Orale medicatie bij LRS
Uitgangsvraag
Wat is de effectiviteit van orale medicatie bij LRS?
Aanbeveling
Streef naar pijnstilling volgens de WHO-pijnladder. Het parenteraal toedienen van opioïden is voorbehouden aan de spoedeisende hulp setting.
Voorschrijven van NSAID’s kent geen grote bezwaren.
Gebruik bij voorkeur geen COX-2 selectieve NSAID’s vanwege het verhoogde cardiovasculaire gezondheidsrisico, zonder aangetoonde voordelen ten opzichte van klassieke NSAID’s.
Behandeling middels opioïden kan worden overwogen bij LRS en dient kortdurend (max. twee weken) en tijdcontingent te zijn (concreet: 2 maal daags langwerkende preparaten afgewisseld met kortwerkende preparaten bij doorbraakpijn).
Adequate monitoring door voorschrijvers en voorlichting aan patiënten over bijwerkingen en langetermijneffecten van opioïden is van belang.
Schrijf geen alpha2liganden, TCA, SNRI of benzodiazepines voor in de (sub)acute fase van LRS.
Voor gepast gebruik van opioïden, zie Generieke richtlijnmodule gepast opioïdengebruik.
Overwegingen
De onderstaande overwegingen en aanbevelingen gelden voor het overgrote deel van de populatie waarop de uitgangsvraag betrekking heeft.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
In de klinische praktijk wordt pijnmedicatie bij het LRS veelvuldig toegepast. Hoeveel precies is onbekend. De indruk bestaat bij veel patiënten en zorgverleners dat pijnmedicatie een positieve bijdrage kan leveren bij patiënten met ernstige pijnklachten bij een acuut LRS. Door adequate pijnstilling in de acute fase hoopt men zo de patiënt conservatief te kunnen blijven behandelen en operatieve interventies zoveel mogelijk te vermijden. Immers, het spontane beloop is gunstig bij een LRS.
Uit literatuurstudie lijken 1) er onvoldoende aanwijzingen dat NSAID’s/ opioïden/ neuropathische pijnmedicatie/ benzodiazepines een positief effect hebben op pijn en functioneren bij een LRS ten opzichte van placebo met uitzondering van een klein positief effect van oxicams (meloxicam en lornoxicam); 2) er onvoldoende aanwijzingen dat NSAID’s/ opioïden/ neuropathische pijnmedicatie een positief effect hebben op pijn en functioneren wanneer medicatie onderling vergeleken wordt.
Er lijkt dus sprake van een discrepantie tussen het waargenomen effect in de praktijk en beperkte conclusies uit de beschikbare evidence. Daarbij moet worden opgemerkt dat de kwaliteit van het bewijs laag tot zeer laag is met weinig klinisch relevante verschillen.
De lage kwaliteit van bewijs heeft te maken met 1) beperkingen in de onderzoeksopzet (risk of bias), 2) imprecisie (kleine patiëntaantallen & niet passeren grens van klinische relevantie).
Toekomstige onderzoeken zouden zich vooral moeten richten op 1) acute LRS-patiënten, 2) het vergelijken van algoritmes. Immers, in de praktijk wordt nooit één middel voorgeschreven, maar altijd combinaties van medicatie waarbij de WHO pijnladder leidend is. De eerste kennislacune betreft de acute fase. Er zijn weinig studies die deze onderzocht hebben, terwijl het LRS normaliter een kort beloop heeft: de meeste patiënten (75%) herstellen binnen 3 maanden. De tweede kennislacune betreft het vergelijken van combinaties van medicijnen, strategieën of ‘cocktails’ die in de praktijk toegepast worden in plaats van afzonderlijke medicatie.
Op basis van de onderzoeken moet worden geconcludeerd dat de bewijskracht van het effect van pijnmedicatie bij het LRS gering is, en het effect beperkt lijkt. De vraag is wel of de meest relevante subgroep in deze richtlijn namelijk patiënten met een acuut LRS (< 6 tot 12 weken) wel voldoende onderzocht is. Hier ligt mogelijk nog een hiaat.
De meest voorkomende bijwerkingen zijn (per groep):
- bij NSAID’s: zuurbranden, misselijkheid, diarree en buikpijn (voor overige bijwerkingen zie het farmacotherapeutisch kompas);
- bij opioïden: constipatie, droge mond, hoofdpijn, versuft zijn en duizeligheid, misselijkheid, vermoeidheid, slapeloosheid, verminderde eetlust en wazig zien (voor overige bijwerkingen zie het farmacotherapeutisch kompas);
- bij alpha2delta liganden: duizeligheid, rugpijn, zweten en algehele malaise;
- bij TCA’s: droge mond, constipatie, vermoeidheid en slaperigheid (voor overige bijwerkingen zie het farmacotherapeutisch kompas).
Waarden en voorkeuren van patiënten (en eventueel hun verzorgers)
Het doel van de patiënt is om snel pijnverlichting te krijgen en beter te functioneren. Op zich is pijnmedicatie veilig en ze leiden zelden tot ernstige complicaties, echter is de effectiviteit op basis van wetenschappelijk onderzoek minimaal. Pijnmedicatie kan laagdrempelig worden verkregen ofwel door zelf kopen (paracetamol en NSAID’s) ofwel op voorschrift van huisarts of specialisten in de tweede lijn.
Kosten (middelenbeslag)
De kosten van de onderzochte middelen, per tablet/capsule, zijn gemiddeld genomen redelijk vergelijkbaar, waarbij de NSAID’s relatief goedkoper, en morfine en topiramaat relatief duurder. De totaalkosten worden bepaald door de sterkte van het medicijn en het aantal giften per dag. Alle onderzochte middelen worden vergoed.
Aanvaardbaarheid voor de overige relevante stakeholders
De afgelopen jaren neemt het gebruik van opioïden toe en lijkt het erop dat de drempel om voor te schrijven is verlaagd. Veel patiënten gebruiken deze medicatie bovendien chronisch en lopen het risico op afhankelijkheid. Hoewel de bewijskracht laag is, lijkt er onvoldoende positief effect van opioïden op pijn en functioneren bij acuut LRS. Het voorschrijven van opioïden bij LRS, maar ook in algemene zin, vraagt om voorzichtigheid (zie nadere toelichting hiervan onder het kopje aanbevelingen).
Haalbaarheid en implementatie
Pijnmedicatie bij het LRS wordt in eerste instantie vaak voorgeschreven door huisartsen, waarna in het ziekenhuis een bijstelling van het medicatiebeleid kan plaatsvinden, bijvoorbeeld door medicatie op te hogen of medicatie te staken/starten. Ofschoon de WHO-pijnladder voor veel zorgprofessionals een goede ‘kapstok’ is wanneer het gaat om voorschrijven van medicatie, is er veel onderlinge praktijkvariatie. Pijnmedicatie beleid is binnen de Nederlandse gezondheidszorg voldoende geïmplementeerd, echter, gaat het er vooral om een ‘best practice’ of strategie te adviseren.
Rationale/ balans tussen de argumenten voor en tegen de interventie
Voordelen van de interventies zijn dat er mogelijk een beperkt gunstig effect op pijn en functioneren is in de acute fase. De kans op bijwerkingen en complicaties is al met al zeer gering. Er lijkt een discrepantie tussen waargenomen effect in de praktijk en de conclusies uit de (laag kwalitatieve) beschikbare evidence.
Aangezien er weinig wetenschappelijke onderbouwing is van pijnmedicatie bij het LRS worden hier een aantal aanbevelingen gedaan op basis van algemene inzichten over medicamenteuze pijnbestrijding, niet specifiek voor rug- of radiculaire pijn (de Jong, 2018; WHO, 1986). Een veel toegepast algoritme is de WHO-pijnladder. Deze is afkomstig uit de oncologie, maar wordt inmiddels breed toegepast (de Jong, 2018; WHO, 1986; Eisenberg, 2005).
Paracetamol is voor patiënten van alle leeftijden eerste keus, omdat dit middel van de beschikbare pijnstillers het breedste veiligheidsprofiel heeft en er zeer ruime ervaring mee is opgedaan. Het is vooral geschikt voor lichte en matige nociceptieve pijn (VAS < 4). De kans op ernstige bijwerkingen is minimaal in vergelijking met andere pijnstillers. Het maximale analgetische effect na orale toediening treedt op na 30 min tot 2h. Rectale toediening werkt langzamer dan orale toediening, intraveneuze toediening (SEH-setting) juist sneller. Bij voldoende hoge dosering (tot 4 gram per dag) is het pijnstillend effect van paracetamol vergelijkbaar met dat van een NSAID (Woo, 2005; Drendel, 2009)
NSAID’s (inclusief COX-2 selectieve NSAID’s of oxicams) zijn net als paracetamol geschikt voor lichte en matige nociceptieve pijn (VAS < 4) en hebben ook een plafondeffect. Het maximale analgetische effect treedt 20 min tot 2 uur na orale toediening op. Rectale toediening werkt langzamer en intramusculaire of intraveneuze toediening sneller dan orale. Absolute contra-indicaties voor NSAID’s zijn ernstig hartfalen, bronchiale hyperreactiviteit en nierfunctiestoornissen. In geval van relatieve contra-indicaties, zoals leeftijd vanaf 60 jaar, gebruik van anticoagulantia of trombocytenaggregatieremmers acetylsalicylzuur, ernstige invaliderende reumatoïde artritis, hartfalen, diabetes mellitus, gebruik van glucocorticoïden en gebruik van SSRI’s, kan een protonpompremmer worden toegevoegd of worden gekozen voor een alternatief. Op basis van een recente Deense studie wordt Diclofenac afgeraden vanwege het verhoogde cardiovasculaire risicoprofiel (Schmidt, 2018). COX-2 selectieve NSAID’s worden niet aanbevolen in verband met hoger risico cardiovasculaire complicaties zonder aangetoonde voordelen ten opzichte van klassieke NSAID’s (Coxib, 2013). Gecombineerd gebruik van paracetamol met NSAID verbetert het analgetisch effect.
Opioïden hebben een centrale rol bij de behandeling van acute pijn. Bij acute ernstige pijn (VAS > 6) worden opioïden in opbouwende dosering geadviseerd. Dit houdt in dat men begint met tweemaal daags langwerkende preparaten (bijvoorbeeld morfine retard 2x 10 mg of oxycodon met gereguleerde afgifte 2x 5 mg) aangevuld met viermaal daags een kortwerkend preparaat tegen doorbraakpijn (bijvoorbeeld morfine 4x 5 mg zo nodig).
Opioïden zijn vooral geschikt voor ernstige nociceptieve pijn en viscerale pijn, maar zijn ook een goed alternatief voor NSAID’s bij lichte of matige pijn. Elk opioïd heeft een eigen profiel voor het intreden van het effect, de eliminatiehalfwaardetijd en bijwerkingen. De werking van een opioïd wordt versterkt door combinatie met paracetamol of een NSAID, waardoor een opioïdsparend effect optreedt.
Gezien de forse stijging van het gebruik in de afgelopen jaren (SFK, 2019), worden hier 3 principes voor omgang met opioïden in de klinische praktijk gegeven (Brouwer, 2019):
- De arts die een opioïd voorschrijft, moet de patiënt vervolgen of de behandeling in goed overleg overdragen aan de huisarts of collega medisch specialist.
- Geef een goede voorlichting over de opioïden. Bespreek met de patiënt de mogelijke bijwerkingen en langetermijneffecten, zoals afhankelijkheid en gewenning.
