Algemene ziekteactiviteit
Uitgangsvraag
Wat zijn de behandelopties met csDMARDs/bDMARDs bij patiënten SLE?
- Wat is de effectiviteit van csDMARDs op ziekteactiviteit?
- Wat is de effectiviteit van bDMARDs op ziekteactiviteit?
Aanbeveling
Start met een csDMARD bij een patiënt met aanhoudende of frequente flares van ziekteactiviteit, ondanks behandeling met hydroxychloroquine (en eventueel een kortdurende behandeling met glucocorticoïden).
Maak in overeenstemming met de patiënt de keuze voor een csDMARD, waarbij
- Methotrexaat en azathioprine een vergelijkbaar profiel hebben wat betreft effectiviteit en bijwerkingen.
- Mycofenolaat mofetil eventueel een vervolgkeuze is op basis van een ernstiger bijwerkingenprofiel; mogelijk is het effect van mycophenolaat mofetil krachtiger.
- Tacrolimus, leflunomide en ciclosporine A zijn opties indien bovenstaande csDMARDs niet verdragen worden of ineffectief zijn.
Overweeg een switch te maken van csDMARD of het toevoegen van belimumab of anifrolumab indien het behandeldoel niet bereikt wordt na het voldoende lang, voldoende hoog gedoseerde toevoeging van csDMARD (en hydroxychloroquine).
Indien bovenstaande stappen niet hebben geleid tot het halen van het behandeldoel, overweeg een behandeling middels cyclofosfamide of rituximab bij refractaire ziekteactiviteit.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
In de samenvatting van de literatuur zijn de resultaten per uitkomstmaat beschreven voor csDMARDs, bDMARDs en andere medicatie. Voorafgaande aan het literatuurselectieproces is met de werkgroep afgestemd welke medicamenten veelal gebruikt worden in de Nederlandse praktijk, zie ‘search and select’. Gezien het groot aantal middelen heeft de werkgroep eerst gezocht naar recente systematische reviews. Deze artikelen zijn gebruikt als handvat.
Een korte samenvatting van de resultaten wordt hieronder beschreven. Het is belangrijk om te benoemen dat de evolutie van SLE-studies resulteert in ‘betere’ studies. De recente studies (d.w.z. bDMARDs) zijn beoordeeld middels de GRADE methodiek, wat resulteert in een (hogere) bewijskracht. In de huidige samenvatting van de literatuur zijn verschillende SLE-indices gebruikt als uitkomstmaat voor het meten van effectiviteit. Deze indices bevatten allen aspecten betreffende verschillende manifestaties. De scores van de verschillende manifestaties zijn gecombineerd tot één totaalscore. Om deze reden is het belangrijk in acht te nemen dat een verbetering van de totaalscore niet direct een verbetering van de score in alle categorieën m.b.t. de specifieke manifestaties betekent. Medicamenteuze behandelingen bij deze specifieke manifestaties worden in de volgende sub-modules beschreven; Cutane-, Gewrichts-, Cardiopulmonale - en Hematologische manifestaties.
Noot: niet alle middelen die staan beschreven in de samenvatting van de literatuur zijn geregistreerd voor de behandeling van SLE.
In totaal worden 7 vergelijkingen beschreven waarin verschillende soorten csDMARDs (methotrexaat (MTX), azathioprine (AZA), mycofenolaatmofetil (MMF), cyclofosfamide (CYC), tacrolimus (TAC), leflunomide (LEF) en ciclosporine A (CSA)) worden vergeleken met placebo/controlebehandeling bij patiënten met SLE. Tijdens de studies werd de standaardbehandeling, meestal met hydroxychloroquine (HCQ) (en/of glucocorticoïden (GC’s)), gehandhaafd in beide groepen.
De resultaten voor de uitkomstmaat ‘SLE-scores on disease activity indices’ beschrijven dat behandelingen met csDMARDs (d.w.z., MTX, AZA, MMF, LEF, CSA) effectiever zijn in vergelijking met een controlebehandeling. Dit gaat ook gepaard met een gereduceerde dosering van GC’s. Er worden mogelijk iets vaker bijwerkingen gerapporteerd bij behandelingen met een additionele csDMARD in vergelijking met een controlebehandeling.
Wanneer de csDMARDs met elkaar vergeleken worden zijn er geen evidente verschillen in effectiviteit m.b.t de uitkomstmaat ‘SLE-scores on disease activity indices’. Zo wordt beschreven dat MMF niet inferieur is aan een behandeling met CYC, CSA niet minder effectief is dan AZA en de effectiviteit van een hoge dosering CYC gelijk is aan een traditionele dosering. Ook worden er mogelijk geen verschillen gevonden voor de uitkomstmaten ‘glucocorticoïddosering’ en ‘bijwerkingen’. Gezien de aard van de SR kan er geen uitspraak worden gedaan over de bewijskracht, omdat de resultaten van de samenvatting van de literatuur niet middels GRADE beoordeeld kunnen worden.
Noot: In de review van Pego-Reigosa (2013) wordt mycofenolaatmofetil (mycophenolate mofetil) beschreven. Mycofenolaat mofetil is de prodrug van het werkzame mycofenolzuur, beiden kunnen worden ingezet.
In totaal worden 3 vergelijkingen beschreven waarin verschillende soorten bDMARDs (belimumab (BEL), rituximab (RTX), anifrolumab) worden vergeleken met een placebo/controlebehandeling bij patiënten met SLE. Ook in deze studies werd de standaardbehandeling, meestal met HCQ (en/of GC’s, csDMARDs), gehandhaafd in beide groepen.
Een behandeling met BEL resulteert in een verlaging van de ziekteactiviteit en een gereduceerde dosering GC’s. Er worden geen verschillen gevonden voor de uitkomstmaat bijwerkingen. De bewijskracht voor de verschillende uitkomstmaten varieert van hoog tot laag. Een belangrijke limitatie is dat voor de uitkomstmaat gereduceerde dosering GC’s, alleen is gekeken naar een dosering van >50%. Er zijn ook studies die hebben gekeken naar een reductie van >25%. Twee van de drie studies laten in tegenstelling tot de beschreven studies, geen verschil zien (Furie 2011; Stohl 2017; Zhang 2018.). Ook worden niet alle relevante bijwerkingen gerapporteerd.
Er worden geen verschillen gevonden voor alle uitkomstmaten bij de vergelijking RTX vs. controlebehandeling. De bewijskracht voor de verschillende uitkomstmaten varieert van laag tot zeer laag.
Een behandeling met anifrolumab resulteert in een verlaging van de ziekteactiviteit en een gereduceerde dosering GC’s. Echter worden bijwerkingen vaker gerapporteerd bij een behandeling met anifrolumab in vergelijking met een controlebehandeling. De bewijskracht voor de verschillende uitkomstmaten varieert van redelijk tot zeer laag. Mede door het studie design en de evolutie van SLE-studies is het mogelijk de ‘nieuwere’ studies te graderen middels GRADE en een hogere bewijskracht.
Anifrolumab is een relatief nieuw geneesmiddel voor de behandeling van SLE. Ten tijde van het richtlijnontwikkelingstraject zijn artikelen gepubliceerd (m.n. post-hoc analyses voor verschillende uitkomstmaten). Deze artikelen zijn nu niet meegenomen in de samenvatting van de literatuur. Wanneer de richtlijnmodule een update krijgt, wordt deze literatuur mogelijk toegevoegd. Noot: De review van Lee (2020) beschrijft slechts enkele uitkomstmaten voor effectiviteit gemeten als ziekteactiviteit score (d.w.z. ‘SLE-scores on disease activity indices’). In de huidige samenvatting van de literatuur is deze data aangevuld a.d.h.v. data uit de individuele studies (Furie, 2017; 2019; Morand, 2020). Er is alleen gekeken naar een dosering van 300 mg anifrolumab (i.v.) elke 4 weken. Deze dosering wordt ook gehanteerd in Nederland.
Er kan geen uitspraak worden gedaan over de effectiviteit en veiligheid van het middel intraveneuze immunoglobulinen (IVIg) vs. een controlebehandeling bij patiënten met SLE. Dit wordt veroorzaakt door de bewijskracht van de conclusies van de samenvatting van de literatuur. Deze is zeer laag.
Internationale richtlijnen en overige literatuur:
In de richtlijn van de ‘British Society for Rheumatology’ voor volwassenen met SLE uit 2018 wordt een behandeling met additioneel gebruik van MTX aanbevolen voor controle van de ziekteactiviteit bij patiënten met een mild actieve niet-orgaanbedreigende SLE (Gordon, 2018). Bij een matig actieve SLE wordt aanbevolen een behandeling met additioneel gebruik van MTX, AZA, MMF of CSA te overwegen bij aanwezigheid van artritis, huidaandoeningen, serositis, vasculitis of cytopenie. Voor refractaire gevallen wordt een additionele behandeling met BEL of RTX overwogen (Gordon, 2018). Wanneer er sprake is van ernstig actieve SLE wordt het volgende aanbevolen; additionele behandeling met MMF of CYC bij refractaire ernstige niet-nierziekten, BEL of RTX als basis wanneer patiënten niet/onvoldoende reageren op andere immunosuppressiva vanwege ineffectiviteit of intolerantie, IVIg kan overwogen bij patiënten met o.a. refractaire cytopenie (Gordon, 2018).
De EULAR-richtlijn voor volwassenen met SLE uit 2019 beveelt aan om immunosuppressieve therapieën (d.w.z. csDMARDs o.a. MTX, AZA, MMF) in te zetten bij patiënten die niet reageren op een behandeling met HCQ of indien het niet mogelijk is de GC-dosering te verlagen. Wanneer er sprake is van orgaanbedreigende SLE kunnen deze therapieën ook worden ingezet als initiële therapie. De csDMARD CYC kan ingezet worden indien er sprake is van ernstig orgaanbedreigende of levensbedreigende SLE, evenals ineffectiviteit bij gebruik van overige csDMARDs. De bDMARD BEL kan als aanvullende behandeling worden overwogen bij onvoldoende effect van een standaardbehandeling. De bDMARD RTX kan worden overwogen bij orgaanbedreigende SLE of intolerantie voor een standaardbehandeling met csDMARDs (Fanouriakis, 2019).
In de bestaande internationale richtlijnen (Gordon, 2018; Fanouriakis, 2019) wordt het medicament ‘anifrolumab’ niet aanbevolen, omdat het middel ten tijde van deze richtlijnen nog niet beschikbaar was. In december 2021 heeft de European Medicines Agency (EMA) geconcludeerd dat anifrolumab, een monoklonale antistof gericht tegen de IFN-1-receptor, van toegevoegde waarde is bij de behandeling van patiënten met SLE en met matig-ernstige (‘moderate’) tot ernstige (‘severe’) SLE ondanks standaardbehandeling (EMA, 2021). Op basis van de resultaten die staan beschreven in de samenvatting van de literatuur, vindt de werkgroep dat anifrolumab van toegevoegde waarde kan zijn voor patiënten met SLE, en in het bijzonder patiënten met cutane -en/of gewrichtsmanifestaties. De werkgroep neemt het advies uit het NVR-standpunt anifrolumab over omtrent de indicatie om anifrolumab te starten: de indicatie te starten met anifrolumab dient te worden besproken in een overleg met specialisten met uitgebreide kennis van het ziektebeeld: bij voorkeur in de vorm van een multidisciplinair overleg (MDO). Een belangrijk aspect om te benoemen is dat er tot op heden geen tot geringe ervaring is met het gebruik van anifrolumab door de medische specialisten in Nederland.
Een review uit 2015 van Mulhearn beschrijft indicaties voor het gebruik van IVIg bij reumatische ziekten. In de review worden specifieke aanbevelingen geschreven voor ‘lupus flare’, lupus nefritis, en lupus bij zwangere vrouwen. Zo wordt er beschreven dat IVIg overwogen kan worden voor gebruik bij refractaire ziekte waar andere behandelingen hebben gefaald (400 mg/kg/5 dagen) en bij acute ernstige opflakkeringen van SLE, in het bijzonder met koorts, pijn en vermoeidheid.
Het beleid van patiënten met SLE rondom zwangerschap is niet opgenomen in de huidige richtlijn. Hiervoor wordt verwezen naar; ‘Medicatiegebruik bij inflammatoire reumatische aandoeningen rondom de Zwangerschap’.
Uit de bestaande literatuur met betrekking tot verlaging van algemene ziekteactiviteitscores bij non-renale SLE is er geen csDMARD met bewezen hogere effectiviteit in vergelijking met een andere csDMARD. Hierbij moet worden opgemerkt dat CYC relatief de meeste ondersteuning vanuit de literatuur heeft betreffende effectiviteit, echter dient dit slechts in specifieke gevallen als eerste keuze ingezet te worden vanwege het bijwerkingenprofiel, een voorbeeld hiervan is ernstige neuropsychiatrische manifestaties, dat buiten de reikwijdte van deze richtlijn valt (https://ard.bmj.com/content/annrheumdis/69/12/2074.full.pdf).
Tevens is er meer literatuur beschikbaar over het effect van csDMARDs en bDMARDs op renale uitkomsten bij renale manifestaties bij SLE. Echter valt dit buiten de scope van de huidige richtlijn. Om deze reden wordt verwezen naar de richtlijnen van de Nefrologie en de EULAR.
Om tot een specifieke voorkeur te komen omtrent de volgorde van inzet van csDMARDs wordt naast de wetenschappelijke literatuur (zeer geringe ondersteuning), gebruik gemaakt van de kennis beschikbaar over de bijwerkingen van de genoemde csDMARDs bij andere reumatische of auto-immuunziektes en de klinische praktijkervaring. De aanbeveling om als eerste keuze csDMARD te kiezen voor methotrexaat wordt ondersteund door zowel de BSR-richtlijn (Gordon, 2018) als de EULAR-richtlijn (Fanouriakis, 2019). De voorkeur voor azathioprine als eerste keuze wordt ondersteund door de EULAR-richtlijn (Fanouriakis, 2019).
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Patiënten geven aan waarde te hechten aan duidelijke en tijdige communicatie over de behandeling en de gevolgen indien de behandeling niet effectief is, ofwel verwachtingsmanagement. Om deze reden kunnen patiënten het belangrijk vinden dat de behandeldoelen (zoals beschreven in module Monitoring) in kaart worden gebracht. Bijv. welke factoren dragen bij aan het aanpassen/switchen van de medicatie? Het ‘samen beslissen’ staat o.a. bij het opstellen van de doelen centraal.
Aanhoudende ziekteactiviteit zorgt voor verminderde kwaliteit van leven en verminderde arbeidsparticipatie. Om deze reden geven patiënten aan dat medisch specialisten/ reumaverpleegkundigen aandacht moeten hebben voor het feit dat de behandeling (en het slagen/falen en eventueel bijstellen van deze behandeling) en de verwachtingen daarover invloed hebben op de privé- en arbeidssituatie van patiënten. Zo is het voor de patiënt en Arboarts belangrijk snel duidelijkheid te hebben over de ontwikkeling van de aandoening wanneer zij bijvoorbeeld in de ziektewet zitten of bezig zijn met een re-integratietraject.
Tegelijkertijd hechten de patiënten ook waarde aan een adequate behandeling uitgevoerd door een specialist deskundig op het gebied van SLE. Indien die niet gewaarborgd kan worden (bijv. door de complexiteit van de situatie), benadrukken patiënten het belang om deze specifieke groep door te verwijzen naar een centrum met expertise.
Kosten (middelenbeslag)
Het gebruik van bDMARDs gaat gepaard met hogere medicatiekosten in vergelijking met het gebruik van csDMARDs. Om deze reden zullen csDMARDs meer kosteneffectief zijn in vergelijking met bDMARDs. Echter is het gebruik van bDMARDs mogelijk kosteneffectief bij een geselecteerde patiëntengroep. Dit kan echter niet worden onderbouwd met wetenschappelijke literatuur.
Aanvaardbaarheid, haalbaarheid en implementatie
Alleen op indicatie worden csDMARDs en/of bDMARDs voorgeschreven. De behandeling van patiënten met SLE dient te worden verzorgd door een specialist met ervaring over het ziektebeeld. Indien ervaring in onvoldoende mate aanwezig is, dient structureel overleg plaats te vinden of kan de patiënt worden doorverwezen naar een centrum met expertise. Voor de behandeling met csDMARDs of bDMARDs is structurele monitoring op het ontstaan van bijwerkingen essentieel. Dit wordt gedaan middels gericht (bloed)onderzoek. Ook dient de patiënt voor start van de behandeling voldoende voorlichting en inspraak te hebben. De patiënt dient te worden geïnformeerd over de (bij)werking, juiste toediening en belang van frequente monitoring. Daarnaast is het geven van adequate instructies vereist bij het geven/voorschrijven van subcutane injecties (bijv. BEL). Bij intraveneuze infusie met BEL, anifrolumab, RTX en CYC zijn adequate voorzieningen en de beschikbaarheid van bekwaam personeel vereist.
Rationale van de aanbeveling
Uit de literatuur is bekend dat zowel aanhoudende ziekteactiviteit als langdurige behandeling met (hoge dosis) GC’s zorgt voor het ontstaan van schade aan organen en een verhoogde mortaliteit bij patiënten met SLE. Daarnaast zorgt aanhoudende ziekteactiviteit voor verminderde kwaliteit van leven en verminderde arbeidsparticipatie. Bij de behandeling van patiënten met SLE wordt gestreefd naar het behalen van remissie of lage ziekteactiviteit (verwijzing module Behandeldoel en -strategie). In het geval van een opvlamming kan de inzet van GC’s noodzakelijk zijn, maar de behandeling is er op gericht het gebruik van GC’s te minimaliseren. Om dit te bewerkstellen en ziekteactiviteit te verminderen, worden csDMARDs ingezet naast een standaardbehandeling met HCQ. Van verschillende csDMARDs is in wetenschappelijk onderzoek aangetoond dat ze een effect hebben op vermindering van ziekteactiviteit (gemeten in verschillende indices zoals SLEDAI en BILAG) en van een aantal is aangetoond dat het gebruik leidt tot reductie van GC’s.
Wetenschappelijk onderzoek naar de effecten en bijwerkingen van csDMARDs bij patiënten met SLE zijn zeer beperkt in absolute en kwalitatieve zin. Onderlinge vergelijkingen (head-to-head studies) zijn insufficiënt. De beschreven literatuur geeft weinig handvatten voor een keuze van een specifieke csDMARD bij de behandeling van patiënten met SLE. Tegelijkertijd is het niet mogelijk om de bewijskracht van de samenvatting van de literatuur m.b.t. de csDMARDs te graderen. Naast de wetenschappelijke literatuur wordt de aanbeveling betreffende de inzet van csDMARDs voor een groot deel gestoeld op ‘klinische praktijkervaring’.
Wat betreft de bDMARDs, zijn er data uit trials bij patiënten met SLE voor BEL, RTX en anifrolumab. De resultaten van de trials met BEL zijn (gematigd) positief. Op basis van deze literatuur en de klinische praktijkervaring heeft BEL een plaats gekregen als ‘add-on’ middel naast een csDMARD. De resultaten vanuit de literatuur betreffende anifrolumab zijn positief. Dit middel wordt naast BEL geplaatst.
De trials naar RTX laten geen overtuigend positief effect zien op reductie van ziekteactiviteit. Op basis van observationele data en ‘praktijkervaring’ is wel een plaats voor RTX bij refractaire ziekteactiviteit (of ziekteactiviteit ondanks behandeling met csDMARDs).
Onderbouwing
Alle patiënten met SLE worden met hydroxychloroquine (HCQ) behandeld, tenzij er een zwaarwegende contra-indicatie is. Bij ziekteactiviteit is er meestal een indicatie voor het starten van een aanvullende behandeling. Vaak worden glucocorticoïden (GC’s) ingezet, in verband met het ongunstige bijwerkingenprofiel van GC’s worden deze bij voorkeur kortdurend ingezet en snel weer afgebouwd. Indien het niet mogelijk is om GC’s voldoende af te bouwen, of er sprake is van ernstige, recidiverende of residuale ziekteactiviteit dan dienen ook andere immunosuppressieve medicamenten gestart te worden. De keuze voor een specifiek medicament wordt bepaald door de mate van de ziekteactiviteit, specifieke orgaanmanifestaties, bijwerkingen en comorbiditeit, eventuele zwangerschapswens en voorkeur van de patiënt. In deze module wordt nagegaan welk bewijs beschikbaar is om de arts en patiënt te steunen bij het maken van een keuze.
csDMARDs
|
- GRADE |
… Azathioprine, cyclosporin, leflunomide, methotrexate, mycophenolate mofetil, and, tacrolimus may be effective treatment options (regarding disease activity indices and flares) in patients with SLE.
In patients with SLE, cyclosporin is not less effective than azathioprine; mycophenolate mofetil is not less effective than IV cyclophosphamide; high-dose IV cyclophosphamide (50 mg/kg x 4 days) has the same effectiveness than a l IV cyclophosphamide regimen according to the NIH scheme ((750 mg/m2/month for 6 months and then 3 months up to 2 years).
Enteric-coated mycophenolate sodium may be more effective than azathioprine in patients with SLE.
