aankleurende laesies; cumulatieve aantal gadolinium aankleurende laesies)
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over het gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 104 weken. Patiënten die dimethylfumaraat kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 1.64 gadolinium aankleurende laesies minder (95% BI: 1.1-2.17) bij een follow-up duur van 104 weken, zie figuur 4.5.
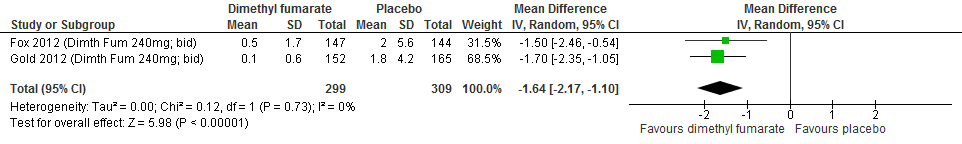
Figuur 4.5 Gadolonium aankleurende laesies dimethylfumaraat versus placebo (104 weeks’ follow-up)
In de Europese richtlijntekst en bijlagen werd niet gerapporteerd over het aantal patiënten zonder gadolinium aankleurende laesies, aantal nieuwe gadolinium aankleurende laesies en het cumulatieve aantal gadolinium aankleurende laesies.
Tolereerbaarheid en veiligheid
4.6 Studieuitval vanwege bijwerkingen of om willekeurige reden (zoals gerapporteerd in Europese richtlijn)
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over studieuitval vanwege bijwerkingen en over studieuitval om een willekeurige reden na een follow-up duur van 104 weken, zie figuur 4.6 en 4.7. De resultaten zijn in tabel 7 samengevat. Volgens deze studies heeft dimethylfumaraat geen effect op stoppen vanwege bijwerkingen en om een willekeurige reden.
Tabel 7. Uitkomsten met betrekking tot studieuitval vanwege bijwerkingen of anderszins (dimethylfumaraat vs. placebo)
|
Uitkomstmaten |
Risicoverschil |
Relatieve risico (RR; 95% BI) |
|
Studieuitval vanwege bijwerkingen |
-0.5% |
0.97 (0.78-1.21) |
|
Studieuitval om willekeurige reden |
-0.7% |
0.97 (0.80-1.16) |
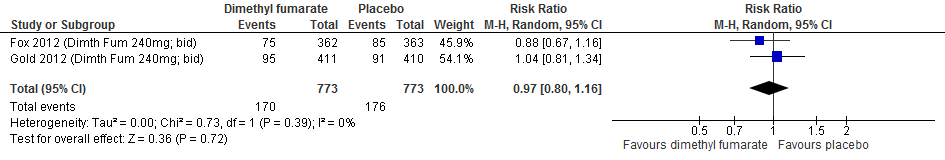
Figuur 4.6 Studieuitval vanwege willekeurige redenen dimethylfumaraat versus placebo (104 weeks’ follow-up)
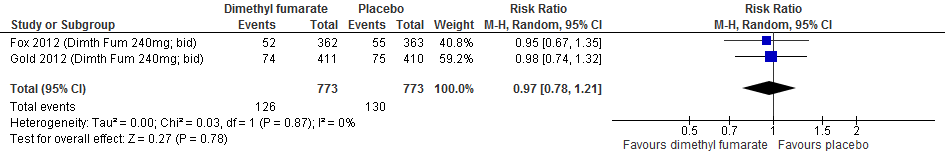
Figuur 4.7 Studieuitval vanwege bijwerkingen dimethylfumaraat versus placebo (104 weeks’ follow-up)
4.7 Infecties
Eén RCT (Fox et al., 2012) rapporteerde volgens de Europese richtlijn over (ernstige) infecties. Volgens Fox et al. verhoogde dimethylfumaraat het risico op ernstige infecties: een risicoverschil van 3.3% (RR: 1.42; 95% BI: 0.55-3.7). Ook het risico op infecties was enigszins verhoogd: een risicoverschil van 0.7% (RR: 1.16; 95% BI: 0.88-1.51).
4.8 Mortaliteit
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over mortaliteit bij een follow-up duur van 104 weken. Dimethylfumaraat had in vergelijking met placebo geen effect op de mortaliteit (RR 1.0; 95% BI: 0.99-1.01).
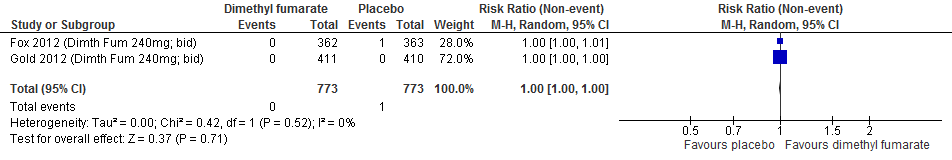
Figuur 4.8 Mortaliteit dimethylfumaraat versus placebo (104 weeks’ follow-up)
4.9 Neoplasmata (alle), maligniteiten
Twee RCTs (Fox et al., 2012; Gold et al., 2012) rapporteerden over neoplasmata bij een follow-up duur van 104 weken. Dimethylfumaraat had in vergelijking met placebo geen effect op het optreden van neoplasmata (RR 1.0; 95% BI: 0.99-1.01).
In de Europese richtlijn en bijlagen wordt niet separaat over maligniteiten gerapporteerd.
Kwaliteit van bewijs
In deze studies waren nog niet de relapsing remitting MS patiënten conform huidige diagnostische criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. Inclusie van deze patiënten zou de uitkomsten annualized relapse rate, progressie van invaliditeit en vrij van relapses in deze studies wellicht hebben beïnvloed. De werkgroep meent dat het effect op deze uitkomsten niet dusdanig groot is dat er sprake is van een ernstige mate van indirect bewijs. De werkgroep heeft om die reden dan ook niet afgewaardeerd.
4.1 “Annualized Relapse Rate” bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
4.2 Proportie vrij van een relapse bij een follow-up duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie), en met één niveau voor inconsistente uitkomsten (I2>50%). Er werd niet afgewaardeerd voor ernstige onnauwkeurigheid want dit is waarschijnlijk een gevolg van inconsistentie.
4.3 Toenemende invaliditeit bij een follow-up duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie) en met één niveau vanwege ernstige onnauwkeurigheid (gering aantal events).
4.4 Nieuwe T2-laesies of groter wordende T2-laesies bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
4.5 Gadolinium aankleurende laesies (gemiddeld aantal) bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
4.6a Studieuitval om een willekeurige reden bij een follow-up duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie) en met één niveau vanwege onnauwkeurigheid (gering aantal events).
4.6b Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie) en met één niveau vanwege onnauwkeurigheid (gering aantal events).
4.7a Infecties bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd voor ernstige onnauwkeurigheid (gering aantal ‘events’).
4.7b Ernstige infecties bij een follow-up -duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in redenen voor uitval tussen behandel- en controlegroep) en met één niveau vanwege onnauwkeurigheid (gering aantal events).
4.8 Mortaliteit bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd voor ernstige onnauwkeurigheid (gering aantal ‘events’).
4.9 Neoplasmata (alle) bij een follow-up duur van 60 maanden
De kwaliteit van bewijs is zeer laag. Er is met één niveau afgewaardeerd voor ernstige risk of bias (30% uitvallers en verschillen in redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie), met één niveau voor ernstige onnauwkeurigheid (gering aantal ‘events’) en met één niveau voor indirect bewijs: de follow-up duur van 104 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk acht voor een beoordeling van het risico op het optreden van maligniteiten.
Conclusies dimethylfumaraat versus placebo
4.1 Annualized relapse rate bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor de annualized relapse rate gemiddeld 0.19 relapses minder (95% BI: -0.25;-0.13) per patiënt per jaar zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.2 Proportie vrij van relapse bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 28% (RR: 1.28; 95% BI: 1.14-1.43) en een risicoverschil van 158 patiënten vrij van relapse meer per 1000 (95% BI: 79 meer tot 242 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.3 Toenemende invaliditeit bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 34% (RR: 0.66; 95% BI: 0.51-0.85) en een risicoverschil van 76 patiënten minder met toenemende invaliditeit (95% BI: 33 minder tot 109 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.4 Nieuwe T2-laesies of groter wordende T2-laesies bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor nieuwe of groter wordende T2-laesies gemiddeld 13.36 laesies minder (95% BI: -16.63;-10.09) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.5 Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor gemiddeld aantal gadolinium aankleurende laesies gemiddeld 1.64 laesies minder (95% BI: -2.17; -1.1) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.6a Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor studieuitval vanwege bijwerkingen een vermindering van het relatieve risico zien met 3% (RR: 0.97; 95% BI: 0.78; 1.21) en een risicoverschil van 5 patiënten minder met studieuitval vanwege bijwerkingen per 1000 (95% BI: 37 minder tot 35 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.6b Studieuitval om een willekeurige reden bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor studieuitval om een willekeurige reden een vermindering van het relatieve risico zien met 3% (RR: 0.97; 95% BI: 0.80; 1.16) en een risicoverschil van 7 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 46 minder tot 36 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.7a Infecties bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor infecties een toename van het relatieve risico zien met 16% (RR: 1.16; 95% BI: 0.88; 1.51) en een risicoverschil van 34 patiënten meer met infecties per 1000 (95% BI: 25 minder tot 108 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Fox et al., 2012 |
4.7b Ernstige infecties bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor ernstige infecties een toename van het relatieve risico zien met 42% (RR: 1.42; 95% BI: 0.55; 3.70) en een risicoverschil van 7 patiënten meer met ernstige infecties per 1000 (95% BI: 8 minder tot 46 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Gold et al., 2012 |
4.8 Mortaliteit bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor mortaliteit geen verschil zien (RR: 1.0; 95% BI: 0.99-1.01) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
4.9 Neoplasmata bij een follow-up duur van 60 maanden
|
Zeer laag
GRADE |
Gebruik van dimethylfumaraat door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor neoplasmata geen verschil zien (RR: 1.0; 95% BI: 0.99-1.01) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect verschilt waarschijnlijk aanzienlijk van het geschatte effect).
Bronnen: Fox et al., 2012; Gold et al., 2012 |
Review 5. Fingolimod versus placebo
Klinische effectiviteit
5.1 “Annualized Relapse Rate”
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over de annualized relapse rate (ARR). Fingolimod verminderde in vergelijking met een placebo de ARR met 0.21 (95% BI: 0.16-0.25) ten opzichte van placebo bij een follow-up duur van 104 weken.
