Keuze van anti-VEGF middel
Uitgangsvraag
Welk anti-VEGF geneesmiddel is eerste keus bij de behandeling van neovasculaire LMD?
Deze uitgangsvraag omvat de volgende onderliggende vragen:
- Wat is de effectiviteit van bevacizumab in vergelijking met andere middelen (ranibizumab, aflibercept (2 mg), brolucizumab, biosimilars voor anti-VEGF)?
- Wat is de effectiviteit van aflibercept (2 mg) in vergelijking met andere middelen (bevacizumab, ranibizumab, brolucizumab, biosimilars voor anti-VEGF)?
Aanbeveling
Geef intravitreaal bevacizumab (Avastin) als middel van eerste keus bij neovasculaire LMD.
Geef als middel van tweede keuze aflibercept (2 mg) of ranibizumab (geen voorkeur).
Geef als middel van derde keuze aflibercept (2 mg) of ranibizumab (geen voorkeur).
Overweeg als middel van vierde keuze brolucizumab. Dit middel heeft echter een minder gunstig bijwerkingen profiel.
Specifieke aanbevelingen bij gebruik van brolucizumab:
- Geef brolucizumab niet in beide ogen op hetzelfde moment.
- Geef brolucizumab niet in ogen met een actieve of doorgemaakte intraoculaire ontsteking of doorgemaakte retinale (occlusieve) vasculitis.
- Bespreek de bijwerkingen met de patiënt voor toediening van brolucizumab en benoem de bijbehorende symptomen.
- Controleer de patiënt vier weken na iedere intravitreale injectie met brolucizumab op intraoculaire ontsteking, retinale (occlusieve) vasculitis, gedurende de eerste zes maanden. Maak bij twijfel een fluorescentie angiogram
- Stop direct de behandeling met brolucizumab in het geval van een intraoculaire ontsteking en/of retinale vasculitis.
Overweeg in het geval van een patiënt met een stabiel klinisch beeld te switchen naar een biosimilar van hetzelfde originele anti-VEGF middel, indien beschikbaar tegen lagere kosten. Een extra controle is niet noodzakelijk, naast de reguliere controle op basis van het behandelinterval.
Overweeg in het geval van non-respons op een bepaald anti-VEGF middel, en anti-VEGF therapie wel de beste behandeloptie is, het gebruik van een biosimilar van een ander anti-VEGF middel.
Informeer de patiënt over de overstap naar een biosimilar. Een informed consent is niet nodig.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
In de afgelopen periode zijn er een aantal nieuwe middelen met de indicatie nLMD op de markt gekomen, waaronder brolucizumab (Beovu) en een aantal biosimilars. Ook is het aantal vergelijkende studies toegenomen. Om deze reden is opnieuw een literatuurstudie verricht met de vraag of er verschillen zijn in visus behoud /verbetering, dikte van de retina en veiligheid tussen verschillende anti-VEGF middelen bij patiënten met neovasculaire LMD.
Twee systematische reviews en twee gerandomiseerde studies vonden geen klinisch relevante verschillen voor de cruciale uitkomstmaat visus behoud/verbetering. De totale bewijskracht, de laagste bewijskracht voor de cruciale uitkomstmaat, is beoordeeld als ‘redelijk’ door het ontbreken van blindering (risk of bias). Op basis van deze resultaten is er geen duidelijke voorkeur voor één anti-VEGF middel.
Er zijn ook andere overwegingen die kunnen bijdragen aan de keuze van het anti-VEGF middel. De financiële kosten zijn hierbij een belangrijke factor gezien de grote verschillen in prijs van de diverse middelen. Ook hierin is de afgelopen jaren geen belangrijke verandering gekomen. De kosten van anti-VEGF middelen zijn in 2022 ongeveer €30 voor bevacizumab, €800 voor aflibercept (2 mg) en brolucizumab en €700 voor ranibizumab (Famacotherapeutisch Kompas, geraadpleegd september 2022). De kosteneffectiviteit van bevacizumab, ranibizumab en aflibercept (2 mg) is goed onderbouwd (Elshout, 2014; Van Asten, 2018; Quist 2022). In de kosteneffectiviteitsanalyse van Van Asten euit 2018 is berekend dat de directe en indirecte kosten van bevacizumab €27.087 per jaar bedragen, ongeveer €4.000 minder dan aflibercept (2 mg) en €6000 minder dan ranibizumab. In een andere Nederlandse studie is gemodelleerd wat de kosten zijn over een periode van 3 jaar van behandeling met bevacizumab, ranibizumab en aflibercept (2 mg). Hierbij is uitgegaan van een treat-and-extend protocol zoals toegepast in de registratie studies. Dit kwam neer op een aantal injecties van 25,4 voor bevacizumab, 23,4 voor ranibizumab en 12,2 voor aflibercept (2 mg). Met deze injectiefrequentie bedroegen de totale kosten per patiënt, inclusief toediening, diagnostiek, aantal consulten en bijwerkingen, over een periode van 3 jaar €14.215, €18.202 en €31.048 bedragen, voor respectievelijk bevacizumab, aflibercept (2 mg) en ranibizumab (Quist, 2022). In een Amerikaanse kostenutiliteitsanalyse vanuit oogheelkundige/medische kosten perspectief waarbij bevacizumab, ranibizumab en aflibercept (2 mg) werden vergeleken, bleek dat alle 3 middelen kosteneffectief waren over een periode van 11 jaar (Brown, 2020). Echter, hetzelfde model toegepast op 2 jaar toonde aan dat alleen bevacizumab kosteneffectief is, en ranibizumab en aflibercept (2 mg) niet.
In september 2022 zijn een aantal biosimilars geautoriseerd door de EMA. Byooviz® is een biosimilar van Ranibizumab en is geautoriseerd door zowel de EMA als de FDA voor de indicatie nLMD en maculaoedeem bij diabetische retinopathie en veneuze occlusies. Voor zover bekend is dit middel echter nog niet beschikbaar in Nederland. Een ander biosimilar van Ranibizumab is Razumab®, wat in 2015 is geautoriseerd in India, maar niet in Europa. Mvasi is een biosimilar voor bevacizumab, geautoriseerd door de EMA voor chemotherapeuticum voor diverse types kanker. Vergelijkende literatuur betreffende de toepassing van biosimilars bij neovasculaire LMD is nog zeer beperkt (Sharma, 2021). Echter, in lijn met het Standpunt Biosimilars van de Federatie Medisch Specialisten heeft het NOG een Addendum geschreven. Hierin wordt gesteld dat in het geval van een patiënt met een stabiel klinisch beeld het te overwegen valt om te switchen naar een biosimilar van hetzelfde originele anti-VEGF middel, indien beschikbaar tegen lagere kosten. Ook kan in het geval van non-respons op een anti-VEGF middel overwogen worden een biosimilar te gebruiken als één van de therapeutische opties bij switch naar een ander anti-VEGF middel.
Uit de literatuuranalyse is niet gebleken dat er subgroepen van patiënten met neovasculaire LMD bestaan die anders reageren op specifieke anti-VEGF middelen. Om deze reden wordt er geen onderscheid gemaakt in onze aanbevelingen tussen subgroepen.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Patiënten zijn gebaat bij een snelle start van de behandeling, met een geneesmiddel wat de grootste kans heeft op succes bij het kleinste aantal behandelingen en controles. Tegelijkertijd moet dit middel zo veilig mogelijk zijn. In deze module onderzoeken we welk geneesmiddel voldoet aan de hiervoor genoemde criteria. Op grond hiervan komen we tot een advies welk middel eerste keus is en welke middel later in de behandeling ingezet kan worden. Dit advies is van toepassing op alle patiënten, met een enkele uitzondering, zoals overgevoeligheid voor een bepaald middel.
De procedure van toediening en de vervolgstappen in de behandeling zijn hetzelfde voor alle hier genoemde geneesmiddelen. De keuzes om te starten met de behandeling of om de behandeling wel of niet voort te zetten komen in andere modules aan de orde.
Bijwerkingen
In een systematische review van niet-industrie gesponsorde RCTs konden geen verschillen worden vastgesteld tussen intravitreaal bevacizumab en ranibizumab voor wat betreft dood en systemic serious adverse events in de eerste twee jaar van behandeling, met uitzondering van gastrointestinale klachten (Moja, 2014).
Ook tussen aflibercept (2 mg) en ranibizumab werd geen verschil gevonden in bijwerkingen, hoewel de betreffende studies strikt genomen onvoldoende statistische power hadden om exacte schattingen te geven over absolute en relatieve risico’s.
Wat betreft brolucizumab lijken er wel problemen te zijn met de oogheelkundige veiligheid. In de registratie studies, HAWK en HARRIOR, werd geconcludeerd dat brolucizumab een overall well-tolerated safety profile heeft. Wel werden hierin al meer iritis/uveitis gevallen gerapporteerd in de brolucizumab groep dan in de aflibercept (2 mg) groep. In een post-hoc analyse van dezelfde studies werd door een onafhankelijk safety committee gevonden dat het risico op drug-related intraoculaire inflammatie (IOI), inclusief retinale vasculitis en retinale vasculaire occlusie (RVO) 4,6% bedraagt (Mones 2021). Dezelfde incidentie bij aflibercept (2 mg) behandelde ogen was 1.1%.
In latere studies is deze bijwerking nader onderzocht, waarbij er inderdaad een verband blijkt te bestaan tussen brolucizumab en het optreden van IOI en RVO. In een grote registratie studie met meer dan 11.000 ogen bleek dat de incidentie van deze bijwerking 2.4% bedroeg (Khanani, 2021). Patiënten met IOI en/of RO in het jaar voor de eerste brolucizumab behandeling hadden het hoogste risico. Ook vrouwen hadden een hoger risico dan mannen. Gebaseerd op deze informatie lijkt er een contra-indicatie te bestaan voor brolucizumab bij patiënten met een actieve IOI en bij patiënten met een voorgeschiedenis van IOI en/of retinale occlusie in de 12 maanden voor de eerste brolucizumab injectie.
In lijn met het Standpunt Werkgroep Medische Retina uit 2020, adviseren wij om de eerste 6 maanden 2-4 weken na iedere injectie (voorafgaand aan de volgende injectie) het oog op IOI te controleren, funduscopie te verrichten om vasculitis uit te sluiten, en bij twijfel een fluorescentie angiogram te verrichten.
Bilaterale injecties op dezelfde dag wordt afgeraden, in ieder geval in de eerste 6 maanden. Het interval tussen 2 doses brolucizumab in de onderhoudsfase, na de eerste 3 injecties, mag niet minder dan 8 weken bedragen.
