Voorbehandeling myomectomie
Uitgangsvraag
LET OP Notificatie
Datum notificatie: 23-7-2021
Hierbij wijzen wij u op berichtgeving van het CBG over het gebruik van Esmya (ulipristalacetaat 5 mg) bij symptomatische myomen.
Esmya is weer beschikbaar en kan als intermitterende behandeling voorgeschreven worden onder bepaalde voorwaarden en aandachtspunten:
- Voorschrijven van ulipristalacetaat 5 mg als voorbehandeling voorafgaand aan chirurgie is niet meer toegestaan.
- Er zijn gevallen van ernstige leverschade (inclusief gevallen leidend tot een levertransplantatie) gemeld bij het gebruik van ulipristalacetaat 5 mg voor de behandeling van matige tot ernstige symptomen van uterusmyomen.
- Het gebruik van ulipristalacetaat 5 mg dient nu alleen overwogen te worden voor de intermitterende behandeling van matige tot ernstige symptomen van uterusmyomen bij premenopauzale vrouwen en bij wie embolisatie en/of chirurgische behandelingsmogelijkheden niet geschikt zijn of gefaald hebben.
- Artsen moeten de risico's en voordelen van beschikbare alternatieve behandelingsmethoden bespreken met hun patiënten zodat deze een goed geïnformeerde beslissing kunnen nemen.
- Alle risico's van ulipristalacetaat 5 mg moeten aan patiënten uitgelegd worden, vooral het risico op leverschade, dat in zeldzame gevallen kan leiden tot een levertransplantatie.
- Patiënten moeten geïnformeerd worden over de mogelijke tekenen en symptomen van leverschade (zoals misselijkheid, braken, epigastrische pijn rechts, anorexia, asthenie, geelzucht). Als deze symptomen optreden, moeten patiënten onmiddellijk contact opnemen met hun arts en moet de behandeling worden gestopt.
- Bij symptomen van leverschade moet de patiënt onmiddellijk worden onderzocht en moeten leverfunctietesten worden uitgevoerd
- Patiënten die ulipristalacetaat 5 mg gaan gebruiken moeten de daarvoor ontwikkelde waarschuwingskaart meekrijgen (pcard_ESMYA_5mg_tbl_NL_K28032-12.indd (geneesmiddeleninformatiebank.nl)).
- Leverfunctietesten (ASAT, ALAT en totaal bilirubine) moeten worden uitgevoerd: 1. Voorafgaand aan start medicatie; 2. vier weken na start medicatie; 3. Acht weken na start medicatie; 4. Twaalf weken na start medicatie; 5. Twee tot vier weken na beëindigen kuur. Dit regime moet elke kuur herhaald worden.
- Meer informatie zie: Gebruik Esmya weer beperkt toegestaan | Nieuwsbericht | College ter Beoordeling van Geneesmiddelen (cbg-meb.nl))
Tevens wijzen wij u op de MYOMEX-2 studie: een gerandomiseerde multicenter trial uitgevoerd in het Nederlandse consortium, waar geschikte patiënten ulipristal in studieverband toegediend krijgen. Er wordt gerandomiseerd tussen intermitterende behandeling met ulipristal en chirurgie (keuze uit embolisatie, myoomenucleatie of hysterectomie). Gedurende het EMA onderzoek heeft de studie niet geïncludeerd. Vanaf 1 juli 2021 is inclusie weer toegestaan. Voor informatie over de studie en deelnemende centra zie: Zorgevaluatie Nederland
Wat is de plaats van GnRH analogen of Ulipristal als voorbehandeling bij patiënten met hevig menstrueel bloedverlies (HMB) die een myomectomie ondergaan?
Aanbeveling
Overweeg een voorbehandeling met GnRH analogen danwel Ulipristal bij vrouwen met hevig menstrueel bloedverlies en uterus myomatosus voorafgaande aan een laparoscopische/ laparotomische myomectomie.
Bespreek met vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus die een laparoscopische/laparotomische myomectomie ondergaan de mogelijke voor- en nadelen van voorbehandeling met GnRH analogen of Ulipristal.
Overwegingen
De onderstaande overwegingen en aanbevelingen gelden voor het overgrote deel van de populatie waarop de uitgangsvraag betrekking heeft.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Op basis van de cruciale uitkomstmaten reductie in (hevig) menstrueel bloedverlies en patiënttevredenheid is er geen voorkeur voor GnRH analogen danwel Ulipristal aan te geven als voorbehandeling bij vrouwen met hevig menstrueel bloedverlies bij laparoscopische / laparotomische myomectomie. Ulipristal vermindert wel het hevig menstrueel bloedverlies ten opzichte van een placebobehandeling. Er zijn geen data beschikbaar van GnRH analogen vergeleken met een placebo of geen voorbehandeling, maar in vergelijking met Ulipristal doen GnRH analogen het even goed in de reductie van het bloedverlies. Dat GnRH analogen het (hevig) menstrueel bloedverlies zouden reduceren is ook geheel in overeenstemming met wat van GnRH analogen verwacht mag worden gezien de werking ervan en is ook in overeenstemming met de ervaring in de praktijk. Van patiënttevredenheid na voorbehandeling met Ulipristal of GnRH analogen zijn geen geschikte studies gevonden.
De belangrijke uitkomstmaten geven ook niet duidelijk richting voor een keuze voor de ene of de andere behandeling. Het myoomvolume neemt af bij beide behandelingen zonder superioriteit van één van beide, alleen het uterusvolume neemt mogelijk iets meer af bij voorbehandeling met GnRH analogen vergeleken met Ulipristal. Het peroperatief bloedverlies neemt af bij beide behandelingen ten opzichte van een placebo, maar ze zijn niet onderling vergeleken. De operatieduur neemt iets af met Ulipristal en van GnRH analogen is het onduidelijk.
De overall bewijskracht is gegradeerd als ‘zeer laag’ doordat er geen studies zijn gevonden waarin de cruciale uitkomstmaat patiënttevredenheid werd bestudeerd. Kwalitatief hoogwaardige RCT’s zijn nodig voor het evidence based beantwoorden van de uitgangsvraag
De behandeling met zowel Ulipristal als GnRH kent nadelen en bijwerkingen, die wat verschillend van aard zijn. Het gebruik van Ulipristal is recentelijk ingeperkt door de EMA (European Medicines Agency) naar aanleiding van enkele patiënten met ernstige leverschade na gebruik van Ulipristal. Een oorzakelijk verband is niet bewezen, maar het houdt onder andere wel in dat leverfuncties vooraf en tijdens het gebruik van Ulipristal getest moeten worden en dat patiënten er over voorgelicht moeten worden. Bij afwijkende leverfuncties kan er niet gestart danwel doorgegaan worden met de behandeling. Voorbehandeling met GnRH analogen kan ernstige overgangsklachten geven, maar ook hoofdpijn, duizeligheid et cetera Ook kan het vaginaal bloedverlies tijdelijk erger worden door een flare up van gonadotrofinen in het begin van het gebruik. Van voorbehandeling met zowel GnRH als Ulipristal wordt door experts wel aangegeven dat het chirurgisch verwijderen van een myoom moeilijker kan worden, doordat de consistentie ervan verandert, waardoor het moeilijker is het kapsel van het myoom te onderscheiden.
Resultaten uit de Nederlandse Myomex-1 studie zijn onlangs gepubliceerd (de Milliano, 2019). Dit is een dubbelblinde, non-inferiority trial waarin Ulipristal wordt vergeleken met GnRH als voorbehandeling van myomen bij laparoscopische myomectomie. De resultaten laten zien dat non-inferiority voor Ulipristal niet kon worden aangetoond voor wat betreft peroperatief bloedverlies, operatieduur en -gemak. Dit betekent dat niet uitgesloten kan worden dat Ulipristal het minder goed doet dan GnRH analogen als voorbehandeling. Sterker nog, uit de resultaten blijkt dat het peroperatief bloedverlies significant meer is bij voorbehandeling met Ulipristal in vergelijking met GnRH (525 ml versus 280 ml; P=0,011), de hechttijd van het eerste myoom langer is (40 versus 20 minuten; P=0,003) en dat er met Ulipristal een kleinere reductie in myoomvolume wordt bereikt dan met GnRH (-7,2% versus -38,4%; P=0.001). Ook werd gerapporteerd dat de myomectomieën na voorbehandeling met Ulipristal technisch moelijker werden gevonden door de operateurs dan na GnRH. Er zijn echter een aantal beperkingen aan de studie die voorzichtigheid bij de interpretatie van deze resultaten vereisen, namelijk dat het beoogde aantal inclusies niet gehaald werd en dat de grootte van de myomen bij baseline niet gelijk was in beide groepen.
