Stopcriteria CFTR-modulatoren bij CF
Uitgangsvraag
Hoe dient de besluitvorming plaats te vinden om een CFTR-modulator al dan niet te continueren of te stoppen bij patiënten met CF?
Aanbeveling
Stop CFTR modulatoren in overleg met de patiënt bij ervaren bijwerkingen, gewogen door de arts en patiënt, die niet opwegen tegen het ervaren klinisch effect.
Ontwikkel criteria om de effecten van behandeling met CFTR-modulatoren te evalueren, waardoor gepast gebruik bevorderd wordt.
Overwegingen
Balans tussen voor- en nadelen en kwaliteit van het bewijs
Analyse van de literatuur naar effectiviteit van CFTR-modulatoren (voor deze analyse ivacaftor en lumacaftor/ ivacaftor) laat een positief effect zien op longfunctie, BMI, kwaliteit van leven en zweettest. De kwaliteit van dit bewijs varieert van hoog tot laag. Op basis van deze analyse zijn er voldoende potentiële voordelen voor de individuele patiënt om een behandeling te starten. De studies die zijn gebruikt voor deze analyse hebben zich gericht op verbetering van verschillende uitkomstmaten. Bij een chronische progressieve ziekte zoals CF is een behandeling die achteruitgang voorkomt ook een effectieve behandeling. Lange termijn vervolgstudies zijn nodig om de effectiviteit van CFTR-modulatoren op voorkomen van progressie van ziekte te evalueren. De mogelijke nadelen van behandeling bestaan uit de gerapporteerde bijwerkingen. De mogelijke bijwerkingen van de nu bekende CFTR-modulatoren zijn uitgebreid maar meest voorkomend zijn benauwdheidsklachten en drukkend gevoel op de borst en gastro-intestinale bijwerkingen (misselijkheid, braken, buikpijn en diarree). Deze bijwerkingen verdwijnen bij een aanzienlijk deel van de gebruikers in de eerste maanden van gebruik. Er zijn geen studies die hebben gekeken naar mogelijke stopcriteria voor gebruik van CFTR-modulatoren. Actuele stopcriteria van behandeling met een CFTR-modulator zijn bijwerkingen (zoals cholestatische hepatitis dan wel persisterende misselijkheid en braken of benauwdheid). Voor de individuele patiënt kunnen de ervaren bijwerkingen in overleg met de behandelend arts aanleiding zijn om de behandeling met CFTR-modulatoren te stoppen.
Wat vinden patiënten: patiëntenvoorkeur
Patiënten zijn enthousiast over CFTR modulatoren, zij voelen zich soms direct heel veel beter. Het gaat met name om minder vermoeidheid en meer energie, waardoor participatie in de maatschappij kan toenemen. Het biedt hen in potentie een beter toekomstperspectief waarbij er sprake kan zijn van een langere levensverwachting met een betere kwaliteit van leven. Patiënten willen graag uitgebreide informatie over te verwachten effectiviteit en risico’s. Bij jonge kinderen is er nog minder bekend over lange termijn effecten en bijwerkingen en dit zorgt ook voor wisselende reacties van ouders. In de praktijk stoppen patiënten soms met CFTR-modulatoren (in overleg met de behandelend arts) als zij geen effecten ervaren. Op dit moment ontbreken echter duidelijke criteria waarop die besluitvorming gebaseerd kan worden.
Wat vinden artsen: professioneel perspectief
Potentiële toepassing van CFTR modulatoren is veelbelovend. Huidige modulatoren laten op groepsniveau een effect zien op verschillende eindpunten zoals longfunctie, lichaamssamenstelling, kwaliteit van leven en zweettest. Individuele respons op de huidige geregistreerde modulatoren is variabel: van bijwerkingen die resulteren in staken, tot zeer goede klinische respons. Er is behoefte aan goede uitkomstmaten voor patiënten die nog milde klachten en geringe afwijkingen hebben maar ook voor patiënten met al vergevorderde ziekte-ernst. Het stabiliseren van de ziekte kan een belangrijke effectmaat zijn. Om deze uitkomstmaat te meten is een langdurige follow-up noodzakelijk. Daarnaast is het vanuit veiligheidsperspectief noodzakelijk om tijdens gebruik van CFTR modulatoren met enige regelmaat te screenen op mogelijke bijwerkingen en bedacht te zijn op interacties met andere medicatie. De beroepsgroep gaat criteria ontwikkelen om doelmatig gebruik van CFTR-modulatoren te bevorderen.
Kosten
De precieze kosten voor de CFTR modulatoren zijn niet bekend vanwege geheimhouding. Bekend is wel dat het zeer dure geneesmiddelen zijn die alleen vergoed worden onder strikte voorwaarden. De mogelijke baten van voorkomen van ziekteprogressie, afname van de zorgconsumptie en verbeterde kwaliteit van leven met ook de mogelijkheid tot verbetering van arbeidsparticipatie van patiënten met CF zijn belangrijke baten van CFTR modulatoren.
Haalbaarheid
Behandeling met CFTR modulatoren is momenteel beschikbaar voor een specifieke groep patiënten met bepaalde genetische mutaties. Voor deze groep is het mogelijk om de betreffende CFTR modulator te gebruiken.
Concluderend laten de huidige beschikbare CFTR modulatoren in de studies maar ook in de dagelijkse praktijk een positief effect zien op longfunctie (FEV1), bij gepoolde analyse een gering effect op aantal pulmonale exacerbaties, een verbetering van lichaamssamenstelling (BMI en gewicht), afname van chloorgehalte in zweet en een verbetering van kwaliteit van leven (respiratoire domein). De bijwerkingen die door patiënten worden ervaren zijn met name benauwdheid/drukkend gevoel op de borst en gastro-intestinale bijwerkingen en leiden slechts in een klein deel van de patiënten tot stoppen met de behandeling.
Er is nu onzekerheid over de precieze bijwerkingen, lange-termijn effecten en responsiviteit van de huidige CFTR modulatoren. In de (nabije) toekomst wordt een nieuwe generatie CFTR modulatoren verwacht, waardoor de overwegingen en aanbevelingen weer kunnen veranderen.
Onderbouwing
CFTR-modulatoren vormen een steeds belangrijker wordend onderdeel in de behandeling van patiënten met CF. Deze modulatoren zijn in staat om het afwezige of gestoorde chloridetransport te herstellen en/of te versterken. Effectiviteit van deze modulatoren verschilt van patiënt tot patiënt waarbij er naast verbetering van verschillende effectparameters ook sprake kan zijn van stabilisatie hiervan en daarmee het stoppen van achteruitgang. Naast relevantie voor de patiënt is het ook vanuit kosten oogpunt relevant om zo goed mogelijk vast te stellen of een CFTR-modulator effectief is.
Indien een CFTR-modulator voor een specifieke genmutatie effectief blijkt, kan deze behandeling beschikbaar komen voor CF-patiënten met de betreffende genmutatie (in Nederland momenteel voor ivacaftor vanaf de leeftijd van 2 jaar en voor een combinatie van lumacaftor/ivacaftor vanaf de leeftijd van 6 jaar).
De klinische langere termijn ervaring met de verschillende CFTR-modulatoren is nog beperkt. Een deel van de patiënten met CF heeft aan de klinische studies meegedaan naar effectiviteit en veiligheid van deze CFTR-modulatoren en konden aansluitend doorgaan met het betreffende middel. Een klein deel van de patiënten met CF heeft toegang gehad tot een compassionate use programma vanwege snelle achteruitgang of zeer slechte longfunctie. Beide groepen gebruiken deze middelen nu maximaal enkele jaren.
CF is een progressieve ziekte en derhalve wordt niet alleen verbetering van verschillende effectparameters maar ook het stoppen van achteruitgang gerekend tot een positief effect van de behandeling met CFTR-modulatoren.
De komende jaren worden nieuwe en effectievere CFTR-modulatoren ontwikkeld en deze middelen hebben een hoge kostprijs. De huidige CFTR-modulatoren en de nieuwe CFTR-modulatoren zijn primair ontwikkeld voor specifieke groepen gebaseerd op onderliggende CFTR-mutatie. Dit betekent dat er voor patiënten met CF met zeldzamer CFTR-mutaties geen mogelijkheid bestaat deel te nemen aan nieuwe studies of gebruik te maken van de modulatoren die mogelijk ook voor hen effectief kunnen zijn. Er wordt druk gezocht naar manieren om ook voor deze patiënten aan te tonen dat behandeling met een CFTR-modulator effectief is. Belangrijke ontwikkeling hierin is een patiënt uniek celmodel zoals het organoid (“mini-darmpje”) model. Hiermee kan op een “mini-darmpje” van de betreffende patiënt worden bekeken welk middel effectief zou kunnen zijn.
De huidige startcriteria voor behandeling met CFTR-modulatoren zijn gebaseerd op CFTR-mutatie, leeftijd en bijkomende morbiditeit zoals CF-gerelateerde leverziekte. Er zijn momenteel geen duidelijke stopcriteria (behoudens bijwerkingen). De meest gerapporteerde bijwerkingen door patiënten in de studies en dagelijkse praktijk zijn buikklachten (misselijkheid, buikpijn en/of diarree) en benauwdheid/ drukkend gevoel op de borst. De meest gevonden afwijkingen in bloed zijn verhoogde transaminasen. Dit kan passagere zijn en dient vervolgd te worden.
De gebruikte effectmaten in de fase 2 en 3 studies zijn mogelijk niet het meest relevant voor de patiënt in de dagelijkse praktijk. Voor jongere patiënten (kinderen) en patiënten die nog relatief weinig last hebben van hun CF zijn de gebruikte effectmaten in de studies niet toereikend, en ook voor volwassen patiënten met een slechtere longfunctie zijn andere effectmaten dan FEV1 zoals kwaliteit van leven relevanter.
(1) De verandering in FEV1 is momenteel de meest gebruikte uitkomstmaat.
De vraag is of de verandering in longfunctie (FEV1) gevoeliger of minder gevoelig is in het aantonen van effectiviteit van de CFTR modulatoren dan:
(2) veranderingen van afwijkingen op een CT-scan.
(3) verandering in exacerbatiefrequentie.
(4) verandering in voedingstoestand (lengte/gewichtverhouding).
(5) verandering in kwaliteit van leven.
(6) verandering in zweettest.
(7) slope of decline (FEV1).
(8) Lung Clearance Index (LCI).
1a. FEV1 % relatieve verandering vanaf baseline bij ivacaftor behandeling
|
Redelijk GRADE |
Een ivacaftorbehandeling geeft waarschijnlijk een relevante relatieve verbetering van FEV1% vergeleken met placebo na 24 weken bij CF-patiënten met een G551D-genmutatie.
Bronnen: (Ramsey, 2011; Davies, 2013; Accurso, 2010) |
|
Redelijk GRADE |
Een ivacaftorbehandeling geeft waarschijnlijk een kleine relatieve verbetering van FEV1% voorspeld vergeleken met placebo na 24 weken bij patiënten met een R117H-genmutatie.
Bronnen: (Moss, 2015) |
1b. FEV1 % relatieve verandering vanaf baseline bij lumacaftor/ivacaftorbehandeling
|
Laag GRADE |
Een lumacaftor/ivacaftor behandeling geeft mogelijk een kleine relatieve verbetering van FEV1% na 8 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Boyle, 2014; Rowe, 2017) |
|
Hoog GRADE |
Een lumacaftor/ivacaftor behandeling geeft een relevante relatieve verbetering van FEV1% na 24 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Wainwright, 2015) |
1c. FEV1 voorspeld absolute verandering vanaf baseline bij ivacaftor behandeling
|
Redelijk GRADE |
Een ivacaftor behandeling geeft waarschijnlijk een relevante absolute verbetering van FEV1 voorspeld na 2 weken vergeleken met placebo bij CF patiënten met een G551D genmutatie.
Bronnen: (Davies, 2013b; Edgeworth, 2017, Davies, 2013; Ramsey, 2011) |
|
Redelijk GRADE |
Een ivacaftor behandeling geeft waarschijnlijk een kleine, mogelijk niet relevante, absolute verbetering van FEV1 voorspeld vergeleken met placebo na 24 weken bij patiënten met een R117H genmutatie.
Bronnen: (Moss, 2015) |
4d. FEV1 % voorspeld absolute verandering vanaf baseline bij lumacaftor/ ivacaftor behandeling
|
Redelijk GRADE |
Een lumacaftor/ ivacaftor behandeling geeft waarschijnlijk een kleine absolute verbetering van FEV1 voorspeld vanaf 8 weken vergeleken met placebo bij CF patiënten met een F508del mutatie.
Bronnen: (Boyle, 2014; Rowe, 2017) |
|
Hoog GRADE |
Een lumacaftor/ ivacaftor behandeling geeft een relevante absolute verbetering van FEV1 voorspeld na 24 weken vergeleken met placebo bij CF patiënten met een F508del mutatie.
Bronnen: (Wainright, 2015) |
3a. Aantal pulmonale exacerbaties bij ivacaftor behandeling
|
Redelijk GRADE |
Een ivacaftor behandeling resulteert waarschijnlijk niet in minder pulmonale exacerbatie episodes na 4 tot 48 weken vergeleken met placebo bij CF patiënten met een G551D, phe508D, R117H of non-G551D gating mutaties.
Bronnen: (Accurso, 2010; Davies, 2013b; De Boeck, 2014; Davies, 2013; Ramsey, 2011; Moss 2015) |
3b. Aantal pulmonale exacerbaties bij lumacaftor/ ivacaftor behandeling
|
Laag GRADE |
Een lumacaftor/ ivacaftor behandeling resulteert mogelijk niet in minder pulmonale exacerbatie episodes na 3, 8 en 24 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Boyle, 2014; Rowe, 2017; Ratjen, 2017; Wainwright, 2017) |
4a. Verandering in BMI en BMI-z-score bij ivacaftor behandeling
|
Redelijk GRADE |
Een ivacaftor behandeling resulteert waarschijnlijk in een kleine relevante verbetering van BMI na 4 en 8 weken vergeleken met placebo bij CF patiënten met een G551D of non-G551D genmutaties.
Bronnen: (De Boeck, 2014; Edgeworth, 2017) |
|
Redlijk GRADE |
Een ivacaftor behandeling resulteert waarschijnlijk in een kleine, mogelijk niet relevante verbetering van BMI na 24 weken vergeleken met placebo bij CF patiënten met een R117H genmutatie.
Bronnen: (Moss, 2015) |
4b. Verandering in gewicht bij ivacaftor behandeling
|
Redelijk GRADE |
Een ivacaftor behandeling resulteert waarschijnlijk in een relevante relatieve verbetering in gewicht vergeleken met placebo na 24 weken bij CF patiënten met een G551D genmutatie.
Bronnen: (Ramsey, 2011; Davies, 2013) |
4c. Verandering in BMI (kg/m2) voor lumacaftor/ ivacaftor behandeling
|
Redelijk GRADE |
Een lumacaftor/ ivacaftor behandeling resulteert waarschijnlijk niet in verbetering van de BMI na 8 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Rowe, 2017) |
|
Redelijk GRADE |
Een lumacaftor/ ivacaftor behandeling resulteert waarschijnlijk in een kleine verbetering van de BMI na 24 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Ratjen, 2017; Wainwright, 2017) |
4d. Verandering in gewicht (kg) voor lumacaftor/ ivacaftor behandeling
|
Redelijk GRADE |
Een ivacaftor/ lumacaftor behandeling resulteert waarschijnlijk niet in verbetering in gewicht na 8 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Rowe, 2017) |
5a. CFQ-R Respiratoir domein bij ivacaftor behandeling
|
Redelijk GRADE |
Een ivacaftor behandeling resulteert waarschijnlijk in een relevante verbetering in CFQ-R vergeleken met placebo vanaf 2 weken bij CF patiënten met een G551D of non-G551D genmutaties.
Bronnen: (Accurso, 2010; Davies, 2013b; De Boeck, 2014; Quittner, 2015; Ramsey, 2011; Davies, 2013) |
5b. CFQ-R Respiratoir domein bij lumacaftor/ ivacaftor behandeling
|
Redelijk GRADE |
Een lumacaftor/ ivacaftor behandeling resulteert waarschijnlijk in een verbetering in de CFQ-R score (respirartoir domein) na 8 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Boyle, 2014; Rowe, 2017) |
|
Redelijk GRADE |
Een lumacaftor/ ivacaftor behandeling resulteert in een kleine, mogelijk niet relevante, verbetering in de CFQ-R score (respirartoir domein) na 24 weken vergeleken met placebo bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Ratjen, 2017; Wainwright, 2017) |
6a. Zweettest chloride concentraties bij lumacaftor behandeling
|
Hoog GRADE |
Een ivacaftor behandeling resulteert in een relevante verbetering in zweettest chloride concentraties vergeleken met placebo vanaf 2 weken bij CF patiënten met G551D of non-G551D genmutaties.
Bronnen: (Davies, 2013b; De Boeck, 2014; Davies, 2013b; Edgeworth, 2017; Ramsey, 2011; Davies, 2013) |
|
Redelijk GRADE |
Een ivacaftor behandeling resulteert waarschijnlijk in een relevante verbetering in zweettest chloride concentraties na 24 weken vergeleken met placebo bij CF patiënten met een R117H genmutatie.
Bronnen: (Moss, 2015) |
6b. Zweettest chloride concentraties bij lumacaftor/ivacaftor behandeling
|
Redelijk GRADE |
Een lumacaftor/ ivacaftor behandeling resulteert waarschijnlijk in een kleine verbetering in zweettest chloride concentraties vergeleken met placebo na 3 en 8 weken bij CF patiënten met een homozygote F508del mutatie.