Vanwege bovengenoemde risico’s op ernstige bijwerkingen moet deze informatie gedeeld worden met patiënten bij de indicatiestelling voor brolucizumab. Tevens moet patiënten worden geadviseerd zich te laten controleren bij klachten, zoals floaters, visusdaling, of fotofobie.
Deze adviezen staan ook in de Direct Healthcare Professional Communication (DHPC), welke in november 2021 is uitgebracht.
Kosten (middelenbeslag)
Voor de maatschappij is kosteneffectiviteit een belangrijk argument. Omdat de behandeling van neovasculaire LMD meestal jaren moet worden voortgezet met vele intravitreale injecties, spelen de kosten van het middel en de procedure een belangrijke rol. Een Budget Impact Analyse is een onderdeel van deze module en kan worden gevonden in de bijlage.
Aanvaardbaarheid, haalbaarheid en implementatie
De aanbevelingen komen grotendeels overeen met de huidige praktijk en zullen niet tot grote veranderingen leiden. Het feit dat het middel van eerste keuze Avastin (bevacizumab) niet is geregistreerd als geneesmiddel voor de indicatie LMD zal geen belemmering vormen, aangezien hiervoor al toestemming is gegeven door de landsadvocaat. Ook voor de maatschappelijke kosten van LMD behandeling heeft onze aanbeveling geen consequenties.
Aanbeveling-1
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Op basis van het literatuuronderzoek concluderen wij dat er gemiddeld genomen geen verschil bestaat in effectiviteit tussen de diverse geneesmiddelen voor subretinale neovascularisaties bij LMD. Wel zijn er verschillen in potentiele bijwerkingen. Om deze reden adviseren wij bij de behandeling van neovasculaire LMD te starten met bevacizumab, het middel met de beste kosteneffectiviteit.
Aanbeveling-2
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Hoewel in een specifieke meta-analyse werd gesuggereerd dat aflibercept (2 mg) geassocieerd is met minder ernstige oogheelkundige bijwerkingen dan ranibizumab (Sarwar, 2016), komt deze conclusie niet overeen met andere literatuur en de eindconclusie van dezelfde auteurs. De bewijslast werd beoordeeld als moderate grade.
Aanbeveling-3
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Bij de beoordeling van de veiligheid van brolucizumab werd in eerste instantie gesuggereerd dat de frequentie van oculaire bijwerkingen lager was dan van aflibercept (2 mg) (Dugel, 2021). Latere studies hebben deze suggestie met goede data weerlegd (Khanani,. 2021). Ook in de post-hoc analyse van de gecombineerde data van HAWK en HARRIER werd een hogere frequentie IOI geconstateerd tijdens brolucizumab-gebruik ten opzichte van aflibercept (2 mg) (Mones, 2021). In lijn met de Direct Healthcare Professional Communication (DHPC) en het advies van de werkgroep Medische Retina van het NOG hebben we onderstaande aanbevelingen geformuleerd bij het gebruik van brolucizumab.
Aanbeveling-4
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Met de huidige en de te verwachten komst van biosimilars komen er nieuwe middelen tot onze beschikking voor de behandeling van nLMD (Kaiser, 2022). Maar ook zullen de kosten van de verschillende anti-VEGF middelen een belangrijkere rol gaan spelen. Deze kosten spelen ook een rol in de aanbevelingen van deze richtlijn, zie aanbeveling 1.
Recent is een Oogheelkundig addendum verschenen op het Standpunt Biosimilars Federatie Medische Specialisten uit 2017 (zie https://www.oogheelkunde.org/wp-content/uploads/2023/03/Oogheelkundig-addendum-biosimilars-22-03-23_final.pdf?rs_file_key=170918432164240a3b7ba8e093700650 , geraadpleegd op 16 april 2023). In deze richtlijn worden dit standpunt, zoals hierboven beschreven, overgenomen.
De ervaring leert dat patiënten verschillend reageren op de diverse anti-VEGF middelen en dat deze respons kan veranderen in de tijd. Het is zoeken naar het juiste middel voor een specifieke patiënt op een specifiek moment in het behandeltraject. Het is daarom belangrijk dat de oogarts gedurende het gehele behandeltraject beschikt over meerdere opties aan anti-VEGF middelen. Als een patiënt goed reageert op een origineel anti-VEGF mag eenzelfde therapeutische respons worden verwacht op de biosimilar van dit middel. Deze verwachting geldt echter niet voor een biosimilar van een ander origineel anti-VEGF middel. Hiervoor moet eerst een nieuwe cyclus van minimaal 3 injecties met controle worden doorlopen voordat kan worden geconcludeerd dat de middelen even goed werken bij deze specifieke patiënt.
Onderbouwing
Patiënten met nLMD worden behandeld met intravitreale injecties met anti-vascular endothelial growth factor (anti-VEGF) middelen. Er zijn steeds meer anti-VEGF middelen beschikbaar, waaronder ook biosimilars.
Op dit moment wordt bevacizumab (Avastin®) als middel van eerste keus aanbevolen op basis van kostenverschillen, “non-inferiority” en huidige inzichten in systemische bijwerkingen Bevacizumab was en is echter niet geregistreerd voor nLMD. Andere anti-VEGF middelen die hiervoor wel zijn geregistreerd zijn aflibercept (2 mg) (Eylea®), ranibizumab (Lucentis®), en brolucizumab (Beovu®). Recent, in september 2022, is Faricimab (Vabysmo®) geregistreerd in Europa voor de behandeling van nLMD. Omdat de registratiestudies zijn gepubliceerd tijdens of na onderstaande literatuur search is faricimab niet meegenomen in de vergelijking. Daarom kan in deze richtlijn geen formele aanbeveling over Faricimab worden geformuleerd. Voor een actueel advies over recent geintroduceerde middelen wordt verwezen naar het standpunt van de NOG Werkgroep Medische Retina. Deze standpunten worden gepubliceerd op de website van het NOG. .
Op het moment van schrijven zijn wel een aantal biosimilars voor anti-VEGF middelen ontwikkeld. Het is daarom opnieuw relevant om te bepalen welk anti-VEGF middel het meest geschikt is als eerste behandelkeuze, met het oog op werkzaamheid, injectie interval, veiligheid en kosten. Een kosten-effectiviteitsanalyse is niet uitgevoerd.
Vision maintenance/improvement
Ranibizumab versus bevacizumab
|
High GRADE |
Ranibizumab results in little to no difference in vision maintenance/improvement when compared with bevacizumab in patients with neovascular age-related macular degeneration.
Sources: Solomon, 2019 |
Ranibizumab versus aflibercept (2 mg)
|
Moderate GRADE |
Ranibizumab likely results in little to no difference in vision maintenance/improvement when compared with aflibercept (2 mg) in patients with neovascular age-related macular degeneration
Sources: Sarwar, 2016 |
Brolucizumab versus aflibercept (2 mg)
|
High GRADE |
Brolucizumab results in little to no difference in vision maintenance/improvement when compared with aflibercept (2 mg) in patients with neovascular age-related macular degeneration.
Sources: Dugel, 2021 |
Retinal thickness
Ranibizumab versus bevacizumab
|
High GRADE |
Ranibizumab results in little to no difference in retinal thickness on OCT macular when compared with bevacizumab in patients with neovascular age-related macular degeneration.
Sources: Solomon, 2019 |
Ranibizumab versus aflibercept (2 mg)
|
Moderate GRADE |
Ranibizumab likely results in little to no difference in retinal thickness on OCT macular when compared with aflibercept (2 mg) in patients with neovascular age-related macular degeneration.
Sources: Sarwar, 2016 |
Brolucizumab versus aflibercept (2 mg)
|
High GRADE |
Brolucizumab results in little to no difference in retinal thickness on OCT macular when compared with aflibercept (2 mg) in patients with neovascular age-related macular degeneration.
Sources: Dugel, 2021 |
Safety
Ranibizumab versus bevacizumab
|
Low GRADE |
Ranibizumab may result in little to no difference in serious systemic adverse events when compared with bevacizumab in patient with neovascular age-related macular degeneration.
Sources: Solomon, 2019 |
Ranibizumab versus aflibercept (2 mg)
|
Moderate GRADE |
Aflibercept (2 mg) likely results in little to no difference in serious systemic adverse events when compared with ranibizumab in patients with neovascular age-related macular degeneration.
Aflibercept (2 mg) likely results in less serious ocular adverse events when compared with ranibizumab in patients with neovascular age-related macular degeneration. Note: The same authors conclude that current available information suggests that the safety profile of aflibercept (2 mg) is comparable with that of ranibizumab.
Sources: Sarwar,2016 |
Brolucizumab versus aflibercept (2 mg)
|
Moderate GRADE |
Brolucizumab likely results in a reduction of ocular adverse events when compared with aflibercept (2 mg) in patients with neovascular age-related macular degeneration.*
Sources: Dugel, 2021 |
*Note: After the literature search, other important studies on the safety of brolucizumab were published, pointing towards an increased risk of ocular adverse events.
Description of studies
Bevacizumab vs. other anti-VEGFs
Bevacizumab versus ranibizumab
Solomon (2019), on behalf of the Cochrane Library, performed a systematic review to compare the relative effects of one of three anti-VEGF agents (pegaptanib, ranibizumab, and bevacizumab) versus another when administered in comparable dosages and regimen. Randomized controlled trials (RCTs) were included that evaluated pegaptanib, ranibizumab, or bevacizumab versus each other or versus a control treatment (e.g., sham treatment, photodynamic therapy), in which participants were followed for at least one year. A search was performed in the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE Ovid, Embase Ovid, the Latin American and Caribbean Health Sciences Literature Database (LILACS), the
International Standard Randomized Controlled Trials Number (ISRCTN) Registry and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) until January 31, 2018. A total of 16 RCTs were included that enrolled 6347 participants with neovascular AMD and one potentially relevant ongoing trial. Of the included trials, 10 trials compared bevacizumab versus ranibizumab, which we included in our analysis. A meta-analysis was performed. The 16 trials were similar in that all enrolled both men and women 50 years of age or older who had subfoveal CNV secondary to AMD.
Bevacizumab versus aflibercept, brolucizumab, or biosimilars for anti-VEGF
No studies were found that compared bevacizumab with aflibercept, brolucizumab, or biosimilars for anti-VEGF.