Sources: Pego-Reigosa (2013); Ordi-Ros (2017); Miyawaki (2013); Islam (2012). |
|
- GRADE |
… Mycophenolate mofetil, cyclosporin and, methotrexate may be used to reduce the glucocorticoid dose in patients with (moderate activity and) nonrenal and/or renal refractory SLE.
In patients with active SLE refractory to steroids, cyclosporin and azathioprine have a similar effect on reduction in glucocorticoid in the medium term.
Sources: Pego-Reigosa (2013). |
|
- GRADE |
… Cyclosporin, mycophenolate mofetil and, tacrolimus may cause adverse events in patients with SLE.
No difference in adverse events were observed between cyclosporin and azathioprine, and between high-dose IV cyclophosphamide (50 mg/kg x 4 days) and cyclophosphamide according to the NIH scheme (750 mg/m2/month for 6 months and then 3 months up to 2 years).
Mycophenolate sodium is a safe alternative therapy in SLE patients with extra-renal involvement.
Low-dose methotrexate appears to have an acceptable toxicity profile, compared with CQ in patients with articular and cutaneous manifestations of SLE.
Similar short-term (6 months) adverse events were reported in patients with mild to moderate SLE activity treated with or without leflunomide.
Sources: Pego-Reigosa (2013); Yahya (2013); Islam (2012). |
|
- GRADE |
… No evidence was found regarding the effect of treatment with cyclosporin, mycophenolate mofetil and, cyclophosphamide on quality of life when compared with control treatment in patients with Systemic Lupus Erythematosus.
No evidence was found regarding the effect of treatment with azathioprine, leflunomide, methotrexate and, tacrolimus on quality of life, reduction in glucocorticoid (except methotrexate), adverse events (except tacrolimus) when compared with control treatment in patients with Systemic Lupus Erythematosus.
Sources: Pego-Reigosa (2013). |
bDMARDs
|
High GRADE |
… Treatment with belimumab reduces disease activity (i.e., SLE scores on disease activity indices), glucocorticoid dose when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Singh, 2021. |
|
Moderate GRADE |
… Treatment with belimumab probably results in little to no difference in quality of life when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Singh, 2021. |
|
Low GRADE |
… Treatment with belimumab may result in little to no difference in adverse events (i.e., SAE, death) when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Singh, 2021. |
|
… Treatment with belimumab probably results in little to no difference in adverse events (i.e., serious infection, withdrawals due to AE) when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Singh, 2021. |
Rituximab
|
Low GRADE |
… Treatment with rituximab may result in little to no difference in improvement of SLE scores on disease activity indices when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Wu, 2020. |
|
- GRADE |
… No evidence was found regarding the effect treatment with rituximab on quality of life, reduction of glucocorticoid when compared with control treatment in patients with Systemic Lupus Erythematosus.
Sources: None. |
|
Low GRADE |
… Treatment with rituximab may result in little to no difference in the occurrence of adverse events (i.e., severe adverse events, deaths, infections, and any infusion related severe adverse events) when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Wu, 2020. |
Anifrolumab
|
Moderate GRADE |
… Treatment with anifrolumab probably reduces SLE scores on disease activity indices when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Lee, 2020. |
|
Very low GRADE |
… The evidence is very uncertain about the effect of treatment with anifrolumab on quality of life when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Lee, 2020. |
|
Moderate GRADE |
… Treatment with anifrolumab probably reduces the glucocorticoid dose when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Lee, 2020. |
|
Moderate GRADE |
… Treatment with anifrolumab probably increases the occurrence of adverse events (i.e., any AE) when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Lee, 2020. |
|
Low GRADE |
… Treatment with anifrolumab may reduce the occurrence of adverse events (i.e., SAE) when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Lee, 2020. |
|
Very low GRADE |
… The evidence is very uncertain about the effect of treatment with anifrolumab on adverse events (i.e., withdrawal due to AE) when compared with placebo in patients with Systemic Lupus Erythematosus.
Sources: Lee, 2020. |
other medication
|
Very low Grade |
… The evidence is very uncertain about the effect of treatment with intravenous Immunoglobulin on SLE scores on disease activity indices, reduction of glucocorticoid when compared with control treatment in patients with Systemic Lupus Erythematosus
Sources: Sakthiswary, 2014. |
|
- GRADE |
… No evidence was found regarding the effect treatment with intravenous Immunoglobulin on quality of life, adverse events when compared with control treatment in patients with Systemic Lupus Erythematosus.
Sources: Sakthiswary, 2014. |
csDMARDs
The systematic review of Pego-Reigosa (2013) investigated the efficacy and safety of nonbiologic immunosuppressants in treatment of non-renal SLE. A sensitive literature search in Medline, Embase and the Cochrane Central Register of Controlled Trials was performed until October 2011. Studies were eligible for inclusion if they included adults with SLE, treatment with a nonbiologic immunosuppressants, a placebo or active comparator group, and outcome measures assessing efficacy and/or safety. Efficacy outcomes were defined as nonrenal manifestations, scores by activity indices, SLE flares, a steroid-sparing effect. Safety outcomes were defined as infections, cardiovascular events, malignancies, etc. The level of evidence and grades of recommendations were based on the Oxford Centre for Evidence-Based medicine, and additionally Jadad-score for RCTs. In total 65 studies were included. Most of them were cohorts, only 11 RCTs were included. In the cohort studies baseline values were used as ‘control’, and post treatment values as ‘intervention’. In the RCT a placebo or control treatment was given in the ‘control’ arm, which was compared with the ‘intervention’ arm. Outcomes were reported per pharmacological therapy (i.e., intervention); methotrexate (MTX), azathioprine (AZA), mycophenolate mofetil (MMF), cyclophosphamide (CYC), tacrolimus (TAC), leflunomide (LEF), and cyclosporin A (CSA). Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. In addition, 2 RCTs and 3 observational studies were performed in patients with neuropsychiatric SLE. Three observational studies were performed in patients with lupus nephritis.
bDMARDs
The Cochrane review by Singh (2021) investigated the benefits and harms of belimumab (BEL) (alone or in combination) in systemic lupus erythematosus (SLE). An information specialist searched with a predefined strategy in relevant databases including; detailed strategies for CENTRAL, MEDLINE, Embase, CINAHL, Web of Science, the World Health Organization (WHO) International Clinical Trials Registry Platform, and clinicaltrials.gov. The search for relevant studies was performed until September, 25th 2019. Randomized controlled trials (RCTs) or controlled clinical trials (CCTs) of BEL (alone or in combination) compared to placebo or control treatment (immunosuppressive drugs, such as azathioprine, cyclosporine, mycophenolate mofetil or another biologic), in adults with SLE were eligible for inclusion. Studies including patients without a diagnosis of SLE were excluded. The standard methodological procedures expected by Cochrane were used for data collection and analysis. The primary outcome was changes in SLE scores on disease activity indices. This was defined as the Systemic Lupus Erythematosus Disease Activity Index Safety of Estrogen in Lupus National Assessment (SELENA) Modification (SELENA-SLEDAI), modified SELENASLEDAI Flare Index (SFI), British Isles Lupus Assessment Group index (BILAG), other similar validated indices. Secondary outcomes included quality of life, reduction in glucocorticoid dose, serious adverse events (SAE), serious infections, withdrawals due to adverse events, and death. The GRADE method was used to assess the quality of evidence. In total 6 RCTs were included. In these trials BEL was compared to placebo. The study duration ranges from 12 to 76 weeks. These trials were performed in North America (i.e., USA/ Canada, n=2), and several countries over the world (n=4). Regarding risk of bias, this was highest for the domain of attrition bias, followed by selection bias. Detailed information is provided in the risk of bias table (Singh, 2021). The trials predominantly included women from outpatient clinics; the mean age range of the participants was 32 to 42 years. Most often the treatment duration was 52 weeks.
The review with meta-analysis of Wu (2020) aimed to investigate the efficacy and safety of rituximab (RTX) in patients with SLE. Therefore, the Cochrane Handbook was followed. A literature research was performed using PubMed, Cochrane Library, EMBASE, Clinicaltrials and CNKI and Chinese database of WanFang databases. The search date was unknown. Articles were eligible for inclusion if they compared RTX with placebo (i.e., control group) in SLE patients in RCTs. In cohort studies the baseline group (i.e., when patients did not receive RTX) was used as control group. Further the studies included efficacy and safety results. Efficacy results were defined as BILAG score, SLEDAI score, complement C3/C4 levels, anti-dsDNA antibodies, peripheral CD19+B cells, serum creatinine, 24-h urinary protein and Up/Ucr. Safety results were defined as the incidence of serious adverse events (SAE), deaths, infections, etc. The methodological quality of the included RCTs was assessed with the Jadad score and for observational studies by the Newcastle-Ottaa Scale (NOS). Results were reported as weighted mean differences, standardized mean differences and relative risks. Heterogeneity was assessed using I². In total two RCTs and 12 observational cohort studies were selected (n=742). In the two RCTs, 241 patients received RTX and 160 patients received placebo treatment for 52 weeks. In total 341 patients were included in the observational studies. In these studies, baseline values were used as ‘control’ group, and post-treatment values as ‘intervention’ group. A limitation of the current review is that one of the RCTs, and 3 of the cohort studies included patients with lupus nephritis.
The systematic review of Lee (2020) aimed to investigate the efficacy and safety of anifrolumab in active SLE. A literature research was performed using MEDLINE, EMBASE, and the Cochrane Controlled Trials Registry databases until August 2020. Articles were eligible for inclusion if they compared anifrolumab with placebo in active SLE patients. The primary outcome was BICLA reaction at week 52. This was defined as all of the following: a reduction of all severe (BILAG-2004 A) or moderately severe (BILAG-2004 B) disease activity at baseline to lower levels (BILAG-2004 B, C, or D and C or D, respectively) and no worsening in other organ systems (with worsening defined as ≥1 new BILAG-2004 A item or ≥2 new BILAG-2004 B items); no worsening in disease activity, as determined by the SLEDAI-2K score (no increase from baseline) and by the PGA score (no increase of ≥0.3 points from baseline); no discontinuation of the trial intervention; and no use of restricted medications beyond protocol-allowed thresholds. Secondary outcomes were loss of the glucocorticoid dosage, and safety (i.e., any adverse events (AE), serious adverse events (SAE), number of patients withdrawn owing to adverse events. The methodological quality of the included studies was assessed with the Jadad-score. The meta-analysis was performed in accordance with the guidance provided by the Preferred Reporting Items for Systematic Reviews and Meta Analysis (PRISMA) statement. Heterogeneity was assessed using I², and funnel plots for publication bias. In total three RCTs were selected with a duration of 52 weeks. A number of 927 patients were included with a mean age range of 39 to 41 years. All studies were performed in several countries in the world and sponsored by the industry. The Jadad score across the studies ranged from 3 to 4, indicating high quality of the included studies. Detailed information is provided in the publication (Lee, 2020).
other medication
Sakthiswary (2014) performed a systematic review with meta-analysis to determine the therapeutic role of intravenous immunoglobin (IVIg) in patients with SLE. A literature research was performed using MEDLINE, Scopus, EMBASE, and Cochrane controlled trials. The search date was unknown, but all included articles were published between 1989 and 2013. Articles (i.e., RCTs, and cohort studies) were eligible for inclusion if they examined the effects of IVIg in patients with SLE. The diagnosis of SLE was based on validated criteria. Further, placebo treatment or standard treatment was used as comparison. Outcome measures were disease activity scores, defined as Systemic Lupus Erythematosus Disease Activity Index (SLEDAI), Systemic Lupus Activity Measure (SLAM), and Lupus Activity Index-Pregnancy (LAI-P), steroid dose reduction, change in the levels of autoantibodies, change in complement 3 (C3) and 4 (C4) levels, and renal function (proteinuria, creatinine). The quality of evidence was not assessed. Results were reported as standardized mean differences and relative risks. Heterogeneity was assessed using I². In total 13 studies were eligible for inclusion; 1 RCT, 2 non-RCTs, 6 prospective cohort studied, and 3 retrospective cohort studies. Of these, 7 were performed in Europa, 5 in Asia, and 1 in the USA. The study duration ranged between 1 and 24 months. In total 443 patients were included in the 13 studies. The current SR is limited by the fact that only the standardized mean differences in a study were reported, and not the individual outcomes per group. In addition, the RCT and one of the controlled trials were performed in patients with lupus nephritis. The other controlled trial was performed in SLE patients with recurrent spontaneous abortions. One prospective cohort study was performed in SLE patients with thrombocytopenia and one in patients with histologically confirmed cutaneous SLE.
Description of additional RCTs
As described above, additional RCTs were searched if the SR was published before 2020 (i.e., csDMARDs and IVIg).
csDMARDs
Islam (2012) performed a prospective open-label study comparing the efficacy and safety of methotrexate (MTX; 10mg weekly) and chloroquine (CQ; 150mg daily) in adults with SLE over 24 weeks. If adults fulfilling American College of Rheumatology (ACR) criteria of SLE
and suffering from arthralgia, or arthritis and active skin lesions, they were eligible for inclusion. Exclusion criteria were involvement of any other organ systems, pregnancy, lactation, any form of eye problems, history of taking antimalarials within the last 4 months or corticosteroids equivalent to > 20 mg of prednisolone per day, raised serum alanine aminotransferase (ALT), and raised serum creatinine. After giving a written informed consent, patients were randomly assigned to treatment with MTX or CQ. Outcome measures were numbers of swollen and tender joints, duration of morning stiffness, visual analog scale (VAS) for articular pain, physician global assessment index, patient global assessment
index, SLE Disease Activity Index (SLEDAI), disappearance of skin rash and erythrocyte sedimentation rate (ESR). In total 13 patients were allocated to the MTX arm, and 24 to the CQ arm. These patients were mostly female (13 in MTX vs. 23 in CQ) and had a mean age of 24 years. No differences were shown between the treatment arms at baseline. The current study was limited due to the open-label design, the limited sample size, and the relative short follow-up of 24 weeks.
Miyawaki (2012) performed a prospective open-label study to determine the efficacy of methotrexate (MTX) for improving serological abnormalities in adults with SLE. Patients with low serum complement and/or high anti-dsDNA levels during prednisone tapering, and meeting the in- and exclusion criteria, were treated with MTX (7.5 mg weekly, the use of other immunosuppressive agents was prohibited during the study) for 18 months. Outcome measures were C3, C4, anti-dsDNA levels, SLEDAI, MCV of red blood cells, and prednisone dose. In total 30 patients received treatment with MTX, and 18 patients were selected as controls. These patients were all female and had a mean age of 47 years in the MTX arm, and 42 years in the control arm, respectively. Baseline differences between the groups were shown regarding disease activity, which was as suspected higher in the MTX arm. The current study was limited due to the open label design, the limited sample size, and the fact that baseline values were different between the arms.
Yahya (2013) performed a prospective open-label study to assess the efficacy of mycophenolate sodium in extra-renal SLE. SLE patients without renal involvement, and meeting the in- and exclusion criteria, were randomized either to receive mycophenolate sodium (i.e., 360 mg twice daily initially, and later escalated to 720 mg twice daily at week 4 if there were no contraindications) or other immunosuppressive agents for 16 weeks. Outcome measures were SLE Disease Activity Index (SLEDAI) score, other organ-specific parameters immunological parameters, including anti-double stranded DNA and C3, and prednisone dose.
In total 8 patients received treatment with MMF, and 6 patients treatment with other immunosuppressive agents. In the control group, 2 patients were lost to follow-up. Regarding the baseline characteristics, only differences were shown in gender as 4 males were included in the MMF group and none in the control group. The current study was limited due to the open-label design, limited sample size and no standardized treatment in the control group.
Ordi-Ros (2017) performed an open-label randomized trial to compare the efficacy and safety of enteric-coated mycophenolate sodium (EC-MPS) versus azathioprine (AZA) in patients with active systemic lupus erythematosus (SLE) disease. SLE patients meeting the in- and exclusion criteria, were randomized and received either EC-MPS (target dose: 1440 mg/day) or AZA (target dose: 2 mg/kg/day) in addition to prednisone and/or antimalarials for 24 months. Outcome measures were proportion of patients achieving clinical remission, assessed by SLE Disease Activity Index 2000 (SLEDAI-2K) and British Isles Lupus Assessment Group (BILAG), at 3 and 24 months. Secondary endpoints included time to clinical remission, BILAG A and B flare rates, corticosteroid reduction and adverse events (AEs). In total 120 patients received treatment with EC-MPS, and 120 patients treatment with AZA. In both arms 2 patients were lost to follow-up and removed from the analysis. Although, a total of 154 patients (64.2%) completed the study: 87 (72.5%) in the EC-MPS arm and 67 (55.8%) in the AZA arm. Regarding the baseline characteristics, no differences were shown between the treatment arms. The current study was limited due to the open-label design.
other medication
No additional RCTs were available, which could be added to the current comparison.
Results
csDMARDs
The results of the systematic review (Pego-Reigos, 2013) are reported at first. Thereafter, additional results from the RCTs (Islam, 2012; Miyawkai, 2012; Yahya, 2013; Ordi-Ros, 2017) are reported per csDMARD.
nonbiologic immunosuppressants
The study of Pego-Reigosa (2013) studies several nonbiologic immunosuppressants namely, cyclophosphamide (CYC), azathioprine (AZA), methotrexate (MTX), leflunomide (LEF), mycophenolate mofetil (MMF), cyclosporin A (CSA), and Tacrolimus (TAC). Outcomes per nonbiologic are described below. The described information is adapted from the study of Pego-Reigosa (2013).
Methotrexate
In total, 7 articles evaluated the efficacy and/or safety of MTX in the treatment of nonrenal manifestations of SLE; 2 were double-blind, placebo-controlled RCTs (Carneiro; 1999; Fortin, 2008), 1 was a crossover open study (Arfi, 1995), and 5 were cohort studies (Pego-Reigosa, 2013) that included 230 patients overall.
The RCT of Carneiro (n=41) compared MTX (20mg/week) treatment with placebo in SLE patients. Mean SLEDAI and VAS scores were significantly lower in MTX patients compared with placebo. In addition, it was possible to decrease the prednisone dose for 13 MTX patients during the study but for only one patient in the placebo group.
The RCT of Fortin (n=86) compared MTX (7.5 mg/week and ↑to 20mg/week) treatment with placebo in SLE patients. Mean SLAM-R and reduction of glucocorticoid dose improved significantly more in the MTX group, compared with placebo. No differences in AEs were reported.
The crossover open study (Arfi, 1995) assessed the efficacy of oral MTX (7.5 mg/week) in SLE patients without major organ involvement and active disease despite >10 mg/day of prednisone. The patients received treatment for 2 periods: 1) 3 months (followed by a 3-month control period without treatment), and then 2) 6 months (followed by a 6-month control period without treatment). In the 13 patients who finished the study, there was a significant reduction of lupus flares during the MTX treatment periods compared with the control phases (P=0.02) without significant differences in the requirements of prednisone.
In summary, the evidence for using MTX in nonrenal SLE treatment is based on high-quality studies, 2 double-blind, placebo-controlled RCTs. However, a small number of patients
were included in these studies (only 61 patients were treated with MTX).
Islam (2012) reported that clinical and laboratory parameters improved significantly over 24 weeks in patients treated with MTX (n=13). Although no statistically significant differences were shown at 24 weeks between patients treated with MTX compared with patients treated with CQ (n=24) for SLEDAI, PGA, VAS pain (i.e., mean (sd) scores for MXT vs. CQ; 2.8 (2.4) vs. 2.5 (2.4); 1.5 (1.1) vs. 1.8 (1.1); 4.2 (2.1) vs.1.8 (1.1)), respectively. The most common side effect ‘anorexia and nausea’ were somewhat more common in patients treated with MTX (54%) compared with CQ (17%).
Miyawaki (2013) reported that SLEDAI significantly reduced in patients treated with MTX (n=30) compared with the control group (n=18) at 18 months. Although, no differences were shown at 18 months as patients in the control group had no active disease at baseline (mean (SD) MTX vs. control at 18 months; 4.9 (0.9) vs. 5.0 (0.9)). However, a statistically significant difference was shown for the outcome ‘prednisone dose’ at 18 months, which was lower in the MTX group (mean 5.8 mg/day) compared with the control group (mean 6.7mg/day). Regarding biomarkers, C3 and/or C4 levels were normalized or elevated at 18 months in 96.7 % of the MTX patients and 33.3 % of the control patients, and anti-dsDNA antibody levels were normalized or lowered in 24 of the 26 MTX patients (92.3 %) and in 50.0 % of the control patients.
1. SLE scores on disease activity indices
Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. Therefore, the review of Pego-Reigosa (2013) reported only conclusions. The following conclusion was reported for disease activity:
- In patients with moderate activity and nonrenal SLE manifestations despite prednisone, NSAIDs, and antimalarials, treatment with MTX (20 mg/day) reduces in the medium term (12 months) the activity of the disease, particularly in patients without damage, with an additional medium-term steroid-sparing effect.
2. Quality of life
No conclusions were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid use
The following conclusion was reported for reduction in glucocorticoid use:
- An additional medium-term steroid-sparing effect was shown in treatment with MTX (20 mg/day), compared with control treatment in patients with moderate activity and nonrenal SLE manifestations.
