Toedieningsroute misoprostol bij medicamenteuze behandeling bij Miskraam
Uitgangsvraag
Wat is de meest passende toedieningsroute van misoprostol bij de medicamenteuze behandeling van een miskraam?
Aanbeveling
Overweeg voor de medicamenteuze behandeling van een miskraam vaginale toediening van 800 microgram misoprostol.
Benadruk dat er een keuze is in de toedieningsvorm (vaginaal, sublinguaal, oraal) van de medicamenten voor de medicamenteuze behandeling van een miskraam.
Overwegingen
De onderstaande overwegingen en aanbevelingen gelden voor het overgrote deel van de populatie waarop de uitgangsvraag betrekking heeft.
Vergelijking 1. Oraal vs. vaginaal
Primaire uitkomsten
De uitkomstmaat succesvolle behandeling, is door vier studies onderzocht (Creinin, 1997; Ngoc, 2014; Rita, 2006; Marwah, 2016). Er lijkt voor de succesvolle medicamenteuze behandeling van een miskraam geen verschil in orale of vaginale toediening van misoprostol.
De resultaten van de vier studies laten zien dat orale misoprostol naar alle waarschijnlijkheid minder effectief is dan vaginale misoprostol voor een succesvolle behandeling van een miskraam. De GRADE-beoordeling liet een groot risico op bias en inconsistenties zien. Dit gezien verschillen tussen de studies in zwangerschapsduur, toegepaste misoprostol regime en timing van uitkomst metingen. In geen van de studies was sprake van adequate blindering. Het mogelijke verschil in effect ten faveure van vaginale misoprostol, wordt vooral gezien in studies waar dosering misoprostol tussen orale en vaginale groep verschilt en erbij de vaginale een hogere dosering wordt gebruikt.
De werkgroep is van mening dat er een voorkeur bestaat voor toediening van vaginale misoprostol bij de medicamenteuze behandeling van een miskraam.
In figuur 3, de forest plot voor de uitkomstmaat pijn, is te zien dat orale misoprostol mogelijk iets vaker pijn veroorzaakt dan vaginale misoprostol hoewel de bewijskracht voor dit minimale verschil gezien de beperkingen in onderzoeksopzet, overlap van het 95% betrouwbaarheidsinterval (imprecisie) en de grote mate van heterogeniteit van de studies uiteindelijk uitkomt op GRADE laag.
Secundaire uitkomsten
Er lijken geen verschillen in de incidentie van bijwerkingen tussen orale of vaginale toediening van misoprostol. Hoewel er geen verschil is in patiënttevredenheid tussen beiden groepen, is de mate van bewijskracht zeer laag aangezien maar twee studies hierover rapporteerden.
De werkgroep is van mening dat er ondanks de beperkte evidence, mogelijk een minimale voorkeur bestaat voor vaginale misoprostol ten opzichte van orale misoprostol ten aanzien van het optreden van bijwerkingen zoals pijn bij de medicamenteuze behandeling van een miskraam.
Vergelijking 2. Sublinguaal vs. vaginaal
Sublinguale toediening van misoprostol lijkt niet te leiden tot een hoger percentage succesvolle behandelingen vergeleken met vaginale toediening bij patiënten met een miskraam (studies van Tanha, 2010; Tang, 2003; Shah, 2010, El Sokkary, 2016; Dehbasi, 2016; Seervi, 2014; Akanksha, 2016, Sonsanoh, 2014). In het algemeen lijken bijwerkingen vaker voor te komen bij sublinguale toediening in vergelijking met vaginale toediening. Dit geld ook voor de voor de uitkomstmaten pijn en diarree.
De huidige richtlijn komt wat betreft geïncludeerde studies in grote lijnen overeen met de recent verschenen Cochrane aangaande deze vergelijking. Beiden hebben de studies van Dehbasi (2016), Shah (2010), Tanha (2010) en Tang geïncludeerd. De huidige richtlijn includeert de studie van Akanksha (2016) en Seervi (2014), beide niet gevonden in de search Cochrane, maar deze studies voldoen wel aan vooraf gestelde inclusie criteria. De studie van El Sokkary uit 2016 is door de Cochrane geëxcludeerd, omdat het randomisatie proces onvoldoende beschreven wordt. De werkgroep is van mening dat het voldoende aannemelijk is dat er is gerandomiseerd is tussen een sublinguale groep en een vaginale groep, hoewel we het er mee eens zijn dat de exacte methode van randomisatie niet wordt beschreven. Opvallend is verder dat er in deze studie een lagere dosering misoprostol dan in overige studies wordt gebruik namelijk viermaal 100 microgram in beide groepen en de follow-up al plaats vindt 24 uur na de laatste dosis. Mogelijk kan dit verklaren waarom er in deze studie in beide groepen een relatief laag percentage succesvolle behandeling wordt gevonden en een voorkeur voor de sublinguale groep.
De werkgroep is van mening dat er op basis van de geïncludeerde studies, voor de medicamenteuze behandeling van een miskraam, een voorkeur bestaat voor vaginale toediening van misoprostol vergeleken met sublinguale toediening.
Vergelijking 3. Oraal vs. sublinguaal
Er lijkt geen verschil in het percentage succesvolle behandeling van een miskraam bij orale toediening van misoprostol in vergelijking met sublinguale toediening. Braken lijkt vaker voor te komen bij orale toediening. Er lijken geen verschillen ten aanzien van de andere secundaire uitkomstmaten.
De studies geïncludeerd voor het beantwoorden van deze vergelijking in de huidige richtlijn en in de recent verschenen Cochrane komen volledig met elkaar overeen. Er zijn hierbij twee studies geïncludeerd. Dit maakt dat de bewijskracht qua gradering laag tot zeer laag is.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Succesvolle behandeling en secundaire uitkomstmaten.
In deze vraag is succesvolle behandeling gedefinieerd als een volledige expulsie van miskraam. Er zijn er drie subvragen gesteld ten aanzien van een toedieningsroute van misoprostol. Het betrof vergelijking van 1) misoprostol toegediend oraal vs. vaginaal, 2) sublinguaal vs. vaginaal en 3) oraal vs. sublinguaal. Na een gerichte search zijn er uiteindelijk vijf studies geïncludeerd, die de orale vs. vaginale route vergeleken, acht studies m.b.t. sublinguaal versus vaginaal toediening en twee studies die naar orale versus sublinguale route keken.
Bij vergelijking 1 (oraal versus vaginaal) liet het gepoolde effect liet dat in de orale groep het percentage succesvolle behandeling 69% (147/213) was en in de vaginale groep 89% (184/207) patiënten. Dit leidde tot een risk ratio van 0,66 (95% BI 0,45 – 0,98, p=0,04) met een random effect model en met een hoge heterogeniteit (I2 90%). Hetgeen inhoudt dat er een verschil is tussen de twee interventiegroepen.
Bij vergelijking 2 (sublinguaal versus vaginaal) liet het gepoolde effect zien dat in de sublinguale groep het percentage succesvolle behandeling 73% (307/423) was en in de vaginale groep 59% (247/419) patiënten (figuur 1). Dit leidde tot een risk ratio van 1,19 (95% BI 0,97 – 1,46, p=0,09) met een random effect model en met een hoge heterogeniteit (I2 79%). Hetgeen inhoudt dat er geen verschil is tussen de twee interventiegroepen.
Bij vergelijking 3 (oraal versus sublinguaal) liet het gepoolde effect zien dat in de orale groep het percentage succesvolle behandeling 51% (59/116) was en in de sublinguale groep 51% (61/120) patiënten. Dit leidde tot een risk ratio van 0,96 (95% BI 0,72 – 1,27, p=0,77) met een random effect model en met een lage heterogeniteit (I2 28%). Hetgeen inhoudt dat er geen verschil is tussen de twee interventiegroepen.
Alle studies lieten geen significant effect zien ten faveure voor een specifieke toedieningsroute met betrekking tot een succesvolle behandeling – volledige expulsie miskraam.
De bewijskracht voor de primaire uitkomstmaat - route voor een succesvolle behandeling heeft een lage gradering van evidence. Dit is te verklaren door inconsistentie in resultaten en gebrek aan blindering in studiedesign van de beschreven studies.
Vaginale en orale toediening van misoprostol leidt tot vergelijkbare (vergelijking 1) algemene complicaties, diarree en pijn lijken vaker voor te komen bij sublinguale toediening dan vaginale toediening (vergelijking 2). Braken lijkt vaker voor te komen bij orale dan bij sublinguale toediening (vergelijking 3). Het is onduidelijk of er meer pijn ontstaat bij orale toediening en er lijkt geen verschil in frequentie optreden van diarree tussen deze twee routes.
De werkgroep is van mening dat er op basis van deze drie vergelijkingen een voorkeur is voor behandeling van een miskraam met misoprostol via een vaginale toedieningsroute.
Dosering misoprostol
De gebruikte doseringen misoprostol in de geïncludeerde studies vertonen een grote variatie. Ditzelfde geldt voor de Nederlandse praktijk. In de studies worden verschillende maximum doseringen gebruikt. Deze varieerde van 400 microgram (El Sokkary, 2016) tot 2400 microgram (Ayudhaya, 2006). Ook het aantal giften varieerde van een tot zes keer net zoals de dosis per gift (100 microgram tot 800 microgram).
In de NICE-richtlijn beveelt men een dosering van 800 microgram en in een uitgebreide netwerk meta-analyse (Wu, 2017) dat zowel 800 microgram vaginaal als 600 microgram sublinguaal kunnen worden gezien als eerste keus.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Het belangrijkste doel van de medicamenteuze behandeling van een miskraam is een effectieve en succesvolle behandeling met zo weinig mogelijk complicaties en noodzaak tot re-interventies met daarbij zo groot mogelijke patiënttevredenheid. De evidence voor deze uitkomstmaat is laag tot zeer laag. Er lijkt geen verschil in patiënttevredenheid te zijn voor patiënten tussen behandeling van miskraam met misoprostol vaginaal of oraal. Ditzelfde geldt voor sublinguaal of vaginale toediening van misoprostol. Het is onduidelijk of er verschil bestaat in patiënttevredenheid tussen orale of sublinguale toediening van misoprostol.
Kosten (middelenbeslag)
De gekozen toedieningsroute van medicamenteuze behandeling van miskraam brengt, naar verwachting, geen verschil in kosten met zich mee.
Aanvaardbaarheid voor de overige relevante stakeholders
In de studies uit de literatuuranalyse zijn geen lange termijn effecten van deze behandelingen beschreven. De medicatie wordt in een tijdsbestek van een paar dagen gebruikt en de van de werkzame stoffen worden geen lange termijn nadelige effecten verwacht.
Haalbaarheid en implementatie
Het huidige behandelbeleid voor medicamenteuze behandeling van een miskraam - ook wel niet-vitale zwangerschap genoemd - kent grote praktijkvariatie. De verschillen zijn onder andere te vinden in toedieningsroute van misoprostol. De werkgroep adviseert bij de medicamenteuze behandeling van een miskraam misoprostol via de vaginale toedieningsroute voor te schrijven. De verwachting is dat de aanbeveling van de werkgroep geen implementatieproblemen met zich mee zal brengen. De werkgroep hoopt juist met deze aanbeveling de bestaande praktijkvariatie te verminderen.
Rationale/ balans tussen de argumenten voor en tegen de interventie
Vanuit gepoolde resultaten lijkt er geen klinisch relevant verschil te zijn met betrekking tot succesvolle expulsie van miskraam tussen drie verschillende toedieningsroutes (oraal versus vaginaal, sublinguaal versus vaginaal en oraal versus sublinguaal). In het geval van bijwerkingen lijkt dat braken, diarree of pijn vergelijkbaar te zijn tussen orale en vaginale route. Echter algemene complicaties, diarree en pijn lijken vaker voor te komen bij sublinguale toediening vergelijkend met vaginale toediening en braken lijkt vaker voor te komen bij een orale toediening versus sublinguaal toediening. Hierdoor lijkt vaginale route mogelijk superieur ten opzichte van orale of sublinguale route. De werkgroep is van mening dat op basis van deze drie vergelijkingen er een voorkeur is voor behandeling van een miskraam met misoprostol via een vaginale route.
De gebruikte doseringen misoprostol in de geïncludeerde studies vertonen grote variatie. Ditzelfde geldt voor de Nederlandse praktijk. Internationaal bestaan er richtlijnen voor de optimale dosering misoprostol. Deze is afhankelijk van de zwangerschapsduur, de toedieningsroute en de indicatie. De internationale richtlijn (Morris, 2017) van de International Federation of Obstetrics and Gynecology (FIGO) en de WHO, maakt een onderscheid gemaakt tussen < 13 weken zwangerschapsduur en 13-26 weken zwangerschapsduur. Voor niet-vitale zwangerschappen met een zwangerschapsduur < 13 weken was de aanbeveling 800 microgram vaginaal elke 3 uur of 600microgram sublinguaal elke 3 uur (2x) te geven.
De NICE-richtlijn (NICE, 2012) adviseert 800 microgram vaginale misoprostol voor de behandeling van een miskraam. Een netwerk meta-analyse (Wu, 2017) over de behandeling van een miskraam met alleen misoprostol laat zien dat zowel 800 microgram vaginaal als 600 microgram sublinguaal op dit moment worden gezien als eerste keus. Lagere doseringen waren minder effectief. De orale toedieningsroute liet meer bijwerkingen zien. Over de optimale dosering en het medicatie-interval is geen consensus, hiernaar is verder onderzoek nodig. Indien er toch voor sublinguale toediening wordt gekozen heeft een dosis van 600 microgram de voorkeur.
Onderbouwing
Misoprostol is een prostaglandine E1 analoog en is geregistreerd als maagbeschermer. Het heeft als belangrijke bijwerkingen rijping van de cervix en uterus contracties. Dit medicament wordt reeds gebruikt voor medicamenteuze behandeling van een miskraam en wordt ook wisselend gebruikt ter voorbereiding van de cervix voor een hysteroscopie. De werkzaamheid en het percentage succesvolle behandelingen van misoprostol is afhankelijk van de gebruikte dosering, aantal herhalingen en het gebruik in combinatie met andere geneesmiddelen zoals mifepriston (beschreven in module 4a Mifepriston + misoprostol vs. misoprostol).
Voor de medicamenteuze behandeling van een miskraam, kan Misoprostol worden gebruikt via verschillende toedieningsroutes zoals oraal, sublinguaal of vaginaal. Het is echter nog steeds niet duidelijk welke route het meest effectief is. Daarbij zijn voor de vrouw gebruiksgemak, het optreden van bijwerkingen en tevredenheid van belang. Meer informatie over misoprostol is te vinden op www.misoprostol.org.
Vergelijking 1. Oraal vs. vaginaal
1.1. Uitkomstmaat succesvolle behandeling (cruciaal)
|
Redelijk GRADE |
Vaginale toediening van misoprostol leidt tot een hoger percentage succesvolle behandelingen vergeleken met orale toediening bij patiënten met een miskraam.
Bronnen: (Creinin, 1997; Marwah, 2016; Ngoc, 2014; Rita, 2006;) |
1.2 Uitkomstmaat bijwerkingen/complicaties: algemeen (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of orale toediening van misoprostol leidt tot meer bijwerkingen/complicaties in het algemeen vergeleken met vaginale toediening bij patiënten met een miskraam.
Bron: (Creinin, 1997) |
1.3. Uitkomstmaat bijwerkingen: braken (cruciaal)
|
Laag GRADE |
Braken lijkt niet vaker op te treden bij orale toediening van misoprostol vergeleken met vaginale toediening bij patiënten met een miskraam.
Bronnen: (Creinin, 1997; Ngoc, 2014; Rita, 2006) |
1.4. Uitkomstmaat bijwerkingen: diarree (cruciaal)
|
Redelijk GRADE |
Diarree treedt waarschijnlijk niet vaker op bij orale toediening van misoprostol vergeleken met vaginale toediening bij patiënten met een miskraam.
Bronnen: (Creinin, 1997; Ngoc, 2014; Rita, 2006; Marwah, 2016, Sonsanoh, 2014) |
1.5. Uitkomstmaat bijwerkingen: pijn (cruciaal)
|
Laag GRADE |
Pijn lijkt even vaak aanwezig te zijn bij orale toediening van misoprostol vergeleken met vaginale toediening bij patiënten met een miskraam.
Bronnen: (Creinin, 1997; Marwah, 2016; Ngoc, 2014; Rita, 2006) |
1.6. Uitkomstmaat patiënttevredenheid (belangrijk)
|
Redelijk GRADE |
Patiënten met een miskraam zijn waarschijnlijk even tevreden bij medicamenteuze behandeling met misoprostol met orale toediening als met vaginale toediening.
Bronnen: (Ngoc, 2014; Marwah, 2016) |
Vergelijking 2. Sublinguaal vs. vaginaal
2.1. Uitkomstmaat succesvolle behandeling (cruciaal)
|
Laag GRADE |
Sublinguale toediening van misoprostol lijkt niet te leiden tot een hoger percentage succesvolle behandelingen vergeleken met vaginale toediening bij patiënten met een miskraam.
