Genetisch onderzoek slechthorend kind
Uitgangsvraag
- Bij welke kinderen met perceptieve slechthorendheid dient genetisch onderzoek uitgevoerd te worden?
- Welke informatie dient besproken te worden met ouders/welke begeleiding is nodig voor en na het genetisch onderzoek?
Aanbeveling
Aanbeveling-1
Bespreek en adviseer genetisch onderzoek, bij voorkeur in een vroeg stadium van het diagnostisch traject, bij elke patiënt met slechthorendheid, in de volgende gevallen:
- bij elke patiënt met dubbelzijdige congenitale/juveniele perceptieve slechthorendheid >30dB (en waarbij infectie als oorzaak is uitgesloten).
- bij unilaterale perceptieve slechthorendheid alleen indien er andere geassocieerde verschijnselen zijn bij de patiënt en/of in de familie.
Verwijs naar de klinisch geneticus als er aanwijzingen zijn voor een meeromvattende aandoening (syndroom).
Aanbeveling-2
Vraag genetisch onderzoek alleen aan na pre-DNA test counseling in samenspraak met ouders/verzorgers. Hiervoor kan verwezen worden naar de klinisch geneticus. Als de KNO-arts danwel kinderarts zelf bekwaam is kan deze zelf het genetisch onderzoek aanvragen.
Laat bij de pre-DNA test counseling de volgende onderwerpen aan bod komen:
- Bespreek dat de opbrengst van genetisch onderzoek afhangt van het type gehoorverlies en andere bijkomende gezondheidsproblemen en belaste familieanamnese
- Bespreek de mogelijke uitkomsten; er kan een erfelijke oorzaak gevonden worden, geen afwijkingen, een onduidelijke uitslag en er is een kleine kans op nevenbevindingen.
- Bespreek dat met DNA-onderzoek er een kans bestaat dat er een syndroom wordt vastgesteld, waarbij verschijnselen nog later kunnen ontstaan. Een voorbeeld is Usher syndroom (waarbij ook slechtziendheid ontstaat).
- Bespreek wat de voor- en nadelen zijn van DNA-onderzoek voor zowel de patiënt als familie. Denk hierbij aan gezinsplanning en mogelijkheden van onderzoek voorafgaand of tijdens een zwangerschap.
Overleg altijd met een counselor van een afdeling Klinische Genetica, bij voorkeur in een Multidisciplinair overleg (MDO) of vergelijkbaar overleg, of verwijs direct naar de Klinische Genetica bij een afwijkende uitslag van het genetisch onderzoek.
Overwegingen
Balans tussen gewenste en ongewenste effecten
Er is systematisch gezocht naar literatuur dat de effecten van genetisch testen vergeleek met niet-genetisch testen in kinderen met perceptief gehoorverlies. Er werden geen artikelen gevonden die voldeden aan de PICO. Er kunnen dan ook geen uitspraken gedaan worden over het effect van genetisch testen in kinderen met sensorisch gehoorverlies. De overwegingen in deze module zijn dan ook gebaseerd op andere relevante literatuur en expert opinie van de werkgroep.
Bij welke kinderen met perceptieve slechthorendheid dient genetisch onderzoek uitgevoerd te worden?
Bij kinderen met een perceptief gehoorverlies >35 dB beiderzijds is er in een aanzienlijk deel een onderliggende genetische oorzaak. 60-70% van het prelinguale gehoorverlies is toe te schrijven aan genetische oorzaken. Van de erfelijke slechthorendheid is 30% syndromaal. Het merendeel (grofweg 70%) betreft niet-syndromale slechthorendheid.
Van de niet-syndromale erfelijke slechthorendheid is weer een onderverdeling te maken: veruit het grootste deel is autosomaal recessief erfelijk (75-85%) (Lieu, 2020; Shearer, 2025).
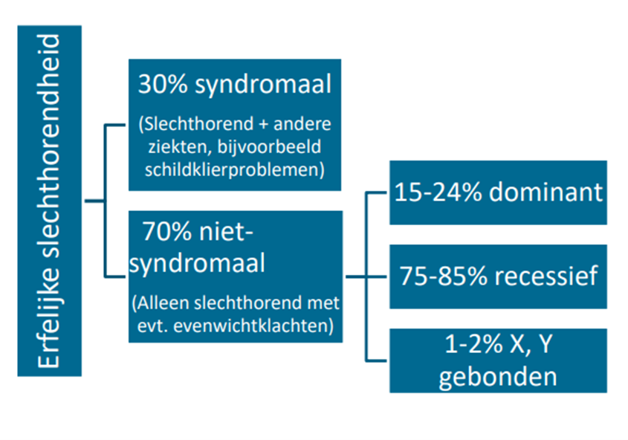
Figuur 1. Oorzaken van erfelijke slechthorendheid bij kinderen
(gebaseerd op figuur van Shearer (2025) Causes of prelingual hearing loss in developed countries (Morton, 2006; Sheffield, 2019))
Syndromale slechthorendheid wil zeggen dat er naast slechthorendheid andere kenmerken aanwezig zijn en hiervoor een gezamenlijke oorzaak is. Een voorbeeld is Waardenburg syndroom, waarbij er naast slechthorendheid o.a. ook hypopigmentatie kan zijn van huid en haar (witte haarlok). Er zijn meer dan 400 genetische syndromen, waarbij gehoorverlies een kenmerk is (Kenna, 2022; Li, 2022). Wanneer er aanwijzingen zijn voor een syndromale vorm van slechthorendheid (bijvoorbeeld na lichamelijk onderzoek of bij een specifiek patroon in de familie), kan in de meeste gevallen gericht DNA-onderzoek worden aangevraagd naar het gen/de enkele genen betrokken bij het betreffende syndroom (bij het genoemde voorbeeld Waardenburg syndroom zijn o.a. het PAX3-gen en MITF-gen betrokken). Als er aanwijzingen zijn voor een syndroom is het advies om de patiënt te verwijzen naar de klinisch geneticus.
Niet-syndromale slechthorendheid wordt gedefinieerd als dubbelzijdig gehoorverlies, waarbij er geen zichtbare afwijkingen aan het uitwendige oor of gerelateerde verschijnselen zijn. Het kan wel geassocieerd zijn met afwijkingen in het middenoor en/of binnenoor. Niet-syndromale slechthorendheid is genetisch zeer heterogeen. Tot nu toe zijn meer dan 150 genen voor niet-syndromale slechthorendheid geïdentificeerd (Molecular Otolaryngoly and Renal Research Laboratories, 2025). Bij het verrichten van genetisch onderzoek is het maar voor een klein deel van de patiënten met een niet-syndromale slechthorendheid mogelijk om op basis van een specifiek fenotype gericht onderzoek aan te vragen (DNA-onderzoek van één of enkele genen). Een specifiek fenotype hierbij kan zijn: de vorm van het audiogram, de leeftijd van onset, de ernst van het gehoorverlies en de mate van progressie. In de meeste gevallen is gericht onderzoek niet mogelijk. Als er geen aanwijzingen zijn voor een specifieke genetische oorzaak dan kan gekozen worden voor breed genetisch onderzoek naar oorzaken van erfelijke slechthorendheid. Dit betreft DNA-onderzoek met een genenfilter (genpanel) slechthorendheid/doofheid.