4. Adverse events
No conclusions were reported for the outcome measure ‘adverse events’.
Azathioprine
Only 2 articles assessed the efficacy and/or safety of AZA in the treatment of nonrenal SLE; 1 was an unblinded RCT (n=24; Hahn, 1975) and 1 was a cohort study (Oelzner, 1996) that included 85 patients overall. The RCT, which compared treatment with AZA 3-4 mg/kg + prednisone vs. only prednisone, reported no differences regarding clinical improvement, mean dose prednisone, and AEs. The retrospective cohort study analyzed the influence of AZA (≥2 mg/kg/day) and prednisolone (7–12 mg/day) on the frequency of SLE flares and evaluated the predictors of these flares in 61 patients (38 without renal disease) over
a mean follow-up period of 7.5 years (Oelzner, 1996). In comparison with a preceding period without AZA, this combined regimen resulted in a significant reduction in flares and an
increase in flare-free patient-years.
In summary, there is little evidence for using AZA in the treatment of nonrenal SLE because there is insufficient data.
1. SLE scores on disease activity indices
Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. Therefore, the review of Pego-Reigosa (2013) reported only conclusions. The conclusion for disease activity is reported as follows:
- The association of AZA with prednisolone treatment might reduce the flare rate.
2. Quality of life
No conclusions were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid
No conclusions were reported for the outcome measure ‘reduction in glucocorticoid’.
4. Adverse events
No conclusions were reported for the outcome measure ‘adverse events’.
Mycophenolate mofetil / enteric-coated mycophenolate sodium
In total, 8 articles evaluated the efficacy and/or safety of MMF in the treatment of nonrenal manifestations of SLE; 1 was an RCT (Ginzler, 2010) and 7 were cohort studies (Pego-Reigoso, 2013) that included 769 patients overall. The RCT by Ginzler (2010) compared patients treated with MMF to treatment with CYC. No differences were shown for efficacy outcomes.
The RCT by Ginzler (2010) explored also as secondary end points the nonrenal findings of the Aspreva Lupus Management Study (ALMS) (8), a prospective, open-label, parallelgroup
RCT that assessed the effect of MMF compared with CYC as induction treatment for lupus nephritis. Some of the cohort studies specifically addressed safety issues.
In summary, the evidence for using MMF in nonrenal SLE treatment is based on studies with a large number of patients and RCT information is available. However, most patients were included in low-quality studies, and the RCT assessed the nonrenal response in patients with lupus nephritis who received induction treatment including high-dose corticosteroids.
Yahya (2013) reported that mycophenolate sodium reduced SLEDAI scores after 16 weeks in 7/8 patients (median score 8), and in 4/4 (median score 5) patients treated with another immunosuppressant. There was a positive trend of steroid dose reduction in both treatment groups (median dose at 16 weeks 3mg/day vs. 2mg/day). Regarding biomarkers, no differences were shown between patients treated with mycophenolate sodium, compared with patients treated with other immunosuppressants.
Ordi-Ros (2017) reported that at least 8 consecutive weeks of clinical remission (i.e., clinical SLEDAI-2K=0, where serology was permitted (maximum SLEDAI=4)) at 24 months was shown in 84/120 (70%) patients treated with enteric-coated mycophenolate sodium, compared with 57/120 (48%) patients treated with AZA. Mean SLEDAI-2K and BILAG-2001 at 24 months was lower in patients treated with enteric-coated mycophenolate sodium (i.e., 2.1 and 1), compared with patients treated with AZA (i.e., 3.3 and 1). BILAG A/B flares over 24 months were observed in 60/120 (50%) of the patients treated with enteric-coated mycophenolate sodium, and 86/120 (72%) of the patients treated with AZA. Mean glucocorticoid dose at 24 months was lower in patients treated with enteric-coated mycophenolate sodium (i.e., 4), compared with patients treated with AZA (i.e., 7). No differences were shown regarding adverse events (i.e., 71/120 vs. 69/120, respectively).
1. SLE scores on disease activity indices
Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. Therefore, the review of Pego-Reigosa (2013) reported only conclusions. The following conclusions were reported for disease activity:
- MMF may prevent short-term (6 months) SLE flares when added to the treatment of patients with increasing anti-dsDNA titer.
2. Quality of life
No conclusions were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid
The conclusion for reduction in glucocorticoid was reported as follows:
- MMF can be used to improve nonrenal activity in patients with nonrenal and/or renal refractory SLE and to reduce the need for corticosteroids.
4. Adverse events
The conclusion for adverse events was reported as follows:
- In patients with renal and/or nonrenal SLE, MMF may cause non–dose-dependent adverse events (particularly in the gastrointestinal system) and drug survival is acceptable with a low withdrawal rate due to adverse events in the medium term (12 months).
Cyclophosphamide
In total, twenty-nine studies evaluated the efficacy and/or safety of CYC in the treatment of nonrenal manifestations of SLE; 4 were unblinded RCTs (Stojanovich, 2003; Gonzalez-Lopez, 2004; Barile-Fabris, 2005; Petri, 2010), 1 was an open prospective study (Boumpas, 1993), and 24 were cohort studies (Pego-Reigosa, 2013) that included 3,742 patients overall. Different nonrenal manifestations were treated, although neuropsychiatric SLE (NPSLE) was studied in a more rigorous way (Stojanovich, 2003; Barile-Fabris, 2005; Petri, 2010). The CYC regimens and duration of CYC treatment and the comedications allowed in those studies varied. The outcome variables used varied, with the most frequent being clinical response to treatment measured by different activity and response indices, serologic response, rate of disease flares, decrease in the dose of prednisone, and adverse events. Some of the studies specifically addressed safety issues such as ovarian failure, neoplasias, or association with damage.
In summary, the evidence for using CYC in the treatment of nonrenal SLE is based on studies of a larger number of patients than those that assessed any other nonbiologic agent and RCT information is available, particularly for NPSLE. However, only a small percentage of patients were included in high-quality studies.
1. SLE scores on disease activity indices
Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. Therefore, the review of Pego-Reigosa (2013) reported only conclusions. Conclusions for disease activity were reported as follows:
- IV CYC is better than methylprednisolone for the long-term treatment of NPSLE and reduction of relapses.
- High-dose IV CYC (i.e., iv CYC 50mg/kg x 4 days) has the same efficacy in the treatment of nonrenal SLE than a traditional IV CYC regimen (i.e., iv CYC 750 mg/ m2/month x 6 months and then every 3 months, up to 2 years).
2. Quality of life
No conclusions were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid
No conclusions were reported for the outcome measure ‘reduction in glucocorticoid’ specifically. In the method section it is described that ‘efficacy’ outcomes were defined as nonrenal manifestations, scores by activity indices, SLE flares, a steroid-sparing effect.
4. Adverse events
The following conclusions were reported for adverse events:
- High-dose IV CYC has the same adverse event rate than a traditional IV CYC regimen in the treatment of nonrenal SLE.
- IV CYC use is associated with development of cervical intraepithelial neoplasia.
- In women with SLE, oral or IV CYC is independently associated in the short term with ovarian failure.
- In women with SLE, the risk of ovarian failure increases with the cumulative dose of oral or IV CYC and is higher with longer IV CYC regimens.
- In women with SLE, the risk of ovarian failure is associated with an older age at commencement of both oral and intravenous CYC; age itself is a risk factor for ovarian failure.
Tacrolimus
Two studies assessed the efficacy and/or safety of TAC in the treatment of nonrenal manifestations of SLE; both were cohort studies (Suzuki, 2011; Kusunoki, 2009) that included 31 patients. In the open-label prospective 24- week study by Suzuki (2011), 21 patients with mild active SLE treated with oral TAC (1–6 mg/day) were studied. The mean SLEDAI score decreased significantly at 24 weeks (P<0.01). In 8 cases, treatment was discontinued within 24 weeks because of inefficacy (6 cases) and adverse effects (2 cases). Nonserious side effects were observed in only 5 cases (23.8%). The retrospective cohort study investigated whether oral TAC (1–3 mg/day) was effective for treating SLE patients without active nephritis (n=10; Kusunoki, 2009). The mean SLEDAI score and the mean dose of prednisolone decreased significantly after 1 year (P<0.05 for both). Four of the 10 patients had adverse events and 2 patients discontinued treatment.
In summary, there is very little evidence for using TAC because only 2 small studies have been reported, neither of which were RCTs, and almost one-third of all patients studied discontinued the drug because of a lack of efficacy or adverse effects.
1. SLE scores on disease activity indices
Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. Therefore, the review of Pego-Reigosa (2013) reported only conclusions. The following conclusion was reported for disease activity:
- In patients with active nonrenal SLE despite conventional treatment, the addition of TAC may be useful to improve disease activity in the medium term.
2. Quality of life
No conclusions were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid
No conclusions were reported for the outcome measure ‘reduction in glucocorticoid’.
4. Adverse events
The conclusion for adverse events was reported as follows:
- In patients with active nonrenal SLE despite conventional treatment, the addition of TAC may cause frequent adverse events.
Leflunomide
In total, 2 articles assessed the efficacy and/or safety of LEF in the treatment of nonrenal SLE; 1 was a double-blind, placebo-controlled RCT (n=12, Tam, 2004) and 1 was a cohort study (Remer, 2001) that included 30 patients overall. The RCT, which compared treatment with LEF vs control treatment without LEF, reported that the reduction in the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) from baseline to 24 weeks was significantly greater in the leflunomide group than the placebo group (mean 11 in the leflunomide group vs. 4.5 in the placebo group). Similar changes in proteinuria, C3, anti-dsDNA, and prednisone dose were reported, and no differences for AEs. The cohort study retrospectively assessed the efficacy and safety of LEF (100 mg/day for 3 days, followed by 20 mg/day) in 18 SLE outpatients (Remer, 2001). After 2–3 months of therapy, most patients had subjective improvement and significantly lower Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores.
In summary, there is very little evidence for use of LEF in nonrenal SLE because the only reported double-blind placebo-controlled RCT included only 6 patients treated with LEF.
1. SLE scores on disease activity indices
Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. Therefore, the review of Pego-Reigosa (2013) reported only conclusions. The conclusion for disease activity was reported as follows:
- In patients with mild to moderate active SLE in spite of prednisolone, the addition of LEF is more effective than placebo in improving disease activity.
2. Quality of life
No conclusions were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid
No conclusions were reported for the outcome measure ‘reduction in glucocorticoid’.
4. Adverse events
The conclusion for adverse events was reported as follows:
- Similar short-term (6 months) side effects were reported in patients with mild to moderate active SLE treated with or without LEF.
Cyclosporin
In total, 8 articles evaluated the efficacy and/or safety of CSA in the treatment of nonrenal
SLE; 2 were unblinded RCTs (Dammacco, 2000; Griffiths, 2010), 1 was a prospective open study (Manger, 1996), and 5 were cohort studies (Pego-Reigosa, 2013) that included 319 patients overall.
In the RCT of Dammacco (2000; n=18) patients were treated with CSA vs. control treatment without CSA. During the trial SLEDAI significantly improved more in the intervention group compared to control. The cumulative dose prednisone was significantly lower in the intervention group compared to control. Further, AEs occurred somewhat less in the intervention group, compared with control (i.e, 60% vs. 62.5%).
In the RCT of Griffiths (2010; n=89) patients were treated with CSA (i.e., initial dose 1-2.5mg/kg/d, maximun 3.5mg/kg/d) vs. AZA (i.e., initial dose0.5-2mg/kg/d, maximum 2.5mg/kg/d). No differences were showed in efficacy and safety outcomes between the treatment arms.
The prospective open study investigated the effect of CSA (2.5–5 mg/ kg/day) in 16 patients with active SLE over an average treatment period of 30.3 months (Manger, 1996). The European Consensus Lupus Activity Measurement score decreased significantly (P=0.005) after 6 months, but not at the end of the observation period. The most frequent side effects were hypertension and deterioration of renal function (3 of 16 patients) and hypertrichosis (5 of 16 patients).
In summary, there is little evidence for using CSA in nonrenal SLE treatment because one of the 2 unblinded, non–placebo controlled RCTs that assessed this drug included only 10 patients treated with CSA, and in the other RCT, almost one-third of all patients discontinued the drug because of adverse events or a lack of efficacy.
1. SLE scores on disease activity indices
Due to important variability in the selected patients, treatment doses, and outcome measures, no meta-analyses were performed. Therefore, the review of Pego-Reigosa (2013) reported only conclusions. The following conclusions were reported for disease activity:
- In patients with renal and/or nonrenal SLE refractory to steroids, the addition of CSA may improve disease activity and induce remission in the short term and long term.
- In patients with active SLE refractory to steroids, CSA is not less effective than AZA in reducing renal and/or nonrenal activity.
2. Quality of life
No conclusions were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid
Conclusions for reduction in glucocorticoid were reported as follows:
-In patients with renal and/or nonrenal SLE refractory to steroids, the addition of CSA has a steroid-sparing effect in the long term.
- In patients with active SLE refractory to steroids, CSA and AZA have both a similar steroid-sparing effect in the medium term.
4. Adverse events
Conclusions for adverse events were reported as follows:
- In patients with renal and/or nonrenal SLE refractory to steroids, the addition of CSA causes frequent adverse events.
- In patients with active SLE refractory to steroids, no significant difference in adverse events were observed between CSA and AZA.
Level of evidence of the literature
As evidence was adapted from the review of Pego-Reigosa (2013), and no detailed information was provided to calculate effect estimates, it was impossible to assess the level of evidence with de GRADE method. Therefore, the conclusion formulated without GRADE. The same method was applied for the literature from the additional RCTs.
Belimumab (10mg/kg)
The current comparison was studied in the Cochrane review of Singh (2021). Data of this review was adapted in the current summary of the literature.
1. SLE scores on disease activity indices
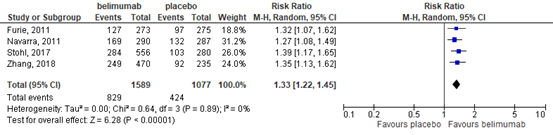
This outcome was reported in four studies at 52 weeks (Furie, 2011; Navarra, 2011; Stohl, 2017; Zhang, 2018). More participants in the belimumab group than in the placebo group showed improvement (at least a 4-point reduction) in SELENA-SLEDAI score. This resulted in a relative risk (RR) of 1.33 (95% confidence interval (CI) 1.22 to 1.45), Figure 1. The absolute difference was 13% (95% CI 8% to 17%) better in the belimumab group with 829 of 1589 (52%) participants versus 424 of 1077 (39%) in the placebo group achieving this reduction.
Figure 1. Forest plot reduction of at least 4 points in SELENA-SLEDAI at 52 weeks; belimumab vs. placebo.
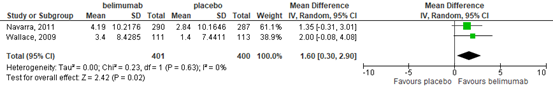
2. Quality of life
This outcome was defined as change in health-related quality of life on SF-36 at 52 weeks in two trials (Navarra, 2011; Wallace, 2009). These studies showed that there was probably no difference in the health-related quality of life as measured by change in SF-36 PCS in participants in the belimumab group compared with the placebo group (mean difference (MD) 1.60, 95% CI 0.30 to 2.90), Figure 2. The absolute risk difference was 1.60%
(95% CI 0.30% to 2.0%) higher (better) in the belimumab group.
Figure 2. Forest plot change in health-related quality of life at 52 weeks; belimumab vs. placebo.
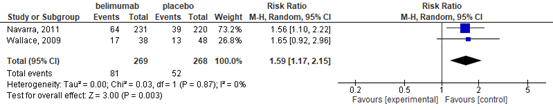
3. Reduction in glucocorticoid
Two of the included trials reported this outcome (Navarro, 2011; Wallace, 2009) defined as reduction of glucocorticoid dose by 50% or more at 52 weeks. The belimumab group showed greater improvement in glucocorticoid dose, with a higher proportion of participants reducing their dose by at least 50%, compared to placebo (RR 1.59, 95% CI 1.17 to 2.15), Figure 3. The absolute risk difference was 11% (95% CI 4% to 18%) better in the belimumab group, with 81 of 269 participants in the belimumab group versus 52 of 268 in the placebo group having at least a 50% reduction in dose.
Figure 3. Forest plot reduction of glucocorticoid dose by 50% or more at 52 weeks; belimumab vs. placebo.
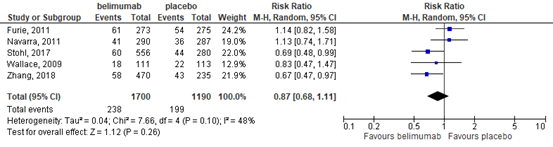
4. Adverse events
Outcome of interest were; serious adverse events, serious infection, withdrawals due to adverse events, and deaths.
4.1 serious adverse events
This outcome was defined was patients with one or more SAE in five trials (Furie, 2011; Navarra, 2011; Stohl, 2017; Wallace, 2009; Zhang, 2018). These trials showed that there may be little or no difference in the number of serious adverse events in the belimumab group compared with the placebo group (RR 0.87, 95% CI 0.68 to 1.11) Figure 4. The absolute risk difference was 2% (95% CI -6% to 2%) less in the belimumab group with 238 of 1700 (14%) participants in the belimumab group versus 199 of 1190 (17%) in the placebo group experiencing at least one serious adverse event.
Figure 4. Forest plot serious adverse event; belimumab vs. placebo.
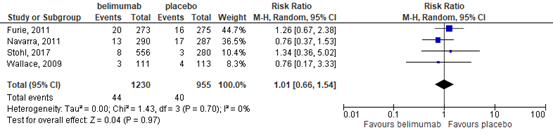
4.2 serious infection
This outcome was studied in four trials (Furie, 2011; Navarra, 2011; Stohl, 2017; Wallace, 2009). These trials showed that there was probably little or no difference in the number of serious infections in the belimumab group compared with the placebo group (RR 1.01, 95% CI 0.66 to 1.54), Figure 5. The absolute risk difference was 0% (95% CI -1% to 1%) with 44 of 1230 (4%) participants in the belimumab group versus 40 of 955 (4%) in the placebo group having at least one serious infection.
Figure 5. Forest plot serious adverse event; belimumab vs. placebo.
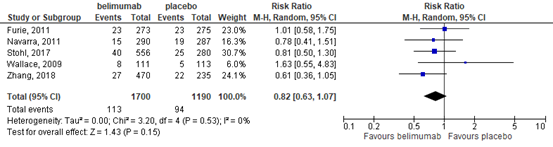
4.3 withdrawals due to adverse events
This outcome was studied in five trials (Furie, 2011; Navarra, 2011; Stohl, 2017; Wallace, 2009; Zhang, 2018). These trials showed that there was probably little or no difference in the proportion of participants who withdrew due to adverse events in the belimumab group compared with the placebo group (RR 0.82 (95% CI 0.63 to 1.07), Figure 6. The absolute risk difference was 1% (95% CI -3% to 1%) fewer in the belimumab group with 113 of 1700 (7%) participants in the belimumab group versus 94 of 1190 (8%) in the placebo group withdrawing due to adverse effects.
Figure 6. Forest plot withdrawals due to adverse events; belimumab vs. placebo
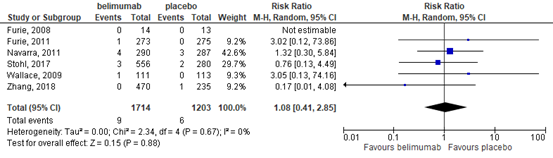
4.4 deaths
This outcome was studied in six trials (Furie, 2008; 2011; Navarra, 2011; Stohl, 2017; Wallace, 2009; Zhang, 2018). These trials showed that there may be little or no difference in the number of deaths in the belimumab group compared with the placebo group (RR 1.08, 95% CI 0.41 to 2.85), Figure 7. The absolute risk difference was 0% (95% CI -1% to 1%) with nine deaths out of 1714 participants in the belimumab group and six deaths out of 1203
participants in the placebo group.
Figure 7. Forest plot deaths; belimumab vs. placebo.
Level of evidence of the literature
The level of evidence (GRADE method) is determined per comparison and outcome measure and is based on results from RCTs and therefore starts at level “high”. Subsequently, the level of evidence was downgraded if there were relevant shortcomings in one of the several GRADE domains: risk of bias, inconsistency, indirectness, imprecision, and publication bias.
The level of evidence regarding the outcome measure SLE scores on disease activity indices, reduction of glucocorticoid dose was not downgraded.
The level of evidence regarding the outcome measure quality of life was downgraded by 1 level because of imprecision (the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference).
The level of evidence regarding the outcome measure adverse events (i.e., SAE) was downgraded by 2 levels because of imprecision (the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference), and inconsistency (I2 = 48%).
The level of evidence regarding the outcome measure adverse events (i.e., serious infection, withdrawals due to AE) was downgraded by 1 level because of imprecision (the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference).
The level of evidence regarding the outcome measure adverse events (i.e., death) was downgraded by 2 levels because of imprecision (the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference, and the total number of events was small (n=15)).
Rituximab
The current comparison was studied in the review of Wu (2020).
1. SLE scores on disease activity indices
This outcome was reported as BILAG score, and clinical response (i.e., combination of complete and partial response) in the two RCTs after a treatment period of 52 weeks. In the observational cohort studies this outcome was reported as BILAG score, and SLEDAI.
