Behandeling therapieresistente hypertensie
Uitgangsvraag
Hoe dient therapieresistente hypertensie te worden behandeld als therapie-ontrouw en secundaire hypertensie zijn uitgesloten?
Aanbeveling
Voeg een aldosteronantagonist (in het bijzonder spironolacton) in lage eenmaal daagse dosering van 25 tot 50 mg toe bij therapieresistente hypertensie.
Doseer spironolacton bij voorkeur niet hoger dan 50 mg om bijwerkingen te voorkomen.
Overweeg bij therapieresistente hypertensie een van de drie medicamenten (bijvoorbeeld een calciumantagonist) in te nemen voor het slapen gaan.
Overweeg vastedosiscombinatietabletten te geven voor het verbeteren van de adherentie en niet zozeer voor verlaging van de bloeddruk.
Voor het bespreken met de patiënt welke mogelijkheid de voorkeur heeft, raadt de werkgroep de Keuzekaart: Hoge bloeddruk die moeilijk te behandelen is, aan.
Overwegingen
In de richtlijnen voor de behandeling van hypertensie van de ESC/ESH (2013) wordt geadviseerd om bij patiënten met therapieresistente hypertensie een vierde middel aan de behandeling toe te voegen uit een klasse van antihypertensiva met een ander (partieel) werkingsmechanisme dan de middelen die de patiënt reeds gebruikt (Sever, 2011). Hierbij worden met name de toevoeging van lage dosis van een aldosteron-receptorblokkers, een α-blokker, maar ook hogere dosis van met name thiazidediuretica (in het bijzonder chloortalidon) genoemd. De recent gepubliceerde RCT (PATHWAY-2) liet evenwel zien dat toevoeging van spironolacton in doseringen tussen 25 en 50 mg het meest effectief was in het verlagen van de thuis gemeten systolische bloeddruk vergeleken met placebo, doxazosine (4 tot 8 mg) of bisoprolol (5 tot 10 mg) (Williams, 2015). Het hier gedocumenteerde literatuuronderzoek heeft alleen bewijskracht gevonden voor de toevoeging van spironolacton aan de medicatie en verandering van het tijdstip van innemen van de medicatie (het derde middel innemen voor het slapen gaan in plaats van in de ochtend). Deze interventies leiden tot een aantoonbaar lagere bloeddruk. Effect op harde eindpunten zijn niet onderzocht.
Toevoeging van een lage dosis spironolacton (25 tot 50 mg) heeft behalve een gunstig effect op de bloeddruk ook het voordeel dat het weinig nadelige bijwerkingen kent. In het algemeen zijn de bijwerkingen van spironolacton (met name hyperkaliemie, en op langere termijn gynaecomastie, impotentie en menstruatiestoornissen) gerelateerd aan de hoogte van de dosering. In meta-analyse van Wang (2016) werd een stijging van het K-gehalte van 0,18 mmol/L (95%BI: 0,04 tot 0,32) gevonden. Dit is conform hetgeen in de literatuur vermeld wordt (incidentie hyperkaliemie 1 tot 7%, [Gwoo, 2014]). De incidentie van de lange termijn bijwerkingen wordt in de literatuur opgegeven variërend tussen de 3 en 6% (Danjuma, 2014).
In het onderzoek over de chronotherapie van Hermida (2008) werden met name de calciumantagonist en de α-blokker uitgewisseld van de ochtend naar de avond. Dit resulteerde vooral in een significante reductie van het aantal non-dippers (<10% daling van de nachtelijke bloeddruk) van 84 naar 43%. Uit andere onderzoeken is bekend dat de nachtelijke bloeddruk de beste voorspeller is van het cardiovasculaire risico (Staessen, 1999). Het effect van het innemen van een medicijn voor het slapen gaan op hart- en vaatziekten is echter bij therapieresistente hypertensie nog nooit onderzocht. Op grond van de bevindingen van de Syst-Eur Trial (Staessen, 1999) en de HOPE Study (Svensson, 2001), waarbij respectievelijk nitrendipine en ramipril voor het slapen werden gegeven, zou men kunnen extrapoleren dat chronotherapie bij resistente hypertensie leidt tot een gunstig effect op cardiovasculaire morbiditeit en mortaliteit. Dit moet evenwel nog worden aangetoond.
Het huidige literatuuronderzoek heeft geen artikelen met betrekking tot vaste-dosis combinaties van medicamenten gevonden die voldeden aan de selectiecriteria. In een recente review over de mogelijke voordelen van vaste-dosis combinaties werd gevonden dat het verschil in systolische en diastolische bloeddruk niet statisch significant verschillend was (respectievelijk -4,1 en -3,1 mmHg) in vergelijking met het tegelijk innemen van de losse componenten, maar wel gepaard ging met een significant betere adherentie (Gupta, 2010). De onderzoeken waren te kort om uit te zoeken of dit soort bloeddrukverschillen op het langetermijneffect zouden kunnen hebben op het voorkomen van intermediaire of harde eindpunten. Bovendien ontbreken de inzichten in kosteneffectiviteit tot op heden.
De genoemde effecten van de verschillende medicamenteuze mogelijkheden bij therapieresistente hypertensie zijn uiteraard gemiddelde waarnemingen en niet op voorhand voorspel bij een individuele patiënt. Dit geldt onder andere bij het gebruik van diuretica en spironolacton in genoemde doseringen, waarbij steeds het evenwicht tussen effectiviteit en bijwerkingen in acht moet worden genomen. Soms zijn lagere, soms hogere dosis nodig dan genoemde in de aanbevelingen.