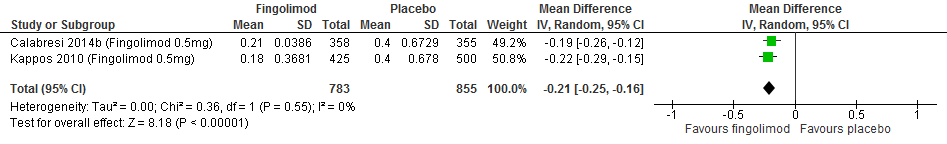
Figuur 5.1 Annualised relapse rate fingolimod versus placebo (104 weeks’ follow-up)
5.2 Proportie vrij van een relapse
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over de proportie patiënten die na 104 weken vrij van een relapse waren gebleven. Patiënten die naar fingolimod waren gerandomiseerd bleven in deze studies in een tijdsbestek van 104 weken meer vrij van een relapse: 23% verschil met patiënten met een placebo (RR: 1.44; 95% BI: 1.28-1.63).
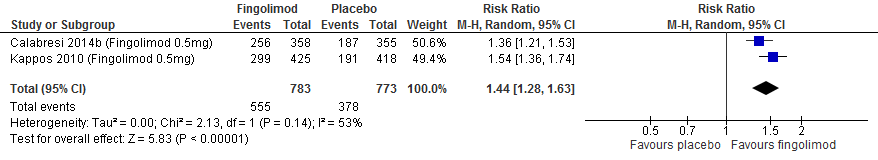
Figuur 5.2 Proportie vrij van een relapse fingolimod versus placebo (104 weeks’ follow-up)
5.3 Toenemende invaliditeit uitgedrukt in Expanded Disability Status Scale (EDSS)
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over de proportie patiënten met toenemende invaliditeit na een follow-up duur van 104 weken. Fingolimod zorgde er in deze studies voor dat bij minder patiënten de invaliditeit toenam: ca. 5-6% minder dan bij patiënten die een placebo kregen (RR: 0.71; 95% BI: 0.56-0.90).
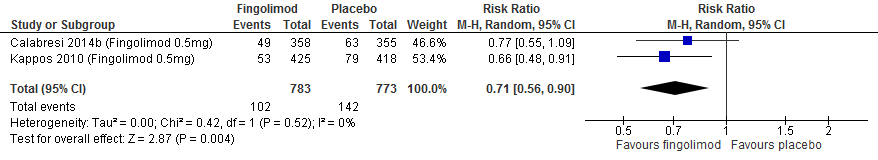
Figuur 5.3 Toenemende invaliditeit fingolimod versus placebo (104 weeks’ follow-up)
Effectiviteit-niet klinisch
5.4 Nieuwe T2-laesies of groter wordende T2-laesies
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over het gemiddelde aantal nieuwe of groter wordende T2 laesies bij een follow-up duur van 104 weken. Fingolimod verminderde dit aantal met 7.03 (95% BI: 5.84-8.22) ten opzichte van placebo.
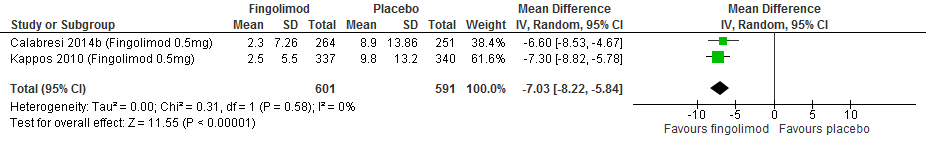
Figuur 5.4 Nieuwe T2-laesies of groter wordende T2-laesies fingolimod versus placebo (104 weeks’ follow-up)
5.5 Aantal patiënten vrij van nieuwe of groter wordende T2 laesies
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over het aantal patiënten vrij van nieuwe of groter wordende T2 laesies bij een follow-up duur van 104 weken. In de groep fingolimod hadden meer patiënten (27.3% meer) geen nieuwe of groter wordende T2 laesies in een tijdsbestek van 104 weken dan patiënten die een placebo kregen (RR 2.16; 95% BI: 1.77-2.63).
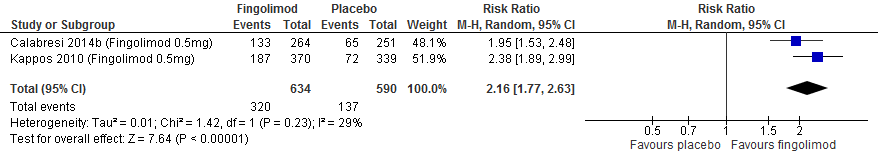
Figuur 5.5 Aantal patiënten vrij van nieuwe of groter wordende T2 laesies fingolimod versus placebo (104 weeks’ follow-up)
5.6 Gadolinium aankleurende laesies (gemiddeld aantal)
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over het gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 104 weken. Patiënten die fingolimod kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 0.87 gadolinium aankleurende laesies minder (95% BI: 0.64-1.1) bij een follow-up duur van 104 weken.
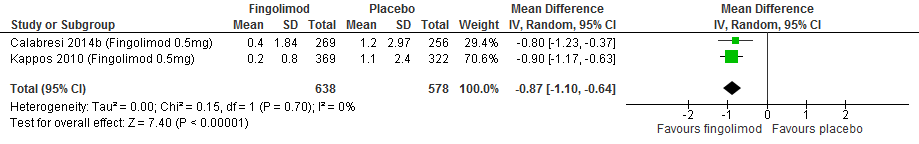
Figuur 5.6 Gadolinium aankleurende laesies (gemiddeld aantal) fingolimod versus placebo (104 weeks’ follow-up)
5.7 Gadolinium aankleurende laesies (aantal patiënten zonder deze laesies)
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over het aantal patiënten zonder gadolinium aankleurende laesies bij een follow-up duur van 104 weken. 23 à 24% patiënten meer die fingolimod kregen, hadden in vergelijking met patiënten die een placebo kregen, geen gadolinium aankleurende laesies (RR 1.36 95% BI: 1.27-1.45) bij een follow-up duur van 104 weken.
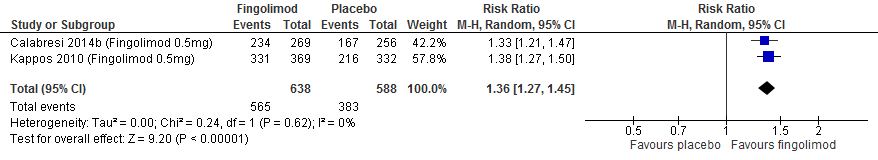
Figuur 5.7 Gadolinium aankleurende laesies (aantal patiënten zonder deze laesies) fingolimod versus placebo (104 weeks’ follow-up)
In de Europese richtlijntekst en bijlage werd niet gerapporteerd over het aantal nieuwe gadolinium aankleurende laesies en het cumulatieve aantal gadolinium aankleurende laesies.
Tolereerbaarheid en veiligheid
5.8 Studieuitval vanwege bijwerkingen of om willekeurige reden (zoals gerapporteerd in Europese richtlijn)
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over studieuitval vanwege bijwerkingen en over studieuitval om een willekeurige reden na een follow-up duur van 104 weken. De resultaten zijn in tabel 8 samengevat. Volgens deze studies verhoogt fingolimod ten opzichte van placebo het risico op stoppen vanwege bijwerkingen met een factor 1.4, en vermindert fingolimod het risico op stoppen om een willekeurige reden.
Tabel 8. Uitkomsten met betrekking tot studieuitval vanwege bijwerkingen of anderszins (fingolimod vs. placebo)
|
Uitkomstmaten |
Risicoverschil |
Relatieve risico (RR; 95% BI) |
|
Studieuitval vanwege bijwerkingen |
4.7% |
1.42 (0.92-2.17) |
|
Studieuitval om willekeurige reden |
-6.0% |
0.75 (0.57-0.99) |
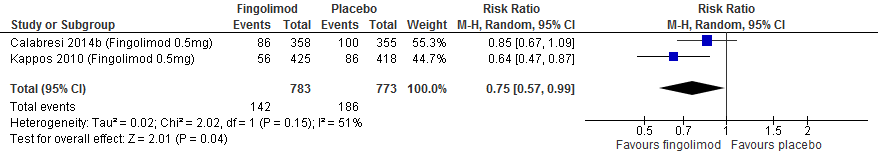
Figuur 5.8 Studieuitval vanwege willekeurige redenen fingolimod versus placebo (104 weeks’ follow-up)
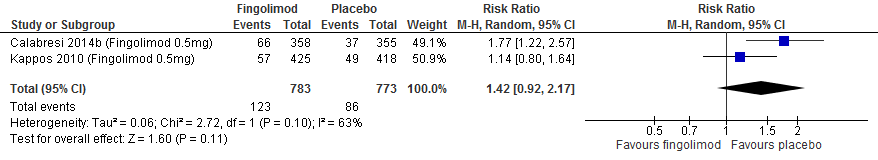
Figuur 5.9 Studieuitval vanwege bijwerkingen fingolimod versus placebo (104 weeks’ follow-up)
5.9 Infecties
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over infecties bij een follow-up duur van 104 weken. In vergelijking met een placebo resulteerde fingolimod in enige toename van infecties: een risicoverschil van 3.2% (RR: 1.04; 95% BI: 0.99 1.09).
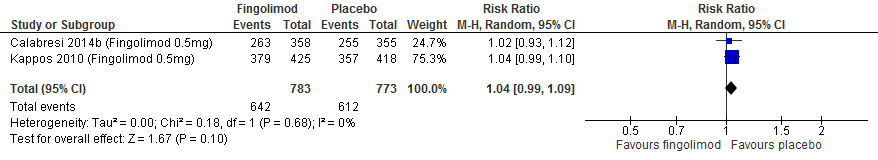
Figuur 5.10 Risico op infecties fingolimod versus placebo (104 weeks’ follow-up)
Mortaliteit
In de Europese richtlijn en bijlagen werd niet over mortaliteit gerapporteerd.
5.10 Neoplasmata (alle), maligniteiten
Twee RCTs (Calabresi et al., 2014b; Kappos et al., 2010) rapporteerden over neoplasmata bij een follow-up duur van 104 weken. In één studie (Calabresi et al., 2014) ging gebruik van fingolimod gepaard met een groter risico op een neoplasma: een risicoverschil van (RR 1.61; 95% BI: 0.68-3.84), in de andere studie (Kappos et al., 2010) juist met een geringer risico (RR 0.39; 95% BI: 0.12-1.24) in vergelijking met een placebo. Het gepoolde effect was een relatief risico van 0.84 (95% BI: 0.21-3.34).