Waarden en voorkeuren van patiënten (en eventueel hun verzorgers)
Het lijkt ten aanzien van het hevig menstrueel bloedverlies, het myoomvolume en het peroperatief bloedverlies wel zinvol een voorbehandeling te geven met GnRH danwel Ulipristal, echter er zijn bij beide behandelingen nadelen en bijwerkingen, waarover de patiënt geïnformeerd moet worden voorafgaand aan het gebruik.
Kosten (middelenbeslag)
De kosten van 3 maanden voorbehandelen met Ulipristal zijn ongeveer een derde hoger dan die van een 3 maanden voorbehandeling met GnRH analogen.
(https://www.farmacotherapeutischkompas.nl/)
Aanvaardbaarheid voor de overige relevante stakeholders
Er zijn geen zaken bekend.
Haalbaarheid en implementatie
Er zijn geen zaken bekend.
Rationale/ balans tussen de argumenten voor en tegen de interventie
Voorbehandeling met Ulipristal of GnRH analogen lijkt mogelijk een verbetering op te leveren in de reductie van het hevig menstrueel bloedverlies, het myoomvolume, het peroperatief bloedverlies en de operatieduur. Er is echter geen overtuigend bewijs welke voorbehandeling hierin effectiever is. Het is belangrijk dat de mogelijke voor- en nadelen van voorbehandeling met Ulipristal, onder andere met betrekking tot mogelijke leverschade, en GnRH analogen, onder andere met betrekking tot overgangsklachten en tijdelijke toename van het bloedverlies, worden besproken met de patiënte.
Onderbouwing
Achtergrond
Op dit moment worden vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus die een myomectomie ondergaan met GnRH analogen of met Ulipristal voorbehandeld. In deze module wordt gekeken naar de bewijskracht voor de effectiviteit van deze behandelingen en of het mogelijk is om een voorkeur aan te gegeven voor een van beide methodes.
Conclusies / Summary of Findings
Deelvraag 1 - Ulipristal versus GnRH analogen
|
Redelijk GRADE |
Voorbehandeling met Ulipristal (5 of 10 mg dagelijks) voor laparoscopische/ laparotomische myomectomie is waarschijnlijk niet effectiever dan voorbehandeling met GnRH analogen in het reduceren van (hevig) menstrueel bloedverlies bij vrouwen met een uterus myomatosus.
Bronnen: (Donnez, 2012b) |
|
Redelijk GRADE |
Voorbehandeling met Ulipristal (5 of 10 mg dagelijks) voor laparoscopische/ laparotomische myomectomie is waarschijnlijk niet effectiever dan voorbehandeling met GnRH analogen in het reduceren van het myoomvolume bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus.
Voorbehandeling met GnRH analogen voor laparoscopische/ laparotomische myomectomie is mogelijk effectiever dan voorbehandeling met Ulipristal (5 of 10 mg dagelijks) in het reduceren van het uterusvolume bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus.
Bronnen: (Donnez, 2012b) |
|
- GRADE |
De bewijskracht voor de uitkomstmaten patiënttevredenheid, bloedverlies peroperatief en operatieduur kon niet worden beoordeeld vanwege van het ontbreken van studies waarin is gekeken naar deze uitkomsten. |
Deelvraag 2 - Ulipristal versus geen behandeling of placebo
|
Redelijk GRADE |
Voorbehandeling met Ulipristal (5 of 10 mg dagelijks) voor laparoscopische/ laparotomische myomectomie is waarschijnlijk effectiever dan een placebo in het reduceren van (hevig) menstrueel bloedverlies bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus.
Bronnen: (Donnez, 2012b) |
|
Redelijk GRADE |
Voorbehandeling met Ulipristal (5 of 10 mg dagelijks) voor laparoscopische/ laparotomische myomectomie is waarschijnlijk effectiever dan een placebo in het reduceren van het myoomvolume bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus.
Bronnen: (Donnez, 2012a; Ferrero, 2016; Nieman, 2011) |
|
Zeer laag GRADE |
Voorbehandeling met Ulipristal (5 mg dagelijks) leidt mogelijk tot minder peroperatief bloedverlies tijdens laparoscopische/ laparotomische myomectomie bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus, vergeleken met een placebo.
Bronnen: (Ferrero, 2016) |
|
Zeer laag GRADE |
Voorbehandeling met Ulipristal (5 mg dagelijks) leidt mogelijk tot een kortere operatieduur van laparoscopische/ laparotomische myomectomie bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus, vergeleken met een placebo.
Bronnen: (Ferrero, 2016) |
|
- GRADE |
De bewijskracht voor de uitkomstmaat patiënttevredenheid kon niet worden beoordeeld vanwege van het ontbreken van studies waarin is gekeken naar deze uitkomst. |
Deelvraag 3 - GnRH analogen versus geen behandeling of placebo
|
Laag GRADE |
Voorbehandeling met GnRH analogen leidt mogelijk tot minder peroperatief bloedverlies tijdens laparoscopische/ laparotomische myomectomie bij vrouwen met een uterus myomatosus, vergeleken met geen behandeling of een placebo.
Bronnen: (Bustos López, 1995; Campo, 1999; Cetin, 1995; De Falco, 2009; Fedele, 1990; Friedman, 1989; Golan, 1993; Hudecek, 2012; Shaw, 1989; Zullo, 1998) |
|
Laag GRADE |
Het is onduidelijk of voorbehandeling met GnRH analogen van invloed is op de operatieduur van laparoscopische/ laparotomische myomectomie bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus, vergeleken met geen behandeling of een placebo.
Bronnen: (Campo, 1999; Cetin, 1995; De Falco, 2009; Fedele, 1990; Friedman, 1989; Golan, 1993; Hudecek, 2012; Zullo, 1998) |
|
- GRADE |
De bewijskracht voor de uitkomstmaten reductie in (H)MB, patiënttevredenheid en afname myoomvolume kon niet worden beoordeeld vanwege van het ontbreken van studies waarin is gekeken naar deze uitkomsten. |
Samenvatting literatuur
Beschrijving studies
Lethaby (2017) verrichtte een systematische review en meta-analyse naar de effectiviteit en veiligheid van voorbehandeling bij vrouwen met myomen. De search werd verricht in juni 2017 in de databases Medline, Embase, CENTRAL, PsycINFO, CINAHL en het Cochrane Gynaecology and Fertility Group specialised register. Daarnaast werden ook trial registers (ClinicalTrials.com; WHO ICTRP), thesissen, dissertaties, grijze literatuur en referentielijsten van alle geïdentificeerde studies gecontroleerd en werden farmaceutische bedrijven benaderd met de vraag of er nog aanvullende onderzoeken bestonden. Het Cochrane review includeerde in totaal 38 RCTs, waarvan 12 zijn opgenomen in de literatuuranalyse van deze module (Bustos López, 1995; Campo, 1999; Cetin, 1995; De Falco, 2009; Donnez, 2012a; Donnez, 2012b; Fedele, 1990; Friedman, 1989; Golan, 1993; Hudecek, 2012; Shaw, 1989; Zullo, 1998).
In totaal beschreven de 12 geïncludeerde studies 1137 vrouwen die een voorbehandeling met Ulipristal of GnRH analogen kregen voor een uterus myomatosus. In vijf studies werden louter myomectomieën uitgevoerd (Bustos López, 1995; Campo, 1999; Friedman, 1989; Hudecek, 2012; Zullo, 1998), in vier studies werden hysterectomieën of myomectomieën uitgevoerd (Donnez, 2012a; Donnez, 2012b; Golan, 1993; Shaw, 1989) en in drie studies was het niet bekend welke chirurgische techniek werd toegepast voor het verwijderen van de myomen (Cetin, 1995; De Falco, 1990; Fedele, 1990).