Op basis van de beschikbare literatuur is het weinig zinvol om uitgebreid genetisch onderzoek te verrichten bij éénzijdig/unilateraal (al dan niet ernstig) gehoorverlies. Uit de literatuur volgt dat bij unilateraal gehoorverlies, zonder gerelateerde bijzonderheden bij patiënt en/of familie, de opbrengst van genetisch onderzoek ligt tussen de 1% tot 5%. Dit is niet kosteneffectief (Li, 2022; Zhu, 2022; Núñez Batalla, 2023; Peart, 2023; Yamamoto, 2023; Clay, 2024). Wel wordt beeldvorming aanbevolen, om onderzoek te doen of er een aanlegstoornis is van het binnenoor. Zie hiervoor module Beeldvormend onderzoek slechthorend kind.
De grootste kans op het vaststellen van een genetische diagnose is bij kinderen met congenitale, dubbelzijdige symmetrische, ernstige perceptieve slechthorendheid, met name in de lage tot middenfrequenties (Liao, 2022; Zhu, 2022; Núñez Batalla, 2023; Peart, 2023; Yamamoto, 2023; Clay, 2024). Met het huidige genpanel in Nederland wordt bij congenitale dubbelzijdige doofheid/ernstige slechthorendheid bij 50-60% een genetische oorzaak gevonden (Zazo Seco, 2017). Dit genpanel wordt regelmatig geüpdatet en ook wordt zorg gedragen dat de genpanels in de verschillende laboratoria voor genoomdiagnostiek in Nederland gelijk zijn.
In dit genpanel zitten ook genen die betrokken zijn bij syndromale vormen van slechthorendheid/doofheid, waarbij de aandoening zich met slechthorendheid kan presenteren. Er bestaat een groep patiënten waarbij er initieel sprake lijkt te zijn van niet-syndromale slechthorendheid en waarbij met genetisch onderzoek toch een syndromale vorm van slechthorendheid vroegtijdig wordt gediagnosticeerd. In dit geval wordt verwijzing naar een klinisch geneticus nadrukkelijk geadviseerd. Geschat wordt dat dit bij ca. 20% van de jonge kinderen (< 1 jaar) met in eerste instantie alleen geïsoleerde bilaterale slechthorendheid het geval is (Downie, 2020; Gooch, 2021). Een voorbeeld hiervan is Usher syndroom, waarbij de visusproblemen pas later ontstaan. Tijdige identificatie van een dergelijk syndroom heeft significante medische en ook (genetische) counseling implicaties. Behalve Usher syndroom zijn andere voorbeelden: Pendred syndroom, Stickler syndroom, BOR syndroom en syndromen met fertiliteitsproblemen (zoals Perrault syndroom bij vrouwen).
Het stellen van een juiste genetische diagnose is niet alleen van belang voor de patiënt, maar kan ook van belang zijn voor de ouders en verdere familie. Het geeft informatie over de prognose (te verwachten progressie en of er nog gezondheidsproblemen bij kunnen komen (syndromaal vs. niet-syndromaal) en kan ook van invloed zijn bij de behandeling en begeleiding. In sommige gevallen is er ziekte specifieke therapie. Hoe een genetische diagnose bij een kind ook voor de ouders van belang kan zijn heeft te maken met hoe de slechthorendheid erfelijk is. Zo is er bij een autosomaal dominante vorm een kans dat één van de ouders ook aangedaan is. Daarnaast kan er een verhoogde kans zijn op een volgend aangedaan kind. De precieze herhalingskans hangt af van het overervingspatroon. De uiting (beginleeftijd, ernst) kan verschillen tussen aangedane personen. Dit kan van invloed zijn op de verdere gezinsplanning: bij een verhoogde kans kunnen ouders deze kans accepteren, maar ook kiezen om af te zien van het krijgen van verdere kinderen. Ook zijn er bij een vastgestelde genetische diagnose mogelijkheden om DNA-onderzoek te doen tijdens een volgende zwangerschap (PND: prenatale diagnostiek) en soms ook voorafgaand aan een zwangerschap (PGT: pre-implantatie genetische test). Afhankelijk van de precieze vorm van erfelijke slechthorendheid kan dit ook voor verdere familieleden gevolgen hebben (voor zichzelf en ook voor hun gezinsplanning) (Shearer, 2025).
Als met genetisch onderzoek geen oorzaak wordt gevonden kan na een aantal jaren een heranalyse van (of hernieuwd) genetisch onderzoek zinvol zijn, omdat de kennis en mogelijkheden t.a.v. genetisch onderzoek steeds verder toenemen. Dit is vooral zinvol als er een sterke verdenking is op een genetische oorzaak of als er een variant is gevonden waarvan de betekenis nog onvoldoende duidelijk is. Binnen het DOOFNL consortium is er een landelijke consensus over het advies voor een dergelijke heranalyse: de termijn voor heranalyse in de toekomst is gesteld op 5-10 jaren. Als er opvallende (bijkomende) gezondheidsklachten zijn kan dit reden zijn voor een eerdere revisie en heranalyse.
Welke informatie dient besproken te worden met ouders/welke begeleiding is nodig voor- en na het genetisch onderzoek?
Er is geen systematisch literatuuronderzoek uitgevoerd naar welke begeleiding nodig is voor ouders van kinderen met slechthorendheid bij wie genetisch onderzoek wordt ingezet, omdat deze vraag niet in een PICO te vatten was.
In samenspraak met de ouders/verzorgers van de patiënt wordt besloten of genetisch onderzoek aangevraagd wordt.
Counseling en het vervolgens aanvragen van genetische testen gebeurt niet altijd door een klinisch geneticus, maar kan ook door bijvoorbeeld een KNO-arts of kinderarts worden gedaan, mits zij bekwaam zijn. Hiervoor is een leidraad opgesteld door de VKGN: “Leidraad aanvragen genetische kiembaandiagnostiek door niet klinisch genetische zorgprofessionals” en bijbehorende bijlage Bijlage Voorwaarden inzetten genetische kiembaandiagnostiek (VKGN, 2024).
De uitslag van genetisch onderzoek kan consequenties hebben voor het kind zelf, maar ook voor de ouders en verdere familieleden. Met het genetisch onderzoek kan een duidelijke oorzaak worden gevonden, maar het niet vinden van een oorzaak sluit een erfelijke oorzaak niet geheel uit. Met de huidige kennis en technieken lukt het (nog) niet om alle erfelijke oorzaken te vinden. Ook kan er een uitslag zijn met een onduidelijke betekenis. Tot slot is er bij iedere genetische test een kleine kans op nevenbevindingen. Vanwege de implicaties die dit kan hebben is het belangrijk om zowel in de pre-test counseling als post-test counseling een aantal punten hierover te bespreken met de ouders/verzorgers van de patiënt.
Samengevat zijn de volgende punten belangrijk om te bespreken:
- De mogelijkheid van genetisch onderzoek.
- Het soort genetisch onderzoek (uitgebreid DNA-onderzoek of juist gericht).
- De uitslagmogelijkheden:
- De oorzaak voor de slechthorendheid wordt gevonden.
- Er wordt geen oorzaak voor de slechthorendheid gevonden.
- Er wordt een verandering in het DNA gevonden, waarvan de betekenis niet (meteen) duidelijk is. Dus een onduidelijke uitslag.
- Er wordt een nevenbevinding gedaan.