Bronnen: (Boyle, 2014; Rowe, 2017; Ratjen, 2017) |
8a. Lung Clearance Index bij ivacaftor behandeling
|
Zeer laag GRADE |
Het is onzeker of een ivacaftor behandeling resulteert in een verbetering in LCI vergeleken met placebo na 2 weken bij CF patiënten met een G551D genmutatie.
Bronnen: (Davies, 2013b) |
8b. Lung Clearance Index bij lumacaftor/ ivacaftor behandeling
|
Redelijk GRADE |
Een lumacaftor/ ivacaftor behandeling resulteert waarschijnlijk in een kleine verbetering in LCI vergeleken met placebo na 24 weken bij CF patiënten met een F508del mutatie.
Bronnen: (Ratjen, 2017) |
Als uitgangspunt is de Cochrane literatuur review van Patel (2015) genomen met daarin drie RCT’s, daar zijn tien losse parallelle en cross-over RCT’s aan toegevoegd.
In acht onderzoeken werd ivacaftor onderzocht in G551D, phe508del, non-G551D en R117H-mutatie patiënten. In de overige vijf onderzoeken kregen de patiënten een combinatie van lumacaftor en ivacaftor in verschillende doseringen, allen bij phe508del mutatie patiënten. In alle studies werd de CFTR-modulator vergeleken met een placebo.
Vijf studies waren placebo gecontroleerde parallelle RCT’s met twee armen. Drie studies waren een placebo gecontroleerde parallelle RCT met 3 of meer armen, waarin verschillende doseringen met de placebo werden vergeleken. Drie studies waren cross-over placebo gecontroleerde RCT’s met een wash-out periode tussen de twee behandelingen. Twee publicaties waren post-hoc analyses van eerdere RCT-studies en beschreven de effecten van een ivacaftorbehandeling voor verschillende FEV1-uitgangswaarden.
In onderstaande tabellen (tabel 1 en 2) staan de studiekarakteristieken en uitkomstmaten per geïncludeerde studie samengevat. De follow-up duur liep uiteen van 3 weken tot en met 48 weken. In vier studies werden patiënten ≥ 18 jaar geïncludeerd, in vijf studies ≥ 12 jaar, in drie studies ≥ 6 jaar en in drie studies patiënten tussen de 6 en 11 jaar. Alle studies hadden een inclusiecriterium van een baseline pp FEV1 van 40% of meer, behalve in de studie van Davies (2013b) waar een inclusiecriterium van pp FEV1 van 90% of meer werd aangehouden.
De volledige in- en exclusiecriteria per studie staan in de evidencetabellen. Alle studies zijn gefinancierd en (deels) opgezet door farmaceutische bedrijven (Vertex).
De resultaten voor ivacaftor en lumacaftor/ivacaftor zijn apart beschreven. De resultaten van Moss (2015) met genmutatie R117H zijn ook apart beschreven en niet gepooled met de overige genmutaties.
Tabel 1 Studie karakteristieken van geïncludeerde studies over CFTR modulatoren
|
Auteur (jaartal) |
CFTR modulator |
Dosering |
Leef tijd |
Gen mutatie |
FEV1 pp baseline, mean (range/±sd) |
Follow-up |
N
|
Design |
|
Accurso, 2010 (CR) |
Iva |
150 mg/12u 250mg/12u |
>18 jr |
G551D |
69% (40-122) |
4 wk |
19 |
RCT 3 arms (part 2) |
|
Edgeworth, 2017 |
Iva |
150mg/12u |
>18 jr |
G551D |
54% (23–110) |
4 wk |
20 |
Cross-over RCT |
|
Ramsey, 2011 (CR) |
Iva |
150mg/12u |
>12 jr |
G551D |
63% (32-98) |
48 wk |
167 |
RCT |
|
Konstan, 2015 |
Iva |
150mg/12u |
>12 jr |
G551D |
Iva: 69% (±18) Pla: 68% (±19) |
48 wk |
209 |
RCT (post-hoc), gepoolde data van Ramsey 2011 en Davies 2013 |
|
Davies, 2013 (CR) |
Iva |
150 mg/ 12u |
6-11 jr |
G551D |
84% (44-134) |
24 wk |
52 |
RCT |
|
Davies, 2013b |
Iva |
150mg/12u |
>6 jr |
G551D |
Iva: 102% (±12) PIa: 93% (±7) |
4 wk |
21 |
Cross-over RCT |
|
De Boeck 2014 |
Iva |
150mg/12u |
>6 jr |
non-G551D |
78% (43-119) |
8 wk |
39 |
Cross-over RCT (part I) |
|
Moss 2015 |
Iva |
150mg/12u |
>6 jr |
R117H |
Iva: 76% (±19) Pla: 70% (±19) |
24 wk |
69 |
RCT |
|
Boyle 2014
|
Luma/iva |
200mg/150mg/12u 200mg/250mg/12u 200mg/250mg/12u 400mg/250mg/12u 600mg/250mg/12u homozygous 400mg/12u/250mg/12u 600mg/250mg/12u –heterozygous |
>18 jr |
phe508del |
|
3 wk/8 wk |
62+109+15 |
RCT 1e cohort, 3 arms RCT 2/ 3e cohort, 6 arms |
|
Rowe 2017 |
Luma/iva |
400mg/12u/250mg/12u |
>18 jr |
phe508del |
|
8 wk |
|
RCT |
|
Elborn 2016 |
Luma/iva |
600mg/250mg/12u 400mg/12u/250 mg/12u |
>12 jr |
phe508del |
|
24 wk |
1108 |
RCT (post-hoc), 3 arms. Gepoolde data van Boyle 2014 en Wainwright 2015 |
|
Wainwright 2015 |
Luma/iva |
400mg/12u/250mg/12u 600mg/250 mg/12u |
>12 jr |
phe508del |
|
24 wk |
|
RCT, 3 arms |
|
Ratjen 2017 |
Luma/iva |
200mg/250mg/12u |
6-11 jr |
phe508del |
|
24 wk |
|
RCT |
CR=Cochrane; Iva=Ivacaftor; Luma=Lumacaftor; RCT=Randomized Controlled Trial
Tabel 2 Gebruikte uitkomstmaten van geïncludeerde studies over CFTR modulatoren
|
|
abs change FEV1% pr |
rel change FEV1% pr |
Number of PEx |
CFQ-R |
Voedings status |
Zweet test |
LCI |
Overig uitkomstmaten (niet beschreven) |
|
Accurso 2010 (CR) |
x
|
x
|
X |
|
|
x
|
|
NPD, FVC |
|
Edgeworth 2017 |
X |
|
|
X |
X |
X |
|
Alfred wellness score, % FFM, minute ventilation, VO2/HR, exercise time, VO2t1/2, Borg scores, spirometry, % VO2max |
|
Ramsey 2011 (CR) |
X |
X |
X |
X |
X |
X |
|
|
|
Konstan 2015 |
X |
|
|
X |
X |
X |
|
|
|
Davies 2013 (CR) |
X |
X |
X |
X |
X |
X |
|
|
|
Davies 2013b |
X |
|
|
X |
|
X |
X |
Percentage of predicted FEF 25%–75% (pp) |
|
De Boeck 2014 |
X |
|
X |
X |
X |
|
|
|
|
Moss 2015 |
X |
X |
X |
X |
X |
X |
|
|
|
Boyle 2014 |
X |
X |
|
X |
|
X |
|
|
|
Rowe 2017 |
X |
X |
|
X |
X |
X |
|
|
|
Elborn 2016 |
X |
X |
X |
X |
X |
|
|
|
|
Wainwright 2015 |
X |
X |
X |
X |
X |
|
|
liver function test elevations (ALT or AST and Total Bilirubin) |
|
Ratjen 2017 |
X |
|
|
X |
X |
X |
X |
Acute change ppFEV1 |
CFQ-R= Cystic Fibrosis Questionnaire Revised (respiratory domain); CR=Cochrane; FEV1% pr=Forced expiratory volume in 1 second, % predicted; LCI=Lung Clearance Index; PEx=Pulmonary Exacerbations.
‘kleine’ x= niet opgenomen in forest plot vanwege onvoldoende/ongeschikte data.
Voedingsstatus= verandering in gewicht, absolute of relatieve verandering in BMI, verandering in height-z-score, weight-z-score, BMI-z-score.
Resultaten
1. Verandering in FEV1 (Forced Expiratory Volume in the 1st second)
1a. FEV1 % relatieve verandering vanaf baseline bij ivacaftor behandeling
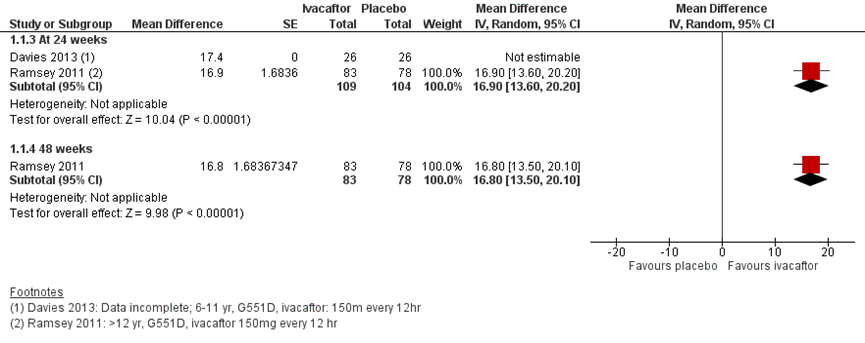
Vier studies rapporteerden over deze uitkomst voor ivacaftorbehandeling vergeleken met placebo. In figuur 1 zijn de resultaten gepresenteerd in een forestplot voor relatieve verandering vanaf baseline van FEV1% voor verschillende follow-upduren.
Na vier weken was er een relatieve maar niet-significant verbetering gevonden in pp FEV1 8,7% (range 2,3 tot 31,3%) voor de 150 mg groep, 4,4% (range 0,0 tot 18,3) in de 250 mg groep en 7,3% (range 5,2 tot 8,2) in de placebogroep (Accurso, 2010). Accurso (2010) rapporteerde medianen (range) en kon daarom niet worden opgenomen in de forest plot.
Na 24 en 48 weken werd een statistisch significant gemiddeld verschil gezien in het voordeel van de ivacaftor 150 mg elke 12 uur behandeling van bijna 17% (Ramsey, 2011) vergeleken met placebo. Ook Davies (2013) vond een statistisch significant verschil in het voordeel van de ivacaftor behandeling: 17,4% (P < 0,0001) na 24 weken, maar presenteerde geen spreidingsmaten.
Moss (2015) onderzocht het effect van ivacaftor vergeleken met placebo in een groep met 69 patiënten met de R117H mutatie met een follow-up duur van 24 weken. Zij vonden een niet significant gemiddeld verschil van 5,0%, p=0,06. (Moss, 2015).
Figuur 1 Forest plot: FEV1 % gemiddelde relatieve verandering vanaf baseline voor ivacaftor behandeling vergeleken met placebo
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat FEV1% relatieve verandering vanaf baseline (24 tot 48 weken, G551D en R117H) voor ivacaftor vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor het niet juist toepassen van een intention-to-treatanalyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
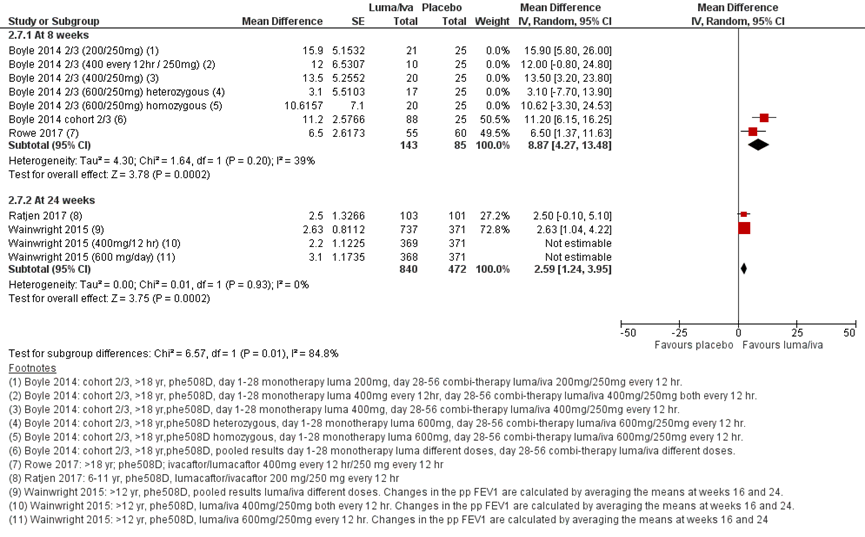
1b. FEV1 % relatieve verandering vanaf baseline bij lumacaftor/ivacaftorbehandeling
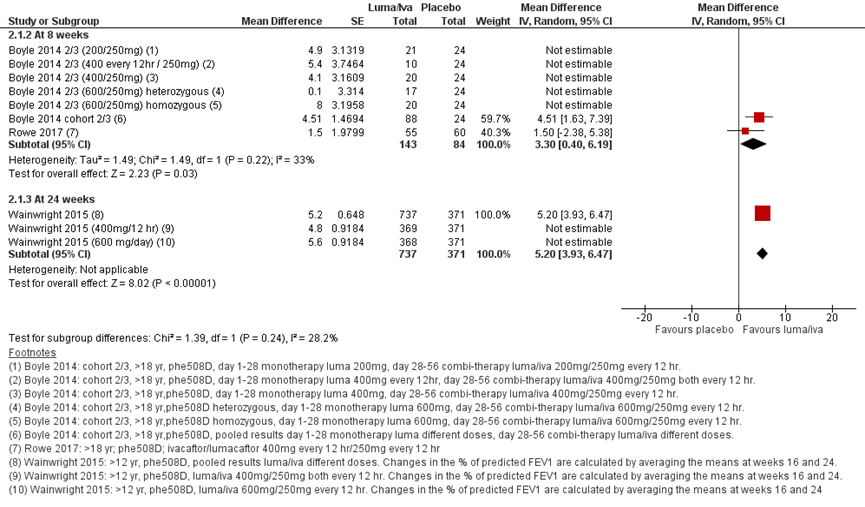
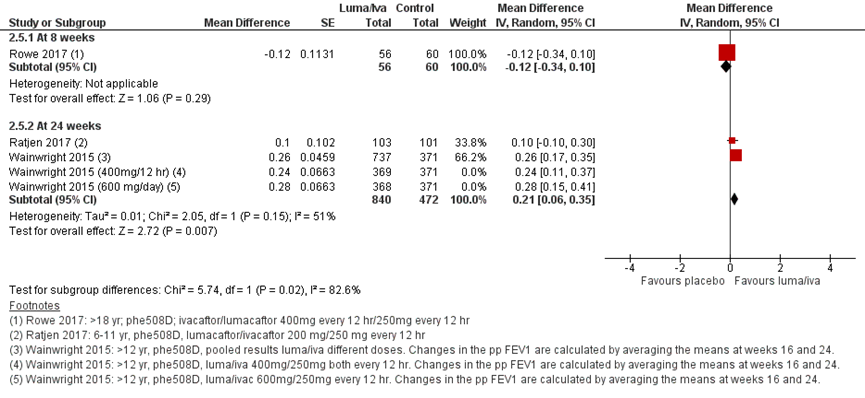
Drie studies rapporteerden over deze uitkomst voor de lumacaftor/ivacaftorbehandeling vergeleken met placebo. Elborn (2016) rapporteerde in nog één additionele publicatie de behandeleffecten van gepoolde data van twee eerder gepubliceerde RCT’s (Boyle, 2014; Wainwright, 2015) voor subgroepen van verschillende pp FEV1-uitgangswaarden. In figuur 2 staan de resultaten gepresenteerd in een forestplot voor relatieve verandering vanaf baseline van FEV1 voor verschillende follow-upduren. Na 8 weken werd een statistisch significant gemiddeld verschil gezien in het voordeel van de lumacaftor/ivacaftor behandeling (Boyle, 2014; Rowe, 2017). Dit verschil was groter na 24 weken (Wainwright, 2015).