- Maak voor het starten van de behandeling afspraken met de patiënt over het doel en het gebruik.
- Realistische en tijd-contingente doelen en afspraken
-
-
- pijnvermindering, maar volledige pijnvermindering wordt zelden bereikt;
- verbetering in functie en stemming;
- verbetering arbeidssituatie.
-
- Afspraken te maken vóór het starten:
-
-
- gronden voor afbouwen;
- vermijden van dosisescalatie. Probeer de morfine-equivalent van 30 tot 50 mg/per dag niet te overschrijden;
- indien mogelijk een stopdatum afspreken, schrijf maximaal voor 2 weken voor;
- herhalingsrecepten en dosisaanpassingen (één voorschrijver);
- wat te doen in noodsituaties.
-
- Multimodale benadering:
- goede diagnostiek;
- proefbehandeling;
- herbeoordeling evaluatie;
- niet-farmacologische behandelingen.
Medicatie tegen neuropathische pijn als alpha2delta (d2) liganden (pregabaline en gabapentine) lijken in de acute fase van een LRS niet effectief (Mathieson, 2017). In het algemeen is de NNT voor medicatie tegen neuropathische pijn erg hoog (Finnerup, 2005). Deze middelen worden in geval van een LRS geadviseerd voor chronische vormen en niet in de acute fase.
Tot slot wordt opgemerkt dat indien pijnmedicatie niet effectief is, aanvullend een wortelblokkade overwogen kan worden (zie de module 'Diagnostische zenuwwortelblokkade') of gekozen kan worden voor een hernia-operatie.
Onderbouwing
Het lumbosacraal radiculair syndroom (LRS) is een veelvoorkomend ziektebeeld met een incidentie van 9/1000 per jaar in Nederland (Van der Linden, 2004; Spijker-Huiges, 2015). Aangezien de meeste patiënten (75%) binnen drie maanden herstellen zonder operatie, ligt de focus van de behandeling gedurende de eerste weken op mobiliseren en pijnstilling. Pijnstilling gebeurt doorgaans door middel van orale medicatie. De vraag is welke orale pijnmedicatie het meest effectief is en de minste bijwerkingen heeft.
1. NSAID’s
1.1 NSAID’s versus placebo:
1.1.1 Uitkomstmaat Pijn (cruciaal)
|
Laag GRADE |
Behandeling met oxicams (meloxicam en lornoxicam) of met diclofenac zou een klein positief effect kunnen hebben op pijn vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Dreisser, 2001; Herrmann, 2009) |
1.1.2 Uitkomstmaat Functioneren (belangrijk)
|
- GRADE |
Er is geen GRADE-beoordeling voor deze uitkomstmaat in verband met het ontbreken van studies. |
1.1.3 Uitkomstmaat Adverse events (belangrijk)
|
Laag GRADE |
Er lijkt geen verschil te zijn in het optreden van adverse events tussen behandeling met oxicams (meloxicam en lornoxicam) of met diclofenac en placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Dreisser, 2001; Hermann, 2009) |
1.2 NSAID’s versus NSAID’s
1.2.1 Uitkomstmaat Pijn direct (< 2 weken) (cruciaal)
|
Redelijk GRADE |
Behandeling met meloxicam of lornoxicam geeft waarschijnlijk niet meer pijnvermindering op korte termijn (binnen 2 weken) vergeleken met diclofenac bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Dreisser, 2001; Hermann, 2009) |
1.2.2 Uitkomstmaat Functioneren (belangrijk)
|
- GRADE |
Er is geen GRADE-beoordeling voor deze uitkomstmaat in verband met het ontbreken van studies. |
1.2.3 Uitkomstmaat Adverse events (belangrijk)
|
Laag GRADE |
Er lijkt geen verschil te zijn in het optreden van adverse events tussen behandeling met oxicams (meloxicam en lornoxicam) en diclofenac bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Dreisser, 2001; Hermann, 2009) |
2. Opioïden
2.1 Morfine versus placebo
2.1.1 Uitkomstmaat Pijn (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met morfine leidt tot een grotere afname in pijn na 2 weken vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007) |
2.1.2 Uitkomstmaat Functioneren (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met morfine leidt tot een verbetering in functioneren (dat wil zeggen het kunnen uitvoeren van algemene dagelijkse activiteiten) op korte termijn (2 weken) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007) |
2.1.3 Uitkomstmaat Adverse events (belangrijk)
|
Laag GRADE |
Er zijn aanwijzingen dat adverse events vaker voorkomen bij behandeling met morfine dan bij placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007) |
2.2. Tapentadol versus oxycodon
2.2.1 Uitkomstmaat Pijn (cruciaal)
|
Laag GRADE |
Behandeling met tapentadol lijkt niet te leiden tot een vermindering in pijn op korte termijn (5 dagen) vergeleken met oxycodon bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Biondi, 2013) |
2.2.2 Uitkomstmaat Functioneren (belangrijk)
|
- GRADE |
Er is geen GRADE-beoordeling voor deze uitkomstmaat in verband met het ontbreken van studies. |
2.2.3 Uitkomstmaat Adverse events (belangrijk)
|
Laag GRADE |
Er lijkt geen verschil te zijn in het optreden van adverse events tussen behandeling met tapentadol en oxycodon bij patiënten met lumbosacraal radiculair syndroom, behalve voor gastro-intestinale adverse events. Behandeling met oxycodon lijkt te leiden tot een hogere kans op gastro-intestinale adverse events (overgeven en constipatie) vergeleken met tapentadol bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Biondi, 2013) |
3. Benzodiazepines
3.1 Diazepam versus placebo
3.1.1 Uitkomstmaat Pijn (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of het gebruik van diazepam leidt tot meer pijnreductie (≥ 50% vermindering in pijn na 7 dagen) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Brötz, 2010) |
3.1.2 Uitkomstmaat Functioneren (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met diazepam leidt tot een verbetering in functioneren (dat wil zeggen het kunnen uitvoeren van algemene dagelijkse activiteiten) op korte termijn (7 dagen) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Brötz, 2010) |
3.1.3 Uitkomstmaat Adverse events (belangrijk)
|
Zeer laag GRADE |
Er is geen GRADE-beoordeling voor deze uitkomstmaat in verband met het ontbreken van studies. |
4. Alpha2delta liganden
4.1 Pregabaline versus placebo
4.1.1a Uitkomstmaat Pijn korte termijn (> 2 weken ≤ 3 maanden) (cruciaal)
|
Laag GRADE |
Behandeling met pregabaline lijkt niet te leiden tot een grotere afname in pijn op korte termijn vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Mathieson, 2017) |
4.1.1b Uitkomstmaat Pijn lange termijn (belangrijk)
|
Laag GRADE |
Behandeling met pregabaline lijkt niet te leiden tot een grotere afname in pijn op lange termijn (na 1 jaar) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Mathieson, 2017) |
4.1.2 Uitkomstmaat Functioneren (belangrijk)
|
Laag GRADE |
Behandeling met pregabaline lijkt niet te leiden tot een verbetering in functioneren (dat wil zeggen het kunnen uitvoeren van algemene dagelijkse activiteiten) op korte termijn (8 weken) en op lange termijn (1 jaar) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Mathieson, 2017) |
4.1.3 Uitkomstmaat Adverse events (belangrijk)
|
Laag GRADE |
Behandeling met pregabaline lijkt te leiden tot een hogere kans op het optreden van adverse events (duizeligheid, rugpijn, zweten en algehele malaise) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Mathieson, 2017) |
4.2 Pregabaline versus gabapentine
4.2.1 Uitkomstmaat Pijn (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met pregabaline leidt tot een grotere afname in pijn op korte termijn vergeleken met gabapentine bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Robertson, 2018) |
4.2.2 Uitkomstmaat Functioneren (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met pregabaline leidt tot een verbetering in functioneren (dat wil zeggen het kunnen uitvoeren van algemene dagelijkse activiteiten) na 8 weken vergeleken met gabapentine bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Robertson, 2018) |
4.2.3 Uitkomstmaat Adverse events (belangrijk)
|
Zeer laag GRADE |
Er zijn voorzichtige aanwijzingen dat adverse events vaker voorkomen bij behandeling met pregabaline dan bij gabapentine bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Robertson, 2018) |
5. Topiramaat
5.1 Topiramaat versus placebo
5.1.1 Uitkomstmaat Pijn (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met topiramaat leidt tot een grotere afname in pijn op korte termijn (2 weken) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2005) |
5.1.2 Uitkomstmaat Functioneren (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met topiramaat leidt tot een verbetering in functioneren (dat wil zeggen het kunnen uitvoeren van algemene dagelijkse activiteiten) na 2 weken vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2005) |
5.1.3 Uitkomstmaat Adverse events (belangrijk)
|
Zeer laag GRADE |
Er zijn voorzichtige aanwijzingen dat adverse events vaker voorkomen bij behandeling met topiramaat dan bij placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2005) |
6. TCA’s
6.1 Nortriptyline/ Amitriptyline versus placebo
6.1.1 Uitkomstmaat Pijn (been) (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met TCA’s (nortriptyline, amitriptyline) leidt tot een grotere afname in pijn op korte termijn (2 weken) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007; Van Elderen, 2015) |
6.1.2 Uitkomstmaat Functioneren (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met nortriptyline leidt tot een verbetering in functioneren (dat wil zeggen het kunnen uitvoeren van algemene dagelijkse activiteiten) na 2 weken vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007) |
6.1.3 Uitkomstmaat Adverse events (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of adverse events vaker optreden bij behandeling met TCA’s (nortriptyline, amitriptyline) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007; Van Elderen, 2015) |
6.2 Combinatie nortriptyline + morfine versus placebo
6.2.1 Uitkomstmaat Pijn (cruciaal)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met een combinatie van nortriptyline en morfine leidt tot een grotere afname in pijn op korte termijn (2 weken) vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007) |
6.2.2 Uitkomstmaat Functioneren (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met een combinatie van nortriptyline en morfine leidt tot een verbetering in functioneren (dat wil zeggen het kunnen uitvoeren van algemene dagelijkse activiteiten) na 2 weken vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007) |
6.2.3 Uitkomstmaat Adverse events (belangrijk)
|
Zeer laag GRADE |
Er zijn voorzichtige aanwijzingen dat adverse events vaker voorkomen bij behandeling met een combinatie van nortriptyline en morfine vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Khoromi, 2007) |
Er zijn voor de uitkomstmaat werkhervatting geen studies behalve voor de vergelijkingen diazepam versus placebo (vergelijking 3.1) en pregabaline versus placebo (vergelijking 4.1)
3.1 Diazepam versus placebo
3.1.4 Uitkomstmaat Werkhervatting (belangrijk)
|
Zeer laag GRADE |
Het is onduidelijk of behandeling met diazepam leidt tot een snellere werkhervatting vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom.
Bronnen: (Brötz, 2010) |
4.1 Pregabaline versus placebo
4.1.4 Uitkomstmaat Werkhervatting (belangrijk)
|
Laag GRADE |
Behandeling met pregabaline lijkt niet te leiden tot minder absentie van werk vergeleken met placebo bij patiënten met lumbosacraal radiculair syndroom gedurende het eerste jaar.