Om de verschillende therapeutische opties met uw patiënt te bespreken is er een Keuzekaart: Hoge bloeddruk die moeilijk te behandelen is, ontwikkeld. Hiermee is in een oogopslag inzichtelijk welke opties met voor- en nadelen voor handen zijn. De richtlijncommissie denkt dat hiermee in dialoog met de patiënt specifiek voor hem/haar toegespitste behandeling beter inzichtelijk gemaakt kan worden.
Onderbouwing
Geschat wordt dat ongeveer 25% van de patiënten met hypertensie drie bloeddrukverlagende medicijnen nodig heeft om op streefwaarde te komen. Therapieresistente hypertensie zien we in toenemende mate in de klinische praktijk. De prevalentie wordt opgegeven tussen de 3 en 30%. Patiënten met therapieresistente hypertensie hebben een veel slechtere cardiovasculaire prognose (Pierdomenico, 2005). In verschillende studies wordt een 2 tot 3 keer verhoogd risico op cardiovasculaire morbiditeit gevonden.
Wat is de beste behandelstrategie van true resistant hypertension als zogenaamd pseudoresistentie (op basis van een witte-jasseneffect en/of therapieontrouw) en secundaire oorzaken zijn uitgesloten? Er lijkt in de literatuur eensgezindheid te bestaan over het gebruik van een thiazidediureticum, in het bijzonder chloorthalidon, als eerste keuze middel. Ook zijn er studies die een gunstig effect van mineralocorticoïdreceptorantagonisten, zoals spironolacton en eplerenone, suggereren om de bloeddruk verder te verlagen. Uiteraard horen calciumantagonisten en remmers van het renine-angiotensine systeem ook bij deze patiënten te worden gegeven, omdat ze waarschijnlijk het cardiovasculaire risico verlagen. Vastedosiscombinatiepreparaten van al deze middelen lijken een lagere bloeddruk en een betere medicatieadherentie te geven. Maar er zijn ook suggesties om het tijdstip van inname van de antihypertensiva in zo'n situatie meer op elkaar af te stemmen (zogenaamd chronotherapie) door bijvoorbeeld middelen voor het slapen te geven in plaats van een extra bloeddrukverlagend medicament toe te voegen.
Bloeddruk
|
Redelijk GRADE |
Bij patiënten met therapieresistente hypertensie resulteert het toevoegen van spironolacton aan de bestaande medicatie in een dagdosering van 25-50 mg in een klinisch relevante verlaging van de systolische en diastolische bloeddruk vergeleken met placebo of toevoeging van ramipril.
Bronnen (Wang, 2016) |
Percentage patiënten op streefwaarde & Hart- en vaatziekten
|
Zeer laag GRADE |
Vanwege het ontbreken van bewijs is het niet mogelijk een conclusie te trekken over het effect van spironolacton op het percentage patiënten op streefwaarde of op het risico op hart- en vaatziekten bij patiënten met therapieresistente hypertensie. |
Combinatietabletten
|
- GRADE |
Vanwege het ontbreken van bewijs is het niet mogelijk een conclusie te trekken over het effect van combinatietabletten op bloeddruk, het percentage patiënten op streefwaarde of het risico op hart- en vaatziekten bij patiënten met therapieresistente hypertensie. |
Chronotherapie
Bloeddruk
|
Laag GRADE |
Chronotherapie (1 medicijn voor het slapen gaan innemen) resulteert in een klinisch relevante verlaging van de systolische en diastolische bloeddruk bij patiënten met therapieresistente hypertensie vergeleken met het innemen van alle medicamenten bij het opstaan.
Bronnen (Hermida, 2008) |
Percentage on target
|
Laag GRADE |
Chronotherapie (1 medicijn voor het slapen gaan innemen) verhoogt bij therapieresistente hypertensie het aantal patiënten dat de streefwaarde van de bloeddruk bereikt vergeleken met het innemen van alle medicamenten bij het opstaan.
Bronnen (Hermida, 2008) |
Hart- en vaatziekten
|
- GRADE |
Vanwege het ontbreken van bewijs is het niet mogelijk een conclusie te trekken over het effect van chronotherapie (1 medicament voor het slapen gaan) op het risico op hart- en vaatziekten bij patiënten met therapieresistente hypertensie. |
Spironolacton of hoge dosering diureticum
Wang (2016) ondernam een systematische zoekactie naar gerandomiseerde, gecontroleerde trials naar de effectiviteit en veiligheid van spironolacton bij patiënten met therapieresistente hypertensie. PubMed, EMBASE en de Cochrane Library werden tot december 2015 doorzocht. Alleen RCT’s die het effect van additionele spironolacton op spreekkamerbloeddruk, ambulante bloeddruk of bijwerkingen onderzochten, werden geïncludeerd. In totaal voldeden vijf trials aan deze criteria. In vier placebogecontroleerde trials werd toevoeging van spironolacton aan de bestaande medicatie onderzocht en in één trial werd de toevoeging van spironolacton of ramipril bestudeerd. De dosering van spironolacton liep uiteen van 25 tot 50 mg per dag en die van ramipril van 5 tot 10 mg. In totaal werden 553 patiënten in de systematische reviews geïncludeerd. De gemiddelde leeftijd van deelnemers varieerde van ongeveer 50 tot 63 jaar en de gemiddelde duur van de follow up was 12 (range 8 tot 16) weken.