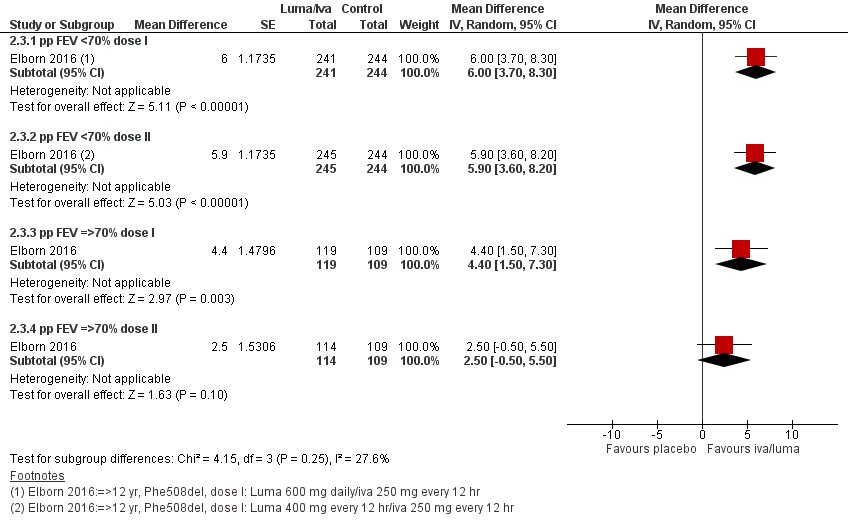
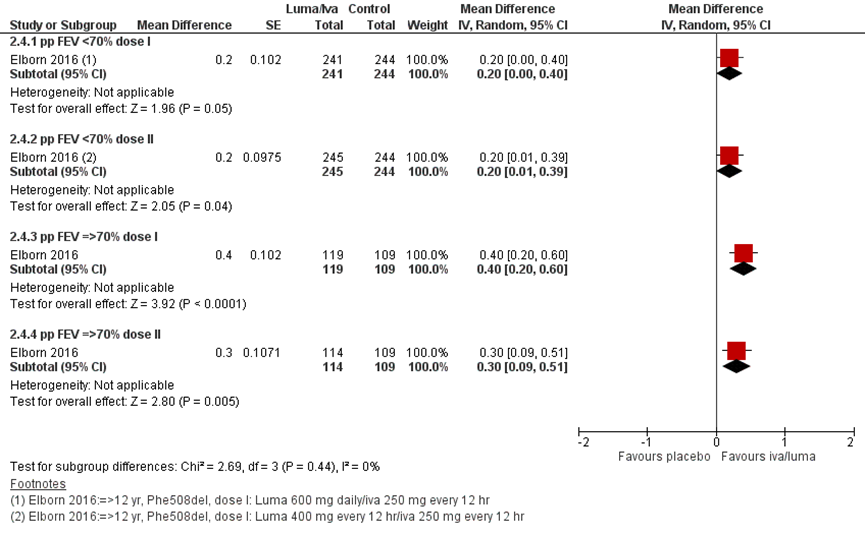
Elborn (2016) liet vergelijkbare resultaten zien in een posthoc-analyse met data van Boyle (2014) en Wainwright (2016) na 24 weken voor de subgroepen met een pp FEV < 70% en ≥ 70%, zoals gepresenteerd in figuur 3. Het verschil was niet meer statistisch significant voor de subgroup met ppFEV1≥70% en de dosering lumacaftor 400 mg/12u.
Figuur 2 Forest plot: FEV1 % gemiddelde relatieve verandering vanaf baseline voor lumacaftor/ivacaftor behandeling vergeleken met placebo
Figuur 3 Forest plot: FEV1 % gemiddelde relatieve verandering vanaf baseline voor lumacaftor/ ivacaftor behandeling vergeleken met placebo per subgroep
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat FEV1% relatieve verandering vanaf baseline (3 tot 8 weken) voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is met twee niveaus verlaagd gezien het geringe aantal patiënten en overlap met de grens voor klinische besluitvorming (imprecisie). Het niveau van de bewijskracht komt uit op GRADE ‘laag’.
De bewijskracht voor de uitkomstmaat FEV1% relatieve verandering vanaf baseline (24 weken) voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is niet in niveau verlaagd. Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘hoog’.
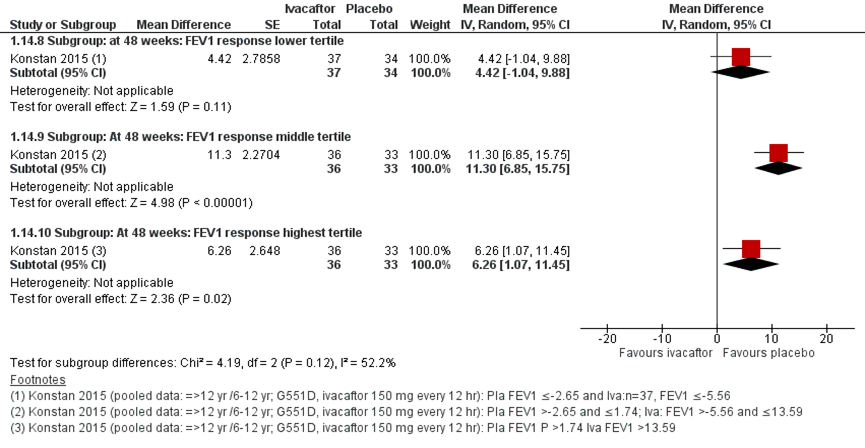
1c. FEV1 voorspeld absolute verandering vanaf baseline bij ivacaftor behandeling
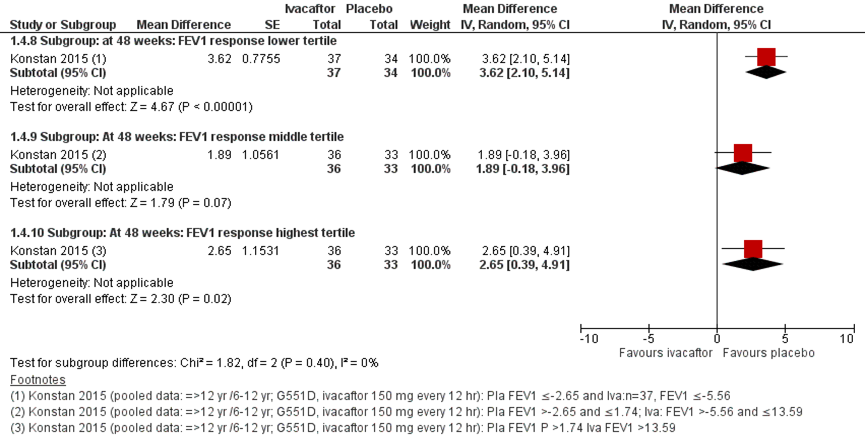
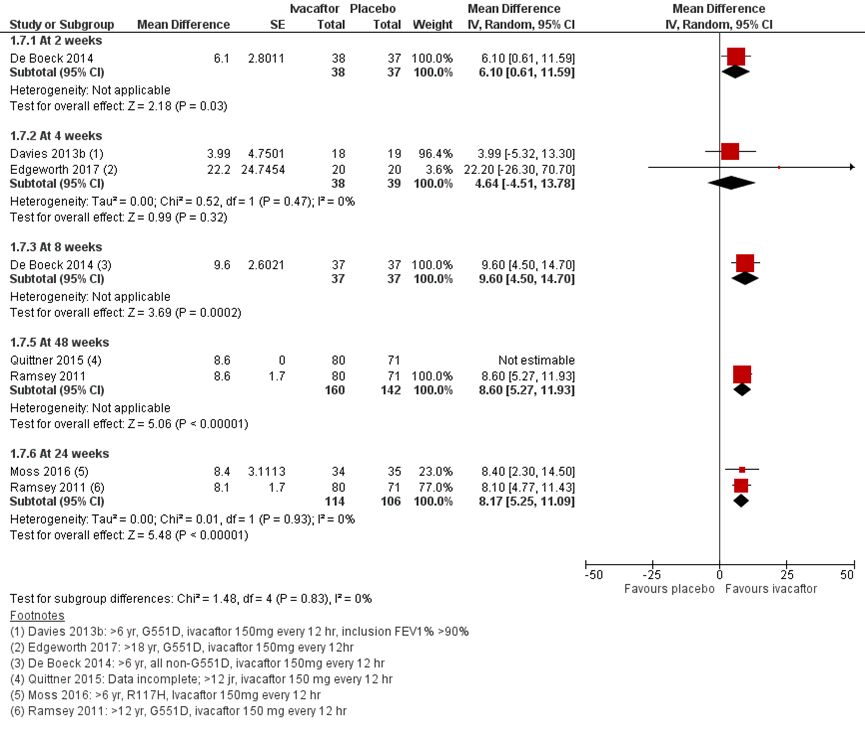
Zes studies rapporteerden over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. Konstan (2015) rapporteerde in nog één additionele publicatie de behandeleffecten van gepoolde data van twee eerder gepubliceerde RCT’s (Ramsey, 2011; Davies, 2013) voor tertielen van respons van FEV1 voorspeld. In figuur 4 staan de resultaten gepresenteerd in een forest plot voor de absolute verandering vanaf baseline van FEV1 voorspeld in percentage punten voor verschillende follow-up duren. Na 2, 4, 8, 24 en 48 weken werd een statistisch significant gemiddeld verschil gezien in patiënten met de G551D mutatie, in het voordeel van de ivacaftor 150 mg elke 12 uur behandeling. Het gemiddelde verschil varieerde van 10,0 tot 10,8 percentage punten. (Davies, 2013b; Edgeworth, 2017; De Boeck, 2014; Davies, 2013; Ramsey, 2011; Konstan, 2015).
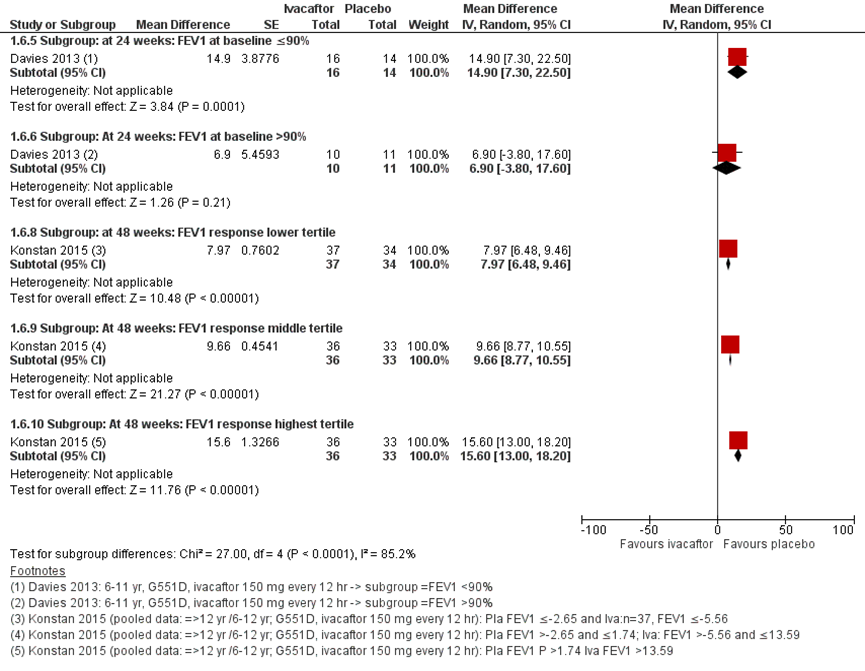
In figuur 5 zijn de resultaten gepresenteerd van subgroepanalyses. Davies (2013) onderzocht twee subgroepen, met een verschillend voorspelde FEV1 als uitgangspunt op baseline. In de groep met een FEV1 ≤ 90% was het gemiddeld verschil tussen de ivacaftor behandeling en placebo statistisch significant na 24 weken. Dit verschil in behandeleffect was niet meer significant in de groep met een pp FEV1 > 90% op baseline (Davies, 2013).
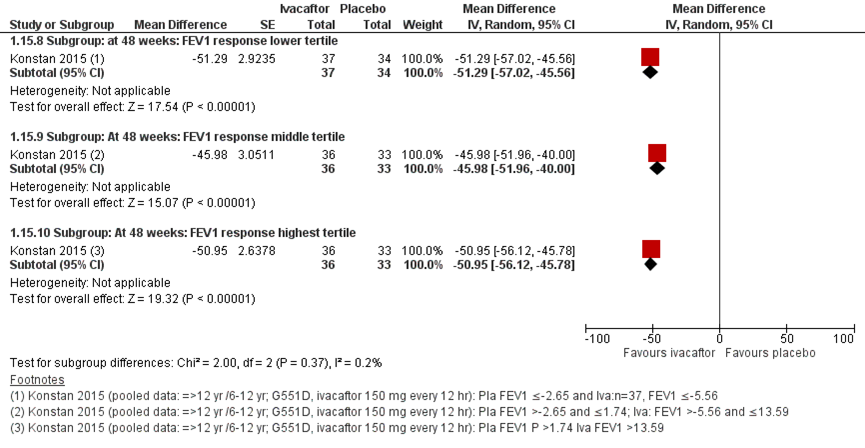
Konstan (2015) onderzocht het behandeleffect na 48 weken in een post-hoc analyse met gepoolede data van de RCT’s van Ramsey (2011) en Davies (2013). De response op voorspelde FEV1 werd in tertielen verdeeld. De absolute verandering in FEV1 voorspeld gaf voor alle tertielen van respons een statistisch significant behandeleffect in het voordeel van de ivacaftor behandeling. Een number needed to treat voor een verhoging van ≥5 percentage punten voor pp FEV1 werd berekend van 1,90 (Konstan, 2015).
Moss (2015) onderzocht het effect van ivacaftor vergeleken met placebo in een groep met 69 patiënten met de R117H mutatie met een follow-up duur van 24 weken. Zij vonden een niet significant gemiddeld verschil van 2,10 percentage punten voor voorspelde FEV1, p=0,20 (Moss, 2011).
Figuur 4 Forest plot: FEV1 gemiddelde absolute verandering in percentage punten vanaf baseline voor ivacaftor behandeling vergeleken met placebo

Figuur 5 Forest plot: FEV1 gemiddelde absolute verandering in percentage punten vanaf baseline na 48 weken voor ivacaftor behandeling vergeleken met placebo per subgroep
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat FEV1 voorspeld absolute verandering vanaf baseline (2 tot 48 weken, G551D en R117H) voor de ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
4d. FEV1 % voorspeld absolute verandering vanaf baseline bij lumacaftor/ ivacaftor behandeling
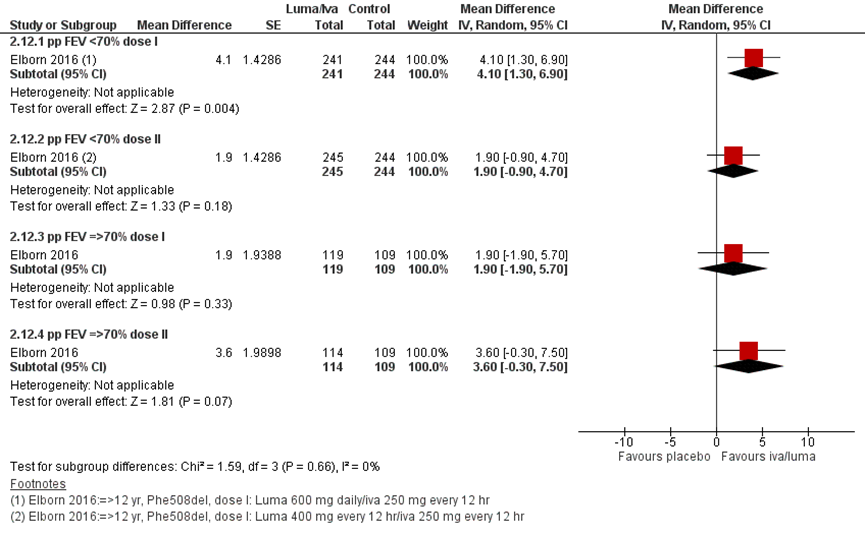
Vier studies rapporteerden over deze uitkomst voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo. Elborn (2016) rapporteerde in nog één additionele publicatie de behandeleffecten van gepoolde data van twee eerder gepubliceerde RCT’s (Boyle, 2014; Wainwright, 2015) voor subgroepen van verschillende pp FEV1 uitgangswaarden. In figuur 6 staan de resultaten gepresenteerd in een forest plot voor de absolute verandering vanaf baseline van FEV1 voorspeld in percentage punten voor verschillende follow-up duren.
Na 8 weken werd een niet significant gemiddeld verschil gezien in het voordeel van de lumacaftor/ ivacaftor behandeling van 1,98 (95% BI -0,65 tot 4,62) percentage punten (Boyle, 2014; Rowe, 2017). Dit verschil was wel significant na 24 weken, 2,98% (95% BI 2,31 tot 3,65) (Wainwright, 2015; Ratjen, 2017).
Elborn (2016) liet vergelijkbare resultaten zien in een posthoc-analyse met data van Boyle (2014) en Wainwright (2016) na 24 weken voor de subgroepen met een pp FEV< 70% en ≥ 70%, zoals gepresenteerd in figuur 7. Het verschil was niet meer statistisch significant voor de subgroup met ppFEV1≥70% en de dosering lumacaftor 400 mg/12u (Elborn, 2016).