Aflibercept vs other anti-VEGFs
Aflibercept versus ranibizumab
Sarwar (2016) performed a systematic review to assess and compare the effectiveness and safety of intravitreal injections of aflibercept versus ranibizumab, bevacizumab, or sham for treatment of patients with neovascular AMD. Randomized controlled trials (RCTs) in which aflibercept monotherapy was compared with ranibizumab, bevacizumab, or sham for participants with neovascular AMD who were treatment-naive were included. A search was performed in the Cochrane Central Register of Controlled Trials (CENTRAL), Ovid MEDLINE, EMBASE, PubMed, Latin American and Caribbean Health Sciences Literature Database (LILACS), the metaRegister of Controlled Trials (mRCT) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) until November 30, 2015. A total of two RCTs were included (VIEW1 and VIEW2), which enrolled 2457 participants with neovascular AMD with active subfoveal choroidal neovascular lesions. Both trials followed the same protocol and compared aflibercept at various doses versus ranibizumab. A meta-analysis was performed. The included studies enrolled both men and women 50 years of age or older who had subfoveal neovascular AMD in the study eye.
Aflibercept versus brolucizumab
Dugel (2021) performed a randomized controlled trial to compare the efficacy and safety of brolucizumab in eyes with neovascular age-related macular degeneration (nAMD). Patients were randomized 1:1:1 to receive brolucizumab 3 mg or 6 mg or aflibercept 2 mg (HAWK) or 1:1 to receive brolucizumab 6 mg or aflibercept 2 mg (HARRIER). HAWK and HARRIER are phase 3, prospective, randomized, double-masked, multicenter studies designed to compare the efficacy and safety of brolucizumab 3 mg (HAWK only) and 6 mg with aflibercept 2 mg in patients with nAMD. Eligible patients were aged ³50 years and had untreated, active choroidal neovascularization lesions secondary to AMD affecting the central subfield (the circular area within 1 mm diameter around the foveal center on imaging); choroidal neovascularization lesions (including classic and occult), as assessed by fluorescein angiography, comprising >50% of total lesion area; IRF and/or SRF affecting the central subfield as assessed on spectral-domain OCT; BCVA between 78 and 23 Early Treatment Diabetic Retinopathy Study (ETDRS) letters (inclusive; Snellen equivalents, approximately 20/32 to 20/400); and no fibrosis or geographic atrophy affecting the central subfield. Patients could not have received any approved or investigational nAMD treatment at any time (study eye). Overall, 1082 patients were randomized in HAWK between December 2014 and May 2016, and 743 patients were randomized in HARRIER between June 2015 and April 2016. Mean baseline BCVA was 60.6 (HAWK) and 61.2 (HARRIER) letters, and approximately 25% of study eyes had BCVA ³71 letters at baseline, which is reflective of current clinical practice and in line with BCVA inclusion criteria.
Aflibercept versus bevacizumab or biosimilars for anti-VEGF
No studies were found that compared aflibercepts with bevacizumab, or biosimilars for anti-VEGF.
Results
Vision maintenance/improvement
Ranibizumab versus bevacizumab
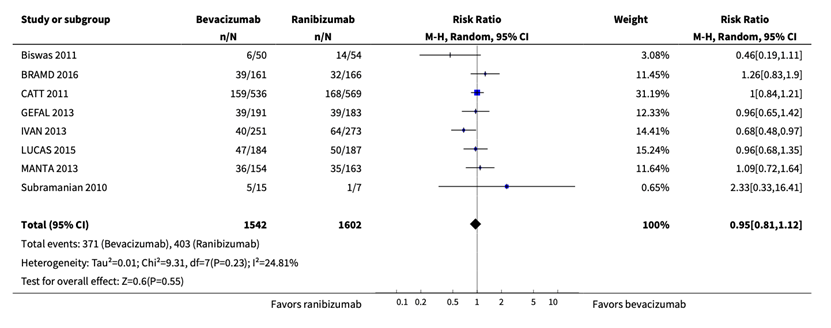
Solomon (2019) described vision maintenance/improvement, which was reported in eight RCTs, as the gain of 15 more letters visual acuity at 1 year (Biswas, 2011; BRAMD, 2016; CATT, 2011; GEFAL, 2013; IVAN, 2013; LUCAS, 2015; MANTA, 2013, Subramanian, 2010). In total, 3144 patients were analyzed, with 1542 in the bevacizumab group and 1602 in the ranibizumab group. The pooled risk ratio (RR) was 0.95 (95% confidence interval (CI): 0.81 to 1.12), towards ranibizumab was reported, which is not clinically relevant.
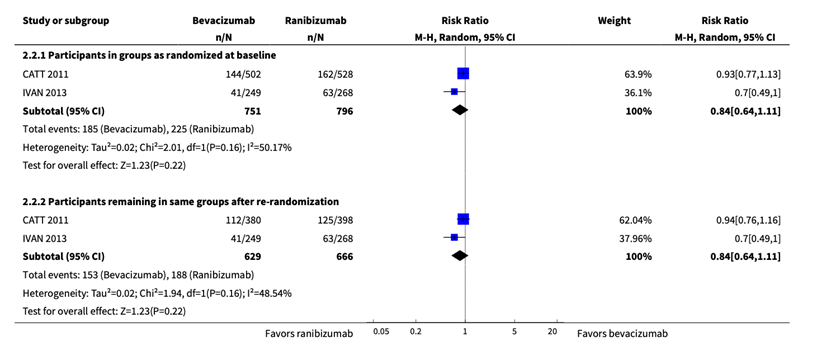
In addition, Solomon described the vision maintenance/improvement at 2 years, which was reported in two studies (CATT, 2011; IVAN, 2013). In total, 1547 patients were analyzed, with 751 in the bevacizumab group and 796 in the ranibizumab group. The pooled RR was 0.84 (95% CI 0.64 to 1.11) towards ranibizumab, which is not statistically significant or clinically relevant.
Ranibizumab versus aflibercept
Two studies reported on vision maintenance/improvement. Sarwar (2016) described vision maintenance/improvement, which was reported in two RCTs, as the proportion of participants who gained 15 or more letters of BCVA at one-year follow-up (VIEW1; VIEW2).
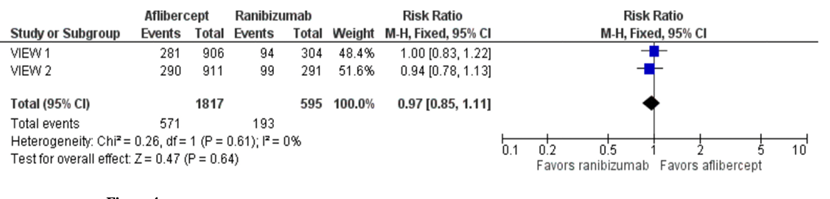
In total, 2412 patients were analyzed, with 1817 in the aflibercept group and 595 in de ranibizumab group. The pooled RR was 0.97 (95% CI 0.85 to 1.11), towards ranibizumab. This result was not clinically relevant.
At two-year follow-up, 562 (30.9%) of 1817 participants in the aflibercept groups and 188 (31.6%) of 595 participants in the ranibizumab groups gained 15 or more letters from baseline. The pooled RR was 0.98 (95% CI 0.85 to 1.12), towards ranibizumab. This result was not clinically relevant.
Brolucizumab versus aflibercept
One study reported on vision maintenance/improvement. Dugel (2021) reported the mean change in BCVA from baseline to week 48. Brolucizumab-treated eyes were noninferior to aflibercept-treated eyes, and these visual gains were maintained to week 96.
HAWK: mean change (least squares [LS] mean ± standard error) in BCVA from baseline to 96w was 5.6 ± 0.79 ETDRS letters for brolucizumab 3 mg, 5.90 ± 0.78 letters for brolucizumab 6 mg, and 5.3 ± 0.78 letters for aflibercept.
HARRIER: mean change was 6.1 ± 0.73 letters for brolucizumab 6 mg and 6.6 ± 0.73 letters for aflibercept. These results were not clinically relevant.
Retinal thickness
Ranibizumab versus bevacizumab
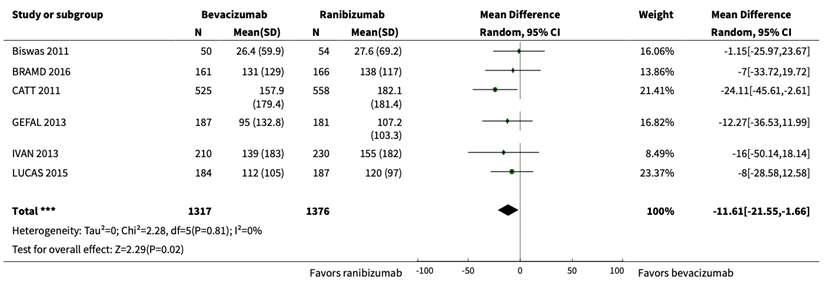
Solomon (2019) described retinal thickness, which was reported in six RCTs (Biswas, 2011; BRAMD, 2016; CATT, 2011; GEFAL, 2013; IVAN, 2013; LUCAS, 2015). In total 2693 patients were analyzed, with 1317 in the bevacizumab group and 1376 in the ranibizumab group. A pooled mean difference of 11.6 μm, 95% CI -21.6 to -1.7), towards ranibizumab was reported, which is not clinically relevant.
In addition, two studies (CATT, 2011; IVAN, 2013) reported the outcome reduction in central retinal thickness at 2 years follow-up. In total 1199 patients were analyzed, with 592 in the bevacizumab group and 607 in the ranibizumab group. The pooled mean difference was -12.4 μm, (95% CI -33.83 to 9.04) towards ranibizumab, which is not clinically relevant.
Ranibizumab versus aflibercept
Two studies reported on retinal thickness. Sarwar (2016) described retinal thickness in two studies (VIEW1; VIEW2). In total, 2412 patients were analyzed, with 1817 in the aflibercept group and 595 in de ranibizumab group. The pooled mean difference was −4.94 μm (95% CI −15.48 to 5.61) towards aflibercept, which is not clinically relevant.
Brolucizumab versus aflibercept
Dugel (2021) reported central subfield thickness for the comparison brolucizumab versus aflibercept.
HAWK reported a mean change of -174.8 μm vs. -148.7 μm (95% CI for treatment difference, -46.2 to -5.9 μm; P = 0.0115) in the brolucizumab 6 mg group and aflibercept group, respectively. The difference is not clinically relevant.
HARRIER reported a mean change of -197.7 μm vs. -155.1 μm; 95% CI for treatment difference, -62.0 to -23.3 μm; P < 0.0001) in the brolucizumab 6 mg group and aflibercept group, respectively. The difference is clinically relevant.
Safety
Ranibizumab versus bevacizumab
Solomon (2019) described that in ten RCTs information about adverse events were provided, but the follow-up, and the types and specificity of the reported adverse events varied between studies.
At one year follow-up, no serious ocular events were reported by four trials (Biswas, 2011; MANTA, 2013; SAVE-AMD, 2017; Subramanian, 2010). Minor adverse events were reported in three of the four trials, including subconjunctival hemorrhage, increased intraocular pressure, transient post injection pain, and mild ocular inflammation. The numbers of participants who experienced these adverse events were not reported.
