Interleukine-1 remmers
Uitgangsvraag
Wat is de plaats van interleukine (IL-)1 remmers bij de behandeling van patiënten met COVID-19?
Aanbeveling
IL-1 remmers worden niet aanbevolen als standaardbehandeling voor opgenomen patiënten met COVID-19
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Er is literatuuronderzoek verricht naar de verschillen in klinische uitkomsten tussen behandeling met en zonder een IL-1 remmer bij patiënten opgenomen in het ziekenhuis met COVID-19. Tot 24 januari 2022 werden er 6 gerandomiseerde gecontroleerde studies (RCT’s) gevonden. Vier studies onderzochten de IL-1 receptor antagonist anakinra (Mariette, 2021; Kyriazopoulou, 2021; Kharazmi, 2021; Declercq, 2021), twee studies bekeken het anti-IL-1-antilichaam canakinumab (Caricchio, 2021; Cremer, 2021). In totaal werden er 1579 patiënten gerandomiseerd (n=847 in de interventiegroep en n=732 in de controlegroep).
Er werden alleen RCT’s geïncludeerd in de analyse, waardoor de kwaliteit van bewijs initieel hoog was. Er waren meerdere studies met een relatief kleine populatie en mede hierdoor een grote spreiding van de puntschatter van de uitkomstmaat (imprecision), waardoor de kwaliteit van dit bewijs naar beneden werd bijgesteld. Daarnaast werden er verschillende uitkomstmaten (heterogenicity) gebruikt bij bijvoorbeeld rapportage van de mortaliteit (14 dagen en 28-30 dagen), waardoor de kwaliteit van dit bewijs waar nodig ook naar beneden werd bijgesteld. De geïncludeerde patiënten verschilden per studie: Mariette (2021) includeerde patiënten met o.a. een CRP boven de 25, terwijl Kyriazopoulou (2021) een verhoogde (≥6 ng ml−1) soluble urokinase plasminogen activator receptor (suPAR) gebruikte. Voor deze verschillen in inclusie werd niet afgewaardeerd in de gradering van de studies, maar zij moeten wel worden meegenomen in de overwegingen.
IL-1 remmers bij patiënten die waren opgenomen in het ziekenhuis
Op basis van de gevonden resultaten kan er met lage zekerheid worden geconcludeerd dat er geen (grote) reductie in de mortaliteit optreedt bij het gebruik van IL-1 remmers (risicoverschil: -2,9%, 95% CI -5,8 tot 0.1; relatief risico: 0.69, 95% CI 0.46 tot 1.03). Deze conclusie komt overeen met de conclusie uit een Cochrane review uit 2022 (Davidson, 2022).
De zes geïncludeerde gerandomiseerde onderzoeken laten tegenstrijdige resultaten zien: 5 RCT’s tonen geen overtuigend voordeel van IL-1 remming, terwijl de studie van Kyriazopoulou (2021) dit wel laat zien.
- Een open-label RCT uit Frankrijk (Mariette, 2021) werd na inclusie van 116 patiënten met mild tot matig ernstige ziekte en PCR-bevestigde SARS-CoV-2 infectie, waarvan 59 in de anakinra groep, voortijdig gestopt omdat uit een tussenanalyse bleek dat geen positief klinisch effect aantoonbaar was (ITT basis). Geïncludeerd werden patiënten die ten minste 3 L/min O2 via een masker of neuscanule nodig hadden, maar geen mechanische beademing, en CRP van meer dan 25 mg/L. Vergeleken werd standaardzorg met of zonder anakinra. De twee primaire uitkomsten waren overlijden op dag 4 of noodzaak tot niet-invasieve of mechanische beademing en overleven zonder behoefte aan mechanische of niet-invasieve beademing (inclusief high flux zuurstof) op dag 14. Gebruik van anakinra leidde hier niet tot betere klinische resultaten.