Bronnen: (Tanha, 2010; Tang, 2003; Shah, 2010, El Sokkary, 2016; Dehbasi, 2016; Seervi, 2014; Akanksha, 2016, Sonsanoh, 2014) |
2.2. Uitkomstmaat bijwerkingen/complicaties: algemeen (cruciaal)
|
Laag GRADE |
Bijwerkingen/complicaties in het algemeen lijken vaker voor te komen bij sublinguale toediening van misoprostol vergeleken met vaginale toediening.
Bronnen: (Shah, 2010; Dehbasi, 2016) |
2.3. Uitkomstmaat bijwerkingen: braken (cruciaal)
|
Laag GRADE |
Braken lijkt niet vaker op te treden bij sublinguale toediening van misoprostol vergeleken met vaginale toediening bij patiënten met een miskraam.
(Tanha, 2010; Tang, 2003; Seervi, 2014; Akanksha, 2016; Dehbasi, 2016) |
2.4. Uitkomstmaat bijwerkingen: diarree (cruciaal)
|
Redelijk GRADE |
Diarree treedt waarschijnlijk vaker op bij sublinguale toediening van misoprostol vergeleken met vaginale toediening bij patiënten met een miskraam.
Bronnen: (Tanha, 2010; Tang, 2003; Dehbasi, 2016; Seervi, 2014; Akanksha, 2016, Sonsanoh, 2014) |
2.5. Uitkomstmaat bijwerkingen: pijn (cruciaal)
|
Laag GRADE |
Pijn lijkt vaker op te treden bij sublinguale toediening van misoprostol vergeleken met vaginale toediening bij patiënten met een miskraam.
Bronnen: (Tanha, 2010; Tang, 2003; El Sokkary, 2016; Dehbasi, 2016; Seervi, 2014, Sonsanoh, 2014) |
2.6. Uitkomstmaat patiënttevredenheid (belangrijk)
|
Laag GRADE |
Patiënten met een miskraam lijken bij behandeling met misoprostol even tevreden te zijn met sublinguale toediening als met vaginale toediening.
Bronnen: (Tanha, 2010; Tang, 2003; Shah, 2010; Seervi, 2014; Akanksha, 2016) |
Vergelijking 3. Oraal vs. sublinguaal
3.1. Uitkomstmaat succesvolle behandelingen (cruciaal)
|
Laag GRADE |
Orale toediening van misoprostol lijkt niet te leiden tot een hoger percentage succesvolle behandelingen vergeleken met sublinguale toediening bij patiënten met een miskraam.
Bronnen: (Kushwah, 2009; Ayudhaya, 2006) |
3.2. Uitkomstmaat bijwerkingen/complicaties: algemeen (cruciaal)
|
- GRADE |
Er is geen GRADE beoordeling in verband met het ontbreken van studies |
3.3. Uitkomstmaat bijwerkingen: braken (cruciaal)
|
Laag GRADE |
Braken lijkt vaker voor te komen bij orale toediening van misoprostol vergeleken met sublinguale toediening bij patiënten met een miskraam.
Bron: (Kushwah, 2009) |
3.4. Uitkomstmaat bijwerkingen: diarree (cruciaal)
|
Laag GRADE |
Diarree lijkt niet vaker op te treden bij orale toediening van misoprostol vergeleken met sublinguale toediening bij patiënten met een miskraam.
Bronnen: (Kushwah, 2009; Ayudhaya, 2006) |
3.5. Uitkomstmaat bijwerkingen: pijn (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of er een verschil is in pijn bij orale toediening van misoprostol vergeleken met sublinguale toediening bij patiënten met een miskraam.
Bronnen: (Kushwah, 2009; Ayudhaya, 2006) |
3.6. Uitkomstmaat patiënttevredenheid (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of patiënten met een miskraam die behandeld worden met misoprostol vaker tevreden zijn bij orale toediening vergeleken met sublinguale toediening.
Bron: (Kushwah, 2009) |
3.7. Uitkomstmaat lange termijn gevolgen (belangrijk)
|
- GRADE |
Er is geen GRADE beoordeling voor deze uitkomstmaat in verband met het ontbreken van studies. |
Er zijn vier studies die het effect van orale toediening van misoprostol vergeleken met vaginale toediening, acht studies die het effect sublinguale toediening vergeleken met vaginale toediening en twee studies die het effect van orale toediening vergeleken met sublinguale toediening. De doseringen misoprostol per studie staan vermeld in tabel 1.
Vergelijking 1. Oraal vs. vaginaal
Creinin (1997) is een gerandomiseerde trial uitgevoerd in de Verenigde Staten waarin patiënten werden geïncludeerd met een vroege miskraam van minder dan 8 weken (N=20). Interventiegroep 1 (N=12) ontving tweemaal een orale dosis van 400 microgram misoprostol. Interventiegroep 2 (N=8) ontving tweemaal een vaginale dosis van 800 microgram (vier tabletten van 200 microgram). Tussen de doseringen zat 24 uur. De groepen hadden vergelijkbare patiëntkarakteristieken maar er werd niet beschreven waarom de aantallen tussen de groepen verschillen. Patiënten en behandelaren waren niet geblindeerd.
In de gerandomiseerde studie van Marwah (2016) uitgevoerd in India werden patiënten met een niet-vitale zwangerschap met een zwangerschapsduur van ≤ 12 weken geïncludeerd (N=100). De interventiegroep 1 (N=50) ontving driemaal een orale dosis van 400 microgram misoprostol. Interventiegroep 2 (N=50) ontving driemaal een vaginale dosis van 400 microgram misoprostol. Tussen de doseringen zat zes uur. Na vaginale toediening werd patiënten gevraagd drie uur in liggende positie te blijven.
In de gerandomiseerde studie van Ngoc (2014) uitgevoerd in Vietnam werden patiënten geïncludeerd met een eerste trimester niet-vitale zwangerschap (N=200). Interventiegroep 1 (N=101) ontving een eenmalige orale dosis van 800 microgram misoprostol (vier tabletten van 200 microgram). De interventiegroep 2 (N=99) ontving een eenmalige vaginale dosis van 800 microgram misoprostol (4 tabletten van 200 microgram). Patiënten en behandelaren waren niet geblindeerd. Daarnaast waren twee patiënten geïncludeerd met een verkeerde diagnose.
In de gerandomiseerde studie van Rita (2006) uitgevoerd in India werden patiënten met een niet-vitale zwangerschap geïncludeerd met een zwangerschapsduur van <13 weken (N=100). Interventiegroep 1 (N=50) ontving driemaal een orale dosis van 400 microgram misoprostol, tussen de doses zat vier uur. Interventiegroep 2 (N=50) ontving tweemaal een vaginale dosis van 600 microgram misoprostol, tussen de doses zat vier uur. Patiënten en behandelaren waren niet geblindeerd. Daarnaast was het niet duidelijk hoe en wanneer bijwerkingen werden beoordeeld.
Vergelijking 2. Sublinguaal vs. vaginaal
In de gerandomiseerde studie van Akanksha (2016) uitgevoerd in India werden patiënten met een niet-vitale zwangerschap geïncludeerd (N=50). Interventiegroep 1 (N=25) ontving vijfmaal een sublinguale dosis van 400 microgram misoprostol. Interventiegroep 2 (N=25) ontving vijfmaal een vaginale dosis van 400 microgram misoprostol. Tussen de vijf dosis zat vier uur. De twee groepen waren vergelijkbaar wat betreft de patiëntkarakteristieken, er was in beide groepen geen loss-to-follow-up. Een beperking van de studie was dat er niets vermeld staat over blindering van patiënten en behandelaren. Een andere beperking is dat uitkomstmaten niet beschreven zijn in de methoden sectie.
In de gerandomiseerde, enkel-geblindeerde studie van Dehbasi (2016) die is uitgevoerd in Iran werden patiënten met een intra-uteriene embryonale dood of niet-vitale zwangerschap in het eerste trimester geïncludeerd (N=52) Interventiegroep 1 (N=27) ontving vijfmaal een sublinguale dosis van 400 microgram misoprostol. Interventiegroep 2 (N=25) ontving vijfmaal een vaginale dosis van 400 microgram misoprostol. Tussen de doses zat vier uur. Bij deze studie werd percentage succesvolle behandeling gedefinieerd als een expulsie die binnen 24 uur plaatsvond. De belangrijkste beperking van de studie was dat twee patiënten uit de sublinguale groep werden geëxcludeerd met als reden dat er een patiënt niet reageerde op de medicatie en een patiënt ernstige buikpijn kreeg.
In de gerandomiseerde studie van El Sokkary (2016), die is uitgevoerd in Egypte, werden patiënten met een niet-vitale zwangerschap geïncludeerd (N=160). Interventiegroep 1 (N=80) ontving viermaal een sublinguale dosis van 100 microgram misoprostol. Interventiegroep 2 (N=80) ontving viermaal een vaginale dosis van 100 microgram misoprostol. Tussen de doses zat 4 uur. Een beperking van de studie is dat patiënten en behandelaren niet geblindeerd waren, de wijze van randomisatie niet is beschreven en er weinig patiëntkarakteristieken worden gepresenteerd.
In de gerandomiseerde studie van Seervi (2014) uitgevoerd in India werden patiënten met een niet-vitale zwangerschap (79,9%) of een lege vruchtzak (20,91%) geïncludeerd (N=110). Interventiegroep 1 (N=54) ontving driemaal een sublinguale dosis van 600 microgram misoprostol. Interventiegroep 2 (N=56) ontving driemaal een vaginale dosis van 800 microgram misoprostol. Tussen de drie dosis zat 6 uur. De twee groepen waren vergelijkbaar wat betreft de patiëntkarakteristieken, ook de loss-to-follow-up was gelijk (interventiegroep 1 - 3,36%, interventiegroep 2 - 4,20%). Een beperking van de studie was dat er niets vermeld was over blindering van patiënten en behandelaren.
In de gerandomiseerde trial van Shah (2010), die is uitgevoerd in Pakistan, werden patiënten met een niet-vitale zwangerschap <20 weken zwangerschap geïncludeerd (N=50). In interventiegroep 1 hadden 22/25 patiënten een zwangerschapsduur van <12 weken en in interventiegroep 2 19/25 patiënten. Interventiegroep 1 (N=25) ontving vijfmaal een sublinguale dosis van 400 microgram misoprostol. Interventiegroep 2 (N=25) ontving vijfmaal een vaginale dosis van 400 microgram misoprostol. Tussen de doses zat 3 uur. Een beperking van de studie is dat een deel van de patiënten een langere zwangerschap had dan 12 weken (indirectness of population). Daarnaast waren patiënten en behandelaren niet geblindeerd.
Tang (2003) is een gerandomiseerde trial die is uitgevoerd in China waarbij patiënten met een niet-vitale zwangerschap in het eerste trimester werden geïncludeerd (N=80). Interventiegroep 1 (N=40) ontving driemaal een sublinguale dosis van 600 microgram misoprostol. Interventiegroep 2 (N=40) ontving driemaal een vaginale dosis van 600 microgram misoprostol. Tussen de drie doses zat 3 uur. De groepen waren vergelijkbaar wat betreft patiëntkarakteristieken. De loss-to-follow up wordt niet vermeld. De belangrijkste beperking van de studie is dat de patiënten en behandelaren niet waren geblindeerd. Daarnaast is niet duidelijk waarom niet alle patiënten participeerden in het tevredenheidsonderzoek.
In de gerandomiseerde studie van Tanha (2010) uitgevoerd in Iran werden patiënten met een niet-vitale zwangerschap in het eerste trimester geïncludeerd (N=220). Interventiegroep 1 (N=110) ontving sublinguaal 400 microgram misoprostol. Interventiegroep 2 (N=110) ontving vaginaal 400 microgram misoprostol. Het aantal doses dat patiënten maximaal kregen staat niet beschreven, wel dat interventiegroep 1 gemiddeld 4,45 tabletten kregen en interventiegroep 2 - 4,85 tabletten. Tussen de doses zat zes uur. De groepen waren alleen verschillend op baseline wat betreft het aantal pariteiten (0,23 in de sublinguale groep versus 0,44 in de vaginale groep, p=0,013).
In de gerandomiseerde studie van Sonsanoh (2014), uitgevoerd in Thailand, werden patiënten met een niet-vitale zwangerschap geïncludeerd. Interventiegroep 1 (N=60) ontving sublinguaal 4 tabletten van 200 microgram misoprostol. Interventiegroep 2 (N=60) ontving vaginaal 4 tabletten van 200 microgram misoprostol. Deze dosering werd na 6 uur herhaald met een maximum van 3 doseringen. De groepen waren verschillend op baseline wat betreft BMI, gewichtstoename en pariteit. Een beperking van de studie is dat in de methode wordt beschreven dat patiënt tevredenheid op een driepuntschaal wordt gescoord terwijl in de resultaten alleen staat beschreven dat er geen verschil was tussen de groepen.
Vergelijking 3. Oraal vs. sublinguaal
In de gerandomiseerde trial van Ayudhaya (2006) uitgevoerd in Thailand werden patiënten met een vroege miskraam en een zwangerschapsduur van 7 tot 12 weken geïncludeerd (N= 138). Interventiegroep 1 (N=68) ontving zesmaal oraal een dosis van 400 microgram misoprostol. Interventiegroep 2 (N=70) ontving zesmaal sublinguaal een dosis van 400 microgram misoprostol. Tussen de doses zat 4 uur. De twee groepen waren vergelijkbaar wat betreft de patiëntkarakteristieken. Een beperking van de studie was dat er niets vermeld is over blindering van patiënten en behandelaren.
In de gerandomiseerde trial van Kushwah (2009) uitgevoerd in India werden patiënten met een vroege miskraam en een zwangerschapsduur van 7 tot 14 weken geïncludeerd (N= 100). Interventiegroep 1 (N=50) ontving eenmalig oraal een dosis van 200 milligram mifepriston gevolgd door 600 microgram misoprostol oraal. Interventiegroep 2 (N=50) ontving eenmalig oraal een dosis van 200 milligram mifepriston gevolgd door 600 microgram misoprostol sublinguaal. De twee groepen waren vergelijkbaar wat betreft de patiëntkarakteristieken. Na 12 uur ontving interventiegroep 1 driemaal oraal een dosis van 400 microgram misoprostol. Interventiegroep 2 ontving driemaal sublinguaal een dosis van 400 microgram misoprostol. Tussen de doses zat 3 uur. Een beperking van de studie was dat er niets vermeld staat over blindering van patiënten en behandelaren.
Tabel 1. Dosering en toediening misoprostol per studie. µg: microgram.
|
Studie |
Toedieningsroute |
Tijd tussen de doseringen |
|||
|
Oraal versus vaginaal |
|||||
|
|
Oraal |
Vaginaal |
|
||
|
|
Max dosering |
Toediening |
Max dosering |
Toediening |
|
|
Creinin, 1997 |
800 µg |
2 x 400 µg |
1600 µg |
2 x 800 µg |
24 uur |
|
Marwah, 2016 |
1200 µg |
3 x 400 µg |
1200 µg |
3 x 400 µg |
6 uur |
|
Ngoc, 2014 |
800 µg |
1 x 800 µg |
800 µg |
1 x 800 µg |
- |
|
Rita, 2006 |
1200 µg |
3 x 400 µg |
1200 µg |
2 x 600 µg |
4 uur |
|
Sublinguaal versus vaginaal |
|||||
|
|
Sublinguaal |
Vaginaal |
|
||
|
|
Max dosering |
Toediening |
Max dosering |
Toediening |
|
|
Akanksha, 2016 |
2000 µg |
5 x 400 µg |
2000 µg |
5 x 400 µg |
4 uur |
|
Dehbasi, 2016 |
2000 µg |
5 x 400 µg |
2000 µg |
5 x 400 µg |
4 uur |
|
El Sokkary, 2016 |
400 µg |
4 x 100 µg |
400 µg |
4 x 100 µg |
4 uur |
|
Seervi, 2014 |
1800 µg |
3 x 600 µg |
2400 µg |
3 x 800 µg |
6 uur |
|
Shah, 2010 |
2000 µg |
5 x 400 µg |
2000 µg |
5 x 400 µg |
3 uur |
|
Sonsanoh, 2014 |
2400 µg |
3 x 800 µg |
2400 µg |
3 x 800 µg |
6 uur |
|
Tang, 2003 |
1800 µg |
3 x 600 µg |
1800 µg |
3 x 600 µg |
3 uur |
|
Tanha, 2010 |
gem 890 µg? |
? (gem. 4,45 tablet) x 400 µg |
gem 970 µg? |
? (gem. 4,85 tablet) x 400 µg |
6 uur |
|
Oraal versus sublinguaal |
|||||
|
|
Oraal |
Sublinguaal |
|
||
|
|
Max dosering |
Toediening |
Max dosering |
Toediening |
|
|
Ayudhaya, 2006 |
2400 µg |
6 x 400 µg |
2400 µg |
6 x 400 µg |
4 uur |
|
Kushwah, 2009 |
1800 µg |
600 µg + (na 12 uur) 3 x 400 µg |
1800 µg |
600 µg + (na 12 uur) 3 x 400 µg |
3 uur |
Resultaten
Vergelijking 1. Oraal vs. vaginaal
1.1. Uitkomstmaat succesvolle behandeling (cruciaal)
De uitkomstmaat succesvolle behandeling, is door vier studies onderzocht (Creinin, 1997; Ngoc, 2014; Rita, 2006; Marwah, 2016). Succesvolle behandeling, werd gedefinieerd als volledige expulsie na volledige dosering. Het gepoolde effect laat zien dat in de orale groep het percentage succesvolle behandeling 69% (147/213) was en in de vaginale groep 89% (184/207) patiënten (figuur 1) Dit leidde tot een risk ratio van 0,66 (95% BI 0,45 – 0,98, p=0,04) met een random effect model en met een hoge heterogeniteit (I2 90%). Hetgeen inhoudt dat er een klinisch relevant verschil is tussen de twee interventiegroepen.