The occurrence of serious systemic adverse events was comparable across anti-VEGF-treated groups and control groups.
Ranibizumab versus aflibercept
Sarwar (2016) described safety in two studies (VIEW1; VIEW2). In total, 2412 patients were analyzed, with 1817 in the aflibercept group and 595 in de ranibizumab group. The pooled RR for serious systemic adverse events was 0.99 (95%CI 0.79 to 1.25), towards aflibercept. This result is not clinically relevant.
The pooled RR for serious ocular adverse events was 0.62 (95%CI 0.36 to 1.07), towards aflibercept. This result is clinically relevant.
Brolucizumab versus aflibercept
Dugel (2021) reported that conjunctival hemorrhage was the most frequently reported ocular adverse event across all treatment groups in HAWK, occurring in 39 (10.9%), 29 (8.1%), and 32 (8.9%) patients in the brolucizumab 3 mg, brolucizumab 6 mg, and aflibercept 2 mg groups, respectively.
The incidence of ocular adverse events (AEs) was similar across all treatment groups in both HAWK and HARRIER up to week 96 (Table 1), with the exception of combined intraocular inflammation (IOI) (iritis and uveitis), which was higher in the brolucizumab 6 mg group of HAWK compared with the aflibercept group (4.7% vs. 0.6%). This result is clinically relevant.
In HARRIER, the most frequently reported ocular adverse events were reduced visual acuity in the brolucizumab 6 mg group (32 patients [8.6%]) and cataract in the aflibercept 2 mg group (43 patients [11.7%]). A RR of 0.73 (95%CI 0.48 to 1.13) towards brolucizumab for safety was calculated. This result is clinically relevant.
Level of evidence of the literature
Vision maintenance/improvement
Ranibizumab versus bevacizumab
The level of evidence regarding the outcome measure “Vision maintenance/improvement” comes from randomized controlled trials and therefore starts at high. The level of evidence was not downgraded, resulting in a level of evidence of high.
Ranibizumab versus aflibercept
The level of evidence regarding the outcome measure “Vision maintenance/improvement” comes from randomized controlled trials and therefore starts at high. The level of evidence was downgraded by one level because exceeding the limits of clinical relevance (imprecision), resulting in a level of moderate.
Brolucizumab versus aflibercept
The level of evidence regarding the outcome measure “Vision maintenance/improvement” comes from a randomized controlled trial and therefore starts at high. The level of evidence was not downgraded, resulting in a level of evidence of high.
Retinal thickness
Ranibizumab versus bevacizumab
The level of evidence regarding the outcome measure “Retinal thickness” comes from randomized controlled trials and therefore starts at high. The level of evidence was not downgraded, resulting in a level of evidence of high.
Ranibizumab versus aflibercept
The level of evidence regarding the outcome measure “Retinal thickness” comes from randomized controlled trials and therefore starts at high. The level of evidence was downgraded by one level because exceeding the limits of clinical relevance (imprecision), resulting in a level of evidence of moderate.
Brolucizumab versus aflibercept
The level of evidence regarding the outcome measure “Retinal thickness on oct macular” comes from a randomized controlled trial and therefore starts at high. The level of evidence was not downgraded, resulting in a level of evidence of high.
Safety
Ranibizumab versus bevacizumab
The level of evidence regarding the outcome measure “safety” comes from a randomized controlled trial and therefore starts at high. The level of evidence was downgraded by two levels due to low numbers of events and participants (imprecision), resulting in a level of evidence of low.
Ranibizumab versus aflibercept
The level of evidence regarding the outcome measure “safety” comes from randomized controlled trials and therefore starts at high. The level of evidence was downgraded by one level due to low numbers of events (imprecision), resulting in a level of evidence of moderate.
Brolucizumab versus aflibercept
The level of evidence regarding the outcome measure “safety” comes from a randomized controlled trial and therefore starts at high. The level of evidence was downgraded by one level due to crossing the lower limit of clinical relevance (imprecision), resulting in a level of evidence of moderate.
What is the effect of bevacizumab and aflibercept compared to any other anti-VEGF in patients with exudative age-related macular degeneration on vision maintenance/improvement, retinal thickness and safety?
Bevacizumab
P: Patients with neovascular age-related macular degeneration
I: Other ant-VEGFs: ranibizumab, aflibercept, brolucizumab, biosimilars for anti-VEGF
C: Bevacizumab
O: Vision maintenance/improvement, retinal thickness, safety
Aflibercept
P: Patients with neovascular age-related macular degeneration
I: Other anti-VEGFs: bevacizumab, ranibizumab, brolucizumab, biosimilars for anti-VEGF
C: Aflibercept
O: Vision maintenance/improvement, retinal thickness, safety
Relevant outcome measures
The guideline development group considered vision maintenance/improvement as a critical outcome measure for decision making; and retinal thickness and safety as an important outcome measure for decision making.
The working group defined the outcome measures as follows:
- Vision improvement: improvement of 15 letters or more from baseline
- Vision maintenance: no loss of letters from baseline
- Retinal thickness: change in central foveal/subfield thickness on OCT
The working group used the GRADE standard limit of 25% as a minimal clinically (patient) important difference for dichotomous outcomes and 10% for continuous variables.
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until June 21st, 2021, for the comparison between bevacizumab and other anti-VEGFs. An additional search was performed until August 2nd, 2021, for the comparison between aflibercept and other anti-VEGFs. The detailed search strategy is depicted under the tab Methods. The systematic literature search resulted in 287 hits. Studies were selected based on the following criteria: study design was a systematic review or randomized controlled trial, included patients with neovascular AMD, compared bevacizumab with other anti-VEGFs or aflibercept vs. other anti-VEGFs, and reported at least one outcome of interest. Twenty-five studies were initially selected based on title and abstract screening. After reading the full text, 21 studies were excluded (see the table with reasons for exclusion under the tab Methods), and three studies were included.
Results
Three studies were included in the analysis of the literature. Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
- Brown G, Brown MM, Rapuano S, Boyer D. Cost-Utility Analysis of VEGF Inhibitors for Treating Neovascular Age-Related Macular Degeneration. Am J Ophthalmol 2020 Oct;218:225-241.
- Dugel PU, Singh RP, Koh A, Ogura Y, Weissgerber G, Gedif K, Jaffe GJ, Tadayoni R, Schmidt-Erfurth U, Holz FG. HAWK and HARRIER: Ninety-Six-Week Outcomes from the Phase 3 Trials of Brolucizumab for Neovascular Age-Related Macular Degeneration. Ophthalmology. 2021 Jan;128(1):89-99. doi: 10.1016/j.ophtha.2020.06.028. Epub 2020 Jun 20. PMID: 32574761.
- Elshout M, van der Reis MI, Webers CAB, Schouten JSAG. The cost-utility of aflibercept for the treatment of age-related macular degeneration compared to bevacizumab and ranibizumab and the influence of model parameters. Graefes Arch Clin Exp Ophthalmol 2014 Dec;252(12):1911-20.
- Gillies MC, Hunyor AP, Arnold JJ, Guymer RH, Wolf S, Pecheur FL, Munk MR, McAllister IL. Macular Atrophy in Neovascular Age-Related Macular Degeneration: A Randomized Clinical Trial Comparing Ranibizumab and Aflibercept (RIVAL Study). Ophthalmology. 2020 Feb;127(2):198-210. doi: 10.1016/j.ophtha.2019.08.023. Epub 2019 Aug 27. PMID: 31619357.
- JAMA Ophthalmol. Published online November 24, 2021. doi:10.1001/jamaophthalmol.2021.4585
- Kaiser PK, Schmitz-Valckenberg MS, Holz FG. ANTI-VASCULAR ENDOTHELIAL GROWTH FACTOR BIOSIMILARS IN OPHTHALMOLOGY. Retina. 2022 Dec 1;42(12):2243-2250. doi: 10.1097/IAE.0000000000003626. Epub 2022 Nov 15. PMID: 36394884; PMCID: PMC9665947.
- Khanani AM, Zarbin MA, Barakat MR, Albini TA, Kaiser PK, B G, Agashivala N, Yu JS, Wykoff CC, MacCumber MW. Safety Outcomes of Brolucizumab in Neovascular Age-Related Macular Degeneration: Results From the IRIS Registry and Komodo Healthcare Map. JAMA Ophthalmol. 2022 Jan 1;140(1):20-28. doi: 10.1001/jamaophthalmol.2021.4585. PMID: 34817566; PMCID: PMC8613703.
- Lorenzo Moja 1, Ersilia Lucenteforte, Koren H Kwag, Vittorio Bertele, Annalisa Campomori, Usha Chakravarthy, Roberto D'Amico, Kay Dickersin, Laurent Kodjikian, Kristina Lindsley, Yoon Loke, Maureen Maguire, Daniel F Martin, Alessandro Mugelli, Bernd Mühlbauer, Isabel Püntmann, Barnaby Reeves, Chris Rogers, Christine Schmucker, Manju L Subramanian, Gianni Virgili. Systemic safety of bevacizumab versus ranibizumab for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2014 Sep 15;9(9):CD011230.
- Moja L, Lucenteforte E, Kwag KH, Bertele V, et al. Systemic safety of bevacizumab versus ranibizumab for neovascular age-related macular degeneration (2014). Cochrane Database Syst Rev 15;9(9): DOI: 10.1002/14651858.CD011230.pub2
- Mones J, Srivastava SK, Jaffe GJ, Tadayuni R, et al. Risk of inflammation, retinal vasculitis, and retinal occlusion-related events with brolucizumab: Post hoc review of HAWK and HARRIER. Ophthalmoly 2021 DOI: 10.1016/j.ophtha.2020.11.011
- Quist SW, de Jong LA, van Asten F, Knoester P, Postma MJ, Freriks RD. Cost-minimisation analysis of a treat-and-extend regimen with anti-VEGFs in patients with neovascular age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2022 Apr;260(4):1083-1095. doi: 10.1007/s00417-021-05359-x. Epub 2021 Oct 13. PMID: 34643793; PMCID: PMC8511619.
- Sarwar S, Clearfield E, Soliman MK, Sadiq MA, Baldwin AJ, Hanout M, Agarwal A, Sepah YJ, Do DV, Nguyen QD. Aflibercept for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2016 Feb 8;2:CD011346. doi: 10.1002/14651858.CD011346.pub2. PMID: 26857947; PMCID: PMC5030844.
- Sharma A, Kumar N, Parachuri N, Bandello F, Kupperman BD, Loewenstein A. Ranibizumab Biosimilar (Razumab) vs Innovator Ranibizumab (Lucentis) in neovascular age-related macular degeneration (n-AMD)- efficacy and safety (BIRA study). 2021 Eye (Lond) Jun 22. DOI: 10.1038/s41433-021-01616-9
- Solomon SD, Lindsley K, Vedula SS, Krzystolik MG, Hawkins BS. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2019 Mar 4;3(3):CD005139. doi: 10.1002/14651858.CD005139.pub4. PMID: 30834517; PMCID: PMC6419319.
