Remdesivir
Uitgangsvraag
Wat is de plaats van remdesivir bij de behandeling van COVID-19 patiënten?
Aanbeveling
Remdesivir wordt niet aanbevolen als standaardbehandeling van opgenomen patiënten met COVID-19
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Er is literatuuronderzoek verricht naar de verschillen in klinische uitkomsten tussen behandeling met en zonder remdesivir bij patiënten met COVID-19. Tot 17 oktober 2021 werden er 6 gerandomiseerde gecontroleerde studies (RCTs) gevonden met in totaal 4289 patiënten in de remdesivir-groep en 3925 in de controle-groep. Een zevende studie (Barratt-Due, 2021) rapporteerde data van een subgroep van Noorse patiënten uit de Solidarity trial (Pan, 2020). In deze studie werd nieuwe data gepubliceerd over de mogelijke antivirale effecten van remdesivir en er werd gedetailleerde klinisch informatie beschreven. De studie van Barratt-Due werd daarom alleen meegenomen bij de evaluatie van specifieke subgroepen.
Omdat de geïncludeerde studies heterogene patiëntgroepen bevatten, zijn de resultaten, waar mogelijk, opgesplitst voor patiënten met milde, matige en ernstige COVID-19 symptomen op basis van respiratoire ondersteuning bij inclusie. Er werden alleen RCTs geïncludeerd in de analyse, waardoor de kwaliteit van bewijs initieel hoog was. Omdat er vijf ongeblindeerde of open-label trials waren, met een mogelijk risico op vertekening van de studieresultaten (risk of bias) bij subjectieve uitkomstmaten, werd de kwaliteit van dit bewijs waar nodig naar beneden bijgesteld. Daarnaast waren er meerdere studies met een relatief kleine populatie en mede hierdoor een grote spreiding van de puntschatter van de uitkomstmaat (imprecision), waardoor de kwaliteit van dit bewijs ook naar beneden werd bijgesteld. Eventueel werd de kwaliteit van het bewijs naar beneden bijgesteld als er veel heterogeniteit tussen de studies was, bijvoorbeeld in rapportage van de uitkomstmaat. De geïncludeerde patiënten verschilden ook per studie (bijvoorbeeld in de ernst van ziekte bij inclusie). Deze verschillen in inclusie werden niet meegenomen in de gradering van de studies, maar zullen wel worden meegenomen in de overwegingen.
Mortaliteit
Op basis van de gevonden resultaten kan worden geconcludeerd dat er voor de cruciale uitkomstmaat ‘mortaliteit’, geen voordeel is voor behandeling van opgenomen patiënten met remdesivir (risicoverschil: 0.01, 95%CI -0.01 tot 0.02; relatief risico 0.93, 95% CI 0.74-1.16). Dit is gebaseerd op 6 studies en heeft bewijskracht moderate.
In de studie van Beigel (2020) werd er wel een voordeel gezien van remdesivir op de mortaliteit na 28-29 dagen in de mensen met matig ernstige ziekte (patiënten met alleen extra zuurstof bij opname, maar geen vorm van invasieve beademing; ziekte ernst ‘moderate’ in Figuur 2). De studie van Ader (2021) en Pan (2020) bevestigden deze bevinding niet. Na het poolen en wegen van de data van de drie studies waren deze resultaten niet statistisch significant (risicoverschil: 3%, 95%CI -1% tot 7% en relatief risico 0.69, 95%CI 0.38 tot 1.23). De studie van Wang (2020) beschrijft geen specifieke subgroepen, maar ruim 80% van de populatie bestond uit patiënten die nog niet ernstig zuurstofbehoeftig waren (wel zuurstoftoediening, maar geen invasieve beademing). In deze studie werd er ook geen voordeel van remdesivir gezien op de mortaliteit. Patiënten in deze studie werden wel relatief laat behandeld: na gemiddeld 10 dagen klachten. In subgroep analyses bij patiënten met een (matig) ernstige COVID-19 infectie werd er ook geen statistisch significant mortaliteitsverschil aangetoond in de verschillende studies.
De publicatie van Pan (2020) was een interim analyse van de Solidarity trial. In mei 2022 verscheen de publicatie waarin de eindresultaten gepubliceerd werden (WHO Solidarity Trial Consortium, 2022). Ook in deze analyse werd er voor opgenomen patiënten met COVID-19 (milde, matige en ernstige COVID-19 samengenomen) geen voordeel gezien van behandeling met remdesivir. Echter, bij een subgroep analyse van patiënten met alleen extra zuurstofbehoefte maar geen noodzaak tot invasieve beademing was er wel een klein voordeel bij analyse van de mortaliteit in het ziekenhuis (14,6% overleed in de groep met remdesivir ten opzichte van 16.3% in de controlegroep; RR 0,87 95% CI 0,76-0,99). Er werd ook een klein en niet statistisch significant voordeel op de mortaliteit in de groep patiënten die zonder extra zuurstofbehoefte werd opgenomen in het ziekenhuis (2,9% overleed in de groep met remdesivir ten opzichte van 3,8% in de controlegroep; RR 0,76 95% CI 0,46-1,28. De mogelijke voordelen van remdesivir die in deze eindresultaten beschreven worden zijn klein. Omdat dit verschil minder was dan de vooraf gedefinieerde grens van klinische relevantie (3% punten verschil), wijzigt dit resultaat niet onze conclusie. Een publicatie van Ali (2022) beschrijft ook een mogelijk lagere mortaliteit bij opgenomen patienten met COVID-19 na behandeling met remdesivir. Dit verschil was niet statistisch significant. Omdat een groot deel van deze patiënten reeds beschreven werd in de Solidarity trial, verandert ook deze studie onze conclusie niet.
Overige uitkomstmaten
De gevonden studies tonen geen consistent positief klinisch effect van remdesivir op duur van ziekenhuisopname of andere uitkomstmaten, bewijskracht laag.
Wel liet de studie van Beigel (2020) zien dat de kans op het starten van beademing of andere vormen van invasieve respiratoire ondersteuning lager was in de remdesivir groep vergeleken met placebo. Ook toonde deze studie een significant sneller klinisch herstel en een kortere opnameduur van 5 dagen in het voordeel van remdesivir. Een voordeel dat ook bij een kleine RCT uit Egypte werd gezien: mediaan 10 dagen bij remdesivir en 16 dagen bij de standaard behandeling (Abd-Elsalam, 2021). Deze RCT is gepubliceerd na het voltooien van het literatuuronderzoek en inmiddels alweer teruggetrokken door het tijdschrift (Abd-Elsalam S. Retraction Notice, 2022). Alle andere studies lieten een dergelijk voordeel niet consistent zien. Een subgroep analyse van de studie van Spinner (2020) bij mensen nog vroeg in het beloop van de ziekte, werd dit positieve effect ook niet gezien: geen statistisch significante kans op een verbetering van de kliniek gezien bij behandeling binnen 9 dagen na ontstaan van symptomen.
Antivirale werking
In vitro en op basis van dieronderzoek waren er aanwijzingen voor een antiviraal effect van remdesivir bij COVID-19. Echter, dit effect werd in een drietal klinisch studies niet aangetoond. De meeste data over effecten op de virale load is afkomstig uit de DisCoVeRy studie (Ader, 2021), waarin bij 677 patiënten op enig moment in hun ziektebeloop een of meerdere nasofarynx monsters werden afgenomen. Behandeling met remdesivir liet ten opzichte van standaard zorg geen verschil zien in daling van de mediane viral load van dag 0 tot dag 3, en ook verder in het beloop (na 15 en 29 dagen) werden er geen verschillen in viral load waargenomen. Subgroep analyses van patiënten vroeg in hun ziektebeloop of met milde of matige ziekte lieten ook geen klinisch relevant antiviraal effect zien. De kleinere studie van Barratt-Due (2021) (n=36 met remdesivir en n=53 met standaardbehandeling) toonde ook geen significante daling in viral load in nasopharynx swabs in eerste 15 dagen na randomisatie (daily viral decrease rate 0.113; 95% CI -0.001-0.227). En Wang (2020) liet ook geen significant verschil zien in de afname van de viral load in de nasopharynx of oropharynx.
Conclusie
Uit de geïncludeerde RCTs blijkt dat er geen klinisch relevant verschil te zien is in de mortaliteit van opgenomen patiënten. De eindresultaten van de Solidarity trial (WHO Solidarity Trial Consortium, 2022) veranderen deze conclusie niet. Bij andere eindpunten (met name opnameduur) zijn er wel verschillen te zien met mogelijke klinische relevantie die door de toediening van remdesivir verklaard zouden kunnen worden. Klinische verbetering lijkt vooral sneller op te treden indien behandeling gestart wordt binnen 10 dagen na aanvang van symptomen bij patiënten met extra zuurstofbehoefte maar zonder indicatie voor (non)invasieve beademing. Deze voordelen worden met name in 1 studie waargenomen (Beigel, 2020). Deze studie werd verricht in een periode waarin corticosteroïd gebruik niet standaard gegeven werd. Of er een aanvullend effect aanwezig is, boven op de huidige standaard behandeling die nu corticosteroïden bevat, is hierdoor onduidelijk. Informatie uit de studie van Wang (2020), Ader (2021) en Barrett-Due (2021) tonen daarnaast geen klinisch relevante antivirale werking aan bij patiënten met zuurstofbehoefte. Het ontbreken van een effect op de virale klaring ontkracht het werkingsmechanisme van remdesivir. Alle bovenstaande redenen bijeengenomen maken dat er geen plaats meer is voor remdesivir in de standaard zorg van patiënten met COVID-19 in Nederland.
Overige overwegingen
In de context
Ook een Cochrane review trekt deze zelfde conclusie en stelt dat er matig-sterk bewijs is dat remdesivir een klein of geen effect heeft op de mortaliteit na 28 dagen (Ansems, 2021). Een mogelijk positief effect van remdesivir bij mensen met een beperkte zuurstofbehoefte wordt wel benoemd. In deze Cochrane review is de DisCoVeRy studie (Ader, 2021) nog niet meegenomen.
De WHO raadt in december 2021 - in tegenstelling tot de IDSA - het gebruik van remdesivir af bij opgenomen patiënten met COVID-19 in alle ziektestadia buiten studieverband (‘weak or conditional recommendation against the use of remdesivir in hospitalised patients with COVID-19’). Dit is een zwakke/voorwaardelijke aanbeveling. Er is daarbij gebruik gemaakt van precies dezelfde gepubliceerde data die in deze richtlijn ook zijn gebruikt, met uitzondering van de DisCoVeRy studie die later is verschenen.
De IDSA richtlijn adviseert in december 2021 behandeling met remdesivir nog wel bij patiënten met zuurstofbehoefte zonder mechanische ventilatie. Ook in dit advies is de meest recente DisCoVeRy studie nog niet meegenomen.
Bijwerkingen
Veiligheidsdata uit fase 1-onderzoek zijn nog niet gepubliceerd, ondanks dat er meerdere fase-1 onderzoeken zijn verricht. Vooral ALAT en ASAT-stijging wordt genoemd als bijwerking in de SmPC (https://lci.rivm.nl/remdesivir). De EMA onderzoekt sinds 2020 of er een verhoogd risico is op nefrotoxiciteit bij gebruik van remdesivir naar aanleiding van berichten hierover bij patiënten met COVID-19 die remdesivir toegediend kregen.
In meerdere studies werd veiligheidsdata verzamelend (Spinner, Wang, Ader, Barratt-Due). De DisCoVeRy studie van Ader (2020) onderzocht de veiligheid van remdesivir in 824 patiënten, waarbij er een vergelijkbaar aantal bijwerkingen werd gezien in de remdesivir groep ten opzichte van de controlegroep (32% versus 31% graad 3 of 4 bijwerkingen). Ook voor ernstige bijwerkingen was dit aantal nagenoeg gelijk. De studies van Spinner en Wang bevestigden deze data en lieten niet significant meer bijwerkingen zien bij het gebruik van remdesivir. In de studie van Barratt-Due werden bijwerkingen bij 42 patiënten met remdesivir bijgehouden. In deze kleine groep werden wat meer bijwerkingen gezien bij remdesivir (38.5% versus 25.3%), deze waren met name respiratoir van aard.
Samenvattend lijkt het gebruik van remdesivir niet gepaard te gaan met veel bijwerkingen.
Andere patiëntengroepen
Ook bij patiënten die niet zijn opgenomen in het ziekenhuis is remdesivir onderzocht (Gottlieb, 2022). In een groep van 562 ambulante patiënten met een hoog risico op een ernstig beloop toonde intraveneus toegediend remdesivir een voordeel: de kans op opname of overlijden binnen 28 dagen was 0,7% in de remdesivir groep versus 5,3% in de controle groep (hazard ratio 0,13 (95% CI 0,03-0,59)). Remdesivir werd in deze studie gedurende 3 opeenvolgende dagen intraveneus toegediend. Dit beperkt de praktische toepasbaarheid. Dit middel wordt daarom op dit moment ook niet aanbevolen in de ambulante setting. Onderzoek naar andere toedieningsvormen van remdesivir wordt op dit moment verricht.