Eén studie vergeleek voorbehandeling met Ulipristal (5 of 10 mg, 1 dd 1 tablet) met GnRH analogen (leuprolide acetaat 3,75 mg, intramusculaire injectie één keer per maand) (Donnez, 2012b). Daarnaast vergeleek één studie voorbehandeling met Ulipristal (5 of 10 mg, 1 dd 1 tablet) met een placebo voorbehandeling (Donnez, 2012a). Tien studies vergeleken voorbehandeling met GnRH analogen met geen behandeling (Bustos López, 1995; Campo, 1999; Cetin, 1995; De Falco, 2009; Fedele, 1990; Golan, 1993; Hudecek, 2012; Shaw, 1989; Zullo, 1998) of een placebo (Friedman, 1989). De duur van de behandeling in alle studies varieerde van 2 tot 4 maanden.
Ferrero (2016) verrichtte een retrospectieve cohortstudie onder premenopauzale vrouwen die laparoscopische myomectomie ondergingen voor hevig menstrueel bloedverlies dat werd veroorzaakt door grote myomen. De geïncludeerde vrouwen hadden minimaal één myoom van ≥ 10 cm (Fédération Internationale de Gynécologie et d'Obstétrique) (FIGO) type 3,4, of 5). In totaal werden 77 vrouwen geïncludeerd, waarvan 34 vrouwen (gemiddelde leeftijd 38,1 jaar (SD 4,6)) voorbehandeling met Ulipristal acetaat ondergingen (5 mg/ dag, oraal) en 43 vrouwen (gemiddelde leeftijd 37,5 jaar (SD 4,0)) geen voorbehandeling kregen en direct geopereerd werden. Vrouwen in de controlegroep hadden ofwel behandeling met Ulipristal geweigerd (n=28) of konden geen behandeling met Ulipristal aangeboden krijgen omdat dit nog niet op de markt was in Italië op dat moment (n=15). Baseline karakteristieken van de interventie- en controlegroep waren niet verschillend van elkaar voor de PBAC score, myoomgrootte en het totale myoomvolume. Voorbehandeling met Ulipristal duurde 3 maanden, de gemiddelde duur van follow-up werd niet gerapporteerd.
Nieman (2011) verrichtte een gerandomiseerde dubbelblind placebo-gecontroleerde klinische fase 2b studie onder premenopauzale vrouwen met symptomatische myomen. In totaal werden 42 vrouwen geïncludeerd, waarvan 14 vrouwen (gemiddelde leeftijd 42,5 jaar (SD 4,3)) een voorbehandeling met CDB10 (Ulipristal acetaat, 10 mg/dag, oraal) kregen, 14 vrouwen (gemiddelde leeftijd 41,3 jaar (SD 5,0)) een voorbehandeling met CDB20 (Ulipristal acetaat, 20 mg/dag, oraal) kregen en 14 vrouwen (gemiddelde leeftijd 43,1 jaar (SD 6,0)) een placebo kregen. In de interventiegroep waren 2 deelnemers lost to follow-up (n=1 CDB10; n=1 CDB20), in de controlegroep waren 2 deelnemers lost to follow-up en 2 deelnemers hadden geen complete uitkomstdata aan het einde van de studie. Voorbehandeling duurde in alle groepen 12 weken, daarna konden vrouwen kiezen of ze een hysterectomie of myomectomie wilden ondergaan, of dat ze nogmaals 3 maanden CDB behandeling met dezelfde dosis wilde ontvangen. Hieronder worden alleen de resultaten gerapporteerd van de eerste 12 weken voorbehandeling.
Resultaten
Deelvraag 1 - Ulipristal versus GnRH analogen
1.1. Reductie in (H)MB
Eén studie in het Cochrane review van Lethaby (2017) beschreef de uitkomstmaat reductie in (H)MB bij vrouwen die een voorbehandeling kregen met Ulipristal vergeleken met GnRH analogen (Donnez, 2012b). Donnez (2012b) rapporteerde het aantal vrouwen bij wie het bloeden preoperatief was gereduceerd met < 75 punten op de PBAC schaal. Er werd preoperatief geen verschil gezien in de reductie in (H)MB tussen vrouwen die met Ulipristal (5 mg of 10 mg) of met GnRH analogen waren voorbehandeld (5 mg: OR 1,42 (95%BI 0,60 tot 3,36); 10 mg: OR 2,57 (95%BI 0,95 tot 6,99)) (Figuur 1).
Figuur 1. Forest plot van reductie in (H)MB) bij vrouwen die een voorbehandeling kregen met Ulipristal vergeleken met GnRH analogen
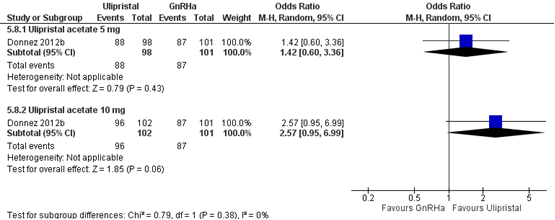
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
1.2. Patiënttevredenheid
Er werden geen studies geïncludeerd die de uitkomst patiënttevredenheid beschreven voor de vergelijking tussen Ulipristal en GnRH analogen.
1.3. Afname myoomvolume
Eén studie in het Cochrane review van Lethaby (2017) beschreef de uitkomstmaat ‘afname myoomvolume’ (Donnez, 2012b). Donnez (2012b) rapporteerde in een per protocol analyse de verandering in het myoomvolume van de drie grootste myomen ten opzichte van baseline. Daarnaast beschreef Donnez (2012b) in deze analyse ook de uitkomstmaat ‘afname van het uterusvolume’.
Na 13 weken voorbehandeling werd bij vrouwen een afname van het myoomvolume van 36% (inter quartile range (IQR) -58 tot -11%) beschreven bij voorbehandeling met Ulipristal 5 mg, een afname van 42% (IQR -69 tot -41%) beschreven bij voorbehandeling met Ulipristal 10 mg en een afname van 53% (IQR -69 tot -36%) beschreven bij voorbehandeling met GnRH analogen. Er werd geen significant verschil in de afname van het myoomvolume gerapporteerd tussenvoorbehandeling met Ulipristal 5 mg versus GnRH analogen (1,23 (95%BI 0,99 tot 1,52)) of Ulipristal 10 mg versus GnRH analogen (1,12 (95%BI 0,91 tot 1,38)). Het was onduidelijk of hier een relatief risico (RR) of odds ratio (OR) werd gerapporteerd door de auteurs).
Na 13 weken voorbehandeling werd een afname van het uterusvolume van 20% (IQR -40 tot -3%) beschreven na voorbehandeling met Ulipristal 5 mg, een afname van 22% (IQR -45 tot 0%) na voorbehandeling met Ulipristal 10 mg en een afname van 47% (IQR -57 tot -35%) na voorbehandeling met GnRH analogen. Er werd een significant grotere afname gerapporteerd bij voorbehandeling met GnRH analogen vergeleken met Ulipristal 5 mg (1,48 (95%BI 1,25 tot 1,74)) of Ulipristal 10 mg (1,41 (95%BI 1,19 tot 1,66)). Het was onduidelijk of hier een relatief risico (RR) of odds ratio (OR) werd gerapporteerd door de auteurs).
1.4. Bloedverlies peroperatief
Er werden geen studies geïncludeerd die de uitkomst bloedverlies peroperatief beschreven voor de vergelijking tussen Ulipristal en GnRH analogen.
1.5. Operatieduur
Er werden geen studies geïncludeerd die de uitkomst operatieduur beschreven voor de vergelijking tussen Ulipristal en GnRH analogen.
Deelvraag 2 - Ulipristal versus geen behandeling of placebo
2.1. Reductie in (H)MB
Eén studie in het Cochrane review van Lethaby (2017) beschreef de uitkomstmaat reductie in (H)MB bij vrouwen die een voorbehandeling kregen met Ulipristal vergeleken met placebo (Donnez, 2012a). Donnez (2012a) rapporteerde het aantal vrouwen bij wie het bloeden preoperatief was gereduceerd met < 75 punten op de PBAC schaal.