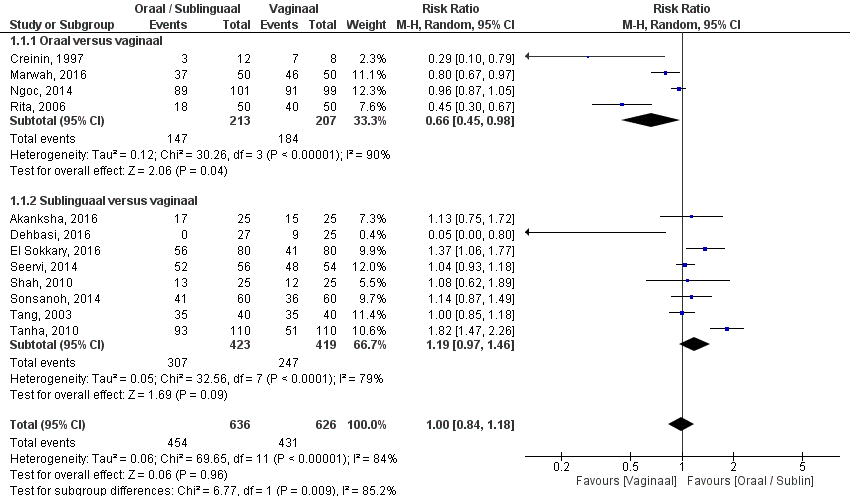
Figuur 1. Uitkomstmaat succesvolle behandeling vergelijking oraal/ sublinguaal versus vaginaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval; O: oraal; S: sublinguaal; V: vaginaal. Let op! Een hoger risico betekent in dit geval meer kans op succes, daarom staat voor deze uitkomstmaat vaginaal aan de linkerkant van de grafiek en oraal/sublinguaal aan de rechterkant van de grafiek.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘succesvolle behandeling’ is met een niveau verlaagd gezien overlap van het 95% BI met de grenzen voor klinische relevantie (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘redelijk’.
1.2. Uitkomstmaat complicaties: algemeen (cruciaal)
Deze uitkomstmaat is door één studie onderzocht. Creinin (1997) liet patiënten noteren of ze last hadden van misselijkheid, braken, diarree, buikpijn en/of bloedingen en vond dat in de orale groep het percentage bijwerkingen/complicaties 67% (8/12) patiënten was en in de vaginale groep 87% (7/8) patiënten, hetgeen inhoudt dat er geen klinisch relevant significant verschil is (p=0,27).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen/complicaties’: algemeen is met drie niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en het geringe aantal patiënten (-2, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
1.3. Uitkomstmaat bijwerkingen: braken (cruciaal)
De uitkomstmaat braken is door vier studies onderzocht (Creinin, 1997; Marwah, 2016; Ngoc, 2014; Rita, 2006). Marwah rapporteerde braken gecombineerd met misselijkheid en vond dat 72% (36/50) patiënten in de orale groep en 60% (30/50) patiënten in de vaginale groep last had van misselijkheid dan wel braken. Het gepoolde effect van de vier studies die braken apart rapporteerden (Creinin, 1997; Ngoc, 2014; Rita, 2006) laat zien dat 8% (13/157) van de patiënten klachten van braken had en in de vaginale groep 11% (18/163) (figuur 2). Dit leidde tot een risk ratio van 0,86 (95% BI 0,16 – 4,52, p=0,85) met een random effect model en met een hoge heterogeniteit (I2 75%). Hetgeen inhoudt dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
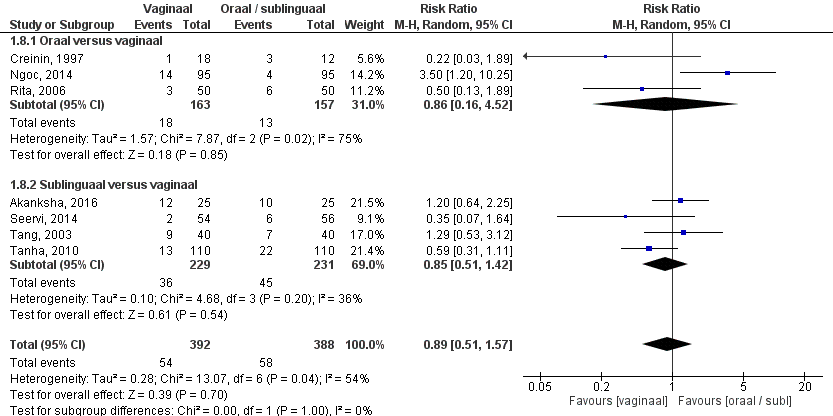
Figuur 2. Uitkomstmaat braken vergelijking oraal/ sublinguaal versus vaginaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval; O: oraal; S: sublinguaal; V: vaginaal.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat braken is met twee niveaus verlaagd gezien tegenstrijdige resultaten (-1, inconsistentie) en gezien overlap van het 95% BI met de grenzen voor klinische relevantie (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
1.4. Uitkomstmaat bijwerkingen: diarree (cruciaal)
De uitkomstmaat diarree is door vier studies onderzocht (Creinin, 1997; Ngoc, 2014; Rita, 2006; Marwah, 2016). Het gepoolde effect laat zien dat in de orale groep het percentage diarree 19% (41/207) patiënten was en in de vaginale groep 18% (37/203) patiënten (figuur 3). Dit leidde tot een risk ratio van 0,94 (95% BI 0,64 – 1,39, p=0,76) met een random effect model en met een lage heterogeniteit (I2 0%). Hetgeen inhoudt dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
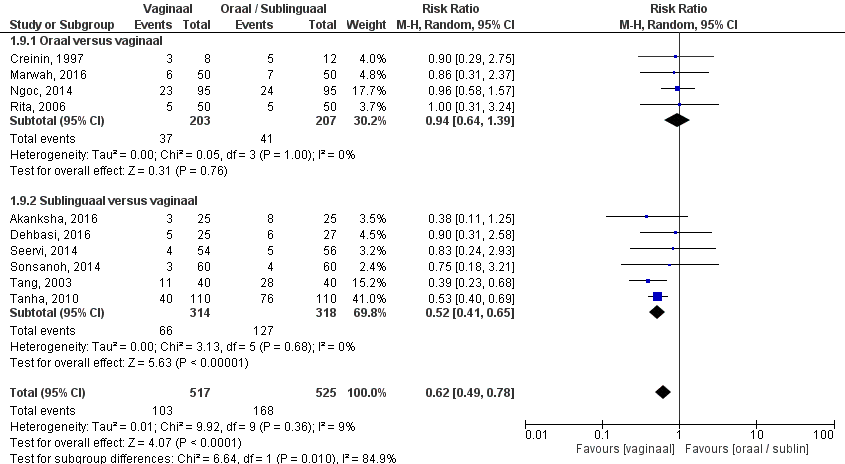
Figuur 3. Uitkomstmaat diarree vergelijking oraal/ sublinguaal versus vaginaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen - diarree’ is met één niveau verlaagd gezien overlap van het 95% BI met de grenzen voor klinische relevantie (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘redelijk’.
1.5 Uitkomstmaat bijwerkingen: pijn (cruciaal)
De uitkomstmaat pijn is door vier studies onderzocht (Creinin, 1997; Marwah, 2016; Ngoc, 2014; Rita, 2006). Creinin (1997) heeft pijn gemeten op de VAS schaal (0-10) en vond een gemiddelde pijnscore van 4,0 ± 3,6 voor de orale groep en 5,9 ± 2,7 voor de vaginale groep, hetgeen niet statistisch significant van elkaar verschilde (p=0,33). Het gepoolde effect van de studies die pijn definieerden als pijn die wel of niet aanwezig was laat zien dat in de orale groep het percentage aanwezigheid van pijn 59% (116/195) was en in de vaginale groep 54% (105/195) patiënten (figuur 4). Dit leidde tot een risk ratio van 0,79 (95% BI 0,46 – 1,35, p=0,39) met een random effect model en met een hoge heterogeniteit (I2 72%). Dit houdt in dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
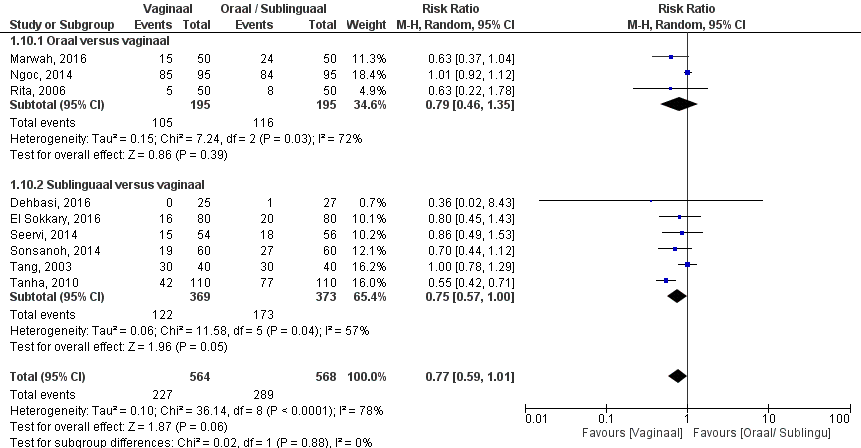
Figuur 4. Uitkomstmaat pijn vergelijking oraal/ sublinguaal versus vaginaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen – pijn’ is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en gezien overlap van het 95% BI met de grenzen voor klinische relevantie (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
1.6 Uitkomstmaat patiënttevredenheid (belangrijk)
De uitkomstmaat patiënttevredenheid is door twee studies onderzocht (Ngoc, 2014; Marwah, 2016). Het aantal patiënten dat tevreden was of erg tevreden was over de behandeling is gescoord. Het gepoolde effect laat zien dat in de orale groep het percentage tevreden patiënten 80,1% (121/150) was en in de vaginale groep 85% (126/148) patiënten (figuur 5). Dit leidde tot een risk ratio van 0,95 (95% BI 0,87 – 1,05, p=0,31) met een random effect model en met een lage heterogeniteit (I2 0%). Dit houdt in dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
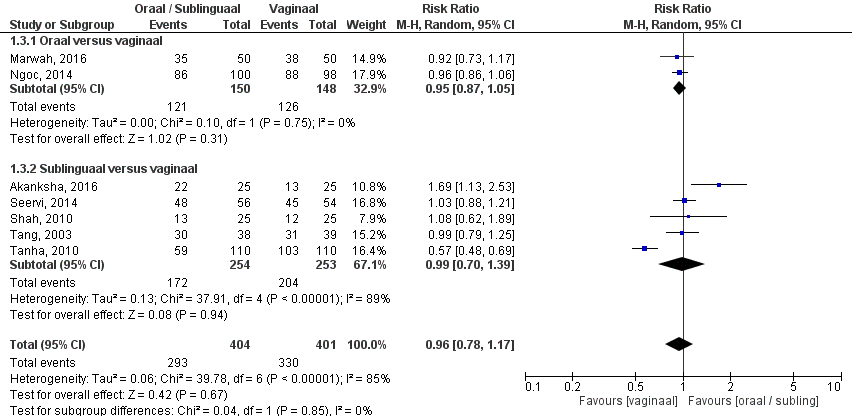
Figuur 5. Uitkomstmaat patiënttevredenheid vergelijking oraal/ sublinguaal versus vaginaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Een hoger risico betekent in dit geval meer kans op patiënttevredenheid, voor vaginaal aan de linkerkant van de grafiek en oraal/sublinguaal aan de rechterkant van de grafiek.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat patiënttevredenheid is met één niveau verlaagd gezien het geringe aantal patiënten (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘redelijk’.
Vergelijking 2. Sublinguaal vs. vaginaal
2.1. Uitkomstmaat succesvolle behandeling (cruciaal)
De uitkomstmaat succesvolle behandeling is door acht studies onderzocht (Tanha, 2010; Tang, 2003; Shah, 2010, El Sokkary, 2016; Dehbasi, 2016; Seervi, 2014; Akanksha, 2016, Sonsanoh, 2014). Succes werd gedefinieerd als volledige expulsie na volledige dosering. Het gepoolde effect laat zien dat in de sublinguale groep het percentage succesvolle behandeling 73% (307/423) was en in de vaginale groep 59% (247/419) patiënten (figuur 1). Dit leidde tot een risk ratio van 1,19 (95% BI 0,97 – 1,46, p=0,09) met een random effect model en met een hoge heterogeniteit (I2 79%). Dit houdt in dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat succesvolle behandeling is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en gezien overlap van het 95% BI met de grenzen voor klinische relevantie (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
2.2. Uitkomstmaat bijwerkingen/complicaties: algemeen (cruciaal)
Deze uitkomstmaat werd door vijf studies gerapporteerd waarbij twee studies (Shah, 2010; Dehbasi, 2016) bijwerkingen in het algemeen beschreven en drie studies complicaties (bloedingen) waarvoor een bloedtransfusie noodzakelijk was noemden (Akanksha 2016, Sonsanoh, 2014 en Seervi 2014). In de sublinguale groep was het percentage algemene bijwerkingen 60% (31/52) en in de vaginale groep 20% (10/50) (figuur 6) (Shah, 2010; Dehbasi, 2016). Dit leidde tot een risk ratio van 0,34 (95% BI 0,18 – 0,61, p=0,0004) met een random effect model en met een lage heterogeniteit (I2 0%). Hetgeen inhoudt dat er een klinisch relevant hogere kans is op bijwerkingen/complicaties bij het gebruik van sublinguale misoprostol. In drie studies werd het aantal bloedingen gerapporteerd waarvoor een bloedtransfusie noodzakelijk was (Akanksha, 2016; Sonsanoh, 2014; Seervi, 2014). Bij de studie van Akanksha, trad bij 1/25 van de patiënten in de sublinguale groep (4%) en 1/25 patiënten van de vaginale groep ruim bloedverlies op waarvoor een bloedtransfusie was vereist. Sonsanoh (2014) rapporteerde dat er 12/60 (20%) patiënten in de sublinguale groep en 3/60 patiënten (5%) in de vaginale groep ruim bloedverlies op trad. Seervi (2014) rapporteert ruim bloedverlies in 2/56 (1,68%) van de patiënten in de sublinguale groep en 4/54 (3,36%) patiënten in de vaginale groep. Er staat bij beide laatste twee studies niet vermeld of er een bloedtransfusie nodig was.
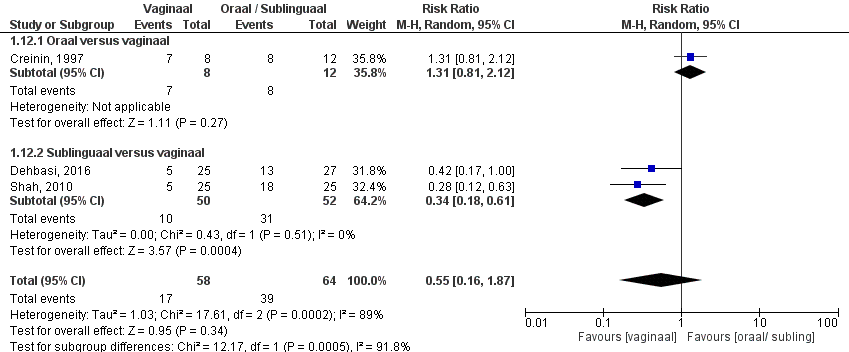
Figuur 6. Uitkomstmaat bijwerkingen/complicaties algemeen vergelijking oraal/ sublinguaal versus vaginaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen/complicaties’ algemeen is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en geringe aantal patiënten (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
2.3. Uitkomstmaat bijwerkingen: braken (cruciaal)
De uitkomstmaat braken is door vijf studies onderzocht (Tanha, 2010; Tang, 2003; Seervi, 2014; Akanksha, 2016; Dehbasi, 2016). Dehbasi (2016) rapporteerde braken gecombineerd met misselijkheid en vond dat 22% (6/27) patiënten in de sublinguale groep en 0% (0/25) patiënten in de vaginale groep last had van misselijkheid dan wel braken. Het gepoolde effect van de vier studies die braken apart rapporteerden (Tanha, 2010; Tang, 2003; Seervi, 2014; Akanksha, 2016) laat zien dat in de orale groep het percentage braken 19% (45/231) patiënten was en in de vaginale groep 16% (36/229) patiënten (figuur 2). Dit leidde tot een risk ratio van 0,85 (95% BI 0,51 – 1,42, p=0,54) met een random effect model en met een lage heterogeniteit (I2 36%). Dit houdt in dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen/braken’ is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en overlap van het 95% BI met de grenzen voor klinische relevantie (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
2.4. Uitkomstmaat bijwerkingen: diarree (cruciaal)
De uitkomstmaat diarree is door zes studies onderzocht (Tanha, 2010; Tang, 2003; Dehbasi, 2016; Seervi, 2014; Akanksha, 2016, Sonsanoh, 2014). Het gepoolde effect laat zien dat in de sublinguale groep het percentage diarree 40% (127/318) patiënten was en in de vaginale groep 21% (66/314) patiënten (figuur 3). Dit leidde tot een risk ratio van 0,52 (95% BI 0,41 – 0,65, p<0,001) met een random effect model en met een lage heterogeniteit (I2 0%). Dit houdt in dat er een klinisch relevant hogere kans is op het krijgen van diarree bij sublinguaal gebruik van misoprostol.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen – diarree’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘redelijk’.
2.5. Uitkomstmaat bijwerkingen: pijn (cruciaal)
De uitkomstmaat pijn is door zes studies onderzocht (Tanha, 2010; Tang, 2003; El Sokkary, 2016; Dehbasi, 2016; Seervi, 2014; Sonsanoh, 2014). Het gepoolde effect van de studies die pijn definieerden als pijn die wel of niet aanwezig was laat zien dat in de sublinguale groep het percentage aanwezigheid van pijn 46% (173/373) was en in de vaginale groep 33% (122/369) patiënten (figuur 4). Dit leidde tot een risk ratio van 0,75 (95% BI 0,57 – 1,00, p=0,05) met een random effect model en met een matige heterogeniteit (I2 57%). Dit houdt in dat dat er een klinisch relevant hogere kans op pijn is in de sublinguale groep.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen – pijn’ is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en gezien overlap van het 95% BI met de grenzen voor klinische relevantie (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
2.6. Uitkomstmaat patiënttevredenheid (belangrijk)
De uitkomstmaat patiënttevredenheid is door vijf studies onderzocht (Tanha, 2010; Tang, 2003; Shah, 2010; Seervi, 2014; Akanksha, 2016). Het aantal patiënten dat tevreden was of erg tevreden was over de behandeling is gescoord. Het gepoolde effect laat zien dat in de sublinguale groep het percentage tevreden patiënten 73% (172/254) was en in de vaginale groep 81% (204/253) patiënten (figuur 5). Dit leidde tot een risk ratio van 0,99 [95% BI 0,70 – 1,39, p=0,94] met een random effect model en met een hoge heterogeniteit (I2 89%). Dit houdt in dat dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat patiënttevredenheid is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en tegenstrijdige resultaten (-1, inconsistentie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
Vergelijking 3. Oraal vs. sublinguaal
3.1. Uitkomstmaat succesvolle behandeling (cruciaal)
De uitkomstmaat succesvolle behandeling is door twee studies onderzocht (Kushwah, 2009; Ayudhaya, 2006). Succes werd gedefinieerd als volledige expulsie na volledige dosering: binnen 22 uur (Kushwah, 2009) en 24 uur (Ayudhaya, 2006). Het gepoolde effect laat zien dat in de orale groep het percentage succesvolle behandeling 51% (59/116) was en in de sublinguale groep 51% (61/120) patiënten (figuur 7). Dit leidde tot een risk ratio van 1,04 (95% BI 0,79 – 1,38, p=0,77) met een random effect model en met een lage heterogeniteit (I2 28%). Dit houdt in dat er geen klinisch relevant verschil is tussen de twee interventiegroepen.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘succesvolle behandeling’ is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en het geringe aantal patiënten (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
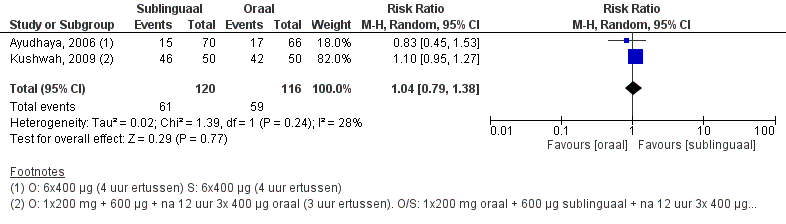
Figuur 7. Uitkomstmaat succesvolle behandeling vergelijking oraal versus sublinguaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval; O: oraal; S: sublinguaal. Een hoger risico betekent in dit geval meer kans op succes, voor vaginaal aan de linkerkant van de grafiek en oraal/sublinguaal aan de rechterkant van de grafiek.
3.2. Uitkomstmaat bijwerkingen/complicaties: algemeen (cruciaal)
Deze uitkomstmaat werd niet gerapporteerd voor deze vergelijking.
Bewijskracht van de literatuur
Er is geen GRADE beoordeling in verband met het ontbreken van studies.
3.3. Uitkomstmaat bijwerkingen: braken (cruciaal)
De uitkomstmaat braken is door twee studies onderzocht (Kushwah, 2009; Ayudhaya, 2006). Ayudhaya (2006) rapporteerde braken gecombineerd met misselijkheid en vond dat 4,5% (3/66) patiënten in de orale groep en 2,9% (2/70) patiënten in de sublinguale groep last had van misselijkheid dan wel braken (p=0,68). Kushwah (2009) rapporteerde alleen braken en vond dat 44% (22/50) patiënten in de orale groep en 22% (11/50) patiënten in de sublinguale groep last had van braken, wat leidde tot een risk ratio van 0,50 (95% BI 0,27 – 0,92, p=0,03). Dit houdt in dat dat er een klinisch relevant hogere kans is op braken bij orale inname.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen – braken’ is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en overlap van het 95% BI met de grenzen voor klinische relevantie en het gering aantal patiënten (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
3.4. Uitkomstmaat bijwerkingen: diarree (cruciaal)
De uitkomstmaat diarree is door twee studies onderzocht (Kushwah, 2009; Ayudhaya, 2006). Het gepoolde effect laat zien dat in de orale groep het percentage diarree 30% (35/116) patiënten was en in de sublinguale groep 25% (30/120) patiënten (figuur 8). Dit leidde tot een risk ratio van 1,17 (95% BI 0,82 – 1,68, p=0,38) met een random effect model en met een lage heterogeniteit (I2 0%). Dit houdt in dat dat er geen klinisch relevant verschil is tussen de groepen.
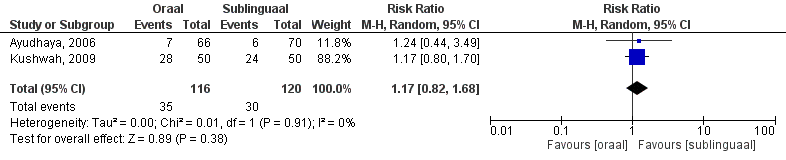
Figuur 8. Uitkomstmaat diarree vergelijking oraal versus sublinguaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval; O: oraal; S: sublinguaal
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen – diarree’ is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en overlap van het 95% BI met de grenzen voor klinische relevantie en het gering aantal patiënten (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
3.5. Uitkomstmaat bijwerkingen: pijn (cruciaal)
De uitkomstmaat pijn is door twee studies onderzocht (Kushwah, 2009; Ayudhaya, 2006). Het gepoolde effect van de studies die pijn definieerden als pijn die wel of niet aanwezig was laat zien dat in de orale groep het percentage aanwezigheid van pijn 72% (84/116) was en in de sublinguale groep 58% (70/120) patiënten (figuur 9). Dit leidde tot een risk ratio van 1,31 (95% BI 0,62 – 2,73, p=0,48) met een random effect model en met een hoge heterogeniteit (I2 92%). Dit houdt in dat dat er geen verschil is tussen de twee interventiegroepen.
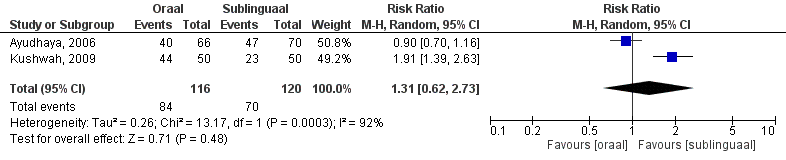
Figuur 9. Uitkomstmaat pijn vergelijking oraal versus sublinguaal
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval; O: oraal; S: sublinguaal
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘bijwerkingen – pijn’ is met drie niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias), tegenstrijdige resultaten (-1, inconsistentie) en het geringe aantal patiënten (-1, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
3.6 Uitkomstmaat patiënttevredenheid (belangrijk)
Patiënttevredenheid werd door Kushwah (2009) onderzocht. In de orale groep was (36/50) 72% van de patiënten tevreden versus (46/50) 91% van de patiënten in de sublinguale groep. Dit leidde tot een risk ratio van 0,78 (95% BI 0,65 – 0,95, p=0,01) met een random effect model. Hetgeen inhoudt dat er een klinisch relevante hogere patiënttevredenheid is in de sublinguale groep.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat patiënttevredenheid is met drie niveaus verlaagd gezien beperkingen in de onderzoeksopzet (-1, risk of bias) en het gering aantal patiënten
(-2, imprecisie). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
1.7, 2.7, 3.7 Uitkomstmaat lange termijn gevolgen (belangrijk)
Deze uitkomstmaat werd voor geen van de vergelijkingen gerapporteerd.
Bewijskracht van de literatuur
Er is geen GRADE beoordeling voor de uitkomstmaat ‘lange termijn gevolgen’ in verband met het ontbreken van studies.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvragen:
Vergelijking 1
Wat zijn de (on)gunstige effecten van orale versus vaginale toediening van misoprostol bij de medicamenteuze behandeling van een miskraam?
P patiënten die medicamenteus behandeld worden voor een miskraam;
I orale toediening van misoprostol;
C vaginale toediening van misoprostol;
O succesvolle behandeling, bijwerkingen/complicaties, patiënttevredenheid, lange termijn gevolgen.
Vergelijking 2
Wat zijn de (on)gunstige effecten van sublinguale versus vaginale toediening van misoprostol bij de medicamenteuze behandeling van een miskraam?
P Patiënten die medicamenteus behandeld worden voor een miskraam.
I Sublinguale toediening van misoprostol.
C Vaginale toediening van misoprostol.
O Succesvolle behandeling, bijwerkingen/complicaties, patiënttevredenheid, lange termijn gevolgen.
Vergelijking 3
Wat zijn de (on)gunstige effecten van sublinguale versus orale toediening van misoprostol bij de medicamenteuze behandeling van een miskraam?
P Patiënten die medicamenteus behandeld worden voor een miskraam.
I Orale toediening van misoprostol.
C Sublinguale toediening van misoprostol.
O Succesvolle behandeling, bijwerkingen/complicaties, patiënttevredenheid, lange termijn gevolgen.
Relevante uitkomstmaten
De werkgroep achtte succesvolle behandeling en bijwerkingen/complicaties voor de besluitvorming cruciale uitkomstmaten; en patiënttevredenheid en lange termijn gevolgen op de fertiliteit voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde niet a priori de genoemde uitkomstmaten succesvolle behandeling, bijwerkingen/complicaties, patiënttevredenheid en lange termijn gevolgen fertiliteit, maar hanteerde de in de studies gebruikte definities.
De werkgroep definieerde voor geen van de uitkomstmaten klinische (patiënt) relevante verschillen, maar hanteerden, indien van toepassing, de onderstaande grenzen voor klinische relevantie voor dichotome uitkomstmaten en vergeleken de resultaten met deze grenzen: RR < 0,80 of > 1,25) (GRADE recommendation) of Standardized mean difference (SMD=0,2 (klein); SMD=0,5 (matig); SMD=0,8 (groot). De interpretatie van continue uitkomstmaten is sterk context gebonden en hiervoor werden a priori geen grenzen voor klinische relevante benoemd.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID) & Embase (via Elsevier is op 2 januari 2019 met relevante zoektermen gezocht naar studies die verschillende toedieningsroutes van misoprostol voor de medicamenteuze behandeling van een miskraam beschreven. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. Voor deze vraag is gebruikt gemaakt van beschikbare literatuur (acht studies) over deze vergelijking uit de NICE-richtlijn ‘Ectopic pregnancy and miscarriage’ (NICE, 2012). In de NICE-richtlijn is tot begin 2012 gezocht naar literatuur. Deze search is aangevuld met een literatuursearch uitgevoerd door de werkgroep. De literatuurzoekactie leverde 474 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria:
- gepubliceerd in het Nederlands of Engels;
- gepubliceerd na 2012;
- studiedesign RCT of SR (voor uitkomstmaat lange termijn fertiliteit cohortstudies);
- studie uitgevoerd in land met vergelijkbare standaard van zorg als Nederland;
- beschrijven verschillende toedieningsvormen misoprostol bij de medicamenteuze behandeling van een miskraam.
Op basis van titel en abstract werden in eerste instantie negen studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens zes studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), en drie studies definitief geselecteerd. In de NICE-richtlijn waren reeds acht studies beschreven en middels cross-referencing zijn daar nog drie studies bijgekomen.
Vijftien onderzoeken zijn opgenomen in de literatuuranalyse. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk of bias tabellen.
- Akanksha Lamba, Kaur R, Sardana P. A study to compare the clinical outcome of sublingual and vaginal misoprostol in the medical management of missed miscarriage in first trimester. Int J Reprod Contracept Obstet Gynecol. 2016 Feb;5(2):491-494
- Ayudhaya OP, Herabutya Y, Chanrachakul B, Ayuthaya NI, O-Prasertsawat P. A comparison of the efficacy of sublingual and oral misoprostol 400 microgram in the management of early pregnancy failure: a randomized controlled trial. J Med Assoc Thai. 2006 Oct;89 Suppl 4:S5-10
- Creinin MD, Moyer R, Guido R. Misoprostol for medical evacuation of early pregnancy failure. Obstet Gynecol. 1997 May;89(5 Pt 1):768-72.
- Dehbashi Z, Moosazadeh M, Afshari M. Comparison between sublingual and vaginal route of misoprostol in management of first trimester miscarriage missing. Mater Sociomed. 2016 Jul 24;28(4):271-273.
- El Sokkary HH. Comparison Between Sublingual and Vaginal Administration of Misoprostol in Management of Missed Abortion. J Obstet Gynaecol India. 2016 Oct;66(Suppl 1):24-9.
- Kushwah B, Singh A. Sublingual versus oral misoprostol for uterine evacuation following early pregnancy failure. Int J Gynaecol Obstet. 2009 Jul;106(1):43-5.
- Lemmers M, Verschoor MA, Kim BV, Hickey M, Vazquez JC, Mol BWJ, Neilson JP. Medical treatment for early fetal death (less than 24 weeks). Cochrane Database Syst Rev. 2019 Jun 17;6:CD002253. doi: 10.1002/14651858.CD002253.pub4. PubMed PMID: 31206170; PubMed Central PMCID: PMC6574399.
- Marwah S, Gupta S, Batra NP, Bhasin V, Sarna V, Kaur N. A Comparative Study to Evaluate the Efficacy of Vaginal vs Oral Prostaglandin E1 Analogue (Misoprostol) in Management of First Trimester Missed Abortion. J Clin Diagn Res. 2016 May;10(5):QC14-8.
- Morris JL, Winikoff B, Dabash R, Weeks A, Faundes A, Gemzell-Danielsson K, Kapp N, Castleman L, Kim C, Ho PC, Visser GHA. FIGO's updated recommendations for misoprostol used alone in gynecology and obstetrics. Int J Gynaecol Obstet. 2017 Sep;138(3):363-366.
- Ngoc NT, Blum J, Westheimer E, Quan TT, Winikoff B. Medical treatment of missed abortion using misoprostol. Int J Gynaecol Obstet. 2004 Nov;87(2):138-42.
- NICE. NICE-guideline ‘Ectopic pregnancy and miscarriage: diagnosis and initial management’. 2012 & Update in 2019.
- Rita S, Kumar S. A randomised comparison of oral and vaginal misoprostol for medical management of first trimester missed abortion. JK Science. 2006 1:35-38
- Seervi N, Hooja N, Rajoria L, Verma A, Malviya K, Mehta N. Comparison of different regimes of misoprostol for the termination of early pregnancy failure. Med J Armed Forces India. 2014 Oct;70(4):360-3.
- Shah N, Azam SI, Khan NH. Sublingual versus vaginal misoprostol in the management of missed miscarriage. J Pak Med Assoc. 2010 Feb;60(2):113-6.