Resultaten
I. Bloeddruk
Als uitkomst is gekozen voor het effect op 24-uur ambulante bloeddruk. Vier trials hadden gegevens over 24-uur ambulante bloeddruk gerapporteerd. Gemiddeld nam de systolische bloeddruk met 10,50 mmHg af in het voordeel van spironolacton (-10,50 mmHg 95%BI: -12,30 tot -8,71) en de diastolische bloeddruk met 4,09 mmHg (-4,09 mmHg 95%BI: -5,28 tot -2,91). De effecten op dag- en nachtbloeddrukwaarden waren vergelijkbaar (tabel 1). De effecten op de spreekkamerbloeddruk (tabel 1) waren het grootst, zoals dat meestal het geval is in vergelijking met 24-uurs bloeddrukmetingen.
Tabel 1 Gemiddelde verschillen in bloeddruk, gemeten met verschillende methoden, bij het gebruik van spironolacton ten opzichte van placebo/ramipril (gebaseerd op Wang, 2016)
|
Meetmethode |
Systolische bloeddruk in mmHg (95%BI) |
Diastolische bloeddruk in mmHg (95%BI) |
|
24-uurs ambulante bloeddruk |
-10,50 (-12,30 tot -8,71) |
-4,09 (-5,28 tot -2,91) |
|
Dagbloeddruk |
-10,20 (-12,41 tot -7,99) |
-4,14 (-5,50 tot -2,78) |
|
Nachtbloeddruk |
-10,02 (-12,62 tot -7,41) |
-3,21 (-4,84 tot -1,58) |
|
Spreekkamerbloeddruk |
-16,99 (-25,04 tot -8,95) |
-6,18 (-9,30 tot -3,05) |
II. Percentage patiënten op streefwaarde
Wang (2016) rapporteerde geen gegevens over het percentage patiënten dat de bloeddrukstreefwaarden haalde bij gebruik van spironolacton.
III. Hart- en vaatziekten
Wang (2016) rapporteerde geen gegevens over het risico op hart- en vaatziekten bij gebruik van spironolacton.
Bewijskracht van de literatuur
Bloeddruk: De bewijskracht voor de uitkomstmaat bloeddruk is met een niveau verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; onduidelijkheid over de blindering van de uitkomst assessor).
Vanwege het ontbreken van bewijs is het niet mogelijk om de bewijskracht voor de uitkomstmaten percentage op bloeddrukstreefwaarden en hart- en vaatziekten te graderen.
Combinatietabletten
Er werden geen studies gevonden die voldeden aan de selectiecriteria waarbij het effect van combinatietabletten op de bloeddruk bij patiënten met therapieresistente hypertensie werd onderzocht.
Chronotherapie
Hermida (2008) ondernam een gerandomiseerde, gecontroleerde trial naar de invloed van het veranderen van het innametijdstip van de medicatie op het circadiane ritme van bloeddruk zonder wijziging van het aantal of de dosering van de bloeddrukverlagende medicatie. Patiënten met ongecontroleerde hypertensie op basis van 24-uurs bloeddrukmetingen (dag >135/85 mmHg, nacht >120/70 mmHg) die drie antihypertensiva in een adequate dosering kregen kwamen in aanmerking. Alle patiënten gebruikten een diureticum in combinatie met een ACE-remmer (43%) of een angiotensine-II receptorblokker (82%). Als derde medicament werd bij 66% een calciumantagonist (dihydropyridine) of een α-blokker (59%) gebruikt. Bij alle patiënten werd het derde medicament veranderd en vervolgens werden de deelnemers gerandomiseerd naar het innemen van alle medicamenten in de ochtend of het innemen van de nieuwe medicatie (1 tablet) vlak voor het naar bed gaan. Het kwam er dus op neer dat met name de calciumantagonist en de α-blokker uitgewisseld werden en al dan niet van een ochtend- naar de avonddosering verschoven. Van de 265 patiënten die voldeden aan de in- en exclusiecriteria waren bij 250 (2 groepen van 125) daarvan na 12 weken valide 48-u ambulante bloeddrukmetingen beschikbaar. Waak- en slaapperioden werden met behulp van een activiteitenmeter, zogenaamde actigrafie, vastgesteld. De gemiddelde leeftijd was 60 jaar.
Resultaten
I. Bloeddruk
Systolische bloeddruk, gemeten tijdens waken, slapen en over 24 uur, was lager bij het innemen van 1 medicament vlak voor het naar bed gaan vergeleken met alle medicatie innemen bij het opstaan (figuur 1). Op de diastolische bloeddruk werd een vergelijkbaar effect gevonden (figuur 1).
