Interleukine-6 remmers
Uitgangsvraag
Wat is de plaats van interleukine (IL-)6 remmers bij de behandeling van COVID-19 patiënten?
Aanbeveling
Behandeling IL-6 remmers (in het bijzonder tocilizumab) blijkt met name bij patiënten met progressieve ziekte en een (matig) ernstige respiratoire insufficiëntie, in combinatie met corticosteroïden, een overlevingsvoordeel te bieden. Op basis van het bewijs is het advies:
Behandel opgenomen patiënten met respiratoire klachten door COVID-19 en een toegenomen zuurstofbehoefte met tocilizumab 600 mg eenmalig i.v. indien zij reeds zijn gestart met dexamethason en een CRP ≥75 mg/L hebben en een persisterend respiratoire verslechtering leidend tot noodzaak tot hoge zuurstofsuppletie - via een venturimasker (≥6 L O2), non-rebreathing masker, NIV of high flow nasal oxygen (Optiflow) - met als meest aannemelijke verklaring de COVID-19 geïnduceerde longinflammatie (niet b.v.: longembolieën of bacteriële pneumonie).
Behandel patiënten met respiratoire insufficiëntie die vanaf de SEH direct op de IC worden opgenomen (en daarom buiten het ziekenhuis al eerder aan de bovengenoemde criteria zouden hebben voldaan) naast dexamethason met tocilizumab. Hierbij wordt geadviseerd de therapie <24 uur na opname op de IC toe te dienen.
Indien tocilizumab niet beschikbaar is of dit om een andere reden niet gegeven kan worden, dan kan andere anti-inflammatoire therapie overwogen worden:
- baricitinib 4 mg dagelijks gedurende 14 dagen of tot ontslag of sarilumab 400 mg eenmalig i.v. (geen voorkeur uitgesproken voor een van beide middelen)
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Er is literatuuronderzoek verricht naar de verschillen in klinische uitkomsten tussen behandeling met en zonder IL-6 remmers bij patiënten met COVID-19. Tot en met 24 januari 2022 werden er 17 gerandomiseerde gecontroleerde studies (RCT’s) gevonden in patiënten die waren opgenomen in het ziekenhuis (n=4774 in de interventiegroep en n=4231 in de controlegroep). De meeste RCT’s onderzochten het middel tocilizumab (10 studies), daarnaast werd er onderzoek gedaan naar sarilumab (4 studies), levilimab (1 studie). Twee studies randomiseerden naar meerdere IL-6 remmers (tocilizumab of siltuximab en tocilizumab of siltuximab) of de standaardbehandeling. De grootste studie die werd meegenomen was de RECOVERY trial (Horby, 2021), met meer dan 4000 geïncludeerde patiënten. Er werden alleen studies gevonden die IL-6 remmers onderzochten bij opgenomen patiënten.
De cruciale uitkomstmaten voor de besluitvorming waren mortaliteit en de noodzaak voor uitgebreide respiratoire ondersteuning. De verschillende IL-6 remmers worden apart besproken.
Er werden alleen gerandomiseerde en gecontroleerde trials geïncludeerd in de analyse, waardoor de kwaliteit van bewijs initieel hoog was. Omdat dat er een aantal open-label trials waren (waaronder de RECOVERY trial), met een mogelijk risico op vertekening van de studieresultaten (risk of bias) bij subjectieve uitkomstmaten, werd de kwaliteit van dit bewijs waar nodig naar beneden bijgesteld. Daarnaast waren er meerdere studies met een relatief kleine populatie en mede hierdoor een grote spreiding van het betrouwbaarheidsinterval rondom de puntschatter van de uitkomstmaat (imprecision), waardoor de kwaliteit van dit bewijs ook naar beneden werd bijgesteld. Er is, waar mogelijk, een sensitiviteitsanalyse gedaan waarin alleen kwalitatief goede studies en studies met meer dan 80% corticosteroïden in de standaard behandeling werden geïncludeerd.
IL-6 remmers bij patiënten die waren opgenomen in het ziekenhuis
Sinds het begin van de COVID-19 pandemie is IL-6 remming in diverse onderzoeken toegepast in de behandeling van patiënten met COVID-19, met tegenstrijdige resultaten. De data betroffen grotendeels patiënten met redelijke tot ernstige COVID-19. De meeste studies onderzochten de IL-6 remmer tocilizumab. Een gepoolde analyse naar het effect van tocilizumab op de mortaliteit binnen 28 dagen laat met een redelijke zekerheid zien dat er een reductie in de mortaliteit optreedt bij het gebruik tocilizumab (risk ratio: 0,90, 95% CI: 0,83 tot 0,98). Het gepoolde (en gewogen) risicoverschil is echter klein (risicoverschil: -0,01, 95% CI: -0,04 tot 0,02). Echter, in deze resultaten werd de studie van Gordon (2021) niet meegenomen, omdat in deze studie niet de 28 dagen mortaliteit maar de 21 dagen mortaliteit werd gerapporteerd. Daarnaast bevat deze analyse een aantal kwalitatief slechtere studies en studies waarin de standaard behandeling vrijwel geen corticosteroïden bevatte.
Als de studies van Horby (2021), Rosas (2021b) en Gordon (2021) gecombineerd worden (die allen van goede kwaliteit waren en waarin de standaard behandeling in meer dan 80% corticosteroïden bevatte), kan er nog steeds met redelijke zekerheid geconcludeerd worden dat er een significante reductie in de mortaliteit optreed, echter, het risicoverschil is dan ook klinisch significant (boven de 4% punten, die vooraf gedefinieerd werden als grens voor klinische significantie).
Hier onder zullen wij de verschillen tussen de studies bespreken en bovenstaande resultaten in een context plaatsen.
De ‘oudere’ studies, die met name in 2020 gepubliceerd werden, toonden wisselende resultaten bij het effect op klinische verbetering. In de CORIMUNO-19 studie (Mariette, 2021) werd aangetoond dat patiënten met tocilizumab minder kans hadden op het gecombineerde eindpunt non-invasieve ventilatie, mechanische ventilatie of overlijden, vergeleken met patiënten die de standaard behandeling ontvingen (24% versus 36%). Deze uitkomst was net statistisch significant (p=0,04). In de EMPACTA-studie van Salama (2020) hadden patiënten in de tocilizumab arm minder kans op het gecombineerde eindpunt mechanische ventilatie of overlijden (12,0% vs 19,3%). Effect op klinische verbetering werd echter niet bevestigd in de andere RCT’s, waaronder de COVACTA studie (Rosas, 2021a). De tijd tot klinische verbetering, ontslag of stoppen van beademing werd onderzocht in de trial van Stone (2020), deze toonde op al deze punten geen verschil aan tussen de tocilizumab-groep en de groep met standaard behandeling. Ook in de studie van Salama (2020) en Salvarani (2020) was er geen verschil in tijd tot klinische verbetering of ontslag tussen beide groepen. Hierbij moet worden vermeld dat de incidentie van overlijden in het onderzoek van Salvarani (2020), laag was. In deze Italiaanse studie kan dit vermoedelijk verklaard worden door exclusie van patiënten die niet geschikt waren voor de intensive care in verband met co-morbiditeiten, waardoor er een minder zieke populatie geselecteerd werd. In de COVACTA studie (Rosas, 2021a) was er wel een statistisch significant effect zien in tijd tot ontslag van 20 versus 28 dagen. Op de overige uitkomsten kon ook in deze studie geen effect worden aangetoond. Toepassing van tocilizumab bleek in de bovengenoemde studies relatief veilig.
Een gerandomiseerde open label trial, waarin de effectiviteit van de behandeling met tocilizumab in patiënten met ernstige COVID-19 werd onderzocht (Veiga, 2021), werd voortijdig beëindigd, omdat er op dag 15 na inclusie een hogere sterfte werd gezien in de tocilizumab groep (11 patiënten [17%]) t.o.v. de controlegroep (2 patiënten [3%]). Tevens waren er meer bijwerkingen gezien in de tocilizumab groep (43% versus 34% in de groep met standaardbehandeling). De populatie bestond uit opgenomen patiënten met bevestigde COVID-19 en extra zuurstofbehoefte of mechanische beademing en met ten minste 2 van 4 verhoogde ‘biomarkers‘ (D dimeer, ferritine, CRP en LDH). De auteurs zelf noemen een aantal beperkingen van deze studie: 1) beperkte statistische power (patiënt aantallen in de studie zijn klein), 2) de groepen lijken niet gelijk aan het begin van de behandeling (m.n. zuurstoftoediening en beademing), 3) er is onduidelijk op welk moment de tocilizumab (en andere medicatie) werd toegediend. Slechts 7% van de patiënten kreeg bij aanvang corticosteroïden.
Op basis van bovenstaande studies was er in 2020 geen onderbouwing voor het adviseren van tocilizumab in de behandeling van patiënten met COVID-19. Echter, begin 2021 verscheen de data van 2 grotere gerandomiseerde onderzoeken, die als eerste een evident effect van tocilizumab op de mortaliteit rapporteerden:
Recovery trial
De grootste studie die geïncludeerd werd, was de RECOVERY trial van Horby (2021). In deze studie werd tocilizumab vergeleken met de standaard behandeling als patiënten voldeden aan de volgende criteria: hypoxie (of SO2 <92% zonder extra O2 toediening, of noodzaak van O2 toediening) en CRP ≥75 mg/L. Uiteindelijk werden er 4116 patiënten in deze studie geïncludeerd, waarvan 562 patiënten (14%) invasief beademend werden. De grote meerderheid (82%) van de patiënten kreeg ook corticosteroïden. Het primaire eindpunt, de 28-dagen sterfte, werd gehaald als volgt: in de tocilizumab groep overleden 621 (31%) patiënten, terwijl in de groep met de standaardbehandeling 729 (35%) patiënten overleden (RR 0,85; 95% CI 0,76-0,94; p=0,0028). Het grootste voordeel werd behaald bij die patiënten die ook met corticosteroïden werden behandeld. Ook werden er significante verschillen in de secondaire eindpunten gevonden. Het percentage patiënten dat met tocilizumab werd behandeld en levend na 28 dagen uit het ziekenhuis werd ontslagen was groter, respectievelijk 57% versus 50% (RR 1,22; 95% CI 1,12-1,33; p<0,0001). De patiënten die initieel niet beademd werden, hadden een grotere kans om niet aan de beademing komen of te overlijden (35% vs. 42%; risk ratio 0,84; 95% CI 0,77-0,92 p<0,0001).
Als de onderzoekspopulatie werd gestratificeerd naar het type ademhalingsondersteuning (enkel toediening van O2, niet-invasieve beademing en invasieve beademing), was er in geen van de afzonderlijke strata een statistisch significant verschil tussen tocilizumab en de standaard behandeling wat betreft het relatieve risico op overlijden binnen 28 dagen. Dit houdt verband met de kleinere groepsgrootte per stratum, maar betekent ook dat het advies - ondanks een trend - niet toegespitst kan worden op de benodigde ademhalingsondersteuning.
REMAP-CAP
Een andere grote studie, de REMAP-CAP studie van Gordon (2021), werd niet meegenomen in de gepoolde resultaten van de (28 dagen) mortaliteit in deze richtlijn omdat zij hun uitkomstmaat na 21 dagen rapporteerden. Ook de REMAP-CAP is een multicenter, open label, adaptive platform trial dat onderzoek doet naar patiënten met ernstige pneumonie, binnen of buiten de pandemische setting. Het COVID-19 immuunmodulatie domein bestaat uit 5 armen: IL-6 remmer (tocilizumab), IL-6 remmer (sarilumab), IL-1 receptor antagonist (anakinra), interferon beta-a1 en een controlegroep (geen immuunmodulatie). Aan 353 patiënten werd tocilizumab toegediend, sarilumab werd gegeven aan 48 patiënten in dit domein. Er zijn 402 patiënten in de controlegroep geïncludeerd. Alle patiënten lagen opgenomen op de intensive care en ontvingen de gift met de IL-6 remmer binnen 24 uur na het starten van orgaanondersteuning. In de groep patiënten met tocilizumab ontving 29% van de patiënten een tweede dosis 12-24u na de eerste gift. De grote meerderheid (80%) van de patiënten kreeg ook dexamethason. De primaire uitkomst was het aantal dagen zonder respiratoire of cardiovasculaire orgaanondersteuning (zogenaamde “organ support free days”) binnen 21 dagen na randomisatie. Alle in het ziekenhuis overleden patiënten kregen hierbij de slechtste score (-1). Van de overige patiënten werd berekend hoeveel dagen zonder orgaanondersteuning zij hadden tot 21 dagen. Meer dagen zonder orgaanondersteuning betekent hierbij dus een sneller herstel. Het aantal (mediaan) dagen zonder orgaanondersteuning was 10 (interquartile range [IQR] -1, 16), 11 (IQR 0, 16) en 0 (IQR -1, 15) voor tocilizumab, sarilumab en de controlegroep respectievelijk. De kans op overlijden in het ziekenhuis was 28,0% (98/350) in de tocilizumab groep, 22,2% (10/45) in de sarilumab groep en 35,8% (142/397) in de controle groep. Vergeleken met de controle groep was de mediane adjusted odds ratio voor ziekenhuisoverleving 1,64 (95% CI 1,14; 2,35) voor tocilizumab en 2,01 (95% CI 1,18; 4,71) voor sarilumab.
Sinds het verschijnen van de bovenstaande grote RCT’s, zijn er twee kleinere onderzoeken gepubliceerd die geen effect van IL-6 remmers laten zien op de mortaliteit na 28 dagen of progressie van de ziekte. De studie van Rosas (2021b) includeerde patiënten met een matige en ernstige COVID-19 pneumonie, maar hield geen rekening met inflammatie bij inclusie. De studie van Sancho-Lopez (2021) includeerde patiënten met een matig-ernstig ziektebeeld, maar mogelijk minder inflammatie dan in de REMAP-CAP studie. Daarnaast werden corticosteroïden korter gegeven in de andere studies (minimaal 3 dagen methylprednisolon). Mogelijk verklaren deze verschillen het ontbreken van een aantoonbaar effect van IL-6 remming.
Bij het formuleren van een advies over tocilizumab is het belangrijk om niet alleen naar de 28 dagen mortaliteit te kijken, omdat studies die andere uitkomstmaten rapporteren (zoals Gordon, 2021) dan niet meegenomen kunnen worden. Ook representeren de meer recente RCT’s waarschijnlijk beter de huidige situatie, waarin corticosteroïden deel uitmaken van de standaardbehandeling.
Als alle bovenstaande overwegingen worden meegenomen, kunnen we de volgende conclusie trekken over het effect van tocilizumab: er is een statistisch significant voordeel te zien van tocilizumab op de mortaliteit, de omvang van het effect is in de twee grootste geïncludeerde studies ook ruim boven de vooraf gestelde definitie van klinische significantie (4% verschil in 28 dagen mortaliteit bij Horby, 2021; 8% verschil in 21 dagen mortaliteit bij Gordon, 2021). Dit effect lijkt bij zowel matig zieke patiënten met inflammatie (een veel gebruikt inclusiecriterium) als bij ernstig zieke patiënten aanwezig te zijn. Ook op de andere uitkomstmaten ‘extensive respiratory support’ en ‘opnameduur’ zijn er voordelen zichtbaar van tocilizumab, al zijn deze niet altijd even groot en statistisch significant.
Soort IL-6 remmer
Verreweg de meeste studies bestudeerden tocilizumab als IL-6 remmer. Sarilumab werd vier studies bestudeerd, met relatief kleine aantallen patiënten. De resultaten van deze studies lijken ook op een mogelijk voordeel te wijzen van sarilumab. Echter, door onder andere de beperkte power van deze studies zijn de resultaten niet significant. Sarilumab werd in de REMAP-CAP studie toegediend als intraveneuze infusie van 400 mg. Er zijn alleen preparaten voor subcutane injectie in Nederland geregistreerd. Uit deze subcutane formulering kan een intraveneuze toediening worden klaargemaakt conform protocol van REMAP-CAP. Tocilizumab is wel als intraveneuze infusie in Nederland geregistreerd, waardoor intraveneuze toediening conform het label kan plaatsvinden. Bovendien is, zowel binnen als buiten dit studieverband, meer ervaring met het gebruik van tocilizumab. Om deze redenen is er bij voldoende beschikbaarheid een voorkeur voor het gebruik van tocilizumab. Bij onvoldoende beschikbaarheid van tocilizumab is toediening van sarilumab intraveneus (uit de subcutane formulering) een optie.
Dosering
Tocilizumab wordt in alle genoemde studies gedoseerd op basis van gewicht. De meeste studies houden een schema aan van 8 mg/kg met een maximum van 800 mg per dag. De RECOVERY studie doseerde volgens gewichtsklassen, waarbij de doseringen tocilizumab werden afgerond. Patiënten van 40-65 kg kreeg 400 mg, patiënten van 65-90 kg kregen 600 mg, en patiënten van >90 kg kregen 800 mg tocilizumab. Dit heeft als voordelen dat het gemakkelijker te doseren is met minder kans op toedienfouten en minder spillage.
Het toedienen van tocilizumab in een ‘fixed-dose’ regime zou dus meer voordelen kunnen hebben, maar tot voor kort waren er geen gegevens beschikbaar over de farmacokinetiek van tocilizumab bij patiënten met ernstige COVID-19. Moes (2021) beschrijft deze farmacokinetiek in een artikel. In 139 plasma monsters van 29 IC patiënten werd de concentratie tocilizumab geanalyseerd en gemodelleerd. De uitkomst was dat tocilizumab doseren op basis van gewicht (8 mg/kg) leidt tot grotere variabiliteit in de gemeten spiegels. De minimaal benodigde plasmaconcentratie voor het behoud van maximale IL-6-receptorbezetting (5 µg/ml) wordt echter bij alle patiënten voor ten minste 15 dagen behouden, onafhankelijk van dosering o.b.v. gewicht. Uit de simulaties wordt duidelijk dat een ‘fixed-dose’ van 600 mg voor alle IC-patiënten minder variatie in blootstelling geeft, en wel de benodigde plasmaconcentratie behaalt en behoudt.
De patiëntenpopulatie met een indicatie voor het gebruik van tocilizumab op de verpleegafdeling en op de IC liggen dusdanig in elkaars verlengde dat hierin eenzelfde advies gevolgd kan worden. Als dosering wordt geadviseerd om tocilizumab in een ‘fixed-dose’ van 600 mg toe te dienen (en is een klinisch gelijk effect te verwachten als bij een dosering van 8 mg/kg).Het is ook mogelijk om het doseerschema van de RECOVERY studie te volgen voor patiënten opgenomen op de verpleegafdeling, maar zal leiden tot meer spillage:
- 40-65 kg: 400 mg tocilizumab
- 65-90 kg: 600 mg tocilizumab
- >90 kg: 800 mg tocilizumab
Sarilumab kan gegeven worden in 400 mg intraveneus eenmalig conform het protocol van REMAP-CAP.
Alternatieve, lagere dosering van tocilizumab bij (dreigend) ernstig tekort aan IL-6 remmers
In de klinische gerandomiseerde studies werd tot heden een dosis tocilizumab gebruikt die ook geregistreerd is voor de behandeling van reumatologische aandoeningen. Een duidelijke rationale voor de initiële keuze van deze dosis voor de specifieke behandeling van COVID-19 ontbrak. In een artikel van Moes (2021) wordt de farmacokinetiek van tocilizumab in patiënten met COVID-19 die op de intensive care afdeling waren opgenomen, beschreven. Hieruit bleek o.a. dat een lagere éénmaal daags gegeven dosis van 400 mg in plaats van 600 mg volgens modelering leidt tot een beperkt verlies van het aantal dagen dat de concentratie tocilizumab zich boven de gestelde doelconcentratie bevindt (plusminus 16.5 versus 19 dagen). Het is onbekend of dit ook leidt tot een relevante afname van het klinisch effect. Op grond van de beschikbare gegevens lijkt het onwaarschijnlijk, dat een potentieel verlies van effect evenredig is aan de dosisverlaging van 600 naar 400 mg.
Om deze reden kan, indien noodzakelijk, bij een (dreigend) tekort aan tocilizumab en afwezige beschikbaarheid van geschikte formuleringen van andere IL-6 remmers, tijdelijk worden overgegaan op een lagere eenmalige vaste dosis van 400 mg tocilizumab i.v., of een begindosis van 200 mg iv gevolgd door een 2e dosis van 200 mg na 7 dagen.
Tweede dosering
In de tocilizumab groep van beide studies (REMAP-CAP en RECOVERY) ontving ongeveer een derde van de patiënten een tweede dosis 12-24u na de eerste dosis op basis van inzichten van de behandelend arts. Het is niet duidelijk wat de criteria voor een tweede toediening zijn geweest. Ook is nog niet helder of er verschil van effect is bij een eenmalige versus tweevoudige gift. Tevens is het ook o.b.v. pathofysiologische en farmacologische overwegingen twijfelachtig of herhaalde toediening toegevoegde waarde zouden kunnen hebben. Vooralsnog is er daarom onvoldoende onderbouwing om een tweede gift te adviseren.
IL-6-remmers en zwangerschap en kinderen
Tocilizumab is het eerste keus middel voor de behandeling van COVID-19 positieve zwangere of lacterende vrouwen indien zij volgens de SWAB leidraad voor anti-IL-6 therapie in aanmerking komen. De indicatiestelling en dosering van tocilizumab gebeurt idealiter in multidisciplinair teamverband waarvan tenminste een longarts en/of internist en/of intensivist en perinatoloog deel uit moeten maken. Alhoewel zwangere en lacterende vrouwen ondervertegenwoordigd waren in studies, zijn er op dit moment geen aanwijzingen dat tocilizumab teratogeen of foetotoxisch is (Jorgenson, 2021). De excretie van tocilizumab in moedermelk is laag. Als tocilizumab niet beschikbaar is, kan overwogen worden om sarilumab te geven, echter, de ervaring met sarilumab gebruik in de zwangerschap is zeer beperkt. Na toediening van elke immuunmodulator in de zwangerschap in verband met COVID-19, zoals tocilizumab en sarilumab, dient kort na de geboorte een kinderarts in consult gevraagd te worden, ook al vindt de bevalling niet direct plaats. Na het gebruik van tocilizumab en sarilumab moet een bloedbeeld worden bepaald bij de neonaat, omdat infectiediagnostiek bij de neonaat minder goed te interpreteren kan zijn. Ook worden vaccinaties bij de neonaat met een levend virus in het eerste levensjaar afgeraden.
Bij kinderen is er geen bewijs dat COVID-19 met meer complicaties gepaard gaat, de ziekte lijkt bij kinderen juist met minder complicaties gepaard te gaan. Dat zou pleiten voor terughoudendheid voor het voorschrijven van IL-6 remmers bij minder zieke pediatrische patiënten (niet op IC opgenomen).
Overige overwegingen
Door gebruik van IL-6 remmers wordt de productie van CRP gedurende 14-28 dagen geremd (dieptepunt van CRP-productie ligt meestal tussen dag 7-15). Het CRP is derhalve geen biomarker meer voor (verdenking op) infecties. Het is mogelijk dat er minder CRP-gestuurde interventies plaatsvonden in de interventie groepen. Mogelijk zou de meting van procalcitonine (PCT) minder onderdrukt worden door IL-6 remmers, maar studies zijn hierover niet eenduidig.
Bijwerkingen
Een Cochrane review uit 2021 beschrijft niet alleen de effectiviteit van tocilizumab bij COVID-19, maar ook data over de veiligheid (Ghosn, 2021). Zij beschrijven 7 RCT’s waarin niet significant vaker ‘adverse events’ worden gezien, vergeleken met de controlegroep (RR 1,23, 95%CI 0,87-1,72). Echter, deze conclusie heeft een bewijskracht van ‘zeer laag’.
Ook bij inflammatoire ziekten zoals reumatoïde artritis is er ervaring met tocilizumab. ‘Adverse events’ die in deze setting werden beschreven zijn onder andere een verhoogd risico op infecties (bij met name bovenste luchtwegen), leverenzymstoornissen, neutropenie en trombocytopenie, hyperlipedemie en gastrointestinale symptomen (Schoels, 2012). Deze werden 1,2 maal vaker gezien dan in de controlegroep (Singh, 2010). In deze setting wordt tocilizumab echter vaak langdurig gegeven. ‘Adverse events’ zijn dan ook niet zonder meer te extrapoleren naar de behandeling van COVID-19, waarbij er een eenmalige gift wordt gegeven.
Virusvarianten
Sinds de opkomst van de omikron variant van SARS-CoV-2 in Nederland eind 2021, is de kans op een ernstig beloop van COVID-19 op populatieniveau zeer sterk gedaald. Het is van belang om op te merken dat de besproken gerandomiseerde studies werden verricht voor de opkomst van de omikron variant. Het is onduidelijk wat de invloed is van deze variant op het effect van anti-inflammatoire therapie, al wordt aangenomen dat patiënten die door de omikron variant een ernstige COVID-19 infectie ontwikkelen nog steeds baat hebben bij anti-inflammatoire therapie. De ‘number needed to treat’ zou wel anders (vermoedelijk hoger) kunnen zijn.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Op grond van de bekende onderzoeksgegevens kunnen IL-6 remmers worden ingezet in de behandeling van COVID-19. In de behandeling van COVID-19, waar deze middelen relatief kortdurend worden voorgeschreven, werden voordelen van IL-6 remmers gevonden (zoals een lagere mortaliteit), maar werden niet meer bijwerkingen gezien. Mogelijk zijn de patiënten aantallen nog niet groot genoeg om alle bijwerkingen in de behandeling van COVID-19 aan te tonen. Een potentieel verhoogd risico op infecties of andere bijwerkingen die bij de behandeling van bijvoorbeeld reumatoïde artritis gezien werden, moet dus meegewogen worden in de keuze om een patiënt met een IL-6 remmer te behandelen. Deze potentiële bijwerkingen spelen overigens ook een rol bij andere anti-inflammatoire middelen die ingezet kunnen worden bij COVID-19.
Kosten (middelenbeslag)
In de huidige studies varieert de gebruikte IL-6 remmer en ook de dosering varieert. De dosering tocilizumab die aanbevolen wordt in deze richtlijn is gebaseerd op gewicht. Indien er 600mg tocilizumab wordt voorgeschreven, dan kost dit rond de 1000 euro. Hierin zijn niet de opname in het ziekenhuis of andere bijkomende kosten meegenomen.
Aanvaardbaarheid, haalbaarheid en implementatie
Op grond van de bekende onderzoeksgegevens kunnen IL-6 remmers worden ingezet in de behandeling van COVID-19. Tocilizumab is beschikbaar in Nederland en patiënten die in aanmerking komen voor deze behandeling liggen opgenomen in het ziekenhuis. De werkgroep voorziet geen problemen qua implementatie.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Interleukine-6 remmers blijken met name bij patiënten met progressieve ziekte en een (matig) ernstige respiratoire insufficiëntie, in combinatie met corticosteroïden, een overlevingsvoordeel te bieden. Een tijdige start van deze geneesmiddelen (zoals in de REMAP-CAP studie) lijkt van belang.
Het brede inclusiecriterium zoals gebruikt in de RECOVERY studie was: progressieve ziekte en een hypoxemie (SpO2 <92% zonder extra O2 toediening, of met noodzaak van O2 toediening), en een CRP ≥75 mg/L. Dit kan bij toepassing in de praktijk tot verwarring leiden. Vanwege deze reden heeft de commissie dit gegeven om praktische redenen vertaald naar de noodzaak tot het gebruik van ≥6L 02. Dit is in de kliniek tevens de overgang van zuurstoftoediening via een neusslang naar een z.g.n. venturimasker, non-rebreathing masker, NIV of high flow nasal oxygen (Optiflow), en voorkomt waarschijnlijk het onnodig gebruik van tocilizumab bij een mildere vorm van de ziekte.
In bovengenoemde patiënten populatie met (matig) ernstige COVID-19, zorgen niet alleen IL-6 remmers (in het bijzonder tocilizumab), maar ook JAK remmers (in het bijzonder baricitinib) voor een klinisch relevante afname van de mortaliteit binnen 28 dagen vergeleken met placebobehandeling. Van beide soorten middelen beschrijven RCT’s geen significante bijwerkingen, al betreft dit een relatief korte follow-up.
Het verwachte effect en de bewijskracht van de literatuur betreffende het effect van tocilizumab en baricitinib op mortaliteit is vergelijkbaar, vooral als er niet alleen gekeken wordt naar de 28 dagen mortaliteit maar ook andere follow-up duur. Er is meer ervaring met tocilizumab in de behandeling van COVID-19 in Nederland en dit middel werd in grotere aantallen patiënten onderzocht: 12 RCT’s onderzochten bij ruim 7000 patiënten het effect van tocilizumab, Ter vergelijking: 3 RCT’s onderzochten bij ruim 2500 patiënten baricitinib. In patiënten met een (matig) ernstige COVID-19 infectie wordt er daarom een voorkeur uitgesproken voor tocilizumab.
Advies:
Behandel opgenomen patiënten met respiratoire klachten door COVID-19 en een toegenomen zuurstofbehoefte met tocilizumab 600 mg eenmalig i.v. indien zij reeds zijn gestart met dexamethason en een CRP ≥75 mg/L hebben en een persisterend respiratoire verslechtering leidend tot noodzaak tot hoge zuurstofsuppletie - via een venturimasker (≥6 L O2), non-rebreathing masker, NIV of high flow nasal oxygen (Optiflow) - met als meest aannemelijke verklaring de COVID-19 geïnduceerde longinflammatie (niet b.v.: longembolieën of bacteriële pneumonie).
Behandel patiënten met respiratoire insufficiëntie die vanaf de SEH direct op de IC worden opgenomen(en daarom buiten het ziekenhuis al eerder aan de bovengenoemde criteria zouden hebben voldaan) naast dexamethason met tocilizumab. Hierbij wordt geadviseerd de therapie <24 uur na opname op de IC toe te dienen.
Indien tocilizumab niet beschikbaar is of dit om een andere reden niet gegeven kan worden, dan kan andere anti-inflammatoire therapie overwogen worden:
- baricitinib 4 mg dagelijks gedurende 14 dagen of tot ontslag of sarilumab 400 mg eenmalig i.v. (geen voorkeur uitgesproken voor een van beide middelen)
Voor de behandeling van kinderen kunnen de doseringen beschreven in het Kinderformularium worden gebruikt.
Onderbouwing
Een infectie met SARS-CoV-2 kan leiden tot ernstige pneumonie en ARDS. De term cytokine storm werd genoemd bij COVID-19 in relatie tot dysregulatie van de immuunrespons. Dit zou wijzen op een verhoging van pro-inflammatoire cytokines, zoals interleukine (IL-)6. IL-6 remmers (b.v. tocilizumab) zijn al geregistreerd voor het cytokine release syndrome dat kan ontstaan ten gevolge van immunotherapie met Chimere Antigeen Receptor (CAR) T-cel therapie. Het toedienen van IL-6 remmers lijkt daarmee een potentiële therapie voor patiënten met COVID-19 en verhoogde IL-6 waarden. Hoewel de IL-6 concentraties bij COVID-19 patiënten verhoogd zijn, zijn deze waarden 10 tot 40 maal lager dan bij patiënten met ARDS (Sinha, 2020; Kox, 2020). Het is lastig om IL-6 waarden te vergelijken, omdat er verschillende bepalingsmethoden zijn die niet goed zijn gestandaardiseerd. Een mogelijk additioneel effect van anti-IL-6 therapie naast anti-inflammatoir effect is remming van de coagulatie-activatie bij COVID-19 (Levi, 2020).
Inmiddels hebben diverse gerandomiseerde gecontroleerde studies (RCT’s) de effectiviteit van IL-6 remmers onderzocht om de plaats van deze middelen bij de behandeling van COVID-19 patiënten te bepalen. Klinische dose-finding studies zijn in deze setting nooit verricht. De groep IL-6 remmers bevat zowel monokloale antistoffen gericht tegen de IL-6 receptor (tocilizumab, sarilumab, levilimab) als anti-IL6 monoklonale antistoffen (siltuximab).