- Een Belgische multicenter RCT (Declercq, 2021) randomiseerde 342 opgenomen patiënten met een hoge zuurstofbehoefte of mechanische ventilatie tweemaal: naar anakinra naast SoC versus SoC, en naar IL-6 blokkade (tocilizumab of siltuximab) naast SoC versus SoC. Door deze dubbele randomisatie ontstonden er relatief kleine groepen patiënten in elk stratum. Er werd geen verschil gezien in de tijd tot klinisch herstel en ook geen verschil in de mortaliteit na 28 dagen.
- Een relatief grote RCT bij 594 patiënten met een ernstige COVID-19 infectie, maar geen noodzaak tot (non-)invasieve beademing, toonde wel een positief resultaat van anakinra (Kyriazopoulou, 2021). In deze trial, genaamd SAVE MORE, werden patiënten geïncludeerd nadat zij gemiddeld 9 dagen klachten hadden en een verhoogd serum suPAR (soluble urokinase plasminogen activator receptor; SuPAR ≥6 ng/ml). Na 28 dagen bleek de kans op een slecht beloop kleiner in de anakinra-groep, gemeten op een 11-punts-schaal (OR 0.36; 95% CI 0.26-0.50) en ook de mortaliteit na 28 dagen was lager (3.2 vs 6.9%).
- Een relatief kleine Iraanse studie met 30 patiënten toonde een (niet statistisch significant) voordeel van anakinra: de 14 dagen mortaliteit was 33% in de anakinra groep vergeleken met 47% in de controlegroep (Kharazmi, 2021).
- Een gerandomiseerde, placebogecontroleerde fase 3 studie naar een anti-IL-1 beta antistof, canakinumab, includeerde 454 opgenomen patiënten met hypoxie (maar zonder invasieve beademing) en tekenen van systemische hyperinflammatie (Caricchio, 2021). De hyperinflammatie werd gedefinieerd als een CRP van 20 mg/L of meer, of een ferritine van 600 ug/L of meer. De geïncludeerde patiënten kregen een eenmalige gift canakinumab (450 tot 750 mg, afhankelijk van het gewicht) of placebo. Er werd geen verschil gezien in de primaire uitkomstmaat, aangezien 88.8% in de canakinumab-groep en 85.7% in de placebo-groep de eerste 29 dagen overleefden zonder mechanische ventilatie (OR 1.39; CI 95% 0.76-2.54). Ook de COVID-19 gerelateerde mortaliteit was niet statistisch significant verschillend (4.9% in de canakinumab-groep versus 7.2% in de placebo-groep), al werd er wel een numeriek voordeel gezien.
- Een relatief kleine Amerikaanse RCT randomiseerde 45 patiënten met COVID-19 en myocardschade naar twee doseringen canakinumab (300 mg of 600 mg) of placebo (Cremer, 2021). Er werd geen voordeel gevonden voor behandeling met canakinumab op de 28 dagen mortaliteit (60% vs 58% vs 58% respectievelijk).
Concluderend laat slechts 1 RCT een statistisch significant positief effect op mortaliteit en snelheid van klinisch herstel zien met IL-1 remming (Kyriazopoulou, 2021). In dit onderzoek werd anakinra toegediend op geleide van suPAR, een biomarker die op het moment van de samenstelling van deze richtlijn niet standaard in de Nederlandse ziekenhuizen beschikbaar is. De verhouding van de suPAR tot het CRP is op dit moment nog onvoldoende duidelijk, waardoor er (nog) geen alternatieve afkapwaarde van het CRP gedefinieerd kan worden. Ook is het nog onduidelijk waarom er in deze ene studie van Kyriazopoulou een voordeel gezien wordt van IL-1 remming, terwijl de andere studies dit niet laten zien.