Figuur 1 Veranderingen van de gemiddelde van systolische en diastolische bloeddruk (mmHg) tijdens waken (diurnal), slapen (nocturnal) en gemiddeld over 24 uur na een interventieperiode van drie maanden (overgenomen uit Hermida, 2008)
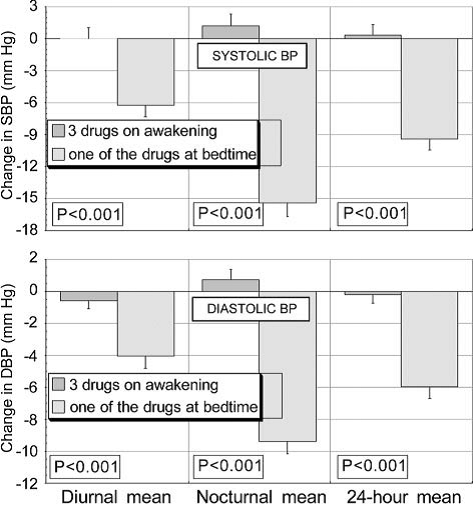
II. Percentage patiënten op streefwaarde
In de groep die alle medicatie ’s morgens nam was na drie maanden bij één (0,8%) patiënt de bloeddruk op streefwaarde. Bij de groep die één middel voor het slapen innam, was dit bij 46/125 (37%) het geval. Het maakte niet uit welke van de vier combinaties van middelen (ACE-remmer of angiotensine II receptorblokker + diureticum + calciumantagonist of α-blokker) werd gegeven.
III. Hart- en vaatziekten
Hermida (2008) rapporteerde geen gegevens over hart- en vaatziekten.
Bewijskracht van de literatuur
Bloeddruk: De bewijskracht voor de uitkomstmaat bloeddruk is met twee niveaus verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias; er was geen placebo gebruikt) en imprecisie (gering aantal patiënten geïncludeerd).
Vanwege het ontbreken van bewijs is het niet mogelijk om de bewijskracht voor de uitkomstmaat hart- en vaatziekten te graderen.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat is de effectiviteit van spironolacton of een hoge dosering diureticum, combinatietabletten en chronotherapie vergeleken met reguliere zorg bij patiënten met therapieresistente hypertensie?
P patiënten met therapieresistente hypertensie;
I 1. spironolacton of een hoge dosering diureticum;
2. combinatietabletten;
3. chronotherapie;
C reguliere zorg;
O bloeddruk, percentage patiënten op streefwaarde, hart- en vaatziekten.
Relevante uitkomstmaten
De werkgroep achtte bloeddruk en hart- en vaatziekten voor de besluitvorming kritieke uitkomstmaten; en percentage patiënten op streefwaarde een voor de besluitvorming belangrijke uitkomstmaat.
Bloeddruk: de werkgroep definieerde een verschil van meer dan 5 mmHg in de gemiddelde systolische bloeddruk 24-uurs ABPM of een verschil van meer dan 10 mmHg de gemiddelde systolische bloeddruk in de spreekkamer als een klinisch relevant verschil.
Percentage patiënten op streefwaarde: de werkgroep definieerde een absoluut verschil van 20% in het voordeel van de interventie als een klinisch relevant verschil.
Hart- en vaatziekten: de werkgroep definieert een NNT over een periode van 10 jaar van 20 als een klinisch relevant verschil.
Zoeken en selecteren (Methode)
In de databases Medline (OVID) en EMBASE (Embase.com) is met relevante zoektermen gezocht naar systematische reviews en gerandomiseerde trials. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 456 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria:
- systematische reviews gezocht in ten minste twee databases met een gedetailleerde zoekstrategie, evidence-tabellen en risk of bias beoordeling en;
- gerandomiseerde trials bij patiënten met therapieresistente hypertensie waarbij de onder de I genoemde interventies werden vergeleken met reguliere zorg.
Op basis van titel en abstract werden in eerste instantie 38 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 36 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording) en twee studies definitief geselecteerd.
Resultaten
Twee onderzoeken zijn opgenomen in de literatuuranalyse; een systematische review naar de effectiviteit van spironolacton bij patiënten met therapieresistente hypertensie en een RCT naar het effect van chronotherapie op bloeddruk bij patiënten met therapieresistente hypertensie. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidence-tabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk of bias tabellen.
- Danjuma MI, Mukherjee I, Kakaronidis J, et al. Converging indications of aldosterone antagonists (spironolactone and eplerenone): a narrative review of safety profiles. Curr Hypertens Rep. 2014:16:1-10.
- Gupta AK, Arshad S, Poulter NR. Compliance, safety and effectiveness of fixed-dose combinations of antihypertensive agents: a meta-analysis. Hypertension. 2010;55:399-407.
- Gwoo S, Kim YN, Shin HS, et al. Predictors of hyperkalemia frisk after hypertension control with aldosteron blockade according to the presence or absence of chronic kidney disease. Nephron Clinical Practice. 2014;381-6.
- Hermida RC, Ayala DE, Fernández JR, et al. Chronotherapy improves blood pressure control and reverts the nondipper pattern in patients with resistant hypertension. Hypertension. 2008;51(1):69-76. Epub 2007 Oct 29. PubMed PMID: 17968001.
- Pierdomenica SD, Lapenna D, Bucci A, et al. Cardiovascular outcome in treated hypertensive patients with responder, masked, false resistant, and true resistant hypertension. Am J Hypertens 2005;18:14221428.
- Sever PS, Messerli FH. Hypertension management 2011: optimal combination therapy. Eur Heart J. 2011;32:2499-2506.
- Staessen JA, Thijs L, Fagard B, et al. For the Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Predicting cardiovascular Risk using conventional vs ambulatory blood pressure in older patients with systolic hypertension. JAMA. 1999;282:539-46.
- Svensson P, de Faire U, Sleight P, et al. Comparative effects of ramipril on ambulatory and office blood pressure. A HOPE substudy. Hypertension. 2001;38:e28-e32.