Mortality (crucial) – Tocilizumab
|
Moderate GRADE |
Based on high quality studies AND studies with more than 80% use of corticosteroids in the standard of care: Treatment with tocilizumab probably reduces mortality when compared with treatment without tocilizumab in hospitalized patients with (moderate and severe) COVID-19.
Source: Hermine, 2020; Gordon, 2021; Horby, 2021; Rosas, 2021a. |
|
Mortality (crucial) in other IL-6 inhibitors than tocilizumab |
|
|
Low GRADE |
Based on high quality studies AND studies with more than 80% use of corticosteroids in the standard of care: Treatment with sarilumab probably reduces mortality when compared with treatment without sarilumab in hospitalized patients with COVID-19.
Source: Gordon, 2021; Sancho-Lopéz, 2021. |
|
Low GRADE |
Treatment with sarilumab may result in little to no clinically relevant difference in mortality when compared with treatment without sarilumab in hospitalized patients with moderate COVID-19.
Source: Lescure, 2021; Mariette, 2021; Sancho-Lopéz, 2021. |
|
Moderate GRADE |
Treatment with sarilumab probably reduces mortality when compared with treatment without sarilumab in hospitalized patients with severe COVID-19.
Source: Gordon, 2021; Lescure, 2021. |
|
- GRADE |
No evidence was found regarding the effect of treatment with levilimab on mortality when compared with treatment without levilimab in hospitalized patients with COVID-19.
Sources: none. |
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with siltuximab on mortality when compared with treatment without siltuximab in hospitalized patients with COVID-19.
Sources: Declercq, 2021. |
Extensive respiratory support (crucial)
|
Low GRADE |
Treatment with tocilizumab may result in little to no difference in need for extensive respiratory support when compared with treatment without tocilizumab in hospitalized patients with COVID-19.
Source: Gordon, 2021; Hermine, 2020; Horby, 2021; Rosas, 2021; Salama, 2021; Salvarani, 2020; Soin, 2021; Stone, 2020; Veiga, 2021. |
|
Extensive respiratory support (crucial) in other IL-6 inhibitors than tocilizumab |
|
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with sarilumab in need for extensive respiratory support when compared with treatment without sarilumab in hospitalized patients with COVID-19.
Sources: Gordon, 2021; Lescure, 2021; Mariette, 2021; Merchante, 2021; Sancho-Lopéz, 2021. |
|
- GRADE |
No evidence was found regarding the effect of treatment with levilimab in need for extensive respiratory support when compared with treatment without levilimab in hospitalized patients with COVID-19.
Sources: none. |
|
Very low GRADE |
Evidence is very uncertain about the effect of treatment with tocilizumab or siltuximab in need for extensive respiratory support when compared with treatment without tocilizumab or siltuximab in hospitalized patients with COVID-19.
Sources: Declercq, 2021 |
Duration of hospitalization (important)
|
Low GRADE |
Treatment with tocilizumab may result in a reduced length of stay compared with treatment without tocilizumab in hospitalized patients with COVID-19.
Source: Gordon, 2021; Hermine, 2020; Horby, 2021; Rosas, 2021a; Rosas, 2021b; Salama, 2021; Salvarani, 2020; Soin, 2021; Stone, 2020; Veiga, 2021; Wang, 2021. |
|
Duration of hospitalization (important) in other IL-6 inhibitors than tocilizumab |
|
|
Low GRADE |
Treatment with sarilumab may result in a reduced length of stay when compared with treatment without sarilumab in hospitalized patients with COVID-19.
Source: Lescure, 2021; Mariette, 2021; Merchante, 2021; Sancho-Lopéz, 2021. |
|
Low GRADE |
Treatment with levilimab may result in little to no difference of length of stay when compared with treatment without levilimab in hospitalized patients with COVID-19.
Source: Lomakin, 2021. |
|
Very low GRADE |
Evidence is very uncertain about the effect of treatment with tocilizumab or siltuximab on length of stay when compared with treatment without tocilizumab or siltuximab in hospitalized patients with COVID-19.
Source: Declercq, 2021. |
Time to clinical improvement (important)
|
Low GRADE |
Treatment with tocilizumab may result in little to no difference in time to clinical improvement when compared with treatment without tocilizumab in hospitalized patients with COVID-19.
Source: Salama, 2021; Soin, 2021; Stone, 2020; Wang, 2021. |
|
Time to clinical improvement (important) in other IL-6 inhibitors than tocilizumab |
|
|
Moderate GRADE |
Treatment with sarilumab probably results in little to no difference in time to clinical improvement when compared with treatment without sarilumab in hospitalized patients with COVID-19.
Source: Lescure, 2021. |
|
- GRADE |
No evidence was found regarding the effect of treatment with levilimab on time to clinical improvement when compared with treatment without levilimab in hospitalized patients with COVID-19.
Source: none. |
|
Very low GRADE |
Evidence is very uncertain about the effect of treatment with tocilizumab or siltuximab on time to clinical improvement when compared with treatment without tocilizumab or siltuximab in hospitalized patients with COVID-19.
Source: Declercq, 2021. |
Description of studies
IL-6 INHIBITORS
Declercq (2021) (COV-AID) reported a multicentre (n=16), open label, randomised controlled phase 3 trial in Belgium. Declercq (2021) aimed to assess whether tocilizumab, siltuximab or anakinra shortened the time to clinical improvement in patients with COVID-19. Patients were eligible for randomization if they met the inclusion criteria (e.g., PaO2/FiO2 < 350 mmHg on room temperature or < 280 mmHg on supplemental oxygen and bilateral pulmonary infiltrates; use invasive mechanical ventilation, OR non-invasive ventilation or continuous use of CPAP for hypoxia, OR oxygen supplementation with an oxygen flow of at least 10 L/min independent of delivery system). In total 342 patients were randomly assigned to treatment with anakinra and standard care (n=112) or only standard care (n=230), and simultaneously randomly assigned to IL-6 blockade (n=227; 114 for tocilizumab and n=113 for siltuximab) or no IL-6 blockade (n=115). Therefore, 44 patients received only anakinra, 32 patients received anakinra + tocilizumab, 36 patients received anakinra + siltuximab, 81 patients received only tocilizumab, 75 patients received only siltuximab, and 74 patients received only standard care. Most patients received hydroxychloroquine (i.e., 42% assigned before August 2020) or dexamethasone (i.e., 84% assigned from August 2020; steroids were used in 60% of the total patientpopulation) as standard care. The median (IQR) age was 65 (54-73) years in the intervention group (i.e., IL-6 blockade group), compared with 64 (55-72) in the control group (i.e., no IL-6 blockade group). In the intervention group 175/227 (77.1%) were males, compared with 90/115 (78.3%) in the control group. The length of the follow-up was 28 days. The following relevant outcome measures were included; duration of hospitalization, time to clinical improvement, respiratory support. The primary outcome was time to clinical improvement. This resulted in a median (range) of 11 days (10-16) in the intervention group, compared with 12 days (11-16) in the control group, HR: 1.00 (95%CI 0.78 to 1.29). There are some concerns regarding risk of bias as the study had an open label design.
Gordon (2021) (REMAP-CAP trial) describes the results of, an international, multifactorial, adaptive platform trial, comparing tocilizumab (n=353) or sarilumab (n=48) in combination with standard of care to standard of care alone (n=402, control group). However, this article is a preliminary report. For this literature summary, the results of sarilumab intervention group and the control group were used. The study included critically ill patients with COVID-19 admitted to ICU and receiving respiratory or cardiovascular organ support. Patients required high-flow nasal cannula, non-invasive ventilation or invasive mechanical ventilation. In total 865 patients were included. The mean (SD) age was 61.4 (12.7) years, and 629/865 (72.7%) of them were males. Patients in the intervention group received sarilumab 400mg (n=48) or tocilizumab (n=353), both in addition to standard care (i.e., no protocol, depending on site, glucocorticoids were used in 93% of the patients). Patients were mostly treated with COVID-19 Immunoglobulin, COVID-19 antiviral, therapeutic anticoagulation, and corticoids at baseline. The control group (n=402) received only standard care. The length of the follow-up was 90 days. The following relevant outcome measures were included; mortality (i.e., in hospital death), duration of hospitalization, need for respiratory support, any serious adverse events. The primary outcome was the number of respiratory and cardiovascular organ support–free days up to day 21. This resulted in a median (IQR) of 11 (0 to 16) days/ 10 (-1 to 16) days in the intervention groups (i.e., sarilumab/tocilizumab), compared with 0 (-1 to 15) days in the control group. There are some concerns regarding risk of bias as the current study had an open-label design.
LEVILIMAB
Lomakin (2021) (CORONA) described a phase III, double-blind, placebo-controlled, randomized trial, which was conducted at 12 hospitals in the Russian Federation. Lomakin (2021) evaluated the efficacy and safety of IL-6 inhibitor levilimab in combination with standard care, compared with only standard care in patients with severe COVID-19. Standard care was in accordance with the National clinical guidelines of the Ministry of Health of the Russian Federation, which included symptomatic treatment, antiviral agents, anticoagulants, supportive care, etc. Hydroxychloroquine, antithrombotic agents, macrolides and lincosamides were mostly used as concomitant therapy. In total 206 patients met the in- and exclusion criteria and were randomized. The mean (SD) age was 59 (13) years in the intervention group (n=103), compared with 58 (11) years in the control group (n=103). In the intervention group 58/103 (56.3%) were males, compared with 51/103 (49.5%) in the control group. The length of the follow-up was 60 days. The following relevant outcome measures were included; mortality, duration of hospitalization, safety. The primary outcome was the proportion of patients with sustained clinical improvement on day 14. This was achieved in 65/103 (63.1%) patients in the intervention group, compared with 44/103 (42.7%) patient in the control group (difference: 20.4%). There are no concerns regarding risk of bias.
Note: The initial primary endpoint was the overall mortality, but the observed mortality rate in the study population was significantly lower than the assumed value. Thus, the study had not enough power to detect the difference between the groups using overall mortality. Therefore, the primary endpoint changed.
SARILUMAB
Lescure (2021) (Sarilumab COVID-19 Global Study Group) described a phase 3, double-blind placebo-controlled, multicenter randomized controlled trial. Lescure (2021) evaluated the clinical efficacy and safety of sarilumab (200 mg or 400mg) in addition to standard of care versus standard of care alone in patients admitted to the hospital with COVID-19 with indication of oxygen or ventilation support. In total 416 patients were included. The median (range) age was 59 (50-68) years, and 216/416 (51.9%) of them were males. Intervention group I (n=159) received sarilumab 200mg, and intervention group II (n=173) received sarilumab 400mg. The control group (n=84) received placebo. All patients received stand care, which included corticosteroids at randomisation in 20%. The length of the follow-up was 60 days. The following relevant outcome measures were included; mortality (i.e., patients alive at day 29), duration of hospitalization (i.e., number of days of hospitalization among patients alive at day 60), time to clinical improvement, need for respiratory support. In the current summary of literature intervention group II (i.e., sarilumab 400mg) was labelled as ‘intervention group, and intervention group I (i.e., sarilumab 200mg) was labelled as ‘intervention group 200mg’. The primary endpoint was time to clinical improvement of two or more points (i.e., estimated based on Kaplan-Meier). This resulted in a median (IQR) 10 (9 to 12) days in intervention group I, 10 (9 to 13) days in intervention group II, and 12 (9 to 15) days in the control group. The HR were 1.03 (95%CI 0.75 to 1.40), and 1.14 (95%CI 0.84 to 1.54), respectively for I-I vs. placebo and II-I vs. placebo.
Mariette (2021) (CORIMUNO-19-SARI-1 Collaborative Group) reported resulted of a multicentre (n=6, France), open-label, randomised controlled phase 2/3 trial, nested within the CORIMUNO-19 cohort. Mariette (2021) assessed the ability of sarilumab to improve the outcome of patients hospitalised with COVID-19 pneumonia. Eligible patients (n=148) were randomized to treatment with sarilumab and standard care (i.e., antibiotic agents, antiviral agents, corticosteroids (15% and 25% of the patients in the intervention and the control group, respectively, were treated with corticosteroids), vasopressor support, anticoagulants at the discretion of the clinicians) or only standard care. Of them, 68 were randomized to the intervention group, and 80 to the control group. In the intervention group, 35/68 (51.4%) received a second injection of sarilumab. Four patients in the control group withdrew consent, and therefore 76 patients were included in the analyses. The median (IQR) age in the intervention group was 62 (53-71) years, compared with 63 (26-72) in the control group. In the intervention group 49/68 (72.1%) were males, compared with 59/76 (77.6%) in the control group. The length of the follow-up was 90 days, and no patients were lost to follow-up. The following relevant outcome measures were included; mortality at day 28 and day 90, duration of hospitalisation, respiratory support, safety. The study reported 2 primary outcomes; proportion of patients who had died or needed non-invasive or mechanical ventilation by day 4 (i.e., 18/68 (26%) patients in the intervention group vs. 20/76 (26%) controls), and survival with no need for mechanical or non-invasive ventilation at day 14 (i.e., 25/68 (37%) patients in the intervention group vs. 26/76 (34%) controls). Importantly the current study was stopped early as no (positive) clinical effect was shown at the interim analysis.
Merchante (2021) (SARICOR) reported resulted of a multicentre (n=10, Spain), open-label, randomised controlled phase 2 trial. Merchante (2021) investigate the efficacy and safety of early treatment with sarilumab in patients hospitalised with COVID-19 pneumonia. Eligible patients (n=115) were randomized to treatment with sarilumab (200mg or 400mg) and standard care (i.e., according to local practice; dexamethasone was used in 90% of the included patients) or only standard care. Of them, 37 were randomized to intervention group I (i.e., sarilumab 200mg), 39 to intervention group II (i.e., 400mg sarilumab), and 39 to the control group. One patient in intervention group I and one patient in intervention group II withdrew consent. The median (IQR) age was 59 (51-70) years, and 78/115 (67.8%) of the patients were males. The length of the follow-up was 28 days, and no patients were lost to follow-up. The following relevant outcome measures were included; mortality at day 28, respiratory support, duration of hospitalisation, need for ICU admission, and time to clinical improvement. The primary outcome was defined as the development of ARDS requiring HFNO, NIMV or IMV during the first 28 days after randomization. This was observed in 10/37 (27.0%) patients in intervention group I, 10/39 (25.6%) patients in intervention group II, and 5/39 (12.8%) in the control group. The HR were 0.87 (95%CI 0.37 to 2.06), and 0.41 (95%CI 0.14 to 1.18), respectively for I-I vs. placebo and II-I vs. placebo. There are some concerns regarding risk of bias as the current study had an open-label design.
Sancho-Lopéz (2021) (SARTRE) reported results of an open-label randomized controlled trial performed in 8 tertiary hospitals in Spain. Sancho-Lopéz (2021) evaluated the clinical efficacy of sarilumab in addition to standard of care versus standard of care alone in patients admitted to the hospital with COVID-19 requiring supplemental oxygen by mask or nasal prongs. Glucocorticoids were given to all patients at a 1 mg/kg/day of methylprednisolone for at least 3 days. In total 201 patients were included. Of them 99 were randomized to the intervention group (i.e., addition of sarilumab (IV) at a single dose), and 102 to the control group. The mean (SD) age in the intervention group was 60 (11.5) years, and 60 (11.8) years in the control group. In the intervention group 71/99 (71.7%) were males, compared to 70/102 (68.6%) in the control group. The length of the follow-up was 28 days. The following relevant outcome measures were included; mortality at day 28, duration of hospitalization (i.e., time to hospital discharge), need for respiratory support, any (serious) adverse events. The primary outcome was the proportion of patients progressing to severe respiratory failure. This was observed in 16/99 (16.2%) patients in the intervention group, compared with 16/102 (15.7%) patient in the control group. This resulted in a RR of 1.03 (95%CI 0.48 to 2.20). There are some concerns regarding risk of bias as the current study had an open-label design.
TOCILIZUMAB
Hermine (2020) (CORIMUNO-19-TOCI-1 trial) compared outcomes of tocilizumab in combination with usual care (n=64) with best supportive care (n=67) in a cohort-embedded multicenter open-label Bayesian RCT in nine university hospitals in France. Hospitalized patients were included with moderate or severe COVID-19 pneumonia and oxygen requirement but not admitted to the intensive care unit. Patients did not require high-flow oxygen by nasal cannula, non-invasive ventilation, or mechanical ventilation (World Health Organization clinical progression scale [WHO-CPS] score of 5). Patients in the tocilizumab group received intravenously administration of tocilizumab of 8 mg/kg on day 1. Administration of an additional fixed doze of tocilizumab, 400 mg IV, on day 3 was recommended if oxygen requirement was not decreased by more than 50%, but decision was left to the treating physician. Usual care (antibiotic agents, antiviral agents, corticosteroids (16% and 18% of the patients in the intervention and the control group, respectively, were treated with corticosteroids prior to randomisation, 30% and 55% respectively after randomisation.), vasopressor support, anticoagulants) was provided at the discretion of the clinicians. The length of follow-up was 28 days. Primary outcomes were scores higher than 5 on the World
Health Organization 10-point Clinical Progression Scale (WHO-CPS) on day 4 and survival
without need of ventilation (including non-invasive ventilation) at day 14. In the tocilizumab group, 12 patients had a WHO-CPS score greater than 5 at day 4 comparted to 19 in the control group (median posterior absolute risk difference [ARD] −9.0%; 90% credible
interval [CrI], −21.0 to 3.1). At day 14, 12% (95%CI −28% to 4%) fewer patients needed non-invasive ventilation (NIV) or mechanical ventilation (MV) or died in the
tocilizumab group than in the control group (24% versus 36%, median posterior HR 0.58; 90% CrI, 0.33 to 1.00).
Horby (2021) (RECOVERY trial) describes the results of the RECOVERY trial, a large open-label trial investigating the effect of multiple drug treatment. The trial was conducted in 131 National Health Service Hospitals in the UK. At first randomization, patients were randomized to usual care or usual care plus either lopinavir–ritonavir, dexamethasone, hydroxychloroquine or azithromycin. For the current study, patients were randomized at a second randomization to receive either tocilizumab and usual care (n=2022) or usual care alone (n=2094). Patients were hospitalized patients with clinical evidence of progressive COVID-19, indicated by an oxygen saturation < 92% on room air or receiving oxygen therapy, and CRP ≥75 mg/L. Of all patients at baseline, 45% received no supplemental oxygen or low-flow oxygen, 41% received high-flow nasal oxygen, continuous positive airway pressure, or other non-invasive ventilation and 14% received invasive mechanical ventilation or ECMO.
Patients in the tocilizumab group received a single intravenous infusion over 60 min. The dose was established by bodyweight and a second dose could be given after 12 to 24 hours if the patient’s condition had not improved. Usual care was provided as the standard of care in the participating hospital. At least 82% of the patients received dexamethasone as part of the usual care. The length of follow-up was 28 days. The primary outcome was 28-day mortality. Overall, 621 (31%) of the 2022 patients allocated tocilizumab and 729 (35%) of the 2094 patients allocated to usual care died within 28 days (RR 0.85; 95% CI 0.76 to 0.94).
Rosas (2021a) (COVACTA trial) conducted a phase 3, international, randomized, double-blind, placebo-controlled trial to assess the efficacy and safety of tocilizumab (n=294) versus placebo (n=144) in hospitalized adult patients with severe COVID-19. Eligible patients were randomly assigned to receive a single intravenous infusion of tocilizumab (at a dose of 8 mg/kg, with a maximum dose of 800 mg) or placebo plus standard care. If clinical signs or symptoms did not improve or worsened, a second infusion of tocilizumab or placebo could be administered 8 to 24 hours after the first dose. Standard care according to local practice (antiviral treatment, low-dose glucocorticoids (used at baseline in 19% of the intervention group and 29% of the placebo group; used during the study in 34% and 52% respectively), convalescent plasma, and supportive care) was provided. However, concomitant treatment with another investigational agent (except antiviral drugs) or any immunomodulatory agent was prohibited. The primary efficacy outcome was clinical status at day 28. Key secondary efficacy outcomes were clinical status at day 14 on the ordinal scale, mortality at day 28, number of ventilator-free days by day 28, the time to improvement from baseline by at least two categories on the ordinal scale, and the time to hospital discharge or readiness for discharge. The median value for clinical status on the ordinal scale at day 28 was 1.0 (95% CI 1.0 to 1.0) in the tocilizumab group and 2.0 (non-ICU hospitalization without supplemental oxygen) (95% CI 1.0 to 4.0) in the
placebo group (between-group difference, −1.0; 95% CI −2.5 to 0).
Rosas (2021b) (REMDACTA trial) conducted a randomized, double-blind, placebo-controlled, multicenter, phase 3 trial, evaluated the efficacy and safety of tocilizumab plus remdesivir versus placebo plus remdesivir in patients aged 12 years and older hospitalized with severe COVID-19 pneumonia. Eligible patients were randomly assigned to receive treatment with tocilizumab plus remdesivir (n=430) or placebo plus remdesivir (n=210). Remdesivir was administered intravenously, followed by a single intravenous dose of tocilizumab 8 mg/kg (maximum, 800 mg) or placebo on day 1. Patients with sustained fever or clinically significant worsening of signs and symptoms of COVID-19 (e.g., increased supplemental oxygen requirement) could receive a second infusion of blinded tocilizumab or placebo within 8 to 24 h of the first infusion. Before randomization, about 19% of the patients in both groups received remdesivir. Corticosteroids were part of the standard of care in most patients (83% of the intervention group and 86% of the placebo group used corticosteroids). At baseline, all patients required >6 L/min supplemental oxygen. Patients were monitored through day 60, and the primary end point (time to hospital discharge or “ready for discharge”) was assessed at day 28. Secondary outcomes include time from randomization to mechanical ventilation or death, clinical status on the ordinal scale at day 14, and time to death. Median time from randomization to hospital discharge or “ready for discharge” was 14 (95% CI 12 to 15) days with tocilizumab plus remdesivir and 14 (95% CI 11 to 16) days with placebo plus remdesivir (Cox proportional HR 0.97 (95% CI 0.78 to 1.19)].
Salama (2020) (EMPACTA trial) describes an international double-blind, placebo-controlled RCT, comparing the effect of tocilizumab (n = 249) with placebo (n = 128) in hospitalized patients with mild to severe COVID-19. Eligible patients were randomly assigned to receive one or two doses of intravenous tocilizumab (8 mg per kilogram of body weight, to a maximum of 800 mg per dose) or placebo plus standard care. Standard of care was provided according to local practice, which could include antiviral treatment, the limited use of systemic glucocorticoids (recommended dose, ≤1 mg per kilogram of body weight of
methylprednisolone or equivalent), and supportive care. In total, 80% of the intervention group and 88% of the control group used corticosteroids in the 7 days before or during the trial (duration not specified). Patients that required mechanical ventilated were not eligible for study participation. At baseline, the majority of the patients received supplemental oxygen (64.2%), some received NIV or high-flow oxygen (26.5%) and a minority did not receive any supplemental oxygen (9.3%). The length of follow-up was 60 days. The primary outcome was mechanical ventilation or death by day 28. The cumulative percentage of patients who had received mechanical ventilation or who had died by day 28 was 12.0% (95% CI 8.5 to 16.9) in the tocilizumab group and 19.3% (95% CI, 13.3 to 27.4) in the placebo group (HR 0.56; 95% CI 0.33 to 0.97).
Salvarani (2020) (RCT-TCZ-COVID-19 trial) conducted a prospective, open-label RCT to evaluate the effect of early tocilizumab administration (n=60) versus standard therapy (n=66) in preventing clinical worsening in patients hospitalized with COVID-19 pneumonia in 24 hospitals in Italy. Eligible patents were randomly assigned to receive intravenous tocilizumab (within 8 hours from randomization (8mg/kg up to a maximum of 800mg, followed by a second dose after 12 hours) or standard of care. Patients in the control arm received supportive care following the protocols of each clinical center. All drugs were
allowed but IL-1 blockers, Jak inhibitors, and tumor necrosis factor inhibitors. Steroids were allowed if already taken before hospitalization (<10% of the patients). Patients at enrolment were allowed to receive oxygen therapy with Venturi mask or high-flow nasal cannula with recorded and present Fio2, but not invasive or non-invasive mechanical ventilation and were not admitted to the intensive care unit. However, patients that were admitted to the ICU were not eligible for study participation. In case of occurrence of documented clinical worsening, patients randomized in both arms could receive any therapy, including steroids (<10% of the patients), and, for patients randomized in the control arm, tocilizumab. The length of follow-up was 14 days for the primary end point (clinical worsening) and 30 days for other outcomes. The primary composite outcome was defined as entry into the intensive care unit with invasive mechanical ventilation, death from all causes, or clinical aggravation documented by the finding of a PaO2/FIO2 ratio less than 150mmHg, whichever came first. In total, 17 of 60 patients (28.3%) in the tocilizumab group and 17 of 63 (27.0%) in the control group showed
clinical worsening within 14 days since randomization (RR 1.05; 95%CI, 0.59 to 1.86).
Two patients in the tocilizumab group and 1 in the control group died before 30 days from
randomization, and 6 and 5 patients were intubated in the 2 groups, respectively. The trial
was prematurely interrupted after an interim analysis for futility.
Soin (2021) (COVINTOC trial) conducted a phase 3, open-label, multicenter, randomized controlled trial to assess the efficacy and safety of tocilizumab plus standard care (n=90) versus standard care alone (n=90) in hospitalized patients with moderate to severe disease. Eligible patients from 12 public and private hospitals across India were randomly assigned to receive a single intravenous tocilizumab infusion at 6 mg/kg up to a maximum dose of 480 mg or standard care. For patients in the intervention group, an additional dose of 6 mg/kg (max 480 mg/kg) could be administered if clinical symptoms worsened or did not show improvement within 12 h to 7 days after administration of the first dose. Standard care was provided according to the protocols at the individual study sites. Standard care included corticosteroids in 91% of the patients. At baseline, 81/91 (89.0%) in the intervention group and 80/88 (90.9%) in the control group received supplemental oxygen and 5/91 (5.5%) in the intervention group and 5/88 (5.6%) in the control group received mechanical ventilation. Supplemental oxygen was recommended to treat hypoxia, and high-flow nasal cannula, non-invasive ventilation, and mechanical ventilation could be considered if hypoxia and respiratory distress progressed. The length of follow-up was 30 days. The primary endpoint was progression of COVID-19 (from moderate to severe or from severe to death) up to day 14. Progression of COVID-19 occurred in eight (8.8%) of 91 patients in the tocilizumab group and 11 (12.5%) of 88 in control group (difference –3.71; 95% CI –18.23 to 11.19).
Stone (2020) (BACC Bay Tocilizumab Trial) performed a randomized, double-blind, placebo-controlled trial to assess the efficacy of tocilizumab plus standard care (n=161) versus placebo plus standard care (n=81) in hospitalized patients with confirmed SARS-CoV-2 infection hyperinflammatory states, and at least two of the following signs: fever (body temperature >38°C), pulmonary infiltrates, or the need for supplemental oxygen in order to maintain an oxygen saturation greater than 92%. Eligible patients from seven Boston hospitals were randomly assigned to receive standard care plus a single dose of either tocilizumab (8 mg per kilogram of body weight administered intravenously, not to exceed 800 mg) or placebo. Antiviral therapy, hydroxychloroquine and steroids were permitted as concomitant therapy. Standard care included corticosteroids in 11% of the intervention group and 6% of the placebo group. Data from patients who were event-free at the end of follow-up were censored at 28 days (for intubation (or death, for patients who died before intubation) and clinical worsening) or at 29 days (for discontinuation of supplemental oxygen among patients who had been receiving it at baseline). Data from patients who could not be reached for 28-day follow-up were censored at hospital discharge. The primary outcome was intubation or death. The HR for intubation or death in the tocilizumab group as compared with the control group was 0.83 (95% CI 0.38 to 1.81).
Veiga (2021) (COVID-19 Coalition Brazil VI (TOCIBRAS)) conducted a multicentre, randomised, open label, parallel group, superiority trial to assess the efficacy of tocilizumab plus standard (n=65) versus standard care alone (n=64) in hospitalized patients with severe or critical coronavirus disease and were receiving supplemental oxygen to maintain oxygen saturation greater than 93% or had been receiving mechanical ventilation for less than 24 hours before analysis. Eligible patients from nine hospitals across Brazil were randomly assigned to receive a single intravenous dose of tocilizumab (8 mg/kg, with a maximum of 800 mg) or standard care alone. The concomitant use of hydroxychloroquine, azithromycin, corticosteroids (69% of the intervention and 73% of the control group used corticosteroids during the study), and antibiotics was allowed according to standard care per local institutional guidelines for patients with covid-19. Remdesivir was not available in Brazil. At baseline, more patients in the tocilizumab group than control group were using supplementary oxygen at enrolment (60% vs 44%), whereas use of non-invasive ventilation or high flow oxygen through a nasal cannula was higher in the control group than in the tocilizumab group (23% vs 41%). The follow-up was 15 days for the primary outcome and 29 days for secondary outcomes. The primary outcome was clinical status at 15 days. This was analyzed as a composite of death or mechanical ventilation because the assumption of odds proportionality was not met. In total, 18 of 65 (28%) patients in the tocilizumab group and 13 of 64 (20%) in the control group were receiving mechanical ventilation or died at day 15 (OR 1.54; 95% CI 0.66 to 3.66). Death at 15 control group (OR 6.42; 95% CI 1.59 to 43.2). The data monitoring committee recommended stopping the trial early, after 129 patients had been enrolled, because of an increased number of deaths at 15 days in the tocilizumab group.
Wang (2021) conducted a randomized, controlled, open-label, multicenter trial to assess efficacy and safety of tocilizumab in addition to standard care (n=34) versus standard care alone (n=31) in hospitalized patients with moderate or severe COVID-19. Eligible patients were randomly assigned to receive tocilizumab (400 mg, diluted in 100 mL of 0.9% saline, and administered intravenously for more than 1 h. A second dose was given if a patient remained febrile for 24 h after the first dose) or standard care alone. Standard care was given in accordance with the “Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia (5th or updated version)” and included corticosteroids in 11%. One patient in the control group that worsened on day 3 after randomization was crossed over to the tocilizumab group. The follow-up was 14 days. The primary end point was the cure rate of the enrolled patients. The cure rate in the tocilizumab group (94.1%) was higher than in the control group (87.1%) (rate difference 95% CI 7.2% to 21.2%).
Table 1. Overview of RCTs comparing IL6-inhibitors with standard care in hospitalized COVID-19 patients.
|
First author, year |
Disease severity, based on need for respiratory support* |
Sample size |
Intervention |
|
Tocilizumab |
|||
|
Declercq (2021) |
Mixed: mild, moderate and severe disease
|
I: N=227 (n=114 for tocilizumab and n=113 for siltuximab) C: N=115 Total: N=342 |
First patients were randomly assigned to receive an IL-1 blockade or not. Thereafter they were randomized to receive an IL-6 blockade or not.