- Sonsanoh A, Chullapram T. Comparison of Sublingual and Vaginal Misoprostol for Termination of Early Pregnancy Failure: A randomized controlled trial. Thai J of Obstet and Gyn. July 2014, Vol. 22, pp. 128-136.
- Tang OS, Lau WN, Ng EH, Lee SW, Ho PC. A prospective randomized study to compare the use of repeated doses of vaginal with sublingual misoprostol in the management of first trimester silent miscarriages. Hum Reprod. 2003 Jan;18(1):176-81.
- Tanha FD, Feizi M, Shariat M. Sublingual versus vaginal misoprostol for the management of missed abortion. J Obstet Gynaecol Res. 2010 Jun;36(3):525-32.
- Wu HL, Marwah S, Wang P, Wang QM, Chen XW. Misoprostol for medical treatment of missed abortion: a systematic review and network meta-analysis. Sci Rep. 2017 May 10;7(1):1664.
Evidence table for intervention studies (randomiz37ed controlled trials and non-randomized observational studies [cohort studies, case-control studies, case series])1
This table is also suitable for diagnostic studies (screening studies) that compare the effectiveness of two or more tests. This only applies if the test is included as part of a test-and-treat strategy – otherwise the evidence table for studies of diagnostic test accuracy should be used.
Research question:
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3 |
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Comparison 1. Oral vs. Vaginal |
|||||||
|
Creinin, 1997 |
Type of study: RCT
Setting and country: USA
Funding and conflicts of interest: Magee Women's Health Foundation |
Inclusion criteria: -Healthy -English speaking -Diagnosis of early pregnancy failure, based on ultrasound demonstration of one of the following: * Embryonic pole 5-14 mm with no embryonic cardiac activity * Irregular intrauterine gestational sac with mean sac diameter of 16 mm or greater and no embryonic pole * Abnormal growth on ultrasound over a minimum of 1 week * Yolk sac present with an abnormal increase in hCG (50% or less) over 48 hours, and an initial value less than 2000 IU/l - At least 18 years old - Vaginal bleeding no more than spotting (not requiring more than one sanitary towel a day) - Gestational age of 8 weeks of less by ultrasound or physical examination - Closed cervical os on bimanual pelvic examination - Haemoglobin of 10mg/dl or more Willingness and ability to sign informed consent - Willingness to abstain from intercourse for the first 3 days of the study and comply with visit schedule - Adequate venous access for phlebotomy - Easy access to a telephone Exclusion criteria: - History of inflammatory bowel disease - Intolerance or allergy to misoprostol N total at baseline: Intervention (Oral): 12 Control (Vaginal): 8
Important prognostic factors2: age ± SD: I:26.3 ± (7.0) C: 29.8 ± (8.0)
Prior miscarriage (number/total (%)) I: 6/12 (50%) C: 4/8 (50%)
Groups comparable at baseline. |
Describe intervention:
400 micrograms of oral misoprostol (repeat dose after 24 hours if needed)
Subjects in the oral arm swallowed misoprostol in the presence of a member of the research staff.
|
Describe control:
800 micrograms of vaginal misoprostol (repeat dose after 24 hours if needed)
Those in the vaginal arm had four 200 micrograms tablets of misoprostol administered |
Length of follow-up: 1 day
Loss-to-follow-up: Not mentioned
Incomplete outcome data: Not mentioned
|
Outcome measures and effect size (include 95%CI and p-value if available):
Treatment success I: 3/12 (25%) C: 7/8 (87.5%) P=0,01
Side effects a. any side effects I: 8/12 (67%) C:7/8 (87%)
b. Nausea I: 6/12 (50%) C: 5/8 (62.5%)
c. Vomiting I: 3/12 (25%) C: 1/8 (12%)
d. Diarrhoea I: 5/12 (42%) C: 3/8 (38%)
Pain Defined as: The maximal amount of pain was assessed using a visual analogue scale, consisting of a 10 mm line with "no pain" at one end and "severe pain" at the other end. Severity of pain/10 (mean (SD)) I: 4.0 (3.6) C: 5.9 (2.7)
|
Aim of the study: To determine whether misoprostol 400 micrograms orally or 800 micrograms vaginally will cause complete uterine evacuation in women with early pregnancy failure. |
|
Ngoc, 2004 |
Type of study: RCT
Setting and country: Vietnam
Funding and conflicts of interest: David and Lucille Packard Foundation |
Inclusion criteria: First trimester, missed miscarriage, defined as: - ultrasound evidence of an intact gestational sac - no evidence of fetal cardiac activity (6 weeks after last menstrual period) - closed cervical os - history of no or minimal bleeding
No known contraindications to misoprostol
General good health
Willingness to attend a follow-up visit
Exclusion criteria: See above
N total at baseline: Intervention (Oral): 101 Control (Vaginal): 99
Important prognostic factors2: For example age range: 19-45
The authors give no further details, but report that there were no significant differences between the two groups, confirming that randomisation was effective.
Groups comparable at baseline |
Describe intervention:
800 micrograms of oral misoprostol (four tablets of 200microgram)
Every woman self-administered their misoprostol in the presence of a study investigator.
|
Describe control):
800 micrograms of vaginal misoprostol (four tablets of 200 microgram)
Every woman self-administered their misoprostol in the presence of a study investigator. |
Length of follow-up: 1 week
Loss-to-follow-up: I: N=1 (1%) C: N=1 (1 %)
Incomplete outcome data: Not mentioned
|
Outcome measures and effect size (include 95%CI and p-value if available):
Treatment success I: 89/101 (88%) C: 91/99 (92%)
Need for further intervention (number/total (%)) a. Total I: 11/100 (11%) C: 7/98 (7%) (NS) 60 503b. Medically indicated before study end I: 5/100 (5%) C: 2/98 (2%) (NS) c. Intervention at patient request I: 6/100 (6%) C: 5/98 (5%) (NS)
Adverse effects of treatment (number of women/total (%))
a. Vomiting I: 4/95 (4%) C: 14/95 (15%) (p=0.023) b. Diarrhoea I: 24/95 (25.3) C: 23/95 (24.2) (NS) c. Fever/chills I: 7/95 (7%) C: 7/95 (7%) (NS)
Measures of pain: Incidence of pain/cramps (number of women/total (%)) I: 84/95 (88%) C: 85/95 (89%) (NS)
Measures of satisfaction a. Satisfied or very satisfied with the method (number of women/total (%)) I: 86/100 (86.0) C: 88/98 (89.8)
b. Would choose the method again (%) I:85.0 % C: 92.9 % (NS) c. Would recommend the method to a friend (%) I: 83.0% C: 90.8% |
Aim of the study To compare the efficacy of two routes of misoprostol administration (oral and vaginal) for the treatment of missed miscarriage. |
|
Rita, 2006 |
Type of study: RCT Setting and country: India
Funding and conflicts of interest:
Not reported |
Inclusion criteria: -Gestation less than 13 weeks -Haemodynamically stable -Haemoglobin more than 10gm% -Closed cervical os -Axillary temperature of < 37.5ºC
Exclusion criteria: -History of inflammatory bowel disease -Allergy to misoprostol
N total at baseline: Intervention: 50 Control:50
Groups comparable at baseline.
|
Describe intervention: 400 micrograms given orally, and repeated every 4 hours up to a maximum of three doses
Over the next 10-12 hours, complete, incomplete, or no expulsion was documented by transvaginal ultrasound. The absence of an echogenic structure measuring less 15 mm in diameter suggested complete miscarriage. Nothing was given by mouth except medication for pain relief until complete expulsion or surgical evacuation. Information was obtained regarding side effects. Rh- women were given anti D immunoglobulin.
|
Describe control):
600 micrograms inserted into the posterior vaginal fornix, with a second dose repeated after 4 hours
Over the next 10-12 hours, complete, incomplete, or no expulsion was documented by transvaginal ultrasound. The absence of an echogenic structure measuring less 15 mm in diameter suggested complete miscarriage. Nothing was given by mouth except medication for pain relief until complete expulsion or surgical evacuation. Information was obtained regarding side effects. Rh- women were given anti D immunoglobulin. |
Length of follow-up: 1 day
Loss-to-follow-up: Not reported
Incomplete outcome data: Not reported
|
Outcome measures and effect size:
Treatment success (number/total (%)) I: 18/50 (36%) C: 40/50 (80%)
Need for further intervention (number/total (%)) I: 32/50 (64%) C: 10/50 (20%)
Adverse effects of treatment (number/total (%)) a. Nausea I: 25/50 (50%) C: 20/50 (40%)
b. Vomiting I: 6/50 (12%) C: 3/50 (6%)
c. Diarrhoea I: 5/50 (10%) C: 5/50 (10%)
d. Hyperpyrexia I: 2/50 (4%) C: 2/50 (4%)
Measures of pain: incidence of severe pain (number/total (%)) I: 8/50 (16%) C: 5/50 (10%) |
Aim of the study To compare the safety and efficacy of oral versus vaginal misoprostol for medical management of missed miscarriage.
Outcomes reported 1. Treatment success: Defined as complete, drug induced expulsion of the products of conception
2. Need for further intervention: The number of women requiring surgical evacuation
3. Adverse effects of treatment: Incidence of nausea, vomiting, diarrhoea and hyperpyrexia are reported.
4. Pain: Incidence of severe pain is reported; however it is unclear what criteria was used to judge severity. |
|
Marwah, 2016 |
Type of study: RCT Setting and country: Single center, India
Funding and conflicts of interest: None
Funding not mentioned |
Inclusion criteria: - Females of age group 18-45 years; - Gestational age ≤ 12 weeks by last menstrual period (LMP); - Diagnosis of missed abortion on USG; -Mild vaginal bleeding or spotting per vaginum; -Closed cervix on bimanual pelvic examination; -Haemoglobin ≥9gm/dl; -Axillary temperature <37.5 degree C; -No history of inflammatory bowel disease, asthma, liver disease or contraindication to use of misoprostol; -Place of residence within 100 km from of the hospital; -Willingness and ability to give informed consent; -Willingness to abstain from intercourse for first 14 days of study; -Willingness to comply with follow-up schedule
Exclusion criteria: -Women <18 years or >45 years of age; - Fetal gestational age >12 weeks; - Any degree of cervical dilatation; -Excessive uterine bleeding; -Haemoglobin concentration <9gm/dl); -Haemodynamic instability; -Blood pressure ≥160/90 mmHg; -Poor general health of any cause; -Deranged coagulation profile (defined as PTI≤85%) -Signs or symptoms of infection; -Maternal history of asthma or cardiac disease or cerebral disease; - Use of anticoagulants or H/O any bleeding disorder; -Known allergy to or C/I to misoprostol use; -Active lactation; -Any prior medical or surgical treatment to interrupt current pregnancy; -Twin gestation sac; - Molar pregnancy; -Inability or refusal of patient to adhere to follow-up.
N total at baseline: Intervention: 50 Control:50
Important prognostic factors2: age ± SD: I:23.9 ± 3.7 C: 23.7 ± 3.9
Previous spontaneous abortion: N (%) I: 17 (34%) C: 18 (36%)% M
Groups comparable at baseline. |
Describe intervention:
Fifty women were dispensed 400μg of oral misoprostol, repeated every six hours for a maximum of three doses. Women swallowed the pills with sips of water in presence of the doctor.
|
Describe control:
Fifty women were administered 400μg of misoprostol intravaginally into posterior fornix (soaked in normal saline solution), repeated six hourly up to a maximum of three doses. Vaginal cleansing had been performed before insertion with 10% povidone iodine (Betadiene). Following insertion, women were asked to remain in fully recumbent position for three hours. |
Length of follow-up: 6 weeks
Loss-to-follow-up: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
Incomplete outcome data: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
|
Outcome measures and effect size:
Treatment success (number/total (%)) I: 37/50 (74%) C: 46/50 (92%)
Adverse events of treatment a. Nausea/vomiting I: 36/50 (72%) C: 30/50 (60%)
b. Diarrhoea I: 7/50 (14%) C: 6/50 (12%)
c. Severe Crampy Pain I: 24 (48%) C: 15 (30%)
d. Fever/Hyperpyrexia I: 4 (8%) C: 2 (4%)
Measures of satisfaction a. Satisfied or very satisfied with the method (number of women/total (%)) I: 35/50 (70%) C: 38/50 (76%)
b. Would choose the method again (%) I: 39/50 (78%) C: 41 (82%)
c. Would recommend the method to a friend (%) I: 37/50 (74%) C: 39/50 (78%)
Time to expulsion/hours (mean) Vaginal: 19.86 Sublingual: 9.53 (p=0.000)
|
Aim of the study
Outcomes Treatment success Success was defined as non-surgical evacuation of POCs confirmed by absence of echogenic structure measuring≥15mm in AP diameter on USG |
|
Comparison 2. Sublingual vs. Vaginal |
|||||||
|
Tanha, 2010 |
Type of study: RCT
Setting and country: Iran, Mirza Kochak Khan Hospital, a premier research and referral facility in Tehran.
Funding: Not reported and conflicts of interest:
|
Inclusion criteria: Silent miscarriage, defined as: - intrauterine gestational sac with a mean sac diameter of at least 2 cm without a fetal pole - presence of a fetal pole with no cardiac activity - gestational sac < 2 cm with no interval growth, or persistent absence of fetal cardiac pulsation on rescanning 7-10 days later < 13 weeks gestation No known contraindications to misoprostol General good health No vaginal bleeding
Exclusion criteria: Incomplete miscarriage Inevitable miscarriage (products of gestation bulging from the cervix) Suspicion of an extra-uterine pregnancy Drug or alcohol abuse as reported by the woman Abnormal blood count tests obtained routinely
N total at baseline: N=220
Intervention: 110 Control:110
Important prognostic factors2: Age/years Vaginal: 29.1 Sublingual: 28.5 (p=0.516) Gestational age/weeks Vaginal: 10.8 Sublingual: 10.6 (p=0.655) Gravidity Vaginal: 2.87 Sublingual: 2.85 (p=0.926) Parity Vaginal: 0.44 Sublingual: 0.23 (p=0.013) Previous miscarriage Vaginal: 0.60 Sublingual: 0.71 (p=0.528) Live children Vaginal: 0.84 Sublingual: 0.96 (p=0.535) (Note: it is not reported what these statistics represent, although the technical team hypothesise that they represent means)
Groups comparable at baseline.
|
Describe intervention:
400 micrograms of sublingual misoprostol every 6 hours (n=110)
Dosage The trial protocol does not state whether there was a maximum number of tablets that a woman could receive. The mean number of tablets was 4.45 in the vaginal group and 4.85 in the sublingual group (p=0.211).
|
Describe control:
400 micrograms of vaginal misoprostol every 6 hours (n=110)
|
Length of follow-up:
Loss-to-follow-up: Not reported
Incomplete outcome not reported
|
Outcome measures and effect size:
Complete evacuation (number/total (%)) Sublingual: 93/110 (84.5) Vaginal: 51/110 (46.4) RR (95% CI): 0.54 (0.442-0.681) (p<0.0001)
Need for further intervention (number/total (%)) Vaginal: 59/110 (53.6) (Note: 15 due to persistent gestational sac after 2 days; 44 due to incomplete miscarriage. Histology from 3 of the patients showed a partial mole) Sublingual: 17/110 (Note: 5 due to persistent gestational sac after 2 days; 12 due to incomplete miscarriage. Histology from 2 of the patients showed a partial mole and complete hydatiditiform mole) RR (95% CI): 3.471 (2.168-5.555) (p<0.0001)
Adverse effects of treatment (number of women/total (%)) a. Vomiting Vaginal: 13/110 (11.8) Sublingual: 22/110 (20) RR (95% CI): 0.591 (0.255-1.128) (p=0.140)
b. Diarrhoea Vaginal: 40/110 (36.4) Sublingual: 76/110 (69.1) RR (95% CI): 0.526 (0.399-0.694) (p<0.0001)
c. Fever Vaginal: 4/110 (3.6) Sublingual: 26/110 (23.6) RR (95% CI): 0.154 (0.056-0.426) (p<0.0001)
Measures of pain (number of women/total (%)) a. Cramp pain Vaginal: 62/110 (56.4) Sublingual: 94/110 (85.5) RR (95% CI): 0.660 (0.550- 0.791) (p<0.0001) b. Severe pain Vaginal: 42/110 (38.2) Sublingual: 77/110 (70) RR (95% CI): 0.859 (0.713-1.036) (p=0.091)
Measures of satisfaction (%) a. Reported being satisfied with treatment Vaginal: 59/110 (53.6%) Sublingual: 103/110 (93.6%) (p=0.001) b. Would recommend method to others Vaginal: 53.6 Sublingual: 84.5 (p=0.004) (Note: satisfaction was also related to success of treatment) |
Aim of the study
To evaluate the efficacy of two routes of misoprostol administration (sublingual and vaginal) for the treatment of missed miscarriage.