Een mogelijke verklaring is het verschil in het gebruik van corticosteroïden in de standaardbehandeling. In de studie van Kyriazopoulou (2021) bevatte de standaardbehandeling in 87% corticosteroïden, hetgeen meer overeenkomt met de huidige standaardbehandeling, terwijl dit bij Declerq (2021), Cremer (2021) en Kharazmi (2021) tussen de 47% en 63% was. In de studies van Mariette (2021) en Caricchio (2021) kreeg minder dan 40% van de patiënten corticosteroïden toegediend in het kader van de standaardbehandeling.
Andere mogelijke verklaringen voor de gevonden verschillen tussen de studies is de studiepopulatie en de timing van de behandeling met IL-1 remmers. De twee kleinere studies onderzochten een relatief zieke populatie met een mortaliteit in de controlegroep boven de 20% (Mariette, 2021; Kharazmi, 2021). De twee grootste studies hadden een vergelijkbare relatief lage mortaliteit in de controlegroep: ongeveer 7% na 28 dagen bij zowel Carrichio (2021) als Kyriazopoulou (2021). Alhoewel alleen de SAVE MORE studie van Kyriazopoulou een statistisch significant voordeel van IL-1 remming op de COVID-19 gerelateerde mortaliteit liet zien, werd er wel in beide studies een numeriek voordeel gezien van behandeling met IL-1 remming. De inzet van IL-1 remming bij patiënten die minder ernstig ziek zijn, gaat mogelijk gepaard met een groter effect van de behandeling. De suggestie dat IL-1 remming met name ingezet zou kunnen worden in het begin van de inflammatoire fase van COVID-19, werd ook gedaan in een review (Veerdonk, 2021). Daarnaast laat een zevende RCT, die nog niet werd meegenomen in de huidige analyse omdat deze alleen als pre-print beschikbaar was, geen effect zien van anakinra bij patiënten op de intensive care, dus in een late fase van de COVID-19 (Derde, 2021).
Naast de onduidelijkheid over de timing van IL-1 remming, is het onduidelijk of de mogelijke winst van IL-1 remming ook behaald zou worden (of in grotere mate) als de standaardbehandeling ook IL-6 remming bevat, zoals tocilizumab. De studie van Declercq (2021) is te klein om hierover een betrouwbare uitspraak te kunnen doen, maar de beperkte data uit deze studie laten geen aanwijzingen zien voor een additioneel voordeel van IL-1 remming plus IL-6 remming.
Naast de cruciale uitkomstmaat ‘mortaliteit’, werd er gekeken naar het effect van IL-1 remmers op de noodzaak tot invasieve respiratoire ondersteuning, de duur van hospitalisatie en de tijd tot symptoomresolutie. Met een lage zekerheid werd geconcludeerd dat er een klein tot geen effect zou kunnen optreden op de duur van de ziekenhuisopname. Op basis van de beschikbare literatuur kon er geen betrouwbare uitspraak gedaan worden over het effect van IL-1 remmers op de noodzaak tot invasieve respiratoire ondersteuning of de tijd tot symptoomresolutie hospitalisatie.
Overige overwegingen
Soort IL-1 remmer
In de huidige richtlijn worden de resultaten van de verschillende middelen (anakinra en canakinumab) zo mogelijk gepoold weergegeven. Deze middelen hebben echter niet precies dezelfde werking: anakinra remt zowel IL-1 alfa als IL-1 beta, terwijl canakinumab alleen op IL-1 beta werkt. Ook als de resultaten per middel weer gegeven worden (wat deels in de resultaten is gepresenteerd), verandert de conclusie niet.
Op dit moment is het bewijs voor de IL-1 remmer anakinra het sterkst.