Of het klinisch relevante effect van remdesivir ook ontbreekt in specifieke patiëntengroepen, zoals patiënten met een verminderd afweersysteem of heel vroeg in het ziektebeloop, is niet onderzocht. Hierover kan dan ook geen advies gegeven worden.
Kosten (middelenbeslag)
Remdesivir wordt niet (meer) aanbevolen bij de behandeling van COVID-19, de kosten zullen hier daarom niet beschreven worden.
Aanvaardbaarheid, haalbaarheid en implementatie
Remdesivir wordt niet (meer) aanbevolen bij de behandeling van COVID-19, dus implementatie is niet van toepassing.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Er werd geen klinisch relevant effect gevonden bij opgenomen patiënten die remdesivir kregen ten opzichte van de controle groepen. De bijwerkingen zijn nog beperkt onderzocht. Omdat het middel niet (meer) wordt voorgeschreven in het kader van de standaardbehandeling van COVID-19, zijn er geen specifieke voorkeuren of waarden beschreven van patiënten.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Van behandeling van opgenomen patiënten met remdesivir is geen winst op harde eindpunten zoals mortaliteit aangetoond. Slechts in enkele studies wordt er op andere eindpunten of in subgroepen een beperkt verschil gezien met mogelijk beperkte klinische relevantie in het voordeel van remdesivir. Hiertegenover staat de observatie van (mogelijke) bijwerkingen en kosten. Op basis hiervan kan geconcludeerd worden dat remdesivir geen plaats heeft bij de standaardbehandeling van COVID-19.
Onderbouwing
Remdesivir (GS-5734) is een nucleoside-analoog (prodrug) met in vitro activiteit tegen verschillende RNA-virussen. Het werd eerder getest als antiviraal middel voor behandeling van Ebola infecties (Warren, 2016), maar bleek niet voldoende klinisch effectief. Remdesivir heeft bij in-vitro onderzoek met onder meer humane longcellijnen activiteit tegen verschillende coronavirussen, onder meer SARS-CoV-1 laten zien (Sheahan, 2017). Voor SARS-CoV-2 is in vitro (Vero E6; niercellijn afkomstig van groene meerapen) antivirale activiteit gezien (Wang M, 2020). In beide studies ligt de EC50 bij lage micromolaire concentraties. Echter het is onduidelijk de of de gebruikte modellen en concentraties representatief zijn voor een infectie bij mensen in de longen. Tevens ontbreken validatie studies. Een studie met MERS-CoV infectie bij muizen lieten een antiviraal effect en minder orgaanschade van remdesivir zien (Sheahan, 2020). Toediening van remdesivir bij resusapen, 12 uur na blootstelling aan SARS-CoV-2, bleek te leiden tot minder longontsteking en een lagere hoeveelheid virus in de longen (Williamson, 2020).
Deze data leidden er toe dat remdesivir al vanaf het begin van de pandemie onderzocht werd voor behandeling van COVID-19. Echter zijn er nooit klinische dose-finding studies gedaan. Inmiddels is in diverse gerandomiseerde gecontroleerde studies (RCT’s) de effectiviteit onderzocht om de plaats van remdesivir bij de behandeling van COVID-19 patiënten te bepalen.
Mortality (crucial)
|
Moderate GRADE |
Treatment with remdesivir probably results in little to no difference in mortality when compared with treatment without remdesivir in hospitalized patients with COVID-19.
Sources: Ader, 2021; Beigel, 2020; Mahajan, 2021; Pan, 2020; Spinner, 2020; Wang, 2020 |
Extensive respiratory support (crucial)
|
Low GRADE |
Treatment with remdesivir may result in little to no difference in the need for extensive respiratory support when compared with treatment without remdesivir in hospitalized patients with COVID-19.
Sources: Barratt-Due, 2021; Beigel, 2020; Pan, 2020 |
Duration of hospitalization (important)
|
Low GRADE |
Treatment with remdesivir may result in little to no difference in the length of stay when compared with treatment without remdesivir in hospitalized patients with COVID-19.
Sources: Ader, 2021; Beigel, 2020; Pan, 2020; Wang, 2020 |
Time to clinical improvement (important)
|
Treatment with remdesivir may result in little to no difference in the time to clinical improvement when compared with treatment without remdesivir in hospitalized patients with COVID-19.
Sources: Ader, 2021; Beigel, 2020; Spinner, 2021; Wang, 2020 |
Ader (2021) (The DisCoVeRy trial) describes a phase 3, open-label, adaptive multicenter randomized controlled trial. This study evaluated the clinical efficacy of remdesivir in addition to standard of care versus standard of care alone in patients admitted to the hospital with COVID-19 with indication of oxygen or ventilation support. Standard of care included anticoagulants and corticosteroids as of a certain time point. Critically ill patients that required intensive care unit admission received 20 mg dexamethasone once daily for 5 days, followed by 10 mg once daily for 5 days at the clinician’s discretion. In total, 40% of the patients received systemic corticosteroids and 7.5% corticosteroids via the inhaled route. Other supportive treatments, such as immunomodulatory agents were given at the investigator’s discretion. None of the participants received a SARS-CoV-2 vaccine during the trial. The intervention group (n=429) received a remdesivir (200 mg loading dose on day 1, followed by 100 mg daily for up to 9 additional days) in addition to standard care. The control group (n=428) received standard of care alone. The length of follow-up was 29 days. The following relevant outcome measures were included: mortality, duration of hospitalization, clinical improvement. The primary outcome was the clinical status at day 15 measured by the WHO seven-point ordinal scale. The difference in clinical status at day 15 between the treatment groups was not statistically significant.
Barratt-Due (2021) (The NOR-Solidarity trial) described an open-label adaptive randomized controlled trial. This was an independent add-on randomized controlled trial to the WHO Solitarity trial (Pan, 2020). Barratt-Due (2021) evaluated the effects of remdesivir in hospitalized adult patients with confirmed COVID-19 infection. The ‘disease severity’ of the patients was not reported based on the requirement of respiratory support but on viral load. The intervention group (n=42) received remdesivir (200 mg loading dose on day 1, followed by 100 mg daily for up to 9 additional days) in addition to standard care. The control group (n=57) received standard of care alone. Standard of care changed during the study as a result of the recovery trial and updated WHO guidelines recommending systemic steroids for severe and critical COVID-19. In total, 2.4% of the patients in the intervention group received steroids, versus 1.8% in the control group. The length of follow-up was three months. The following relevant outcome measures were included: respiratory support. The primary outcome was in-hospital mortality. This outcome measure was also included in the WHO Solidarity trial (Pan, 2020). In total, 7.1% (95%CI 1.8 to 17.5) of the patients in the intervention group died during hospitalization, versus 7.0% (95%CI 2.2 versus 15.6) in the control group. No significant differences were seen between the treatment groups in mortality during hospitalization.
Beigel (2020) (The Adaptive COVID-19 Treatment Trial (ACTT-1)) described a double-blind, multicenter randomized, placebo-controlled trial of intravenous remdesivir in adults who were hospitalized with COVID-19 and had evidence of lower respiratory tract infection. Most patients had either one (25.9%) or two or more (54.5%) of the prespecified coexisting conditions at enrolment, most commonly hypertension (50.2%), obesity (44.8%), and type 2 diabetes mellitus (30.3%). Of all patients, 903 (85.0%) were in the moderate-to-severe disease stratum (requiring invasive or non-invasive mechanical ventilation, or requiring supplemental oxygen, or an SpO2 ≤ 94.0% on room air, or tachypnea (respiratory rate ≥ 24 breaths per minute)) and 159 (15.0%) were categorized as having mild disease (SpO2 > 94.0% and respiratory rate < 24 breaths per minute without supplemental oxygen requirement). The intervention group (n=541) received remdesivir (200 mg loading dose on day 1, followed by 100 mg daily for up to 9 additional days). The control group (n=521) received placebo for up to 10 days. All patients received supportive care according to the standard of care for the trial site hospital. In total, the patients received a variety of concomitant treatments during the study course: 82.3% received antibiotics, 32.2% received vasopressors, 23.0% received corticosteroids, 7.5% received other anti-inflammatory medications (not specified), 4.8% received monoclonal antibodies targeting cytokines. The length of follow-up was 28 days. The following relevant outcome measures were included: mortality, respiratory support, duration of hospitalization, clinical improvement. The primary outcome was time to recovery, defined by either discharge from the hospital or hospitalization for infection-control purposes only. Those who received remdesivir had a significantly shorter median recovery time of 10 days, as compared with 15 days among those who received placebo.
Mahajan (2021) described a randomized controlled trial which evaluated the improvement in clinical outcomes with remdesivir treatment for five days in hospitalized patients who were between 18 and 60 years and had COVID-19 confirmed by PCR assay within the last 4 days. Patients on mechanical ventilation were excluded. The intervention group (n=34) received remdesivir (200 mg loading dose on day 1, followed by 100 mg daily for up to 4 additional days) plus the standard of care. The control group (n=36) received standard care alone. Drugs like corticosteroids and heparin were given as per standard care protocol (exact percentage not mentioned). Standard of care was not further specified in this study. The length of follow-up was 12 days or until discharge/death. The following relevant outcome measure was included: mortality. The primary outcome is the clinical status. Both groups had similar outcomes after adjustment for baseline clinical status.
Pan (2020) (The WHO Solidarity trial) descripted an open-label randomized controlled trial. Pan (2020) compared the efficacy of the addition of remdesivir to standard of care in hospitalized patients with COVID-19. The study included patients who were aged 18 years or older, diagnosed with COVID-19, not known with receiving any trial drug, not expected to be transferred elsewhere within 72 hours, and hand no contraindication to any trial drug. The intervention group (n=2743) received remdesivir (200 mg loading dose on day 1, followed by 100 mg daily for up to 9 additional days) in addition to standard care. The control group (n=2708) received local standard of care alone. In total, the patients received several concomitant drugs during the study course: corticosteroids (intervention group 47.8%, control group 47.6%), convalescent plasma (intervention group 1.9%, control group 2.1%), anti-IL-6 drug (intervention group 4.9%, control group 5.3%). The length of follow-up was 28 days or up to discharge. The following relevant outcome measures were included: mortality, respiratory support, and duration of hospitalization. The primary outcome was in-hospital mortality. Remdesivir did not reduce mortality, overall or in subgroup analyses.
Spinner (2020) describes a randomized open-label multicentre clinical trial of intravenous remdesivir in adults who were hospitalized with COVID-19. The intervention group (n=384) received remdesivir (200 mg loading dose on day 1, followed by 100 mg daily). Remdesivir was provided for either 5 days (intervention group 1, n=193) or 10 days (intervention group 2, n=191). The control group (n=200) received standard of care. Standard of care included steroids in 15-19% of the patients. The original protocol allowed use of other agents with presumptive activity against SARS-CoV-2 if such use was local standard care. This exception was disallowed in a subsequent amendment of the protocol. The length of follow-up was 28 days. The following relevant outcome measures were included: mortality, clinical improvement. The primary outcome was clinical status on day 11 on a 7-point ordinal scale. On day 11, patients in the 5-day remdesivir group had statistically significantly higher odds of a better clinical status distribution than those receiving standard care. The clinical status distribution on day 11 between the 10-day remdesivir and standard care groups was not significantly different.
Wang (2020) describes a randomised, double-blind, placebo-controlled, multicentre trial of intravenous remdesivir in adults who were hospitalized with COVID-19. Almost all patients required a form of supplemental oxygen (supplemental oxygen, high-flow nasal cannula, non-invasive mechanical ventilation, ECMO or invasive mechanical ventilation) at day 1, except 3/78 (3.8%) of patients in the control group. The intervention group (n=158) received remdesivir (200 mg loading dose on day 1, followed by 100 mg daily for up to 9 additional days). The control group (n=78) received a placebo for up to 10 days. Patients received several concomitant drugs during the study course: corticosteroids (intervention group 65%, control group 68%), antibiotics (intervention group 90%, control group 94%), interferon alfa-2b (intervention group 29%, control group 38%), lopinavir-ritonavir (intervention group 28%, control group 29%), and vasopressors (intervention group 16%, control group 17%). The length of follow-up was 28 days. The following relevant outcome measures were included: mortality, duration of hospitalization, clinical improvement. The primary outcome was time to clinical improvement up to day 28, defined as the time (in days) from randomization to the point of a decline of two levels on a six-point ordinal scale of clinical status or discharged alive from hospital, whichever came first. Remdesivir was not associated with a difference in time to clinical improvement.