Outcomes reported 1. Success rate: Defined as the passage of products of conception without needing vacuum aspiration or dilatation and curettage. This was assessed at the follow-up appointment 1-2 days after treatment. 2. Need for further intervention: The number of women requiring surgery, and reasons for the intervention are reported. 3. Adverse effects of treatment: This is reported by the women for the period from 1 hour to 24 hours after every administration of misoprostol, up to the first follow-up visit. Fever is defined as an oral temperature of at least 37.8°C. 4. Measures of pain: The incidence of cramping pain and severe pain are reported by the women for the period from 1 hour to 24 hours after every administration of misoprostol, up to the first follow-up visit. 5. Measures of satisfaction: Assessed by questionnaire at follow-up
|
|
Tang, 2003 |
Type of study: RCT
Setting and country: Hong Kong
Funding and conflicts of interest: Not reported |
Inclusion criteria: First trimester silent miscarriage, defined as: - intrauterine gestational sac with mean sac diameter of ≥ 2 cm without a fetal pole - presence of fetal pole with no cardiac pulsation - gestational sac < 2 cm with no interval growth or persistent absence of fetal cardiac activity on rescanning 7 - 10 days later < 13 weeks gestation Exclusion criteria: -Complete miscarriage - Incomplete miscarriage
N total at baseline: Intervention: 40 Control:40
Important prognostic factors2: age ± SD: I:32.3 ± 7.3 C: 33.6 ± 6.0
Groups comparable at baseline.
|
Describe intervention:
600 micrograms of sublingual misoprostol, every 3 hours up to a maximum of three doses
The sublingual group were instructed to put the tablets under their tongues themselves. They were not allowed any food or drink for 20 minutes to allow complete dissolution of the tablets.
|
Describe control:
600 micrograms of vaginal misoprostol, every 3 hours up to a maximum of three doses
In the vaginal group, the research nurses was responsible for putting the three misoprostol tablets into the vaginal fornix. |
Length of follow-up: 6 weeks
Loss-to-follow-up: Not mentioned)
Incomplete outcome data: Not mentioned
4 |
Outcome measures and effect size:
a. Complete miscarriage Sublingual: 35/40 (87.5) Vaginal: 35/40 (87.5)
Need for further intervention (number of women/total (%)) I: 4/39 (10%) C: 4/39 (10%)
Adverse effects of treatment (number of women/total (%)) a. Nausea I: 24/40 (60%) C: 20/40 (50%)
b. Vomiting I: 7/40 (17%) C: 9/40 (22%)
c. Diarrhoea I: 28/40 (70%) C:11/40 (27%)
d. Fever I: 23/40 (57%) C: 19/40 (47%)
Pain Severe pain I: 30/40 (75%) C: 30/40 (75%)
Measures of satisfaction (number of women/total (%)) a. Satisfied of excellent opinion about treatment I: 30/38 (79%) C: 31/39 (79%)
b. Would recommend treatment to others I: 36/38 (94%) C: 33/39 (85%) |
Aim of the study
Outcomes Complete miscarriage: The outcome of treatment was classified as complete miscarriage if surgical evacuation was not required up to the time of return to normal menstruation.
Need for further intervention
Duration of bleeding Assessed on day 43
Adverse effects Recorded during treatment. Fever was defined as a highest temperature of at least 38ºC.
Pain
Recorded during treatment. Degree of pain was assessed using a questionnaire.
Satisfaction Assessed by questionnaires on days 7 and 43. Unclear which results are reported, or whether they were combined. |
|
Shah, 2010 |
Type of study: RCT
Setting and country: Department of Obstetrics and Gynaecology at Civil Hospital, Karachi. Pakistan
Funding and conflicts of interest: Not reported
|
Inclusion criteria: Ultrasound diagnosis of missed miscarriage < 20 weeks gestation
Exclusion criteria: Incomplete miscarriage Retained products of conception Previous caesarean section scars
N total at baseline: 50 Intervention: 25 Control: 25
Important prognostic factors2: Age/years (mean (SD)) Sublingual: 26.2 (4.2) Vaginal: 26.4 (4.4) (p=0.870) Parity (median (range)) Sublingual: 2 (0-5) Vaginal: 2 (0-5) (p=0.845) Gestational age/weeks (mean (SD)) Sublingual: 10.1 (2.62) Vaginal: 10.6 (2.92) (p=0.480) (Note: 22/25 in the sublingual arm and 19/25 in the vaginal arm had a gestational age of less than or equal to 12 weeks, and hence constitute the main population of interest for this review question) Uterine size/weeks (range) Sublingual: 8-16 Vaginal: 8-18 (p=0.952)
Groups comparable at baseline.
|
Describe intervention:
400 micrograms of sublingual misoprostol, every three hours up to a maximum of 5 doses (n=25)
(Note: those with a gestational age and uterine size of more than 12 weeks were given 200 micrograms of misoprostol instead, in both arms)
|
Describe control):
400 micrograms of vaginal misoprostol, every three hours up to a maximum of 5 doses (n=25)
|
Length of follow-up:
Loss-to-follow-up: Not reported
Incomplete outcome data: Not reported
|
Outcome measures and effect size:
Complete miscarriage rate (number/total (%)) a. In those ≤ 12 weeks Sublingual: 11/22 (50) Vaginal: 10/19 (52.6) (p=0.557) b. In those > 12 weeks Sublingual: 2/3 (66.7) Vaginal: 2/6 (33.3) (p=0.404) c. All women Sublingual: 13/25 (52.0) Vaginal: 12/25 (48.0) (p=0.571)
Need for further intervention number/total (%)) Sublingual: 11/25 (44.0) Vaginal: 13/25 (52.0) (Note: 1 further woman in the sublingual group had an incomplete miscarriage - it is not reported whether she had a surgical evacuation)
Adverse effects of treatment (number/total (%)) a. Any side effects Sublingual: 18/25 (72.0) Vaginal: 5/25 (20.0) (p<0.001) b. Nausea Sublin3g12ual: 5/25 (20.0) Vaginal: 1/25 (4.0) (p=0.094) c. Unpleasant taste Sublingual: 15/25 (60.0) Vaginal: 1/25 (4.0) (p<0.001) d. Shivering Sublingual: 6/25 (24.0) Vaginal: 4/25 (16.0)} (p=0.362)
Satisfaction (%) Sublingual: 13/25 (52%) Vaginal: 12/25 (48%) (NS) (Note: the authors report that this closely followed the success rate of the regimen)
Interval between misoprostol and expulsion/hours (mean (SD)) Sublingual: 13.07 (5.63) Vaginal: 13.29 (5.63) (NS) |
Aim of the study To compare the efficacy of sublingual and vaginal misoprostol in the medical management of missed miscarriage.
Outcomes reported 1. Complete miscarriage rate: This is split by those with a pregnancy of less than or equal to 12 weeks, and those with a pregnancy of more than 12 weeks. 2. Need for further intervention 3. Adverse effects of treatment: The incidence of any side effects, nausea, unpleasant taste and shivering are reported. 4. Satisfaction: Assessed by verbally asking patients whether they were satisfied, dissatisfied or neutral with regards to their treatment regimen.
|
|
El Sokkary, 2016 |
Type of study: RCT
Setting and country: Single center, Egypt
Funding and conflicts of interest: No conflict of interest. Funding not mentioned.
|
Inclusion criteria: missed abortion: – Gestational sac with mean sac diameter of [25 mm, without fetal pole (blighted ovum). – The presence of fetal pole [7 mm, with no cardiac pulsations. – Gestational sac mean diameter \18 mm, with no interval growth being observed on rescanning 10 days later. – The absence of embryo with heartbeat at least 2 weeks after an ultrasound scan that showed a gestational sac without a yolk sac. – The absence of embryo with heartbeat at least 11 days after an ultrasound scan that showed a gestational sac with a yolk sac
Exclusion criteria: - N total at baseline: Intervention: 80 Control: 80
Groups comparable at baseline. |
Describe intervention:
100 mcg of misoprostol (1/2 tablet) was administrated to group 1 cases sublingually and were repeated every 4 h for four doses.
|
Describe control:
(vaginal group); 100mcg of misoprostol (1/2 tablet) was administrated to group 2 vaginally in the posterior fornix, and this was repeated every 4 h for four doses 1 |
Length of follow-up: 1 day
Loss-to-follow-up: Not mentioned
Incomplete outcome data: Not mentioned
|
Outcome measures and effect size:
Treatment success (number/total (%)) I: 56/80 (70%) C: 41/80 (51%)
Need for further intervention (number of women/total (%)) I: 24/80 (30%) C: 39/80 (49%)
Adverse events of treatment a. Nausea I: 44/80 (55%) C: 32/80 (40%)
b. Severe Pain I: 20/80 (25%) C: 16/80 (20%)
c. Hyperpyrexia I: 12/80 (15%) C: 4/80 (5%) |
Aim of the study To compare between sublingual administration of misoprostol and vaginal administration in the management of missed abortion.
Outcome Complete abortion The absence of remnant of conception or endometrial interface thickness less than 15 mm is mandatory to diagnose complete abortion (24h from the beginning of first dose) |
|
Dehbasi, 2016 |
Type of study: RCT
Setting and country: Single center, Iran
Funding and conflicts of interest: None
Funding not mentioned |
Inclusion criteria: - fetal IUFD or missed abortion in sonography reports.
Exclusion criteria: -Patients with external and internal opening of the cervix is open to both symptoms of abortion is spontaneous
N total at baseline: Intervention: 27 Control:25
Important prognostic factors2: age ± SD: I:31.56 ± 5.85 C: 29.48 ± 6.21
Previous spontaneous abortion: N (%) I: 17 (34%) C: 18 (36%)% M
Groups comparable at baseline. |
Describe intervention:
Sublingual Misoprostol (400 μg)
The initial dose repeated every 4 hours t0 termination of pregnancy or maximum dose of 2000 microgram
|
Describe control):
Vaginal Misoprostol (400 μg)
The initial dose repeated every 4 hours t0 termination of pregnancy or maximum dose of 2000 microgram |
Length of follow-up: 6 weeks
Loss-to-follow-up: Not mentioned
Incomplete outcome data: Not mentioned
|
Outcome measures and effect size:
Treatment success (number/total (%)) I: 0/27 (0%) C: 9/25 (36%)
Adverse events of treatment Adverse events total I: 13/27 (48%) C:5/25 (20%)
b. Nausea and vomiting I: 6/27 (22.2%) C: 0/25 (0%)
c. Diarrhoea I: 6/27 (22.2%) C: 5/25 (20%)
d. Abdominal Pain I: 1/27 (3.7%) C: 0/25 (0%) |
Aim of the study
Outcomes Complete abortion Complete abortion was defined as complete expulsion of the pregnancy products or endometrial diameter less than 10 mm and absence of residue in sonographic report, otherwise it was considered as incomplete abortion
The main criterion for effective medical abortion was considered occurring abortion within 24 hours after induction. |
|
Seervi, 2014 |
Type of study: RCT
Setting and country: Department of Obst. & Gynae, S.M.S. Medical College & Hospital, Jaipur, Rajasthan, India
Funding and conflicts of interest: none
|
Inclusion criteria: Women patients with an ultrasound diagnosis of early pregnancy failure, singleton pregnancy, less than 12 weeks gestation, who had not experienced uterine cramping, no active bleeding (os closed on per vaginal examination) and were in a normal frame of mind to give consent and willing for a surgical evacuation in case of failure with medication or active bleeding, were included in the study. embryo with CRL greater than 7mmwith no cardiac activity or irregular gestational sac with mean sac diameter greater than 16 mm or a gestational sac more than 25 mm with no visible fetal pole.5
Exclusion criteria:
N total at baseline: Intervention: 56 Control:54
Important prognostic factors2: age ± SD: I:23.79 ±5.2 C:24.57±5.1
Primigravida I: 21 (37.5%) C: 19 (35.18%)
Gestational age 7.946 ±1.3 7.905±1.2
Anembryonic sac I: 13 (20.91% C: 10 (18.52%)
79.09% of the women patients had fetal pole absent or irregular gestational sac in the ultrasonographic findings. The other finding was a blighted ovum seen 20.91%.
Groups comparable at baseline |
Describe intervention:
sublingual- tablet misoprostol 600 mcg every 6 hourly for 3 doses. The dose was decreased to lessen the side effects. |
Describe control):
vaginal e tablet misoprostol 800 mcg every 6 hourly up to 3 doses. |
Length of follow-up:
Loss-to-follow-up: Intervention: N 4/56 (3.36%) Reasons (surgical evacuation for other causes (4), excessive haemorrhage (2)), sever allergic reaction (1), time regimen not followed (1)
Control: N 5/56 (4.20%) Reasons (absconded (1) surgical evacuation for other causes (4), excessive haemorrhage (4))
Incomplete outcome data: Not reported
|
Outcome measures and effect size:
Treatment success (number/total (%)) I:52/56 (92.9%) C:48/54 (89%)
Satisfaction Highly satisfied: I: 28/56 (50%) C: 27/54 (50%)
Just satisfied I: 20/56 (35.71%) C: 18/54 (33.33%)
Unsatisfied I; 8/56 (14.29% C: 9/54 (16.67%)
Adverse events of treatment Excessive haemorrhage I: 2/56 (1.68% C: 4/54 (3.36)
b. Vomiting I: 6/56 (10.71%) C: 2/54 (3.7%)
C. Diarrhoea I: 5/56 (8.93 %) C: 4/54 (7.41%)5
d. Abdominal pain (requiring analgesics): I: 18/56 (32.14%) C: 15/54 (27.78%)
e. Fever/ chills I: 6/56 (10.71%) C: 2/54 (3.7%)
F. Headache I: 3/56 (5.36%) C: 1/54 (1.85%)
Allergy I: 3/56 (5.36%) C: 2/54 (3.70%)
Induction abortion interval I: 18.241 C: 18.125
|
Aim of the study To compare the efficacy and the side effects of different regimes of misoprostol in causing the complete expulsion of products of conception in early pregnancy failure.
Outcomes Evaluation was done 6 h after 3rd dose of misoprostol, i.e. at 24 h. If the uterus was not felt empty on per vaginal examination and ultrasonography showed products of conception, then dilatation and evacuation was done and was considered a true drug failure.
|
|
Akanksha, 2016; |
Type of study: RCT
Setting and country: Department of Obstetrics & Gynecology, Kasturba Hospital, New Delhi, India
Funding: No funding sources conflicts of interest: None
|
Inclusion criteria: Missed miscarriage is characterized by closed cervix and minimal or no bleeding per vaginum. On ultrasound it is defined as an intrauterine gestational sac with a mean sac diameter of 25 mm or more with no visible embryonic pole, an embryonic pole of more than 7 mm with no cardiac activity or persistent absence of cardiac activity on a second scan at least 7 to 10 days later
Exclusion criteria: Women with suspected ectopic pregnancy, excessive bleeding per vaginum, incomplete miscarriage, hemodynamically unstable, history of asthma, previous cesarean or allergy to prostaglandins were excluded from this study
N total at baseline: 50 Intervention: 25 Control:25
Important prognostic factors2: age ± SD: I: 24.92 ±3.17 C: 24.48 ±3.41
Primigravida I: 12/25 (48%) C: 10/25 (40%)
Gestational age 9.42 ±2.05 9.55±2.43
Groups comparable at baseline.
|
Describe intervention:
400microg of misoprostol sublingually every four hours for a maximum of 5 doses |
Describe control):
400 microgram of misoprostol vaginally every four hours for a maximum of 5 doses |
Length of follow-up:
Loss-to-follow-up: I: 0/25 C: 0/25
Incomplete outcome data: I: 0/25 C: 0/25
|
Outcome measures and effect size:
Treatment success (number/total (%)) I:17/25 (68%) C:15/25 (60%)
Reintervention (surgical) I: 8/25 (32%) C: 10/25 (40%)
Satisfaction: Comfortable I: 22/25 (88%) C: 13/25 (52%)
Discomfort: I:2/25 (12%) C: 12/25 (48%)
Adverse events Excessive bleeding, requiring blood transfusion I: 1/25 (4%) C: 1/25 (4%)
a. Vomiting I: 10/25 (40%) C: 12 / 25 (48%)
Nausea I: 18/25 (72%) C: 20/25 (80%)
Diarrhoea I: 8/25 (32%) C: 03/25 (12%)
Shivering I: 12/25 (24%) C: 5/25 (4%)
Fever I: 10/25 (40%) C: 4/16 (16%)
Unpleasant taste I: 15/25 (60%) C: 1/25 (4%)
Induction abortion interval I: 11.67 ± 6.90 C: 18.33 ±5.60
Mean doses: I:L 3.29 ±1.02 C: 4.2 ± 0.85 |
Aim of the study to compare the efficacy, outcome and side effects of sublingual and vaginal misoprostol for the medical management of missed miscarriage.
Succes: complete expulsion of the products of conception without surgical intervention.
Failure was defined as cases with incomplete or no dilatation within 12 hours of last dose of misoprostol. Surgical evacuation was considered for those who failed to abort after receiving 5 doses of misoprostol and those who had incomplete miscarriage.
|
|
Sonsanoh, 2014 |
Type of study: RCT
Setting and country: Hospital, Thailand
Funding : not reported. and conflicts of interest: none |
Inclusion criteria: as 1) an intrauterine gestational sac with a mean diameter of 25 mm or greater and no visible embryonic pole; 2) an embryonic pole of 5-14 mm with no cardiac activity; and 3) abnormal growth or persistent absence of fetal cardiac activity on a second scan 7-10 days later(16). In addition, all participants should be over 18 years old. Parental consent was required if the participant’s age was less than 18 years old.