Bijwerkingen
Vijf van de geïncludeerde studies rapporteerden het percentage (ernstige) bijwerkingen (Mariette, 2021; Kyriazopoulou, 2021; Kharazmi, 2021; Declercq, 2021; Caricchio, 2021; Cremer, 2021). In geen van de studies werd een significant verschil gevonden. Ook een systematische analyse van Cochrane in patiënten met COVID-19 concludeerde dat IL-1 remming waarschijnlijk geassocieerd is met geen of nauwelijks toename van het aantal bijwerkingen (Davindson, 2022). Een eerder Cochrane studie naar het gebruik van IL-1 remmers voor andere indicaties zoals reumatoide artritis, rapporteerde wel iets vaker (ernstige) infecties, maar dit was niet statistisch significant (Mertens, 2009).
Dosering
De dosering anakinra was niet gelijk in de beschreven studies. Kyriazopoulou (2021) en Kharazmi (2021) en Declercq (2021) bestudeerden een dagelijks dosering van 100 mg (injectie) gedurende 7-10 dagen, 14 dagen en 28 dagen, respectievelijk. Mariette (2020) bestudeerde een dosering van 200 mg 2dd op dagen 1-3, 100 mg 2dd op dag 4 en 100 mg 1dd op dag 5. Alleen in de studie van Kyriazopoulou (2021) werd de anakinra subcutaan gegeven, de andere studies gaven het middel intraveneus. De studie van Declercq (2021) randomiseerde de patiënten twee keer. Bij de eerste randomisatie werden patiënten toegewezen aan een behandeling met een IL-1 remmer of een standaardbehandeling, vervolgens werden dezelfde patiënten nogmaals toegewezen aan een (extra) behandeling met een IL-6 remmer of uitsluitend een standaardbehandeling. De twee studies die een eenmalige toediening canakinumab onderzochten, gebruikten ook verschillende doseringen. Caricchio doseerde op basis van gewicht (doseringen tussen de 450 mg en 750 mg) en Cremer randomiseerde naar 300 mg of 600 mg canakinumab. Om deze reden is in alle analyses afgewaardeerd voor inconsistentie.
Virusvarianten
Sinds de opkomst van de omikron variant van SARS-CoV-2 in Nederland eind 2021, is de kans op een ernstig beloop van COVID-19 op populatieniveau zeer sterk gedaald. Het is van belang om op te merken dat de besproken gerandomiseerde studies werden verricht voor de opkomst van de omikron variant. Het is onduidelijk wat de invloed is van deze variant op het effect van anti-inflammatoire therapie, al wordt aangenomen dat patiënten die door de omikron variant een ernstige COVID-19 infectie ontwikkelen nog steeds baat hebben bij anti-inflammatoire therapie. De ‘number needed to treat’ zou wel anders (vermoedelijk hoger) kunnen zijn.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Op grond van de bekende onderzoeksgegevens worden IL-1 remmers op dit moment niet aanbevolen in de standaardbehandeling van COVID-19. Wel worden deze middelen in verschillende klinische trials onderzocht. De resultaten van deze studies zullen bepalen of er een plek is voor IL-1 remmers in de behandeling van COVID-19 en bij welke patiëntenpopulatie.
Kosten (middelenbeslag)
Gezien het gebrek aan overtuigend bewijs voor effectiviteit wordt IL-1 remming nu niet aanbevolen in de standaardbehandeling van patiënten met COVID-19.
Aanvaardbaarheid, haalbaarheid en implementatie
IL-1 remmers worden niet voorgeschreven in de standaardbehandeling van COVID-19, dus de werkgroep voorziet geen problemen qua implementatie. Wel worden deze middelen in verschillende klinische trials onderzocht. De resultaten van deze studies kunnen in de toekomst de plaatsbepaling van IL-1 remmers voor bepaalde patiëntgroepen veranderen.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Het is nog onvoldoende duidelijk of IL-1 remmers een effectieve behandeling van COVID-19 zijn, in welke fase van het beloop van COVID-19 zij gegeven zouden moeten worden, en op basis van welke beschikbare markers.