Bronnen: (Mathieson, 2017) |
Beschrijving studies
Pinto (2012) is een systematische review waarin onder andere de effectiviteit van NSAID’s, opioïden en benzodiazepines versus placebo of het effect onderling werd onderzocht bij personen met LRS. Er werd een onderscheid gemaakt tussen acute (< 6 weken), subacute (6 tot 12 weken) of chronische (> 12 weken) LRS. Er werden 23 studies geïncludeerd waarvan er twee de effectiviteit van NSAID’s evalueerden. Voor deze richtlijn werden geëxtraheerd: drie placebo-gecontroleerde trials (Dreisser, 2001; Weber, 1993; Grevsten, 1975) en twee trials NSAID versus NSAID (Dreisser, 2001 (twee armen); Herrmann, 2009 (drie armen)). Er werd een meta-analyse verricht voor de NSAID’s versus placebo waarbij er in totaal 460 personen deelnamen in de NSAID groep en 236 personen in de placebogroep (Pinto, 2012). Er werden verschillende typen NSAID’s gepoold namelijk: meloxicam, lornoxicam en diclofenac met een minimum van vier dagen (Herrmann, 2009) en een maximum van 14 dagen toediening (Dreiser, 2001). Pijn werd gemeten aan de hand van de VAS of de NRS en omgerekend naar een 0 tot 100 schaal indien nodig. Functioneren werd gemeten met de Roland Morris disability scale (0 tot 17) en omgerekend naar een 0 tot 100 schaal. Daarnaast onderzochten twee studies de effectiviteit van neuropathische pijnmedicatie; antidepressiva en anti-epileptica (Khoromi, 2005 en Khoromi, 2007). Khoromi (2005) onderzocht het effect van topiramaat versus placebo met klachten gedurende > 3 maanden (mediaan 8 jaar, range 0,5 tot 40 jaar). Er werden 41 patiënten gerandomiseerd waarvan er 29 de studie voltooiden. Pijn en functioneren werden voor de 29 patiënten die de studie voltooiden onderzocht, adverse events werden voor alle 41 patiënten gerapporteerd. In de studie van Khoromi (2007) werden vier armen onderzocht ten opzichte van elkaar: 1) nortiptyline; 2) nortriptyline + morfine; 3) morfine en 4) placebo. Hiervoor werden 55 patiënten gerandomiseerd die de 4 interventies in verschillende volgordes ontvingen waarvan er 28 patiënten alle vier de interventies voltooiden (Khoromi, 2007). Voor deze 28 patiënten werden de uitkomstmaten pijn en functioneren onderzocht, voor alle 55 patiënten werden adverse events gerapporteerd. Pijn (algeheel, been of rug middels de VAS, 0 tot 100) en adverse events werden onderzocht. Functioneren werd in de studies van Khoromi (2005) en Khoromi (2007) onderzocht middels de Oswestry low back pain questionnaire (omgerekend naar een 0 tot 100 schaal).
Biondi (2013) is een gerandomiseerde, dubbel geblindeerde trial waarin het effect van tapentadol werd vergeleken met het effect van oxycodon na 10 dagen bij patiënten met LRS die niet langer dan 30 dagen bestond. Er namen 287 patiënten deel in de interventiegroep en 298 patiënten in de controlegroep. Vermindering van pijn (lage rug en been apart gerapporteerd) op twee, drie, vijf en tien dagen werd gemeten middels de NRS (schaal 0 tot 10). Tevens werden adverse events onderzocht.
Brötz (2010) is een gerandomiseerde, dubbel geblindeerde, placebo gecontroleerde trial waarin het effect van diazepam werd onderzocht (aanvullend aan fysiotherapie en pijnstillers) bij patiënten met LRS met of zonder neurologische uitval wijtende aan lumbale disk prolaps in de leeftijd van 18 tot 75 jaar. Er namen 30 patiënten deel in de interventiegroep en 30 patiënten in de controlegroep. Vermindering van pijn (rug en been gecombineerd) in een percentage op de VAS en functioneren aan de hand van de Roland–Morris disability scale op dag zeven werd gemeten. Daarnaast werd werkhervatting gemeten.
Mathieson (2017) is een gerandomiseerde, dubbelblinde, placebo gecontroleerde trial waarin het effect van pregabaline na acht weken, met een follow-up op 52 weken, werd onderzocht bij patiënten met acute (80,2%) en chronische LRS. Er participeerden 106 patiënten in de pregabaline-groep en 101 patiënten in de placebogroep. De uitkomstmaten in deze studie waren beenpijn, rugpijn (gemeten middels de VAS, schaal 0-100) en lichamelijke beperking (gemeten aan de hand van de Roland-Morris Disability Questionnaire).
Robertson (2018) is een gerandomiseerde dubbelblinde dubbel-dummy cross-over trial waarin het effect van pregabaline na acht weken werd vergeleken met gabapentine na acht weken. Het dubbel-dummy design is gekozen omdat de dosering van beide medicamenten verschilde. Er werd een placebo toegevoegd zodat de frequentie van inname gelijk was waardoor blindering van beide medicamenten behouden bleef. Achttien patiënten met chronische LRS participeerden, waarvan 10 personen eerst pregabaline kregen gevolgd door gabapentine en acht vice versa. Wash-out tussen de twee medicamenteuze behandelingen was één week. Pijn (VAS, schaal 0 tot 10), functioneren (Oswestry Low Back Pain Questionnaire) en adverse events werden onderzocht.
Van Elderen (2015) is een gerandomiseerde dubbel geblindeerde trial waarin onder andere amitriptyline (N=20) met placebo (N=20) werd vergeleken. De patiëntenpopulatie bestond uit patiënten met neuropathische lumbale radiculaire pijn bij lumbale disk hernia (80% versus 80%), spinale kanaalstenose (10% versus 20%) en failed back surgery syndroom (epidurale fibrose (10% versus 20%) in de amitriptyline- versus placebogroep, respectievelijk. Na 14 dagen werd pijn (NRS, schaal 0 tot 10) in het been gemeten en adverse events werden gerapporteerd.
Resultaten
1. NSAID’s
1.1 NSAID’s versus placebo
1.1.1 Uitkomstmaat Pijn (cruciaal)
De systematische review van Pinto (2012) omvat twee studies die NSAID’s evalueerden versus placebo (Dreisser, 2001: meloxicam (7,5 mg en 15 mg) en Herrmann, 2009: lornoxicam en diclofenac). Pijn werd gemeten na acht uur (Herrmann, 2009) en één week (Dreisser, 2001) na het starten van de medicatie. Dreisser (2001) en Herrmann (2009) hebben pijn overall gemeten (figuur 1). Voor beide studies is het verschil in pijn ten opzichte van baseline gepresenteerd. Dreisser (2001) onderzocht twee doseringen van meloxicam en includeerde één controlegroep. In de forest plot is voor deze studie het gewogen gemiddelde weergegeven van de twee doseringen, omdat er voor deze twee armen één controlegroep aanwezig was en de medicamenten niet statistisch significant en niet klinisch relevant van elkaar verschilden. In de studie van Herrmann (2009) zijn drie armen onderzocht: lornoxicam, diclofenac en een controlegroep. In de forest plot is voor deze studie om dezelfde reden als Dreisser (2001) het gewogen gemiddelde weergegeven van het effect van lornoxicam en diclofenac. Dreisser (2001) en Herrmann (2009) vermeldden niets over een advies ten aanzien van bewegen maar Dreisser (2001) vermeldde als uitkomst wel het aantal uren dat patiënten genoodzaakt waren om in bed door te brengen, wat suggereert dat bedrust niet als advies werd gegeven. Het gemiddelde verschil tussen de interventiegroep (N=460) en de controlegroep (N=236) is -6,65 in het voordeel van de NSAID’s op een schaal van 0 tot 100 (95% BI -11,27 tot -2,04, p=0,005). Dit verschil is niet klinisch relevant.
Figuur 1 Uitkomstmaat vermindering van pijn vergelijking NSAID’s versus placebo
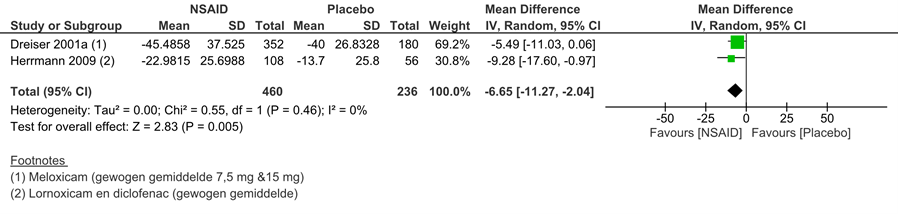
Z: p-waarde van het gepoolde effect: df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval; schaal 0 tot 100
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias: waaronder selectiebias door foutieve randomisatie, zie risk of bias tabel) en met één niveau voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
1.1.2 Uitkomstmaat Functioneren (belangrijk)
Deze uitkomstmaat werd niet gerapporteerd.
Bewijskracht van de literatuur
Er is geen GRADE-beoordeling voor de uitkomstmaat ‘functioneren’ in verband met het ontbreken van studies.
1.1.3 Uitkomstmaat Adverse events (belangrijk)
Dreisser (2001) rapporteerde het aantal personen waarbij ten minste één adverse event optrad: bij 17% in de meloxicam 7,5 mg groep, bij 19% in de meloxicam 15 mg groep en bij 13% in de placebogroep. Deze events leken gerelateerd aan behandeling in 8% van de gevallen bij de meloxicam 7,5 mg groep, 7% bij de meloxicam 15 mg groep en 6% bij de placebogroep. Er was geen statistisch verschil in adverse events tussen de groepen. Eén serieus adverse event leek gerelateerd te zijn aan behandeling en vond plaats in de meloxicam 7,5 mg groep (een anafylactische shock waarbij steroïden toegediend dienden te worden). Herrmann (2009) rapporteerde bij 10,5% van de personen in de lornoxicam groep ten minste één adverse event, bij 12,3% in de diclofenac groep en bij 7% in de placebogroep. Dit was niet klinisch relevant verschillend tussen de groepen. Zuurbranden, misselijkheid, diarree en buikpijn waren de meest voorkomende adverse events in beide studies.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias: waaronder selectiebias door foutieve randomisatie, zie risk of bias tabel) en met één niveau voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
1.2 NSAID’s versus NSAID’s
1.2.1 Uitkomstmaat pijn direct (< 2 weken) (cruciaal)
De review van Pinto (2012) omvat twee studies die NSAID’s (oxicams) versus NSAID’s (diclofenac) evalueerden. Dreisser (2001) onderzocht het effect van meloxicam (7,5 mg en 15 mg) versus diclofenac en Herrmann (2009) het effect van lornoxicam versus diclofenac. Pijn is gemeten na acht uur (Herrmann, 2009) of na een week (Dreisser, 2001) waarbij beide studies pijn overall hebben gemeten. Het gemiddelde verschil tussen de oxicams en diclofenac is 0,89 (95% BI -3,93 tot 5,72, p=0,72 in het voordeel van diclofenac. Dit verschil was niet klinisch relevant.