Table 1. Overview of RCTs comparing remdesivir with standard care in hospitalized COVID-19 patients.
|
Author |
Disease severity, based on need for respiratory support* |
Sample size |
Dosage |
|
Ader (2021) |
Mixed: Mild: n=12 (1%) Moderate: n=492 (59%) Severe: n=326 (40%) |
I: N=429 C: N=428 Total: N=857 |
- 200 mg remdesivir intravenously on day 1. - 100 mg once-daily (on days 2-10) |
|
Barratt-Due (2021) |
Mixed: Disease severity is defined based on viral load.
|
I: 42 C: 57 Total: 99 |
- 200 mg remdesivir intravenously (on day 0). - 100 mg once-daily (on days 1-9). |
|
Beigel (2020) |
Mixed: Mild: n=138 (13%) Moderate: n=435 (41%) Severe: n=478 (45%)
Baseline score was missing in 1% of the patients). |
I: 541 C: 521 Total: 1062 |
- 200 mg remdesivir intravenously (on day 1). - 100 mg once-daily (on days 2-10). |
|
Mahajan (2021) |
Mixed: Mild: 0 Moderate: n=53 (75.7%) Severe: n=17 (24.3%) mostly moderate receiving low flow supplemental oxygen (76%), severe cases received non-invasive ventilation or high-flow oxygen. |
I: 34 C: 36 Total: 70 |
- 200 mg remdesivir intravenously (on day 1). - 100 mg once-daily (on days 2-5). |
|
Pan (2020) |
Mixed: Mild: n= 1325 (24.3%), moderate & severe: n= 4126 (75.7%) |
I: N=2743 C: N=2708 Total: N=5451 |
- 200 mg remdesivir intravenously (on day 0). - 100 mg once-daily (on days 1-9). |
|
Spinner (2020) |
Mixed: Mild: n=491 (84%), Moderate: n=88 (15%) Severe: n=5 (1%) |
I1 (5-day remdesivir): 193 I2 (10-day remdesivir): 191 C: 200 Total: N=584 |
- 200 mg remdesivir intravenously (on day 1). - 100 mg once-daily (on days 2-5 or 2-10). |
|
Wang (2020) |
Mixed: mild: n=3 (1%) moderate: n =194 (82%) severe: n=38 (16%)
1 patient in the remdesivir group died at day 1 |
I: N=158 C: N=78 Total: N=236 |
- 200 mg remdesivir intravenously (on day 1). - 100 mg once-daily (on days 2-10). |
*Disease severity categories:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen [high flow nasal cannula (HFNC)/Optiflow], continuous positive airway pressure [CPAP], non-invasive ventilation [NIV], mechanical ventilation, extracorporeal membrane oxygenation [ECMO or ECLS]).
N: Total sample size
I: Intervention
C: Control
Results
Mortality (crucial)
Mortality in hospitalized patients with COVID-19 was reported in six studies (Ader, 2021; Beigel, 2020; Mahajan, 2021; Pan, 2020; Spinner, 2020; Wang, 2020). The results are presented separately for a follow-up of 28-30 days and other length of follow-up. Barratt-Due (2020) reported mortality, however as these patients were already included in the WHO Solidarity trial (Pan, 2020). To prevent double representation, these results were not included in the meta-analysis.
Mortality, 28-30 days
The pooled incidence of mortality in hospitalized patients in the intervention group was 421/4240 (9.9%), compared to 431/3925 (11.0%) in the control group. The pooled RR was 0.93 (95% CI 0.82 to 1.06; Figure 1), in favour of the intervention group. The pooled RD was 0.01 (95%CI -0.01 to 0.02), in favour of the intervention group. This is not considered clinically relevant.
Figure 1: Mortality (28-30days) in hospitalized patients
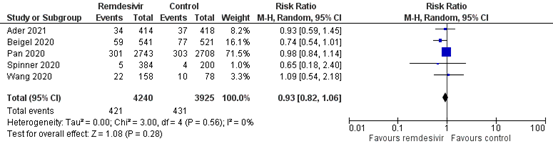
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Subgroup analysis according to disease severity
Mild disease
Two studies reported the mortality specifically for patients with a mild disease severity (Beigel, 2020; Pan, 2020). The mortality was 14/736 (1.9%) in the intervention group, compared to 16/727 (2.3%) in the control group (Figure 2). The pooled RR was 0.85 (95% CI 0.42 to 1.72), in favour of the intervention group. The pooled RD was 0.00 (95% CI -0.02 to 0.01). This is not considered clinically relevant.
Moderate disease
Three studies reported the mortality for patients with a moderate disease severity (Ader, 2021; Beigel, 2020; Pan, 2020). The mortality in the intervention group was 216/2313 (9.3%), compared to 259/2265 (11.4%) in the control group (Figure 2). The RR was 0.69 (95%CI 0.38 to 1.23) in favour of the intervention group. The RD was just below 0.03 (95%CI -0.01 to 0.07), in favour of the intervention group. This is considered clinically relevant.
Severe disease
Two studies reported the mortality for patients with a severe disease severity (Ader, 2021; Beigel, 2020). The mortality was 164/641 (25.6%) in the intervention group, compared to 142/652 (21.8%) in the control group (Figure 2). The pooled RR was 1.16 (95% CI 0.96 to 1.41), in favour of the control group. The RD was 0.02 (95% CI -0.03 to 0.08). This is not considered clinically relevant.
Mixed: mild & moderate disease
Spinner (2020) reported the mortality for patients with a mild and moderate disease severity. The mortality in the intervention group was 5/384 (1.3%), compared to 4/200 (2.0%) in the control group (Figure 2). The RR was 0.65 (95% CI 0.18 to 2.40) in favour of the intervention group. The RD was 0.01 (95% CI -0.02 to 0.03). This is not considered clinically relevant.
Mixed: moderate & severe disease
Wang (2020) reported the mortality for patients with a moderate and severe disease severity. The mortality in the intervention group was 22/158 (13.9%), compared to 10/78 (12.8%) in the control group (Figure 2). The RR was 1.09 (95% CI 0.54 to 2.18) in favour of the intervention group. The RD was 0.01 (95% CI -0.01 to 0.02). This is not considered clinically relevant.
Figure 2: Mortality (28-30days) in hospitalized patients by disease severity
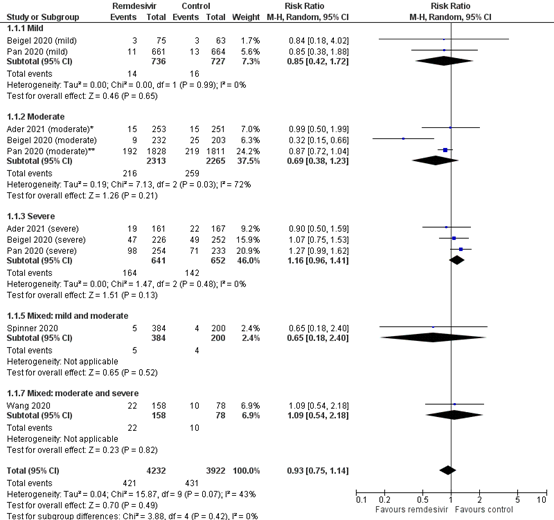
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval. *1% of the patients in the study of Ader (2021) had a mild disease severity. **Pan (2020) included patients who received high-flow oxygen in the moderate subgroup.
Mortality, other follow-up
Mahajan (2021) reported mortality during the 12-day follow up. The mortality was the 14.7% (5/34) in the intervention group, and 8.3% (3/36) in the control group (RR 1.76; 95%CI 0.46 to 6.82). The RD was 0.06 (95%CI -0.09 to 0.21), in favour of the control group. This means that patients in the intervention group have a higher risk of mortality. This is considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because there is a difference in effect estimates among the studies due to heterogeneity in study population (inconsistency, -1). The level of evidence for the outcome mortality in hospitalized patients is considered moderate.
Extensive respiratory support (crucial)
Initiation of mechanical respiratory support in hospitalized patients with COVID-19 was reported in two studies (Beigel, 2020; Pan, 2020). The results were not pooled as only two studies could be included in the meta-analysis. Ader (2021) reported a combined outcome measure new mechanical ventilation, ECMO or death within 29 days and was therefore not included in the meta-analysis. Barratt-Due (2020) reported the initiation of mechanical respiratory support, however as these patients were already included in the WHO Solidarity trial (Pan, 2020), the results were not included in the meta-analysis.
Pan (2020) reported 295/2743 (10.8%) patients in the intervention group in whom respiratory support was initiated after randomization, compared to 284/2709 (10.5%) in the control group. This results in a RR of 1.03 (95% CI 0.88 to 1.20). The RD was 0.00 (95%CI -0.01 to 0.02). This means that there is no difference between the intervention and control group. Ventilation included invasive or non-invasive mechanical ventilation or extra-corporal membrane oxygenation.
Beigel (2020) reported that the number of patients that started non-invasive ventilation or high-flow oxygen during the study was 52/307 (16.9%) in the intervention group, compared to 64/266 (24.16%) in the placebo group. This results in a RR of 0.70 (95% CI 0.51 to 0.98), in favour of the intervention group. The RD was 0.07 (95%CI 0.00 to 0.14), in favour of the intervention group. This is considered clinically relevant. It is unclear whether these patients also started mechanical ventilation or ECMO during the study period.
Beigel (2020) also reported that the number of patients that started using mechanical ventilation or ECMO during the study was 52/402 (12.9%) in the intervention group, compared to 82/364 (22.5%) in the placebo group. This results in a RR of 0.57 (95% CI 0.42 to 0.79). The RD was 0.10 (95%CI 0.04 to 0.15), in favour of the intervention group. This is considered clinically relevant. It is unclear whether these patients also received non-invasive ventilation or high-flow oxygen before the start of mechanical ventilation or ECMO.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because of heterogeneity in study results (inconsistency, -1) and because of a low number of events (imprecision, -1). The level of evidence for the outcome ‘respiratory support’ in hospitalized patients is considered low.
Duration of hospitalization (important)
Duration of hospitalization in hospitalized patients with COVID-19 was reported in three studies (Ader, 2021; Beigel, 2020; Wang, 2020). Due to differences in reporting, data were not pooled. Pan (2020) did not report the duration of hospitalization, but they rather reported the percentage at 7, 14 and 21 days. Therefore, the study was not included.
Ader (2021) reported that the median (IQR) number of days until hospital discharge within 29 days was 15 days (10 to 29) in the intervention group (n=253), compared to 13 days (8 to 29) in the control group (n=251). The difference was 2 days, this is not considered clinically relevant.
Beigel (2020) reported that the median (IQR) duration of initial hospitalization was 12 days (6 to 28) in the intervention group (n=541), compared to 17 days (8 to 28) in the control group. The median duration of initial hospitalization among those who did not die was 10 days (5 to 21) in the intervention group, and 14 days (7 to 27) in the control group. The difference is 5 or 4 days, which was both considered clinically relevant.
Wang (2020) reported that the median (IQR) duration of hospital stay was 25 days (16 to 38) in the intervention group (n=158), compared to 24 days (18 to 36) in the control group. The difference is 1 day, this is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because the studies were either not blinded or because of high numbers lost to follow-up (risk of bias, -1) and variance of point estimates across studies (inconsistency, -1). The level of evidence for the outcome ‘duration of hospitalization’ is considered low.
Time to clinical improvement (important)
Time to clinical improvement was reported in four studies for hospitalized patients with COVID-19 (Ader, 2021; Beigel, 2020; Spinner, 2021; Wang, 2020). Due to differences in reporting, data were not pooled.
Ader (2021) reported that the median (IQR) days to NEWS-2 of 2 or lower or hospital discharge within 29 days was 9 days (5 to 14) in the intervention group (n=253), compared to 8 days (5 to 13) in the control group (n=251). The difference is 1 day, in favour of the control group. In addition, they reported that the median (IQR) time to improvement of two categories of the 7-point ordinal scale or hospital discharge within day 29 was 11 days (8 to 20) in the intervention group, compared to 9 days (6 to 15) in the control group. The difference is 2 days, in favour of the control group. This is not considered clinically relevant.
Beigel (2020) reported that the median time (95% CI) to discharge or NEWS ≤2 for 24 hours was 8 days (7 to 9) in the intervention group (n=541), compared to 12 days (10 to 15) in the control group (n=521). The difference was 4 days, in favour of the intervention group. This is considered clinically relevant.
Spinner (2021) reported the number of patients who showed clinical improvement at day 28. Clinical improvement was defined as an improvement of at least 2 points from baseline on the 7-point ordinal scale. Spinner (2021) reported that 345 (89.8%) of the patients in the intervention group (n=384) showed clinical improvement, compared to 166 (83.0%) of the control group (n=200). This results in a RR of 1.08 (95%CI 1.01 to 1.16). The RD is 0.07 (95%CI 0.01 to 0.13) in favour of the intervention group. This is considered clinically relevant.