- Van Asten, CT J Michels 2, Carel B Hoyng 1, Gert Jan van der Wilt 2, B Jeroen Klevering 1, Maroeska M Rovers 2, Janneke P C Grutters. The cost-effectiveness of bevacizumab, ranibizumab and aflibercept for the treatment of age-related macular degeneration-A cost-effectiveness analysis from a societal perspective. PLoS One 2018 May 17;13(5):e0197670.
- Woo SJ, Veith M, Hamouz J, Ernest J, Zalewski D, Studnicka J, Vajas A, Papp A, Gabor V, Luu J, Matuskova V, Yoon YH, Pregun T, Kim T, Shin D, Bressler NM. Efficacy and Safety of a Proposed Ranibizumab Biosimilar Product vs a Reference Ranibizumab Product for Patients With Neovascular Age-Related Macular Degeneration: A Randomized Clinical Trial. JAMA Ophthalmol. 2021 Jan 1;139(1):68-76. doi: 10.1001/jamaophthalmol.2020.5053. PMID: 33211076; PMCID: PMC7677876.
Evidence tables
Systematic reviews
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Solomon, 2019
[individual study characteristics deduced from Solomon, 2019]
PS., study characteristics and results are extracted from the SR (unless stated otherwise) |
SR and meta-analysis of RCTs
Literature search up to January 2018
A: CATT, 2011 B: IVAN, 2013 C: Biswas, 2011 D: BRAMD, 2016 E: GEFAL, 2013 F: LUCAS,2015 G: MANTA, 2013 H: SAVE-AMD, 2017 I: Scholler, 2014 J: Subramian, 2010
Study design: RCT
Setting and Country: -
Source of funding and conflicts of interest: This review update was supported by the NIHR, via Cochrane Infrastructure funding to the CEV UK Editorial base |
Inclusion criteria SR: - RCT - compared anti-VEGF treatment versus another treatment, sham treatment, or no treatment.
Exclusion criteria SR: - compared different doses of one anti- VEGF treatment against another, studies that included no control or comparator group, or studies that used anti-VEGF agents in combination with other treatments - studies of aflibercept (VEGF Trap-Eye/EYLEA solution) or studies that compared diJerent treatment schedules (e.g. monthly vs as needed dosing)
10 studies included
Important patient characteristics at baseline: See Characteristics of Studies in the article
Groups comparable at baseline? Yes |
Describe intervention:
A: 1.25 mg bevacizumab monthly for 1 year; at 1 year, re-randomization to ranibizumab monthly or variable dosing / 1.25 mg bevacizumab as needed after first injection for 2 years B: 1.25 mg bevacizumab monthly for 2 years/ 1.25 mg bevacizumab monthly for 3 months, then as needed in 3-month cycles C: 1.25 mg bevacizumab monthly for 3 months, then as needed D: 1.25 mg bevacizumab monthly for 1 year E: 1.25 mg bevacizumab; maximum of 1 injection per month F: 1.25 mg bevacizumab; treat and extend protocol G: 1.25 mg bevacizumab monthly for 3 months, then as needed H: 1.25 mg bevacizumab at day 1 and at week 4, then as needed I: 1.25 mg bevacizumab for 3 months, at 30-day intervals, then as needed J: 0.05 mL bevacizumab monthly for 3 months, then as needed
|
Describe control:
A: 0.5 mg ranibizumab monthly for 1 year; at 1 year, re-randomization to ranibizumab monthly or variable dosing / 0.5 mg ranibizumab as needed after first injection for 2 years B: 0.5 mg ranibizumab monthly for 2 years / 0.5 mg Ranibizumab monthly for 3 months, then as needed in 3-month cycles C: 0.5 mg ranibizumab monthly for 3 months, then as needed D: 0.5 mg ranibizumab monthly for 1 year E: 0.5 mg ranibizumab; maximum of 1 injection per month F: 0.5 mg ranibizumab; treat and extend protocol G: 0.5 mg ranibizumab monthly for 3 months, then as needed H: 0.5 mg ranibizumab at day 1 and at week 4, then as needed I: 0.5 mg ranibizumab for 3 months, at 30-day intervals, then as needed J: 0.05 mL ranibizumab monthly for 3 months, then as needed
|
End-point of follow-up:
A: 24 months B: 2 years C: 18 months D: 1 year E: 1 year F: 12 months G: 12 months H: 12 months I: 12 months J: 12 months
For how many participants were no complete outcome data available? See Characteristics of Studies in the article
|
1. Vision maintenance/improvement at one year:
Effect measure: RR [95% CI]: A: 1 [0.84, 1.21] B: 0.68 [0.48, 0.97] C: 0.46 [0.19, 1.11] D: 1.26 [0.83, 1.9] E: 0.96 [0.65, 1.42] F: 0.96 [0.68, 1.35] G: 1.09 [0.72, 1.64] H: Not reported I: Not reported J: 2.33 [0.33, 16.41]
Pooled effect (random effects model): 0.95 [95% CI 0.81 to 1.12] favoring ranibizumab Heterogeneity (I2): 24.81%
2. Retinal thickness at one year: Effect measure: RR [95% CI]: A: -24.11 [-45.61, -2.61] B: -16 [-50.14, 18.14] C: -1.15 [-25.97, 23.67] D: -7 [-33.72, 19.72] E: -12.27 [-36.53, 11.99] F: -8 [-28.58, 12.58] G: Not reported H: Not reported I: Not reported J: Not reported
Pooled effect (random effects model): -11.61 [95% CI -28.55 to 12.58] favoring ranibizumab Heterogeneity (I2): 0%
3. Safety: At one year follow-up, no serious ocular events were reported by four trials (Biswas 2011; MANTA 2013; SAVE-AMD 2017; Subramanian 2010). In four other trials (CATT 2011, GEFAL 2013, IVAN 2013, and LUCAS 2015) less than 1% of participants had endophthalmitis, retinal detachment, retinal pigment epithelial tear, traumatic cataract, or uveitis. Scholler 2014 reported two eyes with subretinal bleeding, both treated with ranibizumab. BRAMD 2016 did not mention ocular adverse events. |
Facultative:
Brief description of author’s conclusion
Personal remarks on study quality, conclusions, and other issues (potentially) relevant to the research question
Level of evidence: GRADE (per comparison and outcome measure) including reasons for down/upgrading
Sensitivity analyses (excluding small studies; excluding studies with short follow-up; excluding low quality studies; relevant subgroup-analyses); mention only analyses which are of potential importance to the research question
Heterogeneity: clinical and statistical heterogeneity; explained versus unexplained (subgroupanalysis) |
|
Sarwar, 2016
[individual study characteristics deduced from Sarwar, 2016]
PS., study characteristics and results are extracted from the SR (unless stated otherwise) |
SR and meta-analysis of RCTs
Literature search up to November 2015
A: VIEW1 B: VIEW2
Study design: RCT
Setting and Country: multicentre, Canada and United States
Source of funding and conflicts of interest: Funding from Genentech and Regeneron
|
Inclusion criteria SR:
Exclusion criteria SR: aflibercept was evaluated as part of combination therapy versus other active treatments, such as laser photocoagulation
2 studies included
Important patient characteristics at baseline: See Characteristics of Studies
Groups comparable at baseline? Yes |
Describe intervention: A: intravitreal aflibercept 0.5 mg every 4 weeks
B: intravitreal aflibercept 0.5 mg every 4 weeks
|
Describe control: A: intravitreal ranibizumab 0.5 mg every 4 weeks
B: intravitreal ranibizumab 0.5 mg every 4 weeks
|
End-point of follow-up: A: 1 year B: 2 years
For how many participants were no complete outcome data available? See Characteristics of Studies
|
1. Vision maintenance/improvement at one year:
Effect measure: RR [95% CI]: A: 1.00 [0.83, 1.22] B: 0.94 [0.78, 1.13]
Pooled effect (random effects model): 0.97 [95% CI 0.85 to 1.11] favoring ranibizumab Heterogeneity (I2): 0%
2. Retinal thickness at one year: Pooled effect (random effects model): -4.94 [95% CI -15.48 to 5.61] favoring aflibercept
3. Safety: Pooled effect (random effects model) for serious systemic adverse events: 0.99 [95% CI 0.79 to 1.25] favoring aflibercept
Pooled effect (random effects model) for serious ocular adverse events: 0.62 [95% CI 0.36 to 1.07] favoring aflibercept
|
Facultative:
Brief description of author’s conclusion Results of this review document the comparative effectiveness of aflibercept versus ranibizumab for visual acuity and morphological outcomes in eyes with neovascular AMD. Current available information on adverse effects of each medication suggests that the safety profile of aflibercept is comparable with that of ranibizumab; however, the number of participants who experienced adverse events was small, leading to imprecise estimates of absolute and relative effect sizes. The eight-week dosing regimen of aflibercept represents reduced treatment requirements in comparison with monthly dosing regimens and thus has the potential to reduce treatment burden and risks associated with frequent injections.
|
Randomized controlled trials
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3
|
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Dugel, 2021 |
Type of study: RCT
Setting and country: multicenter trials conducted in 408 sites in North, Central, and South America; Europe; Asia; Australia; and Japan.
Funding and conflicts of interest: Supported by Novartis Pharma AG, Basel, Switzerland. The sponsor or funding organization participated in the design of the study; management, analysis, and interpretation of the data; and preparation, review, and approval of the manuscript. |
Inclusion criteria: patients were aged ³50 years and had untreated, active choroidal neovascularization lesions secondary to AMD affecting the central subfield (the circular area within 1 mm diameter around the foveal center on imaging); choroidal neovascularization lesions (including classic and occult), as assessed by fluorescein angiography, comprising >50% of total lesion area; IRF and/or SRF affecting the central subfield as assessed on spectral-domain OCT; BCVA between 78 and 23 Early Treatment Diabetic Retinopathy Study (ETDRS) letters (inclusive; Snellen equivalents, approximately 20/32 to 20/400); and no fibrosis or geographic atrophy affecting the central subfield.