Figuur 6 Forest plot: FEV1 gemiddelde absolute verandering in percentage punten vanaf baseline voor lumacaftor/ivacaftor behandeling vergeleken met placebo
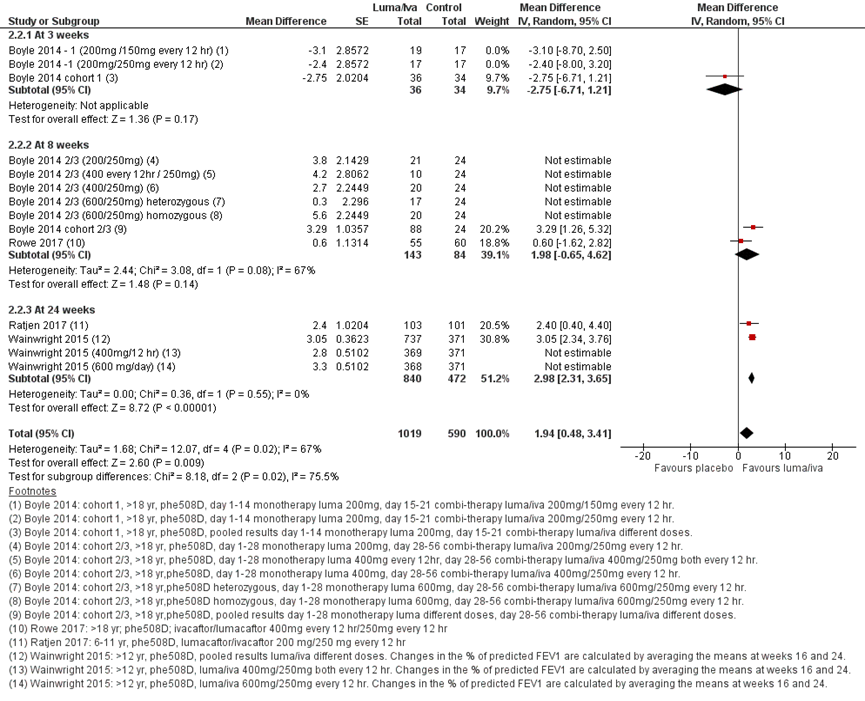
Figuur 7 Forest plot: FEV1 gemiddelde absolute verandering in percentage punten vanaf baseline voor lumacaftor/ivacaftor behandeling vergeleken met placebo per subgroep
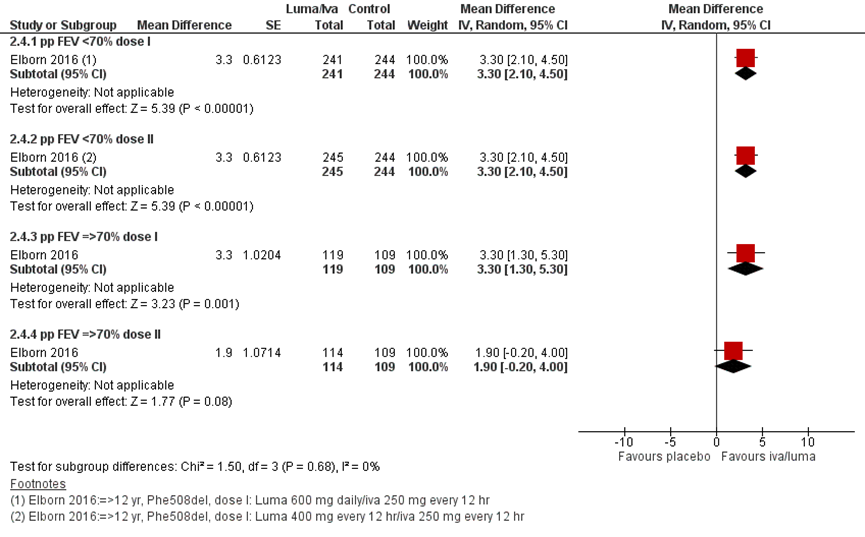
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat FEV1 voorspeld absolute verandering vanaf baseline (3 tot 8 weken) voor de ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
De bewijskracht voor de uitkomstmaat FEV1 voorspeld absolute verandering vanaf baseline (24 weken) voor de ivacaftor behandeling vergeleken met placebo is niet in niveau verlaagd. Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘hoog’.
2. Veranderingen van afwijkingen op een CT-scan
Voor deze uitkomstmaat hebben we geen artikelen gevonden. CT-scan veranderingen is in de studies niet als effectmaat gebruikt.
3. Aantal pulmonale exacerbaties
3a. Aantal pulmonale exacerbaties bij ivacaftor behandeling
Vijf studies rapporteerden over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. In figuur 8 staan de resultaten gepresenteerd in een forest plot voor het aantal pulmonale exacerbaties voor verschillende follow-up duren.
Er zijn voor alle follow-up duren 4, 8 en 48 weken geen significante verschillen te zien in het relatief risico op het aantal pulmonale exacerbaties bij een ivacaftor behandeling vergeleken met een placebo.
Moss (2015) onderzocht het effect van ivacaftor vergeleken met placebo in een groep met 69 patiënten met de R117H mutatie met een follow-up duur van 24 weken. Zij vonden ook geen significant verschil in het relatieve risico voor aantal pulmonale exacerbaties (Moss 2015).
Figuur 8 Forest plot: Het aantal participanten die een episode van een pulmonale exacerbatie had bij ivacaftor vergeleken met placebo behandeling
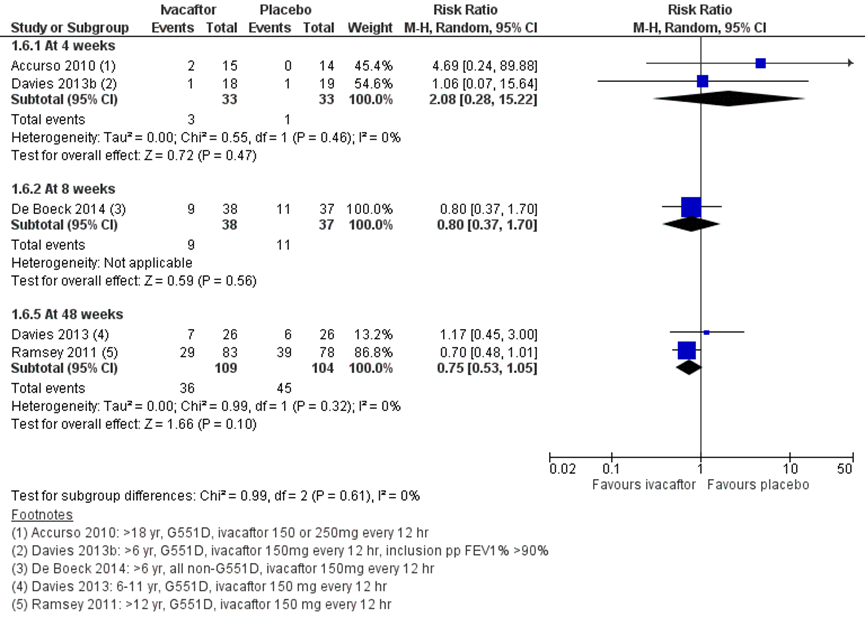
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat aantal pulmonale exacerbaties (4 tot 48 weken) voor de ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
3b. Aantal pulmonale exacerbaties bij lumacaftor/ ivacaftor behandeling
Vier studies rapporteerden over deze uitkomst voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo. Elborn (2016) rapporteerde in nog één additionele publicatie de resultaten van gepoolde data van twee eerder gepubliceerde RCT’s (Boyle, 2014; Wainwright, 2015) voor subgroepen van verschillende pp FEV1 uitgangswaarden. In figuur 9 staan de resultaten gepresenteerd in een forest plot voor het aantal pulmonale exacerbaties voor verschillende follow-up duren.
Na 3, 8 en 24 weken zijn er geen significante verschillen te zien in het relatief risico op het aantal pulmonale exacerbaties bij een lumacaftor/ ivacaftor behandeling vergeleken met een placebo.
Elborn (2016) presenteerde resultaten in een posthoc-analyse met data van Boyle (2014) en Wainwright (2016) na 24 weken voor de subgroepen met een pp FEV< 70% en ≥ 70%. Hier werden wel statistisch significante verschillen gezien tussen een lumacaftor/ ivacaftor behandeling en placebo met minder pulmonale exacerbaties in de lumacaftor/ivacaftor behandelgroep voor zowel de laagste en hoogste dosering met een risk ratio van respectievelijk 0,74 (95% BI 0,57 tot 0,95) en 0,65 (95% BI 0,50 tot 0,84) voor pp FEV<70% en een risk ratio van respectievelijk 0,55 (95% BI 0,35 tot 0,85) en 0,51 (95% BI 0,32 tot 0,80) voor ppFEV≥70%. Het aantal events per groep werd niet gepresenteerd (Elborn, 2016).
Figuur 9 Forest plot: Het aantal participanten die een episode van een pulmonale exacerbatie had bij lumacaftor/ ivacaftor vergeleken met placebo behandeling
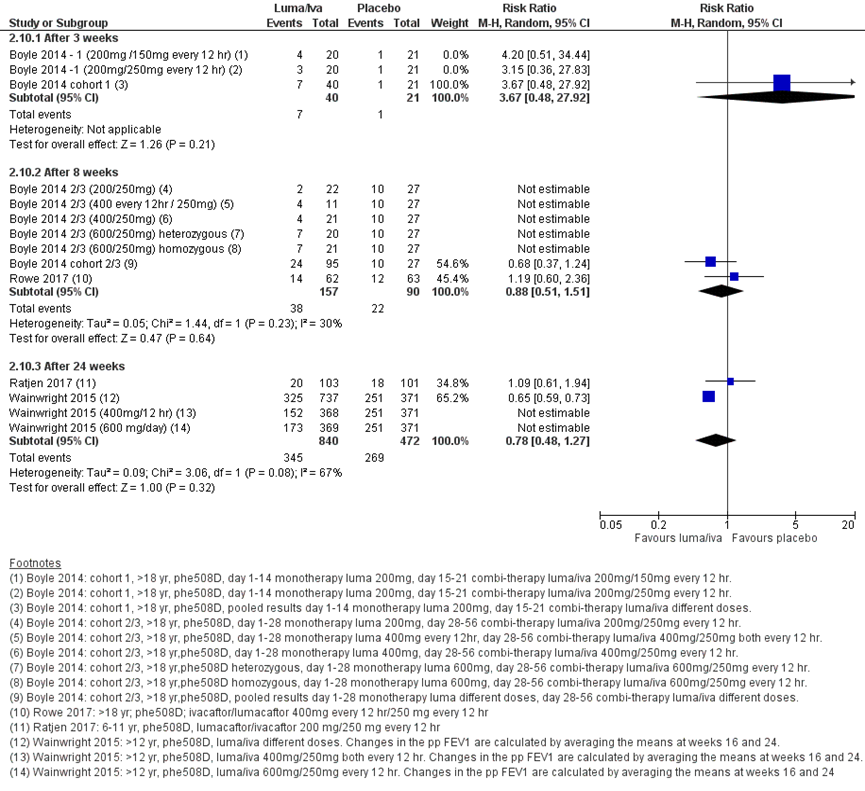
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat aantal pulmonale exacerbaties (3, 8 en 24 weken) voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is met twee niveaus verlaagd gezien het geringe aantal patiënten (imprecisie) en heterogeniteit van de resultaten. Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘laag’.
4. Voedingsstatus
4a. Verandering in BMI en BMI-z-score bij ivacaftor behandeling
Drie studies rapporteerden over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. Vanwege de verschillen in genmutaties en presentatie van uitkomstmaat was de data niet geschikt om te poolen en is geen forest plot gepresenteerd.
Edgeworth (2017) liet in een cross-over trial een statistisch significant verschil zien in gemiddelde percentage verandering in BMI van 1.2% (0,1 tot 2,3), p=0,039, na vier weken. (Edgeworth, 2017).
Na 8 weken liet De Boeck (2014) een statistisch significante verandering zien in gemiddeld BMI (kg/m2) verschil in het voordeel van de ivacaftor-groep vergeleken met de placebogroep van 0,68 kg/m2 (95% BI 0,34 tot 1,02). De Boeck (2014) liet een zelfde significante verandering zien voor het gemiddelde absolute verschil in verandering vanaf baseline voor BMI-for-age-z-score na 8 weken van 0,28 punten (95% BI 0,12 tot 0,45), p=0,001 (De Boeck, 2014).
Moss (2015) onderzocht het effect van ivacaftor vergeleken met placebo in een groep met 69 patiënten met de R117H mutatie met een follow-up duur van 24 weken. Zij vonden een niet significant gemiddeld verschil van 0,26 BMI (kg/m2), p=0.78 (Moss, 2015).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verandering in BMI voor de ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
4b. Verandering in gewicht bij ivacaftor behandeling
Twee studies rapporteerden over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. Nog één additionele studie rapporteerde behandeleffecten voor tertielen van respons van FEV1% voorspeld (Konstan, 2015). In figuur 10 staan de resultaten gepresenteerd in een forest plot voor de gemiddelde verschillen in veranderingen in gewicht (in kg) voor verschillende follow-up duren.
Na 24 en 48 weken werd een significant gemiddeld verschil gezien van 2,4 kg (95% BI 1,5 tot 3,2) en 2,8 kg (95% BI 1,7 tot 3,8) respectievelijk in het voordeel van de ivacaftor behandeling (Ramsey, 2011; Davies, 2013).
Konstan (2015) onderzocht het behandeleffect na 48 weken in een post-hoc analyse met gepoolde data van de RCT’s van Ramsey (2011) en Davies (2013) zoals gepresenteerd in figuur 11. De response op voorspelde FEV1% werd in tertielen verdeeld. De absolute verandering in gewicht gaf voor het laagste en hoogste tertiel een significant behandeleffect in het voordeel van de ivacaftor behandeling. Een number needed to treat voor een verhoging van het lichaamsgewicht van ≥ 5% werd berekend van 5,75.
Figuur 10 Forest plot: Verandering in gewicht (kg) vanaf baseline bij ivacaftor behandeling vergeleken met placebo

Figuur 11 Forest plot: Verandering in gewicht (kg) vanaf baseline bij ivacaftor behandeling vergeleken met placebo per subgroep
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verandering in gewicht (G551D) voor de ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
4c. Verandering in BMI (kg/m2) voor lumacaftor/ ivacaftor behandeling
Drie studies rapporteerden over deze uitkomst voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo. Elborn (2016) rapporteerde in nog één additionele publicatie de behandeleffecten van gepoolde data van twee eerder gepubliceerde RCT’s (Boyle, 2014; Wainwright, 2015) voor subgroepen van verschillende pp FEV1 uitgangswaarden. In figuur 12 staan de resultaten gepresenteerd in een forest plot voor de gemiddelde verschillen in veranderingen in BMI (kg/m2) voor verschillende follow-up duren.
Na 8 weken werd er geen statistisch significant verschil gevonden tussen beide behandelgroepen (Rowe, 2017). Na 24 weken werd er een klein statistisch significant verschil gevonden in het voordeel van de lumacaftor/ ivacaftor behandeling vergeleken met placebo: 0,21 (95% BI 0,06 tot 0,35) (Ratjen, 2017; Wainwright, 2015).
Elborn (2016) liet vergelijkbare resultaten zien in een posthoc-analyse met data van Boyle (2014) en Wainwright (2016) na 24 weken voor beide subgroepen met een pp FEV< 70% en ≥ 70%, zoals gepresenteerd in figuur 13.
Figuur 12 Forest plot: Verandering in BMI (kg/m2) vanaf baseline bij lumacaftor/ivacaftor behandeling vergeleken met placebo
Figuur 13 Forest plot: Verandering in BMI (kg/m2) vanaf baseline bij lumacaftor/ivacaftor behandeling vergeleken met placebo per subgroep
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat BMI (na 8 weken) voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
De bewijskracht voor de uitkomstmaat BMI (na 24 weken) voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd vanwege de overlap met de grens voor klinische besluitvorming (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
4d. Verandering in gewicht (kg) voor lumacaftor/ ivacaftor behandeling
Eén studie rapporteerde over deze uitkomst voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo. Na 8 weken werd er geen statistisch significant verschil gevonden tussen beide behandelgroepen: -0,12 (95% BI -0,34 tot 0,10) (Rowe, 2017).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat verandering in gewicht (na 8 weken) voor de ivacaftor/ lumacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
5. Kwaliteit van leven
5a. CFQ-R Respiratoir domein bij ivacaftor behandeling
Zeven studies rapporteerden over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. Nog één additionele studie rapporteerde behandeleffecten voor tertielen van respons van FEV1% voorspeld (Konstan, 2015). In figuur 14 en 15 staan de resultaten gepresenteerd in een forest plot voor de gemiddelde verschillen in veranderingen in gewicht (in kg) voor verschillende follow-up duren voor respectievelijk de CFQ-R respiratoir domein gepoolde data (kind/ouder + adolescent/volwassene versie) en de CFQ-R respiratoir domein aparte data voor kind en ouder versie.
Vanaf 2 weken zijn er statistisch significante verbeteringen in de CFQ-R score gerapporteerd in het voordeel van de ivacaftor behandeling, gemiddelde verschillen tussen de 5.9 en 9.6 zijn gerapporteerd (Davies, 2013b; De Boeck, 2014; Quittner, 2015; Ramsey, 2011; Davies, 2013). De resultaten van Davies (2013) waren niet alle statistisch significant. Vier punten verschil op de CFQ-R schaal is gedefinieerd als een minimaal klinisch belangrijk verschil.
Accurso (2010) rapporteerde medianen (range) en vond na 2 en 4 weken een niet significante verbetering in CFQ-R (Accurso, 2010).
Konstan (2015) onderzocht het behandeleffect na 48 weken in een post-hoc analyse met gepoolede data van de RCT’s van Ramsey (2011) en Davies (2013) zoals gepresenteerd in figuur 16. De response op pp FEV1% werd in tertielen verdeeld. De verandering in CFQ-R gaf voor het middelste en hoogste tertiel een significant effect in het voordeel van de ivacaftor behandeling.