- Ouders kunnen verwezen worden naar websites/andere informatiebronnen over genetisch onderzoek en uitslagen, zoals de website www.erfelijkheid.nl.
Als ouders afzien van genetisch onderzoek dient dit goed gedocumenteerd te worden in het patiënt dossier. Er is altijd een mogelijkheid om later in de toekomst alsnog genetisch onderzoek te verrichten. Hiervoor kan de patiënt (opnieuw) worden verwezen naar de klinisch geneticus.
Zoals in de leidraad van de VKGN vermeld wordt is er is een strikte indicatie voor overleg met een counselor van een afdeling Klinische Genetica, bij voorkeur in een multidisciplinair- of vergelijkbaar overleg, of directe verwijzing naar de afdeling genetica NA de uitslag van genetische kiembaan/DNA-diagnostiek. Bij voorkeur wordt dan ook afgestemd wie de uitslag bespreekt.
Dit is een strikte indicatie bij de volgende uitslagen:
- Bij een aangetoonde (waarschijnlijk) pathogene variant, ook in geval van dragerschap bij een recessieve aandoening, of vrouwen met een variant in een gen op het X-chromosoom.
- Bij een onduidelijke bevinding (VUS).
- Bij een nevenbevinding.
- Bij een blijvende sterke verdenking op een genetische aandoening bij een patiënt, ondanks een niet afwijkende uitslag van het genetisch onderzoek.
- Bij vragen over adviezen betreffende medische controles of een preventieve behandeling voor patiënt of familieleden.
Praktische aanbevelingen bloedafname:
Voor genetisch onderzoek is het wenselijk om snel bloed af te nemen, waarbij getracht kan worden dit te combineren met andere diagnostiek (zoals beeldvorming onder narcose of onderzoek naar (CMV-)infecties).
Kwaliteit van bewijs
Er is geen literatuur gevonden dat voldeed aan de PICO. De bewijskracht kon daarom niet worden vastgesteld.
Waarden en voorkeuren van patiënten (en eventueel hun naasten/verzorgers)
Er zijn diverse onderzoeken gedaan naar de voor- en nadelen van genetisch onderzoek. De voor- en nadelen worden samengevat door Blesson (2020).
In het algemeen zijn er de volgende voordelen van genetisch onderzoek voor de patiënt en/of familieleden:
- Verklaring/oorzaak vinden voor het gehoorverlies (en eventueel bijkomende problemen).
- Kunnen geven van een ‘naam’ aan de aandoening.
- Vermijden van lange zoektocht naar de oorzaak voor de problemen (zogenoemde diagnostische odyssee).
- Mogelijke aanleiding tot meer specifieke controle- en gerichte behandeladviezen.
- Helpen om een globale inschatting te maken wat betreft de prognose.
- Mogelijkheid ondersteuning te krijgen bij het inschakelen van hulp of ondersteunende middelen.
- Mogelijkheid om sociale ondersteuning en aansluiting te vinden via bijv. ouderverenigingen/patiëntengroepen.
- Informatie geven over herhalingskans voor ouders en andere familieleden.
- Mogelijkheid kunnen geven om keuzes te maken met betrekking tot kinderwens en onderzoek tijdens een zwangerschap (prenatale diagnostiek) of soms voorafgaand aan een zwangerschap (pre-implantatie genetische test (PGT)).
- Schuldgevoelens kunnen wegnemen ten aanzien van de oorzaak.
- Mogelijkheid deelname aan wetenschappelijk onderzoek wat kan helpen bij het verzamelen van meer informatie maar ook voor bijv. het ontwikkelen van toekomstige therapieën.
Nadelen genetisch onderzoek voor de patiënt en/of familieleden:
- Kans op het vinden van een onduidelijke uitslag (variant van onzekere betekenis, VUS) wat kan leiden tot meer onzekerheid.
- Kans op nevenbevindingen.
- Onverwacht vinden van een genetische afwijking bij een ouder.
- Angst voor een “negatief” label bij het vinden van een genetische oorzaak/syndroom.
- Kan leiden tot schuldgevoelens als er bijv. een overgeërfde genetische variant wordt gevonden die de oorzaak is.
- Kan leiden tot teleurstelling als er geen genetische oorzaak gevonden wordt.
- Bloedafname kan belastend zijn.
- Kosten.
De waarde die de patiënt aan deze aspecten hecht verschilt per persoon. Het is daarom van belang om de voor- en nadelen van genetisch onderzoek vooraf te bespreken (pre-test counseling).
Kostenaspecten
Het stellen van een genetische diagnose in een vroeg stadium kan kosten verminderen door het vermijden van onnodige andere diagnostiek en de zogenoemde diagnostische odyssee (Olde Keizer, 2021).
Het testen van een enkel gen is goedkoper dan een brede test met analyse van een genpanel. Echter, omdat voor de meeste patiënten niet van tevoren voorspeld kan worden in welk gen de genetische oorzaak gezocht moet worden (heterogene aandoening), is het noodzakelijk om heel veel genen te testen (Zazo Seco, 2017). Door de jaren heen zijn de mogelijkheden van DNA-testen sterk toegenomen en is een brede test met analyse van een genpanel (middels genoom-sequencing) al gauw goedkoper dan het testen van enkele genen. Daarnaast bespaart het inzetten van een brede test tijd in het diagnostische traject vergeleken met het sequentieel inzetten van enkele genen. Het voordeel van een brede test is bovendien dat er een heranalyse van bestaande data kan plaatsvinden wanneer er extra klinische kenmerken zijn, of wanneer er bijvoorbeeld een nieuw gen voor erfelijke slechthorendheid is geïdentificeerd, dat eerder nog niet in het analysepanel was opgenomen.
Gelijkheid ((health) equity/equitable)
De interventie draagt bij aan gelijkheid in de zorg, omdat het duidelijke indicaties stelt voor genetisch onderzoek bij kinderen met slechthorendheid. Waar specifiek aanvullend onderzoek nodig is, wordt dit bepaald op basis van duidelijke klinische indicaties.
Genetisch onderzoek is voor elke patiënt evenredig beschikbaar. Erfelijkheidsonderzoek valt onder de basis verzekerde zorg, maar mensen zijn wel hun eigen bijdrage en eventueel eigen risico kwijt als zij geen andere medische kosten hebben.
Voor sommige mensen kan het moeten betalen van eventueel eigen risico een reden zijn om af te zien van erfelijkheidsonderzoek. Omdat deze richtlijn gericht is op kinderen is deze beperking er niet.
Aanvaardbaarheid:
In Nederland zijn er in meerdere genoomlaboratoria waarbij er een mogelijkheid is om een genpanel slechthorendheid te verrichten.
Zoals benoemd in de “Leidraad aanvragen genetische kiembaandiagnostiek door niet klinisch genetische zorgprofessionals” en bijbehorende bijlage Bijlage Voorwaarden inzetten genetische kiembaandiagnostiek (VKGN, 2024), is het belangrijk dat de genetisch onderzoek (genenpanel slechthorendheid inclusief CNV-analyse) wordt aangevraagd door een klinisch geneticus of een andere bekwame zorgprofessional (KNO-arts of kinderarts).
Het informed consent dient te worden vastgelegd in het medisch dossier. Er kan voor het vastleggen van informed consent gebruik worden gemaakt van het “toestemmingsformulier uitgebreid genetisch onderzoek” dat te vinden is op de website van de VKGN (www.vkgn.org).