Figuur 2 Uitkomstmaat pijn vergelijking oxicams (meloxicam/lornoxicam) versus diclofenac
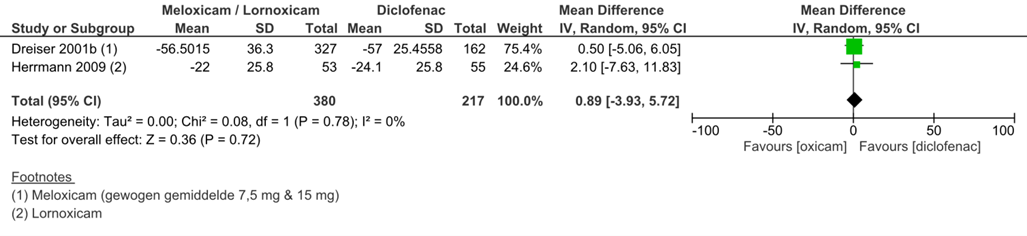
Z: p-waarde van het gepoolde effect: df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval; schaal van 0 tot 100
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias: waaronder selectiebias door foutieve randomisatie, zie risk of bias tabel). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘redelijk’.
1.2.2 Uitkomstmaat Functioneren (belangrijk)
Deze uitkomstmaat werd niet gerapporteerd.
Bewijskracht van de literatuur
Er is geen GRADE-beoordeling voor de uitkomstmaat ‘functioneren’ in verband met het ontbreken van studies.
1.2.3 Uitkomstmaat Adverse events (belangrijk)
Dreisser (2001) rapporteerde het aantal personen waarbij ten minste één adverse event optrad: bij 26% in de meloxicam 7,5 mg groep, 30% in de meloxicam 15 mg groep en 28% in de diclofenac-groep. Deze adverse events leken gerelateerd aan behandeling te zijn bij 13% in de meloxicam 7,5 mg groep, 17% bij de meloxicam 15 mg groep en 17% bij de diclofenac-groep. Er was geen statistisch verschil in adverse events tussen de groepen. Er was één serieus adverse event in de meloxicam 15 mg groep (acute erythemateuze cutaneuze reactie) en één in de diclofenac-groep (verergering van asthenie). Herrmann (2009) rapporteerde bij 10,5% van de personen in de lornoxicam-groep ten minste één adverse event en bij 12,3% in de diclofenac-groep. Dit was niet klinisch relevant verschillend tussen de groepen. In één geval in de diclofenac-groep werd het adverse event als serieus gerapporteerd (buikpijn en misselijkheid). De meest voorkomende adverse events bij beide studies waren zuurbranden, misselijkheid, diarree en buikpijn.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias: waaronder selectiebias door foutieve randomisatie, zie risk of bias tabel) en met één niveau voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
2. Opioïden
2.1 Morfine versus placebo
2.1.1 Pijn (cruciaal)
Khoromi (2007) onderzocht pijn in het been waarbij de gemiddelde pijnscore iedere avond werd uitgevraagd en het gemiddelde over twee weken werd gerapporteerd. De uitkomstmaat werd uitgedrukt als het verschil tussen de twee groepen met een VAS (0 tot 100). Het gemiddelde verschil tussen morfine en placebo was -3,0 (95% BI -16,6 tot 10,6). Dit verschil was niet klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design, incomplete uitkomst data en niet voldoen aan intention-to-treat analyse) en met twee niveaus voor imprecisie (gering aantal patiënten). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
2.1.2 Uitkomstmaat Functioneren (belangrijk)
Khoromi (2007) onderzocht functioneren gemeten met de Oswestry Low Back Pain Questionnaire schaal 0 tot 100). Het gemiddelde verschil tussen morfine en placebo was -4,8 (95% BI -13,2 tot 3,7) in het voordeel van morfine. Dit verschil was niet klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘functioneren’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (incomplete uitkomst data en niet voldoen aan intention-to-treat analyse) en twee niveaus vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
2.1.3 Uitkomstmaat Adverse events (belangrijk)
Khoromi (2007) rapporteerde dat in de morfinegroep twee patiënten zich terugtrokken uit de studie in verband met verminderd bewustzijn: één in verband met misselijkheid, braken en ernstige constipatie, één in verband met uitslag en één in verband met een droge mond. In de placebogroep trok één patiënt zich terug uit de studie in verband met verminderd bewustzijn (Khoromi, 2007). De number needed to harm (NNH) was 10. Deze werd berekend over alle patiënten die werden gerandomiseerd (N=55) en zich terugtrokken uit de studie in verband met adverse events. Van de 28 patiënten die de studie voltooiden werden er bij 93% (N=26/28) in de interventiegroep adverse events gerapporteerd, vergeleken met 50% (N=14/28) in de placebogroep, wat leidde tot een risk ratio van 1,86 (95% BI 1,25 tot 2,73, p=0,002). Dit verschil was klinisch relevant. De meest voorkomende adverse events in de morfinegroep waren: constipatie (64 keer), droge mond (21 keer), hoofdpijn (14 keer), versuft zijn (25 keer) en duizeligheid (14 keer), misselijkheid (7 keer) vermoeidheid (7 keer), slapeloosheid (7 keer), verminderde eetlust (7 keer) en wazig zien (7 keer). De meest voorkomende adverse events in de placebogroep waren: droge mond (21 keer), vermoeidheid (18 keer) en hoofdpijn (14 keer), verminderde eetlust (7 keer) en constipatie (7 keer).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design, incomplete uitkomst data en niet voldoen aan intention-to-treat analyse) en met één niveau voor imprecisie (geringe aantal patiënten). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
2.1 Tapentadol versus oxycodon
2.2.1 Uitkomstmaat Pijn (cruciaal)
Biondi (2013) onderzocht het effect van tapentadol ten opzichte van oxycodon op pijn na vijf dagen op een schaal van 0 tot 10. Het gemiddelde verschil tussen tapentadol en oxycodon was 0,1 (95% BI -0,3 tot 0,5; p=0,63). Dit verschil was niet klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias: incomplete uitkomst data) en met één niveau voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
2.2.2 Uitkomstmaat Functioneren (belangrijk)
Biondi (2013) heeft deze uitkomstmaat niet gerapporteerd.
Bewijskracht van de literatuur
Er is geen GRADE-beoordeling voor de uitkomstmaat ‘functioneren’ in verband met het ontbreken van studies.
2.2.3 Uitkomstmaat Adverse events (belangrijk)
Voor de tapentadol-groep werden geen serieuze adverse events ten gevolge van de behandeling gerapporteerd terwijl in de oxycodon-groep bij drie van de patiënten (0,9%) serieuze adverse events (flauwvallen, insult, angst) ten gevolge van de behandeling optraden (Biondi, 2013). Biondi (2013) rapporteerde bij 52,3% (N=168/321) van de patiënten in de tapentadol-groep en 58,0% (N=188/324) van de patiënten in de oxycodon-groep op zijn minst één adverse event die gerelateerd was aan de behandeling. Gastro-intestinale adverse events kwamen klinisch relevant minder vaak voor in de tapentadol-groep versus de oxycodon-groep (odds ratio voor overgeven: -1,74 (95% BI -2,57 tot -1,17) en voor constipatie: -3,43 (95% BI -8,11 tot -1,45).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; incomplete uitkomstdata) en met één niveau vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
3. Benzodiazepines
3.1 Diazepam versus placebo
3.1.1 Uitkomstmaat Pijn (cruciaal)
Brötz (2010) onderzochten pijn, gemeten met de VAS (0 tot 10), na 7 dagen en rapporteerde het percentage patiënten dat ≥ 50% pijn reductie ervaarde op de VAS. 12/29 patiënten (41%) uit de diazepamgroep en 23/29 patiënten (79%) uit de placebo groep ervaarden ≥ 50% reductie in pijn. Het relatieve risico bedroeg 0,52 in het voordeel van placebo (95% BI 0,33 tot 0,84, p=0,007). Dit verschil is klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; selectieve outcome reporting) en met twee niveaus vanwege imprecisie (gering patiënten aantal). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
3.1.2 Uitkomstmaat Functioneren (belangrijk)
Brötz (2010) heeft functioneren gemeten door middel van de Roland–Morris disability scale (schaal 0 tot 24) na zeven dagen en rapporteerde de vermindering in disability en vond geen klinisch relevant verschil tussen de diazepam en de placebo groep: mediaan (interkwartiele range): diazepam: 3,0 (3-7) en placebo: 5,0 (1-8), p= 0,67.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; incomplete uitkomstdata) en met twee niveaus vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
3.1.3 Uitkomstmaat Adverse events (belangrijk)
Brötz (2010) heeft deze uitkomstmaat niet gerapporteerd.
Bewijskracht van de literatuur
Er is geen GRADE-beoordeling voor de uitkomstmaat ‘adverse events’ in verband met het ontbreken van studies.