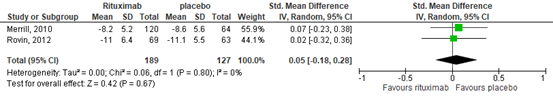
1.1. BILAG score
Changes in BILAG score did not differ between the RTX group and the control group in both RCTs over 52 weeks. This resulted in a standardized mean difference of 0.05 (95% confidence interval (CI) -0.18 to 0.28), see Figure 8.
Figure 8. Forest plot change in BILAG score at 52 weeks; rituximab vs. placebo
This outcome was also reported in 5 observational studies. Of them, 2 reported outcomes after 52 weeks of RTX treatment (Liu, 2015; Jiang, 2016), one after 64 weeks (Qiu, 2013), one after 40 weeks (Vital, 2011), and one after 24 weeks (Leonardo, 2005). The BILAG score improved after treatment with RTX. This resulted in a standardized mean difference of -2.92 (95% confidence interval (CI) -3.49 to -2.34), see Figure 9, in favor of RTX.
Figure 9. Forest plot BILAG score after – before (i.e., control); rituximab vs. placebo
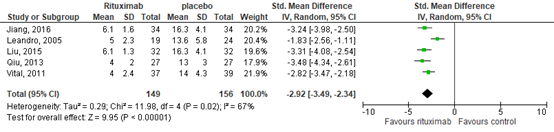
1.2. clinical response
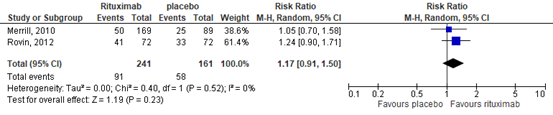
In the RTX group 91/241 (38%) achieved the outcome measure ‘clinical response’, compared with 58/161 (36%) in the placebo group. This resulted in a relative risk (RR) of 1.17 (95%CI 0.91 to 1.50), in favor of the RTX group, see Figure 10. The risk difference was 0.05 (95%CI -0.05 to 0.14).
Figure 10. Forest plot clinical response at week 52; rituximab vs. placebo
1.3. SLEDAI
This outcome was also reported in 6 observational studies. Of them, 2 reported outcomes after 52 weeks of RTX treatment (Liu, 2015; Jiang, 2016), one after 64 weeks (Qiu, 2013), one after 24 weeks (Zhang, 2014), and two after 48 weeks (Tamimoto, 2008; Ortega, 2010). The SLEDAI score improved after treatment with RTX. This resulted in a standardized mean difference of -2.45 (95% confidence interval (CI) -3.24 to -1.67), see Figure 11, in favor of RTX.
Figure 11. Forest plot SLEDAI score after – before (i.e., control); rituximab vs. placebo
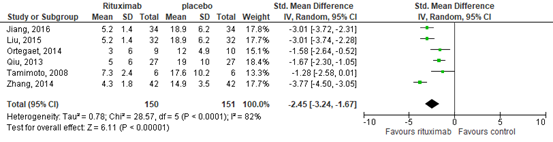
2. Quality of life
This outcome was not reported in the study of Wu (2020).
3. Reduction in glucocorticoid
This outcome was not reported in the study of Wu (2020).
4. Adverse events
Data for the outcome measure ‘adverse events’ was only reported for the RCTs. Outcome of interest were; severe adverse events, deaths, infections, and any infusion related severe adverse events.
4.1. severe adverse events
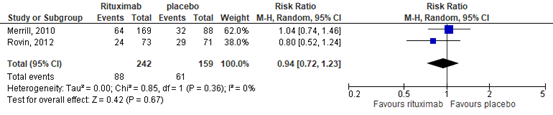
In the RTX group 88/242 (36%) achieved the outcome measure ‘severe adverse events’, compared with 61/159 (38%) in the placebo group. This resulted in a relative risk (RR) of 0.94 (95%CI 0.72 to 1.23), in favor of the RTX group, see Figure 12. The risk difference was -0.02 (95%CI -0.12 to 0.08).
Figure 12. Forest plot severe adverse events after 52 weeks; rituximab vs. placebo
4.2. deaths
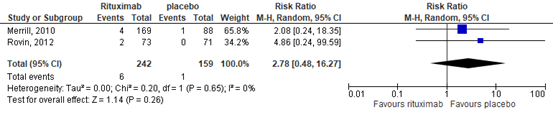
In the RTX group 6/242 (2%) died, compared with 1/159 (1%) in the placebo group. This resulted in a RR of 2.74 (95%CI 0.48 to 16.27), in favor of the placebo group, see Figure 13. The risk difference was 0.02 (95%CI -0.02 to 0.04).
Figure 13. Forest plot deaths after 52 weeks; rituximab vs. placebo
4.3. infections
In the RTX group 30/242 (12%) achieved the outcome measure ‘infection’, compared with 29/159 (18%) in the placebo group. This resulted in a RR of 0.73 (95%CI 0.42 to 1.27), in favor of the RTX group, see Figure 14. The risk difference was -0.05 (95%CI -0.13 to 0.02).
Figure 14. Forest plot infections after 52 weeks; rituximab vs. placebo

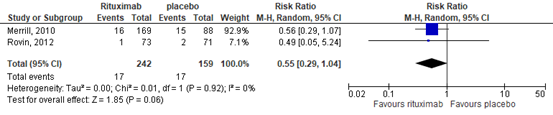
4.4. any infusion related severe adverse events.
In the RTX group 17/242 (3%) achieved the outcome measure ‘any infusion related severe adverse events’, compared with 17/159 (11%) in the placebo group. This resulted in a RR of 0.55 (95%CI 0.29 to 1.04), in favor of the RTX group, see Figure 15. The risk difference was -0.04 (95%CI -0.12 to 0.04).
Figure 15. Forest plot any infusion related severe adverse events after 52 weeks; rituximab vs. placebo
Level of evidence of the literature
The level of evidence (GRADE method) is determined per comparison and outcome measure and is based on results from RCTs or observational studies and therefore starts at level “high” (i.e., RCTs) or “low” (i.e., observational studies). Subsequently, the level of evidence was downgraded if there were relevant shortcomings in one of the several GRADE domains: risk of bias, inconsistency, indirectness, imprecision, and publication bias. For observational studies, it was upgraded if there was a strong association, or a dose-response relation, and/or plausible (residual) confounding.
Based on RCTs:
The level of evidence regarding the outcome measure quality of life, reduction of glucocorticoid could not be assessed with GRADE. The outcome measures were not studied in the included studies.
The level of evidence regarding the outcome measure SLE scores on disease activity indices was downgraded by 2 levels due to imprecision (95%CI of the mean difference includes no significant effect (mean difference=0 or RR=1), no clinically relevant effect (SMD<0.5 or RR 0.75-1.25), and not meeting optimal information size).
The level of evidence regarding the outcome measure adverse events (i.e., severe adverse events, deaths, infections, and any infusion related severe adverse events) was downgraded by 2 levels because of imprecision (2 levels; the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference, and not meeting the optimal information size).
The level of evidence regarding the outcome measure biomarkers was downgraded by 2 levels because of imprecision (2 levels; the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference, and not meeting the optimal information size).
Based on observational studies:
The level of evidence regarding the outcome measure quality of life, reduction of glucocorticoid, adverse events could not be assessed with GRADE. The outcome measures were not studied in the included studies.
The level of evidence regarding the outcome measure SLE scores on disease activity indices was downgraded by 1 level due to imprecision (not meeting optimal information size).
Anifrolumab
The current comparison was studied in the review of Lee (2020). In this review patients were administrated to placebo or 300 mg anifrolumab via intravenous infusion every 4 weeks.
1. SLE scores on disease activity indices
This outcome was reported as BICLA response in all three studies (Furie, 2017;2019; Morand, 2020). In the individual studies outcomes as BILAG, SLEDAI, and physician’s global assessment were reported as well.
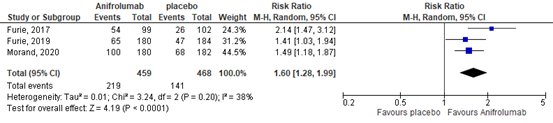
1.1. BICLA
In total 219/459 (48%) patients in the anifrolumab group, compared with 141/468 (30%) in the placebo group achieved BICLA response. This resulted in a relative risk (RR) of 1.60 (95% confidence interval (CI) 1.28 to 1.19), see Figure 25, favoring the anifrolumab group. The risk difference was 0.18 (95%CI 0.95 to 0.28).
Figure 25. Forest plot BICLA response; anifrolumab vs. placebo.
1.2. BILAG
The study of Furie (2017) reported the number of patients who developed new BILAG A flares. This occurred in 12/99 (12%) of the patients in the anifrolumab group, compared with 17/102 (17%) in the control group. This resulted in an RR of 0.73 (95% 0.37 to 1.44).
The study of Furie (2019) reported mean changes from baseline to week 52 in BILAG global score. The mean change (SD) was -13.0 (8.01) in the 143 patients in the anifrolumab group, compared with -10.7 (7.72) in the 147 patients in the control group. This resulted in a standardized mean difference of -0.29 (95%CI -0.52 to -0.06)
1.3. SLEDAI
The study of Furie (2017) reported the number of patients achieving clinical SLEDAI (i.e., ≥4-point reduction in clinical components (no laboratory components) of the SLEDAI). This was achieved in 62/99 (63%) of the patients in the anifrolumab group, compared with 44/102 (43%) in the control group. This resulted in an RR of 1.45 (95% 1.11 to 1.90).
The study of Furie (2019) reported mean changes from baseline to week 52 in SLEDAI-2K. The mean change (SD) was -6.0 (0.34) in the 143 patients in the anifrolumab group, compared with -5.3 (0.33) in the 147 patients in the control group. This resulted in a standardized mean difference of -2.08 (95%CI -2.37 to -1.80)
1.3. Physician’s global assessment
The study of Furie (2017) reported the number of patients achieving an improvement of ≥0.3 points improvement from baseline on physician’s global assessment. This was achieved in 74/99 (75%) of the patients in the anifrolumab group, compared with 55/102 (54%) in the control group. This resulted in an RR of 1.39 (95% 1.12 to 1.71).
The study of Furie (2019) reported mean changes from baseline to week 52 in physician’s global assessment. The mean change (SD) was -1.11 (0.05) in the 143 patients in the anifrolumab group, compared with -0.89 (0.05) in the 147 patients in the control group. This resulted in a standardized mean difference of -4.39 (95%CI -4.82 to -3.96)
2. Quality of life
This outcome was not reported in the study of Lee (2020). But one of the individual studies reported this outcome as ≥3.1-point improvement from baseline in health-related quality of life on SF-36 (PCS; Furie, 2017). This was achieved in 48/99 (49%) in the anifrolumab group, compared with 40/102 (39%) in the placebo group. This resulted in a RR of 1.24 (95% 0.90 to 1.70) in favor of the anifrolumab group.
3. Reduction in glucocorticoid dose
Only an overall outcome was reported for the secondary outcome ‘loss of the glucocorticoid dosage’ (i.e., Reduction of oral corticosteroid dosage to <7.5 mg/day in patients who were receiving >10 mg/day at baseline). This outcome showed a RR of 1.81 (95%CI 1.31 to 2.51), suggesting that the glucocorticoid dosage was more often reduced in the anifrolumab group compared with placebo (Furie, 2017;2019; Morand, 2020).
4. Adverse events
Outcome of interest were; adverse events, serious adverse events, and withdrawals due to adverse events.
4.1 adverse events
Only an overall outcome was reported for this secondary outcome based on data of three studies (Furie, 2017;2019; Morand, 2020). The RR was of 1.82 (95%CI 1.26 to 2.61), suggesting that any AEs occurred more often in the anifrolumab group compared with placebo.
4.2 serious adverse events
This outcome was reported in all three studies (Furie, 2017;2019; Morand, 2020). A SAE occurred in 56/459 (12%) patients treated with anifrolumab, compared with 80/468 (17%) patients treated with placebo. This resulted in a RR of 0.72 (95%CI 0.50 to 1.03), suggesting less SAE in the anifrolumab group, see Figure 26. The risk difference was -0.05 (95%CI -0.10 to -0.01).
Figure 26. Forest plot serious adverse events; anifrolumab vs. placebo.
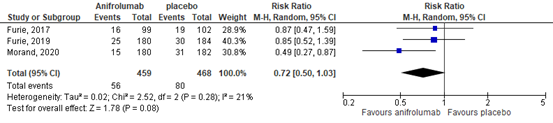
4.3 withdrawal due to adverse event
Only an overall outcome was reported for this secondary outcome based on date of three studies (Furie, 2017;2019; Morand, 2020). The RR was of 0.70 (95%CI 0.20 to 2.44), suggesting that withdrawal due to AE occurred less often in the anifrolumab group compared with placebo.
Level of evidence of the literature
The level of evidence (GRADE method) is determined per comparison and outcome measure and is based on results from RCTs and therefore starts at level “high”. Subsequently, the level of evidence was downgraded if there were relevant shortcomings in one of the several GRADE domains: risk of bias, inconsistency, indirectness, imprecision, and publication bias.
The level of evidence regarding the outcome measure SLE scores on disease activity indices, reduction of glucocorticoid dose, adverse events (i.e., any AE) was downgraded by 1 level due to risk of bias (all included studies were sponsored by the industry).
The level of evidence regarding the outcome measure quality of life was downgraded by 3 levels because of imprecision (2 levels; the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference, and not meeting the optimal information size), and risk of bias (all included studies were sponsored by the industry).
The level of evidence regarding the outcome measure adverse events (i.e., SAE) was downgraded by 2 levels because of imprecision (the 95% confidence interval includes no effect (RR=1)), and risk of bias (all included studies were sponsored by the industry).
The level of evidence regarding the outcome measure adverse events (i.e., withdrawal due to AE) was downgraded by 3 level because of imprecision (the 95% confidence interval includes both no effect and appreciable benefit/harm exceeding a minimal clinically important difference), and risk of bias (all included studies were sponsored by the industry).
Intravenous Immunoglobulin
The current comparison was studied in the review of Sakthiswary (2014). The described information is adapted from the study of Sakthiswary (2014). As only the standardized mean differences in the study were reported, and not the individual outcomes per group, it was not possible to illustrate forest plots if applicable.
1. SLE scores on disease activity indices
Six studies including four cohort studies and 2 nonrandomized RCTs, with a total of 261 subjects, investigated the effect of IVIg on disease activity scores. The disease activity scores used as outcome measures included SLEDAI (in 2 studies), SLAM, (in 2 studies) or LAI-P (in 1 study). The disease activity scores significantly decreased in all studies. An appreciable decline in the scores was seen as early as 6 weeks following the IVIg therapy. The forest plot in the paper reported a standardized mean difference of 0.584 (95% confidence interval (CI) 0.221 to 0.947). This outcome suggests that IVIg is associated with significant reduction in disease activity scores.
2. Quality of life
No data were reported for the outcome measure ‘quality of life’.
3. Reduction in glucocorticoid
Three studies, with a total of 45 subjects, investigated the effect of IVIg as a steroid-sparing agent. Levy (1999) and Zandman-Goddard (2012) reported a significant reduction in the average daily dose of corticosteroids. The pooled data from the above studies demonstrated a mean reduction of 17.95 mg/d in the dose of corticosteroids with IVIg therapy. Boletis (1999; RCT) compared the cumulative steroid dose between the IVIg and the cyclophosphamide-treated patients. The cyclophosphamide arm tended to have a higher dose (4719 vs 3334 mg), but this difference, however, did not reach statistical significance.
4. Adverse events
No data were reported for the outcome measure ‘adverse events’.
Level of evidence of the literature
The level of evidence (GRADE method) is determined per comparison and outcome measure and is based on results from RCTs or observational studies and therefore starts at level “high” (i.e., RCTs) or “low” (i.e., observational studies). Subsequently, the level of evidence was downgraded if there were relevant shortcomings in one of the several GRADE domains: risk of bias, inconsistency, indirectness, imprecision, and publication bias. For observational studies, it was upgraded if there was a strong association, or a dose-response relation, and/or plausible (residual) confounding.
Based on RCTs:
The level of evidence regarding the outcome measure disease activity, quality of life, adverse events could not be assessed with GRADE. The outcome measures were not studied in the included study.
The level of evidence regarding the outcome measure reduction of glucocorticoid was downgraded by 3 levels due to imprecision (2 levels; the 95%CI includes no effect, not meeting optimal information size), and indirectness (patients with lupus nephritis were studied in the RCT).
Based on observational studies:
The level of evidence regarding the outcome measure quality of life, adverse events could not be assessed with GRADE. The outcome measures were not studied in the included study.
The level of evidence regarding the outcome measure disease activity was downgraded by 2 levels due to imprecision (2 levels; the 95%CI includes no clinically relevant effect, not meeting optimal information size).
The level of evidence regarding the outcome measure reduction of glucocorticoid was downgraded by 1 level due to imprecision (1 level; not meeting optimal information size).
A systematic review of the literature was performed to answer the following question:
What are the benefits and harms of treatment with csDMARDs or bDMARDs compared to placebo or control treatment in adults with Systemic Lupus Erythematosus on SLE scores on disease activity indices, quality of life, reduction in glucocorticoid dose, adverse events?
P: adults with Systemic Lupus Erythematosus (SLE)
I: medical treatment with csDMARDs or bDMARDs
C: placebo or control treatment
O: SLE scores on disease activity indices, quality of life, reduction in glucocorticoid dose, adverse events
Relevant outcome measures
The guideline development group considered SLE scores on disease activity indices, as a critical outcome measure for decision making; and quality of life, reduction in glucocorticoid dose, adverse events as an important outcome measure for decision making.
A priori, the working group did not define the outcome measures listed above but used the definitions used in the studies.
A difference of 25% in the relative risk for dichotomous outcomes (i.e., RR 0.80-1.25) and 0.5 standard deviation (reported as SMD) for continuous outcomes was taken as a minimal clinically important difference.
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until November 28th 2020 for systematic reviews and randomized controlled trials (RCTs). The detailed search strategy is depicted under the tab Methods. After removing duplicates the combined systematic literature search resulted in 10067 hits (2020 systematic review, 8047 RCT). Considering the total amount of hits, it was decided to first only use the systematic review (SR) selection. Studies were selected based on the following criteria;
- adults with Systemic Lupus Erythematosus (SLE),
- medical treatment with csDMARDs (i.e., cyclophosphamide, azathioprine, methotrexate, leflunomide, mycophenolate mofetil (MMF), cyclosporin A, and tacrolimus) or bDMARDs (i.e., anifrolumab, belimumab (BEL), rituximab (RTX)) compared to placebo of control treatment,
- outcome according to the PICO was studied.
In total 145 studies were initially selected based on title and abstract screening. After reading the full text 121 studies were excluded and 24 eligible systematic reviews for the present guideline were included. After reading 24 articles full text, 21 SR’s described relevant outcomes for the current clinical question. Sixteen of them were excluded (see the table with reasons for exclusion under the tab Methods). The most recent and complete SR per medication category was selected for the literature analysis. Two SR were published before 2020, and therefore additional RCTs were added if these were available. In total four RCTs were relevant for the current question.
Results
Five SRs were included in the analysis of the literature, and four additional studies. Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
- Castrejón I, Tani C, Jolly M, Huang A, Mosca M. Indices to assess patients with systemic lupus erythematosus in clinical trials, long-term observational studies, and clinical care. Clin Exp Rheumatol. 2014 Sep-Oct;32(5 Suppl 85):S-85-95. Epub 2014 Oct 30. PMID: 25365095.
- EMA, 2021; https://www.ema.europa.eu/en/medicines/human/EPAR/saphnelo
- Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, Cervera R, Doria A, Gordon C, Govoni M, Houssiau F, Jayne D, Kouloumas M, Kuhn A, Larsen JL, Lerstrøm K, Moroni G, Mosca M, Schneider M, Smolen JS, Svenungsson E, Tesar V, Tincani A, Troldborg A, van Vollenhoven R, Wenzel J, Bertsias G, Boumpas DT. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019 Jun;78(6):736-745. doi: 10.1136/annrheumdis-2019-215089. Epub 2019 Mar 29. PMID: 30926722.
- Fanouriakis A, Kostopoulou M, Cheema K, Anders HJ, Aringer M, Bajema I, Boletis J, Frangou E, Houssiau FA, Hollis J, Karras A, Marchiori F, Marks SD, Moroni G, Mosca M, Parodis I, Praga M, Schneider M, Smolen JS, Tesar V, Trachana M, van Vollenhoven RF, Voskuyl AE, Teng YKO, van Leew B, Bertsias G, Jayne D, Boumpas DT. 2019 Update of the Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis. 2020 Jun;79(6):713-723. doi: 10.1136/annrheumdis-2020-216924. Epub 2020 Mar 27. PMID: 32220834.
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S; CD1013 Study Investigators. Anifrolumab, an Anti-Interferon-? Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017 Feb;69(2):376-386. doi: 10.1002/art.39962. PMID: 28130918; PMCID: PMC5299497.