Exclusion criteria: Patients could not have received any approved or investigational nAMD treatment at any time (study eye)
N total at baseline: brolucizumab 3 mg (HAWK): 358 brolucizumab 6 mg (HARRIER) :370 aflibercept 2 mg (HAWK/HARRIER): 360/369
Important prognostic factors2: See supplementary data
Groups comparable at baseline? yes
|
Describe intervention (treatment/procedure/test):
brolucizumab 3 mg (HAWK) brolucizumab 6 mg (HARRIER)
|
Describe control (treatment/procedure/test):
aflibercept 2 mg (HAWK/HARRIER)
|
Length of follow-up: 96 weeks
Loss-to-follow-up: Not reported
Incomplete outcome data: Not reported
|
Outcome measures and effect size (include 95%CI and p-value if available):
1. Vision maintenance/improvement: HAWK: mean change (least squares [LS] mean ± standard error) in BCVA from baseline to 96w was 5.6 ± 0.79 ETDRS letters for brolucizumab 3 mg, 5.90 ± 0.78 letters for brolucizumab 6 mg, and 5.3 ± 0.78 letters for aflibercept.
HARRIER: mean change was 6.1 ± 0.73 letters for brolucizumab 6 mg and 6.6 ± 0.73 letters for aflibercept.
2. Retinal thickness at 24 months: HAWK: mean change of -174.8 μm vs. -148.7 μm (95% CI for treatment difference, -46.2 to -5.9 μm; P = 0.0115) in the brolucizumab 6 mg group and aflibercept group, respectively.
HARRIER: mean change of -197.7 μm vs. -155.1 μm; 95% CI for treatment difference, -62.0 to -23.3 μm; P < 0.0001) in the brolucizumab 6 mg group and aflibercept group, respectively.
3. Safety: HAWK: occurring in 39 (10.9%), 29 (8.1%), and 32 (8.9%) patients in the brolucizumab 3 mg, brolucizumab 6 mg, and aflibercept 2 mg groups, respectively.
HARRIER: the most frequently reported ocular adverse events were reduced visual acuity in the brolucizumab 6 mg group (32 patients [8.6%]) and cataract in the aflibercept 2 mg group (43 patients [11.7%]). A RR of 0.73 (95%CI 0.48 to 1.13) in favor of brolucizumab was calculated.
|
Authors conclusion:
Visual outcomes from 48w to 96w confirm the efficacy achieved at 48w. Brolucizumab demonstrated greater fluid resolution compared with aflibercept. The q12w potential for brolucizumab observed at 48w was maintained to 96w.
|
Risk of bias tables
Systematic reviews
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Solomon, 2019 |
Yes |
Yes |
Yes |
Yes |
Not apllicable |
Yes |
Yes |
Yes |
Yes |
|
Sarwar, 2016 |
Yes |
Yes |
Yes |
Yes |
Not applicable |
Yes |
Yes |
Yes |
Yes |
Randomized controlled trials
|
Study reference
(first author, publication year) |
Was the allocation sequence adequately generated? a
Definitely yes Probably yes Probably no Definitely no |
Was the allocation adequately concealed?b
Definitely yes Probably yes Probably no Definitely no |
Blinding: Was knowledge of the allocated interventions adequately prevented?c
Were patients blinded?
Were healthcare providers blinded?
Were data collectors blinded?
Were outcome assessors blinded?
Were data analysts blinded?
Definitely yes Probably yes Probably no Definitely no |
Was loss to follow-up (missing outcome data) infrequent?d
Definitely yes Probably yes Probably no Definitely no |
Are reports of the study free of selective outcome reporting?e
Definitely yes Probably yes Probably no Definitely no |
Was the study apparently free of other problems that could put it at a risk of bias?f
Definitely yes Probably yes Probably no Definitely no |
Overall risk of bias If applicable/necessary, per outcome measureg
LOW Some concerns HIGH |
|
Dugel, 2021 |
Definitely yes;
Reason: Eyes were randomized (Interactive Response Technology [IRT])1:1:1 to brolucizumab 3 mg, brolucizumab 6 mg, or aflibercept 2mg (HAWK) or 1:1 to brolucizumab 6 mg or aflibercept 2 mg(HARRIER) |
Definitely yes;
Reason: The randomization number was not communicated to unmasked staff. The randomization numbers were generated using the following procedure to ensure that treatment assignment was unbiased and concealed from patients and masked study center personnel. |
Definitely yes;
Reason: Double-Masked Trials |
Probably yes
Reason: Loss to follow-up was infrequent in intervention and control group. |
Definitely yes
Reason: All relevant outcomes were reported |
Definitely yes;
Reason: No other problems noted |
LOW
|
Table of excluded studies
|
Author and year |
Reason for exclusion |
|
Kodjikian, 2014 |
Study included in Solomon (2019) |
|
Solomon, 2016 |
Study included in Solomon (2019) |
|
Moja, 2014 |
Study included in Solomon (2019) |
|
Ye, 2020 |
Does not match our PICO |
|
Plyukhova, 2020 |
Study included in Solomon (2019) |
|
Ba, 2015 |
Study included in Solomon (2019) |
|
Mitchell, 2011 |
Does not match our PICO |
|
Nguyen, 2018 |
Study included in Solomon (2019) |
|
Wang, 2015 |
Study included in Solomon (2019) |
|
Chen, 2015 |
Study included in Solomon (2019) |
|
Zhao, 2021 |
Does not match our PICO |
|
Dugel, 2017 |
Same data as Dugel (2021) |
|
Dugel, 2020 |
Same data as Dugel (2021) |
|
Heier, 2012 |
Same data as Sarwar (2016) |
|
Schmidt-Erfurth, 2014 |
Study included in Sarwar (2016) |
|
Kaiser, 2017 |
Same data as Sarwar (2016) |
|
Mones, 2021 |
Same data as Dugel (2021) |
|
Gillies, 2019 |
Same data as Gillies (2020) |
|
Carrasco, 2020 |
Does not match our PICO |
|
Arnold, 2016 |
Does not match out PICO |
|
Gillies,2020 |
Does not match our PICO |
Beoordelingsdatum en geldigheid
Publicatiedatum : 07-08-2023
Beoordeeld op geldigheid : 17-07-2023
Algemene gegevens
De herziening van deze richtlijn werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Werkgroep
- Dr. R. van Leeuwen, oogarts, Universitair Medisch Centrum Utrecht, Utrecht (NOG, voorzitter)
- Prof. dr. C.C.W. Klaver, oogarts en hoogleraar, Erasmus Medisch Centrum, Rotterdam (NOG)
- Dr. P.H.B. Kok, oogarts, Bergman Clinics, Amsterdam (NOG)
- Dr. J.J.C. van Lith-Verhoeven, oogarts, Elisabeth-TweeSteden Ziekenhuis, Tilburg (NOG)
- Dr. F.D. Verbraak, oogarts, Amsterdam Universitair Medisch Centrum, Amsterdam (NOG)
- Dr. A.C. Lambooij, oogarts, Reinier de Graaf Gasthuis, Delft (NOG)
- Drs. E.A. Huiskamp, oogarts, Universitair Medisch Centrum Groningen, Groningen (NOG)
- Drs. O.A.M. Tigchelaar-Besling, oogarts, Amphia Ziekenhuis, Breda (NOG)
- Drs. L.J. Noordzij, oogarts, Oog Op Zuid, Rotterdam (NOG)
Klankbordgroep
- M.E. Diepman-Leerdam, optometrist, Bergman Clinics, Doetichem (OVN)
- H.J. Jansen-Molenaar, adviseur oogzorg (Oogvereniging) (tot 01-09-2022)
- P. Kortenhoeven, waarnemend coördinator oogzorg (Oogvereniging) (vanaf 01-09-2022)
- H.M.M.J. Schoots, voorzitter MaculaVereniging (MaculaVereniging)
Met ondersteuning van
- Dr. A. van der Hout, adviseur Kennisinstituut van de Federatie Medisch Specialisten
- Drs. B.L. Gal-de Geest, adviseur Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen acties |
|
van Leeuwen * |
Oogarts, netvliesspecialist, in het UMC Utrecht. |
Bestuur Vitreoretinale werkgroep, onbetaald Voorzitter Projectgroep Duurzame Oogheelkunde Vertegenwoordiger namens NOG in Landelijke Netwerk Groene OK, onbetaald |
* Ik doe op dit moment niet mee aan door industrie gesponsorde studies. Wel begeleid ik op dit moment wetenschappelijk onderzoek naar LMD welke betaald wordt door een particulier fonds (Stichting AmphoraEst). Deze stichting heeft geen belang bij het advies of de richtlijn, anders dan optimale zorg voor LMD patiënten. Ik heb geen intellectuele of reputatie belangen bij deze richtlijn commissie, anders dan erkenning als professional. * Ik zet mij actief en publiekelijk in voor verduurzaming van de gezondheidszorg, zowel binnen mijn eigen ziekenhuis als landelijk. Hierbij zijn er raakvlakken met de onderhavige richtlijn. * In het verleden heb ik een financiële vergoeding voor presentaties op gesponsord symposium of nascholing ontvangen. Sinds 2018 niet meer. |
Geen restricties |
|
Noordzij |
Werkzaam als oogarts o.a. in het zelfstandig behandelcentrum Oog op Zuid Oogkliniek. Lid van de Coöperatie Oogheelkunde op Zuid U.A. en voorzitter van het bestuur van de Coöperatie Oogheelkunde op Zuid U.A. Bestuurder bij de Stichting Oogheelkunde op Zuid. Medisch directeur bij het zelfstandig behandel Oog op Zuid Oogkliniek. |
De afgelopen maanden in samenwerking met Novartis een enquete opgesteld voor uirvragen hoe er in Nederland intravitreaal geïnjecteerd wordt. Names het Maasstad Ziekenhuis lid van de werkgroep "Santeon dure geneesmiddelen maculadegeneratie". Lid FMS/ NOG werkgroep Cluster Oog (onbetaald). |
Het Maasstad Ziekenhuis nam deel aan de Raven studie van Novartis to 2020. Ik was daarvan voor de locatie Maasstad ziekenhuis de principal investigator. |
Geen restricties |
|
Verbraak |
Oogarts, Amsterdam Universitair Medisch Centrum |
Voorzitter stuurgroep FRB!NL, niet betaald.Ik ben onbezoldigd voorzitter FRB!NL, een project dat gesteund wordt door Bayer, waar nu alleen nog financiering gaat naar advocaten kantoor (via Oogfonds) voor uitwerking agreement tussen deelnemende NL centra en universiteit van Sydney (SSR project). |
Betaald adviseur: Bayer, Novartis, IDxDR, UCB. vergoeding voor deelname (voorzitter) aan werkgroep die de toekomst van oogheelkunde in kaart wil brengen, project van Novartis, dit beslaat gehele oogheelkunde. Ontwikkeling Qualiteit van Visueel Functioneren questionair, AUMC, grant van Bayer. Ik werk mee aan project om een computer assisted test toe te passen als maat voor kwaliteit van leven/visus bij patiënten, die anti-VEGF injecties krijgen, dit wordt gesteund door Bayer, PI is Ruth van Nispen, onderzoekster wordt (deels) betaald. |
Uitgesloten van besluitvorming bij modules over anti-VEGF. Novartis project over toekomst oogheelkunde: geen belangenverstrengeling. |
|
Van Lith-Verhoeven |
Oogarts ETZ (medisch manager oogheelkunde ETZ) |
Lasik centrum Boxtel: ooglidcorrecties |
Adviescommissie Novartis, Bayer en allergan Organisatie congres Novartis en Bayer Honarium voor eigen presentatie Bayer. 1 advies bijeenkomst oogvitaal bv 1 advies bijeenkost horus pharma (betaald) 1 advies bijeenkomst Roche 1 advies bijeenkomst Roche en co-auteur aan expert opinion T & E.