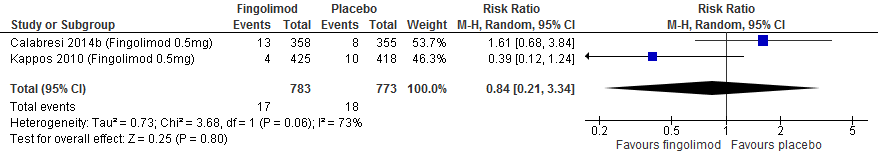
Figuur 5.11 Neoplasmata fingolimod versus placebo (104 weeks’ follow-up)
In de Europese richtlijn en bijlagen wordt niet separaat over maligniteiten gerapporteerd.
Kwaliteit van bewijs
In deze studies waren nog niet de relapsing remitting MS patiënten conform huidige diagnostische criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. Inclusie van deze patiënten zou de uitkomsten annualized relapse rate, progressie van invaliditeit en vrij van relapses in deze studies wellicht hebben beïnvloed. De werkgroep meent dat het effect op deze uitkomsten niet dusdanig groot is dat er sprake is van een ernstige mate van indirect bewijs. De werkgroep heeft om die reden dan ook niet afgewaardeerd.
5.1 “Annualized Relapse Rate” bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
5.2 Proportie vrij van een relapse bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
5.3 Toenemende invaliditeit bij een follow-up duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie), en met één niveau vanwege ernstige onnauwkeurigheid (gering aantal events).
5.4 Nieuwe T2-laesies of groter wordende T2-laesies bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
5.5 Aantal patiënten vrij van nieuwe of groter wordende T2 laesies
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduide-lijke blindering van randomisatie).
5.6 Gadolinium aankleurende laesies -laesies (gemiddeld aantal) bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
5.7 Aantal patiënten zonder gadolinium aankleurende laesies bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
5.8a Studieuitval om een willekeurige reden bij een follow-up duur van 104 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie) en met één niveau vanwege onnauwkeurigheid (gering aantal events).
5.8b Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 weken
De kwaliteit van bewijs is zeer laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie), met één niveau vanwege inconsistentie (I2 = 63%), en met één niveau vanwege onnauwkeurigheid (gering aantal events).[3]
[3] De inconsistentie is daarin gelegen dat de ene studie een groter effect laat zien dan de andere studie, namelijk een relatief risico van 1.77 en een van 1.14.
5.9 Infecties
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie).
5.10 Neoplasmata (alle) bij een follow-up duur van 104 weken
De kwaliteit van bewijs is zeer laag. Er is met één niveau afgewaardeerd vanwege ernstige risk of bias (verschillen in mate van en redenen voor uitval tussen behandel- en controlegroep; onduidelijke blindering van randomisatie), met één niveau vanwege inconsistentie (I2 = 63%), en met één niveau vanwege onnauwkeurigheid (gering aantal events). Er is niet verder afgewaardeerd voor indirect bewijs, de follow-up duur was namelijk aanzienlijk korter dan de minimale follow-up duur van 60 maanden, omdat de kwaliteit van bewijs al zeer laag is.
Conclusies
5.1 Annualized relapse rate bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor de annualized relapse rate gemiddeld 0.21 relapses minder (95% BI: -0.25;-0.16) per patiënt per jaar zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.2 Proportie vrij van relapse bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 44% (RR: 1.44; 95% BI: 1.28-1.63) en een risicoverschil van 215 patiënten vrij van relapse meer per 1000 (95% BI: 137 meer tot 308 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.3 Toenemende invaliditeit bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 29% (RR: 0.71; 95% BI: 0.56-0.90) en een risicoverschil van 53 patiënten minder met toenemende invaliditeit (95% BI: 18 minder tot 81 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.4 Nieuwe T2-laesies of groter wordende T2-laesies bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor nieuwe of groter wordende T2-laesies gemiddeld 7.03 laesies minder (95% BI: -8.22;-5.84) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.5 Aantal patiënten vrij van nieuwe of groter wordende T2 laesies
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken bij meer patiënten (27.3% meer) geen nieuwe of groter wordende T2 laesies zien (RR 2.16; 95% BI: 1.77-2.63) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.6 Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het gemiddeld aantal gadolinium aankleurende laesies gemiddeld 0.87 laesies minder (95% BI: -1.10;-0.64) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.7 Aantal patiënten zonder gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het aantal patiënten zonder gadolinium aankleurende laesies een toename van het relatieve effect zien met 36% (RR: 1.36; 95% BI: 1.27-1.45) en een risicoverschil van 234 patiënten meer zonder laesies (95% BI: 176 meer tot 293 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.8a Studieuitval om een willekeurige reden bij een follow-up duur van 104 weken
|
Laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor studieuitval om een willekeurige reden een vermindering van het relatieve risico zien met 25% (RR: 0.75; 95% BI: 0.57; 0.99) en een risicoverschil van 60 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 2 minder tot 103 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.8b Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 weken
|
Zeer laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 42% (RR: 1.42; 95% BI: 0.92; 2.17) en een risicoverschil van 47 patiënten meer studieuitval vanwege bijwerkingen per 1000 (95% BI: 9 minder tot 130 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect verschilt waarschijnlijk aanzienlijk van het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.9 Infecties bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor infecties een toename van het relatieve risico zien met 4% (RR: 1.04; 95% BI: 0.99; 1.09) en een risicoverschil van 32 patiënten meer met infecties per 1000 (95% BI: 8 minder tot 71 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
5.10 Neoplasmata (alle) bij een follow-up duur van 104 weken
|
Zeer laag
GRADE |
Gebruik van fingolimod door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor neoplasmata een vermindering van het relatieve risico zien met 16% (RR: 0.84; 95% BI: 0.21-3.34) en een risicoverschil van 4 patiënten minder met neoplasmata per 1000 (95% BI: 18 minder tot 54 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect verschilt waarschijnlijk aanzienlijk van het geschatte effect).
Bronnen: Calabresi et al., 2014b; Kappos et al., 2010 |
Review 6. Natalizumab versus placebo
Klinische effectiviteit
6.1 “Annualized Relapse Rate”
Een RCT (Polman et al., 2006) rapporteerde over de annualized relapse rate (ARR) na 52 en 104 weken. Natalizumab verminderde in vergelijking met een placebo de ARR met 0.51 (95% BI: 0.35-0.67) respectievelijk 0.50 (95% BI: 0.37-0.63) ten opzichte van placebo bij een follow-up duur van 52 en 104 weken.
6.2 Proportie vrij van een relapse
Een RCT (Polman et al., 2006) rapporteerde over de proportie patiënten die na 52 en 104 weken vrij van een relapse waren gebleven. Patiënten die naar natalizumab waren gerandomiseerd bleven in deze studie in een tijdsbestek van 52 tot 104 weken meer vrij van een relapse: 20 tot 27% verschil met patiënten met een placebo (RR: 1.33, 95% BI: 1.21-1.47 respectievelijk RR: 1.59, 95% BI: 1.40-1.81).
6.3 Toenemende invaliditeit uitgedrukt in Expanded Disability Status Scale (EDSS)
Een RCT (Polman et al., 2006) rapporteerde over de proportie patiënten met toenemende invaliditeit na een follow-up duur van 104 weken. Natalizumab zorgde er in deze studie voor dat bij minder patiënten de invaliditeit toenam: ca.12% minder dan bij patiënten die een placebo kregen (RR: 0.59; 95% BI: 0.46-0.75).
Effectiviteit-niet klinisch
6.4 Nieuwe T2-laesies of groter wordende T2-laesies
Een RCT (Polman et al., 2006) rapporteerde over het gemiddelde aantal nieuwe of groter wordende T2 laesies bij een follow-up duur van 52 tot 104 weken. Natalizumab verminderde dit aantal met 4.9 (95% BI: 3.84-5.96) respectievelijk 9.1 (95% BI: 7.22-10.98) ten opzichte van placebo.
6.5 Gadolinium aankleurende laesies (gemiddeld aantal)
Een RCT (Polman et al., 2006) rapporteerde over het gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 104 weken. Patiënten die natalizumab kregen, hadden in vergelijking met patiënten die een placebo kregen, gemiddeld 1.10 gadolinium aankleurende laesies minder (95% BI: 0.66-1.54) bij een follow-up duur van 104 weken.[4]
In de Europese richtlijntekst en bijlage werd niet gerapporteerd over het aantal nieuwe gadolinium aankleurende laesies, aantal patiënten zonder gadolinium aankleurende laesies en het cumulatieve aantal gadolinium aankleurende laesies.
[4] In bijlage 5 van de Europese richtlijn worden voor het gemiddeld aantal gadolinium aankleurende laesies standardized mean differences berekend voor de follow-up periodes van 52 en 104 weken. In de richtlijntekst worden negatieve relatieve risico’s gepresenteerd voor de follow-up van 104 weken. Dit kan niet juist zijn; relatieve risico’s kunnen niet negatief zijn. Vergelijking met tabel 2 in Polman et al. (2006) leert dat het gaat om een mean difference in plaats van een relatief risico; het getal zelf in de richtlijntekst is correct.
Tolereerbaarheid en veiligheid
6.6 Studieuitval vanwege bijwerkingen of anderszins (zoals gerapporteerd in Europese richtlijn)
Een RCT (Polman et al., 2006) rapporteerde over studieuitval vanwege bijwerkingen en over studieuitval om een willekeurige reden na een follow-up duur van 104 weken. De resultaten zijn in tabel 9 samengevat. Volgens deze studies verhoogt natalizumab het risico op stoppen vanwege bijwerkingen met een factor 1.3, en vermindert natalizumab het risico op stoppen om een willekeurige reden.
Tabel 9. Uitkomsten met betrekking tot studieuitval vanwege bijwerkingen of anderszins (natalizumab vs. placebo)
|
Uitkomstmaten |
Risicoverschil |
Relatieve risico (RR; 95% BI) |
|
Studieuitval vanwege bijwerkingen |
0.5% |
1.26 (0.49-3.21) |
|
Studieuitval om willekeurige reden |
-1.6% |
0.84 (0.55-1.29) |
6.7 Infecties
Een RCT (Polman et al., 2006) rapporteerde over infecties bij een follow-up duur van 104 weken. Natalizumab verhoogde ten opzichte van placebo het risico op infecties: een risicoverschil van 15.7% (RR: 1.23; 95% BI: 1.13-1.34).