Bij vrouwen die een voorbehandeling met Ulipristal 5 of 10 mg kregen werd een sterkere reductie in (H)MB gezien, vergeleken met vrouwen die een placebo kregen (5 mg: OR 41,41 (95%BI 15,26 tot 112,38); 10 mg: OR 78,83 (95%BI 24,02 tot 258,74)) (Figuur 2).
Figuur 2. Forest plot van de reductie in (H)MB bij vrouwen die een voorbehandeling
kregen met Ulipristal vergeleken met placebo
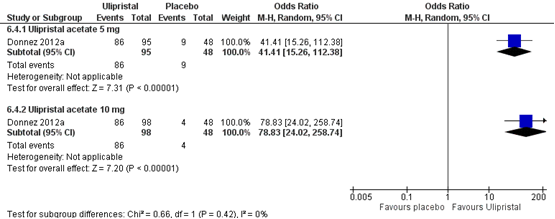
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
2.2. Patiënttevredenheid
Er werden geen studies geïncludeerd die de uitkomst patiënttevredenheid beschreven voor de vergelijking tussen Ulipristal en geen behandeling of placebo.
2.3. Afname myoomvolume
Drie studies beschreven de uitkomstmaat afname myoomvolume (Donnez, 2012a uit Lethaby, 2017; Ferrero, 2016; Nieman, 2011). Alle studies beschreven de afname in het totale myoomvolume, daarnaast beschreef Ferrero (2016) ook de afname in het volume van het grootste myoom. De data konden niet worden gepoold omdat het mediane verschil werd gerapporteerd per studie, de resultaten worden daarom beschrijvend gepresenteerd (Tabel 3).
Alle studies rapporteerden een significante afname van het totale myoomvolume bij voorbehandeling met Ulipristal vergeleken met een placebo (Donnez, 2012a; Ferrero, 2016; Nieman, 2011). Daarnaast rapporteerde Ferrero (2016) ook een significante afname van het volume van het grootste myoom bij vrouwen die voorbehandeld werden met Ulipristal vergeleken met een placebo.
Tabel 1. Het verschil in de afname van het myoomvolume bij voorbehandeling met Ulipristal vergeleken met placebo
|
Auteur (jaartal) |
Ulipristal (dosering) |
Placebo |
Verschil |
|
Donnez (2012a) (intention to treat, sensitiviteits analyse) |
Ulipristal (5 mg): Mediaan -18,9% (IQR -38,6 tot 0%)
Ulipristal (10 mg): Mediaan -6.2% (IQR -35.4 to 0.0) |
Placebo: Mediaan 1,9% (IQR -17,9% tot 19,8%) |
Ulipristal 5 mg versus. placebo: -19,6% (95%BI -31,25 tot -6,5%); P=0.002
Ulipristal 10 mg versus. placebo: -14,2% (95%BI -25,9 tot -2,42%); P=0,010 |
|
Ferrero (2016) |
Ulipristal (5 mg): data niet getoond |
Placebo: data niet getoond |
“Voorbehandeling met Ulipristal leidde tot een lager volume van het grootste myoom (P=0,020) en een lager totaal myoomvolume (P=0,015) ten opzichte van de placebogroep” |
|
Nieman (2011) |
CDB10 (Ulipristal 10 mg): -17% (range niet gerapporteerd)
CDB20 (Ulipristal 20mg): -24% (range niet gerapporteerd) |
Placebo: 7% (range niet gerapporteerd) |
P=0.003 |
2.4. Bloedverlies peroperatief
Eén studie beschreef de uitkomstmaat peroperatief bloedverlies bij vrouwen die een voorbehandeling kregen met Ulipristal vergeleken met een placebo (Ferrero, 2016). De uitkomstmaat was gedefinieerd als de hoeveelheid bloed die was achtergebleven na afloop van de operatie in de zuigpot, berekend als het totale volume in de afzuigfles minus het irrigatievolume.
Vrouwen die voorbehandeld werden met Ulipristal (5 mg) hadden minder peroperatief bloedverlies (mediaan 470,0 mL (IQR 330,0)) vergeleken met vrouwen die een placebo kregen (mediaan 650,0 (IQR 420,0); (P=0,012).
2.5. Operatieduur
Eén studie beschreef de uitkomstmaat operatieduur bij vrouwen die een voorbehandeling kregen met Ulipristal vergeleken met een placebo (Ferrero, 2016). De uitkomstmaat was gedefinieerd als de tijd vanaf de eerste incisie tot het sluiten van de laatste hechting.
Vrouwen die voorbehandeld werden met Ulipristal (5 mg) hadden een kortere operatieduur (gemiddelde 137,6 minuten (SD 26,8)) vergeleken met vrouwen die een placebo kregen (gemiddelde 159,7 minuten (SD 26,8); P<0,001).
Deelvraag 3 - GnRH analogen versus geen behandeling of placebo
3.1. Reductie in (H)MB
Er werden geen studies geïncludeerd die de uitkomst reductie in HMB beschreven voor de vergelijking tussen GnRH analogen en geen behandeling of placebo.
3.2. Patiënttevredenheid
Er werden geen studies geïncludeerd die de uitkomst patiënttevredenheid beschreven voor de vergelijking tussen GnRH analogen en geen behandeling of placebo.
3.3. Afname myoomvolume
Er werden geen studies geïncludeerd die de uitkomst afname myoomvolume beschreven voor de vergelijking tussen GnRH analogen en geen behandeling of placebo.
3.4. Bloedverlies peroperatief
Tien studies uit het Cochrane review van Lethaby (2017) beschreven de uitkomstmaat peroperatief bloedverlies bij vrouwen die voorbehandeld werden met GnRH analogen vergeleken met geen behandeling (Bustos López, 1995; Campo, 1999; Cetin, 1995; De Falco, 2009; Fedele, 1990; Golan, 1993; Hudecek, 2012; Shaw, 1989; Zullo, 1998) of vergeleken met een placebo (Friedman, 1989). De resultaten konden niet worden gepoold door substantiële heterogeniteit tussen de studies en worden daarom alleen beschrijvend gepresenteerd.
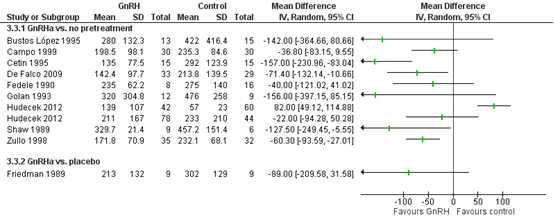
De meeste studies rapporteerden dat voorbehandeling met GnRH analogen leidde tot een afname van het gemiddelde peroperatieve bloedverlies, variërend van een afname van 22 tot 157 ml (Figuur 3). Sensitiviteitsanalyses waarin studies met louter myomectomieën werden vergeleken met studies waarin naast myomectomieën ook hysterectomieën werden uitgevoerd of studies waarin het niet duidelijk was welke chirurgische techniek werd toegepast lieten eenzelfde resultaat zien (data niet getoond).
Figuur 3. Forest plot van het peroperatief bloedverlies bij vrouwen die een voorbehandeling kregen met GnRH analogen vergeleken met een placebo
3.5. Operatieduur
Acht studies uit het Cochrane review van Lethaby (2017) beschreven de uitkomstmaat operatieduur bij vrouwen die voorbehandeld werden met GnRH analogen vergeleken met geen behandeling (Campo, 1999; Cetin, 1995; De Falco, 2009; Fedele, 1990; Golan, 1993; Hudecek, 2012; Zullo, 1998) of vergeleken met een placebo (Friedman, 1989). De resultaten konden niet worden gepoold door substantiële heterogeniteit tussen de studies en worden daarom alleen beschrijvend gepresenteerd.