Extern gefinancierd onderzoek: Novartis, Roche, Chengdu Kanghong biological science, Bayer. Het zijn studies van verschillende sponsoren met verschillende medicijnen, zowel op gebied AMD, DME en RVO. We hebben maar enkele deelnemers per studie. Het geld wat we ontvangen gedurende de studieduur gebruiken we om de studie coordinator (trial nurse) en de medewerkers (TOA's, optometristen) te betalen voor de tijd die ze in het onderzoek stoppen. Dat verekenen we dus met het ziekenhuis.
1 advies bijeenkomst oogvitaal bv 1 advies bijeenkost horus pharma (betaald) 1 advies bijeenkomst Roche 1 advies bijeenkomst Roche en co-auteur aan expert opinion T & E. |
Uitgesloten van besluitvorming bij modules over anti-VEGF.
|
|
Huiskamp |
Oogarts, netvliesspecialist in het Universitair Medisch Centrum Groningen |
Geen |
*Annexin Pharmaceuticals, Onderzoek naar moleculair imaging bij patiënten met retinale veneuze occlusie of diabetische retinopathie. SIGHT studie. Kosten voor de afdeling oogheelkunde worden gefinancierd door Annexin Pharmaceuticals. Deelname als onderzoeker aan deze studie (geen projectleider). * Bayer, Multicenter onderzoek naar medicijn voor diabetische retinopathie. NEON-NPDR studie. Kosten voor de afdeling oogheelkunde worden gefinancierd door Bayer. Deelname als onderzoeker aan deze studie (geen projectleider). * Heidelberg Engineering, Onderzoek naar moleculair imaging bij patiënten met neovasculaire LMD. LEAF studie. Financiering door het UMCG zelf. Apparatuur is voor deze studie ter beschikking gesteld door Heidelberg Engineering. Deelname als onderzoeker aan deze studie (geen projectleider). |
Geen restricties |
|
Kok |
Oogarts, medisch retina speciliast Bergamn Clinics Ogen Amsterdam UMC |
Geen |
Geen |
Geen restricties |
|
Lambooij |
Oogarts, Reinier de Graaf ziekenhuis Delft |
Gastdocent TOA opleiding Dutch Health Tec Academy Utrecht, betaald |
Geen |
Geen restricties |
|
Tigchelaar |
Oogarts |
Consultent oogarts bij visio R&A, betaald Consultent oogarts bij visio school, betaald Werkgroep FRB, onbetaald Eenmalige meet the expert bijeenkomst, uren vergoeding Voor novartis voorafgaand introductie van broculizumab. Broculizumab wordt gebruikt bij amd, in het voorstadium van de introductie was een meet the expert bijeenkomst om van een oogarts in het veld te horen wat overwegingen zijn voor beslissingen in de praktijk. Introductie in nederland in 2020. Honorering was alleen uren vergoeding. |
Geen |
Uitgesloten van besluitvorming bij modules over anti-VEGF, mocht wel meelezen als er geen andere experts waren. |
|
Klaver |
Erasmus MC, 0.6FTE, Hoogleraar Radboudumc, 0.4FTE, Oogarts University of Basel (locatie IOB, Basel), 0.15FTE, Hoogleraar |
Lid bestuur van Euretina, Europese vereniging voor retina specialisten, onbetaald Lid bestuur Landelijke Stichting voor Blinden en Slechtzienden (LSBS), onbetaald Lid bestuur Rotterdamse Oogheelkundig Onderzoek Stichting (ROOS), onbetaald Lid bestuur Collaborative Ophthalmic Research Rotterdam (CORR), onbetaald,
Betaald op projectbasis: Consultant voor TheaPharma Consultant voor Bayer |
* Ik maak geen deel uit van onderzoek geïnitieerd door het bedrijfsleven. Wel financiert Bayer een deel van de ontwikkeling van AI algoritmen die wij gebruiken voor het beoordelen van oogheelkundige beelden in ons EyeNED reading center. Bayer heeft geen invloed op deze algoritmen. Algoritmen hebben deels betrekking op LMD. Echter, Bayer is geen producent/leverancier van AI software. * Ik heb geen persoonlijk gewin bij, ander dan persoonlijke erkenning van mijn expertise als clinicus en als wetenschappelijk onderzoeker in dit vakgebied. De LSBS is een slechtziendenstichting die weinig patiënten met maculadegenratie als lid heeft. Mijn grootste inbreng voor de commissie is mijn expertise op het gebied van genetica, voeding en leefstijl. * Ik organiseer 1x per jaar nascholingsdagen voor de werkgroep Medische Retina. Deze nascholingen worden door Bayer gefinancierd. Bayer heeft geen invloed op de inhoud van deze nascholing. Ik geef 1x per jaar lezingen die georganiseerd worden door de firma Thea Pharma voor Europese oogartsen. Deze lezingen gaan over dieet adviezen voor maculadegeneratie. *Het IOB instituut in Basel is opgericht als een samenwerking tussen de Universiteit van Basel, het universiteitsziekenhuis van Basel, en Novartis. Novartis heeft geen zeggenschap over de wetenschappelijke koers en de onderzoeken die IOB uitvoert. Werknemers van IOB hebben geen financiele relatie met Novartis en leggen geen verantwoording af aan deze partij. |
Uitgesloten van besluitvorming bij modules over anti-VEGF.
|
|
Diepman-Leerdam |
Optometrist werkzaam bij Berman Clinics Doetinchem |
Niet van toepassing |
Geen |
Geen restricties |
|
Jansen Molenaar |
Adviseur oogzorg |
Niet van toepassing |
Geen |
Geen restricties |
|
Schoots-Wilke |
Voorzitter MD |
Vrienden van Walstede - onbetaald |
Voorzitter patiëntenvereniging |
Geen restricties |
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
|
Module |
Uitkomst raming |
Toelichting |
|
Module Symptomen van LMD |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Fasering diagnostiek van LMD |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module OCT |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module FAG |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module ICG-angiografie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module OCT-angiografie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Indicaties en contra-indicaties voor anti-VEGF behandeling bij neovasculaire LMD |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Keuze van anti-VEGF middel |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Chirurgie bij submaculaire bloeding |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Photodynamic therapy bij polypoidale choroidale vasculopathie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Photodynamic therapy bij retinale angiomateuze proliferatie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Photodynamic therapy bij chronische centrale sereuze chorioretinopathie met subretinale neovascularisatie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Photodynamic therapy bij non-responders op anti-VEGF therapie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Thermische laserbehandeling |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Cataractextractie bij patiënt met neovasculaire LMD |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Behandelstrategie voor anti-VEGF medicatie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft]. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Instructies aan patiënt voor herkennen reactivatie van maculaire neovascularisatie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Criteria om te stoppen met anti-VEGF behandeling |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Criteria voor het veranderen van anti-VEGF middel |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Voedingsadvies ter preventie van LMD progressie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Leefstijladviezen ter preventie van LMD progressie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Bloedverdunners bij LMD |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Advies aan familieleden van een patiënt met LMD |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Intravitreale injecties |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Verwijzing naar oogarts |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Verwijzing voor hulpmiddelen en revalidatie |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Informed Consent |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Informed consent voor de behandeling met off-label bevacizumab (Avastin®) |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Informatievoorziening aan patiënten en naasten |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
|
Module Rol van de patiëntenvereniging |
geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het overgrote deel (±90%) van de zorgaanbieders en zorgverleners al aan de norm voldoet. Er worden daarom geen substantiële financiële gevolgen verwacht. |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door het uitnodigen van de Patiëntenfederatie Nederland, MaculaVereniging en Oogvereniging voor de schriftelijke knelpuntenanalyse en het afgevaardigde patiëntenverenigingen in de klankbordgroep. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de MaculaVerening en Oogvereniging en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerden de werkgroep de knelpunten in de zorg voor patiënten met leeftijdsgebonden maculadegeneratie (LMD). De werkgroep beoordeelde de aanbevelingen uit de eerdere richtlijnmodule (NOG, 2014]) op noodzaak tot revisie. Tevens zijn er knelpunten aangedragen door de IGJ, Zorginstituut, OVN en Oogvereniging via enquête.
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE-methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zou de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, Glasziou P, Jaeschke R, Vist GE, Williams JW Jr, Kunz R, Craig J, Montori VM, Bossuyt P, Guyatt GH; GRADE Working Group. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008 May 17;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008 May 24;336(7654). doi: 10.1136/bmj.a139.
Schünemann, A Holger J [corrected to Schünemann, Holger J]. PubMed PMID: 18483053; PubMed Central PMCID: PMC2386626.
Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
Zoekverantwoording
Literature search strategy
Embase
|
No. |
Query |
Results |
|
#10 |
#8 AND #9 |
152 |
|
#9 |
'meta analysis'/de OR 'meta analysis (topic)'/exp OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de |
612349 |
|
#8 |
#1 AND #6 AND #7 AND ([english]/lim OR [dutch]/lim) AND [2011-2021]/py NOT ('conference abstract'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) |
1670 |
|
#7 |
'bevacizumab'/exp OR 'bevacizumab':ti,ab,kw OR 'abevmy':ti,ab,kw OR 'ainex':ti,ab,kw OR 'altuzan':ti,ab,kw OR 'ankeda':ti,ab,kw OR 'avastin':ti,ab,kw OR 'aybintio':ti,ab,kw OR 'bevax':ti,ab,kw OR 'byvasda':ti,ab,kw OR 'bryxta':ti,ab,kw OR 'cizumab':ti,ab,kw OR 'equidacent':ti,ab,kw OR 'krabeva':ti,ab,kw OR 'kyomarc':ti,ab,kw OR 'onbevzi':ti,ab,kw OR 'pusintin':ti,ab,kw OR 'versavo':ti,ab,kw OR 'zirabev':ti,ab,kw OR 'rhumab-vegf':ti,ab,kw |
63965 |
|
#6 |
#2 OR #3 OR #4 OR #5 |
24292 |
|
#5 |
'biosimilar agent'/exp OR biosimilar*:ti,ab,kw |
9287 |
|
#4 |
'brolucizumab'/exp OR 'brolucizumab':ti,ab,kw OR 'beovu':ti,ab,kw OR 'dlx 1008':ti,ab,kw OR 'dlx1008':ti,ab,kw OR 'esba 1008':ti,ab,kw OR 'esba1008':ti,ab,kw OR 'rth 258':ti,ab,kw OR 'rth258':ti,ab,kw |
194 |
|
#3 |
'aflibercept'/exp OR aflibercept:ti,ab,kw OR 'vegf trap':ti,ab,kw OR 'ave 0005':ti,ab,kw OR 'ave0005':ti,ab,kw OR 'bay 86 5321':ti,ab,kw OR 'bay 865321':ti,ab,kw OR 'bay865321':ti,ab,kw OR 'eylea':ti,ab,kw OR 'vascular endothelial growth factor trap':ti,ab,kw OR 'vasculotropin trap':ti,ab,kw OR 'zaltrap':ti,ab,kw |
7271 |
|
#2 |
'ranibizumab'/exp OR 'ranibizumab':ti,ab,kw OR 'lucentis':ti,ab,kw OR 'lucentris':ti,ab,kw |
10865 |
|
#1 |
'age related macular degeneration'/exp OR 'wet macular degeneration'/exp OR (('macula* degeneration' NEAR/3 ('age-related' OR exsudative)):ti,ab,kw) OR amd:ti,ab,kw OR wamd:ti,ab,kw OR warmd:ti,ab,kw OR armd:ti,ab,kw OR namd:ti,ab,kw OR 'subretinal neovascularization'/exp OR ((neovasculari*ation NEAR/3 (choroid* OR subretinal)):ti,ab,kw) OR cnv:ti,ab,kw |
55050 |
Ovid/Medline
|
# |
Searches |
Results |
|
7 |
5 and 6 |
84 |
|
6 |
meta-analysis/ or meta-analysis as topic/ or (metaanaly* or meta-analy* or metanaly*).ti,ab,kf. or systematic review/ or cochrane.jw. or (prisma or prospero).ti,ab,kf. or ((systemati* or scoping or umbrella or "structured literature") adj3 (review* or overview*)).ti,ab,kf. or (systemic* adj1 review*).ti,ab,kf. or ((systemati* or literature or database* or data-base*) adj10 search*).ti,ab,kf. or ((structured or comprehensive* or systemic*) adj3 search*).ti,ab,kf. or ((literature adj3 review*) and (search* or database* or data-base*)).ti,ab,kf. or (("data extraction" or "data source*") and "study selection").ti,ab,kf. or ("search strategy" and "selection criteria").ti,ab,kf. or ("data source*" and "data synthesis").ti,ab,kf. or (medline or pubmed or embase or cochrane).ab. or ((critical or rapid) adj2 (review* or overview* or synthes*)).ti. or (((critical* or rapid*) adj3 (review* or overview* or synthes*)) and (search* or database* or data-base*)).ab. or (metasynthes* or meta-synthes*).ti,ab,kf. |
526510 |
|
5 |
limit 4 to (english language and yr="2011 -Current") |
897 |
|
4 |
1 and 2 and 3 |
1148 |
|
3 |
Bevacizumab/ or 'bevacizumab'.ti,ab,kf. |
19512 |
|
2 |
Ranibizumab/ or 'ranibizumab'.ti,ab,kf. or aflibercept.ti,ab,kf. or 'brolucizumab'.ti,ab,kf. or exp Biosimilar Pharmaceuticals/ or biosimilar*.ti,ab,kf. |
10890 |
|
1 |
(exp Macular Degeneration/ and 'age related'.ti,ab,kf.) or Wet Macular Degeneration/ or ('macula* degeneration' adj3 ('age-related' or exsudative)).ti,ab,kf. or AMD.ti,ab,kf. or WAMD.ti,ab,kf. or WARMD.ti,ab,kf. or armd.ti,ab,kf. or namd.ti,ab,kf. or Choroidal Neovascularization/ or (neovasculari*ation adj3 (choroid* or subretinal)).ti,ab,kf. or CNV.ti,ab,kf. |
34776 |
Zoekopbrengst
|
|
Embase |
OVID/MEDLINE
|
Ontdubbeld (ook tegenover eerdere set van 162 SR)
|
|
SRs |
94 |
50 |
21 |
|
RCT |
93 |
64 |
104 |
|
Totaal |
187 |
114 |
125 |
Zoekstrategie
Embase
|
No. |
Query |
Results |
|
#12 |
#8 AND #11 NOT #10 =RCT |
93 |
|
#11 |
'randomized controlled trial'/exp OR random*:ti,ab OR (((pragmatic OR practical) NEAR/1 'clinical trial*'):ti,ab) OR ((('non inferiority' OR noninferiority OR superiority OR equivalence) NEAR/3 trial*):ti,ab) |
1794324 |
|
#10 |
#8 AND #9 =SR |
94 |
|
#9 |
'meta analysis'/de OR 'meta analysis (topic)'/exp OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de |
612349 |
|
#8 |
#1 AND #2 AND #7 AND ([english]/lim OR [dutch]/lim) AND [2011-2021]/py NOT ('conference abstract'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) |
1280 |
|
#7 |
#3 OR #4 OR #5 OR #6 |
24292 |
|
#6 |
'biosimilar agent'/exp OR biosimilar*:ti,ab,kw |
9287 |
|
#5 |
'brolucizumab'/exp OR 'brolucizumab':ti,ab,kw OR 'beovu':ti,ab,kw OR 'dlx 1008':ti,ab,kw OR 'dlx1008':ti,ab,kw OR 'esba 1008':ti,ab,kw OR 'esba1008':ti,ab,kw OR 'rth 258':ti,ab,kw OR 'rth258':ti,ab,kw |
194 |
|
#4 |
'bevacizumab'/exp OR 'bevacizumab':ti,ab,kw OR 'abevmy':ti,ab,kw OR 'ainex':ti,ab,kw OR 'altuzan':ti,ab,kw OR 'ankeda':ti,ab,kw OR 'avastin':ti,ab,kw OR 'aybintio':ti,ab,kw OR 'bevax':ti,ab,kw OR 'byvasda':ti,ab,kw OR 'bryxta':ti,ab,kw OR 'cizumab':ti,ab,kw OR 'equidacent':ti,ab,kw OR 'krabeva':ti,ab,kw OR 'kyomarc':ti,ab,kw OR 'onbevzi':ti,ab,kw OR 'pusintin':ti,ab,kw OR 'versavo':ti,ab,kw OR 'zirabev':ti,ab,kw OR 'rhumab-vegf':ti,ab,kw |
64487 |
|
#3 |
'ranibizumab'/exp OR 'ranibizumab':ti,ab,kw OR 'lucentis':ti,ab,kw OR 'lucentris':ti,ab,kw |
10865 |
|
#2 |
'aflibercept'/exp OR aflibercept:ti,ab,kw OR 'vegf trap':ti,ab,kw OR 'ave 0005':ti,ab,kw OR 'ave0005':ti,ab,kw OR 'bay 86 5321':ti,ab,kw OR 'bay 865321':ti,ab,kw OR 'bay865321':ti,ab,kw OR 'eylea':ti,ab,kw OR 'vascular endothelial growth factor trap':ti,ab,kw OR 'vasculotropin trap':ti,ab,kw OR 'zaltrap':ti,ab,kw |
7363 |
|
#1 |
'age related macular degeneration'/exp OR 'wet macular degeneration'/exp OR (('macula* degeneration' NEAR/3 ('age-related' OR exsudative)):ti,ab,kw) OR amd:ti,ab,kw OR wamd:ti,ab,kw OR warmd:ti,ab,kw OR armd:ti,ab,kw OR namd:ti,ab,kw OR 'subretinal neovascularization'/exp OR ((neovasculari*ation NEAR/3 (choroid* OR subretinal)):ti,ab,kw) OR cnv:ti,ab,kw |
55050 |
Ovid/Medline
|
# |
Searches |
Results |
|
9 |
(5 and 8) not 7 = RCT |
64 |
|
8 |
exp randomized controlled trial/ or random*.ti,ab,kf. or ((pragmatic or practical) adj clinical trial*).ti,ab,kf. or ((non-inferiority or noninferiority or superiority or equivalence) adj3 trial*).ti,ab,kf. |
1372546 |
|
7 |
5 and 6 = SR |
50 |
|
6 |
meta-analysis/ or meta-analysis as topic/ or (metaanaly* or meta-analy* or metanaly*).ti,ab,kf. or systematic review/ or cochrane.jw. or (prisma or prospero).ti,ab,kf. or ((systemati* or scoping or umbrella or "structured literature") adj3 (review* or overview*)).ti,ab,kf. or (systemic* adj1 review*).ti,ab,kf. or ((systemati* or literature or database* or data-base*) adj10 search*).ti,ab,kf. or ((structured or comprehensive* or systemic*) adj3 search*).ti,ab,kf. or ((literature adj3 review*) and (search* or database* or data-base*)).ti,ab,kf. or (("data extraction" or "data source*") and "study selection").ti,ab,kf. or ("search strategy" and "selection criteria").ti,ab,kf. or ("data source*" and "data synthesis").ti,ab,kf. or (medline or pubmed or embase or cochrane).ab. or ((critical or rapid) adj2 (review* or overview* or synthes*)).ti. or (((critical* or rapid*) adj3 (review* or overview* or synthes*)) and (search* or database* or data-base*)).ab. or (metasynthes* or meta-synthes*).ti,ab,kf. |
534738 |
|
5 |
limit 4 to (english language and yr="2011 -Current") |
629 |
|
4 |
1 and 2 and 3 |
674 |
|
2 |
Bevacizumab/ or 'bevacizumab'.ti,ab,kf. or Ranibizumab/ or 'ranibizumab'.ti,ab,kf. or 'brolucizumab'.ti,ab,kf. or exp Biosimilar Pharmaceuticals/ or biosimilar*.ti,ab,kf. |
27397 |
|
3 |
aflibercept.ti,ab,kf. |
2383 |
|
1 |
(exp Macular Degeneration/ and 'age related'.ti,ab,kf.) or Wet Macular Degeneration/ or ('macula* degeneration' adj3 ('age-related' or exsudative)).ti,ab,kf. or AMD.ti,ab,kf. or WAMD.ti,ab,kf. or WARMD.ti,ab,kf. or armd.ti,ab,kf. or namd.ti,ab,kf. or Choroidal Neovascularization/ or (neovasculari*ation adj3 (choroid* or subretinal)).ti,ab,kf. or CNV.ti,ab,kf. |
35128 |