Figuur 14 CFQ-R Respiratory Domain pooled data (child/parent/adolescent/adult version) change from baseline ivacaftor
Figuur 15 CFQ-R Respiratory Domain separate data (child and parent/caregiver version) change from baseline ivacaftor
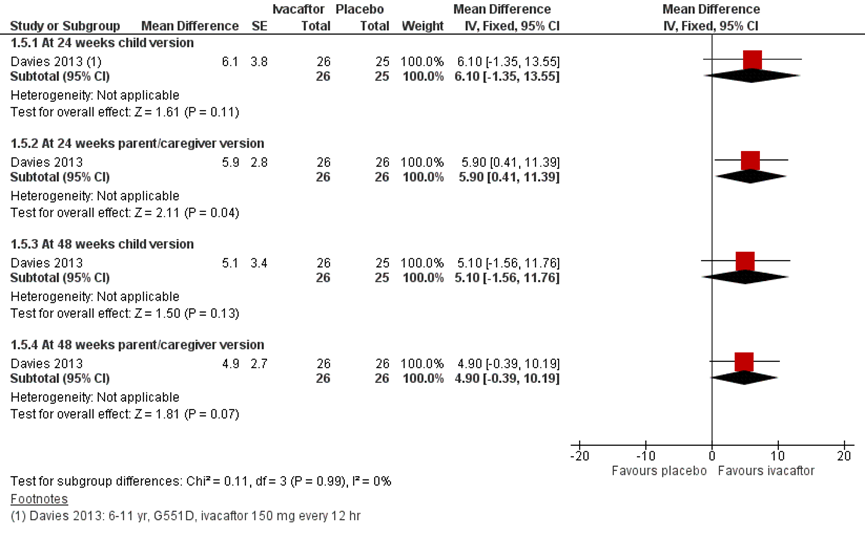
Figuur 16 CFQ-R Respiratory Domain pooled data (child/parent/adolescent/adult version) change from baseline ivacaftor per subgroep
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat CFQ-R (na 2 weken, G551D en non-G551D) voor de ivacaftor/ lumacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
5b. CFQ-R Respiratoir domein bij lumacaftor/ ivacaftor behandeling
Vier studies rapporteerden over deze uitkomst voor de lumacaftor/ivacaftor behandeling vergeleken met placebo. Elborn (2016) rapporteerde in nog één additionele publicatie de behandeleffecten van gepoolde data van twee eerder gepubliceerde RCT’s (Boyle, 2014; Wainwright, 2015) voor subgroepen van verschillende pp FEV1 uitgangswaarden. In figuur 17 staan de resultaten gepresenteerd in een forest plot voor de gemiddelde verschillen in CFQ-R (gepoolde data) voor verschillende follow-up duren.
Na 8 en 24 weken zijn er statistisch significante verbeteringen in de CFQ-R score gerapporteerd in het voordeel van de ivacaftor behandeling, van respectievelijk 8.9 en 2.6 punten (Boyle, 2014; Rowe, 2017; Ratjen, 2017; Wainwright, 2015). Vier punten verschil op de CFQ-R schaal is gedefinieerd als een minimaal klinisch belangrijk verschil.
In de post-hoc analyse van Elborn (2016) data van Boyle (2014) en Wainwright (2016) was de verbetering in CFQ-R minder duidelijk na 24 weken voor beide subgroepen met een pp FEV< 70% en ≥ 70%, zoals gepresenteerd in figuur 18.
Figuur 17 CFQ-R Respiratory Domain pooled data (child/parent/adolescent/adult version) change from baseline lumacaftor/ivacaftor
Figuur 18 CFQ-R Respiratory Domain pooled data (child/parent/adolescent/adult version) change from baseline lumacaftor/ivacaftor per subgroup
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat CFQ-R (na 8 weken) voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
De bewijskracht voor de uitkomstmaat CFQ-R (na 24 weken) voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is niet verlaagd in niveau. Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘hoog’.
6. Zweettest
6a. Zweettest chloride concentraties bij lumacaftor behandeling
Acht studies rapporteerden over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. In figuur 19 staan de resultaten gepresenteerd in een forest plot voor de gemiddelde verschillen in concentraties van zweet chloride (mmol/L) voor verschillende follow-up duren. Nog één additionele studie rapporteerde behandeleffecten voor tertielen van respons van FEV1% voorspeld (Konstan, 2015).
Vanaf 2 weken zijn er statistisch significante verbeteringen in de CFQ-R score gerapporteerd in het voordeel van de ivacaftor behandeling, gemiddelde verschillen tussen de -45 en -49 mmol/L zijn gerapporteerd (Davies, 2013b, De Boeck, 2014; Davies, 2013b; Edgeworth, 2017; Ramsey, 2011; Davies, 2013).
Accurso (2010) rapporteerde weer medianen (range) en vond na 4 weken ook een statistisch significante verbetering in concentraties van zweet chloride (mmol/L) (Accurso, 2010).
Konstan (2015) onderzocht het behandeleffect na 48 weken in een post-hoc analyse met gepoolede data van de RCT’s van Ramsey (2011) en Davies (2013) zoals gepresenteerd in figuur 20. De response op pp FEV1% werd in tertielen verdeeld. De verandering in de zweettest chloride concentratie gaf voor alle tertielen een significant behandeleffect in het voordeel van de ivacaftor behandeling. Een number needed to treat voor een verhoging van de zweettest chloride concentratie ≥ 20 mmol/L werd berekend van 1,03.
Moss (2015) onderzocht het effect van ivacaftor vergeleken met placebo in een groep met 69 patiënten met de R117H mutatie met een follow-up duur van 24 weken. Zij vonden een significant gemiddeld verschil van -24.0 mmol/L , p<0.001 (Moss, 2015).
Figuur 19 Zweettest chloride level change from baseline bij ivacaftor behandeling
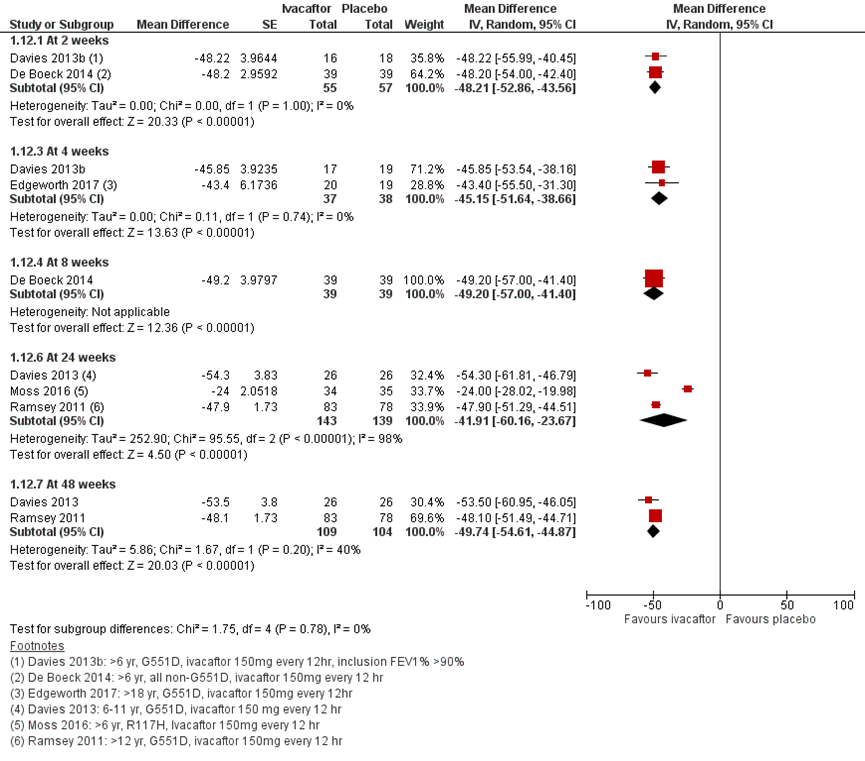
Figuur 20 Zweettest chloride level change from baseline bij ivacaftor behandeling per subgroep
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat zweettest chloride concentratie (2 tot 48 weken, G551D) voor de ivacaftor behandeling vergeleken met placebo is niet verlaagd in niveau. Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘hoog’.
De bewijskracht voor de uitkomstmaat zweettest chloride concentratie (R117H) voor de ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
6b. Zweettest chloride concentraties bij lumacaftor/ivacaftor behandeling
Drie studies rapporteerden over deze uitkomst voor de lumacaftor/ivacaftor behandeling vergeleken met placebo. In figuur 21 staan de resultaten gepresenteerd in een forest plot voor de gemiddelde verschillen in zweettest chloride concentraties voor verschillende follow-up duren.
Na 3 en 8 weken zijn er statistisch significante verbeteringen in de zweettest chloride concentraties gerapporteerd in het voordeel van de ivacaftor behandeling, van respectievelijk -14.6 en -10.2 mmol/L (Boyle, 2014; Rowe, 2017; Ratjen, 2017).
Figuur 21 Zweettest chloride level change from baseline bij lumacaftor/ ivacaftor behandeling
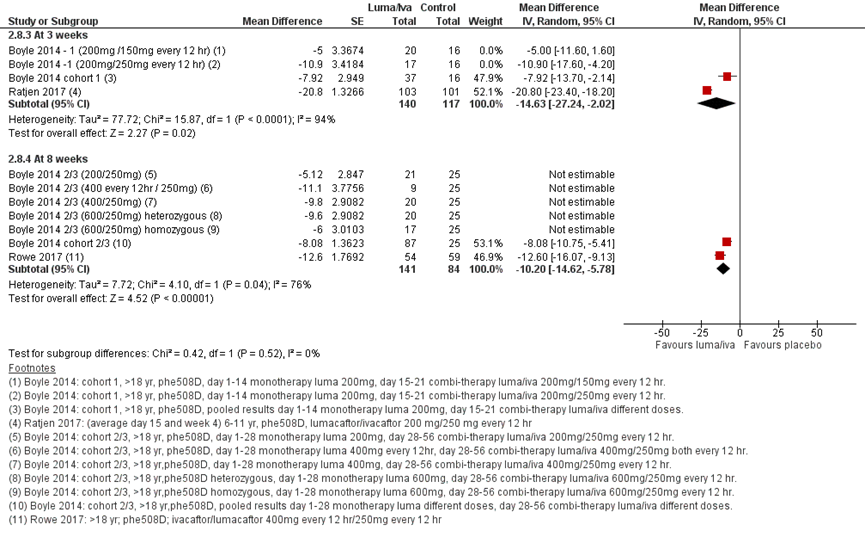
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat zweettest chloride concentraties voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
7. Slope of decline (FEV1)
Voor deze uitkomstmaat hebben we geen artikelen gevonden.
8. Lung Clearance Index
8a. Lung Clearance Index bij ivacaftor behandeling
Eén studie rapporteerde over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. Na 2 en 4 weken zijn er statistisch significante verbeteringen in LCI gerapporteerd in het voordeel van de ivacaftor behandeling: respectievelijk -2,19 (-2,96 tot -1,42), P<0,0001 en -2.07 (95% BI -2,98 tot -1,16), P<0.001, bij een groep met een baseline waarde van FEV1 van > 90% (Davies, 2013b).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat LCI voor de ivacaftor behandeling vergeleken met placebo is met drie niveaus verlaagd gezien het zeer geringe aantal patiënten (imprecisie) (twee niveau’s) en indirectheid, in een groep met FEV1 van > 90%. Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘laag’.
8b. Lung Clearance Index bij lumacaftor/ ivacaftor behandeling
Eén studie rapporteerde over deze uitkomst voor de ivacaftor behandeling vergeleken met placebo. Na 24 weken zijn er statistisch significante verbeteringen in LCI gerapporteerd in het voordeel van de ivacaftor behandeling: -1,10 (95% BI -1,38 tot -0,82) (Ratjen, 2017).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat LCI voor de lumacaftor/ ivacaftor behandeling vergeleken met placebo is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Er is niet afgetrokken voor niet juist toegepaste intention-to-treat analyse, omdat er geen redenen zijn om aan te nemen dat dit bias heeft veroorzaakt. Het niveau van de bewijskracht komt uit op GRADE ‘redelijk’.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat zijn de (on)gunstige effecten van CFTR modulatoren ten opzichte van een placebo of geen behandeling bij patiënten met cystic fibrosis?
P (Patiënten): Cystic Fibrosis patiënten;
I (Interventie): CFTR-modulatoren;
C (Comparison): placebo of geen behandeling met CFTR-modulatoren;
O (Outcomes): FEV1, CT-score, pulmonaire exacerbaties, voedingsstatus (gewicht, lengte, gewicht voor lengte, BMI), kwaliteit van Leven, zweet chloride test, slope of decline FEV1, Lung Clearance Index (LCI).
Relevante uitkomstmaten
De werkgroep definieerde niet a priori de genoemde uitkomstmaten, maar hanteerde de in de studies gebruikte definities. Tenzij anders vermeld werden de door de internationale GRADE-workinggroup voorgestelde default grenzen gehanteerd voor klinische relevantie: een verschil in relatief risico van 25% bij dichotome uitkomstmaten, en een verschil van een halve standaarddeviatie voor continue uitkomstmaten. Vier punten verschil op de CFQ-R schaal is gedefinieerd als een minimaal klinisch belangrijk verschil.
Zoeken en selecteren (Methode)
In de databases Medline (via OVID) en Embase (via Embase.com) is 20 september 2017 met relevante zoektermen gezocht naar Engelstalige systematische reviews en gerandomiseerde gecontroleerde studies (RCT’s) gepubliceerd vanaf 1987 gericht op de (on)gunstige effecten van een behandeling met CFTR modulatoren in CF patiënten. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 455 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: gerandomiseerde gecontroleerde studies die een CFTR-behandeling vergelijken met een placebobehandeling en ten minsten één van de volgende uitkomstmaten hanteren: FEV1, CT-scan, exacerbatie frequentie, voedingstoestand, kwaliteit van Leven, zweettest, slope of decline FEV1, Lung Clearance Index (LCI). Een exclusiecriterium was ingesteld specifiek voor studies met een behandeling met ivacaftor bij phe508del mutaties.
Op basis van titel en abstract werden in eerste instantie 99 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 85 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), en 11 studies definitief geselecteerd.
Elf onderzoeken zijn opgenomen in de literatuuranalyse, waarvan één Cochrane literatuurreview met daarin drie RCT’s en tien losse parallelle en cross-over RCT’s. De Cochrane review bevatte vier RCT’s, maar één RCT is niet opgenomen omdat deze voldeed aan het exclusiecriterium. Deze studie onderzocht de effectiviteit van ivacaftor bij een phe508del genmutatie. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk of bias tabellen.
- Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D; VX09-809-102 study group. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014 Jul;2(7):527-38. doi:10.1016/S2213-2600(14)70132-8.
- Davies J, Sheridan H, Bell N, Cunningham S, Davis SD, Elborn JS, Milla CE, Starner TD, Weiner DJ, Lee PS, Ratjen F. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med. 2013 Oct;1(8):630-638. doi: 10.1016/S2213-2600(13)70182-6.
- De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, Higgins M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014 Dec;13(6):674-80. doi: 10.1016/j.jcf.2014.09.005.
- Edgeworth D, Keating D, Ellis M, Button B, Williams E, Clark D, Tierney A, Heritier S, Kotsimbos T, Wilson J. Improvement in exercise duration, lung function and well-being in G551D-cystic fibrosis patients: a double-blind, placebo-controlled, randomized, cross-over study with ivacaftor treatment. Clin Sci (Lond). 2017 Jul 16;131(15):2037-2045. doi: 10.1042/CS20170995.
- Elborn JS, Ramsey BW, Boyle MP, Konstan MW, Huang X, Marigowda G, Waltz D, Wainwright CE; VX-809 TRAFFIC and TRANSPORT study groups. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet Respir Med. 2016 Aug;4(8):617-626. doi: 10.1016/S2213-2600(16)30121-7.
- Konstan MW, Plant BJ, Elborn JS, Rodriguez S, Munck A, Ahrens R, Johnson C. Efficacy response in CF patients treated with ivacaftor: post-hoc analysis. Pediatr Pulmonol. 2015 May;50(5):447-55. doi: 10.1002/ppul.23173.
- Patel S, Sinha IP, Dwan K, Echevarria C, Schechter M, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst Rev. 2015 Mar 6;(3):CD009841. doi: 10.1002/14651858.CD009841.pub2.
- Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, Milla CE, Robinson PD, Waltz D, Davies JC; VX14-809-109 investigator group. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017 Jul;5(7):557-567. doi: 10.1016/S2213-2600(17)30215-1.
- Richard B. Moss, MD1, Patrick A. Flume, MD2, J. Stuart Elborn, MD3, Jon Cooke4, Steven M. Rowe, MD5, Susanna A. McColley, MD6, Ronald C. Rubenstein, MD7, and Mark Higgins, MD4. Efficacy and safety of ivacaftor treatment: randomized trial in subjects with cystic fibrosis who have an R117H-CFTR mutation. Lancet Respir Med. 2015 July ; 3(7): 524–533. doi:10.1016/S2213-2600(15)00201-5.
- Rowe SM, McColley SA, Rietschel E, Li X, Bell SC, Konstan MW, Marigowda G, Waltz D, Boyle MP; VX09-809-102 Study Group. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann Am Thorac Soc. 2017 Feb;14(2):213-219. doi: 10.1513/AnnalsATS.201609-689OC.
- Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP; TRAFFIC Study Group; TRANSPORT Study Group. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015 Jul 16;373(3):220-31. doi:10.1056/NEJMoa1409547.