6.8 Mortaliteit
Een RCT (Polman et al., 2006) rapporteerde over mortaliteit bij een follow-up duur van 104 weken. Gebruik van natalizumab ging in deze studie gepaard met een groter risico op mortaliteit, namelijk 0.3% meer dan in de placebogroep.
6.9 Neoplasmata (alle), maligniteiten
Een RCT (Polman et al., 2006) rapporteerde over neoplasmata bij een follow-up duur van 104 weken. Gebruik van natalizumab ging in deze studie gepaard met een groter risico op een neoplasma, namelijk 0.5% meer (5 patiënten kregen kanker in de natalizumab-groep en 1 patiënt in de placebogroep) dan in de placebogroep.
In de Europese richtlijntekst en bijlagen wordt niet separaat over maligniteiten gerapporteerd.[5]
[5] De in bijlage 5 van de Europese richtlijn gepresenteerde relatieve risico’s t.a.v. neoplasmata en mortaliteit zijn niet juist en derhalve niet overgenomen.
Kwaliteit van bewijs
In deze studies waren nog niet de relapsing remitting MS patiënten conform huidige diagnostische criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. Inclusie van deze patiënten zou de uitkomsten annualized relapse rate, progressie van invaliditeit en vrij van relapses in deze studies wellicht hebben beïnvloed. De werkgroep meent dat het effect op deze uitkomsten niet dusdanig groot is dat er sprake is van een ernstige mate van indirect bewijs. De werkgroep heeft om die reden dan ook niet afgewaardeerd.
6.1 “Annualized Relapse Rate” bij een follow-up duur van 52 en 104 weken
De kwaliteit van bewijs is hoog.
6.2 Proportie vrij van een relapse bij een follow-up duur van 52 en 104 weken
De kwaliteit van bewijs is hoog.
6.3 Toenemende invaliditeit bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal events).
6.4 Nieuwe T2-laesies of groter wordende T2-laesies bij een follow-up duur van 52 en 104 weken
De kwaliteit van bewijs is hoog.
6.5 Gadolinium aankleurende laesies (gemiddeld aantal) bij een follow-up duur van 52 en 104 weken
De kwaliteit van bewijs is hoog.
6.6a Studieuitval om een willekeurige reden bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd ernstige onnauwkeurigheid (gering aantal events).
6.6b Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege onnauwkeurigheid (gering aantal events).
6.7 Infecties
De kwaliteit van bewijs is hoog.
6.8 Mortaliteit bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (<300 events).
6.9 Neoplasmata (alle) bij een follow-up duur van 104 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege onnauwkeurigheid (gering aantal events).
Conclusies
6.1 Annualized relapse rate bij een follow-up duur van 52 en 104 weken
|
Hoog
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 52-104 weken voor de annualized relapse rate gemiddeld 0.51 relapses minder (95% BI: -0.67;-0.35) per patiënt per jaar zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.2 Proportie vrij van relapse bij een follow-up duur van 52 en 104 weken
|
Hoog
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 en 104 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 33% (RR: 1.33; 95% BI: 1.21-1.47) respectievelijk 59% (RR: 1.59; 95% BI: 1.4; 1.81) en een risicoverschil van 198 patiënten meer (95% BI: 126 meer tot 282 meer) respectievelijk 273 patiënten vrij van relapse meer per 1000 (95% BI: 185 meer tot 375 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.3 Toenemende invaliditeit bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor toenemende invaliditeit een vermindering van het relatieve risico zien met 41% (RR: 0.59; 95% BI: 0.46-0.75) en een risicoverschil van 118 patiënten minder met toenemende invaliditeit (95% BI: 72 minder tot 156 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.4 Nieuwe T2-laesies of groter wordende T2-laesies bij een follow-up duur van 52 en 104 weken
|
Hoog
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 52 en 104 weken voor nieuwe of groter wordende T2-laesies gemiddeld 4.9 laesies minder (95% BI: -5.96;-3.84) respectievelijk 9.1 leasies minder (95% BI: -10.98; -7.22) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.5 Gemiddeld aantal gadolinium aankleurende laesies bij een follow-up duur van 104 weken
|
Hoog
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor het gemiddeld aantal gadolinium aankleurende laesies gemiddeld 1.10 laesies minder (95% BI: -1.54;-0.66) zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.6a Studieuitval vanwege bijwerkingen bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor studieuitval vanwege bijwerkingen een toename van het relatieve risico zien met 26% (RR: 1.26; 95% BI: 0.49; 3.21) en een risicoverschil van 5 patiënten meer met studieuitval vanwege bijwerkingen per 1000 (95% BI: 10 minder tot 42 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.6b Studieuitval om een willekeurige reden bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor studieuitval om een willekeurige reden een vermindering van het relatieve risico zien met 16% (RR: 0.84; 95% BI: 0.55; 1.29) en een risicoverschil van 16 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 44 minder tot 29 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.7 Infecties bij een follow-up duur van 104 weken
|
Hoog
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor infecties een toename van het relatieve risico zien met 23% (RR: 1.23; 95% BI: 1.13; 1.34) en een risicoverschil van 157 patiënten meer met infecties per 1000 (95% BI: 89 meer tot 232 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.8 Mortaliteit bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor infecties geen verschil zien (RR: 1.0; 95% BI: 0.99; 1.0) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Polman et al., 2006 |
6.9 Neoplasmata bij een follow-up duur van 104 weken
|
Redelijk
GRADE |
Gebruik van natalizumab door patiënten met relapsing remitting MS laat bij een follow-up duur van 104 weken voor neoplasmata een risicoverschil van 4.8 patiënten meer met neoplasmata per 1000 zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Polman et al., 2006 |
Review 7. Wat zijn bij patiënten met relapsing remitting MS de netto-baten van het starten van een behandeling met alemtuzumab in vergelijking met een placebo?
Geen RCT heeft dit onderzocht.
Review 8. Wat zijn bij patiënten met relapsing remitting MS de netto-baten van het starten van een behandeling met ocrelizumab/ofatumumab in vergelijking met een placebo?
Geen RCT heeft dit onderzocht.
Review 9. Wat zijn bij patiënten met relapsing remitting MS de netto-baten van het starten van een behandeling met cladribine in vergelijking met een placebo?
Klinische effectiviteit
9.1“Annualized Relapse Rate”
Een RCT (Giovannoni et al., 2010) rapporteerde over de annualized relapse rate (ARR) na 96 weken. Cladribine verminderde in vergelijking met een placebo de ARR met 0.19 (95% BI: 0.14-0.23) ten opzichte van placebo bij een follow-up van 96 weken.
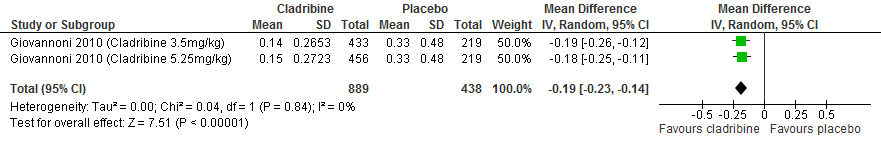
Figuur 9.1 Annualised relapse rate cladribine versus placebo (96 weeks’ follow-up)
9.2 Proportie vrij van een relaps
Een RCT (Giovannoni et al., 2010) rapporteerde over de proportie patiënten die na 96 weken vrij van een relapse waren gebleven. Patiënten die naar cladribine waren gerandomiseerd bleven in deze studie in een tijdsbestek van 96 weken meer vrij van een relapse: 79,7% voor 3,5mg/kg cladribine, in vergelijking met placebo (60,9%) (P<0,001). Dit komt neer op ca. 19% verschil met patiënten met een placebo (RR: 1,31, 95% BI: 1,20-1,42).
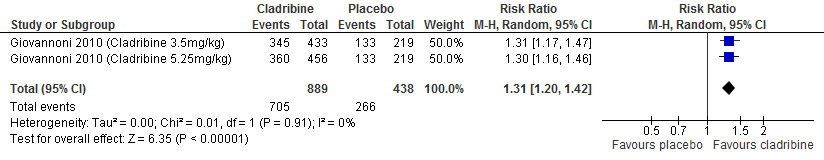
Figuur 9.2 Annualised relapse rate cladrbine versus placebo (96 weeks’ follow-up)
Toenemende invaliditeit uitgedrukt in Expanded Disability Status Scale (EDSS)
Hierover werd niet gerapporteerd in de Europese richtlijntekst en daarbij behorende bijlagen.
Effectiviteit-niet klinisch
9.3 Nieuwe T2-laesies of groter wordende T2-laesies, gemiddeld aantal gadolinium aankleurende laesies, aantal nieuwe gadolinium aankleurende laesies, aantal patiënten zonder gadolinium aankleurende laesies en het cumulatieve aantal gadolinium aankleurende laesies
In de bijlagen van de Europese richtlijn wordt hierover niet gerapporteerd. In de richtlijntekst wordt gerapporteerd: ‘The authors reported statistically significant reductions in GAD lesions, active T2 lesions and combined unique lesions in the intervention group compared with placebo (P < 0.0001)’.[6] De studie zelf rapporteert de volgende getallen zonder betrouwbaarheidsintervallen:
[6] In Giovannoni et al.(2010) staan geen standaarddeviaties bij de gemiddelden gerapporteerd zodat geen betrouwbaarheidsinterval kon worden berekend door de Europese werkgroep en Nederlandse werkgroep.
|
Uitkomstmaat (gemiddelde per patiënt per scan) |
Placebo |
3.5-mg cladribine (relatieve risico reductie) |
5.25-mg cladribine (relatieve risico reductie) |
|
gadolinium aankleurende laesies |
0.91 |
0.12 (85.7%) |
0.11 (87.9%) |
|
nieuwe T2-laesies of groter wordende T2-laesies |
1.43 |
0.38 (73.4%) |
0.33 (76.9%) |
|
gecombineerde unieke laesies |
1.72 |
0.43 (74.4%) |
0.38 (77.9%) |
Tolereerbaarheid en veiligheid
9.4 Studieuitval vanwege bijwerkingen of om willekeurige reden (zoals gerapporteerd in de Europese richtlijn)
Een RCT (Giovannoni et al., 2010) rapporteerde over studieuitval vanwege bijwerkingen en over studieuitval om een willekeurige reden bij een follow-up duur van 96 weken. De resultaten zijn in tabel 10 samengevat. Volgens deze studies verhoogt cladribine het risico op stoppen vanwege bijwerkingen met een factor van ruim 1,1, echter gezien de brede betrouwbaarheidsintervallen is het onzeker wat het effect van cladribine is studieuitval vanwege bijwerkingen.