- Furie, R. A., Morand, E. F., Bruce, I. N., Manzi, S., Kalunian, K. C., Vital, E. M., ... & TULIP-1 study investigators. (2019). Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. The Lancet Rheumatology, 1(4), e208-e219.
- Gordon C, Amissah-Arthur MB, Gayed M, Brown S, Bruce IN, D'Cruz D, Empson B, Griffiths B, Jayne D, Khamashta M, Lightstone L, Norton P, Norton Y, Schreiber K, Isenberg D; British Society for Rheumatology Standards, Audit and Guidelines Working Group. The British Society for Rheumatology guideline for the management of systemic lupus erythematosus in adults. Rheumatology (Oxford). 2018 Jan 1;57(1):e1-e45. doi: 10.1093/rheumatology/kex286. PMID: 29029350.
- Lee YH, Song GG. Anifrolumab for the treatment of active systemic lupus erythematosus: a meta-analysis of randomized controlled trials. Z Rheumatol. 2020 Nov 20. English. doi: 10.1007/s00393-020-00928-7. Epub ahead of print. PMID: 33216191.
- Mosca M, Tani C, Filice ME, Carli L, Delle Sedie A, Vagnani S, Della Rossa A, Baldini C, Bombardieri S. TNF-alpha inhibitors in Systemic Lupus Erythematosus. A case report and a systematic literature review. Mod Rheumatol. 2015 Jul;25(4):642-5. doi: 10.3109/14397595.2013.844306. Epub 2013 Nov 4. PMID: 24252029.
- Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, Tummala R; TULIP-2 Trial Investigators. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020 Jan 16;382(3):211-221. doi: 10.1056/NEJMoa1912196. Epub 2019 Dec 18. PMID: 31851795.
- Mulhearn B, Bruce IN. Indications for IVIG in rheumatic diseases. Rheumatology (Oxford). 2015 Mar;54(3):383-91. doi: 10.1093/rheumatology/keu429. Epub 2014 Nov 17. PMID: 25406359; PMCID: PMC4334686.
- Pego-Reigosa JM, Cobo-Ibáñez T, Calvo-Alén J, Loza-Santamaría E, Rahman A, Muñoz-Fernández S, Rúa-Figueroa Í. Efficacy and safety of nonbiologic immunosuppressants in the treatment of nonrenal systemic lupus erythematosus: a systematic review. Arthritis Care Res (Hoboken). 2013 Nov;65(11):1775-85. doi: 10.1002/acr.22035. PMID: 23609987.
- Said JT, Elman SA, Merola JF. Evaluating safety and compatibility of anti-tumor necrosis factor therapy in patients with connective tissue disorders. Ann Transl Med. 2021 Mar;9(5):430. doi: 10.21037/atm-20-5552. PMID: 33842651; PMCID: PMC8033307.
- Sakthiswary R, D'Cruz D. Intravenous immunoglobulin in the therapeutic armamentarium of systemic lupus erythematosus: a systematic review and meta-analysis. Medicine (Baltimore). 2014 Oct;93(16):e86. doi: 10.1097/MD.0000000000000086. PMID: 25310743; PMCID: PMC4616295.
- Singh JA, Shah NP, Mudano AS. Belimumab for systemic lupus erythematosus. Cochrane Database Syst Rev. 2021 Feb 25;2(2):CD010668. doi: 10.1002/14651858.CD010668.pub2. PMID: 33631841; PMCID: PMC8095005.
- Wu S, Wang Y, Zhang J, Han B, Wang B, Gao W, Zhang N, Zhang C, Yan F, Li Z. Efficacy and safety of rituximab for systemic lupus erythematosus treatment: a meta-analysis. Afr Health Sci. 2020 Jun;20(2):871-884. doi: 10.4314/ahs.v20i2.41. PMID: 33163054; PMCID: PMC7609121.
Evidence tables – systematic reviews
Evidence table for systematic review of RCTs and observational studies (intervention studies)
Table of quality assessment for systematic reviews of RCTs and observational studies
Based on AMSTAR checklist (Shea et al.; 2007, BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher et al 2009, PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Lee, 2020 |
Unclear, inclusion criteria are predefined but PICO not. |
Yes, clearly described in the review. |
Yes, described in the text of the review. |
Yes, clearly described in the review. |
N.a. all RCTs |
Yes, Jadad method. |
Yes, heterogeneity is assessed. |
Yes, Funnel plots are usually used to detect biases in publications |
Yes, source of funding or support are indicated for the systematic review AND for each of the included studies. |
|
Pego-Reigosa, 2013 |
Unclear, inclusion criteria are predefined but PICO not. |
Yes, clearly described in supplementary file of the review |
Yes, clearly described in supplementary file of the review |
Yes, clearly described in supplementary file of the review |
N.A. outcomes were not pooled, only described per comparison. No overall effect estimates were calculated. |
Yes, clearly described in supplementary file of review. |
Yes, studies are descripted per medical treatment, but outcomes are not pooled due to variation. |
n.a. no statistical analyses are performed. |
Unclear, the authors of the SR reported no conflict of interest, but no information is provided for included studies |
|
Sakthiswary, 2014 |
Unclear, inclusion criteria are predefined but PICO not. |
Unclear, information about databases is provided, but no search date. |
Yes, a figure, including this information, is provided. |
Unclear, some characteristics are included but not all relevant baseline characteristics. |
NN.a. baseline values are used as control (not of interest) in the observational studies. Regarding RCTs, no differences at baseline were shown. |
No, no information was provided about an assessment of scientific quality. |
Yes, heterogeneity was assessed with I² test, but I² % was high. |
Unclear |
Unclear, the authors of the SR reported no conflict of interest, but no information is provided for included studies. |
|
Singh, 2013 |
Yes, PICO question and inclusion criteria are clearly defined. |
Yes, clearly described in Cochrane Review. |
Yes, clearly described in Cochrane Review. |
Yes, clearly described in Cochrane Review. |
N.A. all RCTs |
Yes, clearly described in Cochrane Review, according to RoB table |
Yes, heterogeneity was assessed with I² test. |
Yes, publication bias could not be assessed because there were fewer than 10 included studies. |
Yes, source of funding or support are indicated for the systematic review AND for each of the included studies. |
|
Wu, 2020 |
Unclear, inclusion criteria are predefined but PICO not. |
Unclear, information about databases is provided, but no search date. |
Yes, a figure, including this information, is provided. |
Yes, two tables are included for the RCTs and observational studies, respectively. |
N.a. baseline values are used as control (not of interest) in the observational studies. Regarding RCTs, no differences at baseline were shown. |
Yes, Jadad for RCTs and Newcastle-ottawa for observational studies. |
Yes, heterogeneity is assessed with I². |
Unclear |
Unclear, the authors of the SR reported no conflict of interest, but no information is provided for included studies. |
- Research question (PICO) and inclusion criteria should be appropriate and predefined
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs)
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table etc.)
- Clinical and statistical heterogeneity should be assessed; clinical: enough similarities in patient characteristics, intervention and definition of outcome measure to allow pooling? For pooled data: assessment of statistical heterogeneity using appropriate statistical tests (e.g. Chi-square, I2)?
- An assessment of publication bias should include a combination of graphical aids (e.g., funnel plot, other available tests) and/or statistical tests (e.g., Egger regression test, Hedges-Olken). Note: If no test values or funnel plot included, score “no”. Score “yes” if mentions that publication bias could not be assessed because there were fewer than 10 included studies.
- Sources of support (including commercial co-authorship) should be reported in both the systematic review and the included studies. Note: To get a “yes,” source of funding or support must be indicated for the systematic review AND for each of the included studies.
Evidence table for intervention studies (randomized controlled trials and non-randomized observational studies [cohort studies, case-control studies, case series])1
This table is also suitable for diagnostic studies (screening studies) that compare the effectiveness of two or more tests. This only applies if the test is included as part of a test-and-treat strategy – otherwise the evidence table for studies of diagnostic test accuracy should be used.
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors [(potential) confounders]
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
Risk of bias table for intervention studies (randomized controlled trials; based on Cochrane risk of bias tool and suggestions by the CLARITY Group at McMaster University)
|
Study reference
(first author, publication year) |
Was the allocation sequence adequately generated? a
Definitely yes Probably yes Probably no Definitely no |
Was the allocation adequately concealed?b
Definitely yes Probably yes Probably no Definitely no |
Blinding: Was knowledge of the allocated interventions adequately prevented?c
Were patients blinded?
Were healthcare providers blinded?
Were data collectors blinded?
Were outcome assessors blinded?
Were data analysts blinded?
Definitely yes Probably yes Probably no Definitely no |
Was loss to follow-up (missing outcome data) infrequent?d
Definitely yes Probably yes Probably no Definitely no |
Are reports of the study free of selective outcome reporting?e
Definitely yes Probably yes Probably no Definitely no |
Was the study apparently free of other problems that could put it at a risk of bias?f
Definitely yes Probably yes Probably no Definitely no |
Overall risk of bias If applicable/necessary, per outcome measureg
LOW Some concerns HIGH
|
|
Islam, 2012 |
Definitely yes;
Reason: The patients were randomly allocated to either MTX or CQ groups. Randomization was performed following a random number table without considering their presentation. We followed the vertical series of odd and even numbers. |
Definitely yes;
Reason: The patients were randomly allocated to either MTX or CQ groups. Randomization was performed following a random number table without considering their presentation. We followed the vertical series of odd and even numbers. |
Definitely no
Reason: open-label trial |
Probably yes
Reason: in both groups 2 patients were excluded from the analyses due to discontinuation of therapy or excluded from therapy |
Definitely yes;
Reason: All relevant outcomes were reported |
probably no
Reason: the relative small sample size is a limitation. |
High
Reason: the study was an open-label randomized controlled trials with a relative low sample size. |
|
Miyawaki, 2013 |
Definitely no
Reason: patients with an active disease were selected for the intervention and patients with a inactive disease for the control group. |
Definitely no
Reason: patients with an active disease were selected for the intervention and patients with a inactive disease for the control group. |
Definitely no
Reason: open-label trial |
Probably yes
Reason: one patient discontinued MTX |
probably yes;
Reason: All relevant outcomes were reported, but only in figures. |
probably no
Reason: the relative small sample size is a limitation. |
High
Reason: groups are not comparable at baseline, open label design, low sample size. |
|
Yahya, 2013 |
Definitely yes;
Reason: All subjects underwent block randomization 1:1 into two treatment groups, either receiving mycophenolate sodium or other standard immunosuppressive therapy other than MMF. Randomization was done by a computer- generated program which provided a random number generation service. |
…Definitely yes;
Reason: All subjects underwent block randomization 1:1 into two treatment groups, either receiving mycophenolate sodium or other standard immunosuppressive therapy other than MMF. Randomization was done by a computer- generated program which provided a random number generation service. |
Definitely no
Reason: open-label trial |
Probably yes
Reason: no patients in the intervention group dropped out, but 2 in the control group. |
probably yes;
Reason: All relevant outcomes were reported, but only in figures. |
probably no
Reason: the relative small sample size is a limitation. |
High
Reason: the study was an open-label randomized controlled trials with a relative low sample size. |
|
Ordi-Ros, 2017 |
Definitely yes
Reason: The randomised list, stratified by centre and SLEDAI-2K score (6–9 vs ≥10), was created using computer-generated randomnumber sequences in blocks of 10 (C4 Study Design Pack Software, GlaxoSmithKline) by the Vall d’Hebrón Hospital investigational pharmacist, who was blind to patient enrolment. Sequentially numbered, concealed envelopes containing group assignment were provided to the investigators. |
Definitely yes
Reason: The randomised list, stratified by centre and SLEDAI-2K score (6–9 vs ≥10), was created using computer-generated randomnumber sequences in blocks of 10 (C4 Study Design Pack Software, GlaxoSmithKline) by the Vall d’Hebrón Hospital investigational pharmacist, who was blind to patient enrolment. Sequentially numbered, concealed envelopes containing group assignment were provided to the investigators. |
Definitely no
Reason: open-label trial |
Probably no
Reason: Fifty-three patients in the AZA group and 33 in the EC-MPS group discontinued the study. The main reasons for early withdrawal were treatment failure and AEs |
probably yes;
Reason: All relevant outcomes were reported, but some of them only in figures. |
Probably yes
Reason: |
Some concerns
Reason: the study was an open-label randomized controlled trials . |
- Randomization: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomization process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomization (performed at a site remote from trial location). Inadequate procedures are all procedures based on inadequate randomization procedures or open allocation schedules..
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments, but this should not affect the risk of bias judgement. Blinding of those assessing and collecting outcomes prevents that the knowledge of patient assignment influences the process of outcome assessment or data collection (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is usually not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary. Finally, data analysts should be blinded to patient assignment to prevents that knowledge of patient assignment influences data analysis.
- If the percentage of patients lost to follow-up or the percentage of missing outcome data is large, or differs between treatment groups, or the reasons for loss to follow-up or missing outcome data differ between treatment groups, bias is likely unless the proportion of missing outcomes compared with observed event risk is not enough to have an important impact on the intervention effect estimate or appropriate imputation methods have been used.
- Results of all predefined outcome measures should be reported; if the protocol is available (in publication or trial registry), then outcomes in the protocol and published report can be compared; if not, outcomes listed in the methods section of an article can be compared with those whose results are reported.
- Problems may include: a potential source of bias related to the specific study design used (e.g. lead-time bias or survivor bias); trial stopped early due to some data-dependent process (including formal stopping rules); relevant baseline imbalance between intervention groups; claims of fraudulent behavior; deviations from intention-to-treat (ITT) analysis; (the role of the) funding body. Note: The principles of an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
- Overall judgement of risk of bias per study and per outcome measure, including predicted direction of bias (e.g. favors experimental, or favors comparator). Note: the decision to downgrade the certainty of the evidence for a particular outcome measure is taken based on the body of evidence, i.e. considering potential bias and its impact on the certainty of the evidence in all included studies reporting on the outcome.
Evidence tables – additional RCTs
Table of excluded studies
|
Author and year |
Reason for exclusion |
|
Alshaiki, 2018 |
Other SR more recent |
|
Bela, 2021 |
Other SR more recent |
|
Borba, 2014 |
Other SR more recent |
|
Cobo-Ibáñez, 2014 |
Other SR more recent |
|
Duxbury, 2013 |
Other SR more recent |
|
Koh, 2020 |
Other SR more recent |
|
Lan, 2012 |
Other SR more recent |
|
Maclsaac, 2018 |
Other SR more recent |
|
Madhok, 2009 |
Other SR more recent |
|
Mok, 2007 |
Other SR more recent |
|
Mosca, 2015 |
Other SR more recent |
|
Murray, 2010 |
Other SR more recent |
|
Ramos-Casals, 2009 |
Other SR more recent |
|
Sakthiswary, 2014 |
Other SR more recent |
|
Shamliyan, 2017 |
Other SR more recent |
|
Tao, 2019 |
Other SR more recent |
Beoordelingsdatum en geldigheid
Publicatiedatum : 15-02-2023
Beoordeeld op geldigheid : 15-02-2023
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2019 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de zorg voor patiënten met systemische lupus erythematosus.
Werkgroep
- Dr. E. Zirkzee, reumatoloog, werkzaam in Maasstad ziekenhuis Rotterdam, NVR, voorzitter van de werkgroep.
- Dr. M. van Onna, reumatoloog, werkzaam in Amsterdam UMC, NVR.
- Dr. C. Magro Checa, reumatoloog, werkzaam in Zuyderland Medisch Centrum, NVR.
- Dr. R. Luijten, reumatoloog, werkzaam in ETZ Tilburg, NVR (t/m 10-2021).
- Drs. R.J. Goekoop, internist-reumatoloog, werkzaam in Hagaziekenhuis, NVR.
- Dr. R. Klaasen, reumatoloog, werkzaam in Meander MC, NVR (vanaf 10-2021).
- Dr. K. de Leeuw, internist-klinisch immunoloog, werkzaam in UMC Groningen, NIV.
- Dr. M. Limper, internist-klinisch immunoloog, werkzaam in UMC Utrecht, NIV.
- Mw. L. Beaart-van de Voorde, MSc, verpleegkundig specialist AGZ, expertisegebied reumatologie, werkzaam in Leids Universitair Medisch Centrum, V&VN.
- Dr. J.R. Miedema, longarts, werkzaam in Erasmus MC, NVALT.
- Drs. M.J.R. Quanjel, longarts, werkzaam in St. Antonius ziekenhuis Nieuwegein, NVALT.
- Prof Dr. B.J.F van den Bemt, apotheker/klinisch farmacoloog, werkzaam in St. Maartenskliniek/RadboudUMC, NVZA.
- Dr. A. Berden, reumatoloog, werkzaam in Maasstad Ziekenhuis Rotterdam, NVR.
- Mw. W. Zacouris-Verweij, patiëntvertegenwoordiger, NVLE.
- Mw. G. Brandts, patiëntvertegenwoordiger, NVLE.
Klankbordgroep
- Dr. H.B. Thio, dermatoloog, werkzaam in Erasmus MC, NVDV.
- Dr. N. Ajmone Marsan, cardioloog, werkzaam in Leiden UMC, NVvC.
Met ondersteuning van
- Drs. I. van Dusseldorp, Literatuurspecialist, Van Dusseldorp, Delvaux & Ket.
- Dr. A. Claassen, senior beleidsmedewerker, NVR.
- Dr. M. van Vilsteren, senior beleidsmedewerker, NVR.
- Dr. M.M.A. Verhoeven, adviseur, Kennisinstituut van de Federatie Medisch Specialisten.
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
E.J.M. Zirkzee |
Reumatoloog Maasstad ziekenhuis Rotterdam |
2018 Adviesraad SLE GSK (betaald, eenmalig) |
|
Geen |
|
M. van Onna |
Reumatoloog Amsterdam UMC, locatie AMC |
n.v.t. |
|
Geen |
|
C. Magro Checa |
Reumatoloog Zuyderland Medisch Centrum, Heerlen en Sittard-Geleen |
n.v.t. |
Deelname SLE-BRAVE I en II studies (Lilly) |
Geen |
|
R. Luijten |
Reumatoloog ETZ Tilburg |
Medical Information Officer binnen het ETZ (onbetaald) |
|
Geen |
|
R.J. Goekoop |
Internist-Reumatoloog CMIO Haga ziekenhuis 1,0fte |
Voorzitter SANL (onbetaald) / advies raad FMS t.a.v. kwaliteits aanleveringen |
Deelname Bliss-Beieve Studie (GSK), res centrum |
Geen |
|
K. de Leeuw |
Internist-Klinisch immunoloog bij de afdeling Reumatoloog & Klinische Immunologie van het Universitair Medisch Centrum Groningen (UMCG) |
n.v.t. |
Adviesraad GSK, Adviesraad Otsuka, deelname BLISS BELIEVE-studie (GSK), deelname Topaz studie (Biogen) |
Geen |
|
M. Limper |
internist - klinisch immunoloog UMC Utrecht |
lid wetenschappelijke adviesraad Farmacotherapeutisch Kompas; onkostenvergoeding |
Consultancy GSK, Roche, Novartis; Unrestricted grant van Thermo Fisher, unrestricted grant van GSK; deelname BLISS BELIEVE-studie (GSK) en deelname JAK/TK-studie (AbbVie); Lid van de medische adviesraad verbonden aan de NVLE. |
Geen |
|
L. Beaart-van de Voorde |
Verpleegkundig specialist, LUMC - afdeling Reumatologie / Docent Master Advanced Nursing Practice, Hogeschool Leiden |
Voorzitter V&VN-VS Netwerk Reumatologie (onbetaald) / Redactielid Nurse Academy Ouderen & Thuiszorg (vergoeding) |
|
Geen |
|
W. Zacouris-Verweij |
Financieel Adviseur Emuraal Advies B.V. te Rotterdam |
Voorzitter NVLE onbetaald ARCH werkgroep SLE, onbetaald |
Patient Advocate bij UCB voor interne opleiding |
Geen |
|
J.R. Miedema |
Longarts Erasmus MC |
Longarts is regulier betaald |
Adviesraad Beuringer Ingelheim nationaal/internationaal t.a.v. nintedanib voor systemische sclerose en progressieve fibrose. / patent JAK remmer voor pulmonale sarcoidose (eigendom van Erasmus MC, niet individueel); deelname onderzoek / pirfenidon bij asbestose (Roche - NVALT). Rest onderzoek n.v.t. voor SLE richtlijn |
Niet meeschrijven aan aanbevelingen Nintedanib. |
|
M. Quanjel |
Longarts Antonius ziekenhuis Nieuwegein |
Longarts vast in dienst |
Adviesraad Beuringer Ingelheim nationaal/internationaal t.a.v. nintedanib voor systemische sclerose en progressieve fibrose |
Geen |
|
B. van den Bemt |
apotheker/klinisch farmacoloog, werkzaam in St. Maartenskliniek/RadboudUMC |
Incidentele nascholingen gedaan voor Pfizer, Novartis, Sandoz en Bayer |
Adviesraad UCB tav farmaceutische zorg; Onderzoek naar therapietrouw gesponsored door Abbvie |
Geen |
|
A. Berden |
Reumatoloog (sinds eind 2020; voorheen AIOS Leiden UMC) Reumatologie Maasstad Ziekenhuis Rotterdam |
n.v.t. |
|
Geen |
|
R. Klaasen |
Reumatoloog, werkzaam in Meander MC |
Arch werkgroep SLE (niet betaald) Penningmeester Nederlandse vereniging voor reumatologie (betaald) Geneesmiddelen commissie Meander Medisch Centrum (niet betaald) |
Vakgroep reumatologie in Meander Medisch centrum heeft meegedaan (afgerond 2021) aan Bliss-Believe studie (GSK): Wereldwijd opgezet onderzoek naar de combinatie behandeling van Rituximab + Belimumab vergeleken met Placeob + Belimumab en alleen Belimumab in SLE patiënten. (doelgroep: SLE ). |
Geen |
|
G. Brandts |
Patiëntvertegenwoordiger |
Geen |
Geen |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door patiëntenverenigingen uit te nodigen voor de schriftelijke knelpuntenanalyse en meerdere leden van de patiëntenvereniging af te vaardigen in de werkgroep. Het verslag hiervan is besproken in de werkgroep. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen. De conceptrichtlijn is tevens voor commentaar voorgelegd aan meerdere patiëntenverenigingen en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Wkkgz & Kwalitatieve raming van mogelijke substantiële financiële gevolgen
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
|
Module |
Uitkomst raming |
Toelichting |
|
Module monitoring |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module medicamenteuze behandeling - basis |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module medicamenteuze behandeling - DMARD |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
Methode ontwikkeling
Evidence based
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerde de werkgroep de knelpunten in de zorg voor patiënten met de schriftelijk knelpuntenanalyse. Tevens zijn er knelpunten aangedragen door Inspectie voor de Gezondheidszorg en Jeugd, Zorginstituut Nederland, NVZ, ZKN, VIG, NVR, V&VN, KNMP, NVZA, NVN, ReumaNederland, Nationale Vereniging ReumaZorg Nederland, Nationale Vereniging voor Lupus, APS, Sclerodemie, MCTD (NVLE) via de schriftelijke knelpuntenanalyse. Een verslag hiervan is opgenomen onder aanverwante producten (Bijlage I).