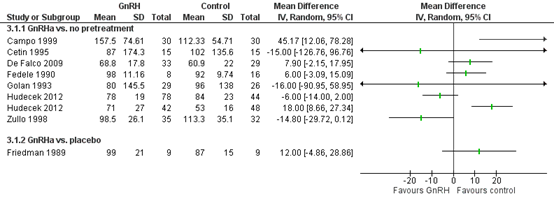
In het Cochrane review van Lethaby (2017) werden geen significante verschillen in de gemiddelde operatieduur gerapporteerd tussen de groepen, behalve voor twee studies over laparoscopische myomectomie: Campo (1999) en Hudecek (2012) rapporteerden dat voorbehandeling met GnRH analogen geassocieerd was met een significante toename van de operatieduur van laparoscopische myomectomie (Figuur 4). Sensitiviteitsanalyses waarin studies met louter myomectomieën werden vergeleken met studies waarin naast myomectomieën ook hysterectomieën werden uitgevoerd of studies waarin het niet duidelijk was welke chirurgische techniek werd toegepast lieten eenzelfde heterogeen beeld zien (data niet getoond).
Figuur 4. Forest plot van de operatieduur bij vrouwen die een voorbehandeling kregen met GnRH analogen vergeleken met een placebo
Bewijskracht van de literatuur
Deelvraag 1 - Ulipristal versus GnRH analogen
RCTs beginnen op een hoog niveau van bewijskracht. De bewijskracht voor de uitkomstmaten reductie in (H)MB, afname myoomvolume en afname uterusvolume is met één niveau verlaagd doordat de studie gesponsord was door de fabrikant van Ulipristal (Donnez, 2012b). De bewijskracht is daarmee ‘redelijk’.
Er werden geen studies geïncludeerd die de uitkomstmaten patiënttevredenheid, bloedverlies peroperatief of operatieduur beschreven voor de vergelijking tussen voorbehandeling met Ulipristal en GnRH analogen voor myoomchirurgie bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus. Er werd daarom geen GRADE-beoordeling uitgevoerd.
Deelvraag 2 - Ulipristal versus geen behandeling of placebo
RCT’s beginnen op een hoog niveau van bewijskracht. De bewijskracht voor de uitkomstmaten reductie in (H)MB en afname myoomvolume is met één niveau verlaagd doordat de studie van Donnez (2012b) gesponsord was door de fabrikant van Ulipristal en doordat de studie van Ferrero (2016) een risico op selectiebias heeft (retrospectieve analyse van database). De bewijskracht is daarmee ‘redelijk’.
Observationele studies beginnen op een laag niveau van bewijskracht. De bewijskracht voor de uitkomstmaten bloedverlies peroperatief en operatieduur is met één niveau verlaagd doordat de studie van Ferrero (2016) een risico op selectiebias heeft (controlegroep bestond uit vrouwen die behandeling met Ulipristal weigerden of geen Ulipristal kregen aangeboden omdat het nog niet op de markt was; daarnaast was het niet bekend hoeveel patiënten met incomplete data waren geëxcludeerd). De bewijskracht is daarmee ‘zeer laag’.
Er werden geen studies geïncludeerd die de uitkomstmaat patiënttevredenheid beschreven voor de vergelijking tussen voorbehandeling met Ulipristal en geen behandeling of placebo voor myoomchirurgie bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus. Er werd daarom geen GRADE-beoordeling uitgevoerd.
Deelvraag 3 - GnRH analogen versus geen behandeling of placebo
RCT’s beginnen op een hoog niveau van bewijskracht. De bewijskracht voor de uitkomstmaten bloedverlies peroperatief en operatieduur is met twee niveaus verlaagd door substantiële heterogeniteit tussen de studies en risk of bias in de studies (Lethaby, 2017). De bewijskracht is daarmee ‘laag’.
Er werden geen studies geïncludeerd die de uitkomstmaten patiënttevredenheid, bloedverlies peroperatief of operatieduur beschreven voor de vergelijking tussen voorbehandeling met Ulipristal en geen behandeling of placebo voor myoomchirurgie bij vrouwen met hevig menstrueel bloedverlies en een uterus myomatosus. Er werd daarom geen GRADE-beoordeling uitgevoerd.
Zoeken en selecteren
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvragen:
Deelvraag-1: Wat zijn de (on)gunstige effecten van voorbehandeling met GnRH analogen in vergelijking tot voorbehandeling met Ulipristal bij myomen voor myomectomie (laparoscopisch/laparotomisch) bij behandeling van hevig menstrueel bloedverlies?
P: patiënten met HMB en een uterus myomatosus;
I: myomectomie (laparoscopisch / laparotomisch) met voorbehandeling met Ulipristal;
C: myomectomie(laparoscopisch/laparotomisch) met voorbehandeling middels GnRH analogen;
O: reductie in (H)MB, patiënttevredenheid (na ingreep), afname myoomvolume, bloedverlies peroperatief, operatieduur.
Deelvraag-2: Wat zijn de (on)gunstige effecten van voorbehandeling met Ulipristal in vergelijking tot géén voorbehandeling of placebo bij myomen voor myomectomie bij behandeling van hevig menstrueel bloedverlies?
P: patiënten met HMB en een uterus myomatosus;
I: myomectomie (laparoscopisch/laparotomisch) met voorbehandeling met Ulipristal;
C: myomectomie (laparoscopisch/laparotomisch) met voorbehandeling middels placebo of geen voorbehandeling;
O: reductie in (H)MB, patiënttevredenheid (na ingreep), afname myoomvolume, bloedverlies peroperatief, operatieduur.
Deelvraag-3: Wat zijn de (on)gunstige effecten van voorbehandeling met GnRH analogen in vergelijking tot géén voorbehandeling of placebo bij myomen voor myomectomie bij behandeling van hevig menstrueel bloedverlies?
P: patiënten met HMB en een uterus myomatosus;
I: myomectomie (laparoscopisch/laparotomisch) met voorbehandeling met GnRH analogen;
C: myomectomie (laparoscopisch/laparotomisch) met voorbehandeling middels placebo of geen voorbehandeling;
O: reductie in (H)MB, patiënttevredenheid (na ingreep), afname myoomvolume, bloedverlies peroperatief, operatieduur.
Relevante uitkomstmaten
De werkgroep achtte reductie in (H)MB en patiënttevredenheid voor de besluitvorming cruciale uitkomstmaten; en afname myoomvolume, bloedverlies per-operatie en operatieduur voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde de uitkomstmaten als volgt:
Reductie in (hevig) menstrueel bloedverlies wordt gemeten aan de hand van PBAC (pictorial blood loss assessment chart) score (Janssen, 1995). Patiënttevredenheid moest gemeten zijn met een gevalideerde schaal. Voor de overige uitkomstmaten definieerde de werkgroep niet a priori de uitkomstmaten, maar hanteerde de in de studies gebruikte definities.
De werkgroep hield de GRADE-default grenzen (25% voor dichotome uitkomstmaten en 0,5 SD voor continue uitkomstmaten) aan als een klinisch (patiënt) relevant verschil (Schünemann, 2013). Voor reductie menstrueel bloedverlies werd een afname van 22% als klinisch (patiënt) relevant verschil gedefinieerd (Lukes, 2010).
Zoeken en selecteren (Methode)
In de databases Medline (via OVID) en Embase (via Embase.com) is op 23 januari 2019 met relevante zoektermen gezocht naar systematisch reviews, randomized controlled trials (RCT’s) en observationeel onderzoek waarin was gekeken naar voorbehandeling bij TCRM of myomectomie bij patiënten met HMB en een uterus myomatosus. Deze search betrof een update van de richtlijnmodules ‘GnRH versus placebo bij de behandeling van hevig menstrueel bloedverlies’ en ‘Ulipristal/GnRH analogen voor TCRM’ uit 2013. De zoekverantwoording is weergegeven onder het tabblad Verantwoording.
De literatuurzoekactie leverde 171 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: 1) patiëntpopulatie betrof vrouwen met HMB en een uterus myomatosus; 2) voorbehandeling met GnRH analogen en/of Ulipristal werd vergeleken met geen voorbehandeling of placebo; en 3) alle vrouwen ondergingen myomectomie na voorbehandeling of placebo/geen voorbehandeling. Op basis van titel en abstract werden in eerste instantie 12 studies voorgeselecteerd. Op basis van referentielijsten van deze 12 geselecteerde studies werden nog 2 additionele studies toegevoegd ter beoordeling. Na raadpleging van de volledige tekst, werden vervolgens 11 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording) en 3 studies definitief geselecteerd.