- Wang C, Xiong B, Huang J. Efficacy and Safety of Spironolactone in Patients with Resistant Hypertension: A Meta-analysis of Randomised Controlled Trials. Heart Lung Circ. 2016;25(10):1021-30. doi: 10.1016/j.hlc.2016.02.016. Epub2016 Apr 11. PubMed PMID: 27118266.
- Williams B, MacDonald TM, Morant S, et al. For The British Hypertension Society's PATHWAY Studies Group. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomized, double-blind, cross-over trial. Lancet. 2015;386:2059-68.
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Wang, 2016
[individual study characteristics deduced from Wang, 2016 ]
PS., study characteristics and results are extracted from the SR (unless stated otherwise) |
SR and meta-analysis of RCTs
Literature search up to December, 2015
A: Abolghasmi, 2011 B: Bobrie, 2012 C: Oxlund, 2013 D: Vaclavik, 2014 E: Ni, 2014
Study design: All Five are RCT
Setting and Country: A: Iran B: France C: Denmark D: Czech Republic E: China
Source of funding: Not stated; authors did not receive any funding for the review |
Inclusion criteria SR:
“Randomised controlled trials investigating the effect of additional spironolactone on office blood pressure (BP), ambulatory BP or adverse events in resistant hypertension patients were included.”
Five studies included
Important patient characteristics at baseline:
N, mean age (yrs) A: 41 patients; I: 49, C: 50 B: 167 patients; I: 56, C: 55 C: 119 patients; I: 62, C: 53 D: 150 patients; I: 60, C: 59 E: 76 patients; I: 55, C: 54
Sex: A: 45% Male B: 75% Male C: 77% Male D: 65% Male E: 59% Male
Groups comparable at baseline? No, differences in age |
A: Spironolactone mean dose of 25-50 mg; once daily B: Spironolactone mean dose of 25 mg; once daily C: Spironolactone mean dose of 35 mg; once daily D: Spironolactone mean dose of 25 mg; once daily E: Spironolactone mean dose of 42.5 mg; once daily |
A: Placebo B: Ramipril C: Placebo D: Placebo E: Placebo |
End-point of follow-up:
Not stated
For how many participants were no complete outcome data available? (intervention/control)
Not stated |
Outcome measure-1 Defined as blood pressure, measured as 24-h ambulatory BP
Systolic BP Effect measure: mean difference [95% CI]: B: -10.00 (-13.11 to -6.89) C: -8.90 (-13.17 to -4.63) D: -10.50 (-14.63 to -6.37) E: -12.00 (-15.26 to -8.74)
Pooled effect (fixed effects model): -10.50 [95% CI -12.30 to -8.71] favoring spironolactone Heterogeneity (I2): 0.0%, p=0.693
Diastolic BP Effect measure: mean difference [95% CI]: B: -4.00 (-5.97 to -2.03) C: -3.90 (-6.11 to -1.69) D: -3.50 (-5.95 to -1.04) E: -6.00 (-9.44 to -2.56)
Pooled effect (fixed effects model): -4.09 [95% CI -5.28 to -2.91] favoring spironolactone Heterogeneity (I2): 0.0%, p=0.696
Outcome measure-2 Defined as percentage on target
Not reported
Outcome measure-3 Defined as CVD
Not reported |
Facultative:
Risk of bias was assessed with the Cochrane risk of bias tool.
Brief description of author’s conclusion: “[…], spironolactone combined with triple-drug therapy significantly decreases ambulatory and office BP […].”
Personal remarks on study quality, conclusions, and other issues (potentially) relevant to the research question
Level of evidence: GRADE (per comparison and outcome measure) including reasons for down/upgrading
Outcome-1: MODERATE Downgraded one level because of unclear blinding of outcome assessors (risk of bias)
Outcome-2 & 3: No GRADE |
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C)3 |
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
CHRONOTHERAPY |
|||||||
|
Hermida, 2008 |
Type of study: RCT [parallel]
Setting: Hypertensive patients
Country: Spain
Source of funding: Non-commercial |
Inclusion criteria:
Exclusion criteria:
N total at baseline: Intervention: 125 Control: 125
Important prognostic factors2: Age ± SD: I: 60 (12) C: 60 (12)
Sex: I: 56% M C: 53% M
Groups comparable at baseline? Yes |
All 3 drugs on awakening
The basic therapeutic schemes where a combination of a diuretic with either an angiotensin-converting enzyme inhibitor (34.4% of the patients) or an angiotensin II receptor blocker (65.6% of the patients). The third drug was a dihydropyridine calcium channel blocker (CCB; 52.8% of the patients; mainly amlodipine or nifedipine gastrointestinal therapeutic system) or an _-blocker (always doxazosin gastrointestinal therapeutic system; 47.2% of the patients).
Change of the third of the drugs mentioned above to a different one, thus maintaining a synergic combination2 and still administering all 3 drugs on awakening or the same approach but administering the new drug at bedtime. |
One drug at bedtime
|
Length of follow-up: 3 months
Loss-to-follow-up: N = 15 (6%) out of 265* Reasons: unclear
Incomplete outcome data: N = 15 (6%) out of 265* Reasons: unclear |
1. Blood pressure Measured as diurnal, nocturnal and 24-h, mean (SD)
Systolic Diurnal I: 137.3 (14.5) C: 133.4 (15.9) p=0.024 Nocturnal I: 134.1 (16.9) C: 120.7 (17.2) p<0.001 24-h I: 136.3 (14.3) C: 129.2 (15.8) p<0.001
Diastolic Diurnal I: 80.3 (12.5) C: 79.3 (10.6) p=0.508 Nocturnal I: 73.8 (11.9) C: 67.2 (9.4) P<0.001) 24-h I: 78.1 (12.0) C: 75.3 (9.8) p=0.046
2. Percentage on target Data not reported
3. CVD Data not reported |
* Data on 250 patients was reported. |
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors [(potential) confounders]
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Wang, 2016 |
Yes |
Yes, gezocht in drie databases |
Yes |
Yes |
NA |
Yes, Cochrane tool was used |
Yes |
Yes |
No, not done for the individual studies |
Notes:
- Research question (PICO) and inclusion criteria should be appropriate and predefined
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs)
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table etc.)