4. Alpha2delta liganden
4.1 Gabapentine / Pregabaline versus placebo
4.1.1a Uitkomstmaat Pijn korte termijn (> 2 weken ≤ 3 maanden) (cruciaal)
Mathieson (2017) rapporteerde een beenpijnscore na acht weken, aan de hand van een NRS-score op een schaal van 0 tot 10. Het gemiddelde verschil tussen de interventiegroep (N=106) en de controlegroep (N=101) is 0,5 in het nadeel van de alpha2delta liganden, hetgeen niet klinisch relevant van elkaar verschilde (95% BI -0,2 tot 1,2, p=0,19).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn korte termijn’ is met twee niveaus verlaagd vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
4.1.1b Uitkomstmaat Pijn lange termijn (cruciaal)
Mathieson (2017) onderzocht het effect van pregabaline versus placebo op pijn in het been met een VAS-score op een schaal van 0 tot 100 na 52 weken. Het gemiddelde verschil was -3,0 in het voordeel van pregabaline, hetgeen niet klinisch relevant was (95% BI -5,0 tot 10,0, p=0,46).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn lange termijn’ is met twee niveaus verlaagd vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
4.1.2 Uitkomstmaat Functioneren (belangrijk)
Mathieson (2017) onderzocht het effect van pregabaline ten opzichte van placebo op functioneren gemeten met de Roland Disability questionnaire (0 tot 23) op acht weken en op 52 weken. Het gemiddelde verschil op acht weken was 0,1 in het voordeel van pregabalin, hetgeen niet statistisch significant was (95%BI -1,8 tot 2,0, p=0,96) en op 52 weken was dat 0,2 in het voordeel van pregabalin, hetgeen ook niet klinisch relevant was (95% BI -1,8 tot 2,2, p=0,85).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘functioneren’ is met twee niveaus verlaagd vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
4.1.3 Uitkomstmaat Adverse events (belangrijk)
Mathieson (2017) rapporteerde serieuze adverse events in 1,9% van de interventiegroep versus 5,9% van de controlegroep, hetgeen niet statistisch significant verschilde (p=0,16). De serieuze adverse events voor de interventiegroep waren: ziekenhuisopname voor kortademigheid en misselijkheid (één patiënt) en suïcidale gedachten (één patiënt). De serieuze adverse events in de controlegroep waren: ziekenhuisopname voor pijn op de borst (één patiënt), ziekenhuisopname voor verergerde rug of beenpijn (drie patiënten), ziekenhuisopname voor psychologische problemen (één patiënt), ziekenhuisopname voor suïcidepoging (één patiënt). Adverse events traden op bij 64% van de interventiegroep en bij 43% van de controlegroep, wat leidde tot een risk ratio van 1,51 (95% BI 1,15 tot 1,97, p=0,002). Dit verschil was klinisch relevant. De meest voorkomende adverse events in de pregabalin-groep waren: duizeligheid (42 patiënten), dorsalgie (19 patiënten), zweten (negen patiënten) en algehele malaise (negen patiënten). De meest voorkomende adverse events in de controlegroep waren: duizeligheid (13 patiënten), dorsalgie (10 patiënten), zweten (acht patiënten) en algehele malaise (drie patiënten).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met twee niveaus verlaagd vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
4.2 Pregabaline versus gabapentine
4.2.1. Uitkomstmaat Pijn (cruciaal)
Robertson (2018) rapporteerde pijnreductie op een VAS schaal van 0-10 na 8 weken van -0,94±1,09) in de pregabaline-groep versus -1,72±1,17 in de gabapentine-groep (p=0,035). Het verschil tussen de groepen (0,78) in het voordeel van gabapentine is niet klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
4.2.2 Uitkomstmaat Functioneren (belangrijk)
Robertson (2018) onderzocht functioneren gemeten met de Oswestry low back pain disability questionary (schaal van 0 tot 100) en rapporteerde geen klinisch relevant verschil tussen de groepen: pregabaline: -8,78±18,86 versus gabapentine: -10,66±9,90; p=0,63.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘functioneren’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
4.2.3 Uitkomstmaat Adverse events (belangrijk)
Robertson (2018) rapporteerde in de pregabaline-groep 31 adverse events (in 81% van de patiënten) ten opzichte van zeven events (in 19% van de patiënten) in de gabapentine-groep (p=0,002). Dit is een klinisch relevant verschil. De meest voorkomende adverse events in de pregabaline-groep waren misselijkheid, overgeven, hoofdpijn, darmproblemen, dubbelzien, duizeligheid en versuft zijn en in de gabapentine-groep versuft zijn, duizeligheid, misselijkheid, overgeven en hoofdpijn.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
5. Topiramaat
5.1 Topiramaat versus placebo
5.1.1 Uitkomstmaat Pijn (cruciaal)
Khoromi (2005) onderzocht pijn in het been en pijn in de rug waarbij de pijnscore iedere avond werd uitgevraagd en het gemiddelde over 2 weken werd gerapporteerd. De uitkomstmaat werd uitgedrukt als het verschil tussen de 2 groepen op de VAS (0 tot 100). Het gemiddelde verschil voor pijn in het been was -7,4 in het voordeel van topiramaat, hetgeen niet statistisch significant was (95% BI -21,2 tot 6,4). Het gemiddelde verschil voor pijn in de rug was -8,7, in het voordeel van topiramaat, hetgeen niet klinisch relevant was (95% BI -21,4 tot 4,0).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (cross-over design) en met twee niveaus voor imprecisie (gering patiënten aantal). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
5.1.2 Uitkomstmaat Functioneren (belangrijk)
Khoromi (2005) onderzocht functioneren gemeten met de Oswestry Low Back Pain Disability questionnaire (0 tot 100) en rapporteerde een gemiddeld verschil van -2,0, in het voordeel van topiramaat, hetgeen klinisch relevant was (95% BI -10,0 tot 6,0, p=0,065).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘functioneren’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (cross-over design) en met twee niveaus voor imprecisie (gering patiënten aantal). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
5.1.3 Uitkomstmaat Adverse events (belangrijk)
Van de 40 patiënten die topiramaat hebben gebruikt, trokken zich twee patiënten terug uit de studie in verband met hinderlijke tintelingen, twee in verband met misselijkheid en anorexia, drie in verband met verminderd bewustzijn en geheugenverlies, één in verband met depressie en angst en één in verband met huiduitslag (Khoromi, 2005). In de placebogroep trok één patiënt zich terug in verband met verminderd bewustzijn. De number needed to harm (NNH) was 4,4 berekend over deze 40 patiënten. Van de 28 patiënten die de studie voltooiden werden er bij 24/28 patiënten (86%) in de topiramaat-groep adverse events gerapporteerd versus 20/28 patiënten (72%) in de placebogroep, wat leidde tot een risk ratio van 1,20 (0,91 tot 1,59, p=0,20). Dit verschil was klinisch relevant. De meest voorkomende adverse events waren: paresthesieën, vermoeidheid, zwakte, verminderd bewustzijn en diarree (Khoromi, 2005).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (cross-over design) en met twee niveaus voor imprecisie (gering patiënten aantal). Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
6. Tricyclische antidepressiva (TCA’s)
6.1 Nortriptyline/ Amitriptyline versus placebo
6.1.1 Uitkomstmaat Pijn (been) (cruciaal)
Khoromi (2007) en van Elderen (2015) onderzochten pijn in het been (op een schaal van 0 tot 100) na gebruik van respectievelijk nortriptyline en amitriptyline versus placebo waarbij Khoromi (2007) de pijn iedere avond scoorde en het gemiddelde over 2 weken onderzocht en van Elderen (2015) de pijnscore na twee weken onderzocht. Khoromi (2007) rapporteerde een gemiddeld verschil van -7,0 in het voordeel van nortriptyline, hetgeen niet statistisch significant was (95% BI -21,1 tot 7,1, p=0,33). Van Elderen (2015) rapporteerde een gemiddeld verschil van -14,7 in het voordeel van amitriptyline (95% BI –28,3 tot 1,6; p= 0,035). Dit verschil was klinisch relevant.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; loss to follow up) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
6.1.2 Uitkomstmaat Functioneren (belangrijk)
Khoromi (2007) onderzocht functioneren gemeten met de Oswestry Low Back Pain Disability questionnaire (op een schaal van 0 tot 100) en vond een gemiddeld verschil van -3,0, in het voordeel van nortriptyline, hetgeen niet klinisch relevant was (95% BI -11,5 tot 5,5). Van Elderen (2015) heeft deze uitkomstmaat niet gerapporteerd.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘functioneren’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias: loss to follow up) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
6.1.3 Uitkomstmaat Adverse events (belangrijk)
Khoromi (2007) rapporteerde dat van de patiënten die gerandomiseerd werden (N=55) drie patiënten zich terugtrokken in verband met verminderd bewustzijn waarvan twee patiënten in de amitriptylinegroep en één in de placebogroep. De number needed to harm (NNH) was 30. Deze werd berekend over alle patiënten die werden gerandomiseerd (N=55) en zich terugtrokken uit de studie in verband met adverse events. Van degenen die de studie voltooiden (N=28) werden er bij 68% (19/28) van de patiënten in de interventiegroep adverse events gerapporteerd, ten opzichte van 50% (14/28) in de placebogroep, wat leidde tot een risk ratio van 1,36 (95% BI 0,87 tot 2,13, p=0,18). Dit verschil was klinisch relevant. De meest voorkomende adverse events die werden gerapporteerd in de nortriptyline-groep waren: droge mond (36 keer), constipatie (25 keer), vermoeidheid (11 keer) en slaperigheid (11 keer). De meest voorkomende adverse events in de placebogroep waren: droge mond (21 keer), vermoeidheid (18 keer) en hoofdpijn (14 keer). Van Elderen (2015) rapporteerde in 2/20 (10%) van de patiënten in de interventiegroep adverse events ten opzichte van 0% in de placebogroep. De meest voorkomende adverse events waren misselijkheid, overgeven, een algeheel ziek gevoel en huiduitslag.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias: loss to follow up) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
6.2 Combinatie nortriptyline + morfine versus placebo
6.2.1 Uitkomstmaat Pijn (cruciaal)
Khoromi (2007) rapporteerde pijn in het been (VAS-score op een schaal van 0 tot 100) waarbij de gemiddelde pijn score iedere avond werd uitgevraagd en het gemiddelde over twee weken werd onderzocht. Het gemiddelde verschil was -3,0 in het voordeel van de combinatie nortriptyline + placebo, hetgeen niet klinisch relevant was (95% BI -16,6 tot 10,6).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘pijn’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design, incomplete uitkomst data en niet voldoen aan intention-to-treat analyse) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
6.2.2 Uitkomstmaat Functioneren (belangrijk)
Khoromi (2007) onderzocht functioneren gemeten met de Oswestry Low Back Pain Disability questionnaire (op een schaal van 0 tot 100) en vond een gemiddeld verschil van -3,1, in het voordeel van nortriptyline, hetgeen niet klinisch relevant was (95% BI -11,3 tot 5,1).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘functioneren’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design, incomplete uitkomst data en niet voldoen aan intention-to-treat analyse) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
6.2.3 Uitkomstmaat Adverse events (belangrijk)
Khoromi (2007) rapporteerde dat er van de 55 patiënten die gerandomiseerd werden er twee patiënten zich terugtrokken in de combinatiegroep nortriptyline + morfine in verband met verminderd bewustzijn, één in verband met misselijkheid en braken en één in verband met huiduitslag. De number needed to harm (NNH) was 11. Deze werd berekend over alle patiënten die werden gerandomiseerd (N=55) en zich terugtrokken uit de studie in verband met adverse events. Van degenen die de studie voltooiden (N=28) werden er bij 25/28 personen (89%) in de interventiegroep adverse events gerapporteerd en in 50% (14/28) van de placebogroep, wat leidde tot een risk ratio van 1,79 (95% BI 1,21 tot 2,64). Dit verschil was klinisch relevant. De meest voorkomende adverse events die voorkwamen in de combinatiegroep waren: constipatie (71 keer), droge mond (29 keer), hoofdpijn (14 keer), versuft (11 keer), vermoeidheid (14 keer), slaperigheid (11 keer). De meest voorkomende adverse events in de placebogroep waren: droge mond (21 keer), vermoeidheid (18 keer) en hoofdpijn (14 keer).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘adverse events’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; cross-over design, incomplete uitkomst data en niet voldoen aan intention-to-treat analyse) en met twee niveaus voor imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
3.1 Diazepam versus placebo
3.1.4 Uitkomstmaat Werkhervatting (belangrijk)
Brötz (2010) onderzocht het effect van diazepam versus placebo op werkhervatting en definieerde dit als de duur van arbeidsongeschiktheid na ontslag uit het ziekenhuis (in dagen). De diazepam groep werkte niet gedurende 26 dagen (interkwartiele range 7 tot 42) en de placebogroep gedurende 15 dagen (interkwartiele range 7 tot 41), wat niet statistisch significant verschilde (p=0,73). Voor deze uitkomstmaat is de duur van ziekenhuisopname ook van belang: 10 dagen (interkwartiele range 8 tot 12) versus 8 dagen (interkwartiele range 7 tot 8) in het voordeel van de placebogroep (p=0,0008). De risk ratio voor het onvermogen om te werken na dag 28 was 1,3, dit verschil was klinisch relevant (95% BI 0,7 tot 2,2). Deze uitkomstmaat hebben de overige studies niet gerapporteerd.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘werkhervatting’ is met één niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; selectieve outcome reporting) en met twee niveaus vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘zeer laag’.
4.1 Pregabaline versus placebo
4.1.4 Uitkomstmaat Werkhervatting (belangrijk)
Mathieson (2017) onderzocht absentie van werk over het verloop van één jaar. Het gemiddelde verschil was voor de hele groep −50,6 uur, in het voordeel van pregabaline, hetgeen niet klinisch relevant was (95% BI, −109,5 tot 8,2; p=0,09) en voor de groep die in dienst waren op baseline was het gemiddelde verschil -97,6 uur, in het voordeel van pregabaline, hetgeen niet klinisch relevant significant was (95% BI -213,8 tot 18,6, p=0,10).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat ‘werkhervatting’ is met twee niveaus verlaagd vanwege imprecisie. Vanwege het studiedesign is het startpunt GRADE ‘hoog’, de uiteindelijke bewijskracht komt uit op GRADE ‘laag’.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat zijn de (on)gunstige effecten van orale medicatie ten opzichte van overig conservatief beleid bij patiënten met LRS?