Evidence table for systematic review of RCTs and observational studies (intervention studies)
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Patel, 2015
|
SR and meta-analysis of RCTs
Literature search up to march 2015
A: Accurso 2010 B: Davies 2013 C: Flume 2011 à NIET MEEGENOMEN vanwege phe508del mutatie D: Ramsey 2011
Study design: All RCT [parallel]
Setting and Country: A: Multicenter and -country B: Multicenter -country C: Multicenter -country D: Multicenter -country
Source of funding: Commercial; all by Vertex Pharmaceuticals Incorporated
|
Inclusion criteria SR: -RCTs of parallel design, comparing CFTR potentiators to placebo in children and adults
Exclusion criteria SR: -Cross-over RCT -Trials combining CFTR potentiators with other mutation-specific therapies
4 studies included
Important patient characteristics at baseline:
N, age A: 20, median 21 (range 18-42 yr) B: 52, mean 8.9 (range 6-12) D:161, mean 25.5 (range 12-53)
Sex: A: 53% male B: 48% male D: 48% male
FEV1% at baseline: A: 69% (40-122) B: 84% (44-134) D: 63 (32-98)
Genmutatie: A: G551D B: G551D D: G551D
Groups comparable at baseline? Yes |
Describe intervention: Ivacaftor A arm1: 150 mg every 12hr A arm2: 250mg every 12hr B: 150 mg every 12hr D: 150 mg every 12hr
|
Describe control: Placebo A: placebo every 12hr B: placebo every 12hr D: placebo every 12hr
|
End-point of follow-up: A: 4 wk B: 24 wk D: 48 wk
For how many participants were no complete outcome data available? (intervention/control) A: 20 participants were randomised. 1/5 placebo participants withdrew due to withdrawal of consent prior to dosing. All other participants were accounted for in the analysis B: 4 (7.7%) withdrew, all in the placebogroup, none in iva group. A modified intention-to-treat analysis (not per protocol) was employed D: 6 withdraw in iva group, 10 in placebo group
|
Outcome measure-1 Defined as absolute change in FEV1% percent predicted
Effect measure: Mean Difference [95% CI]: B: At 24 wk 12.5 (6.7-18.3) At 48 wk 10.0 (4.5-15.1) D: At 24 wk 10.6 (8.6-12.6) At 48 wk 10.5 (8.5-12.5)
A: Median (range) of 0.25 L (0.05 to 0.75) in the 150 mg group, 0.17 L (0 to 0.37) in the 250 mg group and 0.20 L (0.12 to 0.33) in the placebo group. Differences were not significant
Outcome measure-2 Defined as relative change in FEV1% percent predicted
Effect measure: Mean Difference [95% CI]: B: At 24 wk 17.4% (P < 0.0001) C: At 16 wk 2.4% (-0.95-5.75) D: At 24 wk 16.9% (13.6-20.2)
A: Median (range) 8.7% (2.3 to 31.3) in the 150 mg group, 4.4% (0.0 to 18.3) in the 250 mg group and 7.3% (5.2 to 8.2) in the placebo group. Differences were not significant.
Outcome measure-3 Number of pulmonary exacerbations (protocol defined)
Effect measure: OR [95% CI]: A: At 4 wk 1.67 (0.1-41.6) B: At 48 wk 1.23 (0.35-4.32) D: At 48 wk: 0.54 (0.29-1.01)
Outcome measure-4 CFQ-R Respiratory Domain
Effect measure: Mean Difference [95% CI]: B: 24 wk children: 6.10 (95% CI -1.35 to 13.55) 24 wk caregivers: 5.90 (95% CI 0.41 to 11.39) At 48 wk children: 5.10 (95% CI -1.56 to 11.76) At 48 wk caregivers:4.90 (95% CI -0.39 to 10.19) D: At 24 wk: 8.10 (95% CI 4.77 to 11.43) At 48 wk: 8.60 (95% CI 5.27 to 11.93)
A: At 2 wk, a median (range) increase of 5.6 (0.0-16.7) in the 150 mg ivacaftor group and 5.6 (-11.1-11.1) in the 250 mg ivacaftor group. The median change of 2.8 (-5.6-11.1). This was not sign. At 4 wk, in the 150 mg ivacaftor group median improvement of 8.3 (0.0-16.7) points, in the 250 mg group a median improvement of 11.1 (-5.6-33.3) and the placebo group a median improvement of 2.8 (-5.6-11.1) points. This was not sign. Accurso presented also CFQ-R scores fort he other domains.
Outcome measure-5a BMI in kg/m2 change from baseline
Effect measure: Mean Difference [95% CI]: B: At 48 wk 1.09 (p<0.001) D: At 48 wk 0.93 (p<0.001)
Outcome measure-5b Weight in kg absolute change from baseline
Effect measure: Mean Difference [95% CI]: B: At 24 wk 1.9 (0.9-2.9) At 48 wk 2.8 (1.4-4.3) D: At 24 wk 2.8 (1.8-3.8) At 48 wk 2.7 (1.3-4.1)
Outcome measure-6 Sweat chloride change from baseline
Effect measure: Mean Difference [95% CI]: B: At 24 wk -54.3 (-61.8 - -46.8) At 48 wk -53.5 (-60.9- -46.1) D: At 24 wk -47.9 (-51.3 - -44.5) At 48 wk -48.1 (-51.5- -44.7)
A: Median (range) at 4 wk significant difference between the 150 mg group (within group change −59.5 mmol/L (-66.0 to -19.0) and the placebo group (within group change 5.0 mmol/L (-2.0 to 11.0) (P = 0.02); also a significant difference between the 250 mg group (within group change -38.0 mmol/L (-47.0 to -10.5) and placebo group (within group change 5.0 mmol/L (-2.0 to 11.0) (P = 0.03) |
Brief description of author’s conclusion: Both G551D phase 3 trials (n = 219) demonstrated a clinically relevant impact of the potentiator ivacaftor on outcomes at 24 and 48 weeks, providing evidence for the use of this treatment in adults and children (over six years of age) with cystic fibrosis and the G551D mutation (class III). There is no evidence to support the use of ivacaftor in people with the 1F508 mutation (class II) (n = 140). Trials on ivacaftor in people with different mutations are ongoing.
Pooled results are shown in overall analyses with all additional studies
|
Evidence table for intervention studies (randomized controlled trials and non-randomized observational studies (cohort studies, case-control studies, case series))1
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures.
- Provide data per treatment group on the most important prognostic factors ((potential) confounders).
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls.
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders.
Table of quality assessment for systematic reviews of RCTs and observational studies
Based on AMSTAR checklist (Shea,2007; BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher, 2009; PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Patel, 2015 |
Yes |
Yes |
Yes |
Yes |
NA |
Yes |
Yes |
Yes |
Yes |
- Research question (PICO) and inclusion criteria should be appropriate and predefined.
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched.
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons.
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported.
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs).
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table et cetera).
- Clinical and statistical heterogeneity should be assessed; clinical: enough similarities in patient characteristics, intervention and definition of outcome measure to allow pooling? For pooled data: assessment of statistical heterogeneity using appropriate statistical tests (for example Chi-square, I2)?
- An assessment of publication bias should include a combination of graphical aids (for example funnel plot, other available tests) and/or statistical tests (for example Egger regression test, Hedges-Olken). Note: If no test values or funnel plot included, score “no”. Score “yes” if mentions that publication bias could not be assessed because there were fewer than 10 included studies.
- Sources of support (including commercial co-authorship) should be reported in both the systematic review and the included studies. Note: To get a “yes,” source of funding or support must be indicated for the systematic review AND for each of the included studies.
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Edgeworth, 2017 |
A random allocation sequence was generated by a pharmacist and dispensed using sequentially numbered containers. Participants and investigators were blinded until after data were unlocked on completion of all interventions. Patients were assigned (1:1) |
Unlikely |
Unlikely |
Unclear |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
|
Davies, 2013b |
Eligible patients were randomly assigned (ratio 1:1) to one of the two treatment sequences. The patients and all site personnel, including the investigator, the study monitor, and the Vertex study team, were masked to treatment assignment. To protect the scientific integrity of the masking, two iostatisticians were involved in the randomisation process: study biostatistician, masked to the actual treatment code, and an unmasked biostatistician not associated with the study. The study biostatistician created the randomisation specification and dummy randomisation code, which was reviewed and approved by the unmasked biostatistician. After approval, the unmasked statistician generated the final randomisation list. We used a block randomisation scheme with block sizes of 4. |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely
1 patient was excluded after randomisation and before start of intervention, also not included in analyses
|
|
De Boeck, 2014 |
Eligible patients were randomized 1:1 to 1 of 2 treatment sequences |
Unclear |
Unlikely |
Unclear |
Unclear |
Unlikely |
Unlikely |
Unclear
The full analysis and safety sets included all patients randomized to treatment groups who received at least 1 dose of study drug. |
|
Moss, 2015 |
Eligible subjects were randomized to receive placebo or ivacaftor. Randomization was stratified by age and ppFEV1. |
Unclear |
Unlikely |
Unclear |
Unclear |
Unlikely |
Unlikely |
Unlikely |
|
Boyle 2015 |
The randomisation list was generated by computergenerated permuted blocked (cohort 1 block size 6; cohort 2 block size 10; cohort 3 block size 14) randomisation by an independent party. |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
|
Rowe 2017 |
Patients were randomized 1:1 and stratified for sex and ppFEV1 at secreening severity. Details were published previously |
Unclear |
Unlikely |
Unclear |
Unclear |
Unlikely |
Likely |
Unlikely |
|
Elborn 2016 |
Patients were randomly assigned (1:1:1) using an interactive web response system to one of three study Groups. Randomisation was stratified according to age, sex, and ppFEV1 at screening. |
Unlikely |
Unlikely |
Unlikely |
Unclear |
Unlikely |
Unclear |
Unclear |
|
Wainwright 2015 |
Patients were randomly assigned (in a 1:1:1 ratio). Randomization was stratified according to age, sex, and pulmonary function at screening. Details were published previously |
Unclear |
Unlikely |
Unclear |
Unclear |
Unlikely |
Unclear |
Likely |
|
Ratjen 2017 |
Patients were stratified by weight and ppFEV1 severity and then randomly assigned (1:1). Random assignment was determined using an interactive web response system (IWRS), and stratification influenced assignment such that groups were to be balanced for each stratification criterion: within each stratum, the IWRS randomly assigned patients to one of the two treatment groups. Randomisation blocks were assigned within stratum. Randomisation code for the IWRS was prepared by an external qualified randomisation vendor, who reviewed the final randomisation list and transferred it directly to the IWRS vendor. The randomisation vendor had no other involvement in the trial. |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Likely |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear.
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Stalvey 2017 |
Post-hoc analyse van RCT Davies 2013, geen extra relevante informatie |
|
Quittner 2015 |
Post-hoc analyse van RCT Ramsey 2011, geen extra relevante informatie |
|
Borowitz 2016 |
Post-hoc analyse van RCT van Ramsey 2011 en Davies 2013, geen extra relevante informatie (opgesplitst voor leeftijdscategorieën) |
|
Flume 2017 |
Geen RCT: Recovery of lung function following a pulmonary exacerbation, geen biomarker of interest (post-hoc analysis STRIVE study) |
|
Sawicki 2015 |
Geen RCT: Matched case-control study (sustained benefit ivacaftor over 3 years) |
|
Barry 2014 |
Geen RCT: Case-control study in adults with severe lung disease, n=21 in intervention group |
|
Clancy 2012 |
Geen RCT: Short-term fase IIa study: safety, tolerability and pharmacodynamics of VX-809 in adult patients with cystic fibrosis (n=89) |
|
McKone 2014 |
Geen vergelijkend onderzoek (Open-label phase 3 extension study ivacaftor, long-term safet 96 wk) |
|
LaBaste 2017 |
Geen vergelijkend onderzoek: before-after study, lumacaftor/ivacaftor, acute lung function changes |
|
Jennings 2017 |
Geen vergelijkend onderzoek:Retrospective cohort study Lumacaftor/Ivacaftor, 11 months |
|
Hubert 2017 |
Geen vergelijkend onderzoek: retrospective observational study: two year effects n=57 |
|
Hubert 2017 |
Geen vergelijkend onderzoek:short term adverse events and effectiveness 3 months n=53 |
|
Dryden 2016 |
Geen vergelijkend onderzoek: Before-after, n=27 children, retrospective study on ivacaftor |
|
Davies 2016 |
Geen vergelijkend onderzoek: Open-label, single-group study in children 2-5 yr on safety ivacaftor n=34 |
|
Rowe 2014 |
Geen vergelijkend onderzoek: Longitudinale before and after study n=153 biomarkers, follow-up 6 maanden |
|
Stallings 2017 |
Geen vergelijkend onderzoek: Before and after study 3 months n=23 |
|
Popowicz |
Geen vergelijkend onderzoek: before and after study, immediate responses on safety |
|
Gelfond 2017 |
Geen vergelijkend onderzoek: Before and after observational study on alternative clinical outcome n=10 |
|
McKone 2016 |
Congres poster |
|
Ahmad 2016 |
Geen vergelijkend onderzoek: retrospective analyses before-after, n=20 Lum/Iva |
|
Sheikh 2015 |
Geen vergelijkend onderzoek: Prospective study n=10 HRCT imaging 1 year follow-up |
|
Sheikh 2015 |
Geen vergelijkend onderzoek: N=12 observational study; effect on sinus disease |
|
Heltshe 2015 |
Geen vergelijkend onderzoek: Before, after study n=151, follow-up 1 year on P. aeruginosa |
|
Milla 2017 |
Geen vergelijkend onderzoek: Open-label phase 3 one-arm trial lumacaftor/ivacaftor) |
|
Arends 2015 |
Beschrijvende lit review |
|
Whiting 2014 |
Overlap met Cochrane, ongepubliceerde data, we nemen losse studies apart mee |
|
McColey 2013 |
Beschrijvende lit review |
|
Sinha 2013 |
Congresposter |
|
de Boeck 2013 |
Beschrijvende lit review (over klinimetrische eigenschappen van CFTR biomarkers) |
|
Pettit 2012 |
Beschrijvende lit review |
|
Vermeulen 2017 |
Within subject variability of sweat chloride in placebogroep RCT, voldoet niet aan PICO |
|
Talamo 2017 |
Geen syst lit review/expert opinion |
|
Szczesniak 2017 |
Beschrijvende lit review |
|
Schneider 2017 |
Beschrijvende lit review |
|
McGarry 2017 |
N=10 (serie van n=1 cross-over trials) |
|
LeGrys 2017 |
Voldoet niet aan PICO (Analytical and biological variation in repeated sweat chloride concentrations) |
|
Konstan 2017 |
Voldoet niet aan PICO (lumacaftor vs ivacaftor) |
|
Hisert 2017 |
Niet vergelijkend, before after ivacaftor n=12 |
|
Zhang 2017 |
Beschrijvende lit review |
|
Taylor-Cousar 2016 |
Open-label, non-randomized, single-group, assigned treatment study, before-after study, n=44 |
|
Muhlebach 2016 |
Geen systematische lit review (gehele methode en selectieprocedure ontbreekt) |
|
McColley 2016 |
Geen syst lit review over safety findings (also in animal studies) |
|
Mayer Hamblett 2016 |
Beschrijvende lit review |
|
Deeks 2016 |
Beschrijvende review/expert opinion |
|
Collaco 2016 |
Voldoet niet aan PICO: Variability sweat test in twin/sibling study as biomarker for respons to CFTR |
|
Cholon 2016 |
Geen systematische literatuurreview, beschrijvend |
|
Chassagnon 2016 |
Short report/Letter to the editor |
|
Birket 2016 |
In vitro biomarkers |
|
Kuk 2015 |
Beschrijvende lit review |
|
Jones 2015 |
Beschrijvend stuk, expert opinion |
|
Bodewes 2015 |
Beschrijvend stuk, expert opinion over gastrointestinal biomarkers |
|
Clancy 2014 |
Beschrijvende review/expert opinion |
|
Barry 2014 |
Niet vergelijkende cohort studie n=24 zweettest |
|
Accurso 2014 |
Voldoet niet aan PICO (Variatie in zweettest als biomarker binnen trialdata) |
|
Thursfield 2013 |
Beschrijvend stuk, expert opinion |
|
Seliger 2013 |
Voldoet niet aan PICO: Sweat cholrine test as predictor for FEV1% and weight gain |
|
Rowe 2013 |
Voldoet niet aan PICO: Methods for NPD as biomarker |
|
Durmowicz 2013 |
Geen origineel onderzoek (commentary) |
|
Davies 2013 |
Meegenomen in Cochrane Patel |
|
Sousa 2012 |
Voldoet niet aan PICO: human rectal biopsies as biomarker (matched case-control with non-CF patients) |
|
Flume 2012 |
Meegenomen in Cochrane Patel à uiteindelijk geexcludeerd vanwege phe508del genmutatie |
|
Ramsey 2011 |
Meegenomen in Cochrane Patel |
|
Accurso 2010 |
Meegenomen in Cochrane Patel |
|
Egan 2017 |
Editorial |
|
Taylor 2016 |
Congresposter |
|
Sullivan 2016 |
Congresposter |
|
Indahar 2016 |
Congresposter |
|
Hubert 2016 |
Congresposter |
|
Hassan 2016 |
Congresposter |
|
Hassan 2016 |
Congresposter |
|
Bai 2016 |
Congresposter |
|
Aziz 2016 |
Congresposter |
|
Anonymous 2016 |
Congresposter |
|
Sagel 2015 |
Congresposter |
|
Rosenfeld 2015 |
Congresposter |
|
McKone 2015 |
Congresposter |
|
Kane 2015 |
Research Letter |
|
Hathorne 2015 |
Congresposter |
|
de Boeck 2015 |
Symposium summary |
|
Barry 2015 |
Congresposter |
|
Bai 2015 |
Congresposter |
|
Sawicki 2014 |
Congresposter |
|
Pinders-Kessler |
Congresposter |
|
Rowe 2013 |
Congresposter |
|
Hubert 2013 |
Congresposter |
|
Elborn 2013 |
Congresposter |
|
Barry 2013 |
Congresposter |
|
Altes 2011 |
Congresposter |
Beoordelingsdatum en geldigheid
Publicatiedatum : 03-07-2020
Beoordeeld op geldigheid : 28-10-2019
Voor het beoordelen van de actualiteit van deze kwaliteitsstandaard is de werkgroep niet in stand gehouden. Uiterlijk in 2025 bepalen de besturen van de NVALT, NVK en NCFS of de modules van deze kwaliteitsstandaard nog actueel zijn. Op modulair niveau is een onderhoudsplan beschreven. Bij het opstellen van de kwaliteitsstandaard heeft de werkgroep per module een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de kwaliteitsstandaard komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De NVALT, NVK en NCFS zijn regiehouders van deze Kwaliteitsstandaard en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de modules. De andere aan deze kwaliteitsstandaard deelnemende wetenschappelijke verenigingen of gebruikers van de kwaliteitsstandaard delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
|
Module |
Regiehouder(s) |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn |
Frequentie van beoordeling op actualiteit |
Wie houdt er toezicht op actualiteit |
Relevante factoren voor wijzigingen in aanbeveling |
|
Stopcriteria CFTR modulatoren
|
NVK/NVALT |
2020 |
2023 |
Eens in de drie jaar eerder |
NVK/NVALT |
Beschikbaarheid nieuwe literatuur en nieuwe middelen op de markt |
Algemene gegevens
Deze richtlijn is ontwikkeld in samenwerking met:
- Landelijke Vereniging Medische Psychologie
- Nederlands Instituut van Psychologen
- Nederlandse Vereniging van Diëtisten
De ontwikkeling van de medisch inhoudelijke modules binnen deze Kwaliteitsstandaard werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS).