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
Definitie |
|
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, veiligheid aspecten, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE-methodiek.
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Zoekverantwoording
Non-biological csDMARDS
Embase, 5-12-2020
|
N. |
Query |
Results |
|
#10 |
#9 NOT #8 = RCT |
2189 |
|
#9 |
#5 AND #7 |
2420 |
|
#8 |
#5 AND #6 = SR |
470 |
|
#7 |
('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it |
2488699 |
|
#6 |
('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
534524 |
|
#5 |
#4 NOT (('adolescent'/exp OR 'child'/exp OR adolescent*:ti,ab OR child*:ti,ab OR schoolchild*:ti,ab OR infant*:ti,ab OR girl*:ti,ab OR boy*:ti,ab OR teen:ti,ab OR teens:ti,ab OR teenager*:ti,ab OR youth*:ti,ab OR pediatr*:ti,ab OR paediatr*:ti,ab OR puber*:ti,ab) NOT ('adult'/exp OR 'aged'/exp OR 'middle aged'/exp OR adult*:ti,ab OR man:ti,ab OR men:ti,ab OR woman:ti,ab OR women:ti,ab)) |
12248 |
|
#4 |
#3 AND [1-1-1995]/sd NOT ('conference abstract'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
13343 |
|
#3 |
#1 AND #2 |
21597 |
|
#2 |
'antimalarial agent'/exp OR antimalaria*:ti,ab,kw OR 'azathioprine derivative'/exp OR 'azathioprine'/exp OR 'arathioprin':ti,ab,tn,dn OR 'arathioprine':ti,ab,tn,dn OR 'aza-q':ti,ab,tn,dn OR 'azafalk':ti,ab,tn,dn OR 'azahexal':ti,ab,tn,dn OR 'azamedac':ti,ab,tn,dn OR 'azamun':ti,ab,tn,dn OR 'azamune':ti,ab,tn,dn OR 'azanin':ti,ab,tn,dn OR 'azapin':ti,ab,tn,dn OR 'azapress':ti,ab,tn,dn OR 'azaprine':ti,ab,tn,dn OR 'azarex':ti,ab,tn,dn OR 'azasan':ti,ab,tn,dn OR 'azathiodura':ti,ab,tn,dn OR 'azathiopine':ti,ab,tn,dn OR 'azathioprim':ti,ab,tn,dn OR 'azathioprin':ti,ab,tn,dn OR 'azathioprine':ti,ab,tn,dn OR 'azathiopurine':ti,ab,tn,dn OR 'azathropsin':ti,ab,tn,dn OR 'azatioprina':ti,ab,tn,dn OR 'azatox':ti,ab,tn,dn OR 'azatrilem':ti,ab,tn,dn OR 'azopi':ti,ab,tn,dn OR 'azoran':ti,ab,tn,dn OR 'azothioprin':ti,ab,tn,dn OR 'azothioprine':ti,ab,tn,dn OR 'bw 57 322':ti,ab,tn,dn OR 'bw 57-322':ti,ab,tn,dn OR 'bw 57322':ti,ab,tn,dn OR 'bw57-322':ti,ab,tn,dn OR 'bw57322':ti,ab,tn,dn OR 'colinsan':ti,ab,tn,dn OR 'immuran':ti,ab,tn,dn OR 'immurel':ti,ab,tn,dn OR 'immuthera':ti,ab,tn,dn OR 'imunen':ti,ab,tn,dn OR 'imuprin':ti,ab,tn,dn OR 'imuran':ti,ab,tn,dn OR 'imurane':ti,ab,tn,dn OR 'imurek':ti,ab,tn,dn OR 'imurel':ti,ab,tn,dn OR 'imuren':ti,ab,tn,dn OR 'methylnitroimidazolylmercaptopurine':ti,ab,tn,dn OR 'nsc 39084':ti,ab,tn,dn OR 'nsc39084':ti,ab,tn,dn OR 'thioazeprine':ti,ab,tn,dn OR 'thioprine':ti,ab,tn,dn OR 'transimune':ti,ab,tn,dn OR 'zytrim':ti,ab,tn,dn OR 'methotrexate'/exp OR '4 amino 10 methylfolic acid':ti,ab,tn,dn OR '4 amino 10 methylpteroylglutamic acid':ti,ab,tn,dn OR '4 amino n10 methylpteroylglutamic acid':ti,ab,tn,dn OR 'abitrexate':ti,ab,tn,dn OR 'amethopterin':ti,ab,tn,dn OR 'amethopterine':ti,ab,tn,dn OR 'ametopterine':ti,ab,tn,dn OR 'antifolan':ti,ab,tn,dn OR 'biotrexate':ti,ab,tn,dn OR 'canceren':ti,ab,tn,dn OR 'cl 14377':ti,ab,tn,dn OR 'cl14377':ti,ab,tn,dn OR 'emtexate':ti,ab,tn,dn OR 'emthexat':ti,ab,tn,dn OR 'emthexate':ti,ab,tn,dn OR 'emtrexate':ti,ab,tn,dn OR 'enthexate':ti,ab,tn,dn OR 'farmitrexat':ti,ab,tn,dn OR 'farmitrexate':ti,ab,tn,dn OR 'farmotrex':ti,ab,tn,dn OR 'folex':ti,ab,tn,dn OR 'ifamet':ti,ab,tn,dn OR 'imeth':ti,ab,tn,dn OR 'intradose mtx':ti,ab,tn,dn OR 'jylamvo':ti,ab,tn,dn OR 'lantarel':ti,ab,tn,dn OR 'ledertrexate':ti,ab,tn,dn OR 'maxtrex':ti,ab,tn,dn OR 'metex':ti,ab,tn,dn OR 'methoblastin':ti,ab,tn,dn OR 'methohexate':ti,ab,tn,dn OR 'methotrate':ti,ab,tn,dn OR 'methotrexat':ti,ab,tn,dn OR 'methotrexate':ti,ab,tn,dn OR 'methotrexato':ti,ab,tn,dn OR 'methoxtrexate':ti,ab,tn,dn OR 'methrotrexate':ti,ab,tn,dn OR 'methylaminopterin':ti,ab,tn,dn OR 'methylaminopterine':ti,ab,tn,dn OR 'meticil':ti,ab,tn,dn OR 'metoject':ti,ab,tn,dn OR 'metothrexate':ti,ab,tn,dn OR 'metotrexat':ti,ab,tn,dn OR 'metotrexate':ti,ab,tn,dn OR 'metotrexin':ti,ab,tn,dn OR 'metrex':ti,ab,tn,dn OR 'mexate':ti,ab,tn,dn OR 'mexate-aq':ti,ab,tn,dn OR 'mpi 5004':ti,ab,tn,dn OR 'mpi5004':ti,ab,tn,dn OR 'neotrexate':ti,ab,tn,dn OR 'nordimet':ti,ab,tn,dn OR 'novatrex':ti,ab,tn,dn OR 'nsc 740':ti,ab,tn,dn OR 'nsc740':ti,ab,tn,dn OR 'otrexup':ti,ab,tn,dn OR 'rasuvo':ti,ab,tn,dn OR 'reditrex':ti,ab,tn,dn OR 'reumatrex':ti,ab,tn,dn OR 'rheumatrex':ti,ab,tn,dn OR 'rheumatrex dose pack':ti,ab,tn,dn OR 'texate':ti,ab,tn,dn OR 'texorate':ti,ab,tn,dn OR 'trexall':ti,ab,tn,dn OR 'xaken':ti,ab,tn,dn OR 'xatmep':ti,ab,tn,dn OR 'zexate':ti,ab,tn,dn OR 'mtx':ti,ab,tn,dn OR 'cyclosporine'/exp OR 'adi 628':ti,ab,tn,dn OR 'adi628':ti,ab,tn,dn OR 'cequa':ti,ab,tn,dn OR 'cgc 1072':ti,ab,tn,dn OR 'cgc1072':ti,ab,tn,dn OR 'ciclomulsion':ti,ab,tn,dn OR 'cicloral':ti,ab,tn,dn OR 'ciclosporin':ti,ab,tn,dn OR 'ciclosporine':ti,ab,tn,dn OR 'cipol':ti,ab,tn,dn OR 'consupren':ti,ab,tn,dn OR 'cyclasol':ti,ab,tn,dn OR 'cyclokat':ti,ab,tn,dn OR 'cyclosporin':ti,ab,tn,dn OR 'cyclosporine':ti,ab,tn,dn OR 'de 076':ti,ab,tn,dn OR 'de076':ti,ab,tn,dn OR 'deximune':ti,ab,tn,dn OR 'equoral':ti,ab,tn,dn OR 'gengraf':ti,ab,tn,dn OR 'ikervis':ti,ab,tn,dn OR 'iminoral':ti,ab,tn,dn OR 'implanta':ti,ab,tn,dn OR 'imusporin':ti,ab,tn,dn OR 'lx 201':ti,ab,tn,dn OR 'lx201':ti,ab,tn,dn OR 'mc2 03':ti,ab,tn,dn OR 'mc203':ti,ab,tn,dn OR 'mtd 202':ti,ab,tn,dn OR 'mtd202':ti,ab,tn,dn OR 'neoral':ti,ab,tn,dn OR 'neoral-sandimmun':ti,ab,tn,dn OR 'nm 0133':ti,ab,tn,dn OR 'nm 133':ti,ab,tn,dn OR 'nm0133':ti,ab,tn,dn OR 'nm133':ti,ab,tn,dn OR 'nova 22007':ti,ab,tn,dn OR 'nova22007':ti,ab,tn,dn OR 'ol 27400':ti,ab,tn,dn OR 'ol27400':ti,ab,tn,dn OR 'olo 400':ti,ab,tn,dn OR 'olo500':ti,ab,tn,dn OR 'opph 088':ti,ab,tn,dn OR 'opph088':ti,ab,tn,dn OR 'opsisporin':ti,ab,tn,dn OR 'otx 101':ti,ab,tn,dn OR 'otx101':ti,ab,tn,dn OR 'p 3072':ti,ab,tn,dn OR 'p3072':ti,ab,tn,dn OR 'padciclo':ti,ab,tn,dn OR 'papilock':ti,ab,tn,dn OR 'pulminiq':ti,ab,tn,dn OR 'restasis':ti,ab,tn,dn OR 'restaysis':ti,ab,tn,dn OR 'sanciclo':ti,ab,tn,dn OR 'sandimmun':ti,ab,tn,dn |
481751 |
|
#1 |
'systemic lupus erythematosus'/exp OR 'erythematodes visceralis':ti,ab,kw OR 'lupovisceritis':ti,ab,kw OR 'libman sacks disease':ti,ab,kw OR 's.l.e.':ti,ab,kw OR 'sle':ti,ab,kw OR lupus:ti,ab,kw |
143148 |
PubMed, 5-12-2020
|
Search |
Query |
Results |
|
#11 |
Search: #10 NOT #9 = RCT |
196 |
|
#9 |
Search: #6 AND #8 |
272 |
|
#10 |
Search: #7 AND #8 = SR |
249 |
|
#8 |
Search: #5 NOT (("Adolescent"[Mesh] OR "Child"[Mesh] OR "Infant"[Mesh] OR adolescen*[tiab] OR child*[tiab] OR schoolchild*[tiab] OR infant*[tiab] OR girl*[tiab] OR boy*[tiab] OR teen[tiab] OR teens[tiab] OR teenager*[tiab] OR youth*[tiab] OR pediatr*[tiab] OR paediatr*[tiab] OR puber*[tiab]) NOT ("Adult"[Mesh] OR adult*[tiab] OR man[tiab] OR men[tiab] OR woman[tiab] OR women[tiab])) |
4,545 |
|
#7 |
Search: ((random*[tiab] AND (controlled[tiab] OR control[tiab] OR placebo[tiab] OR versus[tiab] OR vs[tiab] OR group[tiab] OR groups[tiab] OR comparison[tiab] OR compared[tiab] OR arm[tiab] OR arms[tiab] OR crossover[tiab] OR cross-over[tiab]) AND (trial[tiab] OR study[tiab])) OR ((single[tiab] OR double[tiab] OR triple[tiab]) AND (masked[tiab] OR blind*[tiab]))) |
763,584 |
|
#6 |
Search: ("Meta-Analysis" [Publication Type] OR "Meta-Analysis as Topic"[Mesh] OR metaanaly*[tiab] OR meta-analy*[tiab] or metanaly*[tiab] OR "Systematic Review" [Publication Type] OR systematic[sb] OR "Cochrane Database Syst Rev"[Journal] or prisma[tiab] OR preferred reporting items[tiab] OR prospero[tiab] OR ((systemati*[ti] OR scoping[ti] OR umbrella[ti] OR structured literature[ti]) AND (review*[ti] OR overview*[ti])) OR systematic review*[tiab] OR scoping review*[tiab] OR umbrella review*[tiab] OR structured literature review*[tiab] OR systematic qualitative review*[tiab] OR systematic quantitative review*[tiab] OR systematic search and review[tiab] OR systematized review[tiab] OR systematised review[tiab] OR systemic review[tiab] OR systematic literature review*[tiab] OR systematic integrative literature review*[tiab] OR systematically review*[tiab] OR scoping literature review*[tiab] OR systematic critical review[tiab] OR systematic integrative review*[tiab] OR systematic evidence review[tiab] OR Systematic integrative literature review*[tiab] OR Systematic mixed studies review*[tiab] OR Systematized literature review*[tiab] OR Systematic overview*[tiab] OR Systematic narrative review*[tiab] OR ((systemati*[tiab] OR literature[tiab] OR database*[tiab] OR data-base*[tiab] OR structured[tiab] OR comprehensive*[tiab] OR systemic*[tiab]) AND search*[tiab]) OR (Literature[ti] AND review[ti] AND (database*[tiab] OR data-base*[tiab] OR search*[tiab])) OR ((data extraction[tiab] OR data source*[tiab]) AND study selection[tiab]) OR (search strategy[tiab] AND selection criteria[tiab]) OR (data source*[tiab] AND data synthesis[tiab]) OR medline[tiab] OR pubmed[tiab] OR embase[tiab] OR Cochrane[tiab] OR ((critical[ti] OR rapid[ti]) AND (review*[ti] OR overview*[ti] OR synthes*[ti])) OR (((critical*[tiab] OR rapid*[tiab]) AND (review*[tiab] OR overview*[tiab] OR synthes*[tiab]) AND (search*[tiab] OR database*[tiab] OR data-base*[tiab]))) OR metasynthes*[tiab] OR meta-synthes*[tiab]) NOT ("Comment" [Publication Type] OR "Letter" [Publication Type] OR "Editorial" [Publication Type] OR (("Animals"[Mesh] OR "Models, Animal"[Mesh]) NOT "Humans"[Mesh])) |
494,686 |
|
#5 |
Search: #4 NOT ("Comment" [Publication Type] OR "Letter" [Publication Type] OR "Editorial" [Publication Type] OR (("Animals"[Mesh] OR "Models, Animal"[Mesh]) NOT "Humans"[Mesh])) |
5,030 |
|
#4 |
Search: #1 AND #2 Filters: from 1995 - 2021 |
5,636 |
|
#3 |
Search: #1 AND #2 |
7,614 |
|
#2 |
Search: "Antimalarials"[Mesh] OR "Antimalarials" [Pharmacological Action] OR antimalaria*:ti,ab,kw OR "azathioprine"[Mesh] OR "arathioprin"[tiab] OR "arathioprine"[tiab] OR "aza-q"[tiab] OR "azafalk"[tiab] OR "azahexal"[tiab] OR "azamedac"[tiab] OR "azamun"[tiab] OR "azamune"[tiab] OR "azanin"[tiab] OR "azapin"[tiab] OR "azapress"[tiab] OR "azaprine"[tiab] OR "azarex"[tiab] OR "azasan"[tiab] OR "azathiodura"[tiab] OR "azathiopine"[tiab] OR "azathioprim"[tiab] OR "azathioprin"[tiab] OR "azathioprine"[tiab] OR "azathiopurine"[tiab] OR "azathropsin"[tiab] OR "azatioprina"[tiab] OR "azatox"[tiab] OR "azatrilem"[tiab] OR "azopi"[tiab] OR "azoran"[tiab] OR "azothioprin"[tiab] OR "azothioprine"[tiab] OR "bw 57 322"[tiab] OR "bw 57-322"[tiab] OR "bw 57322"[tiab] OR "bw57-322"[tiab] OR "bw57322"[tiab] OR "colinsan"[tiab] OR "immuran"[tiab] OR "immurel"[tiab] OR "immuthera"[tiab] OR "imunen"[tiab] OR "imuprin"[tiab] OR "imuran"[tiab] OR "imurane"[tiab] OR "imurek"[tiab] OR "imurel"[tiab] OR "imuren"[tiab] OR "methylnitroimidazolylmercaptopurine"[tiab] OR "nsc 39084"[tiab] OR "nsc39084"[tiab] OR "thioazeprine"[tiab] OR "thioprine"[tiab] OR "transimune"[tiab] OR "zytrim"[tiab] OR "methotrexate"[Mesh] OR "4 amino 10 methylfolic acid"[tiab] OR "4 amino 10 methylpteroylglutamic acid"[tiab] OR "4 amino n10 methylpteroylglutamic acid"[tiab] OR "abitrexate"[tiab] OR "amethopterin"[tiab] OR "amethopterine"[tiab] OR "ametopterine"[tiab] OR "antifolan"[tiab] OR "biotrexate"[tiab] OR "canceren"[tiab] OR "cl 14377"[tiab] OR "cl14377"[tiab] OR "emtexate"[tiab] OR "emthexat"[tiab] OR "emthexate"[tiab] OR "emtrexate"[tiab] OR "enthexate"[tiab] OR "farmitrexat"[tiab] OR "farmitrexate"[tiab] OR "farmotrex"[tiab] OR "folex"[tiab] OR "ifamet"[tiab] OR "imeth"[tiab] OR "intradose mtx"[tiab] OR "jylamvo"[tiab] OR "lantarel"[tiab] OR "ledertrexate"[tiab] OR "maxtrex"[tiab] OR "metex"[tiab] OR "methoblastin"[tiab] OR "methohexate"[tiab] OR "methotrate"[tiab] OR "methotrexat"[tiab] OR "methotrexate"[tiab] OR "methotrexato"[tiab] OR "methoxtrexate"[tiab] OR "methrotrexate"[tiab] OR "methylaminopterin"[tiab] OR "methylaminopterine"[tiab] OR "meticil"[tiab] OR "metoject"[tiab] OR "metothrexate"[tiab] OR "metotrexat"[tiab] OR "metotrexate"[tiab] OR "metotrexin"[tiab] OR "metrex"[tiab] OR "mexate"[tiab] OR "mexate-aq"[tiab] OR "mpi 5004"[tiab] OR "mpi5004"[tiab] OR "neotrexate"[tiab] OR "nordimet"[tiab] OR "novatrex"[tiab] OR "nsc 740"[tiab] OR "nsc740"[tiab] OR "otrexup"[tiab] OR "rasuvo"[tiab] OR "reditrex"[tiab] OR "reumatrex"[tiab] OR "rheumatrex"[tiab] OR "rheumatrex dose pack"[tiab] OR "texate"[tiab] OR "texorate"[tiab] OR "trexall"[tiab] OR "xaken"[tiab] OR "xatmep"[tiab] OR "zexate"[tiab] OR "mtx"[tiab] OR "Cyclosporins"[Mesh] OR "Cyclosporine"[Mesh] OR "adi 628"[tiab] OR "adi628"[tiab] OR "cequa"[tiab] OR "cgc 1072"[tiab] OR "cgc1072"[tiab] OR "ciclomulsion"[tiab] OR "cicloral"[tiab] OR "ciclosporin"[tiab] OR "ciclosporine"[tiab] OR "cipol"[tiab] OR "consupren"[tiab] OR "cyclasol"[tiab] OR "cyclokat"[tiab] OR "cyclosporin"[tiab] OR "cyclosporine"[tiab] OR "de 076"[tiab] OR "de076"[tiab] OR "deximune"[tiab] OR "equoral"[tiab] OR "gengraf"[tiab] OR "ikervis"[tiab] OR "iminoral"[tiab] OR "implanta"[tiab] OR "imusporin"[tiab] OR "lx 201"[tiab] OR "lx201"[tiab] OR "mc2 03"[tiab] OR "mc203"[tiab] OR "mtd 202"[tiab] OR "mtd202"[tiab] OR "neoral"[tiab] OR "neoral-sandimmun"[tiab] OR "nm 0133"[tiab] OR "nm 133"[tiab] OR "nm0133"[tiab] OR "nm133"[tiab] OR "nova 22007"[tiab] OR "nova22007"[tiab] OR "ol 27400"[tiab] OR "ol27400"[tiab] OR "olo 400"[tiab] OR "olo500"[tiab] OR "opph 088"[tiab] OR "opph088"[tiab] OR "opsisporin"[tiab] OR "otx 101"[tiab] OR "otx101"[tiab] OR "p 3072"[tiab] OR "p3072"[tiab] OR "padciclo"[tiab] OR "papilock"[tiab] OR "pulminiq"[tiab] OR "restasis"[tiab] OR "restaysis"[tiab] OR "sanciclo"[tiab] OR "sandimmun"[tiab] OR "sandimmune"[tiab] OR "sandimun"[tiab] OR "sandimune"[tiab] OR "sang 35"[tiab] OR "sang35"[tiab] OR "sangcya"[tiab] OR "sp 14019"[tiab] OR "sp14019"[tiab] OR "sti 0529"[tiab] OR "sti0529"[tiab] OR "t 1580"[tiab] OR "t1580"[tiab] OR "vekacia"[tiab] OR "verkazia"[tiab] OR "voclosporin" [Supplementary Concept] OR "isa 247"[tiab] OR "isa247"[tiab] OR "luveniq"[tiab] OR "lx 211"[tiab] OR "lx211"[tiab] OR "voclosporin"[tiab] OR "Leflunomide"[Mesh] OR "arabloc"[tiab] OR "arava"[tiab] OR "hwa 486"[tiab] OR "hwa486"[tiab] OR "leflunomide"[tiab] OR "repso"[tiab] OR "rs 34821"[tiab] OR "rs34821"[tiab] OR "su 101"[tiab] OR "su101"[tiab] OR "Cyclophosphamide"[Mesh] OR "alkyroxan"[tiab] OR "b 518"[tiab] OR "b518"[tiab] OR "b518 asta"[tiab] OR "carloxan"[tiab] OR "ciclofosfamida"[tiab] OR "ciclolen"[tiab] OR "cicloxal"[tiab] OR "clafen"[tiab] OR "cyclo-cell"[tiab] OR "cycloblastin"[tiab] OR "cycloblastine"[tiab] OR "cyclofos amide"[tiab] OR "cyclofosfamid"[tiab] OR "cyclofosfamide"[tiab] OR "cyclophar"[tiab] OR "cyclophosphamid"[tiab] OR "cyclophosphamide"[tiab] OR "cyclophosphamides"[tiab] OR "cyclophosphan"[tiab] OR "cyclophosphane"[tiab] OR "cyclostin"[tiab] OR "cyclostin n"[tiab] OR "cycloxan"[tiab] OR "cyphos"[tiab] OR "cytophosphan"[tiab] OR "cytophosphane"[tiab] OR "cytoxan"[tiab] OR "endocyclo phosphate"[tiab] OR "endoxan"[tiab] OR "endoxan asta"[tiab] OR "endoxan-asta"[tiab] OR "endoxana"[tiab] OR "endoxon-asta"[tiab] OR "enduxan"[tiab] OR "genoxal"[tiab] OR "ledoxan"[tiab] OR "ledoxina"[tiab] OR "lyophilized cytoxan"[tiab] OR "mitoxan"[tiab] OR "neosan"[tiab] OR "neosar"[tiab] OR "noristan"[tiab] OR "nsc 26271"[tiab] OR "nsc 2671"[tiab] OR "procytox"[tiab] OR "procytoxide"[tiab] OR "semdoxan"[tiab] OR "sendoxan"[tiab] OR "syklofosfamid"[tiab] OR "Mycophenolic Acid"[Mesh] OR "cell cept"[tiab] OR "cellcept"[tiab] OR "cellmune"[tiab] OR "cellsept"[tiab] OR "munoloc"[tiab] OR "myclausen"[tiab] OR "mycophenolic acid 2 morpholinoethyl ester"[tiab] OR "mycophenolic acid mofetil"[tiab] OR "myfenax"[tiab] OR "rs 61443"[tiab] OR "rs61443"[tiab] OR "mycophenolate mofetil"[tiab] OR "erl 080"[tiab] OR "erl080"[tiab] OR "erl080a"[tiab] OR "melbex"[tiab] OR "mycophenolate"[tiab] OR "mycophenolic acid"[tiab] OR "myfortic"[tiab] OR "nsc 129185"[tiab] OR "nsc129185"[tiab] OR "sodium mycophenolate"[tiab] |
284,053 |
|
#1 |
Search: "Lupus Erythematosus, Systemic"[Mesh:NoExp]OR "erythematodes visceralis"[tiab] OR "lupovisceritis"[tiab] OR "libman sacks disease"[tiab] OR "s.l.e."[tiab] OR lupus[tiab] |
89,232 |
BDMARDS, biologicals, TNF inhibitors, NON TNF
Embase, 5-12-2020
|
No. |
Query |
Results |
|
#23 |
#22 NOT #21 = RCT |
1972 |
|
#22 |
#18 AND #20 |
2169 |
|
#21 |
#18 AND #19 = SR |
384 |
|
#20 |
('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it |
2488699 |
|
#19 |
('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
534524 |
|
#18 |
#17 NOT (('adolescent'/exp OR 'child'/exp OR adolescent*:ti,ab OR child*:ti,ab OR schoolchild*:ti,ab OR infant*:ti,ab OR girl*:ti,ab OR boy*:ti,ab OR teen:ti,ab OR teens:ti,ab OR teenager*:ti,ab OR youth*:ti,ab OR pediatr*:ti,ab OR paediatr*:ti,ab OR puber*:ti,ab) NOT ('adult'/exp OR 'aged'/exp OR 'middle aged'/exp OR adult*:ti,ab OR man:ti,ab OR men:ti,ab OR woman:ti,ab OR women:ti,ab)) |
7441 |
|
#17 |
#16 AND [1-1-1995]/sd NOT ('conference abstract'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
7967 |
|
#16 |
#14 AND #15 |
12212 |
|
#15 |
'systemic lupus erythematosus'/exp OR 'erythematodes visceralis':ti,ab,kw OR 'lupovisceritis':ti,ab,kw OR 'libman sacks disease':ti,ab,kw OR 's.l.e.':ti,ab,kw OR 'sle':ti,ab,kw OR lupus:ti,ab,kw |
143148 |
|
#14 |
#11 OR #12 OR #13 |
338816 |
|
#13 |
'anakinra'/exp OR 'anakinra':ti,ab,tn,dn OR 'kineret':ti,ab,tn,dn OR 'recombinant interleukin 1 receptor antagonist':ti,ab,tn,dn OR 'recombinant interleukin 1 receptor blocker':ti,ab,tn,dn OR 'recombinant interleukin 1 receptor blocking agent':ti,ab,tn,dn OR 'rituximab'/exp OR 'abp 798':ti,ab,tn,dn OR 'abp798':ti,ab,tn,dn OR 'blitzima':ti,ab,tn,dn OR 'ct p10':ti,ab,tn,dn OR 'ctp10':ti,ab,tn,dn OR 'gp 2013':ti,ab,tn,dn OR 'gp2013':ti,ab,tn,dn OR 'hlx 01':ti,ab,tn,dn OR 'hlx01':ti,ab,tn,dn OR 'idec 102':ti,ab,tn,dn OR 'idec c2b8':ti,ab,tn,dn OR 'idec102':ti,ab,tn,dn OR 'idecc2b8':ti,ab,tn,dn OR 'mabthera':ti,ab,tn,dn OR 'mk 8808':ti,ab,tn,dn OR 'mk8808':ti,ab,tn,dn OR 'monoclonal antibody idec c2b8':ti,ab,tn,dn OR 'pf 05280586':ti,ab,tn,dn OR 'pf 5280586':ti,ab,tn,dn OR 'pf05280586':ti,ab,tn,dn OR 'pf5280586':ti,ab,tn,dn OR 'r 105':ti,ab,tn,dn OR 'r105':ti,ab,tn,dn OR 'reditux':ti,ab,tn,dn OR 'rg 105':ti,ab,tn,dn OR 'rg105':ti,ab,tn,dn OR 'ritemvia':ti,ab,tn,dn OR 'ritumax':ti,ab,tn,dn OR 'rituxan':ti,ab,tn,dn OR 'rituximab':ti,ab,tn,dn OR 'rituxin':ti,ab,tn,dn OR 'rituzena':ti,ab,tn,dn OR 'rixathon':ti,ab,tn,dn OR 'riximyo':ti,ab,tn,dn OR 'ro 452294':ti,ab,tn,dn OR 'ro452294':ti,ab,tn,dn OR 'ruxience':ti,ab,tn,dn OR 'truxima':ti,ab,tn,dn OR 'tuxella':ti,ab,tn,dn OR 'abatacept'/exp OR 'ctla4 ig':ti,ab,tn,dn OR 'abatacept':ti,ab,tn,dn OR 'bms 188667':ti,ab,tn,dn OR 'bms188667':ti,ab,tn,dn OR 'orencia':ti,ab,tn,dn OR 'ctla4ig':ti,ab,tn,dn OR 'tocilizumab'/exp OR 'actemra':ti,ab,tn,dn OR 'atlizumab':ti,ab,tn,dn OR 'lusinex':ti,ab,tn,dn OR 'r 1569':ti,ab,tn,dn OR 'r1569':ti,ab,tn,dn OR 'roactemra':ti,ab,tn,dn OR 'tocilizumab':ti,ab,tn,dn OR 'tofacitinib'/exp OR 'cp 690 550':ti,ab,tn,dn OR 'cp 690550':ti,ab,tn,dn OR 'cp 690550 10':ti,ab,tn,dn OR 'cp690 550':ti,ab,tn,dn OR 'cp690550':ti,ab,tn,dn OR 'cp690550 10':ti,ab,tn,dn OR 'tasocitinib':ti,ab,tn,dn OR 'tofacitinib':ti,ab,tn,dn OR 'tofacitinib citrate':ti,ab,tn,dn OR 'xeljanz':ti,ab,tn,dn OR 'baricitinib'/exp OR 'baricitinib':ti,ab,tn,dn OR 'incb 028050':ti,ab,tn,dn OR 'incb 28050':ti,ab,tn,dn OR 'incb028050':ti,ab,tn,dn OR 'incb28050':ti,ab,tn,dn OR 'ly 3009104':ti,ab,tn,dn OR 'ly3009104':ti,ab,tn,dn OR 'olumiant':ti,ab,tn,dn OR 'tacrolimus'/exp OR 'advagraf':ti,ab,tn,dn OR 'astagraf':ti,ab,tn,dn OR 'envarsus':ti,ab,tn,dn OR 'fk 506':ti,ab,tn,dn OR 'fk-506':ti,ab,tn,dn OR 'fk506':ti,ab,tn,dn OR 'fr 900506':ti,ab,tn,dn OR 'fr900506':ti,ab,tn,dn OR 'fujimycin':ti,ab,tn,dn OR 'hecoria':ti,ab,tn,dn OR 'modigraf':ti,ab,tn,dn OR 'mustopic oint':ti,ab,tn,dn OR 'prograf':ti,ab,tn,dn OR 'prograft':ti,ab,tn,dn OR 'protopic':ti,ab,tn,dn OR 'protopy':ti,ab,tn,dn OR 'tacforius':ti,ab,tn,dn OR 'tacrolimus':ti,ab,tn,dn OR 'tsukubaenolide':ti,ab,tn,dn OR 'rapamycin'/exp OR 'ay 22989':ti,ab,tn,dn OR 'ay22989':ti,ab,tn,dn OR 'de 109':ti,ab,tn,dn OR 'de109':ti,ab,tn,dn OR 'opsiria':ti,ab,tn,dn OR 'perceiva':ti,ab,tn,dn OR 'rapammune':ti,ab,tn,dn OR 'rapamune':ti,ab,tn,dn OR 'rapamycin':ti,ab,tn,dn OR 'sirolimus':ti,ab,tn,dn OR 'wy 090217':ti,ab,tn,dn OR 'wy090217':ti,ab,tn,dn OR 'efavaleukin alfa'/exp OR 'amg 592':ti,ab,tn,dn OR 'amg592':ti,ab,tn,dn OR 'efavaleukin alfa':ti,ab,tn,dn OR 'efavaleukin alpha':ti,ab,tn,dn OR efavaleukin:ti,ab,tn,dn OR 'intravenous immunoglobulin'/exp OR 'intravenous immunoglobulin*':ti,ab,kw OR (('interleukin 2'/exp OR 't cell growth factor':ti,ab,kw OR 't lymphocyte growth factor':ti,ab,kw OR 't lymphocyte growth factor 2':ti,ab,kw OR 'bioleukin':ti,ab,kw OR 'il 2':ti,ab OR 'interleukin 2':ti,ab,kw OR 'interleukin ii':ti,ab,kw OR 'interleukin-2':ti,ab,kw OR 'lymphocult t hp':ti,ab,kw OR 'lymphocyte mitogenic factor':ti,ab,kw OR 't cell growth factor 2':ti,ab,kw) AND ('low drug dose'/exp OR 'drug low dose':ti,ab,kw OR 'low dosage':ti,ab,kw OR 'low dose':ti,ab,kw OR 'low drug dosage':ti,ab,kw OR 'low drug dose':ti,ab,kw)) |
268676 |
|
#12 |
'etanercept'/exp OR 'avent':ti,ab OR 'benepali':ti,ab OR 'brenzys':ti,ab OR 'chs 0214':ti,ab OR 'chs0214':ti,ab OR 'embrel':ti,ab OR 'enbrel':ti,ab OR 'enerceptan':ti,ab OR 'enia 11':ti,ab OR 'enia11':ti,ab OR 'erelzi':ti,ab OR 'etanercept':ti,ab OR 'etanercept-ykro':ti,ab OR 'gp 2015':ti,ab OR 'gp 2015c':ti,ab OR 'gp2015':ti,ab OR 'gp2015c':ti,ab OR 'hd 203':ti,ab OR 'hd203':ti,ab OR 'infinitam':ti,ab OR 'lbec 0101':ti,ab OR 'lbec0101':ti,ab OR 'lifmior':ti,ab OR 'nepexto':ti,ab OR 'opinercept':ti,ab OR 'sb 4':ti,ab OR 'sb4':ti,ab OR 'tnr 001':ti,ab OR 'tnr001':ti,ab OR 'tunex':ti,ab OR 'ylb 113':ti,ab OR 'ylb113':ti,ab OR 'infliximab'/exp OR 'abp 710':ti,ab,tn,dn OR 'abp710':ti,ab,tn,dn OR 'avakine':ti,ab,tn,dn OR 'avsola':ti,ab,tn,dn OR 'flixabi':ti,ab,tn,dn OR 'gp 1111':ti,ab,tn,dn OR 'gp1111':ti,ab,tn,dn OR 'inflectra':ti,ab,tn,dn OR 'infliximab':ti,ab,tn,dn OR 'ixifi':ti,ab,tn,dn OR 'pf 06438179':ti,ab,tn,dn OR 'pf 6438179':ti,ab,tn,dn OR 'pf06438179':ti,ab,tn,dn OR 'pf6438179':ti,ab,tn,dn OR 'remicade':ti,ab,tn,dn OR 'remsima':ti,ab,tn,dn OR 'renflexis':ti,ab,tn,dn OR 'revellex':ti,ab,tn,dn OR 'ta 650':ti,ab,tn,dn OR 'ta650':ti,ab,tn,dn OR 'zessly':ti,ab,tn,dn OR 'adalimumab'/exp OR 'abp 501':ti,ab,tn,dn OR 'abp501':ti,ab,tn,dn OR 'abrilada':ti,ab,tn,dn OR 'abt d2e7':ti,ab,tn,dn OR 'abtd2e7':ti,ab,tn,dn OR 'adalimumab':ti,ab,tn,dn OR 'adaly':ti,ab,tn,dn OR 'amgevita':ti,ab,tn,dn OR 'amjevita':ti,ab,tn,dn OR 'amsparity':ti,ab,tn,dn OR 'avt 02':ti,ab,tn,dn OR 'avt02':ti,ab,tn,dn OR 'bat 1406':ti,ab,tn,dn OR 'bat1406':ti,ab,tn,dn OR 'bax 2923':ti,ab,tn,dn OR 'bax 923':ti,ab,tn,dn OR 'bax2923':ti,ab,tn,dn OR 'bax923':ti,ab,tn,dn OR 'bi 695501':ti,ab,tn,dn OR 'bi695501':ti,ab,tn,dn OR 'chs 1420':ti,ab,tn,dn OR 'chs1420':ti,ab,tn,dn OR 'cinnora':ti,ab,tn,dn OR 'ct p17':ti,ab,tn,dn OR 'ctp17':ti,ab,tn,dn OR 'cyltezo':ti,ab,tn,dn OR 'da 3113':ti,ab,tn,dn OR 'da3113':ti,ab,tn,dn OR 'dmb 3113':ti,ab,tn,dn OR 'dmb3113':ti,ab,tn,dn OR 'exemptia':ti,ab,tn,dn OR 'fkb 327':ti,ab,tn,dn OR 'fkb327':ti,ab,tn,dn OR 'fyzoclad':ti,ab,tn,dn OR 'gp 2017':ti,ab,tn,dn OR 'gp2017':ti,ab,tn,dn OR 'hadlima':ti,ab,tn,dn OR 'halimatoz':ti,ab,tn,dn OR 'hefiya':ti,ab,tn,dn OR 'hlx 03':ti,ab,tn,dn OR 'hlx03':ti,ab,tn,dn OR 'hulio':ti,ab,tn,dn OR 'humira':ti,ab,tn,dn OR 'hyrimoz':ti,ab,tn,dn OR 'ibi 303':ti,ab,tn,dn OR 'ibi303':ti,ab,tn,dn OR 'idacio':ti,ab,tn,dn OR 'imraldi':ti,ab,tn,dn OR 'kromeya':ti,ab,tn,dn OR 'lu 200134':ti,ab,tn,dn OR 'lu200134':ti,ab,tn,dn OR 'm 923':ti,ab,tn,dn OR 'm923':ti,ab,tn,dn OR 'mabura':ti,ab,tn,dn OR 'monoclonal antibody d2e7':ti,ab,tn,dn OR 'msb 11022':ti,ab,tn,dn OR 'msb11022':ti,ab,tn,dn OR 'ons 3010':ti,ab,tn,dn OR 'ons3010':ti,ab,tn,dn OR 'pf 06410293':ti,ab,tn,dn OR 'pf 6410293':ti,ab,tn,dn OR 'pf06410293':ti,ab,tn,dn OR 'pf6410293':ti,ab,tn,dn OR 'raheara':ti,ab,tn,dn OR 'sb 5':ti,ab,tn,dn OR 'sb5':ti,ab,tn,dn OR 'solymbic':ti,ab,tn,dn OR 'trudexa':ti,ab,tn,dn OR 'zrc 3197':ti,ab,tn,dn OR 'zrc3197':ti,ab,tn,dn OR 'golimumab'/exp OR 'cnto 148':ti,ab,tn,dn OR 'cnto148':ti,ab,tn,dn OR 'golimumab':ti,ab,tn,dn OR 'simponi':ti,ab,tn,dn OR 'certolizumab pegol'/exp OR 'cdp 870':ti,ab,tn,dn OR 'cdp870':ti,ab,tn,dn OR 'cimzia':ti,ab,tn,dn OR 'pha 738144':ti,ab,tn,dn OR 'pha738144':ti,ab,tn,dn OR certolizumab:ti,ab,tn,dn |
78678 |
|
#11 |