- Clinical and statistical heterogeneity should be assessed; clinical: enough similarities in patient characteristics, intervention and definition of outcome measure to allow pooling? For pooled data: assessment of statistical heterogeneity using appropriate statistical tests (e.g. Chi-square, I2)?
- An assessment of publication bias should include a combination of graphical aids (e.g., funnel plot, other available tests) and/or statistical tests (e.g., Egger regression test, Hedges-Olken). Note: If no test values or funnel plot included, score “no”. Score “yes” if mentions that publication bias could not be assessed because there were fewer than 10 included studies.
- Sources of support (including commercial co-authorship) should be reported in both the systematic review and the included studies. Note: To get a “yes,” source of funding or support must be indicated for the systematic review AND for each of the included studies.
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Hermida, 2008 |
“[…] patients were randomly assigned to 1 of 2 groups […]. One member of the research team, according to the order of recruitment, following an allocation table constructed by a computerized random-number generator, did assignment of subjects to treatment groups.” |
Unclear, not enough information. “The assignment of subjects to the respective treatment groups was blinded from the research team member conducting the clinic BP measurements and from those performing the statistical analysis of the data.” |
Likely, patients were not blinded |
Likely, no placebo was used |
Unlikely, trial was blinded endpoint |
Unclear. Trial was not registered. However, outcome data reported in the methods was also reported in the result section. |
Unlikely. Although the drop-out was not reported per arm, only 15 (6%) participants dropped out. |
Unlikely. Cannot be deduced. |
Notes:
- Randomisation: generation of allocation sequences must be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, per case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignment influences the process of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Exclusietabel
|
Auteur en jaartal |
Redenen van exclusie |
|
Review filter |
|
|
Stranges, 2015 |
Narratieve review |
|
Liu, 2015 |
Data niet te herleiden uit individuele trials |
|
Guo, 2015 |
Data niet te herleiden uit individuele trials |
|
Verdecchia, 2010 |
Narratieve review |
|
Marrs, 2010 |
Reeds alle artikelen geïncludeerd door Guo, 2015 |
|
Zaslavskaya, 2004 |
Narratieve review |
|
Rodgers, 1998 |
Narratieve review |
|
RCT filter |
|
|
Tasic, 2016 |
Pilotstudie met 12 patiënten |
|
Hu, 2016 |
Patiënten met meer dan 1 medicament werden geëxcludeerd |
|
Hermida, 2016 |
Narratieve review |
|
Gottwald-Hostalek, 2016 |
Patiënten op monotherapie met een verhoogde bloeddruk kwamen in aanmerking |
|
Smolensky, 2015 |
Narratieve review |
|
Rakugi, 2015 |
Patiënten die max twee antihypertensiva gebruikte waren geïncludeerd |
|
Okamura, 2015 |
Observationele studie; niet gerandomiseerd |
|
Imbalzano, 2015 |
Vergelijking tussen 1 type antihypertensiva versus een ander type |
|
Vaclavik, 2014 |
Spironolacton; reeds geïncludeerd in Wang, 2016 |
|
Sakima, 2014 |
Alle patiënten kregen de interventie; niet alleen patiënten met TRH geïncludeerd |
|
Tanaka, 2013 |
Voldoet niet aan de patiëntenpopulatie |
|
Riksen, 2013 |
Narratieve review |
|
Oxlund, 2013 |
Spironolacton; reeds geïncludeerd in Wang, 2016 |
|
Karns, 2013 |
Vergelijking tussen 1 type antihypertensiva versus een ander type |
|
Hermida, 2013 |
Niet gerandomiseerd (chronotherapie) |
|
Farah, 2013 |
Patiënten behandeld met maximaal twee medicamenten |
|
Ding, 2013 |
Vergeleken combinatie tabletten versus placebo (geen medicamenten) |
|
Derosa, 2013 |
Vergeleken combinatie tabletten versus placebo (geen medicamenten); voldoet niet aan de patiëntenpopulatie |
|
Derosa, 2013 |
Patiënten met stage I essentiële hypertensie |
|
Volpe, 2012 |
Narratieve review |
|
Toh, 2012 |
Patiënten op 1 medicament werden geïncludeerd; patiënten met ongecontroleerde hypertensie werd geëxcludeerd |
|
Park, 2012 |
Niet alleen patiënten met therapieresistente hypertensie geïncludeerd |
|
Hosoya, 2012 |
Alle patiënten ontvingen de interventie |
|
Vaclavik, 2011 |
Trial reeds geïncludeerd in Wang, 2016 |
|
Makita, 2009 |
Voldoet niet aan de patiëntenpopulatie |
|
Neldam, 2008 |
Niet alleen patiënten met therapieresistente hypertensie geïncludeerd |
|
Black, 2007 |
Interventie: entholine receptor antagonist |
|
De Alvaro, 2006 |
Alle patiënten ontvingen de interventie |
|
Holzgreve, 2005 |
Patiënten die twee medicamenten kregen werden geïncludeerd; interventie betrof niet een combinatietablet |
Beoordelingsdatum en geldigheid
Publicatiedatum : 04-12-2017
Beoordeeld op geldigheid : 04-12-2017
Voor het beoordelen van de actualiteit van deze richtlijn is de werkgroep in stand gehouden. Het bestuur van de Nederlandse Internisten Vereniging bepaalt uiterlijk na de autorisatie van de multidisciplinaire richtlijn Cardiovasculair risicomanagement (CVRM) of de modules van deze richtlijn nog actueel zijn. Aangezien deze richtlijn bij de huidige CVRM-richtlijn uit 2011 aansluit, zal bij een herziene CVRM-richtlijn moeten worden gecontroleerd of de hier beschreven richtlijn nog steeds aansluit bij de nieuwe CVRM-richtlijn. De geldigheid van deze richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De Nederlandse Internisten Vereniging is regiehouder van deze richtlijn en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
Algemene gegevens
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van Medisch Specialisten (www.kennisinstituut.nl) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS).