Wang (2020) reported the median time to clinical improvement defined as a two-point reduction in patient’s admission status on a six-point ordinal scale, or live discharge from the hospital, whichever came first. The intervention group (n=158) had a median (IQR) time to improvement of 21 days (13 to 28 days), compared to 23 days (15 to 28) in the control group (n=78). The difference is 2 days, in favour of the intervention group. This is not considered clinically relevant.
Ader (2021) reported that the median (IQR) days to NEWS-2 of 2 or lower or hospital discharge within 29 days was 20 days (12 to 29) in the intervention group (n=161), compared to 26 days (12 to 29) in the control group (n=167). The difference is 6 days, in favour of the intervention group. The difference is considered clinically relevant. In addition, they reported that the median (IQR) time to improvement of two categories of the 7-point ordinal scale or hospital discharge within day 29 was 16 days (10 to 29) in the intervention group, compared to 17 days (10 to 29) in the control group. The difference is 1 day, in favour of the intervention group. This is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because of the studies were not blinded (risk of bias, -1), heterogeneity in reporting ‘clinical improvement’ and variance of point estimates across studies (inconsistency, -1). The level of evidence for the outcome ‘time to clinical improvement’ is considered low.
A systematic review of the literature was performed to answer the following question:
What is the effectivity of treatment with remdesivir compared to treatment without remdesivir in patients with COVID-19?
P: hospitalized with COVID-19 (subgroups mild, moderate, severe)
I: remdesivir + standard care
C: standard care only / placebo treatment + standard care
O: 28-30 day mortality (if not available, any other reports of mortality), extensive respiratory support, duration of hospitalization, time to clinical improvement
Relevant outcome measures
For hospitalized COVID-19 patients, mortality and need for extensive respiratory support were considered as crucial outcome measures for decision making. Duration of hospitalization and time to clinical improvement were considered as important outcome measures for decision making.
Extensive respiratory support was defined as high flow nasal cannula (HFNC)/Optiflow, continuous positive airway pressure (CPAP), non-invasive ventilation (NIV), mechanical ventilation or extracorporeal membrane oxygenation (ECMO or ECLS).
The working group defined 3% points absolute difference as a minimal clinically important difference for mortality (resulting in a NNT of 33), 3 days difference for duration of hospitalization and time to clinical improvement, 5% points absolute difference need for respiratory support and ICU admission (resulting in a NNT of 20).
Studies of hospitalized patients were categorized on based on the respiratory support that was needed at baseline (preferably based on patient inclusion/exclusion criteria; otherwise on baseline characteristics). The following categories were used:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen [high flow nasal cannula (HFNC)/Optiflow], continuous positive airway pressure [CPAP], non-invasive ventilation [NIV], mechanical ventilation, extracorporeal membrane oxygenation [ECMO or ECLS]).
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until 14 October 2021. The detailed search strategy is outlined under the tab Methods. Studies were selected based on the following criteria: randomized controlled trial, peer reviewed and published in indexed journal, comparing treatment with remdesivir and standard care to standard care alone or treatment remdesivir and standard care to placebo and standard care in patients with COVID-19.
The systematic literature search resulted in 77.987 hits. Studies were selected based on the following criteria: systematic review or randomized controlled trials. Eventually, seven studies were included.
Statistical methods
Statistical analyses were conducted using Review Manager (RevMan) software 5.4. For dichotomous outcomes, Mantel Haenszel random‐effects risk ratios (RRs) and risk differences (RDs) were calculated. For continuous outcomes, a random‐effects mean difference (MD) weighted by the inverse variance was calculated. The random-effects model estimates the mean of a distribution of effects.
Results
In total, seven RCTs were included in the analysis. Important study characteristics and results are summarized below. Studies are presented in alphabetical order, only results of the primary outcome is reported in the summary of literature. Additionally, studies are summarized in the evidence tables. The assessment of the risk of bias is summarized separately in the risk of bias tables.
- Abd-Elsalam S, Ahmed OA, Mansour NO, Abdelaziz DH, Salama M, Fouad MHA, Soliman S, Naguib AM, Hantera MS, Ibrahim IS, Torky M, Dabbous HM, El Ghafar MSA, Abdul-Baki EAM, Elhendawy M. Remdesivir Efficacy in COVID-19 Treatment: A Randomized Controlled Trial. Am J Trop Med Hyg. 2021 Sep 10:tpmd210606. doi: 10.4269/ajtmh.21-0606. Epub ahead of print. PMID: 34649223.
- Abd-Elsalam S. Retraction Notice. Am J Trop Med Hyg. 2022 Sep 2;107(3):1. doi: 10.4269/ajtmh.1073ret. Epub ahead of print. PMID: 36099166; PMCID: PMC9490653.
- Ader F, Bouscambert-Duchamp M, Hites M, Peiffer-Smadja N, Poissy J, Belhadi D, Diallo A, Lê MP, Peytavin G, Staub T, Greil R, Guedj J, Paiva JA, Costagliola D, Yazdanpanah Y, Burdet C, Mentré F; DisCoVeRy Study Group. Remdesivir plus standard of care versus standard of care alone for the treatment of patients admitted to hospital with COVID-19 (DisCoVeRy): a phase 3, randomised, controlled, open-label trial. Lancet Infect Dis. 2021 Sep 14:S1473-3099(21)00485-0. doi: 10.1016/S1473-3099(21)00485-0. Epub ahead of print. PMID: 34534511; PMCID: PMC8439621.
- Ali, K., Azher, T., Baqi, M., Binnie, A., Borgia, S., Carrier, F. M., Cavayas, Y. A., Chagnon, N., Cheng, M. P., Conly, J., Costiniuk, C., Daley, P., Daneman, N., Douglas, J., Downey, C., Duan, E., Duceppe, E., Durand, M., English, S., Farjou, G., … Association of Medical Microbiology and Infectious Disease Canada (AMMI) Clinical Research Network and the Canadian Critical Care Trials Group (2022). Remdesivir for the treatment of patients in hospital with COVID-19 in Canada: a randomized controlled trial. CMAJ : Canadian Medical Association journal = journal de l'Association medicale canadienne, 194(7), E242–E251. https://doi.org/10.1503/cmaj.211698
- Ansems K, Grundeis F, Dahms K, Mikolajewska A, Thieme V, Piechotta V, Metzendorf MI, Stegemann M, Benstoem C, Fichtner F. Remdesivir for the treatment of COVID-19. Cochrane Database Syst Rev. 2021 Aug 5;8(8):CD014962. doi: 10.1002/14651858.CD014962. PMID: 34350582; PMCID: PMC8406992.
- Barratt-Due A, Olsen IC, Nezvalova-Henriksen K, Kåsine T, Lund-Johansen F, Hoel H, Holten AR, Tveita A, Mathiessen A, Haugli M, Eiken R, Kildal AB, Berg Å, Johannessen A, Heggelund L, Dahl TB, Skåra KH, Mielnik P, Le LAK, Thoresen L, Ernst G, Hoff DAL, Skudal H, Kittang BR, Olsen RB, Tholin B, Ystrøm CM, Skei NV, Tran T, Dudman S, Andersen JT, Hannula R, Dalgard O, Finbråten AK, Tonby K, Blomberg B, Aballi S, Fladeby C, Steffensen A, Müller F, Dyrhol-Riise AM, Trøseid M, Aukrust P; NOR-Solidarity trial. Evaluation of the Effects of Remdesivir and Hydroxychloroquine on Viral Clearance in COVID-19 : A Randomized Trial. Ann Intern Med. 2021 Sep;174(9):1261-1269. doi: 10.7326/M21-0653. Epub 2021 Jul 13. PMID: 34251903; PMCID: PMC8279143.
- Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, Lopez de Castilla D, Finberg RW, Dierberg K, Tapson V, Hsieh L, Patterson TF, Paredes R, Sweeney DA, Short WR, Touloumi G, Lye DC, Ohmagari N, Oh MD, Ruiz-Palacios GM, Benfield T, Fätkenheuer G, Kortepeter MG, Atmar RL, Creech CB, Lundgren J, Babiker AG, Pett S, Neaton JD, Burgess TH, Bonnett T, Green M, Makowski M, Osinusi A, Nayak S, Lane HC; ACTT-1 Study Group Members. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med. 2020 Nov 5;383(19):1813-1826. doi: 10.1056/NEJMoa2007764. Epub 2020 Oct 8. PMID: 32445440; PMCID: PMC7262788.
- Gordon CJ, Tchesnokov EP, Woolner E, Perry JK, Feng JY, Porter DP, Götte M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J Biol Chem. 2020 May 15;295(20):6785-6797. doi: 10.1074/jbc.RA120.013679. Epub 2020 Apr 13. PMID: 32284326; PMCID: PMC7242698.
- Gottlieb RL, Vaca CE, Paredes R, Mera J, Webb BJ, Perez G, Oguchi G, Ryan P, Nielsen BU, Brown M, Hidalgo A, Sachdeva Y, Mittal S, Osiyemi O, Skarbinski J, Juneja K, Hyland RH, Osinusi A, Chen S, Camus G, Abdelghany M, Davies S, Behenna-Renton N, Duff F, Marty FM, Katz MJ, Ginde AA, Brown SM, Schiffer JT, Hill JA; GS-US-540-9012 (PINETREE) Investigators. Early Remdesivir to Prevent Progression to Severe Covid-19 in Outpatients. N Engl J Med. 2022 Jan 27;386(4):305-315. doi: 10.1056/NEJMoa2116846. Epub 2021 Dec 22. PMID: 34937145; PMCID: PMC8757570.
- Mahajan L, Singh AP, Gifty. Clinical outcomes of using remdesivir in patients with moderate to severe COVID-19: A prospective randomised study. Indian J Anaesth. 2021 Mar;65(Suppl 1):S41-S46. doi: 10.4103/ija.IJA_149_21. Epub 2021 Mar 20. PMID: 33814589; PMCID: PMC7993042.
- Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, Leist SR, Pyrc K, Feng JY, Trantcheva I, Bannister R, Park Y, Babusis D, Clarke MO, Mackman RL, Spahn JE, Palmiotti CA, Siegel D, Ray AS, Cihlar T, Jordan R, Denison MR, Baric RS. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017 Jun 28;9(396):eaal3653. doi: 10.1126/scitranslmed.aal3653. PMID: 28659436; PMCID: PMC5567817.
- Spinner CD, Gottlieb RL, Criner GJ, Arribas López JR, Cattelan AM, Soriano Viladomiu A, Ogbuagu O, Malhotra P, Mullane KM, Castagna A, Chai LYA, Roestenberg M, Tsang OTY, Bernasconi E, Le Turnier P, Chang SC, SenGupta D, Hyland RH, Osinusi AO, Cao H, Blair C, Wang H, Gaggar A, Brainard DM, McPhail MJ, Bhagani S, Ahn MY, Sanyal AJ, Huhn G, Marty FM; GS-US-540-5774 Investigators. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19: A Randomized Clinical Trial. JAMA. 2020 Sep 15;324(11):1048-1057. doi: 10.1001/jama.2020.16349. PMID: 32821939; PMCID: PMC7442954.
- Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, Fu S, Gao L, Cheng Z, Lu Q, Hu Y, Luo G, Wang K, Lu Y, Li H, Wang S, Ruan S, Yang C, Mei C, Wang Y, Ding D, Wu F, Tang X, Ye X, Ye Y, Liu B, Yang J, Yin W, Wang A, Fan G, Zhou F, Liu Z, Gu X, Xu J, Shang L, Zhang Y, Cao L, Guo T, Wan Y, Qin H, Jiang Y, Jaki T, Hayden FG, Horby PW, Cao B, Wang C. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020 May 16;395(10236):1569-1578. doi: 10.1016/S0140-6736(20)31022-9. Epub 2020 Apr 29. Erratum in: Lancet. 2020 May 30;395(10238):1694. PMID: 32423584; PMCID: PMC7190303.
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020 Mar;30(3):269-271. doi: 10.1038/s41422-020-0282-0. Epub 2020 Feb 4. PMID: 32020029; PMCID: PMC7054408.
- WHO Solidarity Trial Consortium, Pan H, Peto R, Henao-Restrepo AM, Preziosi MP, Sathiyamoorthy V, Abdool Karim Q, Alejandria MM, Hernández García C, Kieny MP, Malekzadeh R, Murthy S, Reddy KS, Roses Periago M, Abi Hanna P, Ader F, Al-Bader AM, Alhasawi A, Allum E, Alotaibi A, Alvarez-Moreno CA, Appadoo S, Asiri A, Aukrust P, Barratt-Due A, Bellani S, Branca M, Cappel-Porter HBC, Cerrato N, Chow TS, Como N, Eustace J, García PJ, Godbole S, Gotuzzo E, Griskevicius L, Hamra R, Hassan M, Hassany M, Hutton D, Irmansyah I, Jancoriene L, Kirwan J, Kumar S, Lennon P, Lopardo G, Lydon P, Magrini N, Maguire T, Manevska S, Manuel O, McGinty S, Medina MT, Mesa Rubio ML, Miranda-Montoya MC, Nel J, Nunes EP, Perola M, Portolés A, Rasmin MR, Raza A, Rees H, Reges PPS, Rogers CA, Salami K, Salvadori MI, Sinani N, Sterne JAC, Stevanovikj M, Tacconelli E, Tikkinen KAO, Trelle S, Zaid H, Røttingen JA, Swaminathan S. Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N Engl J Med. 2021 Feb 11;384(6):497-511. doi: 10.1056/NEJMoa2023184. Epub 2020 Dec 2. PMID: 33264556; PMCID: PMC7727327.