Drie onderzoeken zijn opgenomen in de literatuuranalyse. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk-of-biastabellen.
Resultaten
In totaal werd één systematische review en meta-analyse (Lethaby, 2017) en twee individuele studies die niet waren opgenomen in het Cochrane review van Lethaby (2017) geïncludeerd in de literatuuranalyse (Ferrero, 2016; Nieman, 2011).
Referenties
- EMA/482522/2018. Esmya: new measures to minimise risk of rare but serious liver injury.
- Ferrero S, Alessandri F, Vellone VG, Venturini PL, Leone Roberti Maggiore U. Three-month treatment with ulipristal acetate prior to laparoscopic myomectomy of large uterine myomas: a retrospective study. Eur J Obstet Gynecol Reprod Biol.2016 Oct;205:43-7.
- Janssen, C. A., Scholten, P. C., & Heintz, A. P. M. (1995). A simple visual assessment technique to discriminate between menorrhagia and normal menstrual blood loss. Obstetrics & Gynecology, 85(6), 977-982.
- Lethaby A, Puscasiu L, Vollenhoven B. Preoperative medical therapy before surgery for uterine fibroids. Cochrane Database Syst Rev. 2017 Nov 15;11:CD000547.
- Lukes, A. S., Muse, K., Richter, H. E., Moore, K. A., & Patrick, D. L. (2010). Estimating a meaningful reduction in menstrual blood loss for women with heavy menstrual bleeding. Current medical research and opinion, 26(11), 2673-2678.
- de Milliano I, Huirne JAF, Thurkow AL, Radder C, Bongers MY, van Vliet H, van de Lande J, van de Ven PM, Hehenkamp WJK. Ulipristal acetate versus gonadotropin-releasing hormone agonists prior to laparoscopic myomectomy (MYOMEX trial): Short-term results of a double-blind randomized controlled trial. Acta Obstet Gynecol Scand. 2019 Aug 29.
- Nieman LK, Blocker W, Nansel T, Mahoney S, Reynolds J, Blithe D, Wesley R, Armstrong A. Efficacy and tolerability of CDB-2914 treatment for symptomatic uterine fibroids: a randomized, double-blind, placebo-controlled, phase IIb study. Fertil Steril. 2011 Feb;95(2):767-72.e1-2.
- Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html
Evidence tabellen
Evidence table for systematic review of RCTs and observational studies (intervention studies)
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Lethaby, 2017
PS., study characteristics and results are extracted from the SR (unless stated otherwise) |
SR and meta-analysis of RCTs.
Literature search up to June 2017.
A: Donnez, 2012b B: Donnez, 2012a C: Bustos López 1995 D: Campo, 1999 E: Cetin, 1995 F: De Falco, 2009 G: Fedele, 1990 H: Golan 1993 I: Hudecek, 2012 J: Shaw 1989 K: Zullo 1998 L: Friedman 1989
Study design: all studies were parallel group RCTs.
Setting and Country: not reported per included study
Source of funding and conflicts of interest: See Lethaby, 2017 |
Inclusion criteria SR: Premenopausal women, without any other underlying uterine pathology, intending to undergo any surgery for uterine fibroids: either myomectomy (laparotomy or laparoscopy) or resection for uterine fibroids.
Exclusion criteria SR: Trials of medical therapies used as sole treatment for uterine fibroids, without the expectation of subsequent surgery, were not included
12 studies included
Important patient characteristics at baseline: Number of patients; characteristics important to the research question and/or for statistical adjustment (confounding in cohort studies); for example, age, sex, bmi, ...
N, mean age Mean age was not reported per study. A: 307 B: 242 C: 20 D: 60 E: 30 F: 62 G: 24 H: 53 I: 212 J: 32 K: 75 L: 20
Sex: All studies included 100% female participants.
Groups comparable at baseline?
A: Yes B: Yes C: No, there appear to be differences D: Unclear, not reported. E: Unclear F: Yes G: Unclear H: Unclear I: Unclear J: Unclear K: Yes L: Yes
|
Describe intervention:
Ulipristal versus. GnRH: A: ulipristal acetate (SPRM) 5 mg or 10 mg oral tablet daily + intramuscular saline injection once monthly
Ulipristal versus. no treatment/placebo B: Ulipristal acetate 5.mg or 10.mg orally once per day
GnRH versus. no treatment/placebo: C: Nafarelin intranasal spray 200 µg twice daily before myomectomy D: Decapeptyl 3.75 mg intramuscularly every 28 days for 3 months before surgery E: Buserelin intranasally 900 µg/day in 3 doses for 3 months F: 3.75 mg triptorelin subcutaneous depot injection, once a month for 3 months. G: Intranasal buserelin 1200 µg/day before myomectomy H: Intramuscular D-Trp LHRH 3.2 mg micro capsules (Decapeptyl) monthly before surgery (hysterectomy, N = 17; myomectomy, N = 12). I: Goserelin acetate 3.6 mg SC 3 times once every 4 weeks J: Goserelin depot 3.6 mg before surgery (myomectomy and hysterectomy). K: Intramuscular leuprolide acetate depot 3.75 mg followed by laparoscopic myomectomy L: Intramuscular leuprolide acetate depot 3.75 mg monthly for 4 injections before myomectomy,
Surgical intervention after pre-treatment was: A: laparotomic, laparoscopic or vaginal hysterectomy or myomectomy B: laparotomic, laparoscopic or vaginal hysterectomy or myomectomy C: myomectomy (unspecified) D: laparoscopic myomectomy E: unspecified F: unspecified G: unspecified H: hysterectomy or myomectomy I: laparoscopic or laparoscopic myomectomy J: hysterectomy or myomectomy K: laparoscopic myomectomy L: myomectomy (unspecified)
|
Describe control:
Ulipristal versus. GnRH: A: daily oral placebo + Intramuscular injection of 3.75 mg leuprolide acetate(GnRHa) once monthly Ulipristal versus. no treatment/placebo B: placebo (identical pill) orally once per day
GnRH versus. no treatment: C: no treatment D: no treatment E: no treatment F: no treatment, immediate surgery during follicular phase of menstruation. G: no treatment H: no treatment I: no treatment J: no treatment K: no treatment L: Intramuscular placebo monthly for 4 injections before myomectomy
|
End-point of follow-up:
A: 13 weeks (before surgery) with follow up at weeks 17, 26 and 38 Iron supplementation could be used at the discretion of the physician B: 13 weeks of treatment (before surgery) with follow up at weeks 17, 26, and 38, N = 48 C: 3 months D: 3 months E: 3 months F: 3 months G: 3 months H: 2 months I: not reported J: 4 months K: 2 months L: 12 treatment weeks before myomectomy (follow up 27 to 38 months after surgery)
For how many participants were no complete outcome data available? (intervention/control) A: 3/2 B: 1/0 C: 0/0 D: 0/0 E: 0/0 F: 0/0 G: 0/0 H: 0/0 I: 0/0 J: not reported K: 7/0 L: not reported.
|
Ulipristal versus. GnRH:
Outcome measure-1: reduction in HMB One non-inferiority trial, with 307 participants, comparing GnRHa to ulipristal acetate assessed the proportion of women whose bleeding reduced to < 75 units by the PBAC as a result of presurgical treatment (Donnez 2012b). There was no evidence of a difference in bleeding ratesbetween the 2 groups leading the authors to conclude that ulipristal acetate was non inferior to GnRHa in controlling uterine bleeding (ulipristal acetate 5 mg: OR 0.71, 95% CI 0.3 to 1.7; moderate-quality evidence; ulipristal acetate 10 mg: OR 0.39, 95% CI 0.1 to 1.1; moderate-quality evidence; Analysis 5.5).
Outcome 2 – reduction in fibroid volume: Per protocol results: Percentage change from baseline in 3 largest fibroids (IQ range) Ulipristal acetate 5mg: -36% (-58 to -11) Ulipristal acetate 10mg: -42% (-69 to -41) Leuprolide acetate 3.75mg: -53% (-69 to -36) Difference in % points: UA 5mg versus LA 3.75mg: 1.23 (0.99 to 1.52) UA 10mg versus LA 3.75mg: 1.12 (0.91 to 1.38) Authors reported that all 3 treatments reduced the volume of the 3 largest fibroids. Analysis 5.3.