PICO
P: patiënten met LRS (acuut en subacuut);
I: orale medicatie (NSAID’s, benzodiazepines, opioïden, neuropathische pijnmedicatie);
C: overige conservatieve behandelingen (niet zijnde medicatie);
O: pijn, functioneren, adverse events, werkhervatting.
Relevante uitkomstmaten
De werkgroep achtte pijn (in rug/been of gecombineerd) een voor de besluitvorming cruciale uitkomstmaat; en adverse events, functioneren en werkhervatting voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde a priori de Oswestry low back pain disability questionary (waaruit de oswestry disability (ODI) index (schaal 0 tot 100) berekend kan worden) en de Roland Morris disability questionnaire (met 11, 18 of 24 items) voor de uitkomstmaat functioneren of een alternatieve voor functioneren gevalideerde vragenlijst. Deze vragenlijsten richten zich op mate van functioneren door intensiteit van pijn tijdens ADL activiteiten. Voor de uitkomstmaat pijn werd de visual analog scale (VAS, schaal 0 tot 10 of 0 tot 100) of de numeric rating scale (NRS, schaal 0 tot 10 of 0 tot 100) aangehouden. De werkgroep definieerde niet a priori de uitkomstmaat werkhervatting, maar hanteerde de in de studies gebruikte definities.
Minimaal klinisch relevant verschil
Voor het vaststellen van klinische relevantie, de zogenaamde minimal clinically important difference (MCID), heeft de werkgroep zich gebaseerd op het artikel van Ostelo (2008), waar een 20% tot 30% verbetering ten opzichte van de baseline meting als minimaal klinisch relevant verschil wordt aangehouden (within-group change; verschil binnen een groep). De werkgroep is echter geïnteresseerd in between-group changes (verschil tussen twee groepen). Een verschil van 20-30 % tussen groepen werd door de werkgroep als (te) groot bestempeld. De werkgroep heeft besloten om een verschil van 10% aan te duiden als minimaal klinisch relevant verschil. Dit komt grofweg overeen met een verschil van één op de visuele analoge schaal (VAS-schaal: 0 tot 10), één op de numeric rating scale (NRS-schaal: 0 tot 10), drie op de Roland Disability Questionnaire (0 tot 24) en 10 op de oswestry disability (ODI) index (schaal 0 tot 100). Bij risk ratio’s en odds ratio’s worden de grenzen 0,91 en 1,1 aangehouden.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID) en Embase (via Embase.com) is op 17 december 2018 met relevante zoektermen gezocht naar gerandomiseerde studies die orale medicatie (NSAID’s, benzodiazepines, opioïden of neuropathische pijnmedicatie) evalueerden bij patiënten met LRS. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 299 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria:
- systematische reviews of RCT’s;
- volwassen patiënten met LRS en;
- NSAID’s, benzodiazepines, opioïden of neuropathische pijnmedicatie.
Op basis van titel en abstract werden in eerste instantie 34 studies voorgeselecteerd. Via snowballing werden er twee extra artikelen gevonden en 36 studies werden full tekst geraadpleegd. Na raadpleging van de volledige tekst, werden vervolgens 30 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording) en zes studies definitief geselecteerd.
Resultaten
Er is één systematische review (met zeven relevante RCT’s) en vijf RCT’s in de literatuuranalyse opgenomen. In totaal worden er elf vergelijkingen gepresenteerd. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk-of-biastabellen.
- Biondi D, Xiang J, Benson C, Etropolski M, Moskovitz B, & Rauschkolb C. Tapentadol immediate release versus oxycodone immediate release for treatment of acute low back pain. Pain Physician, 16(3), E237-46. 2013
- Brötz D, Maschke E, Burkard S, Engel C, Mänz C, Ernemann U et al. Is there a role for benzodiazepines in the management of lumbar disc prolapse with acute LRS?. Pain, 149(3), 470-475. 2010
- Brouwer BA, Boerman D, Groeneveld GJ. Opiaten: tips & tricks voor de neuroloog. De Neuroloog, juni 2019.
- Coxib and traditional NSAID Trialists' (CNT) Collaboration, Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013 Aug 31;382(9894):769-79. doi: 10.1016/S0140-6736(13)60900-9. Epub 2013 May 30. PubMed PMID: 23726390; PubMed Central PMCID: PMC3778977.
- Dreiser RL, Le Parc JM, Velicitat P et al. Oral meloxicam is effective in acute LRS: two randomised, double-blind trials versus placebo or diclofenac. Inflammation research, 50(1), 17-23. 2001
- Drendel AL, Gorelick MH, Weisman SJ, Lyon R, Brousseau DC, Kim MK. A randomized clinical trial of ibuprofen versus acetaminophen with codeine for acute pediatric arm fracture pain. Ann Emerg Med. 2009 Oct;54(4):553-60. doi: 10.1016/j.annemergmed.2009.06.005. Epub 2009 Aug 19. PubMed PMID: 19692147.
- Eisenberg E, Marinangeli F, Birkhahn J, et al. Time to modify the WHO analgesic ladder? Pain. Clinical Updates 2005:8(5).
- Herrmann WA, & Geertsen MS. Efficacy and safety of lornoxicam compared with placebo and diclofenac in acute LRS/lumbo‐LRS: an analysis from a randomised, double‐blind, multicentre, parallel‐group study. International journal of clinical practice, 63(11), 1613-1621. 2009
- De Jong L, Janssen PGH, Keizer D, Köke AJA, Schiere S, Van Bommel M, Van Coevorden RS, Van de Vusse A, Van den Donk M, Van Es A, Veldhoven CMM, Verduijn MM. NHG-Standaard Pijn. 2018.
- Ostelo RW, Deyo RA, Stratford P, et al. Interpreting change scores for pain and functional status in low back pain: towards international consensus regarding minimal important change. Spine (Phila Pa 1976). 2008;1;33(1):90-4.
- Khoromi S, Patsalides A, Parada S, Salehi V, Meegan JM et al. Topiramate in chronic lumbar radicular pain. The Journal of pain, 6(12), 829-836. 2005
- Khoromi S, Cui L, Nackers L et al. Morphine, nortriptyline and their combination versus. placebo in patients with chronic lumbar root pain. Pain, 130(1-2), 66-75. 2007
- Van der Linden MW, Westert GP, Bakker DH, Schellevis FG. Tweede nationale studie naar ziekten en verrichtingen in de huisartspratijk: klachten en aandoeningen in de bevolking en in de huisartspraktijk. Utrecht/Bilthoven. NIVEL/RIVM, 2004.
- Mathieson S, Maher CG, McLachlan AJ, Latimer J, Koes BW, Hancock MJ, Harris I, Day RO, Billot L, Pik J, Jan S, Lin CC. Trial of Pregabalin for Acute and Chronic Sciatica. N Engl J Med. 2017 Mar 23;376(12):1111-1120. doi: 10.1056/NEJMoa1614292. PubMed PMID: 28328324.
- Pinto RZ, Maher CG, Ferreira ML, Ferreira PH, Hancock M, Oliveira VC et al. Drugs for relief of pain in patients with LRS: systematic review and meta-analysis. Bmj, 344, e497. 2012
- Robertson K, Marshman LA, Plummer D, & Downs E. Effect of Gabapentin versus Pregabalin on Pain Intensity in Adults With Chronic LRS: A Randomized Clinical Trial. JAMA neurology, 76(1), 28-34. 2019
- Schmidt M, Sørensen HT, Pedersen L. Diclofenac use and cardiovascular risks: series of nationwide cohort studies. BMJ. 2018 Sep 4;362:k3426. doi: 10.1136/bmj.k3426. PubMed PMID: 30181258; PubMed Central PMCID: PMC6122252.
- Spijker-Huiges A, Groenhof F, Winters JC, van Wijhe M, Groenier KH, van der Meer K. Radiating low back pain in general practice: incidence, prevalence, diagnosis, and long-term clinical course of illness. Scand J Prim Health Care. 2015 Mar;33(1):27-32.
- Stichting Farmaceutische Kengetallen (SFK). Aantal oxycodongebruikers vrijwel ongewijzigd in 2018. Maart 2019. https://www.sfk.nl/publicaties/PW/2019/aantal-oxycodongebruikers-vrijwel-ongewijzigd-in-2018
- Van Elderen, P., Van Zundert, J., Kozicz, T., Puylaert, M., De Vooght, P., Mestrum, R., & Vissers, K. (2015). Effect of Minocycline on Lumbar Radicular Neuropathic PainA Randomized, Placebo-controlled, Double-blind Clinical Trial with Amitriptyline as a Comparator. Anesthesiology: The Journal of the American Society of Anesthesiologists, 122(2), 399-406.
- Woo WW, Man SY, Lam PK, Rainer TH. Randomized double-blind trial comparing oral paracetamol and oral nonsteroidal antiinflammatory drugs for treating pain after musculoskeletal injury. Ann Emerg Med. 2005 Oct;46(4):352-61. PubMed PMID: 16187469.
- World Health Organization (WHO). Cancer pain relief. 1986.