- WHO Solidarity Trial Consortium. Remdesivir and three other drugs for hospitalised patients with COVID-19: final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet. 2022 May 21;399(10339):1941-1953. doi: 10.1016/S0140-6736(22)00519-0. Epub 2022 May 2. PMID: 35512728; PMCID: PMC9060606.
- Williamson BN, Feldmann F, Schwarz B, Meade-White K, Porter DP, Schulz J, van Doremalen N, Leighton I, Yinda CK, Pérez-Pérez L, Okumura A, Lovaglio J, Hanley PW, Saturday G, Bosio CM, Anzick S, Barbian K, Cihlar T, Martens C, Scott DP, Munster VJ, de Wit E. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature. 2020 Sep;585(7824):273-276. doi: 10.1038/s41586-020-2423-5. Epub 2020 Jun 9. PMID: 32516797; PMCID: PMC7486271.
PICO: What is the (in)effectivity and safety of treatment with remdesivir compared to treatment without remdesivir in patients with COVID-19?
|
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
|
1. Remdesivir |
|||||||
|
Ader, 2021 |
Type of study: phase 3, open-label, adaptive, multicentre, randomised, controlled trial
Setting: 48 sites in Europe, Between March 22, 2020, and Jan 21, 2021
Country: France, Belgium, Austria, Portugal, Luxembourg
Source of funding: European Union’s Horizon 2020 research and innovation programme (Europe); Austrian Group Medical Tumor (Austria); Belgian Health Care Knowledge Centre (Belgium); Fonds Erasme-COVID-Université Libre de Bruxelles (Belgium); REACTing, a French multi-disciplinary collaborative network working on emerging infectious diseases (France); Ministry of Health (France); Domaine d’intérêt majeur One Health Île-de-France (France); European Regional Development Fund (Luxembourg); Ministry of Health (Portugal); Agency for Clinical Research and Biomedical Innovation (Portugal). We thank all participants who consented to enrol in the trial, as well as all study and site staff whose indispensable assistance made the conduct of the DisCoVeRy trial possible (all listed in the appendix 2 pp 35–47).
Conflicts of interest: DC reports grants and lecture fees from Janssen and lecture fees from Gilead, outside the submitted work….
Please refer to full text for the full conflicts of interest. |
Hospitalized patients with confirmed SARS-CoV-2
Inclusion criteria:
Exclusion criteria:
N total at baseline: Total:= 857 Intervention: = 429 Control: N = 428
ITT population: I: 414 C: 418
Important characteristics: Age, median (IQR): I: 63 (55–73) C: 64 (54–72)
Sex, n/N (%) male: I: 291/414 (70%) C: 288/418 (69%)
7-point ordinal scale at baseline: 3: hospitalised, not requiring supplemental oxygen: I:8 (2%) C: 8 (2%) 4: hospitalised, requiring supplemental oxygen I: 241 (58%) C: 244 (58%) 5: hospitalised, on non-invasive ventilation or high flow oxygen devices I: 90 (22%) C: 93 (22%) 6: hospitalised, on invasive mechanical ventilation or ECMO I: 75 (18%) C: 73 (18%)
Groups were comparable at baseline.
|
Remdesivir was administered intravenously at a loading dose of 200 mg on day 1 followed by a 100 mg, 1-h infusion once-daily for a total duration of 10 days |
standards of care |
Length of follow-up: 29 days
Loss-to-follow-up or incomplete data: Intervention: N = 0
Control: N = 0
|
Clinical outcomes
Death within 28 days I: 34 (8%) C: 37 (9%) OR 0·93 (95% CI: 0·57 to 1·52); p=0·77
7-point ordinal scale at day 15 (Primary Outcome) OR 0·98 (95% CI: 0·77 to 1·25); p=0·85
7-point ordinal scale at day 29 OR 1·11 (95% CI: 0·87 to 1·42); p=0·39
Duration of hospitalisation Days to hospital discharge within 29 days I: 15 (10 to 29) C: 13 (8 to 29) HR 0·94 (95% CI: 0·80 to 1·11); p=0·49
New mechanical ventilation, ECMO, or death within 29 days* I: 60/339 (18%) C: 87/344 (25%) HR 0·66 (95% CI: 0·47 to 0·91); p=0·010
Time to symptom resolution
Respiratory support Oxygenation-free days until day 29 I: 17 (2 to 22) C: 17 (0 to 23) Least-square mean difference (LSMD) 0·35 (–0·90 to 1·60); p=0·59
Ventilator-free days until day 29 I: 29 (20 to 29) C: 29 (16 to 29) LSMD 1·08 (–0·15 to 2·30); p=0·080
Safety Any Serious adverse events I: 135 (33%) C: 130 (31%) OR 1·11 (0·83–1·50); p=0·48
Any adverse events I: 241 (59%) C: 236 (57%) OR 1·14 (0·86–1·50); p=0·37
Virological outcomes The median decrease in viral loads between baseline and day 3 was similar in the remdesivir and control groups (appendix 2 pp 12–13). There was no significant effect of remdesivir on the viral kinetics (figure 3; appendix 2 pp 14, 29). |
Definitions: WHO Master Protocol:(1) not hospitalised, no limitation on activities; (2) not hospitalised, limitation on activities; (3) hospitalised, not requiring supplemental oxygen; (4) hospitalised, requiring supplemental oxygen; (5) hospitalised, on non-invasive ventilation or high flow oxygen devices; (6) hospitalised, on invasive mechanical ventilation or ECMO; and (7) dead.
Remarks: It was open-label and not placebo-controlled. Indeed, several treatments were concomitantly evaluated at the beginning of the trial, and masking was thus impossible due to the different modes of administration (intravenous, subcutaneous, or oral) of the different treatment groups.
Authors conclusion: In this randomised controlled trial, the use of remdesivir for the treatment of hospitalised patients with COVID-19 was not associated with clinical improvement at day 15 or day 29, nor with a reduction in mortality, nor with a reduction in SARS-CoV-2 RNA. |
|
Barratt-Due, 2021 |
Type of study: Independent, add-on RCT to WHO Solidarity trial, an open-label, adaptive RCT
Setting: 23 hospitals, between 28 March and 5 October 2020
Country: Norway
Source of funding: National Clinical Therapy Research, no role in RCT
Conflicts of interest: JTA reports grant from South-Eastern Norway Regional Health Authority. ABD reports paid lecture by Allergan Norden AB and participation in advisory board meeting SANOFI-AVENTIS Norwy. SGD reports grant from Research Council of Norway. FLJ reports funding from Oslo University Hospital and Helse SørØst. MT reports participation in European advisory board for Eli Lilly and coordinator of National reference group on COVID-19 treatment in Norway.
|
Hospitalized patients with confirmed SARS-CoV-2 infection
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 181 Remdesivir: n=42 Hydroxychloroquine: n=52 Control remdesivir: n=57 Control HSQ: n=54 (some controls were in both groups)
Important characteristics: Age, mean (SD): 59.8 (15.3) R: 59.7 y (16.5) CR: 58.1 y (15.7) H: 60.3 y (13.3) CH: 59.2 y (16.4)
Sex, n/N (%) male: R: 29/42 (69%) CR: 43/57 (75%) H: 31/52 (60%) CH: 34/54 (63%)
Disease severity, mean (SD): Defined by Viral load (log10 count/1000 cells) R: 1.6 (1.6) CR: 2.3 (1.8) H: 2.3 (1.5) CH: 2.0 (1.5)
Anti-SARS-CoV-2 antibodies, seroconverted (RDB ≥5) n/N (%) R: 14/42 (42.4%) CR: 18/57 (46.2%) H: 15/52 (42.9%) CH: 20/54 (54.1%)
Groups are not comparable on baseline regarding sex, comorbid conditions, ICU admission, P-F ratio less than 40 kPa, ACE and ARB medication, LDH level, D-dimer level, AST level, ALT, level. |
R: standard of care plus 200 mg intravenous remdesivir on day 1, 100 mg daily up to day 9
H: standard of care plus 800 mg oral hydroxychloroquine 2x day on day 1, 400 mg 2x day up to day 9
All study treatment were discontinued at discharge. |
Standard of care according to WHO guidelines recommending systemic steroids |
Length of follow-up: 3 months
Loss-to-follow-up: R: 9 (21%)
CR: 9 (16%)
H: 13 (24%)
CH: 8 (15%)
Incomplete outcome data: Missing data due to discharge or participant withdrawal were imputed with best outcome. Not reported how many.
|
Clinical outcomes Mortality (28 day) R: 2.4 (0.1; 10.1) CR: 5.2 (1.3-13.1) RD: -2.9 (-10.3; 4.5) H: 7.5 (2.4; 16.7) CH: 1.8 (0.1; 7.6) RD: 5.8 (-2.2; 13.7) Mortality (60 day) R: 7.1 (1.8; 17.5) CR: 5.3 (1.3; 13.1) RD: 1.9 (-7.8; 11.6) H: 7.5 (2.4; 16.7) CH: 1.8 (0.1; 7.6) RD: 5.8 (-2.2; 13.7) In-hospital mortality R: 7.1 (1.8; 17.5) CR: 7.0 (2.2; 15.6) RR: 1.0 (0.2; 4.6) H: 7.5 (2.4; 16.7) CH: 3.6 (0.6; 10.6) RR: 2.2 (0.4; 10.8)
Duration of hospitalization Not reported Admission to ICU during hospitalization R: 19.0 (9.2; 32.6) CR: 19.3 (10.5; 30.8) RD: -0.3 (-15.9; 15.4) H: 22.6 (12.8; 35.0) CH: 16.1 (8.1; 27.1) RD: 6.6 (-8.2; 21.4)
Time to symptom resolution Not reported
Respiratory support Mechanical ventilation R: 9.5 (3.1; 20.8) CR: 7.0 (2.2; 15.6) RD: 2.5 (-8.6; 13.6) H: 15.1 (7.2; 26.3) CH: 10.7 (4.4; 20.5) RD: 4.4 (-8.2; 17.0) Time to receipt mechanical ventilation RR R vs CR: 1.4 (0.4; 5.8) RR H vs CH: 2.1 (0.7; 6.2) Duration of mechanical ventilation Reported in appendix figure 1
Safety Adverse events n/N (%) R: 34/42 (81%) H: 26/52 (50%) C: 33/87 (n=38%) Serious adverse events n/N (%) R: 13/42 (31%) H: 12/52 (23%) C: 20/87 (n=23%)
Virological outcomes Viral clearance Not reported Viral load Reported in a figure 2 and appendix figure 2 Subgroup analysis based on symptom duration before hospitalization, the presence of ARS-CoV-2 antibodies, high or low viral load at hospital admission, degree of inflammation, and age were performed. |
Definitions: not applicable
Remarks:
Authors conclusion: The overall lack of effect of remdesivir and HCQ on the clinical course of patients hospitalized for COVID-19 was accompanied by a paucity of effect on SARS-CoV-2 viral clearance in the oropharynx. Our findings question the antiviral potential of these drugs in hospitalized patients with COVID-19.
|
|
Mahajan, 2021 |
Type of study: RCT; not blinded
Setting: June to December 2020;
Country: India
Source of funding: “Financial support and sponsorship Nil. Conflicts of interest There are no conflicts of interest.”