'belimumab'/exp OR 'belimumab':ti,ab,kw OR 'benlysta':ti,ab,kw OR 'lymphostat b':ti,ab,kw OR 'blisibimod'/exp OR 'blisibimod':ti,ab,kw OR 'sifalimumab'/exp OR 'medi 545':ti,ab,kw OR 'medi545':ti,ab,kw OR 'sifalimumab':ti,ab,kw OR 'ustekinumab'/exp OR 'cnto 1275':ti,ab OR 'cnto1275':ti,ab,kw OR 'stelara':ti,ab,kw OR 'ustekinumab':ti,ab,kw OR 'anifrolumab'/exp OR 'anifrolumab':ti,ab,kw OR 'medi 546':ti,ab,kw OR 'medi546':ti,ab,kw OR 'canakinumab'/exp OR 'acz 885':ti,ab,kw OR 'acz885':ti,ab,kw OR 'canakinumab':ti,ab,kw OR 'ilaris':ti,ab,kw OR 'secukinumab'/exp OR 'ain 457':ti,ab,kw OR 'ain457':ti,ab,kw OR 'cosentyx':ti,ab,kw OR 'secukinumab':ti,ab,kw OR 'ixekizumab'/exp OR 'ixekizumab':ti,ab,kw OR 'ly 2439821':ti,ab,kw OR 'ly2439821':ti,ab,kw OR 'taltz':ti,ab,kw OR 'ocrelizumab'/exp OR 'monoclonal antibody 2h7':ti,ab,kw OR 'monoclonal antibody pro 70769':ti,ab,kw OR 'monoclonal antibody pro70769':ti,ab,kw OR 'ocrelizumab':ti,ab,kw OR 'ocrevus':ti,ab,kw OR 'pro 70769':ti,ab,kw OR 'pro70769':ti,ab,kw OR 'rhumab 2h7':ti,ab,kw OR 'obinutuzumab'/exp OR 'afutuzumab':ti,ab,kw OR 'ga 101':ti,ab,kw OR 'ga101':ti,ab,kw OR 'gazyva*':ti,ab,kw OR 'obinutuzumab':ti,ab,kw OR 'r 7159':ti,ab,kw OR 'r7159':ti,ab,kw OR 'rg 7159':ti,ab,kw OR 'rg7159':ti,ab,kw OR 'ro 5072759':ti,ab,kw OR 'ro5072759':ti,ab,kw OR 'veltuzumab'/exp OR 'ha 20':ti,ab,kw OR 'ha20':ti,ab,kw OR 'immu 106':ti,ab,kw OR 'immu106':ti,ab,kw OR 'veltuzumab':ti,ab,kw OR 'ofatumumab'/exp OR 'humax-cd20':ti,ab,kw OR 'humaxcd20':ti,ab,kw OR 'arzerra':ti,ab,kw OR 'gsk 1841157':ti,ab,kw OR 'gsk1841157':ti,ab,kw OR 'humac cd20':ti,ab,kw OR 'ofatumumab':ti,ab,kw OR 'omb 157':ti,ab,kw OR 'omb157':ti,ab,kw OR 'apremilast'/exp OR 'apremilast':ti,ab,kw OR 'cc 10004':ti,ab,kw OR 'cc10004':ti,ab,kw OR 'otezla':ti,ab,kw OR 'forigerimod'/exp OR 'cep 33457':ti,ab,kw OR 'cep33457':ti,ab,kw OR 'forigerimod':ti,ab,kw OR 'ipp 201101':ti,ab,kw OR 'ipp201101':ti,ab,kw OR 'lupuzor':ti,ab,kw OR 'filgotinib'/exp OR 'filgotinib':ti,ab,kw OR 'g 146034':ti,ab,kw OR 'g146034':ti,ab,kw OR 'glpg 0634':ti,ab,kw OR 'glpg0634':ti,ab,kw OR 'gs 6034':ti,ab,kw OR 'gs6034':ti,ab,kw |
24266 |
PubMed, 5-12-2020
|
Search |
Query |
Results |
|
#24 |
Search: #23 NOT #22 = RCT |
189 |
|
#22 |
Search: #21 AND #6 |
209 |
|
#23 |
Search: #21 AND #7 = SR |
231 |
|
#7 |
Search: ((random*[tiab] AND (controlled[tiab] OR control[tiab] OR placebo[tiab] OR versus[tiab] OR vs[tiab] OR group[tiab] OR groups[tiab] OR comparison[tiab] OR compared[tiab] OR arm[tiab] OR arms[tiab] OR crossover[tiab] OR cross-over[tiab]) AND (trial[tiab] OR study[tiab])) OR ((single[tiab] OR double[tiab] OR triple[tiab]) AND (masked[tiab] OR blind*[tiab]))) |
763,584 |
|
#6 |
Search: ("Meta-Analysis" [Publication Type] OR "Meta-Analysis as Topic"[Mesh] OR metaanaly*[tiab] OR meta-analy*[tiab] or metanaly*[tiab] OR "Systematic Review" [Publication Type] OR systematic[sb] OR "Cochrane Database Syst Rev"[Journal] or prisma[tiab] OR preferred reporting items[tiab] OR prospero[tiab] OR ((systemati*[ti] OR scoping[ti] OR umbrella[ti] OR structured literature[ti]) AND (review*[ti] OR overview*[ti])) OR systematic review*[tiab] OR scoping review*[tiab] OR umbrella review*[tiab] OR structured literature review*[tiab] OR systematic qualitative review*[tiab] OR systematic quantitative review*[tiab] OR systematic search and review[tiab] OR systematized review[tiab] OR systematised review[tiab] OR systemic review[tiab] OR systematic literature review*[tiab] OR systematic integrative literature review*[tiab] OR systematically review*[tiab] OR scoping literature review*[tiab] OR systematic critical review[tiab] OR systematic integrative review*[tiab] OR systematic evidence review[tiab] OR Systematic integrative literature review*[tiab] OR Systematic mixed studies review*[tiab] OR Systematized literature review*[tiab] OR Systematic overview*[tiab] OR Systematic narrative review*[tiab] OR ((systemati*[tiab] OR literature[tiab] OR database*[tiab] OR data-base*[tiab] OR structured[tiab] OR comprehensive*[tiab] OR systemic*[tiab]) AND search*[tiab]) OR (Literature[ti] AND review[ti] AND (database*[tiab] OR data-base*[tiab] OR search*[tiab])) OR ((data extraction[tiab] OR data source*[tiab]) AND study selection[tiab]) OR (search strategy[tiab] AND selection criteria[tiab]) OR (data source*[tiab] AND data synthesis[tiab]) OR medline[tiab] OR pubmed[tiab] OR embase[tiab] OR Cochrane[tiab] OR ((critical[ti] OR rapid[ti]) AND (review*[ti] OR overview*[ti] OR synthes*[ti])) OR (((critical*[tiab] OR rapid*[tiab]) AND (review*[tiab] OR overview*[tiab] OR synthes*[tiab]) AND (search*[tiab] OR database*[tiab] OR data-base*[tiab]))) OR metasynthes*[tiab] OR meta-synthes*[tiab]) NOT ("Comment" [Publication Type] OR "Letter" [Publication Type] OR "Editorial" [Publication Type] OR (("Animals"[Mesh] OR "Models, Animal"[Mesh]) NOT "Humans"[Mesh])) |
494,686 |
|
#21 |
Search: #20 NOT (("Adolescent"[Mesh] OR "Child"[Mesh] OR "Infant"[Mesh] OR adolescen*[tiab] OR child*[tiab] OR schoolchild*[tiab] OR infant*[tiab] OR girl*[tiab] OR boy*[tiab] OR teen[tiab] OR teens[tiab] OR teenager*[tiab] OR youth*[tiab] OR pediatr*[tiab] OR paediatr*[tiab] OR puber*[tiab]) NOT ("Adult"[Mesh] OR adult*[tiab] OR man[tiab] OR men[tiab] OR woman[tiab] OR women[tiab])) |
3,165 |
|
#20 |
Search: #18 AND #1 Filters: from 1995 - 2021 |
3,395 |
|
#19 |
Search: #18 AND #1 |
3,485 |
|
#1 |
Search: "Lupus Erythematosus, Systemic"[Mesh:NoExp]OR "erythematodes visceralis"[tiab] OR "lupovisceritis"[tiab] OR "libman sacks disease"[tiab] OR "s.l.e."[tiab] OR lupus[tiab] |
89,232 |
|
#18 |
Search: #14 OR #15 OR #17 |
153,344 |
|
#17 |
Search: "canakinumab" [Supplementary Concept] OR "secukinumab" [Supplementary Concept] OR "ixekizumab" [Supplementary Concept] OR "ocrelizumab" [Supplementary Concept] OR "obinutuzumab" [Supplementary Concept] OR "veltuzumab" [Supplementary Concept] OR "ofatumumab" [Supplementary Concept] OR "apremilast" [Supplementary Concept] OR "GLPG0634" [Supplementary Concept] OR "belimumab" [Supplementary Concept] OR "belimumab"[tiab] OR "benlysta"[tiab] OR "lymphostat b"[tiab] OR "AMG623 peptibody" [Supplementary Concept] OR "blisibimod"[tiab] OR "sifalimumab" [Supplementary Concept] OR "medi 545"[tiab] OR "medi545"[tiab] OR "sifalimumab"[tiab] OR "Ustekinumab"[Mesh] OR "cnto 1275":ti,ab OR "cnto1275"[tiab] OR "stelara"[tiab] OR "ustekinumab"[tiab] OR "anifrolumab" [Supplementary Concept] OR "anifrolumab"[tiab] OR "medi 546"[tiab] OR "medi546"[tiab] OR "acz 885"[tiab] OR "acz885"[tiab] OR "canakinumab"[tiab] OR "ilaris"[tiab] OR "ain 457"[tiab] OR "ain457"[tiab] OR "cosentyx"[tiab] OR "secukinumab"[tiab] OR "ixekizumab"[tiab] OR "ly 2439821"[tiab] OR "ly2439821"[tiab] OR "taltz"[tiab] OR "monoclonal antibody 2h7"[tiab] OR "monoclonal antibody pro 70769"[tiab] OR "monoclonal antibody pro70769"[tiab] OR "ocrelizumab"[tiab] OR "ocrevus"[tiab] OR "pro 70769"[tiab] OR "pro70769"[tiab] OR "rhumab 2h7"[tiab] OR "afutuzumab"[tiab] OR "ga 101"[tiab] OR "ga101"[tiab] OR "gazyva*"[tiab] OR "obinutuzumab"[tiab] OR "r 7159"[tiab] OR "r7159"[tiab] OR "rg 7159"[tiab] OR "rg7159"[tiab] OR "ro 5072759"[tiab] OR "ro5072759"[tiab] OR "ha 20"[tiab] OR "ha20"[tiab] OR "immu 106"[tiab] OR "immu106"[tiab] OR "veltuzumab"[tiab] OR "humax-cd20"[tiab] OR "humaxcd20"[tiab] OR "arzerra"[tiab] OR "gsk 1841157"[tiab] OR "gsk1841157"[tiab] OR "humac cd20"[tiab] OR "ofatumumab"[tiab] OR "omb 157"[tiab] OR "omb157"[tiab] OR "apremilast"[tiab] OR "cc 10004"[tiab] OR "cc10004"[tiab] OR "otezla"[tiab] OR "cep 33457"[tiab] OR "cep33457"[tiab] OR "forigerimod"[tiab] OR "ipp 201101"[tiab] OR "ipp201101"[tiab] OR "lupuzor"[tiab] OR "filgotinib"[tiab] OR "g 146034"[tiab] OR "g146034"[tiab] OR "glpg 0634"[tiab] OR "glpg0634"[tiab] OR "gs 6034"[tiab] OR "gs6034"[tiab] |
7,002 |
|
#15 |
Search: "Rituximab"[Mesh] OR "Interleukin 1 Receptor Antagonist Protein"[Mesh] OR "Abatacept"[Mesh] OR "tocilizumab" [Supplementary Concept] OR "tofacitinib" [Supplementary Concept] OR "baricitinib" [Supplementary Concept] OR "Tacrolimus"[Mesh] OR "Sirolimus"[Mesh] OR "Immunoglobulins, Intravenous"[Mesh] OR "anakinra"[tiab] OR "kineret"[tiab] OR "recombinant interleukin 1 receptor antagonist"[tiab] OR "recombinant interleukin 1 receptor blocker"[tiab] OR "recombinant interleukin 1 receptor blocking agent"[tiab] OR "abp 798"[tiab] OR "abp798"[tiab] OR "blitzima"[tiab] OR "ct p10"[tiab] OR "ctp10"[tiab] OR "gp 2013"[tiab] OR "gp2013"[tiab] OR "hlx 01"[tiab] OR "hlx01"[tiab] OR "idec 102"[tiab] OR "idec c2b8"[tiab] OR "idec102"[tiab] OR "idecc2b8"[tiab] OR "mabthera"[tiab] OR "mk 8808"[tiab] OR "mk8808"[tiab] OR "monoclonal antibody idec c2b8"[tiab] OR "pf 05280586"[tiab] OR "pf 5280586"[tiab] OR "pf05280586"[tiab] OR "pf5280586"[tiab] OR "r 105"[tiab] OR "r105"[tiab] OR "reditux"[tiab] OR "rg 105"[tiab] OR "rg105"[tiab] OR "ritemvia"[tiab] OR "ritumax"[tiab] OR "rituxan"[tiab] OR "rituximab"[tiab] OR "rituxin"[tiab] OR "rituzena"[tiab] OR "rixathon"[tiab] OR "riximyo"[tiab] OR "ro 452294"[tiab] OR "ro452294"[tiab] OR "ruxience"[tiab] OR "truxima"[tiab] OR "tuxella"[tiab] OR "ctla4 ig"[tiab] OR "abatacept"[tiab] OR "bms 188667"[tiab] OR "bms188667"[tiab] OR "orencia"[tiab] OR "ctla4ig"[tiab] OR "actemra"[tiab] OR "atlizumab"[tiab] OR "lusinex"[tiab] OR "r 1569"[tiab] OR "r1569"[tiab] OR "roactemra"[tiab] OR "tocilizumab"[tiab] OR "cp 690 550"[tiab] OR "cp 690550"[tiab] OR "cp 690550 10"[tiab] OR "cp690 550"[tiab] OR "cp690550"[tiab] OR "cp690550 10"[tiab] OR "tasocitinib"[tiab] OR "tofacitinib"[tiab] OR "tofacitinib citrate"[tiab] OR "xeljanz"[tiab] OR "baricitinib"[tiab] OR "incb 028050"[tiab] OR "incb 28050"[tiab] OR "incb028050"[tiab] OR "incb28050"[tiab] OR "ly 3009104"[tiab] OR "ly3009104"[tiab] OR "olumiant"[tiab] OR "advagraf"[tiab] OR "astagraf"[tiab] OR "envarsus"[tiab] OR "fk 506"[tiab] OR "fk-506"[tiab] OR "fk506"[tiab] OR "fr 900506"[tiab] OR "fr900506"[tiab] OR "fujimycin"[tiab] OR "hecoria"[tiab] OR "modigraf"[tiab] OR "mustopic oint"[tiab] OR "prograf"[tiab] OR "prograft"[tiab] OR "protopic"[tiab] OR "protopy"[tiab] OR "tacforius"[tiab] OR "tacrolimus"[tiab] OR "tsukubaenolide"[tiab] OR "ay 22989"[tiab] OR "ay22989"[tiab] OR "de 109"[tiab] OR "de109"[tiab] OR "opsiria"[tiab] OR "perceiva"[tiab] OR "rapammune"[tiab] OR "rapamune"[tiab] OR "rapamycin"[tiab] OR "sirolimus"[tiab] OR "wy 090217"[tiab] OR "wy090217"[tiab] OR "amg 592"[tiab] OR "amg592"[tiab] OR "efavaleukin alfa"[tiab] OR "efavaleukin alpha"[tiab] OR efavaleukin[tiab] OR "intravenous immunoglobulin*"[tiab] OR "t cell growth factor"[tiab] OR "t lymphocyte growth factor"[tiab] OR "t lymphocyte growth factor 2"[tiab] OR "bioleukin"[tiab] OR "il 2":ti,ab OR (("interleukin 2"[tiab] OR "interleukin ii"[tiab] OR "interleukin-2"[tiab] OR "lymphocult t hp"[tiab] OR "lymphocyte mitogenic factor"[tiab] OR "t cell growth factor 2"[tiab]) AND ("drug low dose"[tiab] OR "low dosage"[tiab] OR "low dose"[tiab] OR "low drug dosage"[tiab] OR "low drug dose"[tiab])) |
123,907 |
|
#14 |
Search: "Etanercept"[Mesh] OR "Infliximab"[Mesh] OR "Adalimumab"[Mesh] OR "golimumab" [Supplementary Concept] OR "Certolizumab Pegol"[Mesh] OR 'avent'[tiab] OR 'benepali'[tiab] OR 'brenzys'[tiab] OR 'chs 0214'[tiab] OR 'chs0214'[tiab] OR 'embrel'[tiab] OR 'enbrel'[tiab] OR 'enerceptan'[tiab] OR 'enia 11'[tiab] OR 'enia11'[tiab] OR 'erelzi'[tiab] OR 'etanercept'[tiab] OR 'etanercept-ykro'[tiab] OR 'gp 2015'[tiab] OR 'gp 2015c'[tiab] OR 'gp2015'[tiab] OR 'gp2015c'[tiab] OR 'hd 203'[tiab] OR 'hd203'[tiab] OR 'infinitam'[tiab] OR 'lbec 0101'[tiab] OR 'lbec0101'[tiab] OR 'lifmior'[tiab] OR 'nepexto'[tiab] OR 'opinercept'[tiab] OR 'sb 4'[tiab] OR 'sb4'[tiab] OR 'tnr 001'[tiab] OR 'tnr001'[tiab] OR 'tunex'[tiab] OR 'ylb 113'[tiab] OR 'ylb113'[tiab] OR 'abp 710'[tiab] OR 'abp710'[tiab] OR 'avakine'[tiab] OR 'avsola'[tiab] OR 'flixabi'[tiab] OR 'gp 1111'[tiab] OR 'gp1111'[tiab] OR 'inflectra'[tiab] OR 'infliximab'[tiab] OR 'ixifi'[tiab] OR 'pf 06438179'[tiab] OR 'pf 6438179'[tiab] OR 'pf06438179'[tiab] OR 'pf6438179'[tiab] OR 'remicade'[tiab] OR 'remsima'[tiab] OR 'renflexis'[tiab] OR 'revellex'[tiab] OR 'ta 650'[tiab] OR 'ta650'[tiab] OR 'zessly'[tiab] OR 'abp 501'[tiab] OR 'abp501'[tiab] OR 'abrilada'[tiab] OR 'abt d2e7'[tiab] OR 'abtd2e7'[tiab] OR 'adalimumab'[tiab] OR 'adaly'[tiab] OR 'amgevita'[tiab] OR 'amjevita'[tiab] OR 'amsparity'[tiab] OR 'avt 02'[tiab] OR 'avt02'[tiab] OR 'bat 1406'[tiab] OR 'bat1406'[tiab] OR 'bax 2923'[tiab] OR 'bax 923'[tiab] OR 'bax2923'[tiab] OR 'bax923'[tiab] OR 'bi 695501'[tiab] OR 'bi695501'[tiab] OR 'chs 1420'[tiab] OR 'chs1420'[tiab] OR 'cinnora'[tiab] OR 'ct p17'[tiab] OR 'ctp17'[tiab] OR 'cyltezo'[tiab] OR 'da 3113'[tiab] OR 'da3113'[tiab] OR 'dmb 3113'[tiab] OR 'dmb3113'[tiab] OR 'exemptia'[tiab] OR 'fkb 327'[tiab] OR 'fkb327'[tiab] OR 'fyzoclad'[tiab] OR 'gp 2017'[tiab] OR 'gp2017'[tiab] OR 'hadlima'[tiab] OR 'halimatoz'[tiab] OR 'hefiya'[tiab] OR 'hlx 03'[tiab] OR 'hlx03'[tiab] OR 'hulio'[tiab] OR 'humira'[tiab] OR 'hyrimoz'[tiab] OR 'ibi 303'[tiab] OR 'ibi303'[tiab] OR 'idacio'[tiab] OR 'imraldi'[tiab] OR 'kromeya'[tiab] OR 'lu 200134'[tiab] OR 'lu200134'[tiab] OR 'm 923'[tiab] OR 'm923'[tiab] OR 'mabura'[tiab] OR 'monoclonal antibody d2e7'[tiab] OR 'msb 11022'[tiab] OR 'msb11022'[tiab] OR 'ons 3010'[tiab] OR 'ons3010'[tiab] OR 'pf 06410293'[tiab] OR 'pf 6410293'[tiab] OR 'pf06410293'[tiab] OR 'pf6410293'[tiab] OR 'raheara'[tiab] OR 'sb 5'[tiab] OR 'sb5'[tiab] OR 'solymbic'[tiab] OR 'trudexa'[tiab] OR 'zrc 3197'[tiab] OR 'zrc3197'[tiab] OR 'cnto 148'[tiab] OR 'cnto148'[tiab] OR 'golimumab'[tiab] OR 'simponi'[tiab] OR 'certolizumab pegol'/exp OR 'cdp 870'[tiab] OR 'cdp870'[tiab] OR 'cimzia'[tiab] OR 'pha 738144'[tiab] OR 'pha738144'[tiab] OR certolizumab[tiab] |
27,604 |