Onderbouwing
Een infectie met SARS-CoV-2 kan leiden tot een ernstige pneumonie en ARDS. Een dysregulatie van de immuunrespons lijkt bij COVID-19 een belangrijke rol in de pathofysiologie te spelen (Veerdonk, 2021). Er zijn verschillende middelen onderzocht met anti-inflammatoire werking, waaronder IL-1 remmers. De IL-1 receptor antagonist (IL-1RA) anakinra, dat zowel IL-1 alfa als IL-1 beta remt, werd al vroeg onderzocht bij patiënten met bacteriële sepsis (Opal, 1997). Alleen in een post-hoc analyse bij de subgroep van patiënten met een bacteriële sepsis en leverfunctiestoornissen en tekenen van diffuse intravasale stolling als uiting van macrofagen activatie syndroom (MAS, 6% van totale groep), werd een gunstig effect op overleving gezien (Shakoory, 2016). In een studie met long-epitheelcellen die geïnfecteerd werden met rat-specifieke Corona-virussen was de chemokine-expressie minder na blootstelling aan anakinra. Een eerste patiëntenserie uit Italië toonde aan dat er mogelijk ook klinische winst te bereiken was ten opzichte van historisch controles (Cavalli, 2020).
Inmiddels is in verschillende gerandomiseerde studies (RCT’s) de effectiviteit van IL-1 remmers onderzocht om de plaats van deze middelen bij de behandeling van COVID-19 patiënten te bepalen. Klinische dose-finding studies zijn in deze setting nooit verricht. De groep IL-1 remmers bevat zowel IL-1 receptor antagonisten (IL-1RA; anakinra) als anti-IL-1 beta antistoffen (canakinumab).
Mortality (crucial)
|
Low GRADE |
Treatment with an IL-1 inhibitor may result in reduced mortality when compared with treatment without an IL-1 inhibitor in hospitalized patients with COVID-19.
Source: Caricchio, 2021; Cremer, 2021; Declerq, 2021; Kharazmi, 2021; Kyriazopoulou, 2021; Mariette, 2021. |
Extensive respiratory support (crucial)
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with an IL-1 inhibitor on the need for extensive respiratory support when compared with treatment without an IL-1 inhibitor in hospitalized patients with COVID-19.
Source: Caricchio, 2021; Cremer, 2021; Declercq, 2021; Kharazmi, 2021; Kyriazopoulou, 2021; Mariette, 2021. |
Duration of hospitalization (important)
Time to clinical improvement (important)
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with an IL-1 inhibitor on time to clinical improvement when compared with treatment without an IL-1 inhibitor in hospitalized patients with COVID-19.
Source: Cremer, 2021; Declercq, 2021. |
Anakinra
Declercq (2021) (COV-AID) reported a multicentre (n=16), open label, randomised controlled phase 3 trial in Belgium. They aimed to assess whether tocilizumab, siltuximab or anakinra shortened the time to clinical improvement in patients with COVID-19. Patients were eligible for randomization if they met the inclusion criteria (e.g., PaO2/FiO2 < 350 on room temperature or < 280 on supplemental oxygen and bilateral pulmonary infiltrates; use invasive mechanical ventilation, OR non-invasive ventilation or continuous use of CPAP for hypoxia, OR oxygen supplementation with an oxygen flow of at least 10 L/min independent of delivery system). In total 342 patients were randomly assigned to treatment with anakinra and standard care (n=112) or only standard care (n=230), and simultaneously randomly assigned to IL-6 blockade (n=227; 114 for tocilizumab and n=113 for siltuximab) or no IL-6 blockade (n=115). Therefore, 44 patients received only anakinra, 32 patients received anakinra + tocilizumab, 36 patients received anakinra + siltuximab, 81 patients received only tocilizumab, 75 patients received only siltuximab, and 74 patients received only standard care. Most patients received hydroxychloroquine (i.e., 42% assigned before August 2020) or dexamethasone (i.e., 84% assigned from August 2020) as standard care. In total, 60% used corticosteroids as part of the standard care. The median (IQR) age was 67 (56-74) years in the intervention group (i.e., IL-1 blockade group), compared with 64 (54-72) in the control group (i.e., no IL-1 blockade group). In the intervention group 87/112 (77.7%) were males, compared with 178/230 (77.3%) in the control group. The length of the follow-up was 28 days. The following relevant outcome measures were included; duration of hospitalization, time to clinical improvement, respiratory support. The primary outcome was time to clinical improvement. This resulted in a median (range) of 12 days (10-16) in the intervention group, compared with 12 days (10-15) in the control group, HR: 0.94 (95%CI 0.73 to 1.21). There are some concerns regarding risk of bias as the study had an open label design.