-for the second randomization, a subgroup received siltuximab (11 mg/kg i.v. (single injection)) OR tocilizumab (8 mg/kg i.v. (not exceeding 800 mg; single injection)) |
|
Gordon (2021) |
Severe disease |
I: 353 C: 402** Total N=755 |
One dose of 8 mg tocilizumab per kilogram of actual body weight (up to a maximum of 800 mg) administered IV over period of 1 hour. Dose could be repeated 12 to 24 hours later at the discretion of the treating clinician. |
|
Hermine (2020) |
Moderate disease |
I: 63 C: 67 Total N=130 |
Intravenously administration of tocilizumab of 8 mg/kg on day 1. Administration of an additional fixed doze of tocilizumab, 400 mg IV, on day 3 was recommended if oxygen requirement was not decreased by more than 50%, but decision was left to the treating physician. |
|
Horby (2021) |
Mixed: mild, moderate and severe disease
Mortality stratified for respiratory support at baseline |
I: 2022 C: 2094 Total N=4116 |
A single intravenous infusion of tocilizumab over 60 min was provided. The dose was established by bodyweight (800 mg if weight >90 kg; 600 mg if weight >65 and ≤90 kg; 400 mg if weight >40 and ≤65 kg; and 8 mg/kg if weight ≤40 kg). A second dose could be given 12–24 h later if the patient’s condition had not improved. |
|
Rosas (2021a) |
Mixed: mild, moderate and severe disease
|
I: 294 C: 144 Total N=438 |
A single intravenous infusion of tocilizumab (at a dose of 8 mg per kilogram of body weight, with a maximum dose of 800 mg) |
|
Rosas (2021b) |
Mixed: moderate and severe disease |
I: 430 C: 210 Total N=640 |
A single intravenous dose of tocilizumab 8 mg/kg (maximum, 800 mg). Patients with sustained fever or clinically significant worsening of signs and symptoms of COVID-19 (e.g., increased supplemental oxygen requirement) could receive a second infusion tocilizumab within 8 to 24 h of the first infusion. |
|
Salama (2021) |
Mixed: mild, moderate and severe disease; not mechanically ventilated |
I: 249 C: 128 Total N=377 |
One or two doses of intravenous tocilizumab (8 mg per kilogram of body weight, to a maximum of 800 mg per dose) |
|
Salvarani (2020)
|
Mixed: mild, moderate and severe disease
|
I: 60 C: 66 Total N=126 |
Tocilizumab intravenously within 8 hours from randomization at a dose of 8 mg/kg up to a maximum of 800 mg, followed by a second dose after 12 hours. |
|
Soin (2021)
|
Mixed: mild, moderate and severe disease
|
I: 90 C: 90 Total N=180 |
Tocilizumab was administered as a single intravenous infusion at 6 mg/kg up to a maximum dose of 480 mg. An additional dose of 6 mg/kg (max 480 mg/kg) could be administered if clinical symptoms worsened or did not show improvement within 12 h to 7 days after administration of the first dose. |
|
Stone (2020)
|
Mixed: mild, moderate and severe disease
|
I: 161 C: 81 Total N=242 |
Tocilizumab (8 mg per kilogram of body weight administered intravenously, not to exceed 800 mg) |
|
Veiga (2021)
|
Mixed: moderate and severe |
I: 65 C: 64 Total N=129 |
A single intravenous dose of tocilizumab (8 mg/kg, with a maximum of 800 mg) |
|
Wang (2021)
|
Mixed: moderate and severe |
I: 34 C: 31 Total N=65 |
The first dose of tocilizumab was 400 mg, diluted in 100 mL of 0.9% saline, and administered intravenously for more than 1 h. A second dose was given if a patient remained febrile for 24 h after the first dose. |
|
Sarilumab |
|||
|
Gordon (2021) |
Severe disease** |
I: 48 C: 402** Total: 450 |
Sarilumab, at a dose of 400 mg, was administered as an intravenous infusion once only. |
|
Lescure (2021) |
Mixed: the majority of the patients had a moderate disease severity (60%), 40% had severe disease severity.
|
I-I: N=159 I-II: N=173 C: N=84 Total: N=416 |
The hospital pharmacist added the contents of prefilled syringes of sarilumab 200 mg solution for subcutaneous injection supplied by the sponsor into a specified volume of locally sourced 0·9% sodium chloride solution for IV infusion (two syringes for the 400 mg dose, one syringe for the 200mg dose). |
|
Mariette (2021) |
Moderate: receiving supplemental oxygen without ventilation assistance. |
I: 68 C: 76 Total: 144 |
Sarilumab (IV) at a single dose of 400mg. If an additional dose of 400mg was recommended if oxygen requirement had not decreased by more than 50%. |
|
Merchante (2021) |
Mixed: The majority of the patients needed administration of oxygen at baseline. - no oxygen; n=11 - non-rebreather face mask; n=7 - rebreather face mask; n=18 |
I-I: 37 I-II: 39 C: 39 Total: 115 |
I-I: Sarilumab (IV) at a single dose of 200mg.
I-II: Sarilumab (IV) at a single dose of 400mg. |
|
Sancho-Lopéz (2021) |
Moderate: requiring supplemental oxygen by mask or nasal prongs |
I: 99 C:102 Total: 201 |
Sarilumab (IV) at a single dose of 200 mg for patients <75 kg body weight, or 400 mg for patients weighing ≥75 kg. |
|
Levilimab |
|||
|
Lomakin (2021) |
Mixed: 39% did not require oxygen therapy, 60% require oxygen therapy, 1% require high-flow oxygen therapy or non-invasive ventilation |
I: 103 C: 103 Total: 206 |
Patients allocated to the active drug received a single dose of levilimab at day 1. Levilimab 324 mg administration was performed as two SC injections of 162 mg. |
|
Siltuximab |
|||
|
Declercq (2021) |
Mixed: The majority of the patients needed administration of oxygen at baseline. Intervention: oxygen; N=50 - on non-invasive ventilation or high flow oxygen devices; N=44 - on invasive mechanical ventilation; N=17
Control: oxygen; N=119 - on non-invasive ventilation or high flow oxygen devices; N=84 - on invasive mechanical ventilation; N=22 |
I: N=227 C: N=115 Total: N=342 |
First patients were randomly assigned to receive an IL-1 blockade or not. Thereafter they were randomized to receive an IL-6 blockade or not.
- for the second randomization, a subgroup received siltuximab (11 mg/kg i.v. (single injection)) OR tocilizumab (8 mg/kg i.v. (not exceeding 800 mg; single injection)) |
**2/402 patients in the control group had none or supplementary oxygen only.
*Disease severity categories:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen [high flow nasal cannula (HFNC)/Optiflow], continuous positive airway pressure [CPAP], non-invasive ventilation [NIV], mechanical ventilation, extracorporeal membrane oxygenation [ECMO or ECLS]).
N: Total sample size; I: Intervention; C: Control
Results – IL-6 inhibitors
Since the number of studies that was available and the number of patients that were included, was relatively large , we report all outcomes stratified for the different IL-6 inhibitors. Also conclusions were formulated separately for the different IL-6 inhibitors: i.e. conclusions were reported for tocilizumab and sarilumab, instead of all IL-6 inhibitors combined.
Since the quality of the studies was variable and the use of corticosteroids as part of the standard of care varied from <10% to >90%, the committee decided to conduct and report a sensitivity analysis for the crucial outcomes (‘mortality’ and ‘extensive respiratory support’).
Mortality (crucial)
Results - Tocilizumab
mortality at day 21 – 30
Ten studies reported on the outcome measure mortality at day 21 – 30. The pooled incidence was 927/3785 (24.5%) in the tocilizumab group compared to 987/3336 (29.6%) in the control group. The pooled RR from these ten studies was 0.89 (95% CI 0.82 to 0.96; Figure 1), in favour of the tocilizumab group. The pooled RD was -0.01 (95%CI -0.04 to 0.02). This, including patients with different disease severity states, is not considered clinically relevant. Regarding the sensitivity analysis (Figure 1), the pooled RR in the high quality studies AND studies with more than 80% use of corticosteroids in the standard of care, was 0.87 (95%CI 0.80 to 0.94), and the RD was -0.04 (95%CI -0.07 to -0.02). This is considered clinically relevant. The pooled RR in studies with lower quality and/or initiating less than 80% corticosteroids was 1.09 (95%CI 0.83 to 1.43), and the RD was 0.02 (95%CI -0.01 to 0.04). This is not considered clinically relevant.
Figure 1. Mortality 21-30 days in hospitalized patients treated with tocilizumab
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
mortality at day 14 – 15
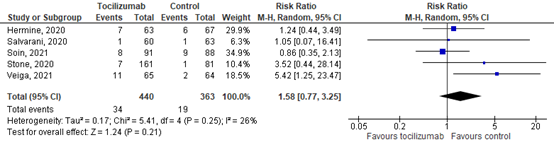
Four studies (Hermine, 2020; Salvarani, 2020; Soin, 2021; Stone, 2020)) reported on mortality at day 14 and one study (Veiga, 2021) reported on mortality at day 15. The pooled incidence of these five studies was 23/375 (6.1%) in the intervention group compared to 17/299 (5.7%) in the control group. The pooled RR from these studies was 1.58 (95% CI 0.77 to 3.25; Figure 2), in favour of the control group. The pooled RD was 0.027 (95% CI -0.013 to 0.067). This is not considered clinically relevant. Patients included in this analysis had a mixed disease state (i.e., mild, moderate or severe). Regarding the sensitivity analysis, all included studies in this analysis were of low quality and/or used <80% corticosteroids.
Figure 2. Mortality 14-15 days in hospitalized patients treated with tocilizumab
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
mortality at day 60
Salama (2021) and Rosas (2021b) reported on mortality at day 60. The pooled mortality rate was 126 out of 680 (18.5%) in the intervention group compared to 69 out of 337 (20.5%) in the control group. The RR is 0.90 (95% CI 0.69 to 1.16). The RD was -0.02 (95% CI -0.07 to 0.03). This is not considered clinically relevant. Patients included in this analysis had a mixed disease state (i.e., mild, moderate or severe).
Declercq (2021) reported the number of deaths in the subgroups at follow-up (i.e., 10-20 weeks). The incidence of mortality was 10/81 (12.3%) in patients treated with only additional tocilizumab, compared with 9/74 (12.2%) in the control group (i.e., receiving only usual care according to the randomizations). This resulted in a RR of 1.02 (95%CI 0.44 to 2.36) and a RD of 0.00 (95%CI -0.10 to 0.11). This is not considered clinically relevant. Patients included in this analysis had a mixed disease state (i.e., mild, moderate or severe). Regarding the sensitivity analysis, all included studies in this analysis were of low quality and/or used <80% corticosteroids.
If possible stratified analyses were performed per disease severity state, see Figure 3.
Moderate disease
Hermine (2020) reported mortality at day 28 – 30 specifically for patients with a moderate disease state. The incidence of mortality was 7/63 (11.1%) in the intervention group, compared with 11/67 (16.4%) in the control group. The RR was 0.68 (95%CI 0.28 to 1.64). The RD was -0.05 (95%CI -0.17 to 0.06). This is considered clinically relevant.
Horby (2021) reported mortality at 28 days specifically for patients with a moderate disease state (e.g., nine patients not receiving any oxygen and 1859 patients receiving simple oxygen only). The incidence of mortality was 180/935 (19.3%) in the intervention group, compared with 214/933 (22.9%) in the control group. The RR was 0.84 (95%CI 0.70 to 1.00). The RD was -0.04 (95CI -0.07 to 0.00). This is considered clinically relevant.
Rosas (2021a) reported mortality at 28 days specifically for patients with a moderate disease state (i.e., patients who received oxygen at baseline but were not mechanically ventilated). The incidence of mortality was 21/183 (14.8%) in the intervention group, compared with 15/90 (16.7%) in the control group. The RR was 0.69 (95%CI 0.37 to 1.27). The RD was -0.05 (95%CI -0.14 to 0.04). This is considered clinically relevant.
The pooled RR in patients with a moderate disease state was 0.82 (95%CI 0.70 to 0.97), and the RD was -0.04 (95%CI -0.07 to -0.01) for mortality at day 28. This is considered clinically relevant.
Severe disease
Horby (2021) reported mortality at 28 days specifically for patients with a severe disease state (e.g., non-invasive ventilation (including high-flow nasal oxygen, continuous positive airway pressure ventilation, and other non-invasive ventilation); and invasive mechanical ventilation (including invasive mechanical ventilation and extracorporeal membranous oxygenation)). The incidence of mortality was 441/1087 (40.6%) in the intervention group, compared with 515/1161 (44.4%) in the control group. The RR was 0.91 (95%CI 0.83 to 1.01). The RD was -0.04 (95%CI -0.08 to 0.00). This is considered clinically relevant.
Rosas (2021a) reported mortality at 28 days specifically for patients with a severe disease state (e.g., patients who were mechanically ventilated at randomization). The incidence of mortality was 31/111 (27.9%) in the intervention group, compared with 19/54 (35.2%) in the control group. The RR was 0.79 (95%CI 0.50 to 1.27). The RD was -0.07 (95%CI -0.22 to 0.08). This is considered clinically relevant.
In addition, Gordon (2021) reported mortality at day 21 in patients with severe disease state. The mortality rate was 98 out of 350 (28.0%) in the intervention group compared to 142 out of 397 (35.8%) in the control group. The RR is 0.78 (95% CI 0.63 to 0.97). The RD was -0.08 (95% CI -0.14 to -0.01). This is considered clinically relevant.
The pooled RR in patients with a severe disease state was 0.89 (95%CI 0.81 to 0.97), and the RD was -0.05 (95%CI -0.08 to -0.02) for mortality at day 21-30. This is considered clinically relevant.
Mixed – moderate & severe disease
Rosas (2021b) reported mortality at 28 days specifically for patients with a moderate or severe disease state. The incidence of mortality was 78/430 (18.1%) in the intervention group, compared with 41/210 (19.5%) in the control group. The RR was 0.79 (95%CI 0.50 to 1.27). The RD was -0.01 (95%CI -0.08 to 0.12). This is not considered clinically relevant.
Veiga (2021) reported mortality at 28 days specifically for patients with a moderate or severe disease state. The incidence of mortality was 14/65 (21.5%) in the intervention group, compared with 6/64 (9.4%) in the control group. The RR was 0.79 (95%CI 0.50 to 1.27). The RD was 0.12 (95%CI -0.00 to 0.24). This is considered clinically relevant (in favour of control).
The pooled RR in patients with a moderate or severe disease state was 1.33 (95%CI 0.56 to 3.16), in favour of the control group, for mortality at day 28. The RD was 0.04 (95%CI -0.09 to 0.18). This is considered clinically relevant (in favour of control).
Mixed – mild, moderate & severe disease
Four studies reported mortality at day 28 – 30 specifically for patients with a mild, moderate or severe disease state. The pooled RR from these studies was 1.12 (95%CI 0.72 to 1.73) for mortality at day 28. The pooled RD was 0.02 (95%CI -0.02 to 0.05). This is not considered clinically relevant.
There were no studies investigating tocilizumab in patients with mild disease only, and this data could not be extracted from the studies that included a mixed population.
Figure 3. Mortality 21-30 days in hospitalized patients with tocilizumab, per disease severity category
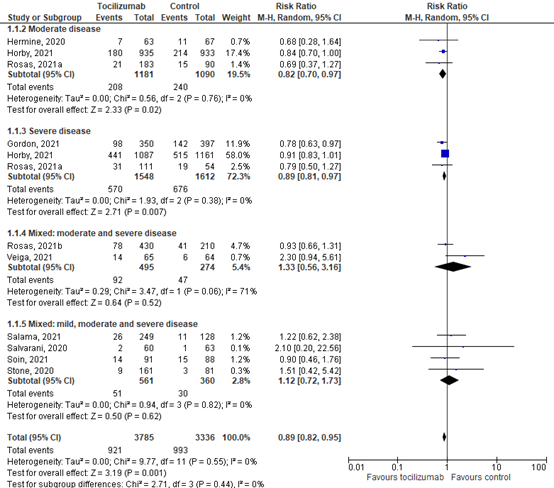
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Importantly, if sensitivity analyses (i.e., including only the high quality studies AND studies with more than 80% use of corticosteroids in the standard of care OR including studies with lower quality and/or initiating less than 80% corticosteroids) were performed for these analyses (i.e., per disease severity category), the same results were shown for (1) moderate disease (Hermine, 2020; Horby, 2021; Rosas2021a), (2) severe disease (Gordon, 2021; Horby, 2021; Rosas, 2021a), and (3) mixed: mild, moderate and severe disease (Salama, 2021; Salvarani, 2021; Soin, 2021; Stone, 2020). Although, except for the disease severity category ‘moderate and severe disease’. The RD of the sensitivity for high quality studies AND studies with more than 80% use of corticosteroids in the standard of care (Rosas, 2021b) was not considered clinically relevant, but the sensitivity analysis based on lower quality and/or initiating less than 80% corticosteroids was considered clinically relevant (Veiga, 2021).
Level of evidence of the literature
The level of evidence regarding the outcome measure mortality started as high, because the studies were RCTs. The level of evidence was downgraded by one level because of inconsistent study results (inconsistency, -1). The level of evidence for the outcome ‘mortality’ is moderate.
Severe disease
The level of evidence regarding the outcome measure mortality started as high, because the studies were RCTs. The level of evidence was downgraded by one level because of imprecision (95% CI crosses threshold of clinically relevance, -1). The level of evidence for the outcome ‘mortality’ is moderate.
Moderate disease
The level of evidence regarding the outcome measure mortality started as high, because the studies were RCTs. The level of evidence was downgraded by one level because of imprecision (95% CI crosses threshold of clinically relevance, -1). The level of evidence for the outcome ‘mortality’ is moderate.
Mortality at day 21 – 30
Gordon (2021) reported the mortality specifically for patients with a severe disease state at day 21. Merchante (2021), Sancho-Lopéz (2021) and Mariette (2021) reported mortality at day 28. Lescure (2021) reported the number of patients a live at day 29. This data was used to calculate mortality at day 29.
The pooled incidence of mortality in hospitalized patients in the intervention group was 32/424 (7.5%), compared to 162/698 (23.2%) in the control group. The pooled relative risk (RR) was 0.72 (95% CI 0.48 to 1.09; Figure 4). The pooled risk difference (RD) was -0.03 (95%CI -0.08 to 0.02), in favour of the intervention group. This is not considered clinically relevant. Regarding the sensitivity analysis (Figure 4), results were in line with the main analysis. The RR in the study with high quality AND the use of more than 80% corticosteroids as part of the standard care was 0.65 (95%CI 0.38 to 1.11), and the RD was -0.06 (95%CI -0.28 to 0.16) (Gordon, 2021; Sancho-Lopéz, 2021). This is considered clinically relevant. The pooled RR in studies with lower quality and/or less than 80% corticosteroids was 0.83 (95%CI 0.44 to 1.59), and the RD was -0.0265 (95%CI -0.0756 to 0.0226). This is not considered clinically relevant.
Figure 4. Mortality 21-30 days in hospitalized patients treated with sariliumab 400mg
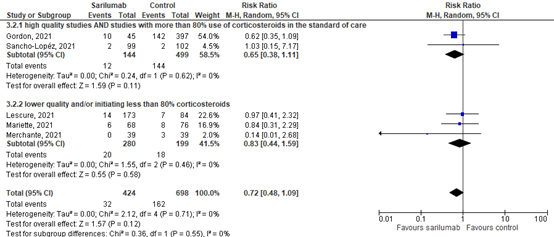
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
If possible stratified analyses were performed per disease severity state, see Figure 5.
Moderate disease
Sancho-Lopéz (2021) reported the mortality specifically for patients with a moderate disease state. The RR was 1.03 (95%CI 0.15 to 7.17) and RD was 0.00 (95%CI -0.04 to 0.04). This is not considered clinically relevant.
Mariette (2021) reported the mortality specifically for patients with a moderate disease state. The RR was 0.84 (95%CI 0.31 to 2.29) and RD was -0.02 (95%CI -0.11 to 0.08) at day 28. This is not considered clinically relevant. Mariette (2021) reported the incidence of mortality at day 90 as well. The incidence was 10/68 (15%) in the intervention group, compared with 16/76 (18%) in the control group. This resulted in a RR of 0.70 (95%CI 0.34 to 1.43) and RD of -0.06 (95%CI -0.19 to 0.06). This is considered clinically relevant.
Lescure (2021) reported the mortality specifically for patients with a moderate disease state (i.e., disease was defined by supplemental oxygen administration by nasal cannula, simple face mask, or another similar device). The incidence of mortality in hospitalized patients in the intervention group 400mg was 6/105 (5.7%), compared to 1/55 (1.8%) in the control group. The RR was 3.14 (95%CI 0.39 to 25.5), and RD was 0.04 (95%CI -0.02 to 0.10). This considered clinically relevant (in favour of the control group).
Sarilumab 200 mg:
The incidence of mortality in hospitalized patients with a moderate disease state in the intervention group 200mg was 5/92 (5.4%), compared to 1/55 (1.8%) in the control group. This resulted in a RR of 2.99 (95%CI 0.36 to 24.92) and a RD of 0.04 (95%CI -0.02 to 0.09). This considered clinically
The pooled RR in patients with a moderate disease state was 1.07 (95%CI 0.47 to 2.43), and the RD was 0.01 (95%CI -0.02 to 0.04) for mortality at day 28. This is not considered clinically relevant.
Severe disease
Lescure (2021) reported the mortality specifically for patients with a severe disease state (i.e., supplemental oxygen delivered by non-rebreather mask or high-flow nasal cannula, use of invasive or non-invasive ventilation, or treatment in an ICU). The incidence of mortality in hospitalized patients in the intervention group 400mg was 8/68 (11.8%), compared to 6/29 (20.7%) in the control group. This resulted in a RR of 0.57 (95%CI 0.22 to 1.49) and a RD of -0.09 (95%CI -0.26 to 0.08) for mortality at day 29. This is considered clinically relevant.
Sarilumab 200 mg:
The incidence of mortality in hospitalized patients with a moderate disease state in the intervention group 200mg was 10/65 (15.4%), compared to 6/29 (20.7%) in the control group. This resulted in a RR of 0.74 (95%CI 0.30 to 1.85) and a RD of -0.05 (95%CI -0.22 to 0.12). This is considered clinically relevant.
Gordon (2021) reported the mortality specifically for patients with a severe disease state at day 21. The incidence of mortality was 10/45 (22.2%) in patients treated with only additional sarilumab, compared with 142/397 (35.8%) in the control group (i.e., receiving only usual care according to the randomizations). The RR was 0.62 (95%CI 0.35 to 1.09), and RD was -0.14 (95%CI -0.27 to -0.01) for mortality at day 21. This is considered clinically relevant.
The pooled RR in patients with a severe disease state was 0.61 (95%CI 0.37 to 0.99), and the RD was -0.12 (95%CI -0.22 to -0.02) for mortality day 21-30. This is considered clinically relevant.
Mild, moderate and severe disease
Merchante (2021) reported the mortality for patients with a mild, moderate or severe disease state. The RR was 0.14 (95%CI 0.01 to 2.68), and RD was -0.08 (95%CI -0.17 to 0.02) for mortality at day 28. This is considered clinically relevant.
Sarilumab 200 mg:
The incidence of mortality in hospitalized patients in the intervention group 200mg was 4/37 (10.1%), compared to 3/39 (7.7%) in the control group. The RR was 1.41 (95%CI 0.34 to 5.86), and RD was 0.03 (95%CI -0.10 to 0.16) was. This is considered clinically relevant (in favour of the control group).
Figure 5: Mortality 21-30 days in hospitalized patients treated with sarilumab 400mg, per disease severity category
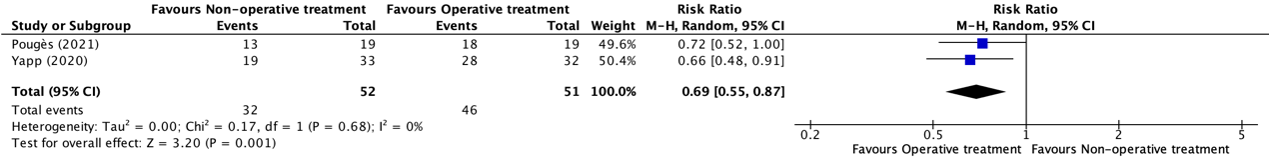
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Importantly, if sensitivity analyses (i.e., including only the high quality studies AND studies with more than 80% use of corticosteroids in the standard of care OR including studies with lower quality and/or initiating less than 80% corticosteroids) were performed for these analyses (i.e., per disease severity category), the same results were shown for (1) moderate disease (Lescure, 2021; Mariette, 2021; Sancho-Lopéz, 2021), (2) severe disease (Gordon, 2021; Lescure, 2021) and (3) mixed: mild, moderate and severe disease (Merchante, 2021).
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 2 levels because of imprecision (95%CI of the mean difference includes no effect (RD=0) and crosses both thresholds of clinical relevance, -2). The level of evidence for the outcome ‘mortality’ is low.
Moderate disease
The level of evidence regarding the outcome measure mortality started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because of inconsistency (-1), and imprecision (95% CI crosses threshold of clinically relevance, -1). The level of evidence for the outcome ‘mortality’ is low.
Severe disease
The level of evidence regarding the outcome measure mortality started as high, because the studies were RCTs. The level of evidence was downgraded by one level because of imprecision (95% CI crosses the thresholds of clinically relevance, -1). The level of evidence for the outcome ‘mortality’ is moderate.
Results - Levilimab
Lomakin (2021) did not report data about mortality. The initial primary endpoint was overall mortality, but because the mortality was significantly lower than assumed, the study did not have enough power to detect a meaningful difference.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence regarding the outcome measures mortality could not be assessed with GRADE. The outcome measures were not studied in the included studies.
Results - Siltuximab
Declercq (2021) reported mortality during the study (i.e., follow-up of 10-20 weeks), and the estimated mortality at day 28 based on Kaplan-Meier
Mortality, 10-20 weeks
Declercq (2021) reported the number of deaths in the subgroups at follow-up (i.e., 10-20 weeks). The incidence of mortality was 15/75 (20.0 %) in patients treated with only additional siltuximab, compared with 9/74 (12.2%) in the control group (i.e., receiving only usual care according to the randomizations). This resulted in a RR of 1.64 (95%CI 0.77 to 3.52) and a RD of 0.08 (95%CI -0.04 to 0.20). This is considered clinically relevant (in favour of the control group).
The incidence of mortality was 5/32 (15.6%) in patients treated with anakinra + tocilizumab, 6/36 (16.7%) in patients treated with anakinra + siltuximab, 10/81 (12.3%) in patients treated with usual care + tocilizumab, and 10/44 (23%) in patients treated with usual care + anakinra. In addition, the estimated mortality at day 28 (i.e., based on Kaplan-Meier) resulted in 13% (95%CI 7 to 23) in the intervention group, compared with 10% (95%CI 5 to 20) in the control group.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 3 levels because of indirectness (other follow-up, -1), and imprecision (95%CI of the mean difference includes no effect (RD=0), crosses both thresholds of clinical relevance, not meeting the optimal information size, -2). The level of evidence for the outcome ‘mortality’ is very low.
Extensive respiratory support (crucial)
Results - Tocilizumab
Nine studies reported on the outcome extensive respiratory support. All studies reported on the outcome mechanical ventilation. Two of these studies also reported on the outcome non-invasive ventilation and high-flow oxygen combined (Hermine, 2020; Veiga, 2021). One study reported outcomes for patients with a moderate disease state only (Hermine, 2021), and one study reported outcomes for patients up to day 21 (Gordon, 2021), respectively.
The pooled incidence was 476/2868 (16.6%) in the intervention group compared to 568/2654 (21.4%) in the control group. The pooled RR from these nine studies was 0.79 (95% CI 0.71 to 0.88, Figure 6), in favour of the tocilizumab group. The pooled RD was -0.04 (95% CI -0.06 to -0.02). This is not considered clinically relevant.
Regarding the sensitivity analyses (i.e., high quality study AND >80% corticosteroids as part of the standard care versus low quality study and/or <80% corticosteroids), results were in line with the main analysis (Figure 6). The pooled RR in the high quality studies AND initiating more than 80% corticosteroids was 0.80 (95%CI 0.71 to 0.90), and the RD was -0.04 (95%CI -0.07 to -0.02). This is not considered clinically relevant. The pooled RR in studies with lower quality and/or initiating less than 80% corticosteroids was 0.74 (95%CI 0.59 to 0.93), and the RD was -0.04 (95%CI -0.07 to 0.00). This is not considered clinically relevant.
Salama (2021) reported results for mechanical ventilation or death by day 28, but no results were reported for the effect on mechanical ventilation alone. After excluding this study from the analyses, the pooled RR for the remaining eight studies is 0.80 (95% CI 0.71 to 0.89). The pooled RD remained -0.04 (95% CI-0.06 to -0.02). This is not considered clinically relevant.
Figure 6. Extensive respiratory support in hospitalized patients treated with tocilizumab
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Hermine (2020) reported on the outcome mechanical ventilation, non-invasive ventilation or high-flow oxygen on day 14, specifically for patients with a moderate disease state. In the tocilizumab group, 11/63 (17.5%) patients received extensive respiratory support (8 patients received non-invasive ventilation or high-flow oxygen and 3 patients received mechanical ventilation). In the control group, 21/67 (31.3%) patients received extensive respiratory support (13 patients received non-invasive ventilation or high-flow oxygen and 8 patients received mechanical ventilation). The RR was 0.56 (95%CI 0.29 to 1.06). The RD was -0.14 (95% CI -0.28 to 0.01). This is considered clinically relevant.
Veiga (2021) reported the need for non-invasive ventilation or high-flow oxygen. In both the tocilizumab group and control group, no patient received non-invasive ventilation or high flow nasal cannula. Veiga (2021) also reported on the outcome mechanical ventilation on day 29 specifically for patients with a moderate or severe disease state. The RR was 0.98 (95%CI 0.26 to 3.77). The RD was -0.00 (95% CI -0.08 to 0.08). This is not considered clinically relevant.
Veiga (2021) also reported on the outcomes on day 8 and 15. The RR for mechanical ventilation on day 8 was 0.78 (95%CI 0.48 to 1.28) and the RD was -0.08 (95%CI -0.24 to 0.08). The RR for mechanical ventilation on day 15 was 0.63 (95% CI 0.26 to 1.51) and the RD was -0.06 (95%CI -0.18 to 0.06). The RR for non-invasive ventilation or high-flow oxygen on day 8 was 0.78 (95%CI 0.48 to 1.28) and the RD was -0.08 (95%CI -0.24 to 0.08). The RR for non-invasive ventilation or high-flow oxygen on day 15 was 0.63 (95% CI 0.26 to 1.51) and the RD was -0.06 (95%CI -0.18 to 0.06). The difference is clinically relevant for 8 and 15 days of follow-up, but not for 29 days.
Gordon (2021) reported progression to invasive mechanical ventilation up to day 21. The incidence was 84/242 (34.7 %) in the intervention group, compared with 116/273 (42.5%) in the control group. The RR was 0.82 (95%CI 0.65 to 1.02). The RD was -0.08 (95% CI -0.16 to 0.01). This is considered clinically relevant.
Level of evidence of the literature
The level of evidence for the outcome measure extensive ventilation started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because of inconsistency (-1), and imprecision (crossing the threshold of clinical relevance, -1). The level of evidence for the outcome ‘mechanical ventilation’ is low.
Results - Sarilumab
Initiation of respiratory support in hospitalized patients with COVID-19 was reported in five studies (Gordon, 2021; Lescure, 2021; Mariette, 2021; Merchante, 2021; Sancho-Lopéz, 2021). This was defined as progression to invasive mechanical ventilation or death, and Brescia-COVID score ≥3 at day 15 and day 28 in the study of Sancho-Lopéz (2021). Mariette (2021) defined this outcome as dead or needing non-invasive ventilation or mechanical ventilation on day 14 (i.e., WHO-CPS score > 5). This outcome measure was defined as initiation of mechanical ventilation, non-invasive ventilation, or use of high-flow nasal cannula in the study of Lescure (2021). In the study of Gordon (2021) this outcome measure was defined as progression to invasive mechanical ventilation up to day 21. Merchante (2021) defined this outcome measure as progression to high-flow nasal oxygenation or (non-)invasive mechanical ventilation.
Sancho-Lopéz (2021) reported progression to invasive mechanical ventilation or death in 4/99 (4%) patients in the intervention group, compared with 9/102 (9%) in the control group. This resulted in a RR of 0.46 (95% 0.15 to 1.48) and a RD of -0.05 (95%CI -0.12 to 0.02). This is considered clinically relevant. In addition, the number of patients with a Brescia-COVID score ≥3 (i.e., the need of high frequency nasal ventilation, CPAP or non-invasive ventilation or mechanical ventilation) at day 15 and 28 was reported in 16/99 (16%) patients in the intervention group, compared with 16/102 (16%) in the control group. This resulted in a RR of 1.03 (95%CI 0.48 to 2.20) and RD of 0.00 (95%CI -0.10 to 0.11). This is not considered clinically relevant.