Tabel 10. Uitkomsten met betrekking tot studieuitval vanwege bijwerkingen of anderszins (cladribine vs. placebo)
|
Uitkomstmaten |
Risicoverschil |
Relatieve risico (RR; 95% BI) |
|
Studieuitval vanwege bijwerkingen |
0.2% |
1.13 (0.43-2.94) |
|
Studieuitval om willekeurige reden |
-3.7% |
0.72 (0.53-0.99) |
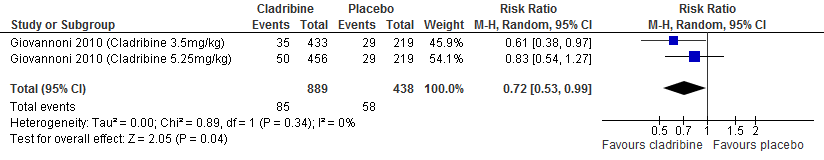
Figuur 9.3 Studieuitval om willekeurige redenen cladribine versus placebo (96 weeks’ follow-up)
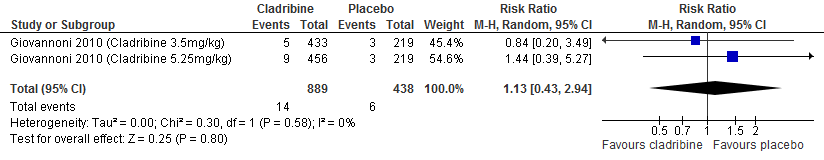
Figuur 9.4 Studieuitval vanwege bijwerkingen cladribine versus placebo (96 weeks’ follow-up)
9.6a Infecties
Een RCT (Giovannoni et al., 2010) rapporteerde over zowel infecties als ernstige infecties bij een follow-up duur van 96 weken. Het risico op een infectie was groter voor patiënten die cladribine gebruikten dan voor patiënten die een placebo gebruikten: een risicoverschil van 5.5% (RR: 1.13; 95% BI: 1-1.29).
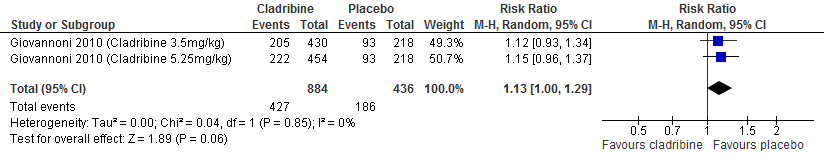
Figuur 9.5 Risico op infectie cladibrine versus placebo (96 weeks’ follow-up)
9.6b Ernstige infecties
Het risico op een ernstige infectie was eveneens groter voor patiënten die cladribine gebruikten dan voor patiënten die een placebo gebruikten: een risicoverschil van 0.8% (RR 1.41; 95% BI: 0.64-3.13).
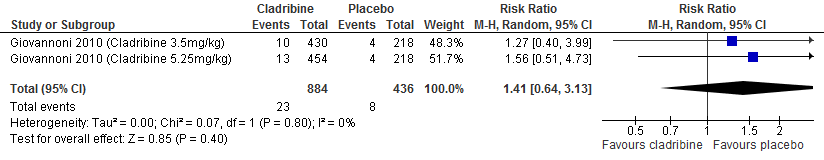
Figuur 9.6 Risico op ernstige infectie cladribine versus placebo (96 weeks’ follow-up)
9.7 Mortaliteit
Een RCT (Giovannoni et al., 2010) rapporteerde over mortaliteit bij een follow-up duur van 96 weken. Cladribine en placebo lieten in deze studie eenzelfde risico op mortaliteit zien: 0.45% (RR 0.99; 95% BI: 0.18-5.36).
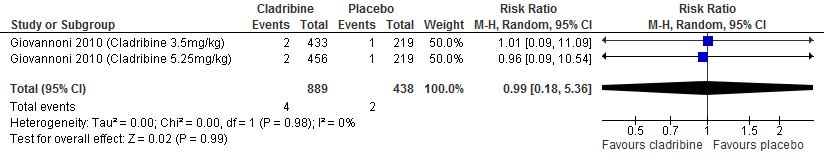
Figuur 9.7 Mortaliteit cladribine versus placebo (96 weeks’ follow-up)
9.8 Neoplasmata, maligniteiten
Een RCT (Giovannoni et al., 2010) rapporteerde over neoplasmata bij een follow-up duur van 96 weken. Het risico op een neoplasma was in deze studie groter voor patiënten die cladribine gebruikten dan voor patiënten met een placebo: een risicoverschil van 1.1% (RR 5.37; 95% BI: 0.69-41.55).
In de Europese richtlijntekst en bijlagen wordt niet separaat over maligniteiten gerapporteerd.
Kwaliteit van bewijs
In deze studies waren nog niet de relapsing remitting MS patiënten conform huidige diagnostische criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. Inclusie van deze patiënten zou de uitkomsten annualized relapse rate, progressie van invaliditeit en vrij van relapses in deze studies wellicht hebben beïnvloed. De werkgroep meent dat het effect op deze uitkomsten niet dusdanig groot is dat er sprake is van een ernstige mate van indirect bewijs. De werkgroep heeft om die reden dan ook niet afgewaardeerd.
9.1 “Annualized Relapse Rate” bij een follow-up duur van 96 weken
De kwaliteit van bewijs is hoog.
9.2 Proportie vrij van een relapse bij een follow-up duur van 96 weken
De kwaliteit van bewijs is hoog.
9.3 Gadolinium aankleurende laesies, actieve T2 laesies, gecombineerde unieke laesies
De kwaliteit van bewijs is redelijk. Er werd met één niveau afgewaardeerd voor ernstige onnauwkeurigheid (geen rapportage van standaarddeviatie of betrouwbaarheidsinterval).
9.4a Studieuitval vanwege bijwerkingen bij een follow-up duur van 96 weken
De kwaliteit van bewijs is laag. Er is met twee niveaus afgewaardeerd vanwege zeer ernstige onnauwkeurigheid (gering aantal events en betrouwbaarheidsinterval sluit zowel een gunstig als een ongunstig relatief en absoluut effect in).
9.4b Studieuitval om een willekeurige reden bij een follow-up duur van 96 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal events).
9.6a Infecties bij een follow-up duur van 96 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal events).
9.6b Ernstige infecties bij een follow-up duur van 96 weken
De kwaliteit van bewijs is redelijk. Er is met één niveau afgewaardeerd vanwege ernstige onnauwkeurigheid (gering aantal events).
9.7 Mortaliteit bij een follow-up duur van 96 weken
De kwaliteit van bewijs is laag. Er is met twee niveaus afgewaardeerd vanwege zeer ernstige onnauwkeurigheid (gering aantal events en betrouwbaarheidsinterval sluit zowel een gunstig als een ongunstig relatief en absoluut effect in).
9.8 Neoplasmata (alle) bij een follow-up duur van 96 weken
De kwaliteit van bewijs is laag. Er is met één niveau afgewaardeerd vanwege onnauwkeurigheid (gering aantal events) en met één niveau voor indirect bewijs: de follow-up duur van 96 weken was aanzienlijk korter dan de minimale follow-up duur van 60 maanden die de Nederlandse werkgroep noodzakelijk acht voor een beoordeling van het risico op het optreden van maligniteiten.
Conclusies
9.1 Annualized relapse rate bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor de annualized relapse rate gemiddeld 0.19 relapses minder (95% BI: -0.23;-0.14) per patiënt per jaar zien in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Giovannoni et al., 2010 |
9.2 Proportie vrij van relapse bij een follow-up duur van 96 weken
|
Hoog
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor de proportie patiënten vrij van relapse een toename van het relatieve effect zien met 31% (RR: 1.31; 95% BI: 1.20-1.42) en een risicoverschil van 188 patiënten vrij van relapse meer per 1000 (95% BI: 121 meer tot 255 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt dicht bij het geschatte effect).
Bron: Giovannoni et al., 2010 |
9.3a Aantal T2-laesies per scan per patiënt bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor het aantal T2-laesies per scan per patiënt een vermindering van het relatieve risico zien met ca. 75% in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Europese richtlijnwerkgroep; Giovannoni et al., 2010 |
9.3b Gadolinium aankleurende laesies per patiënt per scan bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor het aantal gadolinium aankleurende laesies per patiënt per scan een vermindering van het relatieve risico zien met ca. 86% in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Europese richtlijnwerkgroep; Giovannoni et al., 2010 |
9.3c Gecombineerde unieke laesies (nieuwe of grotere T2 laesies en/of gadolinium aankleurende laesies T1 laesies) per patiënt per scan bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor het aantal gecombineerde unieke laesies per patiënt per scan een vermindering van het relatieve risico zien met ca. 76% in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Europese richtlijnwerkgroep; Giovannoni et al., 2010 |
9.4a Studieuitval vanwege bijwerkingen bij een follow-up duur van 96 weken
|
Laag
GRADE |
Het is onzeker wat het effect is van cladribine op de bijwerkingen bij patiënten met relapsing remitting MS, bij een follow-up duur van 96 weken.
Bron: Giovannoni et al., 2010 |
9.4b Studieuitval om een willekeurige reden bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor studieuitval om een willkeurige reden een vermindering van het relatieve risico zien met 28% (RR: 0.72; 95% BI: 0.53; 0.99) en een risicoverschil van 37 patiënten minder met studieuitval om een willekeurige reden per 1000 (95% BI: 1 minder tot 62 minder) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Giovannoni et al., 2010 |
9.6a Infecties bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor infecties een toename van het relatieve risico zien met 13% (RR: 1.13; 95% BI: 1-1.29) en een risicoverschil van 55 patiënten meer met infecties per 1000 (95% BI: 0 meer tot 124 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Giovannoni et al., 2010 |
9.6b Ernstige infecties bij een follow-up duur van 96 weken
|
Redelijk
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor ernstige infecties een toename van het relatieve risico zien met 41% (RR 1.41; 95% BI: 0.64-3.13) en een risicoverschil van 8 patiënten meer met ernstige infecties per 1000 (95% BI: 7 minder tot 39 meer) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect ligt waarschijnlijk dicht bij het geschatte effect).