De module over psychosociale zorg en de organisatie van zorg modules werden ondersteund vanuit de Samenwerkende Ouder- en Patiëntenorganisaties (VSOP) en gefinancierd vanuit ZonMW. De financierders hebben geen enkele invloed gehad op de inhoud van de kwaliteitsstandaard.
Doel en doelgroep
Doel
Het tot stand brengen van een evidence-based kwaliteitsstandaard Cystic Fibrosis, een combinatie van een zorgstandaard en een medisch specialistische richtlijn om patiënten met Cystic Fibrosis optimale afgestemde zorg te bieden.
Doelgroep
De doelgroep zijn alle patiënten met Cystic Fibrosis. De kwaliteitsstandaard behelst de gebieden van diagnostiek, behandeling en follow-up conform de diverse stadia van de aandoening en de verschillende leeftijdscategorieën.
Samenstelling werkgroep
Voor het ontwikkelen van de kwaliteitsstandaard is in 2016 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met CF en vertegenwoordigers van de NCFS.
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname. De werkgroep is verantwoordelijk voor de integrale tekst van deze kwaliteitsstandaard.
Werkgroep
- Drs. G.D. (George) Nossent, longarts, UMCG te Groningen, NVALT (voorzitter)
- Dr. O.W. (Onno) Akkerman, longarts, UMCG te Groningen, NVALT
- S. (Sigrid) Amstelveen-Bökkerink, diëtist, Radboud UMC te Nijmegen, NVD
- Dr. H.G.M. (Bert) Arets, kinderlongarts, Wilhelmina Kinderziekenhuis en UMC Utrecht te Utrecht, NVK
- Dr. M. (Marleen) Bakker, longarts, Erasmus MC te Rotterdam, NVALT
- Drs. J.M.W. (Annemarie) van den Berg, longarts, Hagaziekenhuis te Den Haag, NVALT
- Dr. M.C. (Maaike) Berkhout, KNO arts, OLVG te Amsterdam, NVKNO
- Dr. F.A.J.A. (Frank) Bodewes, kinderarts-MDL, UMCG te Groningen, NVK
- A. (Annet) Bongen, maatschappelijk werker, UMC Utrecht te Utrecht, BPSW
- J. (Jacqueline) Boekhoff, maatschappelijk werker, Amsterdam UMC, locatie VUMC te Amsterdam, BPSW
- Drs. L.H. (Hassan) el Bouzzaoui, longarts, Hagaziekenhuis te Den Haag, NVALT
- D. (Dagmar) Brocke, maatschappelijker werker, UMC Utrecht te Utrecht, BPSW
- Drs. E.J. Brokaar, poliklinisch apotheker, Hagaziekenhuis te Den Haag, NVZA
- Dr. I. (Inez) Bronsveld, longarts, UMC Utrecht te Utrecht, NVALT
- Drs. A. (Agnes) Clement-de Boer, kinderarts (endocrinologie), Hagaziekenhuis te Den Haag, NVK
- W. (Wytze) Doeleman, fysiotherapeut, UMC Utrecht te Utrecht, KNFG
- Dr. M.M. (Menno) van der Eerden, longarts, Erasmus MC te Rotterdam, NVALT
- Prof. Dr. C.K. (Kors) van der Ent, Kinderlongarts, Wilhelmina Kinderziekenhuis en UMC Utrecht te Utrecht, NVK
- Dr. B.C.T. (Boudien) Flapper, kinderarts-sociale pediatrie, UMCG te Groningen, NVK
- Dr. L. (Lianne) van der Giessen, kinderfysiotherapeut, Erasmus MC Sophia Kinderziekenhuis te Rotterdam, KNGF
- Drs. N. (Nanko) de Graaf, kinderradioloog, Erasmus MC te Rotterdam, NVvR
- Dr. V. (Vincent) Gulmans, Hoofd Onderzoek en Kwaliteit van zorg NCFS te Baarn, NCFS
- Prof. dr. H.G.M. (Harry) Heijerman, longarts, UMC Utrecht te Utrecht, NVALT
- Drs. D.M. (Danielle) Hendriks, kinderarts (MDL), Juliana Kinderziekenhuis te Den Haag, NVK
- Dr. J.J.E. Hendriks, kinderlongarts, Zuyderland Medisch Centrum te Heerlen, NVK
- Prof. dr. B. (Bart) van Hoek, MDL-arts, UMC te Leiden, NVMDL
- Drs. R.A.S. (Rogier) Hoek, longarts, Erasmus MC te Rotterdam, NVALT
- Dr. A. (Ageeth) Hofsteenge, kinderdiëtist, Amsterdam UMC, locatie VUMC en AMC, te Amsterdam, NVD
- Drs. C. (Chantal) Hoge, MDL arts, Maastricht UMC+ te Maastricht, NVMDL
- F.M. (Francis) Hollander, diëtist, UMC Utrecht, Utrecht, NVD
- Prof. dr. R.H.J. (Roderick) Houwen, kinderarts MDL, Wilhelmina Kinderziekenhuis en UMC Utrecht te Utrecht, NVK
- Dr. J. (Jakko) van Ingen, arts microbioloog, Radboud UMC te Nijmegen, NVMM
- Dr. H.M. (Hettie) Janssens, (kinder)longarts, Erasmus MC Sophia Kinderziekenhuis te Rotterdam, NVK
- Drs. A.J. (Arjan) Jansz, arts microbioloog, Stichting PAMM te Eindhoven, NVMM
- H.J. (Hetty) van der Kamp, kinderarts-endocrinoloog, HMC Bronovo te Den Haag en Wilhelmina Kinderziekenhuis UMC Utrecht te Utrecht, NVK
- C. (Cora) de Kiviet, verpleegkundig specialist, Wilhelmina Kinderziekenhuis en UMC Utrecht te Utrecht, V&VN
- A. (Annelies) Kok, verpleegkundig consulent, Erasmus MC te Rotterdam, CF Netwerk verpleegkundigen Nederland
- Dr. B.G.P. (Bart) Koot, kinderarts (MDL), Amsterdam UMC, locatie AMC te Amsterdam, NVK
- M.A. (Marian) Kruijswijk, verpleegkundig specialist, UMC Utrecht te Utrecht, V&VN
- Dr. C.J. (Christof) Majoor, longarts, Amsterdam UMC, locatie AMC te Amsterdam, NVALT
- R. (Renske) van der Meer, longarts, Hagaziekenhuis te Den Haag, NVALT
- Dr. P.J.F.M. (Peter) Merkus, kinderlongarts, Radboud MC te Nijmegen, NVK
- Dr. D. (Dick) Mul, kinderarts (endocrinologie), Diabeter te Rotterdam, NVK
- Drs. A.F. (Ad) Nagelkerke, kinderlongarts, Amsterdam UMC, locatie VUmc te Amsterdam, NVK
- drs. J. (Jacquelien) Noordhoek, directeur patiëntenorganisatie NCFS te Baarn, NCFS
- Marit van Oirschot-van de Ven, verpleegkundig specialist, UMC Utrecht te Utrecht, V&VN
- Dr. M (Marianne) Nuijssink, kinderlongarts, Haga ziekenhuis te Den Haag, NVK
- Dr. M.H.E. (Monique) Reijers, longarts, Radboudumc te Nijmegen, NVALT
- Dr. S. (Sietze) Reitsma, KNO-arts, Amsterdam UMC, locatie AMC te Amsterdam, NVKNO
- Prof. dr. Y.B. (Yolanda) de Rijke, afdelingshoofd Klinische Chemie, Erasmus MC te Rotterdam, NVKC
- Dr. B.L. (Bart) Rottier, kinderlongarts, UMCG te Groningen, NVK
- R.A. (Revka) Schrijver, verpleegkundig consulent, Hagaziekenhuis te Den Haag, CF Netwerk verpleegkundigen Nederland
- Drs. L. (Luciënne) Speleman, KNO arts, Wilhelmina Kinderziekenhuis en UMC Utrecht, te Utrecht, NVKNO
- Dr. S.W.J. (Suzanne) Terheggen-Lagro, kinderlongarts, Amsterdam UMC, locatie AMC te Amsterdam, NVK
- Prof. dr. H.A.W.M (Harm) Tiddens, (kinder)longarts, Erasmus MC Sophia Kinderziekenhuis te Rotterdam, NVK
- H. (Hilda) Vale-Eeman, kinderverpleegkundige, Amsterdam UMC, locatie AMC te, Amsterdam, V&VN
- Dr. H. (Hester) van der Vaart, longarts UMCG te Groningen, NVALT
- Dr. H.W. (Harold) de Valk, internist-endocrinoloog UMCU te Utrecht, NIV
- Dr. M. (Marieke) Verkleij, GZ-psycholoog/cognitief gedragstherapeut, Amsterdam UMC, locatie VUMC te Amsterdam, VGCT
- Dr. E.J.M. (Els) Weersink, longarts, Amsterdam UMC, locatie AMC te Amsterdam, NVALT
- M. (Marion) Wessels, Verpleegkundige, UMC Utrecht te Utrecht, V&VN
- Dr. B.J. (Barbara) Wijnberg-Williams, klinisch psycholoog, UMCG te Groningen en Isala Ziekenhuis te Zwolle, NIP
- Dr. K.M. (Karin) de Winter- de Groot, kinderlongarts, Wilhelmina Kinderziekenhuis en UMC Utrecht te Utrecht, NVK
- Dr. P.J.G. (Petra) Zwijnenburg, klinisch geneticus, Amsterdam UMC, locatie VUMC te Amsterdam, VKGN
Met ondersteuning van
- Dr. F. (Floor) Willeboordse, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Ir. T. (Teus) van Barneveld, directeur, Kennisinstituut van de Federatie Medisch Specialisten
- Drs. A. (Anne) Speijer, coördinator kwaliteit van zorg, VSOP
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Drs. G.D. (George) Nossent (voorzitter) |
Longarts UMCG |
Bestuurslid NVALT |
Geen |
Geen actie |
|
O. Akkerman |
Longarts, fulltime, UMCG |
Lid nascholingscommissie VvAwT; onbetaald Lid visitatieteam TBC screening LRCB; onbetaald |
Geen |
Geen actie |
|
S. Amstelveen-Bokkerink |
Diëtist Radboudumc Dekkerswald |
Geen |
Geen |
Geen actie |
|
B. Arets |
Kinderlongarts, UMC Utrecht |
Academisch kinder(long)arts, deels Associate Professor Medisch Onderwijs Universiteit.
|
|
Geen actie
Toelichting: Niet direct betrokken bij module waarop de potentiële belangenverstrengeling van toepassing is. |
|
M. Bakker |
Longarts, Erasmus MC Rotterdam |
Geen |
Deelname aan diverse studies Vertex (chloortransportmodulatoren) |
Geen trekker van module CFTR modulatoren (wel meelezer).
Toelichting op onderzoek gesponsord door Vertex: Betrokken bij inclusie van patiënten en dataverzameling. Niet betrokken bij data-analyse, wel principal investigator, geen auteur. |
|
T. van Barneveld |
Directeur Kennisinstituut Federatie Medisch Specialisten |
|
Geen |
Geen actie |
|
A. van den Berg |
Longarts Hagaziekenhuis Den Haag |
Geen |
Geen |
Geen actie |
|
M. Berkhout |
KNO-arts in opleiding in het Academisch Medisch Centrum Amsterdam. Opleider: Prof. dr. F.G. Dikkers |
Geen |
Geen |
Geen actie |
|
F. Bodewes |
Kinderarts-MDL UMC Groningen |
Geen |
Geen |
Geen actie |
|
J. Boekhoff |
medisch maatschappelijk werker Amsterdam UMC, locatie VUmc |
Geen |
Geen |
Geen actie |
|
A. Bongen |
Medisch maatschappelijk werker UMC Utrecht |
Geen |
Geen |
Geen actie |
|
H. Bouazzaoui |
Longarts Hagaziekenhuis Den Haag |
Geen |
Geen |
Geen actie |
|
D. Brocke |
Medisch maatschappelijk werk UMC Utrecht Divisie Hart en Longen |
Geen |
Geen |
Geen actie |
|
E. Brokaar |
Poliklinisch apotheker in de Haga Apotheek, HagaZiekenhuis |
Geen |
Gedeeltelijke vergoeding voor internationaal CF congres door Novartis (2012 en 2014) |
Geen actie |
|
I. Bronsveld |
Pulmonoloog UMC Utrecht |
Geen |
Geen |
Geen actie |
|
A. Clement - Boers, de |
Kinderarts, diabetes HAGA/Julianaziekenhuis Den Haag |
Voorzitter Stichting D-Support (ondersteuning voor gezinnen met diabetes) onbetaald |
Geen |
Geen actie |
|
W. Doeleman |
Fysiotherapeut UMC-Utrecht |
Geen |
Geen |
Geen actie |
|
H. Eeman |
Kinderverpleegkundig consulent CF Werkgever: Academisch Medisch Centrum Amsterdam |
Geen |
Geen |
Geen actie |
|
M. Eerden |
Longarts Erasmus MC |
Geen |
Geen |
Geen actie |
|
K. van der Ent |
Kinderarts, hoogleraar kinderlongziekten UMC Utrecht |
|
Onderzoeksubsidies afgelopen vijf jaar:
Instituut heeft betalingen ontvangen voor klinische studies en adviesraad bijeenkomsten van Gilead, Vertex, GSK, Nutrica, TEVA, ProQR, Galapagos en Proteastasis.
|
Gemelde belangen en nevenfuncties zijn besproken met initiatief nemende verenigingen NVALT en NVK.
Deelname in werkgroep als meelezer bij alle modules op basis van expertise over CF.
Geen trekker van modules over CFTR modulatoren waarvoor er financiële belangen zijn. |
|
B. Flapper |
Kinderarts, sociale pediatrie |
Geen |
Geen |
Geen actie |
|
L. van der Giessen |
0,8 Kinderfysiotherapeut Erasmus MC Sophia 0,1 Coordinator onderwijs Erasmus MC 0,1 docentbegeleiding Erasmus MC |
B. Gastdocent Hogeschool Rotterdam Post HBO kinderfysio B. Cursusleider NPI B. Gastdocent Maas en Meer (B=betaald) |
Geen |
Geen actie |
|
V. Gulmans |
Hoofd onderzoek en kwaliteit van zorg bij Nederlandse Cystic Fibrosis Stichting |
Geen |
Geen persoonlijke vergoedingen. De NCFS heeft corporate sponsorovereenkomsten met meerdere bedrijven (farmaceuten en hulpmiddelen). De overeenkomsten zijn aangemeld bij het transparantieregister. De totale sponsorinkomsten bedragen niet meer dan 10 % van de totale NCFS-begroting. De sponsors hebben geen invloed op de inhoud van NCFS-uitingen. |
Geen actie |
|
H. Heijerman |
Longarts, hoofd afdeling longziekten UMC Utrecht. |
|
|
Gemelde belangen en nevenfuncties zijn besproken met initiatief nemende verenigingen NVALT en NVK.
Deelname in werkgroep als meelezer bij alle modules op basis van expertise over CF.