Evidence table for systematic review of RCTs and observational studies (intervention studies) (Orale medicatie)
Evidence table for intervention studies (Orale medicatie)
Evidence table for systematic review of RCTs and observational studies (intervention studies) (neuropatische medicatie)
Evidence table for intervention studies (neuropatische medicatie)
Risk of Bias assessment
Table of quality assessment for systematic reviews of RCTs and observational studies (orale medicatie & neuropatische medicatie)
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/ notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
|
Rasmussen-Bar, 2017 |
Yes |
Yes |
Yes |
Yes but minimal |
Not applicable |
Yes |
Yes |
Yes |
Yes |
|
|
Pinto, 2012 |
Yes |
Yes |
Yes |
Yes |
NA |
Yes |
Yes |
Yes |
Yes |
A and B high risk of bias: influence of co-interventions (B) or unclear bias for : selection bias, detection bias, attrition bias (A), group similarity at baseline (A), compliance with interventions (B), funding or other bias (A) C: low risk of bias: (only bias for funding) |
|
Pinto, 2012 |
Yes |
Yes |
Yes |
Yes |
NA |
Yes |
Yes |
Yes |
Yes |
|
Risk of bias table for intervention studies (randomized controlled trials) (orale medicatie)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4 (unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
|
A. Weber, 1993 |
Not described how patients were randomized |
Unlikely |
Unlikely |
Unlikely |
Unclear / unlikely for pain and functioning |
Unlikely |
Likely |
Likely (patient that did not appear for evaluation at 4 wks were excluded). |
Funding bias |
|
B. Dreiser, 2001 |
Not described how patients were randomized |
Unclear |
Unlikely |
Unlikely |
Unclear / unlikely for pain and functioning |
Unlikely |
Unlikely |
Unclear |
|
|
C. Hermann, 2009 |
Block randomization: a seven-digit identification code (containing a three-digit random number) |
Unlikely |
Unlikely |
Unlikely |
Unclear / unlikely for pain and functioning |
Unlikely |
Unlikely |
Unlikely |
Funding bias |
|
Biondi, 2013 |
computer-generated randomization schedule |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Likely |
High loss to follow up due to AE (86,3% versus 82,7%) |
Unlikely |
|
|
Brötz, 2010 |
computerized randomization list |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
|
Risk of bias table for intervention studies (randomized controlled trials) Neuropatische medicatie
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Mathieson, 2017 |
a computer derived random-number sequence |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely (>10% but in both groups) |
Unlikely |
|
Robertson, 2018 |
computer-derived permuted block with varying block size sequence. Cross-over |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
|
Van Elderen, 2015 |
computerized random number generator |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
|
Yildirim, 2003 |
Randomised, not further described how |
Unclear |
Unclear |
Unclear |
Unclear |
Unlikely |
Unlikely |
Likely |
|
Khoromi, 2005 |
Cross-over trial; blocked randomization in blocks of 8 and 4 within a table of 32 random numbers before patient enrollment |
Unclear |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Likely |
Likely |
|
Khoromi, 2007 |
Cross-over trial; by random numbers within blocks of four to one of four treatment sequences specified by a Latin square |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Likely |
Likely |
|
Baron, 2010 |
A central Internet/telephone randomization system (IMPALA) |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Likely |
Likely |
Likely |
Tabel Exclusie na het lezen van het volledige artikel
|
|
Auteur en jaartal |
Redenen van exclusie |
|
1 |
Ansari, 2018 |
Voldoet niet aan PICO, metformin |
|
2 |
Backonja, 2017 |
Voldoet niet aan PICO, intraveneuze injectie |
|
3 |
Baron 2010 |
Voldoet niet aan PICO; ivm design overloop van interventies |
|
4 |
Chou, 2007 |
Studies zijn geïncludeerd in systematische review van Rasmussen, 2017 |
|
5 |
Dosenovic, 2017 |
Voldoet niet aan PICO, studies die wel voldoen zijn los opgenomen of in systematische review Rasmussen, 2017 |
|
6 |
Dworkin, 2007 |
Voldoet niet aan PICO, Khoromi 2005 en 2007 los geïncludeerd |
|
7 |
Ebell, 2017 |
Geen origineel artikel: POEM = patient-oriented evidence that matters |
|
8 |
Enke, 2018 |
Voldoet niet aan PICO; geen subanalyse op LRS alleen |
|
9 |
Hahne, 2010 |
Enige studie waar naar wordt verwezen is Kanayama, 2005 |
|
10 |
Kanayama, 2005 |
Voldoet niet aan PICO; medicatie niet geregistreerd voor deze indicatie |
|
11 |
Holve, 2008 |
Voldoet niet aan PICO; prednison |
|
12 |
Jing, 2017 |
Voldoet niet aan PICO; etanercept |
|
13 |
Khoromi, 2005 |
Opgenomen in Pinto 2012 |
|
14 |
Khoromi, 2007 |
Opgenomen in Pinto 2012 |
|
15 |
Lewis , 2011 |
Voldoet niet aan PICO of al los geïncludeerd |
|
16 |
Luijsterburg, 2007 |
Voldoet niet aan PICO of in Rasmussen, 2017 geïncludeerd |
|
17 |
Mathieson, 2018 |
Voldoet niet aan PICO: artikel dat aan pt inclusie voldiet voldoet niet aan interventie-inclusie (VitB) |
|
18 |
Memeo, 2008 |
Voldoet niet aan PICO, voedingssupplementen |
|
19 |
Onda, 2013 |
Voldoet niet aan PICO, limaprost |
|
20 |
Ostenfeld, 2015 |
Voldoet niet aan PICO, losmapimod |
|
21 |
Pirdubak, 2015 |
Voldoet niet aan PICO, patiënten met steroïde injectie |
|
22 |
Radcliff, 2013 |
Voldoet niet aan selectiecriteria: niet gerandomiseerd |
|
23 |
Rasmussen-Barr, 2016 |
Opgenomen in Rasmussen-Bar, 2017 |
|
24 |
Rasmussen-Bar, 2017 |
Zelfde PICO en studies in Pinto, 2012 en getallen (N) in Rasmussen kloppen niet. |
|
25 |
Robertson, 2016 |
Voldoet niet aan PICO of al los geïncludeerd |
|
26 |
Roncoroni, 2011 |
Voldoet niet aan PICO, systemische steroïden |
|
27 |
Sumracki, 2012 |
Voldoet niet aan PICO, patiënten met capsaicin injectie |
|
28 |
Weber, 1993 |
Voldoet niet aan PICO |
|
29 |
Yildirim, 2003 |
Voldoet niet aan PICO en noodzakelijke informatie ontbreekt. |
|
30 |
Zhou, 2017 |
Voldoet niet aan PICO |
Beoordelingsdatum en geldigheid
Publicatiedatum : 03-08-2020
Beoordeeld op geldigheid : 21-09-2020
Voor het beoordelen van de actualiteit van deze richtlijn is de werkgroep niet in stand gehouden. Uiterlijk in 2025 bepaalt het bestuur van de Nederlandse Vereniging voor Neurologie (NVN) of de modules van deze richtlijn nog actueel zijn. Op modulair niveau is een onderhoudsplan beschreven. Bij het opstellen van de richtlijn heeft de werkgroep per module een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De NVN is regiehouder van deze richtlijnmodules en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
|
Module1 |
Regiehouder(s)2 |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn3 |
Frequentie van beoordeling op actualiteit4 |
Wie houdt er toezicht op actualiteit5 |
Relevante factoren voor wijzigingen in aanbeveling6 |
|
Orale Medicatie |
NVN |
2020 |
2025 |
1x per 5 jaar |
NVN |
- |
|
[1] Naam van de module 2 Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders) 3 Maximaal na vijf jaar 4 (half)Jaarlijks, eens in twee jaar, eens in vijf jaar 5 regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) werkgroep die in stand blijft 6 Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen |
||||||
Algemene gegevens
Deze richtlijn is ontwikkeld in samenwerking met:
-
Nederlands Huisartsen Genootschap
- Nederlandse Vereniging voor Arbeids- en Bedrijfsgeneeskunde
-
Koninklijk Nederlands Genootschap voor Fysiotherapie
- Samenwerkingsverband Pijnpatiënten naar één stem
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijn.
Doel en doelgroep
Doel
Het doel van de richtlijn is tweeledig. Enerzijds is de richtlijn een voortzetting van de richtlijn LRS uit 2008 met herziening op basis van de nieuwe literatuur. Anderzijds dient de richtlijn als handleiding voor de praktijk met betrekking tot de patiënt met rugpijn met uitstraling in een been. Er is namelijk, voorafgaande aan beeldvorming, geen klinische test of vragenlijst waarmee de klinische diagnose LRS met volledige zekerheid kan worden gesteld (zie module diagnostiek). Het kunstmatige onderscheid tussen radiculaire en niet-radiculaire pijn komt hiermee te vervallen.
Doelgroep
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen die betrokken zijn bij de zorg voor patiënten met LRS. De richtlijn is bedoeld voor neurologen, anesthesiologen, radiologen, neurochirurgen, orthopeden, huisartsen, fysiotherapeuten, bedrijfs- en verzekeringsartsen revalidatieartsen, en eventuele andere behandelaars van patiënten zoals Caesar- en Mensendieck therapeuten en ergotherapeuten.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2018 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met een verdenking lumbosacraal radiculair syndroom (LRS) te maken hebben.
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname. De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Werkgroep
- Dr. R.H. Boerman, neuroloog, werkzaam in Rijnstate, Arnhem, NVN (voorzitter)
- Dr. W.J.P. Henneman, radioloog, werkzaam in het Maastricht UMC+, Maastricht, NVvR (vanaf oktober 2019)
- Drs. B.A. Brouwer, neuroloog, werkzaam in het Maastricht UMC+, Maastricht, NVN
- Drs. J. de Haan, orthopedisch chirurg, werkzaam in het Amphia Ziekenhuis, Breda, NOV
- Dr. J.L. Hoving, senior onderzoeker, werkzaam in het Amsterdam UMC, locatie AMC, Amsterdam, NVAB
- Dr. E.M. Kingma, AIOS neurologie, werkzaam in het Universitair Medisch Centrum Groningen, Groningen, NVN
- Dr. B.C. ter Meulen, neuroloog, werkzaam in het Zaans Medisch Centrum, Zaandam, NVN
- Dr. E. de Schepper, huisarts, werkzaam in het Erasmus MC, Rotterdam. NHG
- Prof. dr. R.J.E.M. Smeets, hoogleraar Revalidatiegeneeskunde & revalidatiearts, werkzaam in bij de Universiteit Maastricht & CIR Revalidatie, VRA
- Dr. J. B. Staal, fysiotherapeut n.p. & senior onderzoeker/epidemioloog, werkzaam in het Radboudumc en de Hogeschool van Arnhem en Nijmegen, Nijmegen, KNGF
- Drs. M.A.M.B. Terheggen, anesthesioloog, werkzaam in Rijnstate, Arnhem, NVA
- Drs. I.L. Thomassen, patiëntvertegenwoordiger & voorzitter van Samenwerkingsverband Pijnpatiënten naar één stem.