Bron: Giovannoni et al., 2010 |
9.7 Mortaliteit bij een follow-up duur van 96 weken
|
Laag
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor mortaliteit geen verschil zien (RR 0.99; 95% BI: 0.18-5.36) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Giovannoni et al., 2010 |
9.8 Neoplasmata bij een follow-up duur van 96 weken
|
Laag
GRADE |
Gebruik van cladribine door patiënten met relapsing remitting MS laat bij een follow-up duur van 96 weken voor neoplasmata een toename van het relatieve risico zien met meer dan 400% (RR 5.37; 95% BI: 0.69-41.55) en een risicoverschil van 11 patiënten meer met neoplasmata per 1000 (95% BI: niet te bereken) in vergelijking tot placebo (kwaliteit van bewijs: het werkelijke effect kan aanzienlijk verschillen van het geschatte effect).
Bron: Giovannoni et al., 2010 |
[1] De cijfers kunnen afwijken van de cijfers die in de Europese richtlijntekst worden vermeld. De reden is dat de Europese richtlijntekst geen onderscheid heeft gemaakt tussen placebo gecontroleerde en head-to-head vergelijkingen.
[2] Conform de Europese richtlijn zijn alle soorten interferon samengenomen.
[3] De inconsistentie is daarin gelegen dat de ene studie een groter effect laat zien dan de andere studie, namelijk een relatief risico van 1.77 en een van 1.14.
[4] In bijlage 5 van de Europese richtlijn worden voor het gemiddeld aantal gadolinium aankleurende laesies standardized mean differences berekend voor de follow-up periodes van 52 en 104 weken. In de richtlijntekst worden negatieve relatieve risico’s gepresenteerd voor de follow-up van 104 weken. Dit kan niet juist zijn; relatieve risico’s kunnen niet negatief zijn. Vergelijking met tabel 2 in Polman et al. (2006) leert dat het gaat om een mean difference in plaats van een relatief risico; het getal zelf in de richtlijntekst is correct.
[5] De in bijlage 5 van de Europese richtlijn gepresenteerde relatieve risico’s t.a.v. neoplasmata en mortaliteit zijn niet juist en derhalve niet overgenomen.
[6] In Giovannoni et al.(2010) staan geen standaarddeviaties bij de gemiddelden gerapporteerd zodat geen betrouwbaarheidsinterval kon worden berekend door de Europese werkgroep en Nederlandse werkgroep.
Zoeken en selecteren
Om de uitgangsvraag te kunnen beantwoorden heeft de Nederlandse richtlijnwerkgroep systematische literatuuranalyses (één per geneesmiddel) verricht met de volgende PICO-vraagstelling:
Wat zijn bij patiënten met relapsing remitting MS de netto-baten in vergelijking met een placebo van het starten met:
- interferon-β (interferon-β/peginterferon-β)
- glatirameeracetaat
- teriflunomide
- fumaraatzuur esters (dimethylfumaraat/diroximelfumaraat)
- S1P receptor modulatoren (fingolimod/ozanimod/ponesimod)
- natalizumab (intraveneus of subcutaan)
- anti-CD20 monoclonalen (ocrelizumab/ofatumumab)
- cladribine
- alemtuzumab
Bron voor de systematische literatuuranalyses was de Europese richtlijntekst met bijbehorende bijlagen (adaptatie). De opstellers van de Europese richtlijn zochten daartoe in de databases Medline (OVID), Embase en Cochrane.
In de databases Embase (via embase.com) en Medline (via OVID) is op 27-01-2022 met relevante zoektermen gezocht vanaf 2016 naar systematische reviews, RCT’s en observationele studiedesigns over Ozanimod, Ofatumumab, Natalizumab en Ponesimod bij patiënten met MS. De literatuurzoekactie leverde 707 unieke treffers op.
Selectie- en exclusiecriteria:
|
Type studies |
|
|
Type patiënten |
|
|
Interventie |
|
|
Controle |
|
|
Type uitkomstmaten** |
1. Effectiviteit – klinisch** (cruciaal)
2. Effectiviteit – niet klinisch (belangrijk)
3. Tolereerbaarheid en veiligheid** (cruciaal)
|
|
Type setting |
|
|
Exclusiecriteria |
|
|
* M.b.t. relapsing remitting vermeldt de Europese richtlijn: ‘clinically definite or laboratory-supported definite relapsing-remitting MS according to Poser criteria in the oldest trials and according to the revised McDonald criteria (2001 or 2005) in the most recent trials. Any additional criteria of number of relapses in the years prior to inclusion is valid’. In deze studies waren nog niet de relapsing remitting MS patiënten volgens de 2017 McDonald criteria vertegenwoordigd bij wie MS kan worden gediagnosticeerd door spreiding in tijd te vervangen door aanwezig zijn van unieke oligoclonale banden in liquor. **Bovengenoemde uitkomstmaten zijn voor de Nederlandse richtlijnwerkgroep cruciaal (= beslissend). Alle andere uitkomstmaten die in de Europese richtlijn gerapporteerd worden, vindt de Nederlandse werkgroep minder belangrijk en heeft deze daarom niet geadapteerd. Op deze manier wordt geprobeerd de teksten overzichtelijk te houden. In bijlage 5 van de Europese richtlijn staan alle uitkomstmaten die de Europese richtlijnwerkgroep heeft geïncludeerd, evenals de resultaten per uitkomstmaat en een beoordeling van de kwaliteit van bewijs: http://journals.sagepub.com/doi/suppl/10.1177/1352458517751049. ***De Europese werkgroep heeft met betrekking tot ‘stoppen’ drie uitkomstmaten gehanteerd: discontinuation due to any reason, discontinuation due to side effects, discontinuation of medication due to side effects. In de tekst zijn deze termen vertaald als: studieuitval om willekeurige reden, studieuitval vanwege bijwerkingen, stoppen met medicatie vanwege bijwerkingen. Studieuitval om willekeurige reden of vanwege bijwerkingen heeft dus betrekking op studie-uitvallers van wie geen follow-up gegevens beschikbaar zijn. Dit in tegenstelling tot stoppen met medicatie vanwege bijwerkingen: hier zijn de follow-up gegevens wel beschikbaar. Meestal echter wordt ‘discontinuation of medication due to side effects’ niet en ‘discontinuation due to side effects’ wel gerapporteerd door de Europese werkgroep. |
|
Zestien placebo gecontroleerde RCTs (Calabresi et al., 2014a; Calabresi et al., 2014b; Confavreux et al., 2014; Vollmer et al., 2014; Khan et al., 2013; Fox et al., 2012 [twee behandelarmen]; Gold et al., 2012; O'Connor et al., 2011; Giovannoni et al., 2010; Kappos et al., 2010; Polman et al., 2006; PRISMS, 1998; Jacobs et al., 1996; Johnson et al., 1995; IFNB MS Group, 1993) werden meegenomen in de literatuuranalyse. In appendix 3 van de Europese richtlijn (https://journals.sagepub.com/doi/suppl/10.1177/1352458517751049 staan de redenen van exclusie van andere studies vermeld. Voor alemtuzumab, ocrelizumab, diroximelfumaraat, ponesimod, ozanimod en ofatumumab werden geen placebo gecontroleerde trials uitgevoerd. Deze middelen worden in 5.2. besproken.
Referenties
- Barkhof et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain 1997.
- Ben-Zacharia A, Adamson M, Boyd A, Hardeman P, Smrtka J, Walker B, Walker T. Impact of Shared Decision Making on Disease-Modifying Drug Adherence in Multiple Sclerosis. Int J MS Care. 2018 Nov-Dec;20(6):287-297.
- Berger, J. R., & Markowitz, C. (2018). Deciding on the Best Multiple Sclerosis Therapy: Tough Choices. JAMA neurology, 75(12), 1461-1462.
- Burks J, Marshall TS, Ye X. Adherence to disease-modifying therapies and its impact on relapse, health resource utilization, and costs among patients with multiple sclerosis. Clinicoecon Outcomes Res. 2017 Apr 28;9:251-260.
- Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon beta-1a for relapsing_remitting multiple sclerosis (ADVANCE): a randomised, phase 3, doubleblind study. Lancet Neurol 2014; 13: 657-665.
- Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing_remitting multiple sclerosis (FREEDOMS II): a doubleblind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2014; 13: 545-556.
- Confavreux C, O'Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebocontrolled, phase 3 trial. Lancet Neurol 2014; 13: 247-256.
- Cree BA, Gourraud PA, Oksenberg JR, et al. Long-term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 2016; 80: 499-510.
- Debouverie M, Laforest L, Van Ganse E, Guillemin F; LORSEP Group. Earlier disability of the patients followed in Multiple Sclerosis centers compared to outpatients. Mult Scler. 2009 Feb;15(2):251-7.
- Ebers GC, Traboulsee A, Li D, et al. Analysis of clinical outcomes according to original treatment groups 16 years after the pivotal IFNB-1b trial. J Neurol Neurosurg Psychiatry 2010; 81: 907-912.
- Interferon beta-1b is effective in relapsing_remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology 1993; 43: 655-661.
- EMA confirms recommendations to minimise risk of brain infection PML with Tysabri [press release]. London, UK: European Medicines Agency, 2016. Issued February 26, 2016.
- Eriksson M, Andersen O, Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis. Mult Scler. 2003 Jun;9(3):260-74.
- Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 2012; 367: 1087-1097.
- Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med 2010; 362: 416-426.
- Giovannoni G, Soelberg Sorensen P, Cook S, Rammohan K, Rieckmann P, Comi G, Dangond F, Adeniji AK, Vermersch P. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: Results from the randomized extension trial of the CLARITY study. Mult Scler. 2018 Oct;24(12):1594-1604.
- Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: interim analysis of ENDORSE, a randomized extension study. Mult Scler 2016; 23: 253-265.
- Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 2012; 367: 1098-1107.
- Huygens S, Versteegh M. Modeling the cost-utility of treatment sequences for multiple sclerosis. Value in Health. August 2021; 24: 1612-1619.
- Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol 1996; 39: 285-294.
- Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing_remitting multiple sclerosis: results of a phase III multicenter, double- blind, placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995; 45: 1268-1276.