Doel en doelgroep
Doel
Het doel van dit project is om een richtlijn hypertensie te ontwikkelen ter bevordering van een optimale diagnostiek en behandeling van patiënten met hypertensie in de tweede en derde lijn. Deze richtlijn zal zoveel mogelijk aansluiten bij de CVRM-richtlijn en de ESH/ESC-richtlijn voor hypertensie.
Doelgroep
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen van internisten, cardiologen en neurologen die betrokken zijn bij de zorg voor patiënten met hypertensie in de tweede en derde lijn. Daarnaast is deze richtlijn bedoeld om zorgverleners die anderzijds betrokken zijn bij deze patiënten te informeren, waaronder huisartsen, verpleegkundig specialisten en apothekers.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2015 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met hypertensie (zie hiervoor de samenstelling van de werkgroep). De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname.
- Dr. R.M. (Renske) van den Berg-Vos, neuroloog, OLVG (locatie West), Amsterdam, NVN
- Dr. J. (Jaap) Deinum, internist-vasculaire geneeskunde, Radboud UMC, Nijmegen, NIV, (voorzitter)
- Dr. M.N. (Michiel) Kerstens, internist-endocrinoloog, UMCG, Groningen, NIV
- Dr. R.A. (Roderik) Kraaijenhagen, cardioloog, directeur NIPED, directeur CardioVitaal hartrevalidatie, NIPED, CardioVitaal hartrevalidatie, MC Arterium, Amsterdam, NVvC
- Prof. A.A. (Bram) Kroon, internist-vasculaire geneeskunde, Maastricht UMC, NIV
- Dr. A.T.A. (Ronne) Mairuhu, internist-vasculaire geneeskunde, HagaZiekenhuis, Den Haag, NIV
- Drs. A. (Anne-Margreet) Strijbis, relatiemanager zorg, De Hart&Vaatgroep, Den Haag (De Hart&Vaatgroep)
- Dr. L. (Liffert) Vogt, internist-nefroloog, AMC, Amsterdam, NIV
Meelezers:
- A. (Anna) van Ittersum, verpleegkundig specialist, V&VN
- G. Kuipers, ervaringsdeskundige, Nierpatiënten Vereniging Nederland
- Drs. K. (Karen) Prantl, coördinator Kwaliteit & Onderzoek, Nierpatiënten Vereniging Nederland
- Drs. B.A.D. (Brigit) van Soest-Segers, apotheker, KNMP
- Dr. M. (Mark) van der Wel, huisarts, NHG
Met ondersteuning van:
- N.F. (Natalia) Bullock, projectsecretaresse, Kennisinstituut van de Federatie Medisch Specialisten
- S. (Samara) de Jong-Jaber, MSc, beleidsadviseur, Nederlandse Internisten Vereniging
- G.M. (Maike) van Leeuwen, beleid- en projectondersteuner, Nederlandse Internisten Vereniging
- Dr. M.A. (Margreet) Pols, adjunct-directeur, Kennisinstituut van de Federatie Medisch Specialisten
- Dr. B.H. (Bernardine) Stegeman, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- M.E. (Monique) Wessels, MSc, literatuurspecialist, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De KNMG-Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling” is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of ze in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatie management, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Deinum |
Internist-vasculair geneeskunde |
Voorzitter Nederlandse Hypertensie Vereniging (onbetaald) |
- |
Geen actie |
|
Kroon |
Internist-vasculair geneeskunde |
Penningmeester Nederlandse Hypertensie Vereniging (onbetaald) |
Vascular Dynamics, producent MobiusHD device ten behoeve van barostenting, CALM-FIM study |
Exclusie als opsteller van aanbevelingen over baroreflexactivitietherapie |
|
Mairuhu |
Internist-vasculair geneeskunde |
- |
- |
Geen actie |
|
Kerstens |
Internist-endocrinoloog |
Bestuurslid BijnierNET (onbetaald) |
- |
Geen actie |
|
Vogt |
Internist-nefroloog |
- |
Unilever: investigator-initiated onderzoek |
Geen actie (valt buiten de afbakening van de richtlijn) |
|
Kraaijenhagen |
Cardioloog |
-Medische directeur NIPED |
- |
Geen actie |
|
Van den Berg-Vos |
Neuroloog |
- Lid bestuur Nederlandse Vereniging voor Neurologie (NVN), portefeuillehouder Kwaliteit (deels betaald) - Lid Richtlijncommissie herziening Richtlijn Herseninfarct en hersenbloeding (onbetaald) -Plaatsvervangend opleider afdeling neurologie St. Lucas Andreas Ziekenhuis, Amsterdam (onbetaald) |
- |
Geen actie |
|
Strijbis |
Relatiemanager zorg, De Hart&Vaatgroep |
Voorheen secretaris Platform Vitale Vaten. De Hart&Vaatgroep |
- |
Geen actie |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door afgevaardigde patiëntenvereniging in de werkgroep. De conceptrichtlijn is tevens voor commentaar voorgelegd aan De Hart&Vaatgroep en aan de Nierpatiënten Vereniging Nederland.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn (module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden bij de aanverwante producten. Tijdens de commentaarfase zijn de aanbevelingen nagelopen. Er zijn geen aanbevelingen in deze richtlijn die zich lenen voor ontwikkeling van een indicator. De werkgroep verwijst daarom naar de reeds beschikbare indicatoren op het gebied van CVRM.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van Medisch Specialisten.