Ethische aanvaardbaarheid
De aanbeveling om te werken met een genpanel voorkomt sequentieel testen en scheelt de zorg en de patiënt tijd en kosten. Exoom- of genoombrede sequencing heeft tot gevolg dat het volledige DNA van een individu in kaart wordt gebracht. Het genetisch onderzoek kan in een klein aantal gevallen tot extra onzekerheid leiden als er sprake is van een variant van onbekende betekenis (VUS) of een nevenbevinding.
Niet van alle varianten is meteen duidelijk wat ze betekenen. Varianten kunnen bijvoorbeeld voorkomen in genen met een nog onbekende functie, maar het kunnen ook nieuwe varianten in bekende genen zijn met een nog onbekend effect. In beide gevallen spreekt men van een “variant of unknown significance (VUS)”. Het huidige beleid in Nederland is dat een VUS alleen gemeld wordt indien deze zeer verdacht is en/of nader onderzoek tot meer duidelijkheid en daarmee herclassificatie kan leiden.
In sommige patiënten is het zinvol om, naast de analyse van het genpanel voor slechthorendheid, verder te kijken naar andere mogelijke oorzaken in het DNA (bijvoorbeeld als er een sterke familiaire aanleg is). Bij een dergelijke uitgebreide analyse is er een kans dat er veranderingen in het DNA gevonden worden die een andere erfelijke aandoening veroorzaken dan slechthorendheid. Men spreekt dan van een nevenbevinding. Een nevenbevinding komt in de klinische praktijk bij de analyse van een exoom voor bij ongeveer 1% (van der Schoot, 2021). Terugkoppeling van dergelijke varianten kan leiden tot onzekerheid bij de persoon die de test heeft ondergaan, maar kan gezondheidswinst in de toekomst opleveren (van der Schoot, 2024).
Duurzaamheid
Bij de interventie spelen de duurzaamheidsaspecten een rol aangezien het doen van genetisch onderzoek een energie intensief onderzoek is. Goede criteria bij het aanvragen zorgen voor het doelmatig gebruik van genetisch onderzoek. Het vroegtijdig vaststellen van een oorzaak van de slechthorendheid kan verdere diagnostische onderzoeken voorkomen en daarmee (niet-duurzame) interventies en kosten besparen.
Haalbaarheid
De geformuleerde verwijscriteria zullen naar verwachting niet leiden tot extra belasting voor de zorg, aangezien genpanel diagnostiek over het algemeen al standaard zorg is. De aanbevelingen leiden naar verwachting juist tot meer vroegtijdig en doelmatig gebruik van genetisch onderzoek en betere verwijzingen naar de klinisch geneticus.
Rationale van aanbeveling-1: weging van argumenten voor en tegen de interventies
Bij welke kinderen met perceptieve slechthorendheid dient genetisch onderzoek uitgevoerd te worden?
Er werden geen artikelen gevonden die voldeden aan de PICO bij het systematisch literatuuronderzoek naar het effect van genetisch testen in kinderen met perceptief gehoorverlies. Er zijn maar weinig vergelijkende studies verricht. Dit komt doordat het vrijwel niet mogelijk is om een genetische diagnose te stellen zonder genetisch onderzoek te verrichten. Toch is onze aanbeveling sterk geformuleerd vanwege de klinisch ervaren relevantie.
Het is aan te bevelen om, bij voorkeur in een zo vroeg mogelijk stadium, een genetische oorzaak voor slechthorendheid vast te stellen. Een genetische oorzaak kan bijdragen aan persoonsgerichte zorg voor kinderen met een onderliggende genetische diagnose en daardoor gezondheidswinst opleveren.
In de meeste gevallen wordt gekozen voor een uitgebreid genetisch onderzoek, waarmee onderzoek gedaan wordt naar alle (op dat moment aantoonbare) bekende genetische oorzaken voor slechthorendheid. Genetisch onderzoek van zo’n genpanel slechthorendheid heeft een hoge diagnostische opbrengst. Dit geldt voor dubbelzijdig evident perceptief gehoorverlies (>30 dB beiderzijds) die aangeboren is of ontstaat op jonge kinderleeftijd.
Slechts zelden is er een genetische oorzaak aantoonbaar bij éénzijdig gehoorverlies als er geen bijkomende bijzonderheden zijn. Genetisch onderzoek bij éénzijdige/unilaterale (geïsoleerde) slechthorendheid wordt daarom als niet kosteneffectief en dus niet zinvol beschouwd.
Eindoordeel:
Sterke aanbeveling voor (Doen) en Sterke aanbeveling tegen.
Rationale van aanbeveling-2: weging van argumenten voor en tegen de interventies
Welke informatie dient besproken te worden met ouders/welke begeleiding is nodig voor en na het genetisch onderzoek?
Het is belangrijk dat ouders goed gecounseld worden over de mogelijke uitslagen, omdat sommige uitslagen extra stress, onzekerheid en zorgen bij ouders kunnen opleveren. Ouders kunnen ook een keuze maken over het worden ingelicht over nevenbevindingen, zie hiervoor Uitgebreid DNA-onderzoek en Nevenbevindingen | Richtlijnen en Protocollen | Vakinformatie (vkgn.org).
De aanvraag van genetisch onderzoek kan gedaan worden door een bekwame zorgprofessional. Naast een klinisch geneticus, kan dit ook een KNO-arts of kinderarts zijn (zie bijlage Voorwaarden inzetten genetische kiembaandiagnostiek en de gehele leidraad voor de voorwaarden ten aanzien van inzetten genetische kiembaandiagnostiek door bekwame zorgprofessional).
Eindoordeel:
Sterke aanbeveling voor (Doen).
Onderbouwing
Genetic testing is an integral part of the diagnostic process for children with hearing impairment. Rehabilitation has to start as soon as possible (see “module Diagnostisch traject slechthorend kind”) and a genetic diagnosis can assist in determining the rehabilitation program.
In 60-70% of children with prelingual hearing loss a genetic diagnosis can be established. Apart from gaining insight in e.g. progression of the disease and finetuning rehabilitation, a genetic diagnosis can have implications for parents and other family members. Since it is not always possible to accurately predict beforehand, based on the severity and audiogram, whether the hearing loss is hereditary, the possibility of a genetic cause should always be discussed with parents.
Current practice shows that genetic testing is not always discussed or initiated (in time). Furthermore, not all healthcare providers involved have sufficient knowledge of the possibilities, logistics, and importance of genetic diagnostic testing. Furthermore, not always the right patients are referred for genetic testing. Based on available literature, extensive genetic diagnostics for unilateral hearing loss is generally not very useful, since this is rarely hereditary.
Rapid innovations in genetic testing improve the diagnostic rate, and it is now possible to identify a genetic cause in a large proportion of cases in one single test. Therefore, it is important that healthcare providers involved in the diagnosis of hearing loss are aware of the possibilities and can discuss it with parents.
No summary of literature available.
A systematic review of the literature was performed to answer the following question(s):
What are the favourable and unfavourable effects of diagnostic genetic testing in children with sensorineural hearing loss?