Ulipristal versus. no treatment/placebo Outcome measure-1: reduction in HMB One trial comparing ulipristal acetate with placebo assessed the proportion of women who achieved a reduction in bleeding to < 75 units by PBACafterpresurgical intervention (Donnez 2012a). The odds of bleeding reduction was higher in women receiving both doses of ulipristal acetate compared to placebo (ulipristal acetate 5 mg: OR 41.41, 95% CI 15.3 to 112.4; 1 study; 143 participants; low-quality evidence; Analysis 6.4.1; ulipristal acetate 10 mg: OR 78.83, 95% CI 24.0 to 258.7; 1 study; 146 participants; low-quality evidence; Analysis 6.4.2).
Outcome measure-2: reduction in fibroid volume Donnez 2012a reported a 3% increase with placebo compared to a 21% and 12.3% decrease with ulipristal acetate 5 mg and 10 mg, respectively
GnRH versus. no treatment/ placebo:
Outcome measure-3: duration of operation Eight trials, including 494 participants, assessed this outcome (7 comparing GnRHa to no pretreatment and 1 comparing GnRHa to placebo). Trials could not be pooled due to substantial heterogeneity. There was no evidence that duration of surgery was influenced by whether participants received GnRHa pretreatment or not. Individual studies did not report significant differences between randomised groups, except for two studies comparing GnRHa pretreatment to no pretreatment before laparoscopic myomectomy was undertaken. In these two trials(Campo 1999;Hudecek 2012), GnRHa was associated with a significant increase in the time taken to undertake laparoscopic myomectomy compared to no pretreatment (ranging from an increase of 18 minutes to 45 minutes).
Outcome measure 4: intraoperative blood loss We included 10 studies (549 participants) that assessed this outcome (9 compared GnRHa to no pretreatment and 1 compared GnRHa to placebo). Trials could not be pooled, due to substantial heterogeneity and varied findings. Most trials reported that GnRHa reduced blood loss, ranging from a reduction of 22 mL to 157 mL, although findings were mostly outside the level of significance and the quality of the evidence was very low. Most trials reported that surgery was myomectomy, either unspecified or open. Three trials where laparoscopic myomectomy was performed had mixed results; two trials reported that GnRHa pretreatment reduced blood loss by either37 mLor 60 mL and the other trial reported a greater intraoperative. intra operative blood loss with GnRHa compared to control (82 mL) ( Campo 1999; Hudecek 2012; Zullo 1996). |
For the purpose of this analysis we only report studies included by Lethaby that completely adhere to the PICO of this analysis.
Facultative:
Brief description of author’s conclusion: A rationale for the use of preoperative medical therapy before surgery for fibroids is to make surgery easier. There is clear evidence that preoperativeGnRHareducesuterineandfibroidvolume,andincreasespreoperativehaemoglobin levels, although GnRHa increases the incidence of hot flushes. During hysterectomy, blood loss, operation time and complication rates were also reduced. Evidence suggests that ulipristal acetate may offer similar advantages (reduced fibroid volume and fibroid-related bleeding and increased haemoglobin levels) although replication of these studies is advised before firm conclusions can be made. Future research should focus on costeffectiveness and distinguish between groups of women with fibroids who would most benefit
Personal remarks on study quality, conclusions, and other issues (potentially) relevant to the research question
Level of evidence: GRADE (per comparison and outcome measure) including reasons for down/upgrading
Sensitivity analyses (excluding small studies; excluding studies with short follow-up; excluding low quality studies; relevant subgroup-analyses); mention only analyses which are of potential importance to the research question
Heterogeneity: clinical and statistical heterogeneity; explained versus unexplained (subgroupanalysis) |
Evidence table for intervention studies (randomized controlled trials and non-randomized observational studies (cohort studies, case-control studies, case series))1
This table is also suitable for diagnostic studies (screening studies) that compare the effectiveness of two or more tests. This only applies if the test is included as part of a test-and-treat strategy - otherwise the evidence table for studies of diagnostic test accuracy should be used.
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures.
- Provide data per treatment group on the most important prognostic factors ((potential) confounders).
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls.
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders.
Risk of bias table for intervention studies (observational: non-randomized clinical trials, cohort and case-control studies)
|
Study reference
(first author, year of publication) |
Bias due to a non-representative or ill-defined sample of patients?1
(unlikely/likely/unclear) |
Bias due to insufficiently long, or incomplete follow-up, or differences in follow-up between treatment groups?2
(unlikely/likely/unclear) |
Bias due to ill-defined or inadequately measured outcome ?3
(unlikely/likely/unclear) |
Bias due to inadequate adjustment for all important prognostic factors?4
(unlikely/likely/unclear) |
|
Ferrero, 2016 |
Likely, retrospective cohort study. Patients in the control group consist of patients who refused UPA treatment or were not offered UPA treatment because it was not allowed at that time in Italy. Furthermore, it was unclear whether patients had been excluded because of incomplete data. |
Unclear, duration of follow-up was not reported. Bias will probably be minimal, outcomes of our interest were intraoperatively recorded. |
Unlikely |
Unlikely, although data were not corrected, as groups were similar at baseline for important characteristics bias is unlikely. |
- Failure to develop and apply appropriate eligibility criteria: a) case-control study: under- or over-matching in case-control studies; b) cohort study: selection of exposed and unexposed from different populations.
- 2 Bias is likely if: the percentage of patients lost to follow-up is large; or differs between treatment groups; or the reasons for loss to follow-up differ between treatment groups; or length of follow-up differs between treatment groups or is too short. The risk of bias is unclear if: the number of patients lost to follow-up; or the reasons why, are not reported.
- Flawed measurement, or differences in measurement of outcome in treatment and control group; bias may also result from a lack of blinding of those assessing outcomes (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Failure to adequately measure all known prognostic factors and/or failure to adequately adjust for these factors in multivariate statistical analysis.
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Nieman, 2011 |
Computer-generated blocks of six |
Unclear, not reported |
Unlikely, double blind, placebo controlled trial. |
Unlikely, double blind, placebo controlled trial. |
Unclear, not reported |
Unlikely |
Unlikely, loss to follow-up was equal in both groups, although there were 2 additional dropouts in the control group in phase 2, compared to no drop outs in the intervention groups. Reasons for drop out provided |
Unlikely, patients were analysed as intention to treat. |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules..
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear.
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Risk of bias table for intervention studies (observational: non-randomized clinical trials, cohort and case-control studies)
|
Study reference
(first author, year of publication) |
Bias due to a non-representative or ill-defined sample of patients?1
(unlikely/likely/unclear) |
Bias due to insufficiently long, or incomplete follow-up, or differences in follow-up between treatment groups?2
(unlikely/likely/unclear) |
Bias due to ill-defined or inadequately measured outcome ?3
(unlikely/likely/unclear) |
Bias due to inadequate adjustment for all important prognostic factors?4
(unlikely/likely/unclear) |
|
Ferrero, 2016 |
Likely, retrospective cohort study. Patients in the control group consist of patients who refused UPA treatment or were not offered UPA treatment because it was not allowed at that time in Italy. Furthermore, it was unclear whether patients had been excluded because of incomplete data. |
Unclear, duration of follow-up was not reported. Bias will probably be minimal, outcomes of our interest were intraoperatively recorded. |
Unlikely |
Unlikely, although data were not corrected, as groups were similar at baseline for important characteristics bias is unlikely. |
- Failure to develop and apply appropriate eligibility criteria: a) case-control study: under- or over-matching in case-control studies; b) cohort study: selection of exposed and unexposed from different populations.
- 2 Bias is likely if: the percentage of patients lost to follow-up is large; or differs between treatment groups; or the reasons for loss to follow-up differ between treatment groups; or length of follow-up differs between treatment groups or is too short. The risk of bias is unclear if: the number of patients lost to follow-up; or the reasons why, are not reported.
- Flawed measurement, or differences in measurement of outcome in treatment and control group; bias may also result from a lack of blinding of those assessing outcomes (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Failure to adequately measure all known prognostic factors and/or failure to adequately adjust for these factors in multivariate statistical analysis.