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. Tevens zijn er knelpunten aangedragen door De Hart&Vaatgroep, HAG, NHG, IGZ, NVHVV, Nederlandse Hypertensie Vereniging, VIG (voorheen Nefarma), Nierpatiënten Vereniging Nederland, ZN en V&VN via invitational conference. Een verslag hiervan is opgenomen onder aanverwante producten.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de voorzitter en de adviseur conceptuitgangsvragen opgesteld. Deze zijn met de werkgroep besproken waarna de werkgroep de definitieve uitgangsvragen heeft vastgesteld. Vervolgens inventariseerde de werkgroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als kritiek, belangrijk (maar niet kritiek) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de kritieke uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Er werd voor de afzonderlijke uitgangsvragen aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie voor de oriënterende zoekactie en patiëntenperspectief zijn opgenomen onder aanverwante producten.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR – voor systematische reviews; Cochrane – voor gerandomiseerd gecontroleerd onderzoek; ROBINS-I – voor observationeel onderzoek; QUADAS II – voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidence-tabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A) Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor Grading Recommendations Assessment, Development and Evaluation (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
B) Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008), en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje Overwegingen.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje Overwegingen.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de bijlage Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348.
Medisch Specialistische Richtlijnen 2.0. Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site.html. 2012.
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van Medisch Specialisten.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html. 2013.
Schünemann HJ, Oxman AD, Brozek J, et al. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008;336(7654). doi: 10.1136/bmj.a139. PubMed PMID: 18483053.
Zoekverantwoording
|
Database |
Zoektermen |
Totaal |
|
Medline (Embase) |
1 (((resistant or refractory) adj6 (hypertensi* or 'high blood pressure' or hbp)) or RHTN).ti,ab. (4901) 2 exp Hypertension/ or (hypertensi* or 'high blood pressure').ti. (275222) 3 Drug Resistance/ or (refractory or resistan*).ti,ab. or treatment failure/ or "treatment failure".ti,ab. or "inadequately adj3 controlled".ti,ab. (918192) 4 2 and 3 (21401) 5 "Blood Pressure"/de or "Hypertension"/dt or "Antihypertensive Agents"/tu or "Antihypertensive Agents"/ad (156668) 6 ("true* resistan* hypertensi*" or (uncontrolled adj3 (hypertension or "blood pressure*"))).ti,ab. (2543) 7 1 or 4 or 6 (24485) 8 limit 7 to yr="2000-Current" (14237) 16 fixed dose combination*.ti,ab. (2325) 19 Drug Chronotherapy/ or Chronotherapy/ or "Drug Combinations"/ (65630) 20 (chronotherap* or "treatment schedul*" or night* or nocturnal or combination*).ti,ab. (829071) 21 16 or 19 or 20 (876934) 22 8 and 21 (1375) 23 limit 22 to english language (1197) 24 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (293113) 25 23 and 24 (40) 26 (randomized controlled trial/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (randomized controlled trial or multicenter study).pt. or random*.ti,ab. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1154684) 27 23 and 26 (358) 28 25 or 27 (381) 29 28 not 25 (341) >330 uniek |
402 RCTs
SRs hier 4, vorige search 50. |
|
Embase (Elsevier) |
'resistant hypertension'/exp OR ((resistant OR refractory) NEAR/6 (hypertensi* OR 'high blood pressure' OR hbp)):ab,ti OR ('hypertension'/exp/mj AND [<1966-2013]/py AND (resistant:ab,ti OR refractory:ab,ti)) OR 'true* resistan* hypertensi*':ab,ti OR (uncontrolled NEAR/3 (hypertension OR 'blood pressure*')):ab,ti AND [english]/lim AND [embase]/lim AND ('fixed dose combination*':ab,ti OR 'chronotherapy'/exp/mj OR 'drug combination'/exp/mj OR chronotherap*:ab,ti OR 'treatment schedul*':ab,ti OR night*:ab,ti OR nocturnal:ab,ti OR combination*:ab,ti) AND ('randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it AND [2000-2016]/py >220 (72 uniek) |
- Achtergrond en definities
- Toepassen
- Onderzoek
- Overige