Table 1. PICO
|
Patients |
Children with sensorineural hearing loss (mean hearing loss >35 dB over 1,2 and 4 kHz).
|
|
Intervention |
Performing diagnostic genetic testing |
|
Control |
Not performing diagnostic genetic testing |
|
Outcomes |
|
|
Other selection criteria |
Systematic reviews, randomized controlled trials and observational studies |
Relevant outcome measures
The guideline panel considered syndromal diagnoses, non-syndromal diagnoses and patient satisfaction as a critical outcome measures for decision making; and unexpected findings, incidental findings and quality of life as an important outcome measures for decision making.
A priori, the guideline panel did not define the outcome measures listed above but used the definitions used in the studies.
The guideline panel defined [the following thresholds as a minimal clinically (patient) important difference:
- Syndromal diagnoses: Risk difference (RD) 10%
- Non-syndromal diagnoses: RD 10%
- Patient satisfaction: Mean difference (MD) 10% of maximum score
- Unexpected findings: RD 10%
- Incidental findings: RD 10%
- Quality of life: RD 10% of maximum score
Search and select (Methods)
A systematic literature search was performed by a medical information specialist using the following bibliographic databases: Embase.com and Ovid/Medline. Both databases were searched from 2014 to 11th of July 2025 for systematic reviews, RCTs and observational studies. Systematic searches were completed using a combination of controlled vocabulary/subject headings (e.g., Emtree-terms, MeSH) wherever they were available and natural language keywords. The overall search strategy was derived from four primary search concepts: (1) nonsyndromic; (2) perception deafness; (3) genetic screening; (4) children. Duplicates were removed using EndNote software. After deduplication a total of 197 records were imported for title/abstract screening.
No studies were selected based on title and abstract screening.
- Blesson A, Cohen JS. Genetic Counseling in Neurodevelopmental Disorders. Cold Spring Harb Perspect Med. 2020 Apr 1;10(4):a036533. doi: 10.1101/cshperspect.a036533. PMID: 31501260; PMCID: PMC7117955.
- Clay S, Evans A, Zambrano R, Otohinoyi D, Hicks C, Tsien F. Bioinformatics characterization of variants of uncertain significance in pediatric sensorineural hearing loss. Front Pediatr. 2024 Feb 21;12:1299341. doi: 10.3389/fped.2024.1299341. PMID: 38450295; PMCID: PMC10915201.
- Downie L, Halliday J, Burt R, Lunke S, Lynch E, Martyn M, Poulakis Z, Gaff C, Sung V, Wake M, Hunter MF, Saunders K, Rose E, Lewis S, Jarmolowicz A, Phelan D, Rehm HL; Melbourne Genomics Health Alliance; Amor DJ. Exome sequencing in infants with congenital hearing impairment: a population-based cohort study. Eur J Hum Genet. 2020 May;28(5):587-596. doi: 10.1038/s41431-019-0553-8. Epub 2019 Dec 12. Erratum in: Eur J Hum Genet. 2021 Feb;29(2):363. doi: 10.1038/s41431-020-00750-4. Erratum in: Eur J Hum Genet. 2021 Aug;29(8):1316. doi: 10.1038/s41431-020-00772-y. PMID: 31827275; PMCID: PMC7171096.
- Gooch C, Rudy N, Smith RJ, Robin NH. Genetic testing hearing loss: The challenge of non syndromic mimics. Int J Pediatr Otorhinolaryngol. 2021 Nov;150:110872. doi: 10.1016/j.ijporl.2021.110872. Epub 2021 Aug 16. PMID: 34433113; PMCID: PMC8560556.
- Kenna MA. Genetic testing for pediatric hearing loss: no time to waste. Hum Genet. 2022 Apr;141(3-4):315-317. doi: 10.1007/s00439-021-02333-9. Epub 2022 Mar 30. PMID: 35353226.
- Li MM, Tayoun AA, DiStefano M, Pandya A, Rehm HL, Robin NH, Schaefer AM, Yoshinaga-Itano C; ACMG Professional Practice and Guidelines Committee. Electronic address: documents@acmg.net. Clinical evaluation and etiologic diagnosis of hearing loss: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022 Jul;24(7):1392-1406. doi: 10.1016/j.gim.2022.03.018. Epub 2022 May 10. PMID: 35802133.
- Liao EN, Taketa E, Mohamad NI, Chan DK. Outcomes of Gene Panel Testing for Sensorineural Hearing Loss in a Diverse Patient Cohort. JAMA Netw Open. 2022 Sep 1;5(9):e2233441. doi: 10.1001/jamanetworkopen.2022.33441. PMID: 36166228; PMCID: PMC9516276.
- Lieu JEC, Kenna M, Anne S, Davidson L. Hearing Loss in Children: A Review. JAMA. 2020 Dec 1;324(21):2195-2205. doi: 10.1001/jama.2020.17647. PMID: 33258894.
- Molecular Otolaryngoly and Renal Research Laboratories. 2025. https://hereditaryhearingloss.org/ [cited 12 November 2025].
- Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006 May 18;354(20):2151-64. doi: 10.1056/NEJMra050700. PMID: 16707752.
- Núñez Batalla FJ, Fernández-Cedrón Bermejo C, Guntín García M, Sandoval Menéndez I, Fresno Díaz E, Gómez Martínez JR, Llorente Pendás JL. Universal neonatal hearing screening and delayed hearing loss or late-developmental hearing loss. Acta Otorrinolaringol Esp (Engl Ed). 2023 Sep-Oct;74(5):283-289. doi: 10.1016/j.otoeng.2022.10.007. Epub 2023 May 4. PMID: 37149133.
- Olde Keizer RACM, Henneman L, Ploos van Amstel JK, Vissers LELM, Frederix GWJ. Economic evaluations of exome and genome sequencing in pediatric genetics: considerations towards a consensus strategy. J Med Econ. 2021 Nov;24(sup1):60-70. doi: 10.1080/13696998.2021.2009725. PMID: 34915793.
- Peart L, Gonzalez J, Morel Swols D, Duman D, Saridogan T, Ramzan M, Zafeer MF, Liu XZ, Eshraghi AA, Hoffer ME, Angeli SI, Bademci G, Blanton S, Smith C, Telischi FF, Tekin M. Dispersed DNA variants underlie hearing loss in South Florida's minority population. Hum Genomics. 2023 Nov 24;17(1):103. doi: 10.1186/s40246-023-00556-7. PMID: 37996878; PMCID: PMC10668374.
- Shearer AE, Hildebrand MS, Odell AM, Smith RJH. Genetic Hearing Loss Overview. 1999 Feb 14 [updated 2025 Apr 3]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301607.
- Sheffield AM, Smith RJH. The Epidemiology of Deafness. Cold Spring Harb Perspect Med. 2019 Sep 3;9(9):a033258. doi: 10.1101/cshperspect.a033258. PMID: 30249598; PMCID: PMC6719589.
- van der Schoot V, Haer-Wigman L, Feenstra I, Tammer F, Oerlemans AJM, van Koolwijk MPA, van Agt F, Arens YHJM, Brunner HG, Vissers LELM, Yntema HG. Lessons learned from unsolicited findings in clinical exome sequencing of 16,482 individuals. Eur J Hum Genet. 2022 Feb;30(2):170-177. doi: 10.1038/s41431-021-00964-0. Epub 2021 Oct 25. PMID: 34697415; PMCID: PMC8821629.