|
Hospitalized COVID-19 patients with moderate-to-severe disease
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 82 Randomized: Intervention: 41 Control: 41 Included in analysis: Intervention: 34 (incl. 1 cross over) Control: 36
Important characteristics: Age, mean (SD): I: 58.08±12.1 C: 57.41±14.1 Sex, n/N (%) male: I: 13 (38.3) C: 9 (25.0) Duration of symptoms before involvement in trial (days); mean±SD I: 6.26±2.49 C: 7.38±0.99 Receiving low flow supplemental oxygen I: 27 (79.4) C: 26 (72.2) Receiving non‑invasive ventilation or high‑flow oxygen I: 7 (20.6) C: 10 (27.8) Receiving invasive mechanical ventilation I: 0 (0) C: 0 (0)
Groups comparable at baseline? No, time from symptoms to enrolment longer in the control group, less patient in intervention group received non-invasive ventilation or high-flow oxygen in stead of low flow oxygen compared to the control group. |
Remdesivir + standard of care
IV 200 mg remdesivir on day 1, followed by 100 mg of remdesivir once daily for the subsequent four days
|
Standard of care
supportive therapy throughout the duration of the study. Other drugs used for COVID treatment (off-label use and in the absence of written policy) were not allowed to be administered to the patients in the study period. Drugs like corticosteroids and heparin were given as per standard of care protocol. |
Length of follow up: 12 days, or until discharge of death
Loss to follow-up: I: 8/41 (19.5%) Reasons: 2 patients discharged, 1 patient died, 2 witheld treatment, 3 remdesivir C: 5/41 (12.2%) Reasons: 2 patients discharged, 2 patients died, 1 requested remdesivir treatment |
Clinical outcomes Mortality Reported as ‘death’, score 6, on ordinal scale. Unclear at which moment in time (1 result for 12 to 24 days): Death I: 5 (14.7) C: 3 (8.3)
Duration of hospitalization No data reported
Symptom resolution Nausea, vomiting I: Baseline: 7 After treatment: 3 C: Baseline: 9 After treatment: 2
Need for respiratory support Clinical status from day 12 to 24 Did not require hospitalisation* I: 2 (5.9) C: 3 (8.3) Hospitalised, but did not require supplemental oxygen I: 0 (0) C: 0 (0) Hospitalised, required supplemental oxygen I: 4 (11.8) C: 6 (16.7) Required high‑flow oxygen or non‑invasive ventilation I: 19 (55.9) C: 22 (61.1) Required or received mechanical ventilation I: 4 (11.8) C: 2 (5.6) Death I: 5 (14.7) C: 3 (8.3)
Also reported: AST levels, ALT levels and creatinine levels at baseline and after treatment
Safety Adverse events
Virological outcomes Viral clearance not reported |
Definitions: Clinical status day 1 – 12; assessed on 4-point ordinal scale: 1), receiving low-flow oxygen supplementation; 2), receiving non-invasive ventilation or high-flow oxygen; 3), not receiving supplemental oxygen but requiring medical care; 4), receiving invasive mechanical ventilation
Clinical status day 12 – 24; assessed on 6-point ordinal scale: 1), Do not require hospitalisation, 2), hospitalised, but not requiring supplemental oxygen, 3), hospitalised, requiring supplemental oxygen; 4), Patients requiring high-flow oxygen or non-invasive ventilation; 5), Requiring or receiving mechanical ventilation; 6), Death
Remarks:
Authors conclusion: Remdesivir therapy for five days did not produce improvement in clinical outcomes in moderate to severe COVID‑19 cases.
|
|
Pan, 2020 |
Type of study: RCT (open-label, non-blinded)
Setting & country: 405 hospitals in 30 countries; WHO Solidarity Trial
Source of funding: Funded by the World Health Organization;
ISRCTN Registry nr, ISRCTN83971151; ClinicalTrials.gov nr, NCT04315948.)
|
N total at baseline: N = 11,330
Remdesivir arm I: 2743 C: 2708
Important characteristics: Age, n/N (%): I: <50y: 961/2743 (35%) 50-69y: 1282/2743 (47%) ≥70y: 500/2743 (18%) C: <50y: 952/2708 (35%) 50-69y: 1287/2708 (48%) ≥70y: 469/2708 (17%) Sex, n/N (%) male: I: 1706/2743 (62%) C: 1725/2708 (64%) Respiratory support I: No suppl. Oxygen at entry: 661/2743 (24%) Suppl. Oxygen at entry 1828/2743 (67%) Already receiving ventilation 254/2743 (9%) C: No suppl. Oxygen at entry: 664/2708 (25%) Suppl. Oxygen at entry 1811/2708 (67%) Already receiving ventilation 233/2708 (9%) Previous days in hospital I: 0 days: 724/2743 (26%) 1 day: 917/2743 (33%) ≥2 days: 1102/2743 (40%) C: 0 days: 712/2708 (26%) 1 day: 938/2708 (35%) ≥2 days: 1058/2708 (39%) |
Remdesivir
Intravenous; 200 mg on day 0 and 100 mg on days 1 through 9.
Taking trial drug midway through scheduled duration*: I: 96% C: 2%
Use of non-study drug, n/N (%): Corticosteroids I: 1310 (47.8%) C: 1288 (47.6%) Convalescent plasma I: 52 (1.9%) C: 58 (2.1%) Anti-IL-6 drug I: 133 (4.9%) C: 143 (5.3%) Non-trial interferon I: 3 (0.1%) C: 25 (0.9%) Non-trial antiviral I: 65 (2.4%) C: 152 (5.6%) |
Standard of care
|
Length of follow up: 28 days, or up to discharge
Loss to follow-up: I: 7/2750 (0.3%) Reasons: no or unknown consent C: 17/2725 (0.6%) Reasons: no or unknown consent
|
Clinical outcomes
All-cause in-hospital mortality, regardless of whether death occurred before or after day 28: I: 301/2743 (12.5%) C: 303/2708 (12.7%) RR 0.98 (95% CI 0.84 to 1.14) HR=0.95 (95% CI 0.81-1.11) Adjusted** HR=0.95 (95% CI 0.81-1.11)
All-cause in-hospital mortality, stratified by ventilation at randomization: Ventilated: HR 1.20 (95% CI 0.89-1.64) Not ventilated: HR 0.86 (95% CI 0.72-1.04)
Initiation of mechanical ventilation, in those not receiving ventilation at baseline: I: 295/2489 (11.6%) C: 284/2475 (11.5%) RR 1.03 (95% CI 0.89 to 1.20)
Composite death or initiation ventilation: I: 479/2743 (18.5%) C: 479/2708 (18.9%) RR 0.99 (95% CI 0.88 to 1.11) Publication: RR 0.97 [0.85-1.10]
Hospitalized, not discharged: Percentage of patients (rather than number of patients) ever reported as discharged who were still in the hospital: Day 7, % I: 69% C: 59% Day 14 I: 22% C: 19% Day 21 I: 9% C: 8% |
Definitions/information:
*Taking trial drug midway through scheduled duration, %, calculated only among patients who died or were discharged alive, % patients who were taking the trial drug midway through its scheduled duration (or midway through the time from entry to death or discharge, if this was shorter).
**Adjusted model all-cause mortality: some overlap between the 4 control groups; an exploratory sensitivity analysis used multivariate Cox regression to fit all 4 treatment effects simultaneously; adjusted for several prognostic factors (age, sex, diabetes, bilateral lung lesions at entry (no, yes, not imaged at entry), and respiratory support at entry (no oxygen, oxygen but no ventilation, ventilation).
Authors conclusion: These remdesivir, hydroxychloroquine, lopinavir, and interferon regimens had little or no effect on hospitalized patients with Covid-19, as indicated by overall mortality, initiation of ventilation, and duration of hospital stay. |
|
Beigel, 2020
|
Type of study: Double-blind, randomized placebo-controlled trial
Setting + Country: There were 60 trial sites and 13 subsites in the United States (45 sites), Denmark (8), the United Kingdom (5), Greece (4), Germany (3), Korea (2), Mexico (2), Spain (2), Japan (1), and Singapore (1).
Source of funding: Funded by the National Institute of Allergy and Infectious Diseases and others; ACCT-1 ClinicalTrials.gov number, NCT04280705 |
Inclusion criteria:
[more detailed information can be found in the supplementary file]
Exclusion criteria:
[more detailed information can be found in the supplementary file]
N total at baseline: N = 1062 Intervention: 541 Control: 521
Important characteristics: Intervention group: Age (mean (SD)): 58.6 (14.6) Male: 352 (65.1%)
Control group: Age (mean (SD)): 59.2 (15.4) Male: 332 (63.3%) Groups comparable at baseline. |
Remdesivir: days 2 through 10 or until hospital discharge or death |
Placebo: Matching placebo was administered according to the same schedule and in the same volume as the active drug. |
28 days |
Remdesivir versus control
Median recovery time: Remdesivir: 10 days (95% CI 9 – 11) Placebo: 15 days (95% CI 13 – 18)
Recovery HR for recovery: 1.29; 95% CI, 1.12 to 1.49; P<0.001).
Mortality by 14 days Remdesivir: 6.7% Placebo 11.9% HR for death: 0.55; 95% CI, 0.36 to 0.83) Serious adverse events Remdesivir: 24.6% |
Comments:
Authors conclusion: Remdesivir was superior to placebo in shortening the time to recovery in adults who were hospitalized with Covid-19 and had evidence of lower respiratory tract infection.
|
|
Spinner, 2020 |
Type of study: Randomized open-label multicentre clinical trial
Setting: 105 hospitals
Country: US, Europe and Asia
Source of funding: Gilead Sciences, which designed and conducted the study in consultation with the FDA and the investigators.
|
Inclusion criteria: Willing and able to provide written informed consent; aged ≥ 18 years (at all sites), or aged ≥ 12 and < 18 years of age weighing ≥ 40 kg; SARS-CoV-2 infection confirmed by PCR ≤ 4 days before randomization; currently hospitalized; SpO2 > 94% on room air at screening; radiographic pulmonary infiltrates; men and women of childbearing potential who engage in heterosexual intercourse must agree to use protocol specified method(s) of contraception.
Exclusion criteria: Participation in any other clinical trial of an experimental agent treatment for COVID-19; concurrent treatment with other agents with actual or possible direct acting antiviral activity against SARS-CoV-2 < 24 hours prior to study drug dosing; Requiring mechanical ventilation at screening; ALT or AST > 5 x ULN; creatinine clearance < 50 mL/min, pregnant or breastfeeding woman; known hypersensitivity to the study drug, the metabolites, or formulation excipient.
N total at baseline: N = 596 Intervention1: 197* Intervention2: 199* Control: 200*
Of which respectively 193 (I1), 191 (I2) and 200 (C) patients were included in primary analysis.
Important characteristics: Age, median (IQR): I1: 56 (45-66) I2: 58 (48-66) C: 57 (45-66) P=not reported Sex, n/N (%) male: I1: 118/193 (61) I2: 114/191 (60) C: 125/200 (63) P=not reported
Groups comparable at baseline? Patients in standard care group were more commonly prescribed with other COVID-19 agents. |
I1: Remdesivir for 10 days. 73 (38%) patients completed the assigned treatment duration; the median number of doses for the group was 6 (range, 1-10). Reasons were hospital discharge (n=98), withdrawal (n=8) or adverse events (n=6).
I2: Remdesivir for 5 days. 145 (76%) patients completed the assigned treatment duration (median, 5 doses; range, 1-5). Reasons were hospital discharge (n=35), withdrawal (n=5) or adverse events (n=4).
Remdesivir was dosed intravenously (30-60 minutes) at 200 mg on day 1 followed by 100 mg/d.
Remdesivir treatment was to be discontinued in any patient experiencing severe elevations in liver enzymes or decreases in estimated creatinine clearance to less than 30 mL/min.
|
Standard care (not specified) |
28 days (in person for hospitalized patients or by phone for discharged patients)
|
Difference in clinical status distribution versus standard care (OR (95%CI) I1: not reported * P=0.18 I2: 1.65 (1.09 to 2.48) P=0.02 *The proportional odds assumption was not met.
Clinical improvement day 11 (n/N, %)** I1: 126/193 (65) I2: 134/191 (70) C: 121/200 (61) Difference I1-C (95%CI): 4.8 (−5.0 to 14.4) Difference I2-C (95%CI): 9.7 (0.1 to 19.1) Clinical improvement day 28 (n/N, %)** I1: 174/193 (90) I2: 171/191 (90) C: 166/200 (83) Difference I1-C: not reported Difference I2-C: not reported **Defined as ≥2-point improvement from baseline on the 7-point ordinal scale.
Recovery at day 11 (n/N, %)*** I1: 132 (68) I2: 141 (74) C: 128 (64) Difference I1-C (95%CI): 4.4 (−5.0 to 13.8) Difference I2-C (95%CI): 9.8 (0.3 to 19.0) Recovery at day 28 (n/N, %)*** I1: 178 (92) I2: 175 (92) C: 170 (85) Difference I1-C: not reported Difference I2-C: not reported ***Defined as improvement from a baseline score of 2-5 to a score of 6 or 7 or from a baseline score of 6 to a score of 7, on the 7-point ordinal scale.
All-cause mortality at day 28 (n/N, %) I1: 3/193 (2) I2: 2/191 (1) C: 4/200 (2) HR (95%CI) I1 vs C: 0.76 (0.17 to 3.40) HR (95%CII I1 vs C: 0.51 (0.09 to 2.80)
Adverse events (n/N, %) Any adverse event I1: 113/193 (59) I2: 98/191 (51) C: 93/200 (47) Any serious adverse event I1: 10/193 (5) I2: 9/191 (5) C: 18/200 (9)
There were no significant differences between the remdesivir and standard care groups in duration of oxygen therapy or hospitalization (data not reported). |
Remarks: Patients who had sufficiently improved in the judgment of the investigator could be discharged from the hospital before finishing their assigned course of treatment.