- Johnson KP, Brooks BR, Ford CC, et al. Sustained clinical benefits of glatiramer acetate in relapsing multiple sclerosis patients observed for 6 years. Copolymer 1 Multiple Sclerosis Study Group. Mult Scler 2000; 6: 255-266.
- Jokubaitis et al. Predictors of disability worsening in clinically isolated syndrome, Annals of Clinical and Translational Neurolo-gy 2015.
- Kalb R, Beier M, Benedict RH, Charvet L, Costello K, Feinstein A, Gingold J, Goverover Y, Halper J, Harris C, Kostich L, Krupp L, Lathi E, LaRocca N, Thrower B, DeLuca J. Recommendations for cognitive screening and management in multiple sclerosis care. Mult Scler. 2018 Nov;24(13):1665-1680.
- Kappos L, Radue EW, O'Connor P, et al. A placebo controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010; 362: 387-401.
- Kappos L, O'Connor P, Radue EW, et al. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology 2015; 84: 1582-1591.
- Kappos L, Traboulsee A, Constantinescu C, et al. Long-term subcutaneous interferon beta-1a therapy in patients with relapsing_remitting MS. Neurology 2006; 67: 944-953.
- Kasper J, Kopke S, Muhlhauser I, Nubling M, Heesen C. Informed shared decision making about immunother-apy for patients with multiple sclerosis (ISDIMS): a randomized controlled trial. Eur J Neurol. 2008;15:1345-1352
- Kieseier BC, Arnold DL, Balcer LJ, et al. Peginterferon beta-1a in multiple sclerosis: 2-year results from ADVANCE. Mult Scler 2015; 21: 1025-1035.
- Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R. Three times weekly glatiramer acetate in relapsing_remitting multiple sclerosis. Ann Neurol 2013; 73: 705-713.
- Kremer, I. E. H., Evers, S. M. A. A., Jongen, P. J., & Hiligsmann, M. (2018). Comparison of preferences of healthcare professionals and MS patients for attributes of disease-modifying drugs: A best-worst scaling. Health expectations : an international journal of public participation in health care and health policy, 21(1), 171-180.
- Lorscheider J, Buzzard K, Jokubaitis V, Spelman T, Havrdova E, Horakova D, et al. Defining secondary progressive multiple sclerosis. Brain. 2016 Sep;139(Pt 9):2395-405.
- Morillo Verdugo R, Ramírez Herráiz E, Fernández-Del Olmo R, Roig Bonet M, Valdivia García M. Adherence to disease-modifying treatments in patients with multiple sclerosis in Spain. Patient Prefer Adherence. 2019 Feb 13;13:261-272.
- O'Connor P, Comi G, Freedman MS, et al. Long-term safety and efficacy of teriflunomide: nine-year follow-up of the randomized TEMSO study. Neurology 2016; 86: 920-930.
- O'Connor P, Wolinsky JS, Confavreux C, et al. Randomised trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 2011; 365: 1293-1303.
- Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006; 354: 899-910.
- PRISMS-4: Long-term efficacy of interferon-beta-1a in relapsing MS. Neurology 2001; 56: 1628-1636.
- Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet 1998; 352: 1498-1504.
- Reynders T, D'haeseleer M, De Keyser J, Nagels G, D'hooghe MB. Definition, prevalence and predictive factors of benign multiple sclerosis. eNeurologicalSci. 2017 May 13;7:37-43.
- Sabate E. Adherence to long-term therapies. Evidence for action. Geneva: WHO, 2003
- Stacey D, Bennett CL, Barry MJ, , et al. Decision aids for people facing health treatment or screening decisions. Cochrane Database Syst Rev 2011;10:CD001431.
- Tedeholm H, Skoog B, Lisovskaja V, Runmarker B, Nerman O, Andersen O. The outcome spectrum of multiple sclerosis: disability, mortality, and a cluster of predictors from onset. J Neurol. 2015 May;262(5):1148-63.
- Tintore M, Rovira À, Río J, Otero-Romero S, Arrambide G, Tur C, Comabella M, et al. Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain. 2015 Jul;138(Pt 7):1863-74. doi: 10.1093/brain/awv105. Epub 2015 Apr 21. PMID: 25902415.
- Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, Correale J, Fazekas F, Filippi M, Freedman MS, Fujihara K, Galetta SL, Hartung HP, Kappos L, Lublin FD, Marrie RA, Miller AE, Miller DH, Montalban X, Mowry EM, Sorensen PS, Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2017.
- Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018 Apr 21; 391 (10130) :1622-1636.
- Tremlett H, Zhao Y, Devonshire V. Defining the natural history of MS: the need for complete data and rigorous definitions. Mult Scler. 2008 Sep;14(8):1142-3; author reply 1144-7.
- Trojano M, Pellegrini F, Fuiani A, Paolicelli D, Zipoli V, Zimatore GB, Di Monte E, Portaccio E, Lepore V, Livrea P, Amato MP. New natural history of interferon-beta-treated relapsing multiple sclerosis. Ann Neurol. 2007 Apr;61(4):300-6.
- Tur C, Moccia M, Barkhof F, Chataway J, Sastre-Garriga J, Thompson AJ, Ciccarelli O. Assessing treatment outcomes in multiple sclerosis trials and in the clinical setting. Nat Rev Neurol. 2018 Feb;14(2):75-93.
- Versteegh MM, Huygens SA, Wokke BWH, Smolders S. Effectiveness and cost-effectiveness of 360 disease-modifying treatment escalation sequences in multiple sclerosis. Value in health. 2022; 25: 984-991.
- Vollmer TL, Sorensen SP, Selmaj K, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol 2014; 261: 773-783.
- Wattjes et al. MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis-establishing disease prognosis and monitoring patients, Nat. Rev. Neurol 2015.
Verantwoording
Beoordelingsdatum en geldigheid
Publicatiedatum : 29-01-2024
Beoordeeld op geldigheid : 11-12-2023
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de modules voor het addendum is in 2021 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de indicatiestelling en monitoring van ziektemodulerende therapie bij mensen met MS (zie hiervoor de samenstelling van de werkgroep).
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname. De werkgroep werkte gedurende 2 jaar aan de totstandkoming van de richtlijn. De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Werkgroep
- Mevr. dr. B.A. de Jong, neuroloog, NVN (voorzitter)
- Mevr. B. Bakker, patiëntvertegenwoordiger, MS-patiënten Vereniging Nederland
- Dhr. drs. R. Haenen, revalidatiearts, VRA
- Mevr. K. Harrison, verpleegkundig specialist, V&VN
- Dhr. drs. B.J.A. van Hoeve, neuroloog, NVN
- Mevr. dr. E. Hoitsma, neuroloog, NVN
- Dhr. prof. dr. J. Killestein, neuroloog, NVN
- Dhr. drs. J.B. Masselink, ziekenhuisapotheker, NVZA
- Dhr. dr. B. Moraal, radioloog, NVVR
- Dhr. dr. J. Mostert, neuroloog, NVN
- Dhr. dr. J. Murk, arts-microbioloog, NVMM
- Mevr. M. Pippel, patiëntvertegenwoordiger, MS-patiënten Vereniging Nederland
- Dhr. dr. J. Smolders, neuroloog, NVN
- Dhr. dr. M.T. Wijburg, AIOS neurologie, NVN
- Mevr. Dr. A. Coumans, Gynaecoloog, NVOG
Met dank aan:
- Dhr. Dr. Ir. J.J.A. de Beer, zelfstandig richtlijnmethodoloog
- Mevr. dr. S. Huygens, gezondheidseconoom, Huygensandversteegh.com
- Dhr. dr. M. Versteegh, gezondheidseconoom, Huygensandversteegh.com
Met ondersteuning van:
- Mevr. dr. C.T.J. Michels, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Mevr. dr. M.L. Molag, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Achternaam werkgroeplid |
Hoofdfunctie |
Nevenwerkzaamheden |
Gemelde belangen |
BelandenCie |
|
Bakker |
MS Patiënt vertegenwoordiger MSVN |
Functionel tester software Thunderbyte AI |
geen |
Geen restrictie |
|
Coumans |
Gynaecoloog |
Geen |
geen |
Geen restrictie |
|
De Jong (voorzitter) |
Functie: neuroloog |
Voorzitter commissie Addendum MS richtlijn van NVN (onbetaald). Voorzitter commissie MS registratie van NVN (onbetaald). Lid van wetenschappelijk adviesraad van Prinses Beatrixfonds (onbetaald) Lid van het PROMS initiative, onderdeel van Global MSIF Movement (onbetaald) |
MS Centrum Amsterdam, Amsterdam UMC, locatie VUmc ontvangt financiële ondersteuning van: Bayer AG, Biogen, Roche, GlaxoSmithKline, Merck, Celgene BMS, Sanofi Genzyme, Novartis, TEVA, Stichting MS Research, NWO, ZonMw, Hersenstichting, Nationaal MS Fonds, Zorgverzekeraars Nederland (ZN) en Health~Holland (TKI*).
*TKI: Dit is een studie gesponsord door Sanofi, het gaat om een Bruton’s tyrosine kinase inhibitor: tolebrutinib (komt niet voor in richtlijn).
Dit zijn deels wel en niet persoonsgebonden subsidies (= deels contractonderzoek). Subsidies van instanties zoals MS research, ZonMw, hersenstichting zijn allemaal persoonsgebonden maar hierbij ik ben hierbij geen PI. Ik ben wel PI bij: - subsidie van ZN om onderzoek te doen met de landelijke MS registratie. - fase IV studie waarbij observationeel de kwaliteit en veiligheid van alemtuzumab (Sanofi Genzyme) gemonitord wordt (zit niet in adviesraad, krijg er geen geld voor). Studie gaat over post-surveillance en houdt geen relatie met indicatiestelling. - fase III studie naar tyrosine kinase inhibitors bij MS (dit middel staat niet in de richtlijn en komt naar verwachting niet op korte termijn op de markt).