Mariette (2021) reported needing non-invasive ventilation or mechanical ventilation or dead at day 14. This incidence was 18/68 (18%) in the intervention group, compared with 20/76 (26%) in the control group. This resulted in a RR of 0.89 (95%CI 0.51 to 1.58) and a RD of -0.03 (95%CI -0.17 to 0.11). By excluding the number of patients who died during the first 14 days, the incidence was 12/68 (18%) in the intervention group, compared with 12/76 (16%) in the control group. This resulted in a RR of 1.12 (95%CI 0.54 to 2.32) and a RD of -0.02 (-0.10 to 0.14). This is not considered clinically relevant.
Gordon (2021) reported progression of initiation of respiratory support in 6/37(16%) patients with severe COVID-19 in the intervention group, compared with 116/272 (43%) in the control group. This resulted in a RR of 0.38 (95% CI 0.22 to 2.26) and a RD of -0.26 (95% CI -0.40 to -0.13). This is considered clinically relevant.
Merchante (2021) reported progression to high-flow nasal oxygenation or (non-)invasive mechanical ventilation in 10/39 (25.6%) patients in the intervention group, compared with 5/39 (12.8%) in the control group. This resulted in a RR of 2.00 (95% CI 0.75 to 5.32) and RD of 0.13 (95%CI -0.04 to 0.30). This is considered clinically relevant (in favour of the control group).
Sarilumab 200 mg:
Merchante (2021) reported progression to high-flow nasal oxygenation or (non-)invasive mechanical ventilation in 10/37 (27.0%) patients in the intervention group, compared with 5/39 (12.8%) in the control group. This resulted in a RR of 2.11 (95% CI 0.75 to 5.32) and RD of 0.14 (95%CI -0.04 to 0.32). This is considered clinically relevant (in favour of the control group).
Lescure (2021) reported initiation of respiratory support in 33/173(19%) patients in the intervention group, compared with 13/84 (15%) in the control group. This resulted in a RR of 1.23 (95% CI 0.69 to 2.22) and RD of 0.04 (95% CI -0.06 to 0.13). This is not considered clinically relevant.
Sarilumab 200 mg:
Lescure (2021) reported initiation of respiratory support in 26/159 (16%) patients in the intervention group 200mg, compared with 13/84 (15%) in the control group. This resulted in a RR of 1.06 (95% CI 0.57 to 1.95) and a RD of 0.01 (95% CI -0.09 to 0.11). This is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 3 levels because of inconsistency (-1) and imprecision (95%CI of the mean difference includes no effect (RD=0) and crosses both thresholds of clinical relevance, -2). The level of evidence for the outcome ‘respiratory support’ is very low.
Severe disease
The level of evidence regarding the outcome measure extensive respiratory support started as high, because the studies were RCTs. The level of evidence was downgraded by one level of imprecision (not meeting optimal information size, -1). The level of evidence for the outcome ‘respiratory support’ is moderate.
Results - Levilimab
Initiation of respiratory support in hospitalized patients with COVID-19 was not reported.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence regarding the outcome measures respiratory support could not be assessed with GRADE. The outcome measures were not studied in the included studies.
Results – other (when results of different IL-6 inhibitors were reported combined, i.e., tocilizumab and siltuximab)
Declercq (2021) defined this outcome as number of invasive ventilation days
Declercq (2021) reported the mean number of invasive ventilator days, which was 5 (95%CI 3 to 7) in the intervention group (i.e., IL-6 blockade group), compared with 5 (95%CI 3 to 9) days in the control group (i.e., no IL-6 blockade group). This resulted in a mean difference of 0 days. This is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 3 levels because of indirectness (1 level, different IL-6 blockades were used as intervention) and imprecision (2 levels, 95%CI of the mean difference includes no effect (RD=0) and crosses both thresholds of clinical relevance). The level of evidence for the outcome ‘extensive respiratory support’ is very low.
Duration of hospitalization (important)
Results
In total, five studies reported the number of patients who were discharged from the hospital after 28-30 days. Results will be displayed per IL-6 inhibitor.
Results - Tocilizumab
Seven studies (Horby, 2021; Rosas, 2021a, Rosas, 2021b; Salama, 2021; Stone, 2020; Veiga, 2021; Wang, 2021) reported time to hospital discharge and three studies (Hermine, 2020, Horby, 2021, Salvarani, 2020) reported the incidence of discharge after a follow-up of 28-30 days. One study reported duration of ICU stay, but not of total hospitalization (Soin, 2021).
Three studies (Hermine, 2020, Horby, 2021, Salvarani, 2020) reported the incidence of discharge after a follow-up of 28-30 days. The pooled incidence of hospital discharge after follow-up was 1256 out of 2145 (58.6%) in the intervention group compared to 1151 out of 2224 (51.8%) in the control group. The pooled RR was 1.08 (95%CI 0.96 to 1.21, Figure 7). The pooled RD was 0.053 (95% CI -0.0004 to 0.1099). This is considered clinically relevant.
Figure 7. Incidence of discharge after 28-30 days in hospitalized patients treated with tocilizumab.
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Seven studies (Horby, 2021; Rosas, 2021a, Rosas, 2021b; Salama, 2021; Stone, 2020; Veiga, 2021; Wang, 2021) reported time to hospital discharge. Six of these studies reported the median time to discharge, only Veiga (2021) reported a mean duration. The median time to discharge the studies ranged from 6 to 26 days in the intervention group compared to 6 to >28 days in the control group. In two of the six studies, the difference between intervention and control group was more than 3 days. In the four other studies, the difference between the two groups was 0, 1.5, and 2 days.
One study (Veiga, 2021) reported mean (SD) duration of hospital stay of 11.3 (8.0) days in the tocilizumab group compared to 14.7 (8.2) days in the control group. The mean difference in this study was -3.40 (95%CI -6.20 to -0.60) days. This is considered clinically relevant.
Four studies (Gordon, 2021; Rosas 2021a; Salama, 2021; Stone, 2020) reported the hazard ratio for duration of hospitalization. The HR in the studies ranged from 1.08 (95%CI 0.81 to 1.43) to 1.41 (95%CI 1.18 to 1.70), in favour of the intervention group.
One study (Soin, 2021) reported median time of ICU stay, but not of total hospitalization. The median ICU stay was 7 (IQR 3 to 10) days in the intervention compared to 6 (IQR 3.5 to 11) days in the control group. The difference was 1 day, this is not considered clinically relevant.
Level of evidence of the literature
The level of evidence regarding the outcome measure duration of hospitalization started as high, because the studies were RCTs. The level of evidence was downgraded by 2 levels because of inconsistency (different outcomes between studies, -1), and imprecision (crossing the threshold of clinical relevance, -1). The level of evidence for the outcome ‘duration of hospitalization’ is low.
Results - Sarilumab
Duration of hospitalization in hospitalized patients with COVID-19 was reported in four studies (Lescure, 2021; Mariette, 2021; Merchante, 2021; Sancho-Lopéz, 2021).
Two studies (Mariette, 2021; Merchante, 2021) reported the incidence of discharge after a follow-up of 28-30 days. The pooled incidence of hospital discharge after follow-up was 87 out of 107 (81.3%) in the intervention group compared to 87 out of 115 (75.7%) in the control group. The pooled RR was 1.06 (95%CI 0.94 to 1.20, Figure 8). The pooled RD was 0.052 (95% CI -0.047 to 0.151). This is considered clinically relevant.
Sarilumab 200 mg:
Merchante (2021) reported that the number of patients which discharged from the hospital within 28 days. A number of 30/37 (81.2%) patients in the intervention group discharged, compared with 34/39 (87.2%) in the control group. This resulted in a RR of 0.93 (95%CI 0.76 to 1.13) and a RD of -0.06 (95%CI -0.23 to 0.10). This difference not clinically relevant.
Sancho-Lopéz (2021) reported the time to hospital discharge in days. This was 7 (95% CI 6-8) days in the intervention group, compared with 7 (95% CI 6-8) days in the control group. The difference was 0 days, this is not considered as a clinically relevant difference.
In addition, the number of patients with ICU admission at day 15 and day 28 was reported. In total 7/99 (7%) patients in the intervention group were admitted to ICU, compared with 10/102 (10%) in the control group, both at day 15 and 28. This resulted in a RR of 0.72 (95% CI 0.29 to 1.82) and a RD of -0.03 (95% CI -0.10 to 0.05). This difference is not clinically relevant.
Figure 8. Incidence of discharge after 28-30 days in hospitalized patients treated with sarilumab.
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval.
Lescure (2021) reported that the mean (SE) number of days hospitalization among
patients alive at day 60 was 16 days (0.9) in the intervention group (n=173), compared with 16 days (1.3) in the control group (n=84). The difference was 0 days, this is not considered as a clinically relevant difference.
Sarilumab 200 mg:
Lescure (2021) reported that the mean (SE) number of days hospitalization among
patients alive at day 60 was 16 days (1.0) in the intervention group 200mg (n=159), compared with 16 days (1.3) in the control group (n=84). The difference was 0 days, this is not considered as a clinically relevant difference.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 2 levels because of inconsistency (different outcomes between studies, -1), and imprecision (95%CI of the mean difference includes no effect (RD/MD=0), -1). The level of evidence for the outcome ‘duration of hospitalization’ is low.
Results - Levilimab
Duration of hospitalization in hospitalized patients with COVID-19 was reported in one study (Lomakin, 2021). Lomakin (2021) reported the mean (IQR) duration of hospital stay in days. In addition, the number of patients which were transferred to ICU were reported.
Lomakin (2021) reported that the median (IQR) duration was 11 (8-16) days in the intervention group, compared with 11 (7-18) days in the control group. The difference was 0 days. This is not considered as clinically relevant.
In the intervention group 3/103 (3%) were transferred to ICU, compared with 10/103 (10%) in the control group. This resulted in a RR of 0.30 (95%CI 0.09 to 1.06) and a RD of -0.07 (-0.13 to -0.00). This is considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 2 levels because of imprecision (95%CI of the mean difference includes no effect (RD=0), and not meeting the optimal information size, -2). The level of evidence for the outcome ‘duration of hospitalization’ is low.
Results – other (when results of different IL-6 inhibitors were reported combined, i.e. tocilizumab and siltuximab)
Declercq (2021) defined this outcome as median number of days in the hospital and ICU, respectively.
Declercq (2021) reported that the mean number of days in hospital was 20 (95%CI 18 to 22) in the intervention group (i.e., IL-6 blockade group), compared with 19 (95%CI 16 to 22) days in the control group (i.e., no IL-6 blockade group). This resulted in a mean difference of 1 days, which is not considered clinically relevant.
Regarding the number of days at ICU, this was 11 (95%CI 8 to 14) in the intervention group, compared with 10 (95%CI 7 to 15) in the control group. This resulted in a mean difference of -1 day, which is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 3 levels because of indirectness (different IL-6 blockades were used as intervention. -1) and imprecision (95%CI of the mean difference includes no effect (RD=0) and crosses both thresholds of clinical relevance, -2). The level of evidence for the outcome ‘duration of hospitalization’ is very low.
Time to clinical improvement (important)
Results - Tocilizumab
Four studies (Salama, 2021; Soin, 2021; Stone, 2020; Wang, 2021) reported on the outcome clinical improvement. Three of these studies (Salama, 2021; Soin, 2021; Stone, 2020) reported median time to improvement. Median time to improvement in the studies ranged from 6 to 7 days in the intervention group compared to 5 to 7 days in the control group. The difference of 1 day is not considered clinically relevant.
One study (Wang, 2021) reported the cure rate. In the intervention group, 32/34 (94.1%) were cured compared to 27/31 (87.1%) in the control group. The RR was 1.08 (95% CI 0.92 to 1.27). The RD was 0.07 (95%CI -0.07 to 0.21). This is considered clinically relevant.
Level of evidence of the literature
The level of evidence regarding the outcome measure time to clinical improvement started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because of inconsistency (heterogeneity in reporting ‘time to clinical improvement’, -1) and imprecision (crossing the threshold of clinical relevance, -1). The level of evidence for the outcome ‘time to clinical improvement’ is low.
Results - Sarilumab
Time to clinical improvement for hospitalized patients with COVID-19 was reported in one study (Lescure, 2021).
Lescure (2021) reported that the median time (95% CI) to NEWS2 of 2 or lower for 24 hours was 9 days (8 to 11) in the intervention group (n=173), compared to 11 days (8 to 14) in the control group (n=84). The difference is 2 days, this is not considered clinically relevant. The median time (95%) to resolution of fever was also reported, which was 9 (7 to 10) in the intervention group (n=173), compared with 7 (6 to 12) in the control group (n=84). The difference is 2 days, this is not considered clinically relevant.
Sarilumab 200 mg:
Lescure (2021) reported that the median time (95% CI) to NEWS2 of 2 or lower for 24 hours was 8 days (7 to 9) in the intervention group 200mg (n=159), compared to 11 days (8 to 14) in the control group (n=84). The difference is 3 days, this is considered clinically relevant. The median time (95%) to resolution of fever was also reported, which was 8 (7 to 9) in the intervention group 200mg (n=159), compared with 7 (6 to 12) in the control group (n=84). The difference is 1 days, this is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 1 level because of imprecision (not meeting the optimal information size, -1). The level of evidence for the outcome ‘time to clinical improvement’ is moderate.
Results -Levilimab
Time to clinical improvement in hospitalized patients with COVID-19 was not reported.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence regarding the outcome measures time to clinical improvement could not be assessed with GRADE. The outcome measures were not studied in the included studies.
Results – other (when results of different IL-6 inhibitors were reported combined, i.e. tocilizumab and siltuximab)
Time to clinical improvement in hospitalized patients with COVID-19 was reported in one study (Declercq, 2021).
Declercq (2021) reported the median (95%CI) time to clinical improvement in days. This was 11 (10 to 16) days in the intervention group (i.e., IL-6 blockade group), compared to 12 (11 to 16) days in the control group. This resulted in a HR of 1.00 (95%CI 0.78 to 1.29). The absolute difference of 1 days is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 3 levels because of indirectness (different IL-6 blockades were used as intervention, -1) and imprecision (95%CI of the mean difference includes no effect (RD=0) and crosses both thresholds of clinical relevance, -2). The level of evidence for the outcome ‘time to clinical improvement’ is very low.
A systematic review of the literature was performed to answer the following question:
What is the effectivity of treatment with an interleukin (IL-)6 inhibitor compared to treatment without an interleukin (IL-)6 inhibitor in patients with COVID-19?
PICO
P: hospitalized with COVID-19 (subgroups mild, moderate, severe)
I: IL-6 inhibitor + standard care
C: standard care only / placebo treatment + standard care
O: 28-30 day mortality (if not available, any other reports of mortality), extensive respiratory support, duration of hospitalization, time to clinical improvement
Relevant outcome measures
For hospitalized COVID-19 patients, mortality and need for extensive respiratory support were considered as crucial outcome measures for decision making. Duration of hospitalization, and time to clinical improvement were considered as important outcome measures for decision making.
Extensive respiratory support was defined as high flow nasal cannula (HFNC)/Optiflow, continuous positive airway pressure (CPAP), non-invasive ventilation (NIV), mechanical ventilation or extracorporeal membrane oxygenation (ECMO or ECLS).
The working group defined 3% points absolute difference as a minimal clinically important difference for mortality (resulting in a NNT of 33), 3 days difference for duration of hospitalization and time to clinical improvement, 5% points absolute difference need for respiratory support and ICU admission (resulting in a NNT of 20).
The results of studies in non-hospitalized and hospitalized patients are summarized separately. Studies of hospitalized patients were categorized based on the respiratory support that was needed at baseline (preferably based on patient inclusion/exclusion criteria; otherwise on baseline characteristics). The following categories were used:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen [high flow nasal cannula (HFNC)/Optiflow], continuous positive airway pressure [CPAP], non-invasive ventilation [NIV], mechanical ventilation, extracorporeal membrane oxygenation [ECMO or ECLS]).
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until 20 January 2022. The detailed search strategy is outlined under the tab Methods. Studies were selected based on the following criteria: randomized controlled trial, peer reviewed and published in indexed journal, comparing treatment with anti-IL-6 or anti-IL-6-R and standard care to standard care alone or treatment anti-IL-6 or anti-IL-6-R and standard care to placebo and standard care in patients with COVID-19.
The systematic literature search resulted in 80255 hits. Studies were selected based on the following criteria: systematic review or randomized controlled trials. Eventually, seventeen studies were included.
Statistical methods
Statistical analyses were conducted using Review Manager (RevMan) software 5.4. For dichotomous outcomes, Mantel Haenszel random‐effects risk ratios (RRs) and risk differences (RDs) were calculated. For continuous outcomes, a random‐effects mean difference (MD) weighted by the inverse variance was calculated. The random-effects model estimates the mean of a distribution of effects.
Results
In total, seventeen RCTs were included in the analysis of the literature. Two of the seventeen RCTs studied more than one IL-6 inhibitor; one RCT studied tocilizumab and sarilumab, and one RCT studied tocilizumab and siltuximab. Ten studies described tocilizumab, four studies sarilumab, and one study levilimab. Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
- CORIMUNO-19 Collaborative group. (Mariette, 2021) Sarilumab in adults hospitalised with moderate-to-severe COVID-19 pneumonia (CORIMUNO-SARI-1): An open-label randomised controlled trial. Lancet Rheumatol. 2021 Nov 17. doi: 10.1016/S2665-9913(21)00315-5. Epub ahead of print. PMID: 34812424; PMCID: PMC8598187.
- Declercq J, Van Damme KFA, De Leeuw E, Maes B, Bosteels C, Tavernier SJ, De Buyser S, Colman R, Hites M, Verschelden G, Fivez T, Moerman F, Demedts IK, Dauby N, De Schryver N, Govaerts E, Vandecasteele SJ, Van Laethem J, Anguille S, van der Hilst J, Misset B, Slabbynck H, Wittebole X, Liénart F, Legrand C, Buyse M, Stevens D, Bauters F, Seys LJM, Aegerter H, Smole U, Bosteels V, Hoste L, Naesens L, Haerynck F, Vandekerckhove L, Depuydt P, van Braeckel E, Rottey S, Peene I, Van Der Straeten C, Hulstaert F, Lambrecht BN. Effect of anti-interleukin drugs in patients with COVID-19 and signs of cytokine release syndrome (COV-AID): a factorial, randomised, controlled trial. Lancet Respir Med. 2021 Dec;9(12):1427-1438. doi: 10.1016/S2213-2600(21)00377-5. Epub 2021 Oct 29. PMID: 34756178; PMCID: PMC8555973
- Ghosn, L., Chaimani, A., Evrenoglou, T., Davidson, M., Graña, C., Schmucker, C., Bollig, C., Henschke, N., Sguassero, Y., Nejstgaard, C. H., Menon, S., Nguyen, T. V., Ferrand, G., Kapp, P., Riveros, C., Ávila, C., Devane, D., Meerpohl, J. J., Rada, G., Hróbjartsson, A., … Boutron, I. (2021). Interleukin-6 blocking agents for treating COVID-19: a living systematic review. The Cochrane database of systematic reviews, 3(3), CD013881. https://doi.org/10.1002/14651858.CD013881
- Hermine, O., Mariette, X., Tharaux, P. L., Resche-Rigon, M., Porcher, R., Ravaud, P., ... &
Korganow, A. S. (2021). Effect of tocilizumab vs usual care in adults hospitalized with
COVID-19 and moderate or severe pneumonia: a randomized clinical trial. JAMA
internal medicine, 181(1), 32-40. - Horby, P. W. and RECOVERY Collaborative Group (2021). Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet (London, England), 397(10285), 1637–1645. https://doi-org.saz.idm.oclc.org/10.1016/S0140-6736(21)00676-0
- Jorgensen SCJ, Lapinsky SE. Tocilizumab for coronavirus disease 2019 in pregnancy and lactation: a narrative review. Clin Microbiol Infect. 2022 Jan;28(1):51-57. doi: 10.1016/j.cmi.2021.08.016. Epub 2021 Aug 23. PMID: 34438068; PMCID: PMC8381634.
- Kox M, Waalders NJB, Kooistra EJ, Gerretsen J, Pickkers P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA. 2020 Sep 3;324(15):1565–7. doi: 10.1001/jama.2020.17052. Epub ahead of print. PMID: 32880615; PMCID: PMC7489366.
- Lescure FX, Honda H, Fowler RA, Lazar JS, Shi G, Wung P, Patel N, Hagino O; Sarilumab COVID-19 Global Study Group. Sarilumab in patients admitted to hospital with severe or critical COVID-19: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2021 May;9(5):522-532. doi: 10.1016/S2213-2600(21)00099-0. Epub 2021 Mar 4. PMID: 33676590; PMCID: PMC8078879.
- Levi M. Tocilizumab for severe COVID-19: A promising intervention affecting inflammation and coagulation. Eur J Intern Med. 2020 Jun;76:21-22. doi: 10.1016/j.ejim.2020.05.018. Epub 2020 May 16. PMID: 32425364; PMCID: PMC7229910.
- Lomakin NV, Bakirov BA, Protsenko DN, Mazurov VI, Musaev GH, Moiseeva OM, Pasechnik ES, Popov VV, Smolyarchuk EA, Gordeev IG, Gilyarov MY, Fomina DS, Seleznev AI, Linkova YN, Dokukina EA, Eremeeva AV, Pukhtinskaia PS, Morozova MA, Zinkina-Orikhan AV, Lutckii AA. The efficacy and safety of levilimab in severely ill COVID-19 patients not requiring mechanical ventilation: results of a multicenter randomized double-blind placebo-controlled phase III CORONA clinical study. Inflamm Res. 2021 Dec;70(10-12):1233-1246. doi: 10.1007/s00011-021-01507-5. Epub 2021 Sep 29. PMID: 34586459; PMCID: PMC8479713.
- Merchante N, Cárcel S, Garrido-Gracia JC, Trigo-Rodríguez M, Esteban Moreno MÁ, León-López R, Espíndola-Gómez R, Aguilar Alonso E, García DV, Romero-Palacios A, Pérez-Camacho I, Gutiérrez-Gutiérrez B, Martínez-Marcos FJ, Fernández-Roldán C, Pérez-Crespo PMM, Caño AA, León E, Corzo JE, de la Fuente C, Torre-Cisneros J. Early Use of Sarilumab in Patients Hospitalised with COVID-19 Pneumonia and Features of Systemic Inflammation. Antimicrob Agents Chemother. 2021 Dec 13:AAC0210721. doi: 10.1128/AAC.02107-21. Epub ahead of print. PMID: 34902262.
- Moes DJAR, van Westerloo DJ, Arend SM, Swen JJ, de Vries A, Guchelaar HJ, Joosten SA, de Boer MGJ, van Gelder T, van Paassen J. Towards Fixed Dosing of Tocilizumab in ICU-Admitted COVID-19 Patients: Results of an Observational Population Pharmacokinetic and Descriptive Pharmacodynamic Study. Clin Pharmacokinet. 2022 Feb;61(2):231-247. doi: 10.1007/s40262-021-01074-2. Epub 2021 Oct 11. PMID: 34633645; PMCID: PMC8502793
- REMAP-CAP Investigators, Gordon AC, Mouncey PR, Al-Beidh F, Rowan KM, Nichol AD, Arabi YM, Annane D, Beane A, van Bentum-Puijk W, Berry LR, Bhimani Z, Bonten MJM, Bradbury CA, Brunkhorst FM, Buzgau A, Cheng AC, Detry MA, Duffy EJ, Estcourt LJ, Fitzgerald M, Goossens H, Haniffa R, Higgins AM, Hills TE, Horvat CM, Lamontagne F, Lawler PR, Leavis HL, Linstrum KM, Litton E, Lorenzi E, Marshall JC, Mayr FB, McAuley DF, McGlothlin A, McGuinness SP, McVerry BJ, Montgomery SK, Morpeth SC, Murthy S, Orr K, Parke RL, Parker JC, Patanwala AE, Pettilä V, Rademaker E, Santos MS, Saunders CT, Seymour CW, Shankar-Hari M, Sligl WI, Turgeon AF, Turner AM, van de Veerdonk FL, Zarychanski R, Green C, Lewis RJ, Angus DC, McArthur CJ, Berry S, Webb SA, Derde LPG. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19. N Engl J Med. 2021 Apr 22;384(16):1491-1502. doi: 10.1056/NEJMoa2100433. Epub 2021 Feb 25. PMID: 33631065; PMCID: PMC7953461.
- Rosas, I. O., Bräu, N., Waters, M., Go, R. C., Hunter, B. D., Bhagani, S., ... & Malhotra, A.
(2021). Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. New
England Journal of Medicine, 384(16), 1503-1516. - Rosas, I. O., Diaz, G., Gottlieb, R. L., Lobo, S. M., Robinson, P., Hunter, B. D., ... & Olsson, J. K.
(2021b). Tocilizumab and remdesivir in hospitalized patients with severe COVID-19
pneumonia: a randomized clinical trial. Intensive care medicine, 47(11), 1258-1270. - Salama, C., Han, J., Yau, L., Reiss, W. G., Kramer, B., Neidhart, J. D., ... & Mohan, S. V. (2021).
Tocilizumab in patients hospitalized with Covid-19 pneumonia. New England Journal
of Medicine, 384(1), 20-30. - Salvarani, C., Dolci, G., Massari, M., Merlo, D. F., Cavuto, S., Savoldi, L., ... & Costantini, M.
(2021). Effect of tocilizumab vs standard care on clinical worsening in patients
hospitalized with COVID-19 pneumonia: a randomized clinical trial. JAMA internal
medicine, 181(1), 24-31. - Sancho-López A, Caballero-Bermejo AF, Ruiz-Antorán B, Múñez Rubio E, García Gasalla M, Buades J, González Rozas M, López Veloso M, Muñoz Gómez A, Cuenca Abarca A, Durán Del Campo P, Ibáñez F, Díaz de Santiago A, Romero Y, Calderón J, Pintos I, Ferre Beltrán A, Centeno Soto G, Campos J, Ramos Martínez A, Avendaño-Solá C, Fernández Cruz A; SARTRE-Study Group. Efficacy and Safety of Sarilumab in patients with COVID19 Pneumonia: A Randomized, Phase III Clinical Trial (SARTRE Study). Infect Dis Ther. 2021 Dec;10(4):2735-2748. doi: 10.1007/s40121-021-00543-2. Epub 2021 Oct 17. PMID: 34658006; PMCID: PMC8520759.
- Schoels MM, van der Heijde D, Breedveld FC, Burmester GR, Dougados M, Emery P, Ferraccioli G, Gabay C, Gibofsky A, Gomez-Reino JJ, Jones G, Kvien TK, Murakami M, Nishimoto N, Smolen JS. Blocking the effects of interleukin-6 in rheumatoid arthritis and other inflammatory rheumatic diseases: systematic literature review and meta-analysis informing a consensus statement. Ann Rheum Dis. 2013 Apr;72(4):583-9. doi: 10.1136/annrheumdis-2012-202470. Epub 2012 Nov 10. Erratum in: Ann Rheum Dis. 2013 Jun;72(6):1110. Murikama, Miho M [corrected to Murakami, Miho]. PMID: 23144446; PMCID: PMC3595140.
- Singh JA, Beg S, Lopez-Olivo MA. Tocilizumab for rheumatoid arthritis. Cochrane Database Syst Rev. 2010 Jul 7;(7):CD008331. doi: 10.1002/14651858.CD008331.pub2. PMID: 20614469.
- Sinha P, Matthay MA, Calfee CS. Is a "Cytokine Storm" Relevant to COVID-19? JAMA Intern Med. 2020 Sep 1;180(9):1152-1154. doi: 10.1001/jamainternmed.2020.3313. PMID: 32602883.
- Soin, A. S., Kumar, K., Choudhary, N. S., Sharma, P., Mehta, Y., Kataria, S., ... & Ramanan, A.
V. (2021). Tocilizumab plus standard care versus standard care in patients in India
with moderate to severe COVID-19-associated cytokine release syndrome (COVINTOC): an open-label, multicentre, randomised, controlled, phase 3 trial. The Lancet Respiratory Medicine, 9(5), 511-521. - Stone, J. H., Frigault, M. J., Serling-Boyd, N. J., Fernandes, A. D., Harvey, L., Foulkes, A. S., ... &
Mansour, M. K. (2020). Efficacy of tocilizumab in patients hospitalized with Covid-19.
New England Journal of Medicine, 383(24), 2333-2344. - Veiga, V. C., Prats, J. A., Farias, D. L., Rosa, R. G., Dourado, L. K., Zampieri, F. G., ... &
Scheinberg, P. (2021). Effect of tocilizumab on clinical outcomes at 15 days in
patients with severe or critical coronavirus disease 2019: randomised controlled
trial. Bmj, 372. - Wang, D., Fu, B., Peng, Z., Yang, D., Han, M., Li, M., ... & Xu, X. (2021). Tocilizumab in patients
with moderate or severe COVID-19: a randomized, controlled, open-label, multicenter
trial. Frontiers of medicine, 1-9.
PICO: What is the (in)effectivity and safety of treatment with an IL-6 inhibitor compared to treatment without remdesivir in patients with COVID-19?
|
Tocilizumab (humanized monoclonal antibody against the interleukin-6 receptor) |
|||||||
|
Hermine, 2022
(TOCI-2 and SARI-2 trial, embedded in the CORIMUNO-19 cohort)
|
Type of study: Two cohort-embedded, investigator-initiated, multicenter, open-label, Bayesian randomised controlled clinical trials
Setting: hospital-based, between March 31 to April 18, 2020 (TOCI-2) and from March 27 to April, 2020
Country: France (12 sites, TOCI-2; 8 sites, SARI-2)
Source of funding: This trial was publicly funded (Ministry of Health, Programme Hospitalier de Recherche Clinique [PHRC COVID-19-20-0143, PHRC COVID-19-20-0029], Foundation for Medical Research (FRM), AP-HP Foundation, and the Reacting program).
Conflicts of interest: Roche and Sanofi donated TCZ and SARI.
|
Hospital based COVID-19 patients with moderate, severe pneumonia or critical pneumonia
Inclusion criteria:
Exclusion criteria:
TOCI-2 N total at baseline: N = 97 Intervention: 51 Control: 46
Important characteristics: Age, median (IQR): I: 63.2 y (59.4-70.9) C: 65.4 y (57.6-70.5)
Sex, n/N (%) male: I: 33/49 (67%) C: 33/43 (77%)
Disease severity, n/N (%) Defined by WHO-CPS score ≥7, n/N (%) I: 36/49 (74%)
SARI-2 N total at baseline: N = 91 Intervention: 50 Control: 41
Important characteristics: Age, median (IQR): I: 61.9 y (53.8-66.2) C: 61.2 y (55.3-68.5)
Sex, n/N (%) male: I: 36/48 (75%) C: 26/33 (79%)
Disease severity, n/N (%) Defined by WHO-CPS score ≥7, n/N (%) I: 32/48 (67%)
|
TOCI-2 Tocilizumab was administered intravenously (IV) at 8 mg kg−1 on day 1.