- van der Schoot V, van der Meer E, Hillen MA, Yntema HG, Brunner HG, Oerlemans AJM. Exploring uncertainties regarding unsolicited findings in genetic testing. Patient Educ Couns. 2024 Feb;119:108064. doi: 10.1016/j.pec.2023.108064. Epub 2023 Nov 10. PMID: 37976670.
- Yamamoto N, Balciuniene J, Hartman T, Diaz-Miranda MA, Bedoukian E, Devkota B, Lawrence A, Golenberg N, Patel M, Tare A, Chen R, Schindler E, Choi J, Kaur M, Charles S, Chen J, Fanning EA, Dechene E, Cao K, Jill MR, Rajagopalan R, Bayram Y, Dulik MC, Germiller J, Conlin LK, Krantz ID, Luo M. Comprehensive Gene Panel Testing for Hearing Loss in Children: Understanding Factors Influencing Diagnostic Yield. J Pediatr. 2023 Nov;262:113620. doi: 10.1016/j.jpeds.2023.113620. Epub 2023 Jul 19. PMID: 37473993.
- Zazo Seco C, Wesdorp M, Feenstra I, Pfundt R, Hehir-Kwa JY, Lelieveld SH, Castelein S, Gilissen C, de Wijs IJ, Admiraal RJ, Pennings RJ, Kunst HP, van de Kamp JM, Tamminga S, Houweling AC, Plomp AS, Maas SM, de Koning Gans PA, Kant SG, de Geus CM, Frints SG, Vanhoutte EK, van Dooren MF, van den Boogaard MH, Scheffer H, Nelen M, Kremer H, Hoefsloot L, Schraders M, Yntema HG. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur J Hum Genet. 2017 Feb;25(3):308-314. doi: 10.1038/ejhg.2016.182. Epub 2016 Dec 21. PMID: 28000701; PMCID: PMC5315517.
- Zhu Y, Hu L, Yang L, Wang L, Lu Y, Dong X, Xiao T, Xu Z, Wu B, Zhou W. Association Between Expanded Genomic Sequencing Combined With Hearing Screening and Detection of Hearing Loss Among Newborns in a Neonatal Intensive Care Unit. JAMA Netw Open. 2022 Jul 1;5(7):e2220986. doi: 10.1001/jamanetworkopen.2022.20986. PMID: 35816303; PMCID: PMC9274323.
Risk of Bias tables
Not applicable.
Table of excluded studies
Not applicable.
Beoordelingsdatum en geldigheid
Publicatiedatum : 16-07-2026
Beoordeeld op geldigheid : 16-07-2026
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2024 een multidisciplinair cluster ingesteld. Het cluster Pediatrische KNO bestaat uit meerdere richtlijnen (zie hier de actuele clusterindeling). De stuurgroep bewaakt het proces van modulair onderhoud binnen het cluster. De expertisegroepsleden brengen hun expertise in, indien nodig. De volgende personen uit het cluster zijn betrokken geweest bij de herziening van deze module:
Clusterstuurgroepleden
- Mevr. Dr. J. (Jolijn) Brouwer, voorzitter, KNO-arts, HagaZiekenhuis, Den Haag
- Dhr. Dr. A.J.N. (Joost) Bittermann, KNO-arts, UMC Utrecht
- Mevr. Dr. Y.J.W. (Yvonne) Simis, Klinisch fysicus – audioloog, Amsterdam UMC
- Mevr. H. (Hanneke) van der Hoek-Snieders, logopedist, UMC Utrecht
Betrokken clusterexpertisegroepleden
- Mevr. Dr. J.S. (Jolien) Klein Wassink-Ruiter, klinisch geneticus, UMC Groningen
- Mevr. H.G. (Helger) IJntema, klinisch moleculair geneticus, Radboud UMC, Nijmegen
- Mevr. Dr. J.C.C. (Josine) Widdershoven, KNO-arts, Maastricht UMC
- Mevr. Dr. A.J. (Arlette) van Sorge, Oogarts (kinderoogheelkunde), Koninklijke Visio, Amsterdam
Met ondersteuning van
- Dr. J.C. (José) Maas, senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Drs. E.R.L. (Evie) Verweg, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- L.H.M. (Linda) Niesink-Boerboom, medisch informatiespecialist, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
Een overzicht van de belangen van de clusterleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten via secretariaat@kennisinstituut.nl.
Gemelde (neven)functies en belangen stuurgroepleden
|
Naam |
Hoofdfunctie |
Nevenwerkzaamheden |
Persoonlijke financiële belangen |
Persoonlijke relaties |
Extern gefinancierd onderzoek |
Intellectuele belangen en reputatie |
Overige belangen |
Datum |
Restrictie |
|
Jolijn Brouwer |
KNO arts HagaZiekenhuis |
Lid kerngroep pediatrie van de Nederlandse Vereniging KNO |
Vacatiegeld voor de werkzaamheden in de kerngroep pediatrie |
Geen |
Geen |
Geen |
Geen |
06-05-2024 (Herbevestiging 26-05-2026) |
Geen restrictie |
|
Joost Bittermann |
Ik ben fulltime kinder-KNO-arts in het Wilhelmina Kinderziekenhuis te Utrecht (UMC-Utrecht). Dit is mijn reguliere betaalde vaste baan. |
-Op uitnodiging geef ik uitleg/instructie/presentatie/cursus mbt gebruik coblatie. Coblatie is een techniek om o.a. amandelen mee te behandelen. De naam van het bedrijf is 'Smith & Nephew'
-Ik ben voorzitter van de kerngroep kinder-KNO van de Nederlandse Vereniging KNO |
-Op uitnodiging geef ik uitleg/instructie/presentatie/cursus mbt gebruik coblatie (bedrijf Smith & Nephew): Ik ontvang een financiële vergoeding wanneer ik een dergelijke activiteit heb gedaan.
-Voor mijn werkzaamheden als kerngroep voorzitter ontvang ik vacatiegeld |
Geen |
Geen |
Geen |
Geen |
12-03-2024 (Herbevestiging 17-03-2026) |
Geen restrictie |
|
Hanneke van der Hoek-Snieders |
Medewerkend teamleider paramedici, UMC Utrecht |
Ik geef een post hbo-cursus aan logopedisten over het behandelen van jonge niet/nauwelijks sprekende kinderen, betaald |
Geen |
Geen |
Geen |
Geen |
Geen |
19-01-2025 (Herbevestiging 17-03-2026) |
Geen restrictie |
|
Yvonne Simis |
Klinisch fysicus – audioloog Amsterdam UMC Hoofd audiologisch Centrum Amsterdam UMC |
Externe klachtenfunctionaris voor medewerkers van MOC ’t Kabouterhuis Amsterdam |
Geen |
Geen |
Geen |
Geen |
Geen |
19-12-2023 (Herbevestiging 16-03-2026) |
Geen restrictie |
Gemelde (neven)functies en belangen betrokken expertisegroepleden
|
Naam |
Hoofdfunctie |
Nevenwerkzaamheden |
Persoonlijke financiële belangen |
Persoonlijke relaties |
Extern gefinancierd onderzoek |
Intellectuele belangen en reputatie |
Overige belangen |
Datum |
Restrictie |
|
Jolien Klein Wassink-Ruiter |
Klinisch geneticus UMCG |
lid van de landelijke werkgroep DOOFNL en de landelijke werkgroep schisis |
Geen |
Geen |
Geen |
Nee |
Geen |
07-02-2024 (Herbevestiging 07-05-2026) |
Geen restrictie |
|
Helger IJntema |
Hoofd laboratorium Genoomdiagnostiek, Radboud UMC |
geen |
geen |
geen |
geen |
geen |
geen |
02-04-2025 (Herbevestiging 16-03-2026) |
Geen restrictie |
|
Josine Widdershoven |
KNO-arts Maastricht UMC+ |
KNO-arts Universitair Ziekenhuis Antwerpen (BE) 1 dag/maand onbetaald consulent |
Geen |
Geen |
Geen |
Geen |
Geen |
13-03-2025 (Herbevestiging 16-03-2026) |
Geen restrictie |
|
Arlette van Sorge |
Oogarts Koninklijke Visio Amsterdam |
Voorzitter VRS (Vereniging Revalidatie bij Slechtziendheid), Stuurgroep cluster Oog (NOG), Bestuurslid Oogcontact, Lid van visitatiecommissie NOG |
Geen |
Geen |
Geen |
Geen |
Geen |
24-01-2025 (Herbevestiging 18-03-2026) |
Geen restrictie |
Inbreng patiëntenperspectief
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijnmodule voerden de clusterleden conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uit om te beoordelen of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling is de richtlijnmodule op verschillende domeinen getoetst (zie het stroomschema bij Werkwijze).