Data on clinical improvement and recovery are also reported for other days. Furthermore, data on other exploratory outcome measures and details on adverse events were reported, but data is not shown here.
Peaks in discharge rates were observed in the remdesivir groups following the end of their assigned duration of treatment (i.e., there were increased rates of discharge on day 6 in the 5-day remdesivir group and on day 11 in the 10-day group), suggesting that discharges were delayed for some patients to allow them to complete full courses of assigned remdesivir treatment.
Original protocol was amended on the basis of emerging understanding of the clinical presentation and assessment of COVID-19.
Authors conclusion: Among patients with moderate COVID-19, those randomized to a 10-day course of remdesivir did not have a statistically significant difference in clinical status compared with standard care at 11 days after initiation of treatment. Patients randomized to a 5-day course of remdesivir had a statistically significant difference in clinical status compared with standard care, but the difference was of uncertain clinical importance. |
|
Wang, 2020a |
Type of study: Randomised, double-blind, placebo-controlled, multicentre trial
Setting: 10 hospitals in Wuhan, Hubei, between Feb 6 and March 12, 2020
Country: China
Source of funding: Chinese Academy of Medical Sciences Emergency Project of COVID-19, National Key Research and Development Program of China, the Beijing Science and Technology Project
Conflicts of interest: One author has served as non-compensated consultant to Gilead Sciences on its respiratory antiviral program, outside the submitted work. All other authors declare no competing interests.
|
Inclusion criteria:
Eligible patients of child-bearing age (men and women) agreed to take effective contraceptive measures (including hormonal contraception, barrier methods, or abstinence) during the study period and for at least 7 days after the last study drug administration
Exclusion criteria:
N total at baseline: N = 237 Intervention: 158 Control: 79
Important characteristics: Age, median (IQR) I: 66 (57–73) C: 64 (53–70) Sex, n/N (%) male: I: 89/158 (56%) C: 51/78 (65%) Time from symptom onset to starting study treatment, n/N (%) of ≤10 days I: 71/155 (46%)
Groups comparable at baseline? More patients with hypertension, diabetes, or coronary artery disease in the remdesivir group than the placebo group. More patients in the control group than in the remdesivir group had been symptomatic for 10 days or less at the time of starting remdesivir or placebo treatment, and a higher proportion of remdesivir recipients had a respiratory rate of more than 24 breaths per min. No other major differences in symptoms, signs, laboratory results, disease severity, or treatments were observed between groups at baseline. |
Remdesivir
Treatment regimens Intravenous remdesivir (200 mg on day 1 followed by 100 mg on days 2–10 in single daily infusions) for a total of 10 days (both provided by Gilead Sciences, Foster City, CA, USA). |
Placebo
Treatment regimens The same volume of placebo infusions for a total of 10 days (provided by Gilead Sciences, Foster City, CA, USA) |
Follow-up period: 28 days
|
Primary clinical endpoint Clinical improvement Defined as a 2-point reduction in patients’ admission status on a 6-point ordinal scale, or live discharge from the hospital, whichever came first. n/N (%) Day 7: I: 4/158 (3%) C: 2/78 (3%)
Day 14: I: 42/158 (11%) C: 18/78 (23%)
Day 28: I: 103/158 (65%) C: 45/78 (58%)
Time to clinical improvement Median (IQR) days I: 21 (13–28) C: 23 (15–28) HR 1.23 (95% CI: 0.87 to 1.75)
Subgroup analysis ≤10 days from symptom onset: I: 18 (12–28) C: 23 (15–28) HR 1.52 (95% CI: 0.95 to 2.43)
Secondary outcomes Proportion of patients in each category of the 6-point scale Day 7 OR: 0.69 (0.41─1.17)
Day 14 OR: 1.25 (0.76─2.04)
Day 28 OR 1.15 (0.67─1.96)
All-cause mortality at day 28, n/N (%) I: 22/158 (14%) C: 10/78 (13%) Difference: 1.1% (95% CI: -8.1 to 10.3)
Duration of invasive mechanical ventilation, median (IQR) days I: 7·0 (4·0 to 16·0) C: 15·5 (6·0 to 21·0) Difference: –4·0 (–14·0 to 2·0)
Duration of oxygen support, median (IQR) days I: 19·0 (11·0 to 30·0) C: 21·0 (14·0 to 30·5) Difference: –2·0 (–6·0 to 1·0)
Duration of hospital admission, median (IQR) days I: 25·0 (16·0 to 38·0) C: 24·0 (18·0 to 36·0) Difference: 0·0 (–4·0 to 4·0)
Virological measures Proportions of patients with viral RNA detected and viral RNA load Measured by quantitative RT-PCR Viral load decreased over time similarly in both groups. When adjusted for baseline sputum viral load at enrolment, the remdesivir group showed no significant difference at day 5 from placebo, but a slightly more rapid decline in load (p=0.0672). The cumulative rate of undetectable viral RNA of nasopharyngeal and oropharyngeal swabs by day 28 was 153 (78%) of 196 patients, and the negative proportion was similar among patients receiving remdesivir and those receiving placebo.
Safety outcomes Treatment-emergent adverse events I: 102/155 (66%) C: 50/78 (64%)
Serious adverse events, I: 28/155 (18%) C: 20/78 (26%)
Premature discontinuations of study drug I: 18/155 (12%) C: 4/78 (5%) |
Remarks: - number of patients needed according to power calculation was not reached, because no patients were enrolled after March 12, because of the control of the outbreak in Wuhan. Based on the termination criteria specified in the protocol, the data safety and monitoring board recommended that the study be terminated. - At this stage, the interim analysis was abandoned. When all the other assumptions stayed the same, with the actual enrolment of 236 participants, the statistical power was reduced from 80% to 58%.
Authors conclusion: Our trial found that intravenous remdesivir did not significantly improve the time to clinical improvement, mortality, or time to clearance of virus in patients with serious COVID-19 compared with placebo. We found that this dose regimen of intravenous remdesivir was adequately tolerated but did not provide significant clinical or antiviral effects in seriously ill patients with COVID-19. However, we could not exclude clinically meaningful differences and saw numerical reductions in some clinical parameters.
In this study of adult patients admitted to hospital for severe COVID-19, remdesivir was not associated with statistically significant clinical benefits. However, the numerical reduction in time to clinical improvement in those treated earlier requires confirmation in larger studies.
[See also the study by Shih, 2020: Remdesivir is Effective for Moderately Severe Patients: A Re-Analysis of the First Double-Blind, Placebo-Controlled, Randomized Trial on Remdesivir for Treatment of Severe COVID-19 Patients Conducted in Wuhan City]
Note: 6-point ordinal scale: 6 = death; 5 = hospital admission for extracorporeal membrane oxygenation or mechanical ventilation; 4 = hospital admission for non-invasive ventilation or high-flow oxygen therapy; 3 = hospital admission for oxygen therapy (but not requiring high-flow or non-invasive ventilation); 2 = hospital admission but not requiring oxygen therapy; 1 = discharged or having reached discharge criteria (defined as clinical recovery—ie, normalisation of pyrexia, respiratory rate <24 breaths per minute, saturation of peripheral oxygen >94% on room air, and relief of cough, all maintained for at least 72 h)
|
Risk of bias table for intervention studies (randomized controlled trials).
|
|
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2 (unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4 (unlikely/likely/unclear) |
Bias due to loss to follow-up?5 (unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6 (unlikely/likely/unclear) |
|
|
1. Remdesivir |
|||||||||
|
Ader et al., 2021 |
Computerized block randomization
Participants were randomly assigned 1:1:1:1:1 when five groups were initially implemented, and were then assigned 1:1 to receive either standard of care or standard of care plus remdesivir, once the other three treatment groups had been stopped for futility. Randomisation was done in the electronic case report form to ensure appropriate allocation concealment and used computer-generated blocks of various sizes; it was stratified on severity of disease at inclusion and on European administrative region. |
Likely
Participants allocated to standard of care alone or in combination with remdesivir were recruited contemporaneously.
Allocated treatment was not masked to participants nor study investigator.
|
Likely
Open-label trial |
Likely
Open-label trial |
Likely
Open-label trial |
Unlikely
All outcome measures described in the methods are reported in the results. |
Unlikely
No lost to follow up. |
Unlikely
Participants included in the analysis are not significantly different from those who were randomized into the trial. |
|
|
Barratt-Due et al., 2021
|
Computerized.
Eligible patients were allocated in an equal ratio using computer randomization procedures. There were 2 separate allocation lists. The first was the global list, in which the allocation sequence was prepared by an independent statistician appointed by the international trial steering group. A secondary national list was additionally prepared as a backup if allocation according to the global list was not available. The randomization procedure ensured that a patient could be allocated only to an available treatment. The randomization lists were not stratified or blocked; thus, the randomization can be regarded as simple. |
Unlikely
The first was the global list, in which the allocation sequence was prepared by an independent statistician appointed by the international trial steering group. A secondary national list was additionally prepared as a backup if allocation according to the global list was not available. |
Likely
Open label |
Likely
Open label |
Unclear
Despite being a randomized controlled trial with blinded analyses of all relevant data, it did not include a placebo group. |
Unlikely
All outcomes were reported in main article of appendix
ClinicalTrials.gov: NCT04321616 |
Likely
15 to 24% of participants in study groups were lost to follow-up. Missing data on outcomes due to discharge or participant withdrawal were imputed with best outcome. |
Unlikely
Each pairwise intention-to-treat analysis was between the remdesivir or HCQ group and its respective SoC. Some participants receiving SoC act as controls for both active treatment groups, whereas some act in one or the other, giving a partial overlap of the 2 control groups. |
|
|
Mahajan, 2021
|
Computerized (Excel)
“For administering intervention, random number was generated in Excel to follow simple random sampling technique.”
|
Unclear
Not described at what time the random number was generated and who had knowledge of it |
Likely
“We did not give placebo injection in the no-remdesivir group and did not do blinding.” |
Likely
“We did not give placebo injection in the no-remdesivir group and did not do blinding.” |
Likely
“We did not give placebo injection in the no-remdesivir group and did not do blinding.” |
Unclear
Trial not registered; some results described without data presented
|
Likely
Relatively small sample size and multiple drop outs (I: 19.5%; C: 12.2%) |
Likely
Analysis not performed according to intention-to-treat protocol; drop outs and 1 cross over in relatively small study groups |
|
|
Pan, 2020
|
Computerized
Once a hospital has obtained approval, electronic entry of patients who have given informed consent takes only a few minutes. At the end of it, the randomly allocated treatment is displayed on the screen and confirmed by electronic messaging. |
Unlikely
Allocation displayed on screen after entry of patient in the system |
Likely
Unblinded trial (mortality: unlikely)
|
Likely
Unblinded trial (mortality: unlikely)
|
Likely
Unblinded trial (mortality: unlikely)
|
Unlikely
Protocol published
https://www.who.int/publications/m/item/an-international-randomised-trial-of-additional-treatments-for-covid-19-in-hospitalised-patients-who-are-all-receiving-the-local-standard-of-care |
Unlikely
Low rate of lost-to follow-up and reasons comparable between groups |
Unlikely
analyses performed according to intention-to-treat protocol |
|
|
Beigel, 2020 |
Eligible patients were randomly assigned in a 1:1 ratio to receive either remdesivir or placebo. Randomization was stratified by study site and disease severity at enrollment |
Unlikely
Adequate procedure |
Unlikely
No bias expected
|
Unlikely
No bias expected
|
Unlikely
At the time of the data and safety monitoring board report, which was based on data cutoff date of April 22, 2020, a total of 482 recoveries (exceeding the estimated number of recoveries needed for the trial) and 81 deaths had been entered in the database. At that time, the data and safety monitoring board recommended that the preliminary primary analysis report and mortality data from the closed safety report be provided to trial team members from the National Institute of Allergy and Infectious Diseases (NIAID). |
Unlikely
Primary endpoints relevant and described. |
Unlikely
|
Unlikely
Intention-to-treat analysis performed |
|
|
Spinner, 2020
|
The randomization list was created and validated by the interactive web response system (IWRS) vendor. A dummy randomization list was provided to the biostatistician employed by the study sponsor for review. A separate list of sequential patient numbers within each treatment group was generated by the IWRS vendor. |
Unclear
Patients were not stratified by site at enrolment, which may have led to imbalances in patient care and discharge practices. |
Unclear
It was an open label study. |
Likely
It was an open label study, which might have led to differences in patient care. |
Unclear
It was an open label study, which might have led to differences in reporting the results. |
Unlikely
Outcome measures reported in the method section are also reported.
|
Unlikely
One patient was lost to follow up (I2 group) and was therefore not included in the primary analysis. |
Unlikely
Not all randomized patients were included in analysis, but intention to treat analysis did not reveal different results. |
|
|
Wang, 2020 |
Eligible patients were randomly assigned (2:1) to either the remdesivir group or the placebo group. Randomisation was stratified according to the level of respiratory support as follows: (1) no oxygen support or oxygen support with nasal duct or mask; or (2) high-flow oxygen, non-invasive ventilation, invasive ventilation, or extracorporeal membrane oxygenation. The permuted block (30 patients per block) randomisation sequence, including stratification, was prepared by a statistician not involved in the trial using SAS software, version 9.4. |
Unlikely
Eligible patients were allocated to receive medication in individually numbered packs, according to the sequential order of the randomisation centre. Envelopes were prepared for emergency unmasking. |
Unclear
Double-blind, but method of blinding was not described.
|
Unclear
Double-blind, but method of blinding was not described.
|
Unclear
It was not described whether assessors were blinded. |
Unlikely
The protocol is available online. This trial is registered with ClinicalTrials.gov, before the start of recruitment |
Unlikely
Numbers lost to follow-up were not reported. |
Unlikely
All allocated patients were included in the intention-to-treat analysis. |
|
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Beoordelingsdatum en geldigheid
Publicatiedatum : 07-11-2022
Beoordeeld op geldigheid : 03-10-2022
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut). Deze ondersteuning werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De werkgroep werd gefinancierd uit een VWS subsidie.