+
Usual care
SARI-2 Sarilumab was administrated IV at a fixed dose of 400 mg on day 1
+
Usual care |
Usual care: antibiotic agents, antiviral agents, corticosteroids, vasopressor support, anticoagulants) was provided at the discretion of the clinicians since, at that time, no standard of care (SOC) was defined, including the use of corticosteroids. |
Length of follow-up: 90 days
TOCI-2 Loss-to-follow-up or incomplete data: I: 2/51 (3.9%) Reason
C: 3/46 (6.5%) Reasons
No loss-to-follow up
SARI-2 Loss-to-follow-up or incomplete data: I: 2/50 (4.0%) Reason
C: 8/41 (19.5%) Reasons
No loss-to-follow up |
Clinical outcomes Mortality not reported
Overall survival at D14, estimate (95% CI) TOCI-2 I: 90% (82 to 99) C: 79% (68 to 92) HR: 0.37 (0.12 to 1.15) SARI-2 I: 75% (64 to 88) C: 73% (59 to 90) HR: 0.95 (0.40 to 2.25)
Overall survival at D28, estimate (95% CI) TOCI-2 I: 84% (74 to 95) C: 77% (65 to 90) HR: 0.56 (0.22 to 1.46) SARI-2 I: 71% (59 to 85) C: 67% (52 to 85) HR: 0.89 (0.40 to 1.96) Overall survival at D90, estimate (95% CI) TOCI-2 I: 76% (64 to 89) C: 70% (57 to 85) HR: 0.67 (0.30 to 1.49) SARI-2 I: 71% (59 to 85) C: 61% (46 to 80) HR: 0.74 (0.35 to 1.58)
Duration of hospitalization not reported
Discharge at D28, estimate (95%CI) TOCI-2 I: 55% (40 to 68) C: 42% (27 to 56) HR: 1.45 (0.80 to 2.63) SARI-2 I: 35% (22 to 49) C: 30% (16 to 46) HR: 1.21 (0.55 to 2.66)
Discharge at D90, estimate (95%CI) TOCI-2 I: 70% (54 to 82) C: 60% (44 to 74) HR: 1.35 (0.84 to 2.17) SARI-2 I: 65% (48 to 77) C: 52% (33 to 68) HR: 1.30 (0.71 to 2.37)
ICU discharge at D28, estimate (95%CI) TOCI-2 I: 72% (55 to 84) C: 60% (42 to 74) HR: 1.28 (0.73 to 2.24) SARI-2 I: 60% (43 to 74) C: 71% (50 to 85) HR: 0.78 (0.42 to 1.44)
ICU discharge at D90, estimate (95%CI) TOCI-2 I: 84% (66 to 93) C: 83% (63 to 93) HR: 1.15 (0.73 to 1.18) SARI-2 I: 79% (61 to 89) C: 82% (57 to 93) HR: 0.84 (0.49 to 1.47)
Time to symptom resolution not reported
No improvement in WHO score at D4, n/N (90% CI) TOCI-2 I: 35/49 (71%) C: 30/43 (70%) Median posterior absolute risk difference +1.7% (-13.6+17.1) SARI-2 I: 34/48 (71%) C: 26/33 (79%) Median posterior absolute risk difference -7.3% (-22.5 to +8.7)
WHO-CPS score, D2-D14 (longitudinal analysis) TOCI-2 aOR: 0.76 (0.27 to 2.13) SARI-2 aOR: 0.72 (0.21 to 2.41)
Extubation or removal of HIV at D14 (95%CI) TOCI-2 I: 47% (32 to 60) C: 42% (27 to 56) HR: 1.19 (0.71 to 2.04) SARI-2 I: 38% (24 to 51) C: 33% (18 to 50) HR: 1.05 (0.55 to 2.07)
Oxygen supply independency, estimate at D28 (95% CI) TOCI-2 I: 59% (44 to 72) C: 49% (33 to 63) HR: 1.44 (0.82 to 2.52) SARI-2 I: 44% (29 to 57) C: 36% (20 to 53) HR: 1.20 (0.59 to 2.44)
Oxygen supply independency, estimate at D90 (95% CI) TOCI-2 I: 69% (53 to 80) C: 64% (47 to 77) HR: 1.28 (0.80 to 2.03) SARI-2 I: 71% (52 to 83) C: 56% (35 to 68) HR: 1.29 (0.74 to 2.25)
Invasive respiratory support not reported
Non-invasive respiratory support not reported
Safety Serious adverse events Patients with at least one AE TOCI-2 I: 33/49 (67%) C: 30/43 (70%) SARI-2 I: 32/48 (68%) C: 22/33 (68%)
Virological outcomes Viral clearance not reported
|
Primary outcomes:
Secondary outcome(s):
Definitions:
Remarks: Results on secondary outcomes should be regarded as exploratory (stated by the authors)
Authors conclusion: In critically ill patients with COVID-19, anti-IL-6 Receptors did not significantly increase the number of patients alive without any NIV, MV by D14. |
|
Declercq (2021) |
See evidence table of Declercq (2021) by Anakinra.
|
||||||
|
Rosas, 2021b |
Type of study: Randomized, double-blind, placebo-controlled, multicenter, phase 3 trial.
Setting: Hospital, June 2020-March 2021
Country: Not stated.
Source of funding: This trial was supported by F. Hofmann-La Roche Ltd.
Conflicts of interest: IOR Grant from Roche/Genentech related to the submitted work and grant from Genentech and personal fees from Genentech, Boehringer, and Bristol Myers Squibb outside the submitted work. GD: Grants from Gilead Sciences, Regeneron Inc., Roche, Boehringer Ingelheim, and Edasa Biotech outside the submitted work. Gilead Medical Afairs sentinel panel and Scientifc Advisory Board for Safeology Inc. RLG: Personal fees from Eli Lilly, Gilead Sciences Inc., GlaxoSmithKline, Johnson and Johnson, and Roivant Sciences Inc. and non‑ fnancial support from Gilead Sciences Inc. outside the submitted work. SML: Investigator fees from Roche/Genentech during the conduct of the trial. PR: Nothing to disclose. BDH: Personal fees from Kite Pharma and Novartis outside the submitted work. AWC: Nothing to disclose. JSO: Institutional funding from Roche during conduct of the trial. NAH: Grants from GlaxoSmithKline, Sanof, AstraZeneca, Genentech, Boehringer Ingelheim, Novartis, and Gossamer Bio; personal fees from GlaxoSmithKline, Sanof, AstraZeneca, Genentech, Novartis, Regeneron, Teva, and Amgen outside the submitted work. AS: Personal fees from Genentech, AbbVie, Pharmacyclics, Janssen, Kite Pharma, Celgene, Ver‑ astem, BeiGene, Novartis, TG Therapeutics, Seattle Genetics, Morphosys, Jazz Pharmaceuticals, and Gilead Sciences and nonfnancial support from Bristol Myers Squibb outside the submitted work. JG-D: Nothing to disclose. IG: Nothing to disclose. JC: Grant provided to institution from Roche/Genentech during conduct of the trial. Grants and personal fees from Gilead Sciences Inc. outside the submitted work. OG: Employee of Roche and shareholder of Roche Holding AG. EG: Employee of Roche. NL-K: Employee of Roche/Genen‑ tech. LT: Employee of Roche/Genentech and has an unpublished patent pending, “Tocilizumab and Remdesivir Combination Therapy for COVID-19 Pneumonia.” KT: Former employee of Roche/Genentech and owns stock in Roche/Genentech. HC: Employee of Gilead Sciences Inc. DB: Former employee of and holds stock in Gilead Sciences Inc. JKO: Employee of Roche/Genentech.
|
Hospitalized COVID-19 patients
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 640 Intervention: N = 430 Control: N = 210
Important characteristics: Age, mean (SD): I: 60.1 y (13.3) C: 58.2 y (13.3)
Sex, n/N (%) male: I: 266/434/ (61.9%) C: 139/210 (66.2%)
Disease severity, mean (SD): Defined by ordinal scale for clinical status
3 (Non-ICU hospital ward or ready for hospital ward requiring supplemental oxygen I: 29/434 (6.7%) C: 13/210 (6.2%)
4 (ICU or non-ICU hospital ward, requiring noninvasive ventilation or high-fow oxygen) I: 336/434 (78.1%) C: 175 (210 (83.3%)
5 (ICU, requiring intubation and mechanical ventilation) I: 30/434 (9.1%) C: 9/210 (4.3%)
6 ICU, requiring extracorporeal membrane oxygenation or mechanical ventilation and additional organ support) I: 26/434 (6.0%) C: 13/210 (6.2%)
Groups comparable at baseline? Yes. |
Remdesivir plus tocilizumab
Remdesivir was administered intravenously, followed by a single intravenous dose of tocilizumab 8 mg/kg (maximum, 800 mg)
|
Remdesivir plus placebo
Remdesivir was administered intravenously, followed by a single intravenous dose of placebo 9 mg/kg (maximum, 800 mg).
|
Length of follow-up: 60 days.
Loss-to-follow-up: Intervention: N = 9
Control: N =4
Incomplete outcome data: Intervention: N = 98
Control: N = 50
|
Clinical outcomes Mortality at day 28, n/N (%) I: 78/430 (18.1%) (95% CI 14.5 to 21.8) C: 41/210 (19.5%) (95% CI 14.2 to 24.9) Weighted difference: -1.3 (-7.8 to 5.2) P=0.69
Mortality at day 60, n/N (%) I: 97/430 (22.6%) (95% CI 18.6 to 26.5) C: 54/210 (25.7%) (95% CI 19.8 to 31.6) Weighted difference -3 (-10.1 to 4) P=0.39
Time to death to day 28, median (IQR) I: non evaluable C: non evaluable P=0.79 HR 0.95 (95% CI 0.65 to 1.39)
Duration of hospitalization Time to hospital discharge or ready for discharge to day 28, median (IQR) I: 14 (12-15) days C: 14 (11-16) days P=0.74 HR 0.97 (95% CI 0.78 to 1.19)
Time to symptom resolution Clinical status at day 14 assessed on the 7-category ordinal scale, n/N (%)
1 I: 231/430 (54.0%) C: 110/210 (52.4%)
2 I: 11/430 (2.6%) C: 4/210 (1.9%)
3 I: 38/430 (8.9%) C: 24/210 (11.4%)
4 I: 41/430 (9.6%) C: 14/210 (6.7%)
5 I: 21/430 (4.9%) C: 14/210 (6.7%)
6 I: 43/430 (10%) C: 24/210 (11.4%)
7 I: 43/430 (10%) C: 20/210 (9.5%)
Respiratory support Time to mechanical ventilation or death to day 28, median (IQR) I: non evaluable C: non evaluable P=0.9 HR 0.98 (95% CI 0.72 to 1.34)
Safety Adverse events, N I: N = 1094 C: N = 530
Patients with 1 or more than 1 event, n/N (%) I: 320 (429 (74.6%) C: 147/213 (69.0%)
Patients with 1 or more than 1 serious adverse event, n/N (%) I: 128/429 (29.8%) C: 72/213 (33.8%)
Virological outcomes Viral clearance Not reported |
Definitions: -
Remarks: -
Authors conclusion: In this randomized, double-blind, placebo-controlled trial, tocilizumab plus remdesivir did not shorten time to hospital discharge or “ready for discharge” to day 28 compared with placebo plus remdesivir in patients with severe COVID-19 pneumonia, most of whom received systemic corticosteroids. Serious infections were not more frequent with tocilizumab treatment, and no new safety signals were identified.
|
|
Horby, 2021a
Recovery Collaborative Group |
Type of study: RCT (open-label), part of the RECOVERY trial.
Setting: 131 National Health Service Hospitals participating in the RECOVERY trial, enrolment between April 23, 2020 until Jan 24, 2021; This study was part of the RECOVERY trial, in which patients were randomized to one of the following groups: Part A: 542 dexamethasone, 557 lopinavir− ritonavir, 383 hydroxy-chloroquine, 2041 azithromycin, 3083 colchicine, 8107 usual care; Part B: 5285 convalescent plasma, 2416 REGN−COV2, 6301 usual care; Part C: 4450 aspirin, 4594 usual care; After 21 days, eligible patients were randomised to either tocilizumab + usual care versus usual care alone.
Country: United Kingdom
Source of funding: UK Research and Innovation, National Institute of Health Research, Roche (provision of tocilizumab and reviewed draft publication for factual accuracy on tocilizumab).
Conflicts of interest: The authors have no conflict of interest or financial relationships relevant to the submitted work.
|
Hospitalized, adult patients with clinical evidence of progressive COVID-19.
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 4116 Intervention: 2022 Control: 2094
Important characteristics: Age, mean (SD): I: 63.3 y (13.7) C: 63.9 y (13.6) Sex, n/N (%) male: I: 1337/2022 (66%) C: 1437/2094 (69%) Number of days since symptom onset, median (IQR): I: 9 (7–13) C: 10 (7–14) Number of days since hospitalisation, median (IQR): I: 2 (1–5) C: 2 (1–5)
Disease severity Respiratory support at second randomisation No ventilator support, n/N (%): Defined as patients not receiving any oxygen and patients receiving low-flow oxygen I: 935/2022 (46%) C: 933/2094 (45%) Non-invasive ventilation, n/N (%): Defined as patients receiving high-flow nasal oxygen, continuous positive airway pressure, or other non-invasive ventilation I: 819/2022 (41%) C: 867/2094 (41%) Invasive mechanical ventilation, n/N (%): Defined as patients receiving invasive mechanical ventilation or extracorporeal membranous oxygenation I: 268/2022 (13%) C: 294/2094 (14%)
Oxygen saturation, median (IQR): I: 94% (92–96) C: 94% (91–95)
Groups comparable at baseline? Yes |
Usual care + Tocilizumab
(A single intravenous infusion over 60 min was provided. The dose was established by bodyweight (800 mg if weight >90 kg; 600 mg if weight >65 and ≤90 kg; 400 mg if weight >40 and ≤65 kg; and 8 mg/kg if weight ≤40 kg). A second dose could be given 12–24 h later if the patient’s condition had not improved.)
|
Usual care |
Length of follow up: Until discharge, death or at day 28 after randomisation.
Loss to follow-up: I: 1964/2022 (97%) completed follow-up Reasons: not reported C: 2049/2094 (98%) completed follow-up Reasons: not reported
(for the outcome 28-day mortality 99% completed follow-up)
|
Clinical outcomes Mortality (28-30 day), n/N (%) 28-day mortality, n/N (%) I: 621/2022 (31%) C: 729/729 (35%) RR (95%CI): 0.85 (0.76–0.94) P= 0.0028 Subgroup analyses are available, based on e.g. days since symptom onset, corticosteroid treatment and respiratory support.
Duration of hospitalization Median time to discharge, days I: 19 C: >28 Discharged from hospital <28 days, n/N (%) I: 1150/2022 (57%) C: 1044/2094 (50%) RR (95%CI): 1.22 (1.12–1.33) P<0.0001
Time to symptom resolution Not reported
Respiratory support Receipt of invasive mechanical ventilation or death Invasive mechanical ventilation, n/N (%) I: 265/1754 (15%) C: 343/1800 (19%) RR (95%CI): 0.79 (0.69–0.92) P= 0.0019
Death, n/N (%) I: 490/1754 (28%) C: 580/1800 (32%) RR (95%CI): 0.87 (0.78–0.96) P=0.0055
Receipt of ventilation Non-invasive ventilation, n/N (%) I: 281/935 (30% C: 309/933 (33%) RR (95%CI): 0.91 (0.79–1.04) P= 0.15 Invasive mechanical ventilation, n/N (%) I: 67/935 (7%) C: 86/933 (9%) RR (95%CI): 0.78 (0.57–1.06) P= 0.11
Successful cessation of invasive mechanical ventilation, n/N (%) I: 95/268 (35%) C: 98/294 (33%) RR (95%CI): 1.08 (0.81–1.43) P=0.60
Safety Adverse events Any major cardiac arrhythmia I: 108/2022 (6%) C: 133/2094 (7%)
Authors stated that there were three reports of serious adverse reactions believed to be related to tocilizumab: one each of otitis externa, Staphylococcus aureus bacteraemia, and lung abscess, all of which resolved with standard treatment.
Virological outcomes Viral clearance Not reported
Also reported: Use of haemodialysis or Hemofiltration
|
Definitions: Receipt of invasive mechanical ventilation or death/receipt of ventilation included only those on no ventilator support or non-invasive ventilation at second randomisation (of tocilizumab). Successful cessation of invasive mechanical ventilation included only those on invasive mechanical ventilation at second randomisation
Remarks: 16% of patients in the tocilizumab group reportedly did not receive treatment and reasons were not recorded. Furthermore, 77 patients in the usual care group did receive tocilizumab.
There is high risk of bias, since participants and local study staff were not blinded.
Roche Products supported the study through the supply of tocilizumab and reviewed the draft publication for factual accuracy relating to tocilizumab.
Authors conclusion: The RECOVERY trial has shown that for patients hospitalised with severe COVID-19, treatment with tocilizumab reduces mortality, increases the chances of successful hospital discharge, and reduces the chances of requiring invasive mechanical ventilation. These benefits are additional to those previously reported for dexamethasone. These findings require an update to clinical guidelines, which has already begun, and efforts to increase the global availability and affordability of tocilizumab.
|
|
Wang, 2021 |
Type of study: open-label multicenter RCT
Setting hospital-based, between February 13 and March 13, 2020
Country: 6 hospitals in Anhui and Hubei, China
Source of funding: This work was supported by the Department of Science and Technology of Anhui Province and Health Commission of Anhui Province (No. 202004a07020001) and the China National Center for Biotechnology Development (No. 2020YFC0843800). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflicts of interest: The authors declare that they have no conflicts of interest. |
patients with moderate or severe COVID-19
Inclusion criteria:
Exclusion criteria:
N total at baseline: Randomized: N = 65
Intervention: N = 33 Control: N = 32
One patient in the control group, who worsened severely 3 days after randomization, was crossed over to the tocilizumab group in accordance with the study protocol. However, this patient was erroneously included in the intervention group for the ITT analyses.
Important characteristics: Age, median (IQR): I: 63.5 y (58-71) C: 63 y (54-69)
Sex, n/N (%) male: I: 18/34 (52.94%) C: 15/31 (48.39%)
Groups were comparable at baseline. |
tocilizumab + standard care
(The first dose of tocilizumab was 400 mg, diluted in 100 mL of 0.9% saline, and administered intravenously for more than 1 h. A second dose was given if a patient remained febrile for 24 h after the first dose.) |
standard care |
Length of follow-up: Not specified.
Loss-to-follow-up: None.
Incomplete outcome data: None. |
Subgroup analyses were performed for patients with moderate or severe COVID-19 for primary and secondary outcomes. The primary outcome was the cure rate.
Clinical outcomes Mortality Not reported.
Cure rate** I: 32/34 (94.14%) C: 27/31 (87.10%) rate difference 7,02 (95%CI: -7.19-21.23)
Recovery rate of hypoxia at day 14 I: 22/24 (91.67%) C: 12/20 (60.00%) rate difference 31.67 (95%CI: 7.52-55.82)
Worsening rate of hypoxia during hospitalization Not reported.
Duration of hospitalization Median (IQR) I: 26 (17-27) C: 24 (15-28) median difference 2 (95%CI: -4-2)
Time to symptom resolution Not reported.
Respiratory support Not reported.
Safety Serious adverse events I: 0/34 (0%) C: 1/31 (3.23%)
Non-serious adverse events I: 20/34 (58.82%) C: 4/31 (12.90%)
Also reported are specific treatment-related adverse events.
Virological outcomes Viral clearance (i.e. time to negative virus load) Median (IQR) I: 17 (12-20) C: 16 (12-21.5) median difference 1.5 (95%CI: -4-5) |
Definitions: * The diagnosis of moderate or severe disease was defined according to the Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia (5th or updated version) as follows: moderate disease, fever or other respiratory symptoms, bilateral pulmonary lesions confirmed by chest imaging; severe disease was defined if any of the following conditions was met: (1) respiratory rate ≥ 30 breaths/min; (2) SpO2 ≤ 93% while breathing room air; (3) PaO2/FiO2 ≤ 300 mmHg.
**The definition for cure followed the standard given by the Diagnosis and Treatment Protocol for Novel Coronavirus Pneumonia (5th or updated version): (1) fever attenuated continuously for 7 days, (2) twice negative SARS-CoV-2 nucleic acid detections, (3) CT scan demonstrating chest effusion improved more than 50%. when the patient is discharged from the hospital.
Remarks: The authors did not enrol enough eligible patients in accordance with the previously designed protocol. The time between the onset of disease and randomization was relatively long in some patients.
Authors conclusion: This study showed that tocilizumab treatment is not associated with a significantly higher cure rate among COVID-19 patients. However, it can improve oxygenation, symptoms, and reduce disease worsening with an acceptable side effect profile. Tocilizumab had no significant influence on the time needed for negative viral load. For COVID-19 patients with bilateral pulmonary lesions and elevated IL-6 levels, tocilizumab is recommended for better disease management. |
|
Soin, 2020
COVINTOC trial |
Type of study: RCT; open-label, multicentre
Setting: Public and private hospitals; recruited between May 30, 2020, and Aug 31, 2020.
Country: India
Source of funding: Medanta Institute of Education and Research, Roche India, Cipla India, and Action COVID-19 India; The funder of the study had a role in study design, data collection, data analysis, data interpretation, and writing of the report.
|
Hospitalized COVID-19 patients with moderate to severe illness
Inclusion criteria:
severe: respiratory rate ≥30 per min or SpO2 <90% in ambient air, or ARDS or septic shock)
Exclusion criteria:
N total at baseline: N = 180 Randomized: 90 vs. 90 Analyzed: Intervention: 91 Control: 88
Important characteristics: Age, median (IQR): I: 56 (47–63) y C: 54 (43–63) y Sex, n/N (%) male: I: 76/91 (84%) C: 76 (86%) Disease severity Moderate I: 41 (45%) C: 47 (53%) Severe I: 50 (55%) C: 41 (47%) Respiratory support Supplemental oxygen I: 81 (89%) C: 80 (91%) Non-invasive bilevel positive airway pressure ventilation I: 28 (31%) C: 20 (23%) Mechanical ventilation I: 5 (5%) C: 4 (5%) Intensive care unit I: 64 (70%) C: 54 (61%)
Groups comparable at baseline. |
Tocilizumab
Tocilizumab 6 mg/kg plus standard care
Tocilizumab was administered as a single intravenous infusion at 6 mg/kg up to a maximum dose of 480 mg. An additional dose of 6 mg/kg (max 480 mg/kg) could be administered if clinical symptoms worsened or did not show improvement within 12 h to 7 days after administration of the first dose. The dosing regimen was selected on the basis of the cost and supply considerations in India and because a single dose between 4 mg/kg and 8 mg/kg plus an additional dose to a maximum of 800 mg, if required, has been recommended on the basis of initial reports on the use of tocilizumab in the treatment of COVID-19 in China [ref 21].
|
Standard care |
Length of follow up: 28 days
Loss to follow-up: I: 16/91 (17.6%) (died, n=11; withdrawn consent, n=2; withdrew because of no efficacy, n=2; not willing to visit hospital, n=1)
C: 20/88 (22.7%) (died, n=15; withdrew consent, n=3, discharged against medical advice, n=1; chose to receive tocilizumab, n=1)
143 (79%) of 180 patients completed 28 days of follow-up, 75 (82%) in the tocilizumab group and 68 (76%) in the standard care group |
Clinical outcomes Mortality Day 7 I: 2 (2%) C: 2 (2%) Diff –0·1 (–4·4 to 4·3), p= 0·97 Day 14 I: 8 (9%) C: 9 (10%) Diff –1·4 (–10·0 to 7·2), p= 0·74 Day 21 I: 10 (11%) C: 14 (16%) Diff –4·9 (–14·9 to 5·1), p= 0·33 Day 28 I: 11 (12%) C: 15 (17%) Diff –5·0 (–15·3 to 5·4), p= 0·35
Duration of hospitalization ICU admission I: 71 (78%) C: 64 (73%) Diff 5·3 (–7·3 to 17·9), p= 0·41
Duration of ICU stay, days Mean (SD) I: 8·2 (6·2) C: 8·4 (6·5), p= 0·91 Median (IQR) I: 7·0 (3·0 to 10·0) C: 6·0 (3·5 to 11·0)
Symptom resolution Progression of COVID-19, day 14; defined as progression from moderate to severe or from severe to death up to I: 8 (9%) C: 11 (13%)
At least a one-grade Improvement in cytokine release syndrome up to day 28 [American Society for Transplantation and Cellular Therapy cytokine release syndrome grade ref 22] I: 58 (64%) C: 59 (67%) diff –3·3 (–17·9 to 11·3), p= 0·64
Time to clinical improvement (defined as National Early Warning Score 2 [NEWS2] ≤2 maintained for 24 h)
Organ failure-free days Mean (SD) I: 24·6 (9·2) C: 23·2 (10·6), p= 0·35 Median (IQR) I: 28·0 (28·0 to 28·0) C: 28·0 (28·0 to 28·0)
Time to clinical improvement, median time (days) based on NEWS2 score I: 9·0 (95% CI 8·0–21·0) C: 8·0 (95% CI 6·0–10·0) Log rank p=0·43 Based on COVID-19 grade I: 7·0 (95% CI 5·0–8·0) C: 7·0 (95% CI 5·0–9·0) Log rank p=0·93
Need for respiratory support Ventilator-free days Mean (SD) I: 24·3 (9·2) C: 23·2 (10·6), p= 0·45 Median (IQR) I: 28·0 (28·0 to 28·0) C: 28·0 (28·0 to 28·0)
Duration of supplemental oxygen-free days Mean (SD) I: 17·1 (9·4) C: 18·3 (9·9), p= 0·41 Median (IQR) I: 20·0 (12·0 to 24·0) C: 22·0 (16·0 to 25·0)
Incidence of mechanical ventilation up to day 28 I: 14 (15%) C: 13 (15%) Diff 0·6 (–9·9 to 11·1), p= 0·91
Safety Adverse events, 28 days Adverse events, patients with at least one event I: 33 (36%) patients, 54 events C: 22 (25%) patients, 55 events Infections I: 6 (7%) C: 5 (6%) Serious adverse events* I: 18 (20%) patients, 23 events C: 15 (17%) patients, 24 events Deaths I: 13 (14%) C: 15 (17%) Grade 3 or worse adverse events I: 2 (2%) C: 5 (6%)
Virological outcomes Viral clearance time to negative result on RT-PCR
Also reported: Serum concentrations of IL-6, ferritin, and C-reactive protein (CRP), requirement for renal replacement therapy |
Authors conclusion: Routine use of tocilizumab in patients admitted to hospital with moderate to severe COVID-19 is not supported. However, post-hoc evidence from this study suggests tocilizumab might still be effective in patients with severe COVID-19 and so should be investigated further in future studies.
|
|
Rosas, 2021a |
Type of study: A phase 3, international, randomized, double-blind placebo-controlled trial
Setting: 62 hospitals
Country: Nine countries in Europe and North America (Canada, Denmark, France, Germany, Italy, the Netherlands, the United Kingdom, and the United States).
Source of funding: Supported F. Hoffmann–La Roche and by a grant (HHSO100201800036C) from the Biomedical Advanced Research and Development Authority of the Office of the Assistant Secretary for Preparedness and Response, Department of Health and Human Services. Drs. Cooper and Youngstein were supported in part by the Biomedical Research Centre of the United Kingdom National Institute for Health Research. Dr. Malhotra is supported by the National Institutes of Health.
|
hospitalized patients with severe COVID-19 pneumonia.
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 438 Intervention: 294 Control: 144
Important characteristics: Age, mean (SD): I: 60.9 y (14.6) C: 60.6 y (13.7)
Distribution (age) n/N (%): 18-64: I: 163/297 (55.4%) C: 81/144 (56.2%)
65-84: I: 117/294 (39.8%) C: 60/144 (41.7%)
>84: I: 14/294 (4.8%) C: 3/144 (2.1%)
Sex, n/N (%) male: I: 205/294 (69.7%) C: 101/144 (70.1%)
Illness severity on National Early Warming Score 2, mean (SD): I: 7.1 (3.0) C: 7.0 (3.0)
Ordinal scale for clinical status, n/N (%): 2 I: 9/294 (3.1%) C: 6/144 (4.2%)
3 I: 78/297 (26.5%) C: 44/144 (30.6%)
4 I: 94/294 (32.0%) C: 39/144 (27.1%)
5: I: 45/294 (15.3%) C: 15/144 (10.4%)
6 I: 68/294 (23.1%) C: 40/144 (27.8%)
Groups comparable at baseline? |
Single intravenous infusion of tociluzumab at a dose of 8 mg per kilogram of bodyweight, with a maximum dose of 800 mg plus standard care.
|
Placebo plus standard care |
Length of follow up: The primary analysis was performed at day 28, and the final trial visit occurred at day 60.
Loss to follow-up: Intervention group: - N= 70 (24%) discontinued trial on or before day 28; - N=57 (19%) died; - N=7 (2%) were lost to follow-up; - N=6 (2%) withdrew)
The 28 day follow-up evaluation completed in 224/294 (76.2%) patients.
Control group: - N=36 discontinued trial on or before day 28; - N=27 (19%) died; - N=5 (3%) were lost to follow-up; - N=2 (1%) withdrew; - N=2 (1%) were withdrawn by physician.
The 28 day follow-up evaluation completed in 108/144 (75%) patients.
|
Clinical outcomes Mortality at day 28, n/N (%): I: 58/294 (19.7%) C: 28/144 (19.4%) Difference: 0.3 (95% CI= -7.6 to 8.2) P=0.94
Clinical status on 7-category ordinal scale at day 28, median value (95% CI) I: 1.0 (95% CI= 1.0 to 1.0) C: 2.0 (95% CI= 1.0 to 4.0) Difference: -1.0 (95% CI= -2.5 to 0.00) P=0.31
Clinical status on 7-category ordinal scale at day 14, median value (95% CI) I: 3.0 (95% CI= 2.0 to 4.0) C: 4.0 (95% CI=. 3.0 to 5.0) Difference: -1.0 (95% CI= -2.0 to 0.5)
Days until hospital discharge or readiness for discharge, median number (95% C) I: 20.0 (95% CI= 17.0 to 27.0) C: 28.0 (95% CI= 20.0 to ‘not evaluable’)
Symptom resolution – days until improvement by ≥2 categories on 7-category ordinal scale in clinical status, median number of days (95% CI) I: 14.0 (95% CI= 12.0 to 17.0) C: 18.0 (95% CI= 15.0 to 28.0) HR= 1.26 (95% CI= 0.97 to 1.64)
Day in ICU, median number of days (95% CI) I: 9.8 (95% CI= 7.0 to 15.7) C: 15.5 (95% CI= 8.7 to 25.5) Difference: -5.8 (95% CI= -15.0 to 2.9)
Incidence of ICU stay among patients not in ICU at baseline, n/N (%) I: 27/127 (21.3%) C: 23/64 (35.9%) Difference: -14.8 (95% CI= -28.6 to -1.0)
Ventilator-free days at day 28, median number (95% CI) I: 22.0 (95% CI= 18.0 to 28.0) C: 16.5 (95% CI= 11.0 to 26.0) Difference: 5.5 (95% CI= -2.8 to 13.0)
Subgroup analysis
Incidence of mechanical ventilation among patients not receiving mechanical ventilation at randomization, n/N (%) I: 51/183 (27.9%) C: 33/90 (36.7%) Difference: -8.9% (95% CI= -20.7 to 3.0)
Clinical failure among patients not receiving mechanical ventilation at randomization, n/N (%) I: 53/183 (29.0%) C: 38/90 (42.2%) HR= 0.61 (95% CI= 0.40 to 0.94)
Clinical status on the ordinal scale at day 28 among patients who were receiving mechanical ventilation at randomization, median value (95% CI) I: 5.0 (95% CI= 3.0 to 5.0) C: 5.0 (95% CI= 4.0 to 6.0)
Clinical status on the ordinal scale at day 28 among patients who were not receiving mechanical ventilation at randomization, median value (95% CI) I: 1.0 (95% CI= 1.0 to 1.0) C: 1.0 (95% CI= 1.0 to 1.0)
Safety population I: N = 295 C: N = 143
Any adverse event Patients with ≥1 event, N (%) I: 228/295 (77.3%) C: 116 (81.1)
Total number of events, N I: N = 778 C: N = 360
Any serious adverse event Patients with ≥1 event, N (%) I: 103/295 (34.9%) C: 55/143 (38.5%)
Total number of events, N I: N = 160 C: N = 101
Death N (%) I: 58/295 (19.7%) C: 28/143 (19.6)
Patients with adverse events of special interest, n/N (%) Infection I: 113/295 (38.3%) C: 58/143 (40.6%)
Serious infection I: 62/295 (21.0%) C: 37/143 (25.9%)
Opportunistic infection I: 1/295 (0..3%) C: 1/143 (0.7%)
Virological outcomes Not reported |
Definitions: - National Early Warning Score 2: a standardized assessment for identifying acutely ill patients on the basis of respiration rage, oxygen saturation, systolic blood pressure, pulse rate, level of consciousness, and temperature. Values on this instrument range from 0 to 20, with higher scores indicating greater clinical risk.