Geen trekker van modules over CFTR modulatoren waarvoor advies is gegeven. |
|
D. Hendriks |
kinderarts-maagdarmleverziekten Juliana Kinderziekenhuis/Hagaziekenhuis Den Haag |
Geen |
Geen |
Geen actie |
|
H. Hendriks |
kinderarts-longarts in Zuyderland MC en 0,1 fte gedetachteerd naar MUMC + voor CF zorg. |
Geen |
Geen |
Geen actie |
|
B. Hoek |
gastroenteroloog en hepatoloog, LUMC Hoogleraar |
Geen |
Geen gesponsord onderzoek op het gebied van CF. Bij genoemde studies mogelijk auteur en: Novartis: PI, inclusie patiënten Zambon: PI, design studies, data analyse Chiesi: design studie, patiënt inclusie, data analyse Abbvie, Norgine, Astellas: patiënt inclusie CLIF: patiënt inclusie Perspectum: patiënt inclusie, design deel van studie Organ Assist: PI, patiënt inclusie Novartis - deelname aan internationale medicijnstudie bij auto-immuun hepatitis Zambon-unrestricted grant voor onderzoek naar auto-immuun hepatitis en overlap syndromen Chiesi - investigator-initiated studie naar farmacoklnetiek van Envarsus (tacrolimus) na levertransplantatie Abbvie - studie naar langetermijn effect op leverfibrose na DAA behandeling van hepatitis C Norgine - deelname aan rifaximin bij hepatische encefalopathie studie CLIF consortium - deelname aan internationale studies naar accuut - op chronisch leverfalen Perspectum - studies naar nieuew NRI technieken bij leverziekte en na levertransplantatie Astellas - deelname aan immuunsuppressie studie na levertransplantatie OrganAssist - deelname aan studie met machineperfusie voor levertransplantatie |
Geen actie |
|
R. Hoek |
Longarts Erasmus MC Centrum voor Longtransplantatie Centrum voor Cystic Fibrosis en Recidiverende Luchtweginfecties |
Nederlands Transplantatievereniging
allen onbezoldigd |
Geen |
Geen actie |
|
A. Hofsteenge |
Dietist: CF-team (kindergeneeskunde) (Vumc en AMC) Kindergeneeskunde (polikliniek) (VUmc) |
Geen |
Geen |
Geen actie |
|
C. van Hoge |
Maag- darm- leverarts MUMC+ |
Geen |
Geen |
Geen actie |
|
F. Hollander-Kraaijeveld |
Diëtist UMC Utrecht |
Geen |
Geen |
Geen actie |
|
R. Houwen |
Hoofd afdeling kinder-MDL UMCU/WKZ |
De (poli)klinische zorg voor patienten met MDL problematiek, het geven van onderwijs alsmede het vooruitbrengen van de zorg en kennis op dit gebied, onder meer door (meewerken aan) publicaties. Tevens aansturen van de subafdeling kinder-MDL. Deze taken worden verricht in een 100% dienstverband. |
|
Geen actie |
|
J. van Ingen |
Arts-microbioloog, Radboudumc, Nijmegen |
Geen |
Mede-auteur van de internationale richtlijn voor diagnostiek en behandeling van niet-tuberculeuze mycobacterieën bij pantiënten met CF (Floto, 2016). |
Geen actie |
|
H. Janssens |
Kinderlongarts, Erasmus MC/Sophia Kinderziekenhuis |
Voorzitter CF-team Sophia |
Co-promotor bij meerdere studies gefinancierd door unrestricted grant van Chiesi en Gilead. |
Geen actie, meelezer bij module over behandeling P.Aeruginosa (Gilead, Chiesi), geen trekker.
Toelichting gesponsorde onderzoeken. -Studies van Chiesi en Gilead waren met een unrestricted grant, financieen gerund door Sophia BV. De aard van de studies kon door het onderzoeksteam vrij ingevuld worden en gepubliceerd. -Deelname centrum aan sponsor initiated studies van Vertex, en Gilead. Geen mede-auteur. Als voorzitter van CF-team verantwoordelijk voor uitvoer van de studie, en beoordeling adverse events en lab uitslagen van deelnemende patiënten. |
|
A. Jansz |
arts-microbioloog, St PAMM, streeklaboratorium voor de volksgezondheid Veldhoven |
Coördinerend arts-microbioloog, RIVM/IDS, Bilthoven, een detachering van 20% auditor voor de RvA |
Geen |
Geen actie |
|
H. van der Kamp |
kinderarts-endocrinoloog (0,7 fte) werkzaam in het WKZ Utrecht en in het Bronovo ziekenhuis in Den Haag (0,15 fte). |
Lid bestuur NVE (onbetaald) Lid adviesgroep groeihormoon (onbetaald) Lid adviesgroep neonatale screening AGS & CHT (onbetaald) |
Geen |
Geen actie |
|
C. de Kiviet |
Verpleegkundig Specialist Bij CF-centrum UMC Utrecht, locatie WKZ |
Lid van Accreditatiecommissie Van de RSV (Register Verpleegkundig Specialismen) >Vacatievergoeding per bijeenkomst -Beoordelaar scholing voor herregistratie Verpleegkundig Specialisten > Vergoeding per beoordeelde scholing |
Geen |
Geen actie |
|
A. Kok |
verpleegkundig consulent CF Erasmus MC-Sophia Rotterdam |
Geen |
Geen |
Geen actie |
|
B. Koot |
kinderarts AMC Amsterdam |
Bestuur sectie kinder MDL, NVK, onbetaald |
Geen |
Geen actie |
|
M. Kruiswijk |
verpleegkundig specialist afdeling Cystic Fibrosis |
Geen |
Geen |
Geen actie |
|
C. Majoor |
Longarts, Amsterdam UMC, locatie AMC |
|
Inclusie patiënten aan fase IIa, IIb, III en IV studies van Vertex en Galapagos |
Geen actie
Toelichting: Niet direct betrokken bij module waarop de potentiële belangenverstrengeling van toepassing is. |
|
R. van der Meer |
Longarts, aandachtsgebied Cystic Fibrose. Werkzaam in Hagaziekenhuis. Den Haag. |
Geen |
Geen |
Geen actie |
|
P. Merkus |
Kinderarts-pulmonoloog, Radboudumc Amalia Kinderziekenhuis |
|
Deelname Studies bij CF door farmaceutische bedrijven: Vertex, PTC (PI, betrokken bij inclusie, betrokken bij data-analyse) |
Geen actie
Toelichting: Niet direct betrokken bij module waarop de potentiële belangenverstrengeling van toepassing is. |
|
D. Mul |
Kinderarts-endocrinoloog bij Diabeter |
|
Geen |
Geen actie, trekker module diabetes (screening en behandeling)
Toelichting op deelname adviesraad medisch Novo Nordisk: conflicteert niet met trekkersrol van diabetes modules. |
|
A. Nagelkerke |
Kinderlongarts VU medisch centrum |
Geen |
|
Geen actie
Toelichting: Niet direct betrokken bij module waarop de potentiële belangenverstrengeling van toepassing is.
|
|
J. Noordhoek |
Directeur Nederlandse Cystic Fibrosis Stichting |
|
|
Geen actie |
|
M. Nuijsink |
Kinderarts Juliana Kinderziekenhuis/ HAGA ziekenhuis |
Geen |
|
Geen actie
Toelichting: Niet direct betrokken bij module waarop de potentiële belangenverstrengeling van toepassing is. |
|
M. van Oirschot-van de Ven |
Kinderverpleegkundig consulent CF, Academisch Medisch Centrum Amsterdam |
Geen |
Geen |
Geen actie |
|
M. Reijers |
Longarts, Radboud UMC, Nijmegen |
|
|
Geen actie
Toelichting: Niet direct betrokken bij module waarop de potentiële belangenverstrengeling van toepassing is.
|
|
S. Reitsma |
KNO-arts AMC Amsterdam |
Geen |
Geen |
Geen actie |
|
Y de Rijke |
Afdelingshoofd Klinische Chemie Erasmus MC te Rotterdam |
Geen |
Geen |
Geen actie |
|
B. Rottier |
UMC Groningen, kinderlongarts |
Geen |
Geen |
Geen actie |
|
R. Schrijver |
Verpleegkundig consulent CF Hagaziekenhuis |
Geen |
Geen |
Geen actie |
|
A. Speijer |
Coördinator kwaliteit van zorg |
Lid advies - en expertcommissie AQUA van Zorginstituut Nederland.
|
Geen |
Geen actie |
|
L. Speleman |
paediatrische KNO, UMC Utrecht Wilhelminakinderziekenhuis |
|
Geen |
Geen actie |
|
S. Terheggen-Lagro |
Kinderlongarts AMC |
|
Geen |
Geen actie |
|
H. Tiddens |
Kinderlongarts, Erasmus MC, Sofia kinderziekenhuis
|
|
|
Geen actie, meelezer bij module over behandeling P.Aeruginosa geen trekker.
Toelichting gesponsorde onderzoeken. iABC: Co-investigator, CT aquisitie, analyse van CTs, geen betrokkenheid bij inclusie van patiënten ALPINE II: betrokken bij inclusie van patiënten |
|
H. van der Vaart |
Longarts UMCG |
Geen |
Geen |
Geen actie |
|
H. de Valk |
Internist-endocrinoloog, UMC Utrecht |
|
Geen |
Geen actie |
|
H. Verkade |
Kinderarts MDL, UMCG Groningen |
|
Geen |
Geen actie |
|
M. Verkleij |
GZ-psycholoog/cognitief gedragstherapeut, Amsterdam UMC locatie VUmc |
Geen |
Geen |
Geen actie |
|
E. Weersink |
longarts, AMC Amsterdam |
Geen |
|
Geen actie
Toelichting: Niet direct betrokken bij module waarop de potentiële belangenverstrengeling van toepassing is. |
|
M. Wessels-Bakker |
Verpleegkundig Specialist Longtransplantatie, onderzoeker |
Geen |
Geen |
Geen actie |
|
B. Wijnberg-Williams |
Klinisch psycholoog/medisch psycholoog, richting kinderen en jeugdigen, UMCG (0,4 fte) en Isala ziekenhuis (0,7fte) |
Lid van Accreditatiecommissie Federatie van Gezondheidszorgpsychologen en Psychotherapeuten (FGzPt) (vacatiegelden) |
Geen |
Geen actie |
|
F. Willeboordse |
Adviseur Kennisinstituut Federatie Medisch Specialisten |
Geen |
Via werk partner aandelen bij moederbedrijf Johnson & Johnson. Partner is werkzaam bij Janssen Vaccines BV. |
Geen actie |
|
K. Winter-de Groot |
kinderarts-pulmonoloog WKZ/UMC Utrecht |
|
Geen |
Geen actie |
|
P. Zwijnenburg |
kinderarts-klinisch geneticus VUMC afd. klinische genetica |
Geen |
Geen |
Geen actie |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door deelname aan de werkgroep van de patiëntenorganisatie NCFS en de VSOP voor zeldzame en genetische aandoeningen. De uitgangsvragen zijn getoetst via een online peiling onder patiënten en ouders. Voor sommige onderwerpen is het patiënten-panel van de NCFS geraadpleegd om input te verkrijgen over patiëntenvoorkeuren over een behandeling of screeningsmethode.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de kwaliteitsstandaarden de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de kwaliteitsstandaard in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden bij de aanverwante producten.
Werkwijze
Deze medisch inhoudelijke modules in deze kwaliteitsstandaard is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Voor de modules over psychosociale zorg en de modules over organisatie van zorg is er volgens de AQUA methode gewerkt: GRADE. De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie https://www.gradeworkinggroup.org/docs/Criteria_for_using_GRADE_2016-04-05.pdf).
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de stuurgroep en de adviseur de knelpunten. De werkgroep stelde vervolgens een long list met knelpunten op en prioriteerde de knelpunten op basis van: (1) klinische relevantie, (2) de beschikbaarheid van (nieuwe) evidence van hoge kwaliteit, (3) en de te verwachten impact op de kwaliteit van zorg, patiëntveiligheid en (macro)kosten. Tevens zijn er knelpunten aangedragen door VSOP, NCFS, NVK, NVALT, Patiëntenfederatie, Lareb, NVMDL, ZiN, ZN, NIP, NVKNO, VKGN, Vereniging innovatieve geneesmiddelen, NVKC, V&VN, NVMM, KNGF, NIV, NVvR, NVD, NVAB, NVZA via een invitational conference.
De concept uitgangsvragen zijn door de NCFS online voorgelegd aan mensen met CF en ouders van kinderen met CF. Vrijwel alle uitgangsvragen werden door tenminste 85% van de 68 respondenten als zinvol tot zeer zinvol ervaren. Bij twee vragen was dit percentage lager (56% respectievelijk 72%), veroorzaakt door het feit dat 25% van de respondenten bij deze vragen geen mening had.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de stuurgroep en de adviseur concept-uitgangsvragen opgesteld. Deze zijn met de werkgroep besproken waarna de werkgroep de definitieve uitgangsvragen heeft vastgesteld. Vervolgens inventariseerde de werkgroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Er is voor de afzonderlijke uitgangsvragen aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie voor de oriënterende zoekactie en patiëntenperspectief zijn opgenomen onder aanverwante producten.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR - voor systematische reviews; Cochrane - voor gerandomiseerd gecontroleerd onderzoek; Newcastle-Ottowa - voor observationeel onderzoek; QUADAS II - voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A) Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk* |
|
|
Laag |
|
|
Zeer laag |
|
*in 2017 heeft het Dutch GRADE Network bepaald dat de voorkeursformulering voor de op een na hoogste gradering ‘redelijk’ is in plaats van ‘matig’
B) Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008), en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de cruciale uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Voor deze kwaliteitsstandaard was de Europese Best Practice richtlijn van de ECFS (Castellani, 2018) een belangrijk uitgangspunt voor bijna alle modules.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de kwaliteitsstandaard is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van Zorg.
Indicatorontwikkeling
De werkgroep heeft besloten geen indicatoren te ontwikkelen bij de huidige richtlijn omdat er geen substantiële barrières konden worden geïdentificeerd die implementatie van de aanbeveling zouden kunnen bemoeilijken.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Brouwers, M. C., Kho, M. E., Browman, G. P., Burgers, J. S., Cluzeau, F., Feder, G., ... & Littlejohns, P. (2010). AGREE II: advancing guideline development, reporting and evaluation in health care. Canadian Medical Association Journal, 182(18), E839-E842.
Castellani C, Duff AJA, Bell SC, Heijerman HGM, Munck A, Ratjen F, Sermet-Gaudelus I, Southern KW, Barben J, Flume PA, Hodková P, Kashirskaya N, Kirszenbaum MN, Madge S, Oxley H, Plant B, Schwarzenberg SJ, Smyth AR, Taccetti G, Wagner TOF, Wolfe SP, Drevinek P. ECFS best practice guidelines: the 2018.
revision. J Cyst Fibros. 2018 Mar;17(2):153-178. doi: 10.1016/j.jcf.2018.02.006. Epub 2018 Mar 3. Review.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site.html
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van Medisch Specialisten.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann, H. J., Oxman, A. D., Brozek, J., Glasziou, P., Jaeschke, R., Vist, G. E., ... & Bossuyt, P. (2008). Rating Quality of Evidence and Strength of Recommendations: GRADE: Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ: British Medical Journal, 336(7653), 1106.
Wessels, M., Hielkema, L., & van der Weijden, T. (2016). How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. Journal of the Medical Library Association: JMLA, 104(4), 320.
Zoekverantwoording
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID) 1987 – september 2017
|
1 Cystic Fibrosis/ or (cystic* adj3 fibros*).ti,ab,kf. or mucoviscoidos*.ti,ab,kf. or mucoviscidos*.ti,ab,kf. (46783) 2 ((exp Cystic Fibrosis Transmembrane Conductance Regulator/ or (CF or CFTR).ab,ti.) and (modulat* or potentiator* or corrector*).ab,ti.) or (ivacaftor or lumacaftor or kalydeco or orkambi).ab,ti. (2321) 3 1 and 2 (1516) 4 limit 3 to (english language and yr="1987 -Current") (1490) 5 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (345971) 6 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1789382) 7 4 and 5 (13) 8 4 and 6 (210) 9 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ [Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies] (2988005) 10 4 and 9 (90) 11 7 or 8 or 10 (282)
= 282 (247 uniek) |
455 |
|
Embase (Elsevier) |
'cystic fibrosis'/exp OR ((cystic* NEAR/3 fibros*):ti,ab) OR mucoviscoidos*:ab,ti OR mucoviscidos*:ab,ti
AND ('cystic fibrosis transmembrane conductance regulator mouse'/exp OR cf:ab,ti OR cftr:ab,ti) AND (modulat*:ab,ti OR potentiator*:ab,ti OR corrector*:ab,ti) OR ivacaftor:ab,ti OR lumacaftor:ab,ti OR kalydeco:ab,ti OR orkambi:ab,ti
AND [english]/lim AND [1987-2017]/py
Gebruikte filters: Systematische reviews: ('meta analysis'/de OR cochrane:ab OR embase:ab OR psychlit:ab OR cinahl:ab OR medline:ab OR (systematic NEAR/1 (review OR overview)):ab,ti OR (meta NEAR/1 analy*):ab,ti OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT ('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp NOT 'human'/exp) Randomized controlled trials: 'clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti NOT 'conference abstract':it Observationeel onderzoek: 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR ('prospective study'/de NOT 'randomized controlled trial'/de) OR 'cohort analysis'/de OR (cohort NEAR/1 (study OR studies)):ab,ti OR (case:ab,ti AND (control NEAR/1 (study OR studies)):ab,ti) OR (follow:ab,ti AND (up NEAR/1 (study OR studies)):ab,ti) OR (observational NEAR/1 (study OR studies)):ab,ti OR (epidemiologic NEAR/1 (study OR studies)):ab,ti OR ('cross sectional' NEAR/1 (study OR studies)):ab,ti
= 345 (340 uniek) |