Exclusion criteria: Women with suspected ectopic pregnancy, heavy vaginal bleeding, and maternal history of asthma or cardiac disease, open internal cervical on speculum examination, history allergy to prostaglandins, vital sign unstable and an inability to confirm pregnancy failure or intrauterine gestational sac location were excluded from this study.
N total at baseline: Intervention: 60 Control: 60
Important prognostic factors2: For example age ± SD: I:29.5 ±6.2 C: 29.4 ± 7.3
Type (embryonic death / anembryonic pregnancy I: 24 (40%) / 36 (60%) C: 29 (48.3% / 31 (51.6%)
Groups comparable at baseline? No, different in, BMI. Weight gain and primigravida |
Describe intervention (treatment/procedure/test):
In Group 1 (n = 60), they were given 4 tablets of 200 μg misoprostol with 2-3 drops of normal saline placed in the posterior vaginal fornix by digital insertion. The women remained in the semiprone position for 30 min.
Every 6 hours, maximum of 3 dosis
|
Describe control (treatment/procedure/test):
In Group 2 (n = 60), 4 tablets of 200 μg misoprostol were sublingually given.
Every 6 hours, maximum of 3 dosis
|
Length of follow-up:
2 wk
Loss-to-follow-up: None
Incomplete outcome data: none
|
Outcome measures and effect size (include 95%CI and p-value if available):
Treatment success (number/total (%)) I: 41/60 (68.3%) C: 36/60 (60%), p=0.44
Reintervention (surgical) I: 16/60 (26.7%) C: 24/60 (40%)
Adverse events (bleeding , perforations)
Heavy bleeding: 12/60 (20%) 3/60 (5%)
Side effects: general
Side effects: vomiting -
Side effects: nausea 6/60 (10%) 7/60 (11.7%)
Side effects: diarrhea I: 4/60 (6.7%) C: 3/60 (5.0%)
Side effects: pain I: 27/60 (45%) C: 19/60 (31.7%)
Satisfaction: (The assessment of satisfaction ranged from dissatisfied, satisfied and very satisfied.) Women from both groups were equally satisfied with the treatments that they have received; none of the participants was dissatisfied.
|
Aim: To compare the complete abortion rate, the induction-abortion time and the side effects of 800 μg misoprostol via vaginal and sublingual routes. |
|
Comparison 3. Oral vs. Sublingual |
|||||||
|
Ayudhaya, 2006 |
Type of study: RCT
Setting and country: Department of Obstetrics and Gynaecology at Ramathibodi Hospital, Bangkok, Thailand
Funding and conflicts of interest: not reported
|
Inclusion criteria: Diagnosis of early pregnancy failure, defined as one of the following: - Intrauterine gestational sac with mean sac diameter of >2 cm without a fetal pole (blighted ovum) - Presence of a fetal pole without cardiac pulsations - Gestational sac diameter <2cm with no interval growth or a persistent absence of fetal cardiac pulsation on re-scanning after 7-10 days Gestational age of 7-12 weeks
Exclusion criteria: Abnormal vaginal bleeding Severe abdominal pain
N total at baseline: N=138 (however 2 participants were later excluded) Intervention: Control:
Important prognostic factors2: Age/years (mean (SD)) Oral: 32.0 (5.8) Sublingual: 33.4 (6.2) (NS) Gestational age/weeks (mean (SD)) Oral: 10.7 (1.5) Sublingual: 11.0 (1.4) (NS) Nulliparous (number/total (%)) Oral: 31/68 (45.6) Sublingual: 27/70 (38.6) (NS)
Groups comparable at baseline
|
Describe intervention:
400 micrograms of misoprostol orally every 4 hours, up to a maximum of 6 doses (n=68)
Women in the sublingual group had two tablets placed under the tongue by a nurse. They were not allowed to eat for 20 minutes, to allow complete dissolution. A nurse was also responsible for oral administration to the patients. Patients were allowed 30 ml of water to drink, then misoprostol was given the same way every 4 hours until products of conception were detected.
|
Describe control):
400 micrograms of misoprostol sublingually every 4 hours, up to a maximum of 6 doses (n=70)
|
Length of follow-up:
Loss-to-follow-up: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
Incomplete outcome data: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
|
Outcome measures and effect size:
Clinical outcome of treatment (number/total (%)) a.Complete miscarriage Oral: 17/66 (25.8) Sublingual: 15/70 (21.4) (NS)
b.Incomplete miscarriage Oral: 23/66 (34.8) Sublingual: 27/70 (38.6) (NS)
b.Medical failure Oral: 26/66 (39.4) Sublingual: 28/70 (40.0) (NS)
Adverse effects of treatment (number of women/total (%)) a. Nausea/vomiting Oral: 3/66 (4.5) Sublingual: 2/70 (2.9) b. Diarrhoea Oral: 7/66 (10.6) Sublingual: 6/70 (8.6) c. Fever Oral: 2/66 (3.0) Sublingual: 15/70 (21.4) d. Chills Oral: 0/66 (0) Sublingual: 4/70 (5.7)
(Note: these % have been calculated by the technical team and differ slightly from those reported in the paper, because the authors calculated % using 68 women in the oral arm, despite reporting excluding them, and excluding them for other outcomes. The authors also report that no serious complications occurred in either group.)
Measures of pain: abdominal pain (number of women/total (%)) Oral: 40/66 (60.6) Sublingual: 47/70 (67.1) |
Aim of the study To compare repeated doses of sublingual with oral misoprostol in the medical management of early
Outcomes reported 1. Clinical outcomes: Complete miscarriage, incomplete miscarriage, and medical failure within 24 hours. 2. Adverse effects: Nausea or vomiting, diarrhoea, fever and chills are reported.
3. Abdominal pain
Complete miscarriage was defined as no active bleeding, closed cervical os and endometrial thickness < 1 cm.
Induction to expulsion interval/hours (mean (SD)) Oral: 10.7 (6.6) Sublingual: 8.7 (5.4) Total dosage received/micrograms (mean (SD)) Oral: 1706 (90.1) Sublingual: |
|
Kushwah, 2009 |
Type of study: RCT
Setting and country: prenatal clinic of the Department of Obstetrics and Gynaecology of Sucheta Kriplani Hospital, Delhi, India
Funding and conflicts of interest: Not reported
|
Inclusion criteria: Gestational sac of 25 mm or larger with no embryo present (anembryonic gestation) Presence of a fetal pole without cardiac pulsations (missed miscarriage)
All participants had early pregnancy failure confirmed by ultrasound between the 7th and 14th week of gestation.
Exclusion criteria: Vaginal bleeding Any evidence of infection History of allergy to misoprostol Major medical problems
N total at baseline: 100 Intervention: 50 Control:50
Important prognostic factors2: Age/years (mean (SD)) Sublingual: 26.6 (4.4) Oral: 24.6 (3.8) Parity (mean (SD)) Sublingual: 2.1 (0.9) Oral: 2.1 (0.9) Gestation/days (mean (SD)) Sublingual: 57.7 (7.8) Oral: 59.9 (9.0) (Note: only 1 woman, from the oral arm, had gestation of over 80 days) Type of pregnancy (number/total (%)) Sublingual: - Anembryonic: 23/50 (46) - Missed miscarriage: 27/50 (54) Oral: - Anembryonic: 34/50 (68) - Missed miscarriage: 16/50 (32)
Groups comparable at baseline. |
Describe intervention:
200mg of oral mifepristone + 600 micrograms sublingual misoprostol (n=50)
Women who did not expel products of conception within 12 hours of the first dose of misoprostol were given up to 3 supplemental doses of 400 micrograms at three hour intervals (sublingually or orally depending on their allocation). Those who received the maximum misoprostol allocation and did not expel products of conception within 4 hours of taking the last 400 microgram dose underwent surgical evacuation under intravenous sedation and paracervical block.
|
Describe control):
200mg of oral mifepristone + 600 micrograms of oral misoprostol (n=50)
|
Length of follow-up:
Loss-to-follow-up: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
Incomplete outcome data: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
|
Outcome measures and effect size:
Successful uterine evacuation (number of women/total (%)) Sublingual: 46/50 (92) Oral: 42/50 (84)
Adverse effects of treatment (number of women/total (%))
a. Nausea Sublingual: 17/50 (34) Oral: 26/50 (52) (Note: 1 woman from each arm required medication)
b. Vomiting Sublingual: 11/50 (22) Oral: 22/50 (44) (Note: 3 women from the oral arm required medication)
c. Diarrhoea Sublingual: 24/50 (48) Oral: 28/50 (56) (Note: 5 women in each arm had more than 4 episodes and required medication)
d. Fever: any Sublingual: 10/50 (20) Oral: 26/50 (52)
e. Fever: ≥37.8ºC Sublingual: 0 Oral: 4/50 (8)
Measures of pain (number/total (%)) a. Incidence of pain requiring no analgesia Sublingual: 14/50 (28) Oral: 26/50 (52) b. Incidence of pain requiring analgesia Sublingual: 9/50 (18) Oral: 18/50 (36) c. Incidence of pain: total Sublingual: 23/50 (46) Oral: 44/50 (88)
Reported satisfaction (number/total (%)) Sublingual: 46/50 (92%) Oral: 36/50 (72%)
|
Aim of the study To compare the efficacy of misoprostol administered sublingually or orally for uterine evacuation after early pregnancy failure.
Outcomes reported 1. Successful uterine evacuation: not directly defined, but see criteria for complete evacuation above. 2. Adverse effects of treatment: The incidence of nausea, vomiting, diarrhoea (≤4 episodes and >4 episodes), and fever (both above and below 37.8°C) are reported. It is unclear at what point these outcomes were assessed. 3. Measures of pain: The incidence of abdominal pain requiring no analgesia and requiring analgesia are reported (along with the % of women who had no abdominal pain). It is unclear at what point this outcome was assessed. 4. Satisfaction: The proportion of women reporting being satisfied (phrased as a yes or no question) is reported.
Induction to evacuation interval/hours (mean (SD)) Sublingual: 46 (4.5) Oral: 9.4 (5.6) (Note: this only includes women for whom evacuation was successful) It is not reported how many women required the supplemental doses of misoprostol.
Evacuation was considered incomplete when active vaginal bleeding continued, the cervical os remained open, and products of conception were visible on ultrasound. In this case, women underwent surgical evacuation under paracervical block. |
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors [(potential) confounders]
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
Risk of bias table for intervention studies (randomized controlled trials)
Comparison 1 Oral vs. Vaginal
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Creinin, 1997 |
computer-generated random number table |
Unlikely |
Neither the clinician nor the patient was blinded to the treatment group |
Neither the clinician nor the patient was blinded to the treatment group |
Neither the clinician nor the patient was blinded to the treatment group |
Unlikely |
Unlikely |
Likely |
|
Ngoc, 2004 |
computer-generated code in blocks of 10. |
sequentially numbered randomized envelope |
Neither the investigator nor the woman was blinded to the treatment assignment |
Neither the investigator nor the woman was blinded to the treatment assignment |
Neither the investigator nor the woman was blinded to the treatment assignment |
Unlikely |
Unlikely |
Likely ( 2 women wrongly diagnosed) |
|
Rita, 2006 |
permuted block method |
Not reported |
Not reported |
Not reported |
Not reported |
Unclear how and when "severe pain" and adverse effects were judged. |
Not reported |
Unlikely |
|
Marwah, 2016 |
computer generated sequentially numbered envelopes |
Not reported |
Not reported |
Not reported |
Not reported |
Unlikely |
Unlikely |
Unlikely |
Comparison 2. Sublingual vs. Vaginal
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
El Sokkary, 2016 |
Not reported |
Unlikely |
Not reported |
Not reported |
Not reported |
Unlikely |
Unlikely |
Unlikely |
|
Dehbasi, 2016 |
block randomization was performed according to the time of admission |
Unlikely |
Not blinded |
Single blind allocation and intervention were conducted by a nurse working in the midwifery unit. |
Single blind allocation and intervention were conducted by a nurse working in the midwifery unit. |
Unlikely |
Not described |
Likely, 2 excluded that did not repond or had severe abdominal pain |
|
Tanha, 2010 |
computer-generated code |
Not reported |
Neither the clinician or the patient were blinded to treatment allocation. |
Neither the clinician or the patient were blinded to treatment allocation. |
Those assessing treatment outcome were not blinded to treatment allocation. |
Unlikely |
Unlikely |
5 of the participants were later diagnosed as having a partial or complete mole (however, this represents only 2.3% of the study population) |
|
Tang, 2003 |
computer-generated random numbers |
Not reported |
Blinding is not reported - this would be impossible for the participants, however could have been achieved for those assessing outcomes. |
Blinding is not reported - this would be impossible for the participants, however could have been achieved for those assessing outcomes. |
Blinding is not reported - this would be impossible for the participants, however could have been achieved for those assessing outcomes. |
Likely (satisfaction) |
Unlikely |
Unlikely |
|
Shah, 2010 |
consecutive sealed envelopes.
|
Not reported |
Blinding was not done. Blinding the participants would have been difficult (although not impossible), however they could have blinded the physicians judging treatment success and need for further intervention.
|
Blinding was not done. Blinding the participants would have been difficult (although not impossible), however they could have blinded the physicians judging treatment success and need for further intervention.
|
Blinding was not done. Blinding the participants would have been difficult (although not impossible), however they could have blinded the physicians judging treatment success and need for further intervention.
|
Unlikely |
Unlikely |
Unlikely |
|
Seervi, 2014 |
coin tossing method |
Not reported |
Not reported |
Not reported |
Not reported |
Outcome measures not reported in methods |
Unlikely |
Unlikely |
|
Akanksha, 2016 |
lottery method (random sampling). |
Not reported |
Not reported |
Not reported |
Not reported |
Unlikely |
Unlikely |
Unlikely |
|
Sonsanoh, 2014 |
We used and assigned blocks of four randomizations to two groups of participants. |
Cards labeled with the assigned route were placed in sealed, opaque envelopes which were filled and labeled in accordance with the list of randomizations. The allocation was concealed by the use of sealed number of treatments. |
Not reported |
Not reported |
Not reported |
Unlikely
Note: satisfaction was defined in methods in a 3 point rating scale but is only descriptive in results |
Unlikely |
Unlikely |
Comparison 3. Oral vs. Sublingual
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Kushwah, 2009 |
computer generated random numbers.
|
Not reported |
Not reported |
Not reported |
Not reported |
The point at which adverse effects, pain and satisfaction were assessed is not reported. |
Unlikely |
Unlikely |
|
Ayudhaya, 2006 |
computer generated random numbers
|
Not reported |
Blinding is not reported. It would have been difficult to blind the participants (although not impossible); however those assessing the outcome of the treatment could have been blinded to allocation. |
Blinding is not reported. It would have been difficult to blind the participants (although not impossible); however those assessing the outcome of the treatment could have been blinded to allocation. |
Blinding is not reported. It would have been difficult to blind the participants (although not impossible); however those assessing the outcome of the treatment could have been blinded to allocation. |
Unlikely |
Unlikely |
Unlikely |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules..
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Exclusietabel
|
Auteur en jaartal |
Redenen van exclusie |
|
Bhandekar, 2018 |
APLA, beschrijft niet de juiste studiepopulatie. Voldoet niet aan PICO. |
|
Garg, 2015 |
APLA, beschrijft niet de juiste studiepopulatie. Voldoet niet aan PICO. |
|
Nautiyal, 2015 |
APLA, beschrijft niet de juiste studiepopulatie. Voldoet niet aan PICO. |
|
Saeed, 2018 |
Quasi-experimentele studie, voldoet niet aan inclusiecriteria. |
|
Souza, 2013 |
Beschrijf labour induction, niet de juiste studiepopulatie. Voldoet niet aan PICO. |
|
Yazdani, 2012 |
APLA, beschrijft niet de juiste studiepopulatie. Voldoet niet aan PICO. |
Beoordelingsdatum en geldigheid
Publicatiedatum : 24-07-2020
Beoordeeld op geldigheid : 24-07-2020
Voor het beoordelen van de actualiteit van deze richtlijn is de werkgroep niet in stand gehouden. Uiterlijk in 2025 bepaalt het bestuur van de NVOG of de modules van deze richtlijn nog actueel zijn. Op modulair niveau is een onderhoudsplan beschreven. Bij het opstellen van de richtlijn heeft de werkgroep per module een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De NVOG is regiehouder van deze richtlijn en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de verschillende richtlijnmodules. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
|
Module1 |
Regiehouder(s)2 |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn3 |
Frequentie van beoordeling op actualiteit4 |
Wie houdt er toezicht op actualiteit5 |
Relevante factoren voor wijzigingen in aanbeveling6 |
|
Toedieningsroute misoprostol bij medicamenteuze behandeling |
NVOG |
2020 |
2025 |
1x 5 jaar |
NVOG |
Nvt |
|
[1] Naam van de module 2 Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders) 3 Maximaal na vijf jaar 4 (half)Jaarlijks, eens in twee jaar, eens in vijf jaar 5 regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) werkgroep die in stand blijft 6 Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen |
||||||
Algemene gegevens
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijn.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2018 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van verschillende relevante specialismen die betrokken zijn bij de zorg voor patiënten die een miskraam hebben doorgemaakt. De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname.