- Standard of care: according to local practice (anti-viral treatment, low-dose glucocorticoids, convalescent plasma, and supportive care).
- Opportunistic infections were reported in one patient with candida sepsis in the intervention group and in one patient with respiratory moniliasis in the control group.
Remarks: -
Authors conclusion: Results of this trial must be interpreted in the context of therapies for severe Covid-19 pneumonia. Among the treatments for hospitalized patients with Covid-19 that have been investigated in randomized, controlled trials, dexamethasone was found to reduce mortality only among patients who were receiving mechanical ventilation or supplemental oxygen at randomization.28 Remdesivir shortened the time until recovery but did not have a significant effect on 14-day mortality.27 Clinical trials are under way to investigate many potential treatments, including other antiviral and antiinflammatory drugs, other targeted immunomodulators (e.g., sarilumab, anakinra, baricitinib, and canakinumab), anticoagulants, and antifibrotics (tyrosine kinase inhibitors).29 However, the need for effective treatments for patients with severe Covid-19 pneumonia continues to be a major challenge at this point in the pandemic
|
|
The REMAP-CAP Investigators, 2020 |
See the evidence table of the REMAP-CAP Investigators (2020) by sarilumab.
|
||||||
|
Veiga, 2021 |
Type of study: RCT (open label trial) Setting: Nine hospitals in Brazil, 8 May to 17 July 2020. Follow-up for the last patient was completed on August 11, 2020.
Country: Brazil Source of funding: Trial was funded by the hospitals and research institutes participating in the Coalition covid-19 Brazil. Exploratory laboratory analysis was conducted and funded by Fleury Laboratory in São Paulo, Brazil. Instituto Votorantim provided a donation for tocilizumab. It has no role in the design of the trial, the conduct, the analysis, or the decision to submit the manuscript for publication.
|
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 129 patients Intervention: 65 patients Control: 64 patients (two patients received tocilizumab at discretion of the doctor)
Important characteristics: Age, mean (SD): I: 57.4 years (15.7) C: 57.3 years (13.5)
I: 44/65 (68%) C: 44/64 (69%)
Based on a seven level ordinal clinical status scale (see comments) I: C:
Groups comparable at baseline? Not for respiratory support, baseline clinical status and use of azithromycin. |
Tocilizumab (a single intravenous infusion at a dose of 8 mg/kg (maximum 800 mg) plus standard care. The concomitant use of hydroxychloroquine, azithromycin, corticosteroids, and antibiotics was allowed according to standard care per local institutional guidelines for patients with covid-19. Remdesivir was not available in Brazil. |
Standard care |
Length of follow up: 15 day follow-up
Loss to follow-up: “The 15 day follow-up was completed for all patients.” However, it is unclear if the 28/29 days follow-up also was completed for all patients. |
Clinical outcomes Mortality at day 15:
Duration of hospitalization (days, mean (SD)):
Clinical status (6 level ordinal scale) at day 8:
|
Definitions: Seven level ordinal clinical status scale: 2 – not admitted to hospital but with limitation in activities
Remarks: - Groups were not comparable at baseline for respiratory support, baseline clinical status and use of Azithromycin. - “The 15 day follow-up was completed for all patients.” However, it is unclear if the 28/29 days follow-up also was completed for all patients.
Authors conclusion: In patients with severe or critical covid-19 tocilizumab plus standard care was not superior to standard care alone in improving clinical outcomes at 15 days, and might increase mortality.
|
|
Salama, 2020
EMPACTA trial |
Type of study: RCT; double-blind, placebo-controlled, phase 3 trial
Setting: EMPACTA trial; multi-center
Country: International; United States 45 sites, Mexico 2 sites, Kenya 2 sites, South Africa 3 sites, Peru 5 sites, and Brazil 6 sites
Source of funding: Funded by Genentech; EMPACTA ClinicalTrials.gov number, NCT04372186. “The sponsor designed the trial, conducted it according to the protocol, collected the data, and performed analyses; a contract research organization paid by the sponsor managed and monitored the trial under the direction and supervision of the sponsor.”
|
Hospitalized patients with COVID-19 pneumonia
Inclusion criteria:
Exclusion criteria:
positive airway pressure, bilevel positive airway pressure, or mechanical ventilation
within 24 hours, as determined by treating physician active tuberculosis or
(other than SARS-CoV-2 infection or well controlled HIV infection)
N total at baseline: N = 389 Modified intention-to-treat: Intervention: 249 (259 assigned to this arm; 10 dropped out) Control: 128 (129 assigned to this arm, 1 dropped out)
Important characteristics: Age, mean±SD: I: 56.0±14.3 C: 55.6±14.9 Sex, n/N (%) male: I: 150/249 (60.2%) C: 73/128 (57.0%) BMI, mean±SD: I: 32.0±7.9 C: 33.1±7.2 Days from first Covid-19 symptom at baseline, median (range) I: 8.0 (0.0-31.0; n=248) C: 8.0 (0.0-36.0, n=127) Disease severity, mean (SD): Defined by ordinal scale; see right column for definitions I: 2: 24/249 (9.6%) 3: 161/249 (64.7%) 4: 64/249 (25.7%) C: 2: 11/128 (8.6%) 3: 81/128 (63.3%) 4: 36/128 (28.1%) ICU admission at baseline, no. (%) I: 36 (14.5%) C: 22 (17.2%)
Groups comparable at baseline.
Other drugs - within 7 days prior to first dose study drug or during study Systemic corticosteroid I: 200/249 (80.3%) C: 112/128 (87.5%) Antiviral I: 196/249 (78.7%) C: 101/128 (78.9%) Dexamethasone I: 138/249 (55.4%) C: 86/128 (67.2%) Remdesivir I: 131/249 (52.6%) C: 75/128 (58.6%)
|
Tocilizumab + standard care
Tocilizumab: one or two doses of intravenous tocilizumab (8 mg per kilogram of body weight, to a maximum of 800 mg per dose) |
Standard care + placebo
Standard care: according to local practice, which could include antiviral treatment, the limited use of systemic glucocorticoids (recommended dose, ≤1 mg per kilogram of body weight of methylprednisolone or equivalent), and supportive care.
Placebo: one or two doses of placebo
|
Length of follow up: 60 days; “Efficacy was evaluated by day 28, and patients were followed for a total of 60 days. Patients who were discharged before day 28 were considered to have completed the trial and were followed weekly up to day 28, with a safety follow-up visit conducted by day 60.”
Loss to follow-up: I: 10/259 (3.9 %) Reasons: withdrew, n = 8; withdrawn by physician, n = 1; site unable to confirm administration of study drug, n = 1.
C: 1/129 (0.8%) Reasons: withdraw, n = 1. |
Clinical outcomes
Mortality, day 28: I: 26/ (10.4 [7.2 to 14.9]) C: 11 (8.6 [4.9 to 14.7]) HR 1.04 (95% CI 0.51 to 2.12) Weighted difference, percentage points: 2.0 (95% CI −5.2 to 7.8)
Mechanical ventilation (invasive mechanical ventilation or ECMO) or death by day 28: I: 12.0% (95% CI 8.5 to 16.9) C: 19.3% (95% CI 13.3 to 27.4) HR 0.56 (0.33 to 0.97) Sub group - age ≤60: I: 9/151 events C: 7/76 events HR 0.63 (95% CI 0.23 – 1.71) Subgroup - age >60: I: 20/98 events C: 17/52 events HR 0.53 (95% CI 0.28 to 1.03) Time to hospital discharge or readiness for discharge, day 28; score of 1 on ordinal scale I: 6.0 (6.0 to 7.0) days HR 1.16 (0.91 to 1.48)
Time to clinical improvement, day 28; at least a two-category improvement in clinical status relative to baseline score (for category 2 at baseline, clinical status of category 1 was considered to have met threshold), median: I: 6.0 (6.0 to 7.0) days C: 7.0 (6.0 to 9.0) days HR 1.15 (95% CI 0.90 to 1.48)
Time to clinical failure, day 28; time to death, mechanical ventilation, admission to ICU, or, in patients who were already in ICU at enrollment, worsening by two categories from baseline on the seven-category ordinal scale], or withdrawal [whichever occurred first]); median: HR 0.55 (95% CI 0.33 to 0.93)
Safety Incidence and severity of adverse events [according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0] Total adverse events I: 357 C: 187 Patients with ≥1 adverse event, no. (%) I: 127/250 (50.8%) C: 67/127 (52.8%) Serious event I: 38/250 (15.2%) C: 25/127 (19.7%) of which events related to tocilizumab or placebo, as determined by the investigator I: 3/250 (1.2%) C: 0/127 (0.0%)
|
Definitions: Clinical status - 7-category ordinal scale 1 - discharged (or ready for discharge as evidenced by normal body temp. and respiratory rate + stable oxygen saturation while breathing ambient air or ≤2 liters of suppl. oxygen); 2, hospitalized in non–intensive care unit (ICU) hospital ward (or ready for a hospital ward) and not receiving suppl. oxygen; 3, hospitalized in non–ICU hospital ward (or ready for hospital ward) and receiving suppl. oxygen; 4, hospitalized in ICU or non–ICU hospital ward and receiving noninvasive ventilation or high-flow oxygen; 5, hospitalized in ICU and receiving intubation and mechanical ventilation; 6, hospitalized in ICU and receiving ECMO or mechanical ventilation and additional organ support; 7, died.
Remarks: -
Authors conclusion: In hospitalized patients with Covid-19 pneumonia who were not receiving mechanical ventilation, tocilizumab reduced the likelihood of progression to the composite outcome of mechanical ventilation or death, but it did not improve survival. No new safety signals were identified.
|
|
Stone, 2020 |
Type of study:
Setting: Hospital
Country: Boston, USA
Source of funding: Not mentioned
|
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = Intervention: Control:
Important characteristics: Age, mean (SD): I: C: P=0 Sex, n/N (%) male: I: C:
Groups comparable at baseline? |
Standard care plus a single dose of either tocilizumab (8 mg per kilogram of body weight administered intravenously, not to exceed 800 mg)
|
Placebo |
28 days
|
TIME-TO-EVENT OUTCOMES IN THE MODIFIED INTENTION-TO-TREAT POPULATION
Mechanical ventilation or death
N patients with event within 28 days I: N = 17 C: N = 10
Percentage of patients with event (95 CI) at day 14 I: 9.9 (95% CI= 6.2 to 15.7) C: 10.0 (95% CI= 5.1 to 18.9)
Percentage of patients with event (95 CI) at day 28 I: 10.6 (95% CI= 6.7 to 16.6) C: 12.5 (95% CI= 6.9 to 22.0)
Hazard ratio (95% CI) HR= 0.83 (95% CI= 0.38 to 1.81)
Log rank value 0.64
Clinical worsening on ordinal scale
N patients with event within 28 days I: N = 31 C: N = 14
Percentage of patients with event (95 CI) at day 14 I: 18.0 (95% CI= 12.9 to 24.9) C: 14.9 (95% CI= 8.7 to 24.7)
Percentage of patients with event (95 CI) at day 28 I: 19.3 (95% CI= 14.0 to 26.2) C: 17.4 (95% CI= 10.7 to 27.7)
Hazard ratio (95% CI) HR= 1.11 (95% CI= 0.59 to 2.10)
Log rank value 0.73
Mechanical ventilation
N patients with event within 28 days I: N = 11 C: N = 8
Percentage of patients with event (95 CI) at day 14 I: 6.8 (95% CI= 3.6 to 11.4) C: 10.0 (95% CI= 4.6 to 17.7)
Percentage of patients with event (95 CI) at day 28 I: 6.8 (95% CI= 3.6 to 11.4) C: 10.0 (95% CI= 4.6 to 17.7)
Hazard ratio (95% CI) HR= 0.65 (95% CI= 0.26 to 1.62)
Death
N patients with event within 28 days I: N = 9 C: N = 3
Percentage of patients with event (95 CI) at day 14 I: 4.4 (95% CI= 2.1 to 8.9) C: 1.3 (95% CI= 0.2 to 8.7)
Percentage of patients with event (95 CI) at day 28 I: 5.6 (95% CI= 3.0 to 10.5) C: 3.8 (95% CI= 1.2 to 11.3)
Hazard ratio (95% CI) HR= 1.52 (95% CI= 0.41 to 5.61)
Discontinuation of supplemental oxygen among patients receiving it at baseline
N patients with event within 28 days I: N = 114 C: N = 56
Percentage of patients with event (95 CI) at day 14 I: 75.4 (95% CI= 67.9 to 82.2) C: 78.8 (95% CI= 68.3 to 87.7)
Percentage of patients with event (95 CI) at day 28 I: 82.6 (95% CI= 75.9 to 88.4) C: 84.9 (95% CI= 75.2 to 92.2)
Median N of days to event (95% CI) I: 5.0 (95% CI= 3.8 to 7.6) C: 4.9 (95% CI= 3.8 to 7.8)
Hazard ratio (95% CI) HR= 0.94 (95% CI= 0.67 to 1.30)
Log rank value 0.69
Clinical improvement on ordinal scale
N patients with event within 28 days I: N = 147 C: N = 72
Percentage of patients with event (95 CI) at day 14 I: 86.3 (95% CI= 80.6 to 91.1) C: 81.5 (95% CI= 72.4 to 89.0)
Percentage of patients with event (95 CI) at day 28 I: 91.3 (95% CI= 86.3 to 95.1) C: 88.9 (95% CI= 81.0 to 94.5)
Median N of days to event (95% CI) I: 6.0 (95% CI= 5.0 to 6.0) C: 5.0 (95% CI= 4.0 to 7.0)
Hazard ratio (95% CI) HR= 1.06 (95% CI= 0.80 to 1.41)
Initial discharge
N patients with event within 28 days I: N = 147 C: N = 72
Percentage of patients with event (95 CI) at day 14 I: 86.3 (95% CI= 80.6 to 91.1) C: 81.5 (95% CI= 72.4 to 89.0)
Percentage of patients with event (95 CI) at day 28 I: 91.3 (95% CI= 86.3 to 95.0) C: 88.9 (95% CI= 81.0 to 94.5)
Median N of days to event (95% CI) I: 6.0 (95% CI= 4.0 to 7.0) C: 6.0 (95% CI= 5.0 to 6.0)
Hazard ratio (95% CI) HR= 1.08 (95% CI= 0.81 to 1.43)
DURATION OUTCOMES AND ADMISSION TO THE ICU OR DEATH IN THE MODIFIED INTENTION-TO-TREAT POPULATION
Median duration of receipt of supplemental oxygen (IQR) in days I: 4.0 (1.8 to 11.6) C: 3.9 (1.1 to 9.2)
Median duration of mechanical ventilation (IQR) in days I: 15.0 (12.6 to NR) C: 27.9 (16.3 to NR)
Admission to ICU or death (%) I: 15.9% C: 15.8% Relative risk: 0.97 (0.50 to 1.88)
|
Remarks: -
Authors conclusion: In this randomized, double-blind, placebocontrolled trial, we did not find any efficacy of interleukin-6 receptor blockade for the treatment of hospitalized patients with Covid-19.
|
|
Salvarani, 2020 |
Type of study: RCT (prospective, open-label, randomized clinical trial)
Setting: 24 hospitals; March 31 - June 11, 2020
Country: Italy
Source of funding: “The work at the IRCCS Sacro Cuore Don Calabria Hospital was partly funded by the Italian Ministry of Health “Fondi Ricerca Corrente – Linea 1, progetto 4”. Roche provided the drug and its distribution to the centers.”
|
Inclusion criteria:
Exclusion criteria:
Patients at enrollment were allowed to receive oxygen therapy with Venturi mask or high-flow nasal cannula with recorded and preset Fio2, but not invasive or noninvasive mechanical ventilation. After randomization, patients were allowed to receive supplemental oxygen therapy, including noninvasive ventilation, according to clinical needs.
N total at baseline: N = 126 Intervention: 60 Control: 66
Important characteristics: Age, median (IQR): I: 61.5y (51.5-73.5) C: 60.0y (54.0-69.0) Sex, n/N (%) male: I: 40/60 (66.7%) C: 37/66 (56.1%)
Control group had lower CRP, IL-6, ferritin, and D-dimer levels and were more frequently treated with antivirals compared to intervention group. |
Tocilizumab + supportive care
Tocilizumab intravenously within 8 hours from randomization at a dose of 8 mg/kg up to a maximum of 800 mg, followed by a second dose after 12 hours.
|
Supportive care
Supportive care: following the treatment protocols of each center:
12/66 received 2 doses of tocilizumab IV after clinical worsening |
Length of follow-up: 30 days (primary endpoint 14 days)
Loss to follow-up: 3/66 (4.5%) patients in the control group withdrew consent and were excluded from the analysis. Other patients were all included in the efficacy analysis.
|
Clinical worsening at 14 days; Definition: see information at the right column I: 17/60 (28.3%) C: 17/63 (27.0%) Rate ratio 1.05 (0.59 – 1.86) p=.87
Admission to ICU with mechanical ventilation 14 days I: 6/60 (10%) C: 5/63 (7.9%) Rate ratio 1.26 (0.41 – 3.91) 30 days I: 6/60 (10%) C: 5/63 (7.9%) Rate ratio 1.26 (0.41 – 3.91)
Death from any cause 14 days I: 1/60 (1.7%) C: 1/63 (1.6%) Rate ratio 1.05 (0.07 – 16.4) 30 days I: 2/60 (3.3%) C: 1/63 (1.6%) Rate ratio 2.10 (0.20 – 22.6)
Discharge from hospital 14 days I: 34/60 (56.7%) C: 36/63 (57.1%) Rate ratio 0.99 (0.73 – 1.35) 30 days I: 54/60 (90%) C: 58/63 (92.1%) Rate ratio 0.98 (0.87 – 1.09)
Adverse events, No (%) I: 14/60 (23.3%) 1 patient had 2 events C: 7/63 (11.1) 1 patient had 3 events In publication, also data on 6 sub-categories of AE’s |
Information: The primary outcome ‘clinical worsening’ was defined as occurrence of 1 of the 3 following events:
Remarks:
Authors conclusion: The administration of tocilizumab in patients with COVID-19 pneumonia and a Pao2/ Fio2 ratio between 200 and 300 mm Hg did not reduce the risk of clinical worsening. Further blinded, placebo-controlled randomized clinical trials are needed to confirm the results and to explore possible applications of tocilizumab in different stages of the disease, such as in patients with a Pao2/Fio2 ratio less than 200 mm Hg.
|
|
Hermine, 2020 |
Type of study: Cohort-embedded, investigator-initiated multicenter, open-lael, Bayesian randomized clinical trial.
Setting: Nine university hospitals
Country: France
Source of funding: This trial was publicly funded (Ministry of Health, Programme Hospitalier de Recherche Clinique, Foundation for Medical Research (FRM), AP-HP Foundation and the Reacting program).
The funding agencies had no access to the trial data and had no role in the design, conduct or reporting of the trial. Roche donated TCZ in unrestricted grant, and had no role in the trial design or conduct; the collection, management, analysis, interpretation of the data; or in the preparation, review of the manuscript or the approval of the manuscript for submission.
|
Inclusion criteria group 1:
Inclusion criteria group 2:
Exclusion criteria:
N total at baseline: N = 131 Intervention: N = 64 Control: N = 67
Important characteristics: Age, median (IQR): I: 64.0 (57.1 to 74.3) C: 63.3 (57.1 to 72.3)
Sex, n/N (%) male: I: 44/63 (70%) male C: 44/67 (66%) male
BMI, median (IQR) I: 27.9 (23.3 to 30.8) C: 27.4 (24.5 to 31.3) |
Tocilizumab in combination with usual care
Intravenously administration of Tocilizumab of 8 mg/kg on day 1. Administration of an additional fixed doze of tocilizumab, 400 mg IV, on day 3 was recommended if oxygen requirement was not decreased by more than 50%, but decision was left to the treating physician.
|
Usual care alone
Usual care (antibiotic agents, antiviral agents, corticosteroids, vasopressor support, anticoagulants) was provided at the discretion of the clinicians. |
28 days
|
The 2 primary outcomes were (1) the proportion of patients dead or needing non-invasive or mechanical ventilation on day 4 (>5 on the WHO-CPS); and (2) survival with no need for non-invasive or mechanical ventilation at day 14.
Primary outcome by day 14; N I: 15/63 C: 24/67
Mechanical ventilation or death by day 14; N (95% CI) I: 17% (95% CI= 8 to 26) C: 27% (95% CI= 15 to 37) Difference -9 (95% CI= -24 to 5)
Deaths day 14; N I: 7/63 C: 6/67
Survival day 14; % (95% CI) I: 89% (95% CI= 81 to 97) C: 91% (95% CI= 84 to 98)
Deaths day 28; N I: 7/63 C: 8/67
Survival day 28; % (95% CI) I: 89% (81 to 97) C: 88% (80 to 96)
Patients with at least 1 adverse event; N (%) I: 28 (44%) C: 36 (54%)
Patients with multiple adverse events; N (%) I: 16 (25%) C: 19 (28%)
Patients with at least 1 serious adverse event; N (%) I: 20 (32%) C: 29 (43%) P = 0.21
Patients with multiple serious adverse events; N (%) I: 5 (8%) C: 10 (15%) |
Remarks: TCZ (group 1): receiving tociluzumab
UC (group 2): receiving usual care
Authors conclusion:
|
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Sarilumab |
|||||||
|
Lescure, 2021 |
Type of study: RCT; double-blind, placebo-controlled
Setting: multinational phase 3 trial; 45 hospitals
Country: Argentina, Brazil, Canada, Chile, France, Germany, Israel, Italy, Japan, Russia, and Spain
Source of funding: ‘This study was funded by Sanofi (Paris, France) and Regeneron Pharmaceuticals (Tarrytown, USA). Medical writing assistance was provided by Richard J Hogan, and Vojislav Pejović of Eloquent Scientific Solutions, a division of Envision Pharma Group, and editorial and graphics assistance was provided by Eloquent Scientific Solutions. This support was funded by Sanofi.’ |
Hospitalized COVID-19 patients with severe or critical illness.
Inclusion criteria:
• admitted to hospital • laboratory-confirmed SARSCoV-2 infection in any specimen within 2 weeks before random assignment • evidence of pneumonia by chest imaging or chest auscultation • no alternative explanation for clinical presentation • severe disease (defined as administration of supplemental oxygen by nasal cannula, simple face mask, or another similar device) or critical disease (defined as need for supplemental oxygen delivered by non-rebreather mask or high-flow nasal cannula, use of invasive or noninvasive ventilation, or treatment in ICU).
Exclusion criteria low probability of surviving 48h or remaining at the investigational site > 48h, • dysfunction ≥ 2 organ systems, need for extracorporeal life support, or renal replacement therapy at screening; • absolute neutrophil count < 2000 cells per mm 3; aspartate aminotransferase or alanine aminotransferase (ALT) exceeding 5x upper limit of normal at screening; • < 50 000 platelets per mm 3 at screening; • known active, incompletely treated, suspected or known extrapulmonary tuberculosis; • previous or concurrent use of immunosuppressant drugs at screening, including, but not limited to, IL-6 inhibitors or Janus kinase inhibitors within 30 days of baseline; • anti-CD20 agents without evidence of B-cell recovery to baseline concentrations or IL -1 receptor antagonist within 1 week of baseline; • abatacept within 8 weeks of baseline; tumour necrosis factor α inhibitors within 2 –8 weeks of baseline; • alkylating agents, including cyclophosphamide, within 6 months of baseline; • cyclosporine, azathioprine, mycophenolate mofetil, leflunomide, or methotrexate < 4 weeks of baseline; • or intravenous immunoglobulin < 5 months of baseline; • use of systemic chronic (eg, oral) corticosteroids for a condition not related to COVID -19 at doses higher than prednisone 10 mg/day or equivalent at screening; • suspected or known active systemic bacterial or fungal infections < 4 weeks of screening
N total at baseline: Randomized: N = 420 Intervention-I: 161 Intervention-II: 173 Control: 86
Included in mITT analysis: N = 416 Intervention-I: 159 Intervention-II: 173 Control: 84
Important characteristics: Age, median (IQR): I: 58·0 (51·0–67·0) II: 58·0 (48·0–67·0) C: 60·0 (53·0–69·5) Sex, n/N (%) male: I: 108/159 (68%) II: 99/173 (57%) C: 54/84 (64%)
Severity of illness Severe I: 92/159 (58%) II: 105/173 (61%) C: 55/84 (65%) Critical I: 65/159 (41%) II: 68/173 (39%) C: 29/84 (35%)
Multisystem organ dysfunction II: 0/173 C: 0/84
Clinical status, 7-point scale 2 I: 17/159 (11%) II: 24/173 (14%) C: 9/84 (11%) 3 I: 28/159 (18%) II: 21/173 (12%) C: 11/84 (13%) 4 I: 112/159 (70%) II: 128/173 (74%) C: 64/84 (76%) 5 I: 2/159 (1%) II: 0/173 C: 0/84
Time from dyspnoea onset to baseline, days I: 5·0 (2·0–10·0) II: 4·0 (2·0–9·0) C: 7·0 (3·0–10·0)
Duration of hospital stay before dosing, days I: 3·0 (1·0–4·0) II: 2·0 (2·0–4·0) C: 4·0 (2·0–6·0)
Admitted to ICU before dosing I: 61 (38%) II: 59 (34%) C: 28 (33%)
Duration of ICU stay before dosing, days I: 2·0 (1·0–3·0) II: 2·0 (1·0–3·0) C: 1·0 (1·0 –3·5)
Groups comparable at baseline?
|
I: sarilumab 200 mg II: sarilumab 400 mg
The hospital pharmacist added the contents of prefilled syringes of sarilumab 200 mg solution for subcutaneous injection supplied by the sponsor into a specified volume of locally sourced 0·9% sodium chloride solution for IV infusion (two syringes for the 400 mg dose, one syringe for the 200mg dose).
An option for a second dose existed (within the assigned treatment group) within 24–48 h of the first dose, based on the investigator's benefit-risk assessment (amended protocol 02; April 8, 2020). |
Placebo
The hospital pharmacist prepared the volume of locally sourced 0·9% sodium chloride solution for IV infusion.
An option for a second dose existed (within the assigned treatment group) within 24–48 h of the first dose, based on the investigator's benefit-risk assessment (amended protocol 02; April 8, 2020).
Number of doses received 1 dose: I: 146 (91·8%) II: 162 (93·6%) 2 doses: I: 13 (8·2%) II: 11 (6·4%) |
Length of follow up: 60 days
Loss to follow-up: I: 2/161 (1.2%) Reasons: 2 did not start treatment (randomised twice, n=1; suspected bacterial infection, n=1) II: 0/173 (0%) C: 2/86 (2.3%) Reasons: 2 did not start treatment (improved, n=1; withdrew consent, n=1)
Number of doses received 1 dose: 79 (94·0%) 2 doses: 5 (6·0%) |
Clinical outcomes
Mortality Patients alive at day 29 I: 143 (90%) II: 159 (92%) C: 77 (92%)
Difference vs placebo I: −1·7 (−9·3 to 5·8) II: 0·2 (−6·9 to 7·4)
Patients alive at day 60, n (%) I: 142 (89·3) II: 155 (89·6) C: 75 (89·3)
Difference vs placebo (95% CI) I: 0·0 (−8·2 to 8·2) II: 0·3 (−7·7 to 8·3)
Results also reported for severe vs. critically ill patients separately
Duration of hospitalization Proportion of patients with need for ICU care during study (among patients not in ICU at baseline), n (%) I: 11 (11·2) II: 17 (14·9) C: 7 (12·5)
Difference vs placebo (95%CI) I: −1·3 (−12·0 to 9·4) II: 2·4 (−8·4 to 13·3)
Number of days of hospitalisation among patients alive at day 60, least quares mean (SE), days I: 15·6 (1·0) II: 16·1 (0·9) C: 15·9 (1·3)
Difference vs placebo (95%CI) I: −0·2 (−3·5 to 3·0) II: 0·2 (−3·0 to 3·5)
Time from first dose to discharge due to recovery, Kaplan-Meier estimates, days Median (95% CI) C: 14·0 (11·0–16·0) I: 11·0 (10·0–15·0) II: 13·0 (10·0–15·0)
Hazard ratio vs placebo (95% CI) I: 1·05 (0·79 to 1·40) II: 1·00 (0·76 to 1·33)
Time from first dose to discharge due to recoveTime to ≥2-point improvement on seven -point clinical status scale, median Kaplan Meier estimate, days: I: 10·0 (9·0 to 12·0) II: 10·0 (9·0 to 13·0) C: 12·0 (9·0 to 15·0)
Hazard ratio vs placebo I: 1·03 (0·75 to 1·40) II: 1·14 (0·84 to 1·54)
Results also reported for severe vs. critically ill patients separately
Time to NEWS2 of <2 and maintained for 24 hours; Kaplan -Meier estimates, days, Median (95% CI) I: 9·0 (7·0–10·0) II: 9·0 (8·0–11·0) C: 11·0 (8·0–14·0) Hazard ratio vs placebo (95% CI) I: 1·05 (0·77–1·44) II: 1·09 (0·80–1·48) Analysis of time to resolution of fever Kaplan-Meier estimates, Days Median (95% CI)§ I: 8·0 (7·0–9·0) II: 9·0 (7·0–10·0) C: 7·0 (6·0–12·0) Hazard ratio vs placebo (95% CI) I: 0·91 (0·59–1·40) II: 0·92 (0·60–1·40)
Need for respiratory support Time to improvement in oxygenation (defined as increase in SpO2/FiO2 of ≥50 compared with the nadir SpO2/FiO2 for ≥48 hours), Kaplan-Meier estimates, days, median (95% CI)§
I: 6·0 (5·0–7·0) II: 6·0 (5·0–7·0) C: 7·0 (5·0–8·0) Hazard ratio vs placebo (95% CI) I: 1·17 (0·86–1·58) II: 1·11 (0·83–1·50)
Initiation of mechanical ventilation, non-invasive ventilation, or use of high -flow nasal cannula, n (%) C: 13 (19·1) I: 26 (20·5) II: 33 (23·4)
Difference vs placebo (95%CI) I: 1·4 (−10·3 to 13·0) II: 4·3 (−7·4 to 16·0)
Patients alive off supplemental oxygen at day 29, n (%) I: 135/159 (84·9%) II: 145/173 (83·8%) C: 73/84 (86·9%) Difference vs placebo (95%CI I: −2·0 (−11·1 to 7·1) II −3·1 (−12·2 to 6·0) Percent of ventilator -free days in the first 28 days, least squares mean (SE) I: 74·8 (2·2) II: 75·7 (2·1) C: 77·3 (3·0)
Difference vs placebo (95%CI) I: −2·4 (−10·2 to 5·3) II: −1·6 (−9·2 to 6·1)
Also reported: Percent of days with hypoxaemia / supplemental oxygen / resting respiratory rate >24 breaths/min, time to saturation ≥94% on room air
Safety Adverse events Any treatment-emergent adverse event I: 103 (65%) II: 121 (70%) C: 55 (65%) Any serious treatment emergent adverse event I: 42 (26%) II: 51 (29%) C: 20 (24%) of which: Any serious infection I: 18 (11%) II: 22 (13%) C: 10 (12%) Pneumonia I: 1 (1%) II: 6 (3%) C: 0 COVID-19 pneumonia I: 11 (7%) II: 4 (2%) C: 2 (2%) Bacterial pneumonia I: 1 (1%) II: 3 (2%) C: 1 (1%)
Any treatment-emergent adverse event leading to death I: 17 (11%) II: 18 (10% C: 9 (11%)
Any adverse event of special interest I: 53 (33%) II: 76 (44%) C: 18 (21%) Of which Alanine aminotransferase increase I: 48 (30%) II: 55 (32%) C: 16 (19%) Invasive bacterial or fungal infection I: 8 (5%) II: 15 (9%) C: 3 (4%) Grade ≥2 hypersensitivity reaction I: 1 (1%) II: 7 (4%) C: 0 Grade 4 neutropenia I: 3 (2%) II: 6 (3%) C: 0 Grade ≥2 infusionrelated reaction I: 1 (1%) II: 6 (3%) C: 0
Virological outcomes Viral clearance not reported
Also reported: Sarilumab concentration, pharmacodynamic markers, and laboratory findings potentially related to COVID-19 severity, over time. |
Definitions: 7-point ordinal scale: (1) death; (2) admitted to hospital, on invasive mechanical ventilation or extracorporeal membrane oxygenation; (3) admitted to hospital, on noninvasive ventilation or high-flow oxygen devices; (4) admitted to hospital, requiring supplemental oxygen; (5) admitted to hospital, not requiring supplemental oxygen, requiring ongoing medical care (COVID-19- related or otherwise); (6) admitted to hospital, not requiring supplemental oxygen, no longer requiring ongoing medical care; and (7) discharged from hospital. |
|
Merchante, 2021
|
Type of study: Phase II, open-label, multicentre randomized clinical trial
Setting: hospital-based, between 13 July 2020 and 5 March 2021
Country: 10 clinical sites in Andalusia, Spain
Source of funding: This study was funded by the COVID-19 Research Program of the Regional Government of Andalusia.