Kharazmi (2021) described an open label, randomized controlled trial performed in Iran. Kharazmi (2021) assessed the efficacy and safety of anakinra in patients hospitalised with COVID-19. Patients were eligible for randomization if they met the inclusion criteria (e.g., oxygen saturation less than or equal to 93% measured using a peripheral capillary pulse oximeter; fever (core temperature of 37.8 or more); cough or shortness of breath; PaO2/FiO2 less than 300). In total 30 patients were included and randomized to treatment with anakinra and standard care (i.e., standard protocol for COVID-19, n=15) or only standard care (n=15). Standard medication to treat COVID-19 were corticosteroid (73.3% intervention, 53.3% control), interferon (93.3% intervention, 60.0% control), lopinavir/ritonavir (46.7% intervention, 80.0% control), remdesivir (13.3% intervention, 26.7% control), favipiravir (60.0% intervention, 26.7% control). The mean age (±SD) was 49 (±19) years in the intervention group, compared with 59 (±2) years in the control group. In the intervention group 8/15 (53.3%) patients were males, compared with 11/15 (73%) in the control group. The length of the follow-up was 14 days. The following relevant outcome measures were included; duration of hospitalization. The primary outcome was need for endotracheal intubation. This was reported in 3/15 (20.0%) patients in the intervention group, compared with 10/15 (66.7%) in the control group. The rate difference was 46.7%. There are some concerns regarding risk of bias due to the relative short follow-up of 14 days, the open label design (without placebo), and the fact that the study was a pilot study with a small sample size.
Kyriazopoulou (2021) (SAVE-MORE) described a phase 3, double-blind placebo-controlled, multicenter randomized controlled trial. Kyriazopoulou (2021) evaluated anakinra in addition to standard of care versus standard of care alone in patients admitted to the hospital with COVID-19. Dexamethasone was mostly used as standard care (i.e., 86%). In total 594 patients were included as they met the inclusion criteria (e.g., findings in chest X-ray or chest computed tomography compatible with lower respiratory tract infection; need for hospitalization; plasma suPAR [soluble urokinase-type plasminogen activator receptor] ≥6 ng/ml). At baseline 544/594 (92%) patients needed oxygen administration, 50/594 (8.4%) was diagnosed with a moderate pneumonia (based on WHO classification for COVID-19), and 544/594 (91.6%) with a severe pneumonia. Importantly, patients which needed non-invasive ventilation (NIV) (CPAP or BPAP) or mechanical ventilation were excluded according to the exclusion criteria.
The mean (±SD) age was 62 (±12) years, and 344/594 (57.9%) of them were males. Patients in the intervention group (n=405) received anakinra daily for 7 to 10 days (injection, 100 mg of anakinra at a final volume of 0.67ml). The control group (n=189) received placebo (i.e., daily injection of 0.67 ml of 0.9% sodium chloride). The length of the follow-up was 28 days. The following relevant outcome measures were included; mortality, duration of hospitalization (i.e., number of days of hospitalization, time of ICU stay), need for respiratory support. The primary outcome was overall comparison of the distribution of frequencies of the scores from the 11-point WHO-CPS between the two arms of treatment on day 28. This resulted in a odds ratio of 0.36 (95% CI 0.26 to 0.50), in favour of anakinra.