Ontvangen onderzoekssubsidie van MS Vereniging Nederland in 2022: “Prevalentie van MS in Nederland” met een bedrag €60.167 Ontvangen onderzoekssubsidie van Stichting MS Research in 2016: "Improving cognitieve functioning among patients with multiple sclerosis by cognitive rehabilitation therapy or mindfulness based cognitive therapy: a dual-center, investigator-blinded, parallel group randomized controlled trial" met een bedrag van € 250.000.
|
Restrictie als TKIs ter sprake komt in een module. |
|
Haenen |
Functie: revalidatiearts |
Geen |
Geen |
Geen restrictie. |
|
Harrisson |
Functie: Verpleegkundig Specialist Werkgever: Tergooi ziekenhuis |
Richtlijn Revalidatie bij MS, vergoeding van gemaakte uren. |
Geen |
Geen restrictie. |
|
Hoitsma |
Functie: neuroloog |
Voorzitter landelijke MS werkgroep (onbetaald). |
Lokale onderzoeker bij (geen PI over gehele studies):
Betreft bij allen betaling onderzoeksactiviteiten. |
Geen restrictie. |
|
Killestein |
Functie: neuroloog (hoogleraar MS) |
Adviseur Springer Healthcare - Mednet Neurologie (betaald). Hoofdredacteur Tijdschrift voor Neurologie en Neurochirurgie (TNN), tot februari 2021, (betaald).
Voorzitter Horizonscan geneesmiddelen Zorginstituut Nederland. Werkgroep Neurologische aandoeningen inclusief gedrag (onbetaald). Lid Wetenschappelijke Adviesraad Stichting MS Research (onbetaald). Lid editorial board Neurology (onbetaald). Lid editorial board MS Journal (onbetaald).
|
MS Centrum Amsterdam, Amsterdam UMC, locatie VUmc ontvangt financiële ondersteuning van: Bayer AG, Biogen, Roche, GlaxoSmithKline, Merck, Celgene BMS, Sanofi Genzyme, Novartis, TEVA, Immunic Therapeutics, Stichting MS Research, NWO, ZonMw, Hersenstichting, Nationaal MS Fonds, Zorgverzekeraars Nederland (ZN) en Health~Holland (TKI)
1. Principle investigator NEXT-MS -natalizumab extended dosing bij MS -NCT04225312. Sponsor: Hersenstichting en Stichting MS Research. 2. Principle investigator DOT-MS - discontinuation of therapy in MS, NCT04260711- Sponsor: ZonMW en Stichting MS research 3. Principle investigator BLOOMS trial: efficacy, safety and cost-effectiveness of B cell tailored ocrelizumab versus standard ocrelizumab in RRMS - Sponsor: ZonMW en Treatmeds, moet nog starten. 4. Principle investigator Apps-MS studie - Sponsor: Stichting MS-Research en Health~Holland TKI. 5. Principle investigator Blood platelet-based liquid biopsies for optimization of diagnosis and treatment of patients with multiple sclerosis - Sponsor Merck. à Merck maakt medicatie die gebruikt wordt bij MS, Rebif en Mavencla. Betrfet onderzoek m.b.t. diagnostische biomarker (wel/geen MS). Geen enkele bemoeienis van Merck met de inhoud. 6. Deelname RELAMS - onderzoek naar relapses en pseudorelapses bij MS. 7. Deelname RAM-MS trial: Randomized Autologous heMatopoietic stem cell transplantation versus alemtuzumab, cladribine or ocrelizumab for patients with RRMS. EudraCT Number: 2017-001362-25 Sponsor: Stichting MS Research en Nationaal MS fonds. 8. Lid Steering Committee Ensemble trial Roche, vergoeding naar VUmc. 9. Lid Steering Committee Liberto trial Roche, vergoeding naar VUmc. 10. Lid Steering Committee NOVA trial Biogen, vergoeding naar VUmc. 11. Lid Adjudication Committee Immunic Calliper Phase II trial, vergoeding naar VUmc |
Geen restrictie. |
|
Masselink |
Functie: ziekenhuisapotheker Werkgever: Medisch Spectrum Twente |
Lid werkgroep neurologische aandoeningen (incl. gedrag) Horizonscan Geneesmiddelen, |
Geen |
Geen restrictie. |
|
Mostert |
Functie: neuroloog |
Lid hoofdredactieraad Tijdschrift voor Neurologie en Neurochirurgie (onbetaald).
Tot mei 2021: 1.5 jaar voorzitter en daarvoor 2 jaar secretaris van de landelijke MS werkgroep (onbetaald). |
Lokale onderzoeker bij (geen PI over gehele studies):
De financiering op contractbasis (per verrichting vergoeding). |
Bij wijzigingen, graag contact opnemen met belangencie (n.a.v. non-inferiority study) |
|
Murk |
Functie: arts-microbioloog Werkgever: Elisabeth-Tweesteden ziekenhuis Tilburg |
Redactielid tijdschrift infectieziekten (onbetaald)
Sectielid infectieziektenserologie SKML, coördinator rondzending H. pylori serologie (Nederlandse organisatie die externe kwaliteitsrondzendingen voor laboratoria organiseert) (onbetaald)
|
Ik werk als arts-microbioloog in vrije vestiging. Het verrichten van laboratoriumdiagnostiek is een onderdeel van de werkzaamheden waardoor een vrijgevestigde arts-microbioloog gefinancierd wordt. |
Geen restrictie. |
|
Pippel |
Functie: Gemeentesecretaris Werkgever: Gemeente Zandvoort |
Lid Adviesplatform MS vereniging |
Geen |
Geen restrictie. |
|
Smolders |
Neuroloog, 0.8 FTE afdeling Neurologie, Erasmus Medisch Centrum Rotterdam |
Bestuurslid MS werkgroep (onbetaald).
Kosteneffectiviteitsonderzoek MS in samenwerking met het bedrijf Huygens & Versteegh |
Onderzoek wordt gefinancierd door de MS research stichting (Voorschoten), het Nationaal MS fonds (Rotterdam) en de Erasmus Foundation (Rotterdam) Van de collectebusfondsen-studies ben ik PI. De bedrijven betreft onderzoeksprojecten met het centrum, waarbij ik niet als PI fungeer. Onderzoeksprojecten binnen MS centrum ErasMS worden gefinancierd door Merck (BTK inhibitor), Idorsia (preklinisch onderzoek compound), GSK (preklinisch onderzoek compound) en Roche (federated learning project).
In verleden:
|
Geen restrictie. |
|
Van Hoeve |
Functie: neuroloog Werkgever: ZorgSaam Zeeuws-Vlaanderen, Terneuzen |
Lid van MS zorg Zuid Zuid West, regionaal samenwerkingsverband tussen neurologen uit verschillende ziekenhuis, met MS als aandachtsgebied (onbetaald). |
April 2023: Binnenkort deelnemend centrum aan SPIN-P studie (gericht op o.a. etiologische/ prognostische factoren m.b.t. primair progressieve MS). Deelname Ectrims congres (oktober 2023) op uitnodiging Merck |
Geen restrictie. |
|
Wijburg |
Functie: AIOS Neurologie, Werkgever: Amsterdam UMC |
Geen |
Geen |
Geen restrictie.
|
|
Huygens (extern adviseur) |
Eigenaar Huygens & Versteegh B.V. |
Tijdelijk senior adviseur ziektemodellen Zorginstituut Nederland (betaald) |
Het ErasmuMC/iMTA MS model is ontwikkeld door Simone Huygens & Matthijs Versteegh met een onderzoeksgrant van ErasmusMC tijdens hun dienstverband bij iMTA Erasmus Universiteit Rotterdam. In hun Huygens & Versteegh B.V. maken zij melding van vergoedingen door IQVIA (niet MS gerelateerd), Beigene (niet MS gerelateerd), Optimax (niet MS gerelateerd), ErasmusMC (niet MS gerelateerd), ICER-US (MS gerelateerd, onafhankelijk expert advies of ICER-US rapport kosteneffectiviteit MS geneesmiddelen), EuroQoL (niet MS gerelateerd), iMTA (niet MS gerelateerd), Takeda (niet MS gerelateerd), Chiesi (niet MS gerelateerd) en Merck KGgA (MS gerelateerd, niet gerelateerd aan geneesmiddelen opgenomen in de richtlijn of het model (nieuwe moleculaire entiteit)). De werkzaamheden voor de richtlijn met het ErasmusMC/iMTA model zijn onbezoldigd uitgevoerd. |
Geen restrictie.
|
|
Versteegh (extern adviseur) |
Eigenaar Huygens & Versteegh B.V. |
Tijdelijk senior adviseur ziektemodellen Zorginstituut Nederland (betaald) Editorial board member Medical Decision Making (onbetaald) Lid EuroQoL research foundation (onbetaald maar met vergoeding reiskosten en congresbezoek) Columnist NTvG (onbetaald) |
Het ErasmuMC/iMTA MS model is ontwikkeld door Simone Huygens & Matthijs Versteegh met een onderzoeksgrant van ErasmusMC tijdens hun dienstverband bij iMTA Erasmus Universiteit Rotterdam. In hun Huygens & Versteegh B.V. maken zij melding van vergoedingen door IQVIA (niet MS gerelateerd), Beigene (niet MS gerelateerd), Optimax (niet MS gerelateerd), ErasmusMC (niet MS gerelateerd), ICER-US (MS gerelateerd, onafhankelijk expert advies of ICER-US rapport kosteneffectiviteit MS geneesmiddelen), EuroQoL (niet MS gerelateerd), iMTA (niet MS gerelateerd), Takeda (niet MS gerelateerd), Chiesi (niet MS gerelateerd) en Merck KGgA (MS gerelateerd, niet gerelateerd aan geneesmiddelen opgenomen in de richtlijn of het model (nieuwe moleculaire entiteit)). De werkzaamheden voor de richtlijn met het ErasmusMC/iMTA model zijn onbezoldigd uitgevoerd. |
Geen restrictie.
|
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door in de voorbereidende fase de MS-patiënten Vereniging Nederland te vragen om schriftelijke input omtrent knelpunten en aandachtspunten. Patiënten werden tevens in de werkgroep vertegenwoordigd door twee afgevaardigden van de MS-patiënten Vereniging Nederland. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen. De conceptrichtlijn is tevens voor commentaar voorgelegd aan Patiëntenfederatie Nederland en MS-patiënten Vereniging Nederland. De eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep de knelpunten in de zorg voor patiënten met MS. De werkgroep beoordeelde de aanbeveling(en) uit de eerdere richtlijnmodule (NVN, 2012; NVN, 2020) op noodzaak tot revisie. Tevens zijn er knelpunten aangedragen via een schriftelijke knelpunteninventarisatie. Een rapportage hiervan kan worden aangevraagd via de Richtlijnendatabase. Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.