Conflicts of interest: The authors declare no competing interest. |
hospitalized patients
Inclusion criteria:
Exclusion criteria:
N total at baseline: Randomized: N = 115
Intervention II (400 mg Sarilumab): N = 39
Control: N = 39
Important characteristics: Age, median (IQR): I(I) (200 mg): 65 y (53-72) I (II) (400 mg): 57 y (49-67) C: 57 y (51-71)
Sex, n/N (%) male: I (I) (200 mg): 23/37 (65%) I (II) (400 mg): 29/39 (74%) C: 26/39 (66%)
Diabetes mellitus I (I) (200 mg): 9/37 (24%) I (II) (400 mg): 2/39 (5%) C: 6/39 (15%)
Groups were comparable at baseline. Slightly more patients with DM in de Sarilumab 200 mg group (p=0.06) |
Intervention I single subcutaneous dose of 200 mg sarilumab (Sarilumab-200) + Usual care
Intervention II single subcutaneous dose of 400 mg sarilumab (Sarilumab-400) + Usual care
|
Usual care up to 14 days according to local practice as listed in the protocol of the Spanish Ministry of Health and the Spanish Agency of Medicines and Medical products.
|
Length of follow-up: 28 days
Loss-to-follow-up or incomplete data: One patient withdrew informed consent (intervention I group) and there was a protocol violation (n=1)
One patient withdrew informed consent (intervention II group) and one patient did not receive the allocated intervention.
|
Clinical outcomes Progression to HFNO, NIMV, IMV (event within 28 days) Events, n (%) I(I): 10/37 (27%) I (II): 10/39 (26%) C: 5/39 (13%)
I(I) compared to C HR 0.87 (95%CI: 0.37-2.06), p=0.76
I(II) compared to C HR 0.41 (95%CI: 0.14-1.18), p=0.09
Need for mechanical ventilation Events, n (%) I(I): 6/37 (16%) I (II): 3/39 (8%) C: 4/39 (10%)
I(I) compared to C HR 1.68 (95%CI: 0.47-5.98), p=0.41
I(II) compared to C HR 0.78 (95%CI: 0.17-3.48), p=0.74
Death Events, n (%) I(I): 4/37 (8%) I (II): 0/39 (0%) C: 3/39 (8%)
I(I) compared to C HR 1.41 (95%CI: 0.31-6.31), p=0.64
I(II) compared to C HR 0.01 (95%CI: 0.00-160.68), p=0.07
Also clinical improvement on an ordinal scale, discontinuation of supplemental oxygen in patients receiving it at baseline and discharge alive from hospital were reported as well as a detailed overview of the occurred adverse events. |
Definitions: The primary outcome variable was the development of ARDS requiring HFNO, NIMV 259 or IMV during the first 28 days after randomization.
Remarks: -
Authors conclusion: In patients recently hospitalised with COVID-19 pneumonia and features of systemic inflammation, early IL-6 blockade with a single dose of sarilumab 400 mg was safe and associated with a trend for better outcomes. |
|
The REMAPCAP Investigato rs, 2020 |
Type of study: RCT Open-label design
Setting: Hospitals up to November 19, 2020
Country: International trial.
Source of funding: The trial has multiple international funders. Roche Products and Sanofi supported the Trial through provision of tocilizumab and sarilumab in the United Kingdom. The funders as well as Roche and Sanofi had no role in designing the trial, analyzing the data, writing the manuscript, or making the decision to submit the manuscript for publication. |
Critically ill patients with COVID19 admitted to ICU and receiving respiratory or cardiovascular organ support
Inclusion criteria: • 18 years of age or older • either clinically suspected or microbiologically confirmed Covid-19 • severe disease state: admitted to ICU and receiving respiratory or cardiovascular organ support* • enrolled within 24 hours after starting organ support in the ICU
• presumption that death was imminent with a lack of commitment to full support previously participated in REMAP-CAP within 90 days • Additional exclusion criteria, specific to the Immune Modulation Therapy domain, are listed in a Supplementary Appendix.
N total at baseline: N = 803 Tocilizumab: 353 Sarilumab: 48 Control: 402
Important characteristics: Age, mean±SD: I_toci: 61.5±12.5 I_sari: 63.4±13.4 C: 61.1±12.8
Sex, n/N (%) male: I_toci: 261/353 (74) I_sari: 39/48 (81) C: 283/402 (70) Acute respiratory support: None or supplemental oxygen only, n/N (%) I_toci: 1/353 (<1) I_sari: 0/48 C:2/402 (<1) High-flow nasal cannulae I_toci: 101/353 (29) I_sari: 17/48 (35) C: 110/402 (27) Noninvasive ventilation only I_toci: 147/353 (42) I_sari: 23/48 (48) C: 169/402 (42) Invasive mechanical ventilation I_toci: 104/353 (29) I_sari: 8/48 (17) C: 121/402 (30)
Groups comparable at baseline? Baseline characteristics were balanced across intervention groups and typical of a critically ill population with Covid -19. |
1) tocilizumab or 2) sarilumab Tocilizumab, at a dose of 8 mg per kilogram of actual body weight (up to a maximum of 800 mg), was administered as an intravenous infusion over a period of 1 hour; this dose could be repeated 12 to 24 hours later at the discretion of the treating clinician if clinical improvement was judged insufficient.
Sarilumab, at a dose of 400 mg, was administered as an intravenous infusion once only. All investigational drugs were dispensed by local pharmacies and were open label. |
standard care (no immune modulation) |
Length of follow up: 90 days
Loss to follow-up: Tocilizumab: 16/366 (4.4%) Reasons: -13 Withdrew consent -3 Had outcome that was not available
Sarilumab: 3/48 (6.3%) Reasons: -3 Had outcome that was not available
C: 15/412 (3.6%) Reasons: -10 Withdrew consent -5 Had outcome that Was not available |
Clinical outcomes In-hospital death, n/N (%) I_toci: 98/350 (28) I_sari: 10/45 (22) C: 142/397 (36)
Primary in-hospital survival Adjusted odds ratio Mean I_toci: 1.66±0.31 I_sari: 2.25±0.96 C: 1 Median (95% credible interval) I_toci: 1.64 (1.14 to 2.35) I_sari: 2.01 (1.18 to 4.71) C: 1 Probability of superiority to control — % I_toci: 99.6 I_sari: 99.5 C: -
Time to ICU discharge Adjusted HR - mean (SD) I_toci: 1.43 (0.13) I_sari: 1.69 (0.32) C: 1 median (95% CrI) I_toci: 1.42 (1.18 to 1.70) I_sari: 1.64 (1.21 to 2.45) C: 1 Probability of superiority to control, % I_toci: >99.9 I_sari: 99.9 C: -
Time to hospital discharge Adjusted HR - mean (SD) I_toci: 1.42 (0.13) I_sari: 1.65 (0.31) C: 1 median (95% CrI) I_toci: 1.41 (1.18 to 1.70) I_sari: 1.60 (1.17 to 2.40) C: 1 Probability of superiority to control, % I_toci: >99.9 I_sari: 99.8 C:-
Symptom resolution Not reported.
Number of respiratory and cardiovascular organ support*–free days up to day 21 (primary outcome ) Median (IQR) I_toci: 10 (−1 to 16) I_sari: 11 (0 to 16) C: 0 (−1 to 15) Adjusted odds ratio Mean I_toci: 1.65±0.23 I_sari: 1.83±0.44 C: 1 Median (95% credible interval) I_toci: 1.64 (1.25 to 2.14) I_sari: 1.76 (1.17 to 2.91) C: 1 Probability of superiority to control — % I_toci: >99.9 I_sari: 99.5 C: -
Safety Serious adverse events Patients with >1 serious adverse event, n (%) I_toci: 9/353 (2.5) I_sari: 0/48 (0.0) C: 11/402 (2.7) Adjusted OR - mean (SD) I_toci: 1.22 (0.55) I_sari: 2.99 (2.95) C: 1 median (95% CrI) I_toci: 1.10 (0.48 to 2.58) I_sari: 2.10 (0.51 to 10.77) C: 1 Probability of superiority to control, % I_toci: 59.3 I_sari: 84.0 C: -
Virological outcomes Viral clearance Not reported.
Also reported (secondary outcomes): 90-day Survival (time to event); Respiratory support-free days; Cardiovascular support-free days; WHO scale at day 14; Progression to invasive mechanical ventilation, ECMO or death, restricted to those not intubated at baseline . |
Definitions: Respiratory organ support was defined as invasive or noninvasive mechanical ventilation, including through high-flow nasal cannulae if the flow rate was more than 30 liters per minute and the fraction of inspired oxygen was more than 0.4.
Cardiovascular organ support was defined as the intravenous infusion of any vasopressor or inotrope.
The primary outcome was respiratory and cardiovascular organ support– free days, on an ordinal scale combining inhospital death (assigned a value of −1) and days free of organ support to day 21.
Remarks: -The trial uses an open-label design. -Because this is an early, preliminary report, some data are missing, including 11 outcomes.
Authors conclusion: In critically ill patients with Covid-19 receiving organ support in ICUs, were both effective as compared with the current standard of care, which included glucocorticoids in the majority of patients (>80%). The benefit was consistent across primary and secondary outcomes and across subgroups and secondary analyses, including survival. |
|
Sancho-López, 2021 [SARTRE-trial] |
Type of study: RCT, open-label
Setting: Eight tertiary hospitals
Country: Spain
Source of funding: Biomedical Research Foundation of the Puerta de Hierro Majadahonda University Hospital and Sanofi (provided sarilumab free of charge). None of the funders had any role in the study’s design, collection, management, analysis and interpretation of data, writing of the report and the decision to submit the report for publication.
Conflicts of interest: Authors declared that they have nothing to disclose. |
Hospitalized patients with COVID-19, requiring supplemental oxygen by mask or nasal prongs
Inclusion criteria:
Exclusion criteria: • Admission to the ICU;
N total at baseline: N = 201 Intervention: 99 Control: 102
Important characteristics: Age, mean (SD):
Sex, n/N (%) male:
Disease severity, mean (SD):
Days from symptom onset to randomization, median (IQR)
Ferritin, ng/mL
IL-6, pg/mL
Also available: coexisting conditions and other laboratory values Groups comparable at baseline? |
Sarilumab + usual care
Sarilumab (IV) at a single dose of 200 mg for patients <75 kg body weight, or 400 mg for patients weighing > 75 kg. |
Usual care CS were given to all patients at a 1 mg/ kg/day of methylprednisolone for at least 3 days (background medication). Standard of care also included antibiotic agents, antiviral agents, steroid boluses, vasopressor support, and anticoagulants that were provided at the discretion of the investigators. |
Length of follow-up: 28 days
Loss-to-follow-up: Intervention: 5 (5.1%)
Control: 5 (4.9%) Reasons: loss of FU after discharge (n=4) or after admission at ICU in another hospital (n=1) Another patient did not receive the allocated intervention, due to withdrawing consent to participate.
Incomplete outcome data: Intervention: NR Reasons NA Control: NR Reasons NA |
Clinical outcomes Mortality (28 day) P= 0.4976
Duration of hospitalization
ICU admission Day 15 RR (95%CI): 0.70 (0.25-1.91) P=0.4863
Day 28
Time to symptom resolution
Respiratory support Progression to IMV or death, n/N (%)
Brescia-COVID score ≥3, day 15, n/N (%) C: 16/102 (15.69) RR (95%CI): 1.03 (0.48-2.20) P=0.8874
Brescia-COVID score ≥3, day 28, n/N (%) I: 16/99 (16.16) C: 16/102 (15.69) RR (95%CI): 1.03 (0.48-2.20) P=0.8874
Time to BRESCIA COVID score = 0
Time to respiratory failure
Time to reduction of supplemental oxygen requirements C: 96/102 (94.1) HR (95%CI): 0.9742 (0.73-1.30) P=0.7975
Safety Treatment Emergent Adverse events, n/N (%)
Adverse events, n/N (%) I: 18/99 (18.2) C: 16/102 (15.7) Effect (95%CI): NR P=NR
Virological outcomes Viral clearance Not reported
Also available: (other) Brescia-COVID scores at other days than 15 and 28, subgroup analyses for the group BRESCIA COVID score ≥ 3 by age, sex, sarilumab dose |
Definitions: • Treatment Emergent Adverse Events were defined as CTCAE grade 3 or higher; • Adverse events included following disorders: blood and lymphatic system, Gastro-intestinal, Hepatobiliary, infections, injury, investigations, metabolism and nutrition, musculoskeletal and connective tissue, nervous system, psychiatric, renal and urinary, respiratory, thoracic and mediastinal, skin and subcutaneous tissue and vascular.
Remarks: • The largest part of the participants was recruited in one tertiary centre (n=132). • Study was funded by Sanofi, which had however no role in study’s design, data collection, management, analysis and interpretation nor in writing the report. • Study might have been underpowered. • Patients in the control group progressing to • Brescia-COVID ≥ 2 plus inflammatory parameters were given the option to be rescued with sarilumab at the same weight-adjusted doses (n=18). Patients randomly assigned to sarilumab therapy at baseline progressing to Brescia- COVID ≥ 2 were rescued according to local clinical practice protocol (n=27).
Authors conclusion: Our clinical trial failed to demonstrate any benefits of an early therapeutic intervention with sarilumab when added to an optimized SOC regimen that includes CS in the treatment of hospitalized patients with COVID-19 pneumonia with inflammatory parameters, who were under standard oxygen therapy. No new safety issues were identified. |
|
CORIMUNO-19 Collaborative Group, 2021
(CORIMUNO-SARI-1 trial) |
Type of study: multicentre, open-label, randomized controlled phase 2/3 clinical trial, nested within the CORIMUNO-19 cohort
Setting: hospital-based, between March 27 and April 6, 2020
Country: 6 centres in France
Source of funding: This trial was publicly funded by the Ministry of Health, Programme Hospitalier de Recherche Clinique, and Assistance Publique – Hôpitaux de Paris Foundation and Foundation for Medical Research. Sanofi donated sarilumab as an unrestricted grant and had no role in the study design, no role in the collection, analysis, or interpretation of the data, and no role in the writing of the report.
Conflicts of interest: None to declare. |
hospitalized patients with moderate-to-severe COVID-19 pneumonia
Inclusion criteria:
Exclusion criteria:
N total at baseline: Randomized: N = 148 ITT population: N = 144*
Intervention: N = 68 Control: N = 76
Important characteristics: Age, median (IQR): I: 61.7 y (53.0-71.1) C: 62.8 y (26.0-71.7)
Sex, n/N (%) male: I: 49/68 (72%) C: 59/76 (78%)
Coexisting conditions Chronic cardiac disease I: 17/67 (25%) C: 19/76 (25%)
Diabetes I: 22/68 (32%) C: 22/76 (29%)
Chronic kidney disease (stage 1-3) or dialysis I: 10/67 (15%) C: 7/76 (9%)
Asthma I: 3/67 (4%) C: 8/76 (11%)
Chronic pulmonary disease (not asthma) I: 6/67 (9%) C: 3/76 (4%)
Active malignant neoplasm I: 3/67 (4%) C: 1/76 (1%)
Groups were comparable at baseline. |
sarilumab (fixed dose of 400 mg i.v. on day; additional fixed dose of 400 mg i.v. on day 3 was recommended if O2 requirement had not decreased by more than 50%, but the decision was left to the treating physician) + usual care (antibiotic agents, antiviral agents, corticosteroids, vasopressor support, anticoagulants) |
usual care alone(antibiotic agents, antiviral agents, corticosteroids, vasopressor support, anticoagulants) |
Length of follow-up: 90 days
Loss-to-follow-up or incomplete data: none |
A subgroup analysis according to antiviral drug use at baseline was prespecified in the protocol. Analyses according to the use of corticosteroids and particularly dexamethasone were added post-hoc considering recent publications.
Clinical outcomes Mortality Mortality at day 14 I: 6/68 (9%) C: 8/76 (11%) Adjusted HR 0.68 (95%CI: 0.23-2.03)
Mortality at day 28 I: 8/68 (12%) C: 14/76 (18%) aHR 0.65 (95%CI: 0.27-1.59)
Mortality at day 90 I: 10/68 (15%) C: 16/76 (21%) aHR 0.70 (95%CI: 0.31-1.58)
Duration of hospitalisation Discharged at day 28 I: 51/68 (75%) C: 53/76 (70%) aHR 1.19 (95%CI: 0.81-1.75)
Time to symptom resolution Not reported
Respiratory support Independent from O2 at day 28 I: 50/68 (74%) C: 54/76 (71%) aHR 1.06 (95%CI: 0.72-1.57)
Other Dead or needing non-invasive ventilation or mechanical ventilation on day 4 (i.e. WHO-CPS score > 5; coprimary outcome) I: 18/68 (26%) C: 20/76 (26%) Median posterior ARD 0.2% (90%CrI: -11.7-12.2) Median posterior aOR 1.02 (90%CrI: 0.54-1.94) Posterior probability of any benefit 48.9% Posterior probability of moderate or greater benefit than usual care† 21.6%
Survival with no need for non-invasive ventilation (including high-flow O2) or mechanical ventilation at day 14 (coprimary outcome) I: 25/68 (37%) C: 26/76 (34%) Median posterior aOR 1.10 (90%CrI: 0.69-1.74) Posterior probability of any benefit 37.4% Posterior probability of moderate or greater benefit than usual care† 18.6%
Safety Adverse events Patients with at least one adverse event I: 37/68 (54%) C: 33/76 (43%) p = 0.24
Patients with multiple adverse events I: 17/68 (25%) C: 11/76 (14%)
Number of adverse events I: 77 C: 58
Incidence rate per 1000 patient-day I: 14.6 (95%CI: 11.7-18.2) C: 10.3 (95%CI: 8.0-13.4)
IRR 1.41 (95%CI: 1.00-1.98); control group is reference group p = 0.048
Serious adverse events Patients with at least one serious adverse event I: 27/68 (40%) C: 28/76 (37%) p = 0.73
Patients with multiple serious adverse events I: 10/68 (15%) C: 9/76 (12%)
Number of serious adverse events I: 44 C: 40
Incidence rate per 1000 patient-day I: 8.3 (95%CI: 6.2-11.2) C: 7.1 (95%CI: 5.2-9.7)
IRR 1.16 (95%CI: 0.76-1.79); control group is reference group p = 0.47
Deaths I: 10/68 (15%) C: 16/76 (21%)
Virological outcomes Not reported |
Definitions: † Moderate or greater benefit was defined as an ARD <-5·5% for the day 4 outcome and a HR <·0.85 for the day 14 outcome.
Remarks: * Four patients in the control group withdrew consent after randomization, and asked their personal data to be erased.
Authors conclusion: Sarilumab treatment did not improve early outcomes in patients with moderate-to-severe COVID-19 pneumonia. Further studies are warranted to evaluate the effect of sarilumab on long-term survival. |
|
Levilimab (Monoclonal antibody- IL-6 inhibitor- BCD-089; Ilsira) |
|||||||
|
Lomakin, 2021 |
Type of study: multicenter double-blind, placebo-controlled phase III trial
Setting:
Country: 12 investigational sites in the Russian Federation Source of funding: This study was funded by JSC BIOCAD grant no. BCD-089-4/CORONA. Conflicts of interest: Several authors are JSC BIOCAD employees. The sponsor designed the trial, was responsible for the monitoring, collected the data, and performed the data analysis. Sponsor’s representatives were not IDMC members, had no access to the blinded data, and did not participate in voting. |
hospitalized patients with severe COVID-19 not requiring mechanical ventilation
Inclusion criteria: • age ≥ 18 y • positive for SARS-CoV-2 RNA • hospitalized with radiologically confirmed pneumonia with at least 1 criteria of disease severity: • respiratory rate > 30/min • SpO2 ≤ 93 • PaO2/FiO2 < 300 mmHg • increase of the lung involvement > 50% after 24-48 h • decreased consciousness level • agitation • unstable hemodynamics • arterial blood lactate > 2 mmol/L • qSOFA > 2, defined by the presence of any 2 symptoms (SBP ≤ 100 mmHg, respiratory rate ≥ 22/min, GCS ≤ 14)
Exclusion criteria: • critical form of COVID-19, defined by the presence of respiratory failure and need of the invasive mechanical ventilation, septic shock or multiple organ failure • suspected active bacterial, fungal, viral, or other infection (besides COVID-19) • confirmed active tuberculosis • life expectancy < 24 h, in the opinion of the investigator or who were unlikely to remain at the investigational site beyond 48 h • treated with other monoclonal antibodies, immunosuppressive agents or participating in a clinical trials of other drug • history of allergic reaction to monoclonal antibodies • any illness or laboratory findings that, in the opinion of the study investigator, might pose an additional risk to the patient by their participation in the study • pregnant or breastfeeding • absolute neutrophil count < 1.0 × 109/L
N total at baseline: Randomized: N = 206 ITT population: N = 206 Intervention: N = 103 Control: N = 103
Important characteristics: Age, mean (SD): I: 58.5 y (12.9) C: 58.2 y (10.8)
Sex, n/N (%) male: I: 58/103 (56.3%) C: 51/103 (49.5%)
The proportion of patients aged ≥ 75 y was higher in the intervention group than in the control group (11.7 vs. 3.9%).
Patients in the intervention group more often had cardiac disorders(19.4 vs. 11.7%).
More patients in the control group received corticosteroids (4.9 vs. 8.7%) |
levilimab 324 mg s.c. |
placebo |
Length of follow-up: 60 days
Loss-to-follow-up or incomplete data: Intervention: N =1 (1.0%) Reason • early withdrawal (n = 1) Control: N = 4 (3.9%) Reason • withdrawal before study medication was administered (n = 2)• early withdrawal (n = 1) lost to follow-up (n = 1) |
Clinical outcomes Mortality
Duration of hospitalisation Duration of hospital stay Days, median (IQR)
Time to symptom resolution
Respiratory support
Other Day 7 p = 1.0000
Day 14 I: 65/103 (63.1%) C: 44/103 (42.7%) p = 0.0017
Day 21 I: 79/103 (76.7%) C: 49/103 (47.6%) p < 0.0001
Day 28 I: 87/103 (84.5%) C: 57/103 (55.3%) p < 0.0001
Day 30 I: 87/103 (84.5%) C: 57/103 (55.3%) p < 0.0001
Transfer to ICU I: 3/103 (2.9%) C: 10/103 (9.7%) p = 0.0449
Duration of fever Days, median (IQR) I: 1 (1-3) C: 2 (1-3) p = 0.1065
Change in ESR from baseline mm/h, median (IQR) Day 3 I: 30 (18-44.5) C: 38 (23-56) p = 0.0035
Day 5 I: 25 (15-41) C: 40 (24-55) p = 0.0002
Day 7 I: 23 (15-36) C: 31 (21-45) p = 0.0009
Change in CRP from baseline mg/L, median (IQR) Day 3 I: 14.6 (5.1-29.8) C: 31.7 (12-62.6) p < 0.0001
Day 5 I: 5.3 (1.5-15) C: 17.7 (6.9-44) p < 0.0001
Day 7 I: 3.9 (1.3-8.4) C: 9.2 (4.1-19) p < 0.0001
Change in IL-6 from baseline pg/mL, median (IQR) Day 3 I: 65.9 (16.5-20.1) C: 16.4 (3.9-80.4) p = 0.0017
Day 4 I: 64.2 (18.3-247.1) C: 20.1 (1.5-119.1) p = 0.0121
Day 14 I: 25.4 (12.6-77.8) C: 108.7 (22.1-10.5) p = 0.1000
Safety Adverse drug reactions I: 28/103 (27.2%) C: 24/101 (9.3%) p = 0.5750
Grade ≥ 3 adverse drug reactions I: 10/103 (9.7%) C: 7/101 (6.9%) p = 0.4279
Serious adverse events I: N = 1 C: N = 2
Grade 4 neutropenia I: N = 0 C: N = 0
Hypersensitivity I: N = 0 C: N = 0
Injection site reactions I: N = 0 C: N = 0
Virological outcomes Not reported |
Definitions:
* The 7-category ordinal scale includes the following categories: 1 not hospitalized/discharged; 2 hospitalized, not requiring O2 therapy or other medical care; 3 hospitalized, not requiring O2 therapy, requiring other medical care; 4 hospitalized, requiring O2 therapy; 5 hospitalized, requiring high-flow O2 therapy or non-invasive ventilation; 6 hospitalized, requiring mechanical ventilation or ECMO; 7 death. Remarks: - Authors conclusion: In patients with radiologically confirmed SARS-CoV-2 pneumonia, requiring or not oxygen therapy (but not ventilation) with no signs of other active infection administration of LVL + SOC results in an increase of sustained clinical improvement rate. |
|
siltuximab |
|||||||
|
Declercq, 2021 |
Type of study: Multicentre, open-label, 2x2 factorial, randomised, controlled phase 3 trial
Setting: Hospital-based, between April 4 and December 6, 2020
Country: 16 hospitals in Belgium
Source of funding: Belgian Health Care Knowledge Centre and VIB Grand Challenges program. The funder of the study (Belgian Health Care Knowledge Centre) was involved in purchasing study medication and study design, but was not involved in data collection, data analysis, data interpretation, writing of the manuscript, or the decision to submit. The funder (VIB Grand Challenges) was involved in purchasing reagents for measuring biomarkers.
Conflicts of interest: Conflicts of interest were transparently and extensively reported. |
Hospitalised patients with COVID-19, hypoxia, and signs of a cytokine release syndrome
Inclusion criteria:
Exclusion criteria:
N total at baseline: Randomized: N = 342 ITT population: N = 342
Intervention 1:: N = 112 Control 1: N = 230 Intervention 2a: N = 113 Intervention 2b: N = 114 Control 2: N = 115
Important characteristics: Age, median (IQR): I1: 67 y (56-74) C1: 64 y (54-72)
I2: 59 y (52-74) C2: 62 y (55-71)
Sex, n/N (%) male: I1: 87/112 (78%) C1: 178/230 (77%)
I2: 175/227 (77%) C2: 90/115 (78%)
Mechanical ventilation at day of randomisation Invasive I1: 17/112 (15%) C1: 22/230 (10%)
I2: 22/227 (10%) C2: 17/115 (15%)
Non-invasive or high flow oxygen device I1: 44/112 (39%) C1: 84/230 (37%)
I2: 89/227 (39%) C2: 39/115 (34%)
Groups were comparable at baseline. |
Intervention 1: anakinra 100 mg 1 dd s.c. for 28 days or until hospital discharge + standard care
Intervention 2: siltuximab 11 mg/kg i.v. (single injection) (= intervention 2a)
OR
tocilizumab 8 mg/kg i.v. (not exceeding 800 mg; single injection) (= intervention 2b) |
Control 1: standard care
Control 2: standard care |
Length of follow-up: 28 days
Loss-to-follow-up: I1: 0/112 (0%) Reasons: -
C1: 6/230 (3%) Reasons:
I2a: 2/113 (2%) Reasons:
I2b: 2/114 (2%) Reasons:
C2: 2/115 (2%) Reasons: withdrew consent (n = 2) |
Clinical outcomes Mortality Number of deaths I1: 10/44 (23%) I1+I2a: 5/32 (16%) I1+I2b: 6/36 (17%) I2a: 10/81 (12%) I2b: 15/75 (20%) C: 9/74 (12%)
Estimated mortality at day 28 I1: 16% (95%CI: 8-31) I1+I2a: 13% (95%CI: 5-30) I1+I2b: 17% (95%CI: 8-33) I2a: 11% (95%CI: 6-20) I2b: 13% (95%CI: 7-23) C: 10% (95%CI: 5-20)
Estimated mortality at day 90 I1: 23% (95%CI: 13-38) I1+I2a: 16% (95%CI: 7-34) I1+I2b: 17% (95%CI: 8-33) I2a: 12% (95%CI: 7-22) I2b: 19% (95%CI: 12-30) C: 13% (95%CI: 7-23)
Duration of hospitalization Time until discharge Days, median I1: 14 (95%CI: 11-19) C1: 12 (95%CI: 11-18) HR 0.90 (95%CI: 0.70-1.16)
I2: 12 (95%-CI: 11-18) C2: 13 (95%-CI: 11-19) HR 1.02 (95%CI: 0.80-1.31)
Number of days in hospital Mean I1: 19 (95%CI: 17-22) C1: 19 (95%CI: 17-21) expected count ratio 1.01 (95%CI: 0.85-1.21)
I2: 20 (95%-CI: 18-22) C2: 19 (95%-CI: 16-22) expected count ratio 1.03 (95%CI: 0.86-1.22)
Number of days in ICU Mean I1: 11 (95%CI: 8-15) C1: 10 (95%CI: 8-13) expected count ratio 1.05 (95%CI: 0.69-1.59)
I2: 11 (95%-CI: 8-14) C2: 10 (95%-CI: 7-15) expected count ratio 1.03 (95%CI: 0.68-1.56)
Number of days in ICU in patients ventilated at day of randomisation Mean I1: 20 (95%CI: 15-27) C1: 22 (95%CI: 17-29) expected count ratio 0.89 (95%CI: 0.60-1.32)
I2: 20 (95%-CI: 16-27) C2: 22 (95%-CI: 16-29) expected count ratio 0.94 (95%CI: 0.64-1.40)
Number of days in ICU relative to the number of days alive the first 28 days after randomisation Mean I1: 42% (95%CI: 31-56) C1: 36% (95%CI: 29-46) expected count ratio 1.14 (95%CI: 0.79-1.66)
I2: 38% (95%-CI: 30-48) C2: 40% (95%-CI: 29-54) expected count ratio 0.96 (95%CI: 0.66-1.39)
Time to symptom resolution Time to clinical improvement (primary outcome)† Days, median I1: 12 (95%CI: 10-16) C1: 12 (95%CI: 10-15) HR 0.94 (95%CI: 0.73-1.21)
I2: 11 (95%-CI: 10-16) C2: 12 (95%-CI: 11-16) HR 1.00 (95%CI: 0.78-1.29)
Estimated probability of having experienced clinical improvement at day 28 I1: 75% (95%CI: 67-83) C1: 73% (95%CI: 67-79)
I2: 74% (95%-CI: 68-79) C2: 74% (95%-CI: 66-82)
Respiratory support Median time until independence from supplemental O2 or discharge Days, median I1: 12 (95%CI: 10-20) C1: 12 (95%CI: 10-15) HR 0.91 (95%CI: 0.71-1.17)
I2: 11 (95%-CI: 10-15) C2: 12 (95%-CI: 10-15) HR 1.00 (95%CI: 0.78-1.28)
Median time until independence from invasive ventilation Days, median I1: 21 (95%CI: 8-not estimable) C1: 27 (95%CI: 9-not estimable) HR 1.21 (95%CI: 0.54-2.71)
I2: 23 (95%-CI: 8-not estimable) C2: 54 (95%-CI: 9-not estimable) HR 1.45 (95%CI: 0.63-3.33)
Median time until first use of high-flow oxygen device, ventilation, or death Days, median I1: < 50% reached event C1: < 50% reached event HR 0.97 (95%CI: 0.52-1.82)
I2: < 50% reached event C2: < 50% reached event HR 0.85 (95%CI: 0.47-1.55)
Number of days without supplemental O2 use up to 28 days after randomisation Mean I1: 9 (95%CI: 7-12) C1: 9 (95%CI: 7-12) expected count ratio 0.97 (95%CI: 0.68-1.38)
I2: 10 (95%-CI: 8-12) C2: 8 (95%-CI: 6-11) expected count ratio 1.17 (95%CI: 0.82-1.68)
Number of invasive ventilator days Mean I1: 5 (95%CI: 3-9) C1: 5 (95%CI: 3-7) expected count ratio 1.05 (95%CI: 0.54-2.03)
I2: 5 (95%-CI: 3-7) C2: 5 (95%-CI: 3-9) expected count ratio 0.89 (95%CI: 0.46-1.72)
Number of invasive ventilator days in patients ventilated at day of randomisation Mean I1: 15 (95%CI: 11-20) C1: 16 (95%CI: 13-21) expected count ratio 0.93 (95%CI: 0.63-1.37)
I2: 15 (95%-CI: 12-20) C2: 16 (95%-CI: 12-22) expected count ratio 0.96 (95%CI: 0.65-1.42)
Number of invasive ventilator days relative to the number of days alive the first 28 days after randomisation Mean I1: 23% (95%CI: 14-38) C1: 21% (95%CI: 14-31) expected count ratio 1.08 (95%CI: 0.57-2.05)
I2: 21% (95%-CI: 14-30) C2: 23% (95%-CI: 14-38) expected count ratio 0.89 (95%CI: 0.47-1.70)
Number of invasive ventilator-free days up to 28 days after randomisation Mean I1: 18 (95%CI: 15-21) C1: 18 (95%CI: 16-20) expected count ratio 1.00 (95%CI: 0.84-1.19)
I2: 18 (95%-CI: 17-20) C2: 17 (95%-CI: 15-20) expected count ratio 1.07 (95%CI: 0.90-1.27)
Number of invasive ventilator-free days up to 28 days after randomisation in patients ventilated at day of randomisation Mean I1: 6 (95%CI: 3-14) C1: 6 (95%CI: 3-13) expected count ratio 1.01 (95%CI: 0.33-3.07)
I2: 7 (95%-CI: 4-15) C2: 5 (95%-CI: 2-12) expected count ratio 1.39 (95%CI: 0.46-4.20)
Safety Serious infections Sepsis I1: 5/44 (11%) I1+I2a: 2/32 (6%) I1+I2b: 3/36 (8%) I2a: 4/81 (5%) I2b: 11/75 (15%) C: 6/74 (8%)
Septic shock I1: 5/44 (11%) I1+I2a: 1/32 (3%) I1+I2b: 3/36 (8%) I2a: 3/81 (4%) I2b: 6/75 (8%) C: 3/74 (4%)
Serious adverse events not leading to mortality Infectious disorder (not COVID-19) I1: 1/44 (2%) I1+I2a: 2/32 (6%) I1+I2b: 1/36 (3%) I2a: - I2b: 4/75 (5%) C: 1/74 (1%)
Bleeding I1: 2/44 (5%) I1+I2a: 1/32 (3%) I1+I2b: - I2a: 1/81 (1%) I2b: - C: 1/74 (1%)
Thrombosis I1: 1/44 (2%) I1+I2a: - I1+I2b: 1/36 (3%) I2a: - I2b: 1/75 (1%) C: 1/74 (1%)
Acute kidney injury I1: 1/44 (2%) I1+I2a: - I1+I2b: - I2a: - I2b: 1/75 (1%) C: 1/74 (1%)
Cardiac disorder I1: - I1+I2a: - I1+I2b: - I2a: 1/81 (1%) I2b: 1/75 (1%) C: 1/74 (1%)
Other I1: - I1+I2a: 2/32 (6%) I1+I2b: 2/36 (6%) I2a: 3/81 (4%) I2b: 1/75 (1%) C: 1/74 (1%)
Virological outcomes Viral clearance Not reported.