De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Werkgroep
- Drs. E. Hink, gynaecoloog, werkzaam in het Radboudumc, Nijmegen, Nederlandse Vereniging voor Obstetrie en Gynaecologie (voorzitter).
- M.M.J. van den Berg MSc., fertiliteitsarts, werkzaam in het Academisch Medisch Centrum, Amsterdam, Vereniging van Fertiliteitsartsen.
- Dr. F.M. van Dunné, gynaecoloog, werkzaam in het Haaglanden Medisch Centrum, Den Haag, Nederlandse Vereniging voor Obstetrie en Gynaecologie.
- Prof. dr. M. Goddijn, hoogleraar Voortplantingsgeneeskunde & gynaecoloog, werkzaam in het Academisch Medisch Centrum, Amsterdam, Nederlandse Vereniging voor Obstetrie en Gynaecologie.
- M. Hermus, verloskundige, werkzaam bij Verloskundigen Oosterhout, Oosterhout, Koninklijke Nederlandse Organisatie van Verloskundigen. (tot april 2019)
- Drs. A. B. Hooker, gynaecoloog, werkzaam in het Zaans Medisch Centrum, Zaandam, Nederlandse Vereniging voor Obstetrie en Gynaecologie.
- Dr. S. Kuc, fertiliteitsarts, werkzaam in het St. Antonius Ziekenhuis, Utrecht, Vereniging van Fertiliteitsartsen.
- Dr. M. Lemmers, AIOS gynaecologie, werkzaam in het Academisch Medisch Centrum, Amsterdam, Nederlandse Vereniging voor Obstetrie en Gynaecologie.
- Dr. R.H.F. van Oppenraaij, gynaecoloog, werkzaam in het Maasstad Ziekenhuis, Rotterdam, Nederlandse Vereniging voor Obstetrie en Gynaecologie.
- A.J.Y. van Sluis, echoscopist, oa. werkzaam bij Rivas Zorggroep, Gorinchem, Beroepsvereniging Echoscopisten Nederland.
- K. Spijkers, patiëntvertegenwoordiger, Utrecht, Patiëntenfederatie Nederland.
Meelezer
- A. Beuckens, Beleidsmedewerker KNOV, Koninklijke Nederlandse Organisatie van Verloskundigen. (vanaf mei 2019)
Met ondersteuning van
- A.A. Lamberts MSc., senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten, Utrecht
- Dr. L. Viester, adviseur, Kennisinstituut van de Federatie Medisch Specialisten, Utrecht
- L.H.M. Niesink-Boerboom MSc, literatuurspecialist, Kennisinstituut van de Federatie Medisch Specialisten, Utrecht
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in de tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van Medisch Specialisten.
|
Overzicht Belangenverklaringen |
|||||||||
|
Achternaam |
Hoofdfunctie |
Nevenwerkzaamheden |
Persoonlijke financiële belangen |
Persoonlijke relaties |
Extern gefinancierd onderzoek |
Intellectuele belangen en reputatie |
Overige belangen |
Getekend op |
Actie? |
|
van den Berg |
Fertiliteitsarts, Amsterdam UMC locatie AMC |
ESHRE, Bestuurslid SIG Implantation & Ectopic Pregnancy (onbetaald) Travelgrants t/m juli 2021 |
geen |
geen |
geen |
geen |
geen |
18-7-2018 |
Geen actie nodig |
|
Dunné |
Gynaecoloog, Haaglanden Medisch Centrum (HMC) te Den Haag |
Lid Werkgroep Otterlo (landelijke richtlijnen commissie Obstetrie) (onbetaald) |
geen |
geen |
geen |
geen |
geen |
9-3-2018 |
Geen actie nodig. |
|
Goddijn |
Hoogleraar Voortplantingsgeneeskunde, gynaecoloog, pijlerhoofd Voortplantingsgeneeskunde, Amsterdam UMC |
-Bestuurslid SIG Fertiliteitspreservatie NVOG (vice-voorzitter/ onbetaald) |
geen |
geen |
Onderzoeks financiering vanuit ZonMw en KWF/Pink Ribbon. Niet conflicterend met onderwerp richtlijn. |
geen |
geen |
5-3-2018 |
Geen actie nodig. |
|
Hink |
Gynacoloog perinatoloog Radboudumc te Nijmegen |
geen |
geen |
geen |
geen |
geen |
geen |
19-3-2018 |
Geen actie nodig |
|
Hooker |
Gynaecoloog, Zaans Medisch Centrum (ZMC), te Zaandam. |
Docent 2-daagse cursus Zicht op Ziekenhuizen, studiecentrum voor Bedrijf en Overheid. Onderwerp: kwaliteit en veiligheid in de zorg en veranderde positie van de medisch specialist. (betaald). |
Adviseur firma Fziomed (USA), advies bij ontwikkelen middel ter preventie intra-uteriene verklevingen (betaald). |
geen |
Was hoofdonderzoeker van de Post Abortion Prevention of Adhesion |
Zie kolom extern gefinancierd onderzoek |
geen |
12-5-2018 |
Besloten dat indien er een vraag over dit specifieke onderwerp in het raamwerk wordt opgenomen Angelo Hooker hier geen voortrekker van zal zijn. |
|
Klapwijk-Hermus |
Verloskundigen Oosterhout – eerstelijns verloskundige, eigenaar praktijk |
Promotietraject, onderwerp: evaluatie van geboortecentra in Nederland. Afrondend, (onbetaald) |
geen |
geen |
geen |
geen |
geen |
14-5-2018 |
Geen actie nodig |
|
Kuc |
Fertiliteitsarts , St. Antonius Ziekenhuis, te Nieuwegein |
Bestuurslid vereninging van fertiliteitsartsen VVf (onbetaald) |
geen |
geen |
geen |
geen |
geen |
3-5-2018 |
Geen actie nodig |
|
Lamberts |
Adviseur Kennisinstituut |
Beleidsmedewerker Kwaliteit NVKG |
geen |
geen |
geen |
geen |
geen |
2014 |
Geen actie nodig |
|
Lemmers |
AIOS Gynaecologie VUMC te Amsterdam |
Arts-onderzoeker (onbetaald). Uitvoeren Cochrane review medicamenteuze behandeling miskraam tot 20 weken. |
geen |
geen |
geen |
geen |
geen |
15-3-2018 |
Geen actie nodig. |
|
Oppenraaij |
Gynaecoloog, Maasstad Ziekenhuis, te Rotterdam |
Bestuurslid werkgroep jonge zwangerschap NVOG (onbetaald) |
geen |
geen |
Deelname ahrends studie van Bayer onderzoek naar nieuwe medicatie bij vrouwen met endometriose. Onderzoek is inmiddels gesloten zonder enige inclusie in ons ziekenhuis. |
geen |
geen |
5-5-2018 |
Geen actie nodig. |
|
Sluis |
Obstetrisch en Gynaecologisch echoscopiste bij Screeningscentrum Fara te Ede (8u per week) |
Bestuurslid BEN, (onbetaald) |
geen |
geen |
geen |
geen |
geen |
1-6-2018 |
Geen actie nodig |
|
Spijkers |
Senior Beleidsadviseur/ projectmanager Patiëntenfederatie Nederland |
Voorzitter Stichting Samen voor Duchenne, (onbetaald) |
geen |
geen |
geen |
geen |
geen |
24-5-2018 |
Geen actie nodig |
|
Viester |
Adviseur Kennisinstituut |
geen |
geen |
geen |
geen |
geen |
geen |
2018 |
Geen actie nodig |
|
Beuckens (meelezer) |
Beleidsmedewerker kwaliteit en implementatie KNOV |
Coaching/luisterend oor - betaald (minder dan 5 keer per jaar) |
geen |
geen |
geen |
geen |
geen |
2019 |
Geen actie nodig |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde van de Patiëntenfederatie Nederland zitting te laten nemen in de werkgroep. Patiëntenverenigingen Freya en VSOP zijn eveneens gevraagd zitting te nemen in de werkgroep, zij gaven echter aan de voorkeur te hebben om alleen bij de invitational conference aanwezig te zijn en input te leveren tijdens de commentaarfase. Er is in samenwerking met de Patiëntenfederatie Nederland op 1 mei 2019 een focusgroep georganiseerd, de resultaten hiervan zijn meegenomen in de richtlijntekst. Een verslag van de bijeenkomst is als aanverwant product opgenomen bij de richtlijn. Er heeft een invitational conference plaatsgevonden aan de start van het traject waar verschillende patiëntenorganisaties voor waren uitgenodigd (Freya, VSOP, Patiëntenfederatie Nederland, Stichting Kind&Ziekenhuis). Een verslag hiervan is besproken in de werkgroep en de belangrijkste knelpunten zijn verwerkt in de richtlijn. Tijdens de oriënterende zoekactie werd gezocht op literatuur naar patiëntenperspectief (zie Strategie voor zoeken en selecteren van literatuur). De conceptrichtlijn is tevens voor commentaar voorgelegd aan de VSOP, Freya en de Patiëntenfederatie Nederland.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn (module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden bij de aanverwante producten.
Werkwijze
Voor deze richtlijn is waar mogelijk uitgegaan van de NICE-richtlijn ‘Ectopic pregnancy and miscarriage: Diagnosis and initial management in early pregnancy of ectopic pregnancy and miscarriage’ uit 2012. Bruikbare onderdelen zijn van een update voorzien zodat ook recente evidence is meegenomen.
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van Medisch Specialisten.
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. Tevens zijn er knelpunten aangedragen door verschillende partijen tijdens de invitational conference. Een verslag hiervan is opgenomen onder aanverwante producten.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de voorzitter en de adviseur conceptuitgangsvragen opgesteld. Deze zijn met de werkgroep besproken waarna de werkgroep de definitieve uitgangsvragen heeft vastgesteld. Vervolgens inventariseerde de werkgroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als kritiek, belangrijk (maar niet kritiek) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de kritieke uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Er is op 1 februari 2019 oriënterend gezocht naar literatuur over patiëntenvoorkeuren en patiëntrelevante uitkomstmaten (patiëntenperspectief) in de databases Medline (Ovid) & Embase (Elsevier). Vervolgens werd voor de afzonderlijke uitgangsvragen aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie voor de oriënterende zoekactie en patiëntenperspectief zijn opgenomen onder aanverwante producten.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR – voor systematische reviews; Cochrane – voor gerandomiseerd gecontroleerd onderzoek; ACROBAT-NRS – voor observationeel onderzoek; QUADAS II – voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende hoeveelheid studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A) Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, matig, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Matig |
|
|
Laag |
|
|
Zeer laag |
|
B) Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008), en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module ‘Organisatie van Zorg’ en onder het kopje Patiëntenperspectief.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
NICE. Ectopic pregnancy and miscarriage: Diagnosis and initial management in early pregnancy of ectopic pregnancy and miscarriage. 2012. Update in 2019.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, et al. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008;336(7654). doi: 10.1136/bmj.a139. PubMed PMID: 18483053.
Kennisinstituut van Medisch Specialisten. Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. 2015
Zoekverantwoording
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
1990 – januari 2019
|
1 FETAL VIABILITY/ (1729) 2 exp PREGNANCY OUTCOME/ (52170) 3 ((f?etus$ or f?etal or embryo$ or pregnan$ or gestat$) adj3 (viab?l$ or non?viab?l$ or continu$ or term or outcome$ or live or living or alive)).ti,ab. (72858) 4 ((live or term) adj3 birth$).ti,ab. (26924) 5 exp FETAL DEATH/ (28394) 6 exp ABORTION, SPONTANEOUS/ (33761) 7 miscarr$.ti,ab. (12606) 8 ((spontaneous or threatened or imminent or imminens or missed or delay$ or inevitable or incomplete$ or complete$ or early or silent or quiescent) adj2 abort$).ti,ab. (14638) 9 ((pregnan$ or embryo$ or f?etal or f?etus$) adj3 (loss$ or demise or death$ or resorp$ or disintegrat$ or wast$ or reject$ or fail$)).ti,ab. (32907) 10 (anembryo$ or empty sac$).ti,ab. (188) 11 (blight$ adj2 (ova or ovum)).ti,ab. (203) 12 exp GESTATIONAL TROPHOBLASTIC DISEASE/ (5555) 13 (trophoblast$ adj3 (gestat$ or pregnan$ or disease$ or tumo?r$ or neoplas$)).ti,ab. (5121) 14 ((hydatid$ or invasi$) adj3 mol$).ti,ab. (5364) 15 molar pregnan$.ti,ab. (1239) 16 (placenta$ adj3 (tumo?r$ or neoplas$)).ti,ab. (1003) 17 (chorio carcinoma$ or chorio?carcinoma$).ti,ab. (6232) 18 (non vital* or avital* or first trimester or curettage).ti,ab. (32333) 19 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 (229869) 20 exp *Misoprostol/ or (misoprostol* or cytotec or arthrotec or napratec).ti. (3235) 21 exp *Administration, Oral/ or exp *Administration, Intravaginal/ or (oral* or mouth or buccal* or swallow* or sublingual* or vagina* or intravagina* or genital* or paracervical*).ti. (285130) 22 19 and 20 and 21 (477) 23 limit 22 to english language (467) 24 limit 23 to yr="1990 -Current" (467) 25 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1816404) 26 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (379497) 27 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ [Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies] (3097539) 28 24 and 26 (14) 29 24 and 25 (365) 30 29 not 28 (353) 31 24 and 27 (194) 32 31 not 28 not 30 (61) 33 28 or 30 or 32 (428) = 428 |
474
|
|
Embase (Elsevier) |
('spontaneous abortion'/exp OR 'ectopic pregnancy'/exp OR 'trophoblastic tumor'/exp OR miscarr*:ti,ab OR (((spontaneous OR threatened OR imminent OR imminens OR missed OR delay* OR inevitable OR incomplete* OR early OR silent OR quiescent) NEAR/2 abort*):ti,ab) OR (((pregnan* OR embryo* OR f?etal OR f?etus*) NEAR/3 (loss* OR demise OR death* OR resorp* OR disintegrat* OR wast* OR reject* OR fail* OR viab?l* OR non?viab?l*)):ti,ab) OR anembryo*:ti,ab OR 'empty sac*':ti,ab OR ((blight* NEAR/2 (ova OR ovum)):ti,ab) OR (((ectopic OR 'extra uterine' OR 'extra?uterine' OR tub* OR ampullary OR isthm* OR fimbrial OR cornual OR interstitial OR abdom* OR ovar* OR cervi*) NEAR/3 (pregnan* OR gestat*)):ti,ab) OR ((pregnan* NEAR/3 (unknown OR uncertain) NEAR/3 (location* OR site*)):ti,ab) OR pul:ti,ab OR ((trophoblast* NEAR/3 (gestat* OR pregnan* OR disease* OR tumor* OR tumour* OR neoplas*)):ti,ab) OR (((hydatid* OR invasi*) NEAR/3 mol*):ti,ab) OR 'molar pregnan*':ti,ab OR ((placenta* NEAR/3 (tumor OR tumour OR neoplas*)):ti,ab) OR 'chorio carcinoma*':ti,ab OR chorio?carcinoma*:ti,ab OR 'not vital pregnan*':ti,ab OR 'avital pregnan*':ti,ab OR 'first trimester pregnancy'/exp OR 'first trimester':ti,ab OR curettage:ti,ab) AND ('misoprostol'/exp/mj OR misoprostol*:ti OR cytotec:ti OR arthrotec:ti OR napratec:ti) AND ('oral drug administration'/exp/mj OR 'intravaginal drug administration'/exp/mj OR oral*:ti OR mouth:ti OR buccal*:ti OR swallow*:ti OR sublingual*:ti OR vagina*:ti OR intravagina*:ti OR genital*:ti OR paracervical*:ti) AND [english]/lim AND [1990-2018]/py NOT 'conference abstract':it
Gebruikte filters:
Systematische reviews: ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab ORcinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematicreview'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) = 0
RCT’s: ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/expOR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it = 191
Observationeel onderzoek: 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR ('prospective study'/de NOT 'randomizedcontrolled trial'/de) OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (case:ab,ti AND ((control NEAR/1 (study OR studies)):ab,ti)) OR (follow:ab,ti AND ((up NEAR/1 (study OR studies)):ab,ti)) OR ((observational NEAR/1 (study OR studies)):ab,ti) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti) = 16 = 207 |
- Relevant voor patiënten
- Achtergrond en definities
- Toepassen
- Onderzoek
- Overige