Table of quality assessment for systematic reviews of RCTs and observational studies
Based on AMSTAR checklist (Shea et al.; 2007, BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher et al 2009, PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Lethaby, 2017 |
Yes |
Yes |
Yes |
Yes |
Not applicable |
Yes, Cochrane tool |
Yes |
Yes |
Yes |
- Research question (PICO) and inclusion criteria should be appropriate and predefined.
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched.
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons.
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported.
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs).
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table et cetera).
- Clinical and statistical heterogeneity should be assessed; clinical: enough similarities in patient characteristics, intervention and definition of outcome measure to allow pooling? For pooled data: assessment of statistical heterogeneity using appropriate statistical tests (e.g. Chi-square, I2)?
- An assessment of publication bias should include a combination of graphical aids (e.g., funnel plot, other available tests) and/or statistical tests (e.g., Egger regression test, Hedges-Olken). Note: If no test values or funnel plot included, score “no”. Score “yes” if mentions that publication bias could not be assessed because there were fewer than 10 included studies.
- Sources of support (including commercial co-authorship) should be reported in both the systematic review and the included studies. Note: To get a “yes,” source of funding or support must be indicated for the systematic review AND for each of the included studies.
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Barlow, 2014 |
Voldoet niet aan PICO |
|
Bondi, 2016 |
Congresabstract |
|
Donnez, 2012 |
Artikel is reeds geïncludeerd in Lethaby 2017 |
|
Donnez, 2012 |
Artikel is reeds geïncludeerd in Lethaby 2017 |
|
Kalampokas, 2016 |
Uitgangspunt is review van Lethaby 2017. Eén losse studie die niet in Lethaby 2017 zat gecontroleerd (Nieman, 2011). |
|
Lethaby, 2007 |
Oudere versie van Lethaby 2017. |
|
Lethaby, 2011 |
Oudere versie van Lethaby 2017. |
|
Mavrelos, 2010 |
Voldoet niet aan PICO |
|
Moroni, 2014 |
Geen systematische review (narrative review) |
|
Revazov, 2016 |
Congresabstract |
|
Tsoi, 2015 |
Voldoet niet aan PICO |
Verantwoording
Beoordelingsdatum en geldigheid
Laatst beoordeeld : 22-04-2020
Bij het opstellen van de module heeft de werkgroep een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijnmodule komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
|
Module[1] |
Regiehouder(s)[2] |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn[3] |
Frequentie van beoordeling op actualiteit[4] |
Wie houdt er toezicht op actualiteit[5] |
Relevante factoren voor wijzigingen in aanbeveling[6] |
|
Voorbehandeling myomectomie |
NVOG |
2020 |
Eind 2025 |
Elke 2 jaar |
NVOG |
Ontwikkelingen uit nieuw onderzoek. |
[1] Naam van de module
[2] Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders)
[3] Maximaal na vijf jaar
[4] (half)Jaarlijks, eens in twee jaar, eens in vijf jaar
[5] regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) werkgroep die in stand blijft
[6] Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen
Algemene gegevens
De richtlijnmodule is ter goedkeuring/geen bezwaar voorgelegd aan:
- Nederlands Huisartsen Genootschap
- Stichting Bekkenbodem4All
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit deStichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijn.
Samenstelling werkgroep
Werkgroep
- Dr. A. Timmermans, gynaecoloog, NVOG (voorzitter werkgroep (proces))
- Dr. C.A.H. Janssen, gynaecoloog, NVOG (inhoudelijk voorzitter)
- Dr. S. (M.) van der Kooij, gynaecoloog, NVOG
- M. Bosch, patiëntvertegenwoordiger, Stichting Bekkenbodem4All
Klankbordgroep
- Drs. C.J.H. de Vries, wetenschappelijk medewerker, NHG
Met ondersteuning van
- Dr. E.J.M. den Breejen, senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten (tot 15 juni 2019)
- Dr. A. Bijlsma-Rutte, adviseur, Kennisinstituut van de federatie Medisch Specialisten
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Bosch |
Stichting Bekkenbodem4All: PR/Communicatie en belangenbehartiging |
Fotograaf Bisdom Groningen-Leeuwarden, deels betaald, deels vrijwilliger Bestuur Dutch Foundation for Ladakhi Nuns: onbetaald PR medewerker Stichting Uit De Kunst, Zuidhorn: onbetaald" |
Geen |
Geen |
|
Janssen |
Gynaecoloog Groene hart ziekenhuis |
Geen |
Geen |
Geen |
|
van der Kooij |
Gynaecoloog |
Geen |
Geen |
Geen |
|
Timmermans |
Gynaecoloog werkzaam bij Amsterdam UMC locatie AMC (0.4 fte) en gedetacheerd naar Bergman Vrouwenzorg Amsterdam (0.4 fte) |
Lid commissie kwaliteitsdocumenten NVOG (onbetaald) |
Mede-aanvrager op ZONMW subsidie aanvraag naar effectiviteit van insertie Mirena aansluitend aan novasure endometriumablatie (proces loopt, nog geen bericht of subsidie aanvraag is toegekend)
|
Geen |
|
De Vries (klankbord NHG) |
Wetenschappelijk medewerker, Nederlands Huisartsen Genootschap, afdeling Richtlijn ontwikkeling en Wetenschap. Onder andere betrokken bij de NHG-Standaarden Vaginaal Bloedverlies en Amenoroe |
Geen |
Geen |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een patiëntvertegenwoordiger als werkgroeplid te betrekken bij de ontwikkeling van deze module. De conceptmodule werd tevens voor commentaar voorgelegd aan de patiëntenvereniging.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van het ontwikkelproces is rekening gehouden met de implementatie van de richtlijnmodule en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de module in de praktijk kunnen bevorderen of belemmeren. De implementatietabel is te vinden bij de aanverwante producten.
Werkwijze
AGREE
Deze module is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based module tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse
Uit de inventarisatie van de knelpunten door de werkgroep bleek dat er een noodzaak was voor revisie van deze richtlijnmodule. Tijdens deze inventarisatie zijn er knelpunten aangedragen door relevante partijen via een schriftelijke enquête. Een verslag hiervan is opgenomen onder aanverwante producten.
Uitgangsvraag en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse is door de werkgroepleden en de adviseur een uitgangsvraag opgesteld. Vervolgens inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet kritiek) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Aan de hand van specifieke zoektermen werd gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De geselecteerde databases waarin is gezocht en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie is opvraagbaar bij de Richtlijnendatabase, zie het tabblad ‘Zoekverantwoording’ voor verdere details.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration:
- Cochrane - voor gerandomiseerd gecontroleerd onderzoek;
- ACROBAT-NRS - voor observationeel onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz 2017).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt, kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
Bij de ontwikkeling van de module is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag, randvoorwaarden die van invloed zijn op de implementatie van de aanbeveling zijn opgenomen in de implementatietabel.
Indicatorontwikkeling
Op verzoek van de NVOG werden er geen indicatoren ontwikkeld bij deze module vanwege het beperken van de registratielast van medisch specialisten.
Kennislacunes
Tijdens de ontwikkeling van deze module is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvraag. Er is nagegaan of (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Mocht dit bij deze module het geval zijn, dan is er een aanbeveling voor het doen van onderzoek opgenomen in de Kennislacunes, te vinden onder de aanverwante producten.
Commentaar- en autorisatiefase
De conceptmodule werd aan de betrokken (wetenschappelijke) verenigingen, instanties en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve module werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd. De commentaartabel is op te vragen bij het Kennisinstituut via secretariaat@kennisinstituut.nl.
Literatuur
Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348.
Hultcrantz M, Rind D, Akl EA, et al. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, et al. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008;336(7654). doi: 10.1136/bmj.a139. PubMed PMID: 18483053.
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van de Federatie Medisch Specialisten.
Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
Zoekverantwoording
Zoekacties zijn opvraagbaar. Neem hiervoor contact op met de Richtlijnendatabase.
Bijlagen
- Relevant voor patiënten
- Achtergrond en definities
- Toepassen
- Onderzoek