Several subgroups were prespecified on the basis of allocated treatment for the other randomisation, invasive ventilation and serum concentrations of CRP, IL-1β, and IL-6 on the day of randomisation. Post-hoc subgroup analyses included serum concentrations of IL-1RA, admission status to ICU or concomitant glucocorticoid use at randomisation. |
Definitions: * Patients needed to have either a single ferritin concentration measurement of more than 2000 μg/L at inclusion when they immediately required high flow oxygen or mechanical ventilation, or a ferritin concentration of more than 1000 μg/L, which had been increasing over the previous 24 h, or lymphopenia below 800/mL with two of the following criteria: an increasing ferritin concentration of more than 700 μg/L; an increasing lactate dehydrogenase concentration of more than 300 international units (IU)/L; an increasing CRP concentration of more than 70 mg/L; or an increasing D-dimers concentration of more than 1000 ng/mL. If the patient had three of the previous criteria at hospital admission with lymphopenia of less than 800/μL, there was no need to document an increase over 24 h.
† Defined as the time in days from randomisation until either an increase of at least two points on a 6-category ordinal scale (compared with the worst status at day of randomisation) or to discharge from the hospital alive, whichever occurred first. The 6-category ordinal scale was defined as 1=death; 2=hospitalised, on invasive mechanical ventilation or extracorporeal membrane oxygenation; 3=hospitalised, on non-invasive ventilation or high-flow oxygen devices; 4=hospitalised, requiring supplemental oxygen; 5=hospitalised, not requiring supplemental oxygen; 6=not hospitalised.
Remarks: -
Authors conclusion: Drugs targeting IL-1 or IL-6 did not shorten the time to clinical improvement in this sample of patients with COVID-19, hypoxic respiratory failure, low SOFA score, and low baseline mortality risk. |
Risk of bias table for intervention studies (randomized controlled trials).
|
Declercq (2021) |
See RoB assessment of the Declercq (2021) by Anakinra. |
|||||||
|
Rosas, 2021b
|
Eligible patients were randomly assigned in a 2:1 ratio using an interactive web-based response system and a permuted block method to receive blinded treatment with tocilizumab plus remdesivir or placebo plus remdesivir. Randomization was stratified by geographic region (North America, Europe, other) and by a 2-level factor based on clinical status at screening (ordinal scale categories 4–5, category 6) on a 7-category ordinal scale (additional details are in the Online Resource).
|
Unlikely
|
Unlikely
Double-blind study.
|
Unlikely
Double-blind study
|
Unlikely
Double-blind study
|
Unlikely
All predefined outcome measures were reported.
|
Unlikely
A slightly higher proportion of patients in the placebo plus remdesivir arm than the tocilizumab plus remdesivir arm discontinued remdesivir before completing 10 days of treatment (44.2% vs 39.4%); however, most of these early discontinuations were the result of hospital discharge, consistent with remdesivir use in other trials [17] and in clinical practice
|
Unlikely
Participants included in the analysis are exactly those who were randomized into the trial
|
|
Horby, 2021a
Recovery Collaborative group, 2021
|
Computerized
Patients were assigned to either usual standard of care or usual standard of care plus tocilizumab in a 1:1 ratio by means of web-based simple (unstratified) randomisation. |
Unlikely
Allocation was concealed until after randomisation. |
Likely
It was an open-label trial; participants were not masked to the allocated treatment |
Likely
Local study staff was not masked to the allocated treatment.
(except for mortality) |
Unclear
Local study staff was not masked to the allocated treatment. However, the steering committee, investigators, and all others involved in the trial were masked to the outcome data during the trial. |
Unlikely
Outcome measures described in the protocol were also reported. |
Unlikely
97% versus 98% completed the trial. |
Unlikely
Intention to treat analysis was performed. |
|
Wang, 2021 |
Computerized
Randomization numbers were generated using SAS statistical software package. Each consecutively coded patient was randomly enrolled by the sub-site investigators until the total number of cases allocated to the site was reached. The patients were randomly assigned in a 1:1 ratio. |
Unclear
Not described. |
Likely
open-label RCT |
Likely
open-label RCT |
Unclear
Not described. |
Unlikely
However, the worsening rate of hypoxia during hospitalization was only reported for subgroups of patients with moderate or severe COVID-19. |
Unlikely
No participants were lost to follow-up. |
Unlikely
However, one patient in the control group, who worsened severely 3 days after randomization, was crossed over to the tocilizumab group in accordance with the study protocol. However, this patient was erroneously included in the intervention group for the ITT analyses. |
|
Soin, 2021
|
Computerized
“Eligible patients were randomly assigned using block randomisation in a 1:1 ratio to receive open-label tocilizumab plus current standard care (tocilizumab group) or current standard care alone (standard care group). The randomisation sequence was generated using SAS, version 9.4 and an interactive web response system.”
|
Unclear
Concealment not described
“Randomisation numbers were assigned in sequential order to the study sites according to the pregenerated sequence provided by the investigational medicinal product team at the Medanta Institute of Education and Research.” |
Likely
“After randomisation, none of the study personnel or patients were masked to treatment assignment in this open-label trial.“ |
Likely
“After randomisation, none of the study personnel or patients were masked to treatment assignment in this open-label trial.“ |
Likely
“After randomisation, none of the study personnel or patients were masked to treatment assignment in this open-label trial.“ |
Unlikely
Relevant and announced outcomes reported; registered with the Clinical Trials Registry India (CTRI/2020/05/025369 |
Unlikely
1 cross-over from control to intervention group, 1 withdrawn consent in standard care group; not expected to induce bias in results
|
Unlikely
Analysis not performed according to ITT-protocol; small number of patients not included in the analysis, 1 cross-over; not expected to induce bias in results |
|
Rosas, 2021a
|
Web-based response system and permuted block randomization / Stratification according to use of mechanical ventilation (yes or no).
Eligible patients were randomly assigned in a 2:1 ratio to receive a single intravenous infusion of tocilizumab (at a dose of 8 mg per kilogram of body weight, with a maximum dose of 800 mg) or placebo plus standard care by means of an interactive voice or Web-based response system and permuted-block randomization. Randomization was stratified according to geographic region (North America or Europe) and the use of mechanical ventilation (yes or no).
|
Likely
Patients were stratified according to geographic region and use of mechanical ventilation (yes or no). |
Unlikely
The study was a double-blinded, placebo-controlled study.
|
Unlikely
The study was a double-blinded, placebo-controlled study.
|
Unlikely
The study was a double-blinded, placebo-controlled study.
|
Unlikely
All predefined outcomes were reported.
|
Unlikely
Lost to follow-up did not significantly differ between the two groups.
|
Unlikely
Participants included in the analysis are exactly those who were randomized into the trial and analysed by intention-to-treat analysis.
|
|
The REMAP-CAP Investigators, 2020 |
See RoB assessment of the REMAP-CAP Investigators (2020) by sarilumab.
|
|||||||
|
Veiga, 2021
|
Patients were randomised in a 1:1 ratio to receive either standard care or tocilizumab plus standard care, with random blocks of sizes 2, 4, 6, and 8, and stratified by age (<60 and ≥60 years) and sex, according to a computer-generated schedule using the sample function of software R 3.6.3 (R Foundation). |
Unlikely
Allocation concealment was ensured by a central automated web accessed system (REDCap), developed by CZO. |
Likely |
Likely |
Likely |
Unlikely |
Unclear
|
Unlikely |
|
Salama, 2020
|
Computerized
“Using permuted-block randomization and an interactive voice- or Web-response system, we randomly assigned the patients, in a 2:1 ratio, to receive standard care plus one or two doses of either intravenous tocilizumab (8 mg per kilogram of body weight, to a maximum of 800 mg per dose) or placebo. The randomization was stratified according to country (the United States, Mexico, Kenya, South Africa, Peru, or Brazil) and age (≤60 or >60 years).” |
Unlikely
“After initial written informed consent has been obtained, all screening procedures and assessments have been completed, and eligibility has been established for a patient, the study site will obtain the patient's identification number and treatment assignment from an interactive voice or web-based response system (IxRS).” |
Unlikely
“Study site personnel and patients will be blinded to treatment assignment during the study, with the exception of the study pharmacist.” |
Unlikely
“Study site personnel and patients will be blinded to treatment assignment during the study, with the exception of the study pharmacist.” |
Unlikely
“Study site personnel and patients will be blinded to treatment assignment during the study, with the exception of the study pharmacist.” |
Unlikely
ClinicalTrials.gov number, NCT04372186. Primary outcome slightly different (protocol: proportion of patients requiring mechanical ventilation at day 28; publication: proportion of patients who had received mechanical ventilation or who had died by day 28.
|
Likely
Percentage of loss to follow-up differs between groups (intervention group 3.9%, control group 0.8%). Unclear why more participants of the intervention group withdrew from study participation.
|
Unlikely
Analysis performed according to (modified) intention-to-treat protocol; as indicated by randomized participants minus participants that withdrew or were withdrawn. |
|
Stone, 2020 |
Block randomization
|
Unlikely
Randomization was performed with randomly permuted blocks of sizes 3 and 6. Randomization was stratified according to site.
|
Unlikely
It was a randomized, double-blind, placebo-controlled trial.
|
Unlikely
It was a randomized, double-blind, placebo-controlled trial.
|
Unlikely
A blinded interim analysis for safety was performed. ] |
Unlikely
|
Unlikely
|
Unlikely
Intension-to-treat analysis was performed. |
|
Salvarani, 2020 |
Computerized
“Patients were randomized using a web-based system with a 1:1 allocation ratio. Randomization was stratified by center” |
Unlikely
“allocation concealment was based on a centralized randomization list that was not available to clinicians” |
Unclear
It was an open label study. |
Likely
It was an open label study, which might have led to differences in patient care. |
Unclear
It was an open label study, which might have led to differences in reporting the results. |
Unlikely
Outcomes reported in accordance with study protocol / trial register
Trial Registration: ClinicalTrials.gov Identifier NCT04346355; EudraCT Identifier: 2020-001386-37 |
Unclear
3 patients withdrew consent in control group. |
Unlikely
3 patients withdrew consent in control group. Other patients were included in the analysis according to randomization; adherence to ITT analysis protocol. |
|
Hermine, 2020 |
Participants were randomly assigned in a 1:1 ratio to receive TCZ plus usual care (TCZ group) or usual care alone (UCgroup) via a web-based secure centralized system.
|
Unlikely
An independent statistician provided a computer-generated assignment randomization list stratified by center and blocked with varying block sizes unknown to the investigators |
Likely
The trial was not blinded because it was logistically impossible at the time of the pandemic to set up a double-blind study quickly
|
Likely
|
Likely
|
Unlikely
|
Likely
8 patients were lost to follow-up at day 28 in the TCZ group versus 3 in the UC group |
Unlikely
Analyses were performed on an intention-to-treat basis with no correction for multiplicity for secondary outcomes. Thus, these results are exploratory and reported as point estimates and 95% confidence intervals (CIs) |
|
Sarilumab |
||||||||
|
Lescure, 2020 |
Computerized
Eligible patients were randomly assigned (2:2:1) to one dose of intravenous sarilumab 400 mg, sarilumab 200 mg, or placebo according to a central randomisation scheme using permuted blocks of five and implemented through an interactive response technology. |
Unclear
Concealment was not described |
Unlikely
“Patients, care providers, outcome assessors, and investigators remained masked to assigned intervention throughout the course of the study. An unmasked pharmacist was responsible for the preparation and dispensation of all study interventions.” |
Unlikely
“Patients, care providers, outcome assessors, and investigators remained masked to assigned intervention throughout the course of the study. An unmasked pharmacist was responsible for the preparation and dispensation of all study interventions.” |
Unlikely
“Patients, care providers, outcome assessors, and investigators remained masked to assigned intervention throughout the course of the study. An unmasked pharmacist was responsible for the preparation and dispensation of all study interventions.” |
Unlikely
Elaborate description of outcomes in register; not all FU’s included in the publication Register: ClinicalTrials.gov, NCT04327388; EudraCT, 2020- 001162-12; and WHO, U1111-1249-6021. |
Unlikely |
Unlikely |
|
Merchante, 2021
|
Unclear
|
Unclear
Not reported |
Likely
Open-label trial |
Likely
Open-label trial |
Unclear
Not described |
Likely
Not all outcome measures described in the trial protocol are reported in the results |
Unlikely |
Unlikely
|
|
The REMAP-CAP Investigators, 2020 |
Computerized
randomization Participants were assigned by means of a centralized computer program to each intervention, starting with balanced assignment for tocilizumab, sarilumab, or control, with actual proportions dependent on the number of interventions available at each site |
Unlikely
Centralized randomization was used that was remote from study sites. |
Likely
Open-label trial. |
Likely
Open-label trial. Clinical staff were aware of the intervention assignment of individual patients |
Unclear
Not reported. |
Unlikely
Secondary outcomes were all prespecified, and details are provided in the Supplementary Appendix. |
Unclear
Because this is an early, preliminary report, some data are missing, including 11 outcomes. |
Unclear
Not reported. |
|
Sancho-López, 2021 |
Computerized
Participants were randomly assigned in a 1:1 ratio to receive either sarilumab plus SOC or SOC alone. Randomization codes were produced by means of the RERAND system integrated within the eCRF system based on Oracle, stratified by center and using blocks multiple of 2 elements. |
Unlikely
The randomization schedule was managed through the eCRF in a concealed manner. |
Likely
It is an open label study, which mainly will have an impact on the subjective outcomes. |
Likely
It is an open label study. However, medical decisions to increase oxygen support were taken by different physicians, many of them not investigators of the trial. |
Likely
It is an open label study, which mainly will have an impact on the subjective outcomes. |
Unclear
Some of the outcome measures mentioned in the study protocol were not reported in the study (e.g. duration of hospitalization, non-invasive ventilation) |
Unlikely
Number of lost to follow-up and reasons were not different between the treatment groups. |
Unlikely
ITT-analysis was performed. |
|
CORIMUNO-19 Collaborative Group, 2021 |
Computerized
Participants were randomly assigned in a 1:1 ratio via a web-based secure centralised system. An independent statistician provided a computer-generated randomisation list stratified by centre and blocked with random block size (randomly selected among 2 and 4); the block size was unknown to the investigators and statisticians analysing the data. |
Unlikely
Participants were randomly assigned via a web-based secure centralised system. An independent statistician provided a computer-generated randomisation list. |
Likely
Open-label trial |
Likely
Open-label trial |
Unlikely
Analyses were done by the study statisticians who were blinded to the actual randomisation groups. |
Unlikely
All outcome measures described in the trial protocol are reported in the results. |
Unlikely
No patients were lost to follow-up |
Unlikely
Analyses were done on an intention-to-treat basis with no correction for multiplicity for prespecified secondary outcomes |
|
Levilimab (Monoclonal antibody- IL-6 inhibitor- BCD-089; Ilsira) |
||||||||
|
Lomakin, 2021 |
Computerized Randomization was performed centrally. After the investigator entered the eligibility screening data, the central electronic system generated a unique subject identifier and a unique investigational product lot number. |
Unlikely Randomization was performed centrally |
Unlikely Patients were blinded to the treatment allocation. |
Unclear Not reported. However, levilimab and placebo were provided in identical primary and secondary packages with identical labels. |
Unlikely The investigator was blinded to the treatment allocation. |
Unlikely All outcome measures described in the methods are reported in the results, except for duration of hospital stay. Noteworthy, the initial primary endpoint was overall mortality, but because the mortality was significantly lower than assumed, the study did not have enough power to detect a meaningful difference. |
Unlikely The percentage of patients lost to follow-up is less than 10%, but higher in the control group. |
Unlikely All randomized patients who received levilimab or placebo were included in the ITT (‘as randomized’) analysis. |
|
siltuximab |
||||||||
|
Declercq, 2021 |
Computerised
Included patients were randomly assigned by means of permuted block randomization with varying block size and stratification by center. Patients were allocated in a 1:2 ratio to anakinra or no IL-1 blockade. Simultaneously, patients were randomly assigned in a 1:1:1 ratio to siltuximab, tocilizumab, or no IL-6 blockade. Randomization and subsequent data collection were done by means of the web based system REDCap. |
Unlikely
Randomization and subsequent data collection were done by means of the web based system REDCap. |
Likely
open-label trial |
Likely
open-label trial |
Unclear
not described |
Unlikely
All outcome measures described in the methods are reported in the results. |
Unlikely
The percentage of patients lost to follow-up is less than 10% and is similar between treatment groups. |
Unlikely
The primary and supportive efficacy endpoints were assessed in the intention-to-treat population. Safety was assessed in the safety population. |
|
|
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2 (unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4 (unlikely/likely/unclear) |
Bias due to loss to follow-up?5 (unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6 (unlikely/likely/unclear) |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Beoordelingsdatum en geldigheid
Publicatiedatum : 07-11-2022
Beoordeeld op geldigheid : 03-10-2022
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut). Deze ondersteuning werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De werkgroep werd gefinancierd uit een VWS subsidie.
De financiers hebben geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodules is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de behandeling van patiënten met COVID-19.
In 2020 is een multidisciplinair expertiseteam behandeling ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van het expertiseteam behandeling) die betrokken zijn bij de zorg voor patiënten met COVID-19. Dit expertiseteam fungeerde als stuurgroep, welke opdracht heeft gegeven tot het ontwikkelen van de module, alsmede fungeerde als klankbordgroep.
Werkgroep
- Dr. Marjolein Hensgens, internist-infectioloog, Afdeling Infectieziekten, UMC Utrecht en LUMC Leiden (Stichting Werkgroep Antibiotica Beleid)
- Drs. Emilie Gieling, apotheker, Afdeling Klinische Farmacie, UMC Utrecht.
- Prof. Dr. Dylan de Lange, intensivist, Afdeling Intensive Care, UMC Utrecht.
- Dr. Wim Boersma, longarts, Afdeling Longziekten, Noordwest Ziekenhuisgroep, Alkmaar.
- Dr. Paul van der Linden, apotheker, Afdeling Klinische Farmacie, Tergooi MC, Hilversum (Stichting Werkgroep Antibiotica Beleid).
- Prof. Dr. Bhanu Sinha, arts-microbioloog, Afdeling Medische Microbiologie & Infectiepreventie, UMCG, Groningen (Stichting Werkgroep Antibiotica Beleid).
- Dr. Mark de Boer, internist-infectioloog, Afdelingen Infectieziekten en Klinische Epidemiologie, LUMC, Leiden (Stichting Werkgroep Antibiotica Beleid).
- Tot 1-11-2021 tevens deel van de werkgroep: Dr. Albert Vollaard, internist-infectioloog, LCI, RIVM
Stuurgroep (expertiseteam Behandeling COVID-19)
- Dr. L.M. van den Toorn (voorzitter), longarts, Erasmus Medisch Centrum (Erasmus MC), NVALT
- Dr. M.G.J. de Boer, internist-infectioloog, Leids Universitair Medisch Centrum (LUMC), SWAB/NIV)
- Drs. A.J. Meinders, internist-intensivist, St. Antonius Ziekenhuis, NVIC
- Prof. dr. D.W. de Lange, intensivist-toxicoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), NVIC
- Dr. C.H.S.B. van den Berg, infectioloog-intensivist Universitair Medisch Centrum Groningen (UMCG), NVIC
- Dr. S.U.C. Sankatsing, internist-infectioloog, Diakonessenhuis, NIV
- Dr. E.J.G. Peters, internist-infectioloog, Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. M.S. Boddaert, arts palliatieve geneeskunde, Leids Universitair Medisch Centrum (LUMC), IKNL
- Dr. P.L.A. Fraaij, kinderarts-infectioloog, Erasmus Medisch Centrum (Erasmus MC), Sophia Kinderziekenhuis, NVK
- Dr. E. van Leeuwen, gynaecoloog, Amsterdam University Medical Centers (Amsterdam UMC), NVOG
- Dr. J.J. van Kampen, arts-microbioloog, Erasmus Medisch Centrum (Erasmus MC), NVMM
- Dr. M. Bulatović-Ćalasan, internist allergoloog-immunoloog en klinisch farmacoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. A.F.J. de Bruin, anesthesioloog-intensivist, St. Antonius Ziekenhuis, NVA
- Drs. A. Jacobs, klinisch geriater, Catharina Ziekenhuis, NVKG
- Drs. B. Hendriks, ziekenhuisapotheker, Leids Universitair Medisch Centrum (LUMC), NVZA
- Drs. M. Nijs, huisarts, NHG
- Dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
Meelezer
- Drs. K. (Klaartje) Spijkers, senior adviseur patiëntenbelang, Patiëntenfederatie Nederland, Utrecht
Met ondersteuning van:
- dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
- dr. L.M.P. Wesselman, adviseur, Kennisinstituut van Medisch Specialisten
- dr. D. Nieboer, adviseur, Kennisinstituut van Medisch Specialisten
- drs. A.L.J. (Andrea) Kortlever - van der Spek, adviseur, Kennisinstituut van Medisch Specialisten
-
M. Griekspoor MSc., junior adviseur, Kennisinstituut van Medisch Specialisten
- drs. I. van Dusseldorp, senior literatuurspecialist, Kennisinstituut van Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
De Lange |
1. Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht (0,6 fte) |
Secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE), onbezoldigd. |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, klinisch epidemioloog, senior medisch specialist, Leids Universitair Medisch Centrum, afdeling Infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Sinha |
Arts-microbioloog/hoogleraar, Universitair Medisch Centrum Groningen (voltijd) (zie ook https;//www.rug.nl/staff/b.sinha/) |
- SWAB-bestuur: secretaris [onbetaald; vacatiegeld voor instelling] |
- Projectsubsidie EU (Cofund): deelprojecten, cofinanciering
Mogelijk boedbeeldfunctie SWAB |
Geen actie nodig |
|
Van der Linden |
Ziekenhuisapotheker |
Penningmeester SWAB, vacatiegeld |
Geen |
Geen actie nodig |
|
Vollaard |
Internist-infectioloog, Landelijke Coordinatie Infectieziektebestrijding, RIVM |
Arts voor ongedocumenteerde migranten, Dokters van de Wereld, Amsterdam (onbetaald) |
Geen |
Geen actie nodig |
|
Gieling |
Ziekenhuisapotheker - Klinisch Farmacoloog, UMC Utrecht |
Lid OMT Nederlandse Vereniging voor Ziekenhuisapothekers (onbetaald) |
Geen |
Geen actie nodig |
|
Boersma |
Longarts Noordwest Ziekhuisgroep |
Lid sectie infectieziekten NVALT, onbetaald |
Eenmalige digitale deelname aan adviesraad MSD Pneumovax over Pneumococcal disease, betaald
|
Geen actie nodig |
|
Hensgens |
Internist-infectioloog, UMC Utrecht (0.8 aanstelling, waarvan nu 0.4 gedetacheerd naar LUMC) Internist-infectioloog, LUMC (via detachering, zie boven) |
Geen |
Geen |
Geen actie nodig |
Stuurgroep
|
Achternaam stuurgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Van den Toorn (voorzitter) |
Voorzitter NVALT |
Geen |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, senior medisch specialist, LUMC, afdeling infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Meinders |
Internist-intensivist, St.-Antonius ziekenhuis, Nieuwegein |
commissie werk |
Geen |
Geen actie nodig |
|
De Lange |
Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht |
secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE) (onbetaald) |
Geen |
Geen actie nodig |
|
Van den Berg |
Infectioloog-intensivist, UMCG |
Geen |
Geen |
Geen actie nodig |
|
Sankatsing |
Internist-infectioloog/internist-acute geneeskunde, Diakonessenhuis, Utrecht |
- Bestuurslid Nederlandse Vereniging van Internist-Infectiologen (NVII) (onbetaald). |
Geen |
Geen actie nodig |
|
Peters |
Internist - aandachtsgebieden infectieziekten en Acute Geneeskunde Amsterdam UMC, locatie Vumc |
Wetenschappelijk Secretaris International Working Group on the Diabetic Foot (onbetaald) |
Geen |
Geen actie nodig |
|
Boddaert |
Medisch adviseur bij Integraal Kankercentrum Nederland (IKNL) en Palliatieve Zorg Nederland (PZNL) Arts palliatieve geneeskunde in LUMC |
Geen |
Geen |
Geen actie nodig |
|
Fraaij |
Kinderarts infectioloog- immunoloog, Erasmus MC-Sophia, Rotterdam |
Bestuur Stichting Infecties bij Kinderen (onbetaald) |
deelname aan RECOVER, European Union's Horizon 2020 research |
Geen actie nodig |
|
Van Leeuwen |
Gyaecoloog Amsterdam Universitair Medisch Centra |
Geen |
Geen |
Geen actie nodig |
|
Van Kampen |
Arts-microbioloog, afdeling Viroscience, Erasmus MC |
- associate editor antimicrobial resistance & infection control (onbetaald) - lid antibioticacommissie Erasmus MC (onbetaald) |
1. Mede uitvinder patent: 1519780601-1408/3023503 2. R01AI147330 (NIAID/NH) (HN onderzoek (1+2 niet gerelateerd aan COVID-19)
|
Geen actie nodig |
|
Bulatovic |
Internist allergoloog-immunoloog en klinische farmacoloog, UMC Utrecht en Diakonessenhuis Utrecht |
Functie 1: arts |
Geen |
Geen actie nodig |
|
De Bruin |
Anesthesioloog - Intensivist St. Antonius ziekenhuis Nieuwegein en Utrecht |
Geen |
Geen |
Geen actie nodig |
|
Jacobs |
Klinisch geriater en klinisch farmacoloog |
Geen |
Geen |
Geen actie nodig |
|
Hendriks |
Ziekenhuisapotheker farmaceutische patiëntenzorg, afd. Kiinische Farmacie en Toxicoiogie, Leids Universitair Medisch Centrum |
Lid SWAB werkgroep surveillance antibioticagebruik, onbetaald Lid SWAB richtlijncommissie antibiotica allergie, onbetaald |
Geen |
Geen actie nodig |
|
Nijs |
Huisarts |
Geen |
Geen |
Geen actie nodig |
|
Hofstede |
Senior adviseur Kennisinstituut van Medisch Specialisten |
Geen |
Geen |
Geen actie nodig |
Meelezer
|
Achternaam |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Spijkers |
Senior adviseur patiëntenbelang |
Voorzitter Stichting Samen voor Duchenne |
Geen |
Geen actie nodig |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde patiëntenvereniging in de klankbordgroep. De verkregen input is meegenomen bij het opstellen van de module. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Werkwijze
Van leidraad naar richtlijnmodules
Bij aanvang van de pandemie in 2020 was het onduidelijk of bestaande of nieuwe medicijnen een relevante bijdrage konden leveren aan het herstel van patiënten geïnfecteerd met het SARS-CoV-2. Vandaar dat eind februari 2020 werd aangevangen met de eerste versie van de leidraad ‘Medicamenteuze behandeling voor patiënten met COVID-19 (infectie met SARS–CoV-2)’, welke begin maart 2020 online beschikbaar werd gesteld op de website van de SWAB (https://swab.nl/nl/covid-19). Sindsdien werd het adviesdocument op wekelijkse basis gereviseerd en indien nodig op basis van nieuwe publicaties van onderzoek aangepast. Het initiatief en de coördinatie hiertoe werden genomen door de SWAB Leidraadcommissie, ondersteund door het kennisinstituut van de Federatie Medisch Specialisten en een brede klankbordgroep waarbinnen de betrokken specialisten(verenigingen) zijn vertegenwoordigd. In september 2021 is gestart met het doorontwikkelen van de leidraad naar richtlijnmodules.
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de COVID-19 pandemie zijn knelpunten op verschillende manieren geïnventariseerd:
1. De expertiseteams benoemde de knelpunten in de zorg voor patiënten met COVID-19.
2. Er is een mailadres geopend (covid19@demedischspecialist.nl) waar verschillende partijen knelpunten konden aandragen, die vervolgens door de expertiseteams geprioriteerd werden.
3. Door de Federatie van Medisch Specialisten zijn webinars georganiseerd waarbij vragen konden worden ingestuurd. Deze vragen zijn na afloop van de webinars voorgelegd aan de expertiseteams en geprioriteerd.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. Wanneer mogelijk werd de data uit verschillende studies gepoold in een random-effects model. Review Manager 5.4 werd gebruikt voor de statistische analyses. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
Definitie |
|
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
Bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Zoekverantwoording
On request.