- Dr. C.L.A.M. Vleggeert – Lankamp, Neurochirurg, Leids Universitair Medisch Centrum, Leiden, NVvN
Met ondersteuning van
- Dr. J. Buddeke, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Drs. A.A. Lamberts, senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Drs. L.H.M. Niesink-Boerboom, literatuurspecialist, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u hieronder. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Achternaam werkgroeplid |
Hoofdfunctie |
Nevenwerkzaamheden |
Persoonlijke financiële belangen |
Persoonlijke relaties |
Extern gefinancierd onderzoek |
Intellectuele belangen en reputatie |
Overige belangen |
Getekend op |
Ondernomen actie? |
|
Boerman |
neuroloog staflid Rijnstate ziekenhuis |
lid tuchtcolleges Groningen, Eindhoven, Amsterdam en Zwolle |
geen |
geen |
geen |
geen |
geen |
20-6-2018 |
Geen actie |
|
Brouwer |
Neuroloog afdeling anesthesiologie MUMC+ |
Lid algemeen bestuur P.A.I.N. (onbetaald) |
geen |
geen |
geen |
geen |
Afdeling waar WG-lid werkzaam is heeft van Medtronic een Grant ontvangen. |
12-6-2018 |
Geen actie |
|
Buddeke |
Adviseur, Kennisinstituut van de Federatie Medisch Specialisten |
geen |
geen |
geen |
geen |
geen |
geen |
2018 |
Geen actie |
|
de Haan |
Orthopaedisch chirurg, Amphia breda |
geen |
geen |
geen |
geen |
geen |
geen |
5-6-2018 |
Geen actie |
|
de Schepper |
Onderzoeker (UD), huisarts (np), afdeling Huisartsgeneeskunde, Erasmus MC Rotterdam |
geen |
geen |
geen |
geen |
Promotieonderzoek Beeldvormende diagnostiek |
geen |
1-5-2018 |
Geen actie |
|
Henneman |
Radiolooog MUMC+, Maastricht |
geen |
geen |
geen |
geen |
geen |
geen |
16-12-2019 |
Geen actie |
|
Hoving |
Wetenschappelijk onderzoeker, Coronel Instituut voor Arbeid en Gezondheid, Academisch Medisch Centrum in Amsterdam |
geen |
geen |
geen |
geen |
Betrokken bij richtlijn Arbeid en rugklachten LRS van de NVAB. |
geen |
2-7-2018 |
Geen actie |
|
Kingma |
Arts-assistent in opleiding tot neuroloog, Universitair Medisch Centrum Groningen |
geen |
geen |
geen |
geen |
geen |
geen |
11-6-2018 |
Geen actie |
|
Lamberts |
Senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten |
Beleidsmedewerker Kwaliteit Nederlandse Vereniging voor Klinische Geriatrie |
geen |
geen |
geen |
geen |
geen |
2014 |
Geen actie |
|
Smeets |
Hoogleraar Revalidatiegeneeskunde, Universiteit Maastricht (0.4 fte) |
Eigen bedrijf genaamd RevaXpert; inhuur van mijn expertise voor scholing derden op het gebied van chronische pijn in houding en bewegingsapparaat in de vorm van presentaties, gemiddeld twee keer per jaar. (Betaald) |
geen |
geen |
Niet betrokken bij wetenschappelijk onderzoek dat betrekking heeft op de inhoud van deze richtlijn. |
Geen risico op aantasting van intellectuele belangen of reputatie |
geen |
8-6-2018 |
Geen actie |
|
Staal |
Lector musculoskeletale revalidatie hogeschool, van Arnhem en Nijmegen (0,8 fte), |
-Section editor BMC Musculosceletal disorders (betaald) |
geen |
geen |
geen |
geen |
geen |
14-5-2018 |
Geen actie |
|
ter Meulen |
Neuroloog zaans MC |
Lid Raad van Advies NVvR de Wervelkolom (onbetaald) |
geen |
geen |
geen |
Promotieonderzoek naar Wortelblokkades, Amsterdam UMC, locatie Vumc |
geen |
27-6-2018 |
Geen actie |
|
Terheggen |
Anesthesioloog/pijnspecialist Rijnstate Arnhem/Velp/Zevenaar (1,0 fte) |
Specialist-manager Pijncentrum en Acute PijnService Rijnstate (betaald) |
geen |
Echtgenote is werkzaam bij Pfizer inc. .Geen mogelijke belangen-verstrengeling aangezien Pfizer geen gepatenteerde producten (meer) levert geïndiceerd bij LRS |
geen |
geen |
geen |
3-7-2018 |
Geen actie |
|
Thomassen |
Voorzitter Samenwerkingsverband Pijnpatiënten naar één stem (vacatiegelden) Voorzitter Patiëntenvereniging CRPS (vrijwilliger) |
geen |
geen |
geen |
geen |
geen |
geen |
19-7-2018 |
Geen actie |
|
Vleggeert |
Neurochirurg LUMC, (1,0 fte) |
- Secretaris van de Board van de Cervical Spine Research Society Europe (onbetaald) |
geen |
geen |
Onderzoek naar epiduraalinjecties bij sciatica (Ynske Meyesfonds). |
geen |
geen |
25-4-2018 |
Geen actie |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde van een patiëntenvereniging plaats te laten nemen in de werkgroep. Tijdens de oriënterende zoekactie werd gezocht op literatuur naar patiëntenperspectief (zie Strategie voor zoeken en selecteren van literatuur). De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland, het Samenwerkingsverband Pijnpatiënten naar één stem, de Nederlandse Vereniging van Rugpatiënten 'De Wervelkolom' en de Dwarslaesie Organisatie Nederland.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijnmodules en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden bij de aanverwante producten.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naarhet stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. Tevens zijn er knelpunten aangedragen door verschillende stakeholders tijdens de invitational conference. Een verslag van deze bijeenkomst is opgenomen onder aanverwante producten.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de voorzitter en de adviseur conceptuitgangsvragen opgesteld. Deze zijn met de werkgroep besproken waarna de werkgroep de definitieve uitgangsvragen heeft vastgesteld. Vervolgens inventariseerde de werkgroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Er is op 23 januari 2019 oriënterend gezocht naar bestaande buitenlandse richtlijnen en literatuur over patiëntvoorkeuren en patiëntrelevante uitkomstmaten. Voor de afzonderlijke uitgangsvragen is aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie voor de oriënterende zoekactie en patiëntenperspectief zijn opgenomen onder aanverwante producten.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR - voor systematische reviews; Cochrane - voor gerandomiseerd gecontroleerd onderzoek; Newcastle-Ottowa - voor observationeel onderzoek; QUADAS II - voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A) Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk* |
|
|
Laag |
|
|
Zeer laag |
|
*in 2017 heeft het Dutch GRADE Network bepaalt dat de voorkeursformulering voor de op een na hoogste gradering ‘redelijk’ is in plaats van ‘matig’
B) Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008) en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de cruciale uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Randvoorwaarden conservatief en in de module Randvoorwaarden chirurgische ingreep.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Brouwers, M. C., Kho, M. E., Browman, G. P., Burgers, J. S., Cluzeau, F., Feder, G., ... & Littlejohns, P. (2010). AGREE II: advancing guideline development, reporting and evaluation in health care. Canadian Medical Association Journal, 182(18), E839-E842.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van Medisch Specialisten.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann, H. J., Oxman, A. D., Brozek, J., Glasziou, P., Jaeschke, R., Vist, G. E., ... & Bossuyt, P. (2008). Rating Quality of Evidence and Strength of Recommendations: GRADE: Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ: British Medical Journal, 336(7653), 1106.
Wessels, M., Hielkema, L., & van der Weijden, T. (2016). How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. Journal of the Medical Library Association: JMLA, 104(4), 320.
Zoekverantwoording
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
2000 – december 2018
|
1 (((lumbosacral or lumbar) adj2 radicular adj2 (pain or syndrom*)) or 'lumbosacral radiculopathy').ti,ab,kw. or exp *LRS/ or sciatic*.ti. or exp *Intervertebral Disc Displacement/ or ('lumbar disc hernia*' or 'herniated disc').ti. or ('disc protusion' or 'nerve root pain').ti,ab,kw. (24877) 2 (exp Low Back Pain/ or ('low back' adj2 pain*).ti,ab,kw.) and acute.ti,ab,kw. and leg*.ti,ab,kw. (192) 3 1 or 2 (25028) 4 exp Acetaminophen/ or exp Ibuprofen/ or exp Piroxicam/ or exp Indomethacin/ or exp PHENYLBUTAZONE/ or exp DICLOFENAC/ or exp TRAMADOL/ or exp Morphine/ or exp OXYCODONE/ or exp FENTANYL/ or exp BUPRENORPHINE/ or exp *Anti-Inflammatory Agents, Non-Steroidal/ or exp *Analgesics/ or exp Analgesics, Opioid/ or ('oral medicat*' or paracetamol or ibuprofen or coxib* or piroxicam or tizanidin* or meloxicam or indomethacin* or indometacin* or phenylbutazone or diclofenac or opioid* or tramal or tramadol or ultram or morphin* or oxycodon* or percocet* or oxycontin or fentanyl or fentanil or butrans or buprenorphin* or subutex or palexia or tapentadol or nucynta or tapal or sarpogrelat*).ti,ab,kw. (441622) 5 3 and 4 (767) 6 limit 5 to (english language and yr="2007 -Current") (382) 7 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (377842) 8 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1812521) 9 6 and 7 (40) 10 6 and 8 (154) 11 9 or 10 (160) 12 exp Anticonvulsants/ or exp PREGABALIN/ or exp AMITRIPTYLINE/ or exp NORTRIPTYLINE/ or exp Duloxetine Hydrochloride/ or (pregabalin or lyrica or gabapentin or neurontin or amitriptylin* or elavil or nortriptylin* or allegron or aventyl or noritren or nortrilen or pamelor or duloxetin*).ti,ab,kw. (148641) 13 3 and 12 (175) 14 limit 13 to yr="2000 -Current" (132) 15 7 and 14 (7) 16 8 and 14 (35) 17 15 or 16 (35) 18 11 or 17 (170) = 170 (168 uniek) totaal |
299 |
|
Embase (Elsevier) |
((('lumbosacral radicular pain'/exp OR (((lumbosacral OR lumbar) NEAR/2 radicular NEAR/2 (pain OR syndrom*)):ab,ti)) OR ('lumbosacral radiculopathy'/exp OR 'lumbosacral radiculopathy':ab,ti)) OR ('LRS'/exp/mj OR sciatic*:ti OR 'lumbar disk hernia'/exp/mj OR 'lumbar disc hernia*':ti OR 'herniated disc':ti OR 'disc protusion':ab,ti OR 'nerve root pain':ab,ti) OR (('low back pain'/exp OR (('low back' NEAR/2 pain*):ab,ti)) AND acute:ab,ti AND leg*:ab,ti))
AND ('paracetamol'/exp OR 'cyclooxygenase 2 inhibitor'/exp OR 'piroxicam'/exp OR 'tizanidine'/exp OR 'meloxicam'/exp OR 'indometacin'/exp OR 'phenylbutazone'/exp OR 'diclofenac'/exp OR 'tramadol'/exp OR 'morphine'/exp OR 'oxycodone'/exp OR 'fentanyl'/exp OR 'buprenorphine'/exp OR 'tapentadol'/exp OR 'sarpogrelate'/exp OR 'analgesic agent'/exp/mj OR 'nonsteroid antiinflammatory agent'/exp/mj OR 'opiate'/exp/mj OR 'oral medic*':ab,ti OR paracetamol:ab,ti OR ibuprofen:ab,ti OR coxib*:ab,ti OR piroxicam:ab,ti OR tizanidin*:ab,ti OR meloxicam:ab,ti OR indomethacin*:ab,ti OR indometacin*:ab,ti OR phenylbutazone:ab,ti OR diclofenac:ab,ti OR opioid*:ab,ti OR tramal:ab,ti OR tramadol:ab,ti OR ultram:ab,ti OR morphin*:ab,ti OR oxycodon*:ab,ti OR percocet:ab,ti OR oxycontin:ab,ti OR fentanyl:ab,ti OR fentanil:ab,ti OR butrans:ab,ti OR buprenorphin*:ab,ti OR subutex:ab,ti OR palexia:ab,ti OR tapentadol:ab,ti OR nucynta:ab,ti OR tapal:ab,ti OR sarpogrelat*:ab,ti)
AND (english)/lim AND (2007-2018)/py NOT 'conference abstract':it) = 232 OR (((('lumbosacral radicular pain'/exp OR (((lumbosacral OR lumbar) NEAR/2 radicular NEAR/2 (pain OR syndrom*)):ab,ti)) OR ('lumbosacral radiculopathy'/exp OR 'lumbosacral radiculopathy':ab,ti)) OR ('LRS'/exp/mj OR sciatic*:ti OR 'lumbar disk hernia'/exp/mj OR 'lumbar disc hernia*':ti OR 'herniated disc':ti OR 'disc protusion':ab,ti OR 'nerve root pain':ab,ti) OR (('low back pain'/exp OR (('low back' NEAR/2 pain*):ab,ti)) AND acute:ab,ti AND leg*:ab,ti))
AND ('anticonvulsive agent'/exp/mj OR 'pregabalin'/exp OR 'gabapentin'/exp OR 'amitriptyline'/exp OR 'nortriptyline'/exp OR 'duloxetine'/exp OR pregabalin:ab,ti OR lyrica:ab,ti OR gabapentin:ab,ti OR neurontin:ab,ti OR amitriptylin*:ab,ti OR elavil:ab,ti OR nortriptylin*:ab,ti OR allegron:ab,ti OR aventyl:ab,ti OR noritren:ab,ti OR nortrilen:ab,ti OR pamelor:ab,ti OR duloxetin*:ab,ti)
AND (english)/lim AND (2000-2018)/py NOT 'conference abstract':it)) = 68 = 253 (251 uniek) totaal |
- Achtergrond en definities
- Toepassen
- Onderzoek