De financiers hebben geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodules is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de behandeling van patiënten met COVID-19.
In 2020 is een multidisciplinair expertiseteam behandeling ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van het expertiseteam behandeling) die betrokken zijn bij de zorg voor patiënten met COVID-19. Dit expertiseteam fungeerde als stuurgroep, welke opdracht heeft gegeven tot het ontwikkelen van de module, alsmede fungeerde als klankbordgroep.
Werkgroep
- Dr. Marjolein Hensgens, internist-infectioloog, Afdeling Infectieziekten, UMC Utrecht en LUMC Leiden (Stichting Werkgroep Antibiotica Beleid)
- Drs. Emilie Gieling, apotheker, Afdeling Klinische Farmacie, UMC Utrecht.
- Prof. Dr. Dylan de Lange, intensivist, Afdeling Intensive Care, UMC Utrecht.
- Dr. Wim Boersma, longarts, Afdeling Longziekten, Noordwest Ziekenhuisgroep, Alkmaar.
- Dr. Paul van der Linden, apotheker, Afdeling Klinische Farmacie, Tergooi MC, Hilversum (Stichting Werkgroep Antibiotica Beleid).
- Prof. Dr. Bhanu Sinha, arts-microbioloog, Afdeling Medische Microbiologie & Infectiepreventie, UMCG, Groningen (Stichting Werkgroep Antibiotica Beleid).
- Dr. Mark de Boer, internist-infectioloog, Afdelingen Infectieziekten en Klinische Epidemiologie, LUMC, Leiden (Stichting Werkgroep Antibiotica Beleid).
- Tot 1-11-2021 tevens deel van de werkgroep: Dr. Albert Vollaard, internist-infectioloog, LCI, RIVM
Stuurgroep (expertiseteam Behandeling COVID-19)
- Dr. L.M. van den Toorn (voorzitter), longarts, Erasmus Medisch Centrum (Erasmus MC), NVALT
- Dr. M.G.J. de Boer, internist-infectioloog, Leids Universitair Medisch Centrum (LUMC), SWAB/NIV)
- Drs. A.J. Meinders, internist-intensivist, St. Antonius Ziekenhuis, NVIC
- Prof. dr. D.W. de Lange, intensivist-toxicoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), NVIC
- Dr. C.H.S.B. van den Berg, infectioloog-intensivist Universitair Medisch Centrum Groningen (UMCG), NVIC
- Dr. S.U.C. Sankatsing, internist-infectioloog, Diakonessenhuis, NIV
- Dr. E.J.G. Peters, internist-infectioloog, Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. M.S. Boddaert, arts palliatieve geneeskunde, Leids Universitair Medisch Centrum (LUMC), IKNL
- Dr. P.L.A. Fraaij, kinderarts-infectioloog, Erasmus Medisch Centrum (Erasmus MC), Sophia Kinderziekenhuis, NVK
- Dr. E. van Leeuwen, gynaecoloog, Amsterdam University Medical Centers (Amsterdam UMC), NVOG
- Dr. J.J. van Kampen, arts-microbioloog, Erasmus Medisch Centrum (Erasmus MC), NVMM
- Dr. M. Bulatović-Ćalasan, internist allergoloog-immunoloog en klinisch farmacoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. A.F.J. de Bruin, anesthesioloog-intensivist, St. Antonius Ziekenhuis, NVA
- Drs. A. Jacobs, klinisch geriater, Catharina Ziekenhuis, NVKG
- Drs. B. Hendriks, ziekenhuisapotheker, Leids Universitair Medisch Centrum (LUMC), NVZA
- Drs. M. Nijs, huisarts, NHG
- Dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
Meelezer
- Drs. K. (Klaartje) Spijkers, senior adviseur patiëntenbelang, Patiëntenfederatie Nederland, Utrecht
Met ondersteuning van:
- dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
- dr. L.M.P. Wesselman, adviseur, Kennisinstituut van Medisch Specialisten
- dr. D. Nieboer, adviseur, Kennisinstituut van Medisch Specialisten
- drs. A.L.J. (Andrea) Kortlever - van der Spek, adviseur, Kennisinstituut van Medisch Specialisten
-
M. Griekspoor MSc., junior adviseur, Kennisinstituut van Medisch Specialisten
- drs. I. van Dusseldorp, senior literatuurspecialist, Kennisinstituut van Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
De Lange |
1. Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht (0,6 fte) |
Secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE), onbezoldigd. |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, klinisch epidemioloog, senior medisch specialist, Leids Universitair Medisch Centrum, afdeling Infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Sinha |
Arts-microbioloog/hoogleraar, Universitair Medisch Centrum Groningen (voltijd) (zie ook https;//www.rug.nl/staff/b.sinha/) |
- SWAB-bestuur: secretaris [onbetaald; vacatiegeld voor instelling] |
- Projectsubsidie EU (Cofund): deelprojecten, cofinanciering
Mogelijk boedbeeldfunctie SWAB |
Geen actie nodig |
|
Van der Linden |
Ziekenhuisapotheker |
Penningmeester SWAB, vacatiegeld |
Geen |
Geen actie nodig |
|
Vollaard |
Internist-infectioloog, Landelijke Coordinatie Infectieziektebestrijding, RIVM |
Arts voor ongedocumenteerde migranten, Dokters van de Wereld, Amsterdam (onbetaald) |
Geen |
Geen actie nodig |
|
Gieling |
Ziekenhuisapotheker - Klinisch Farmacoloog, UMC Utrecht |
Lid OMT Nederlandse Vereniging voor Ziekenhuisapothekers (onbetaald) |
Geen |
Geen actie nodig |
|
Boersma |
Longarts Noordwest Ziekhuisgroep |
Lid sectie infectieziekten NVALT, onbetaald |
Eenmalige digitale deelname aan adviesraad MSD Pneumovax over Pneumococcal disease, betaald
|
Geen actie nodig |
|
Hensgens |
Internist-infectioloog, UMC Utrecht (0.8 aanstelling, waarvan nu 0.4 gedetacheerd naar LUMC) Internist-infectioloog, LUMC (via detachering, zie boven) |
Geen |
Geen |
Geen actie nodig |
Stuurgroep
|
Achternaam stuurgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Van den Toorn (voorzitter) |
Voorzitter NVALT |
Geen |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, senior medisch specialist, LUMC, afdeling infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Meinders |
Internist-intensivist, St.-Antonius ziekenhuis, Nieuwegein |
commissie werk |
Geen |
Geen actie nodig |
|
De Lange |
Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht |
secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE) (onbetaald) |
Geen |
Geen actie nodig |
|
Van den Berg |
Infectioloog-intensivist, UMCG |
Geen |
Geen |
Geen actie nodig |
|
Sankatsing |
Internist-infectioloog/internist-acute geneeskunde, Diakonessenhuis, Utrecht |
- Bestuurslid Nederlandse Vereniging van Internist-Infectiologen (NVII) (onbetaald). |
Geen |
Geen actie nodig |
|
Peters |
Internist - aandachtsgebieden infectieziekten en Acute Geneeskunde Amsterdam UMC, locatie Vumc |
Wetenschappelijk Secretaris International Working Group on the Diabetic Foot (onbetaald) |
Geen |
Geen actie nodig |
|
Boddaert |
Medisch adviseur bij Integraal Kankercentrum Nederland (IKNL) en Palliatieve Zorg Nederland (PZNL) Arts palliatieve geneeskunde in LUMC |
Geen |
Geen |
Geen actie nodig |
|
Fraaij |
Kinderarts infectioloog- immunoloog, Erasmus MC-Sophia, Rotterdam |
Bestuur Stichting Infecties bij Kinderen (onbetaald) |
deelname aan RECOVER, European Union's Horizon 2020 research |
Geen actie nodig |
|
Van Leeuwen |
Gyaecoloog Amsterdam Universitair Medisch Centra |
Geen |
Geen |
Geen actie nodig |
|
Van Kampen |
Arts-microbioloog, afdeling Viroscience, Erasmus MC |
- associate editor antimicrobial resistance & infection control (onbetaald) - lid antibioticacommissie Erasmus MC (onbetaald) |
1. Mede uitvinder patent: 1519780601-1408/3023503 2. R01AI147330 (NIAID/NH) (HN onderzoek (1+2 niet gerelateerd aan COVID-19)
|
Geen actie nodig |
|
Bulatovic |
Internist allergoloog-immunoloog en klinische farmacoloog, UMC Utrecht en Diakonessenhuis Utrecht |
Functie 1: arts |
Geen |
Geen actie nodig |
|
De Bruin |
Anesthesioloog - Intensivist St. Antonius ziekenhuis Nieuwegein en Utrecht |
Geen |
Geen |
Geen actie nodig |
|
Jacobs |
Klinisch geriater en klinisch farmacoloog |
Geen |
Geen |
Geen actie nodig |
|
Hendriks |
Ziekenhuisapotheker farmaceutische patiëntenzorg, afd. Kiinische Farmacie en Toxicoiogie, Leids Universitair Medisch Centrum |
Lid SWAB werkgroep surveillance antibioticagebruik, onbetaald Lid SWAB richtlijncommissie antibiotica allergie, onbetaald |
Geen |
Geen actie nodig |
|
Nijs |
Huisarts |
Geen |
Geen |
Geen actie nodig |
|
Hofstede |
Senior adviseur Kennisinstituut van Medisch Specialisten |
Geen |
Geen |
Geen actie nodig |
Meelezer
|
Achternaam |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Spijkers |
Senior adviseur patiëntenbelang |
Voorzitter Stichting Samen voor Duchenne |
Geen |
Geen actie nodig |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde patiëntenvereniging in de klankbordgroep. De verkregen input is meegenomen bij het opstellen van de module. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Werkwijze
Van leidraad naar richtlijnmodules
Bij aanvang van de pandemie in 2020 was het onduidelijk of bestaande of nieuwe medicijnen een relevante bijdrage konden leveren aan het herstel van patiënten geïnfecteerd met het SARS-CoV-2. Vandaar dat eind februari 2020 werd aangevangen met de eerste versie van de leidraad ‘Medicamenteuze behandeling voor patiënten met COVID-19 (infectie met SARS–CoV-2)’, welke begin maart 2020 online beschikbaar werd gesteld op de website van de SWAB (https://swab.nl/nl/covid-19). Sindsdien werd het adviesdocument op wekelijkse basis gereviseerd en indien nodig op basis van nieuwe publicaties van onderzoek aangepast. Het initiatief en de coördinatie hiertoe werden genomen door de SWAB Leidraadcommissie, ondersteund door het kennisinstituut van de Federatie Medisch Specialisten en een brede klankbordgroep waarbinnen de betrokken specialisten(verenigingen) zijn vertegenwoordigd. In september 2021 is gestart met het doorontwikkelen van de leidraad naar richtlijnmodules.
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de COVID-19 pandemie zijn knelpunten op verschillende manieren geïnventariseerd:
1. De expertiseteams benoemde de knelpunten in de zorg voor patiënten met COVID-19.
2. Er is een mailadres geopend (covid19@demedischspecialist.nl) waar verschillende partijen knelpunten konden aandragen, die vervolgens door de expertiseteams geprioriteerd werden.
3. Door de Federatie van Medisch Specialisten zijn webinars georganiseerd waarbij vragen konden worden ingestuurd. Deze vragen zijn na afloop van de webinars voorgelegd aan de expertiseteams en geprioriteerd.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. Wanneer mogelijk werd de data uit verschillende studies gepoold in een random-effects model. Review Manager 5.4 werd gebruikt voor de statistische analyses. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
Definitie |
|
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
Bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Zoekverantwoording
On request.