|
Module |
Uitkomst raming |
Toelichting |
|
Genetisch onderzoek slechthorend kind |
geen financiële gevolgen |
Uit de toetsing volgt dat de aanbeveling(en) niet breed toepasbaar zijn (<5.000 patiënten) en zal daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven. |
Werkwijze
Voor meer details over de gebruikte richtlijnmethodologie verwijzen wij u naar de Werkwijze. Relevante informatie voor de ontwikkeling/herziening van deze richtlijnmodule is hieronder weergegeven.
Zoekverantwoording
Zoekopbrengst - 11 juli 2025
|
|
EMBASE |
OVID/MEDLINE |
Ontdubbeld |
|
SR |
5 |
2 |
6 |
|
RCT |
15 |
0 |
15 |
|
Observationele studies |
157 |
42 |
176 |
|
Totaal |
177 |
44 |
197* |
*in Rayyan
Zoekstrategie - 11 juli 2025
Embase.com
|
No. |
Query |
Results |
|
#1 |
'nonsyndromic hearing loss'/exp OR 'non syndromic hearing loss'/exp OR 'nonsyndromic sensorineural hearing loss'/exp OR 'non syndromic sensorineural hearing loss'/exp OR 'non-syndromal*':ti,ab,kw OR nonsyndromal*:ti,ab,kw OR 'non syndromic*':ti,ab,kw OR nonsyndromic*:ti,ab,kw |
15303 |
|
#2 |
'perception deafness'/exp OR 'congenital deafness'/exp OR 'nonsyndromic hearing loss'/exp OR 'non syndromic hearing loss'/exp OR 'nonsyndromic sensorineural hearing loss'/exp OR 'non syndromic sensorineural hearing loss'/exp OR (((perception OR perceptive OR congenital) NEAR/3 deafness):ti,ab,kw) OR (((sensorineural OR 'sensori neural' OR sensory OR neurosensory OR neurogenic OR neural OR nerve) NEAR/4 (hearing OR hearingloss OR deafness)):ti,ab,kw) OR 'acoustic nerve diseas*':ti,ab,kw OR 'acoustic nerve palsy':ti,ab,kw OR 'auditory nerve palsy':ti,ab,kw OR 'auditory nerve paralysis':ti,ab,kw OR 'auditory nerve paresis':ti,ab,kw OR 'auditory perceptual disorder*':ti,ab,kw OR 'central auditory diseas*':ti,ab,kw OR 'perceptive hearing loss':ti,ab,kw |
59044 |
|
#3 |
'genetic screening'/exp OR 'genetic analysis'/exp OR 'whole exome sequencing'/exp OR 'whole genome sequencing'/exp OR 'copy number variation'/exp OR 'single nucleotide polymorphism'/exp OR 'multiplex ligation dependent probe amplification'/exp OR (((genetic* OR dna) NEAR/3 (test* OR analys* OR screening)):ti,ab,kw) OR (((exome OR genome OR generation) NEAR/3 sequenc*):ti,ab,kw) OR 'copy number variation':ti,ab,kw OR 'single nucleotide variation*':ti,ab,kw OR cnv:ti,ab,kw OR snv:ti,ab,kw OR mlpa:ti,ab,kw OR episignature*:ti,ab,kw |
1218445 |
|
#4 |
'boy'/exp OR 'child'/exp OR 'minors'/exp/mj OR 'pediatric patient'/exp OR 'pediatrics'/exp OR 'schoolchild'/exp OR infan*:ti,ab OR toddler*:ti,ab OR minors*:ti,ab OR boy:ti,ab OR boys:ti,ab OR boyfriend:ti,ab OR boyhood:ti,ab OR girl*:ti,ab OR kid:ti,ab OR kids:ti,ab OR child*:ti,ab OR children*:ti,ab OR schoolchild*:ti,ab OR juvenil*:ti,ab OR youth*:ti,ab OR pediatric*:ti,ab OR paediatric*:ti,ab OR peadiatric*:ti,ab OR school*:ti,ab |
5146428 |
|
#4 |
#1 AND #2 AND #3 AND #4 AND [2014-2025]/py NOT (('animal'/exp OR 'animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) NOT ('conference abstract'/it OR 'conference review'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) |
389 |
|
#5 |
'meta analysis'/de OR 'meta analysis (topic)'/exp OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de |
957678 |
|
#6 |
'clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti |
4580452 |
|
#7 |
'case control study'/de OR 'comparative study'/exp OR 'control group'/de OR 'controlled study'/de OR 'controlled clinical trial'/de OR 'crossover procedure'/de OR 'double blind procedure'/de OR 'phase 2 clinical trial'/de OR 'phase 3 clinical trial'/de OR 'phase 4 clinical trial'/de OR 'pretest posttest design'/de OR 'pretest posttest control group design'/de OR 'quasi experimental study'/de OR 'single blind procedure'/de OR 'triple blind procedure'/de OR (((control OR controlled) NEAR/6 trial):ti,ab,kw) OR (((control OR controlled) NEAR/6 (study OR studies)):ti,ab,kw) OR (((control OR controlled) NEAR/1 active):ti,ab,kw) OR 'open label*':ti,ab,kw OR (((double OR two OR three OR multi OR trial) NEAR/1 (arm OR arms)):ti,ab,kw) OR ((allocat* NEAR/10 (arm OR arms)):ti,ab,kw) OR placebo*:ti,ab,kw OR 'sham-control*':ti,ab,kw OR (((single OR double OR triple OR assessor) NEAR/1 (blind* OR masked)):ti,ab,kw) OR nonrandom*:ti,ab,kw OR 'non-random*':ti,ab,kw OR 'quasi-experiment*':ti,ab,kw OR crossover:ti,ab,kw OR 'cross over':ti,ab,kw OR 'parallel group*':ti,ab,kw OR 'factorial trial':ti,ab,kw OR ((phase NEAR/5 (study OR trial)):ti,ab,kw) OR ((case* NEAR/6 (matched OR control*)):ti,ab,kw) OR ((match* NEAR/6 (pair OR pairs OR cohort* OR control* OR group* OR healthy OR age OR sex OR gender OR patient* OR subject* OR participant*)):ti,ab,kw) OR ((propensity NEAR/6 (scor* OR match*)):ti,ab,kw) OR versus:ti OR vs:ti OR compar*:ti OR ((compar* NEAR/1 study):ti,ab,kw) OR (('major clinical study'/de OR 'clinical study'/de OR 'cohort analysis'/de OR 'observational study'/de OR 'cross-sectional study'/de OR 'multicenter study'/de OR 'correlational study'/de OR 'follow up'/de OR cohort*:ti,ab,kw OR 'follow up':ti,ab,kw OR followup:ti,ab,kw OR longitudinal*:ti,ab,kw OR prospective*:ti,ab,kw OR retrospective*:ti,ab,kw OR observational*:ti,ab,kw OR 'cross sectional*':ti,ab,kw OR cross?ectional*:ti,ab,kw OR multicent*:ti,ab,kw OR 'multi-cent*':ti,ab,kw OR consecutive*:ti,ab,kw) AND (group:ti,ab,kw OR groups:ti,ab,kw OR subgroup*:ti,ab,kw OR versus:ti,ab,kw OR vs:ti,ab,kw OR compar*:ti,ab,kw OR 'odds ratio*':ab OR 'relative odds':ab OR 'risk ratio*':ab OR 'relative risk*':ab OR 'rate ratio':ab OR aor:ab OR arr:ab OR rrr:ab OR ((('or' OR 'rr') NEAR/6 ci):ab))) |
16364227 |
|
#8 |
#4 AND #5 - SR |
5 |
|
#9 |
#4 AND #6 NOT #8 - RCT |
15 |
|
#10 |
#4 AND #7 NOT #8 OR #9 - OBS |
157 |
|
#11 |
#8 OR #9 OR #10 - Totaal |
177 |
Ovid/Medline
|
# |
Searches |
Results |
|
1 |
(non-syndromal* or nonsyndromal or non-syndromic* or nonsyndromic*).ti,ab,kf. |
12248 |
|
2 |
exp Hearing Loss, Sensorineural/ or ((perception or perceptive or congenital) adj3 deafness).ti,ab,kf. or ((sensorineural or 'sensori neural' or sensory or neurosensory or neurogenic or neural or nerve) adj4 (hearing or hearingloss or deafness)).ti,ab,kf. or 'acoustic nerve diseas*'.ti,ab,kf. or 'acoustic nerve palsy'.ti,ab,kf. or 'auditory nerve palsy'.ti,ab,kf. or 'auditory nerve paralysis'.ti,ab,kf. or 'auditory nerve paresis'.ti,ab,kf. or 'auditory perceptual disorder*'.ti,ab,kf. or 'central auditory diseas*'.ti,ab,kf. or 'perceptive hearing loss'.ti,ab,kf. |
44712 |
|
3 |
exp Genetic Testing/ or exp Exome Sequencing/ or exp DNA Copy Number Variations/ or exp Polymorphism, Single Nucleotide/ or (((genetic* or dna) adj3 (test* or analys* or screening)) or ((exome or genome or generation) adj3 sequenc*) or 'copy number variation' or 'single nucleotide variation*' or cnv or snv or mlpa or episignature*).ti,ab,kf. |
625756 |
|
4 |
(child* or schoolchild* or infan* or pediatri* or paediatr* or boy or boys or boyhood or girl or girls or girlhood or youth or youths or toddler* or childhood or preschool* or juvenile?).tw. |
2650219 |
|
5 |
1 and 2 and 3 and 4 |
193 |
|
6 |
limit 5 to yr="2014 -Current" |
124 |
|
7 |
6 not (comment/ or editorial/ or letter/) not ((exp animals/ or exp models, animal/) not humans/) |
121 |
|
8 |
exp Meta-Analysis/ or exp Network Meta-Analysis/ or exp Systematic Review/ or (networkmeta analy* or networkmetaanaly* or metaanaly* or meta analy* or metanaly* or prisma or prospero or metaanali* or meta anali* or metanali*).ti,ab,kf. or ((systemati* or scoping or umbrella or structured literature) adj3 (review* or overview*)).ti,ab,kf. or ((structured or systemic*) adj3 (review* or overview* or synth*) adj3 literature).ti,ab,kf. or (systemic* adj1 review*).ti,ab,kf. or ((systemati* or literature or database* or data base*) adj10 search*).ti,ab,kf. or ((structured or comprehensive* or systemic*) adj3 search*).ti,ab,kf. or ((literature adj3 (review* or overview*)) and (search* or database* or data base*)).ti,ab,kf. or ((data extraction* or data source*) and (study selection* or studies selection*)).ti,ab,kf. or (search strateg* and selection criteria*).ti,ab,kf. or (data source* and data synth*).ti,ab,kf. or (medline* or pubmed* or pub med* or embase or cochrane*).ti,ab,kf. or cochrane.jw. or ((critical* or rapid*) adj2 (review* or overview* or synth*)).ti. or (((critical* or rapid*) adj3 (review* or overview* or synth*)) and (search* or database* or data base*)).ab. or metasynth*.ti,ab,kf. or meta synth*.ti,ab,kf. |
843744 |
|
9 |
exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw. |
2914491 |
|
10 |
Case-control Studies/ or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or comparative study/ or control groups/ or controlled before-after studies/ or controlled clinical trial/ or double-blind method/ or historically controlled study/ or matched-pair analysis/ or single-blind method/ or (((control or controlled) adj6 (study or studies or trial)) or (compar* adj (study or studies)) or ((control or controlled) adj1 active) or "open label*" or ((double or two or three or multi or trial) adj (arm or arms)) or (allocat* adj10 (arm or arms)) or placebo* or "sham-control*" or ((single or double or triple or assessor) adj1 (blind* or masked)) or nonrandom* or "non-random*" or "quasi-experiment*" or "parallel group*" or "factorial trial" or "pretest posttest" or (phase adj5 (study or trial)) or (case* adj6 (matched or control*)) or (match* adj6 (pair or pairs or cohort* or control* or group* or healthy or age or sex or gender or patient* or subject* or participant*)) or (propensity adj6 (scor* or match*))).ti,ab,kf. or (confounding adj6 adjust*).ti,ab. or (versus or vs or compar*).ti. or exp cohort studies/ or epidemiologic studies/ or ((multicenter study/ or observational study/ or seroepidemiologic studies/ or (cohort* or 'follow up' or followup or longitudinal* or prospective* or retrospective* or observational* or multicent* or 'multi-cent*' or consecutive*).ti,ab,kf.) and ((group or groups or subgroup* or versus or vs or compar*).ti,ab,kf. or ('odds ratio*' or 'relative odds' or 'risk ratio*' or 'relative risk*' or aor or arr or rrr).ab. or (("OR" or "RR") adj6 CI).ab.)) or Case control.tw. or cohort.tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ |
8095931 |
|
11 |
7 and 8 - SR |
2 |
|
12 |
(7 and 9) not 11 - RCT |
0 |
|
13 |
(7 and 10) not (11 or 12) - OBS |
42 |
|
14 |
11 or 12 or 13 - Totaal |
44 |