Mariette (2021) (CORIMUNO-ANA-1) described the results of an open-label, Bayesian randomised clinical trial nested within the CORIMUNO-19 cohort. Patients with mild to moderate COVID-19 pneumonia, severe acute respiratory syndrome were selected from 16 hospitals in France. Eligible patients (i.e., C-reactive protein serum concentration > 25 mg/L not requiring admission to ICU at time of admission; mild-to-moderate COVID-19 pneumonia with a WHO-CPS score of 5 points, receiving at least 3 L/min of oxygen but without ventilation assistance (eg, high-flow oxygen, non-invasive ventilation, or mechanical ventilation) were randomized to receive anakinra (200 mg twice a day on days 1–3, 100 mg twice on day 4, 100 mg once on day 5) with standard care or only standard care (i.e., at the discretion of the site clinicians). At baseline patients were mostly treated with anticoagulants (59% intervention, 53% control), Azithromycin (19% intervention, 25% control) or glucocorticoids (12% intervention, 15% control). In total 116 patients were included, but two of them withdrew consent. Therefore 59/114 (51.7%) were randomized to the intervention group, and 55/114 (48.2%) to the control group. The median age (IQR) was 67 (55.5 to 74) and 65 (59.5 to 78) in the intervention and control group, respectively. The length of the follow-up was 90 days. The following relevant outcome measures were included; mortality (i.e., at day 28), duration of hospitalization, respiratory support. The study reported two primary outcomes; proportion of patients who had died or needed non-invasive or mechanical ventilation by day 4 (i.e., 21/59 (35.6%) patients in the intervention group vs. 21/55 (38.2%) controls (difference -2.5% (95%CI -17.1 to 12.0)), and survival with no need for mechanical or non-invasive ventilation at day 14 (i.e., 28/59 (47%) patients in the intervention group vs. 28/55 (51%) controls (RR 0.97 (95%CI 0.62 to 1.52)). Importantly the current study was stopped early as no (positive) clinical effect was shown at the interim analysis.
Canakinumab
Caricchio (2021) (CAN-COVID) described a phase 3, double-blind placebo-controlled, multicenter randomized controlled trial, which was conducted in 39 hospitals in Europe and the US. They evaluated the efficacy and safety of canakinumab in patients hospitalized with severe COVID-19. In total 454 patients were included as they met the severity criteria (e.g., had hypoxemia but did not require invasive mechanical ventilation (IMV); diagnosis of infection with SARS-CoV-2 within 7 days prior to randomization; diagnosis of pneumonia with pulmonary infiltrates on chest x-ray or computed tomographic scan within 5 days prior to randomization; peripheral capillary oxygen saturation of 93% or less on room air or arterial oxygen partial pressure/fraction of inspired oxygen less than 300). These patients were randomized to receive canakinumab or placebo. In total 227 patients were included in the intervention group, but 2 withdrew consent. Also 227 patients were included in the placebo group, but 4 patients did not receive placebo. The median (IQR) age was 59 (49-69) years in the intervention group, compared with 57 (50-68) years in the placebo group. In the intervention group 135/227 (59.4%) were males, compared with 132/227 (58.1%) in the control group. Patients in the intervention group received a single dose of canakinumab (450 mg for body weight of 40-<60 kg, 600 mg for 60-80 kg, and 750 mg for >80 kg, see Table 1), and patients in the control group received placebo. In addition, all patients continued to receive standard care treatment for COVID-19 per local practice. Medication which was initiated prior to day 1 was heparin (any dose, 73% both groups), dexamethasone (≥6 mg/d; 41% intervention, 32% placebo), azithromycin (37% both groups), remdesivir (22% intervention, 20% placebo), hydroxychloroquine (14% intervention, 13% control), and convalescent plasma or serum (3.5% in both groups). The length of the follow-up was 127 days, but an interim analysis is reported with a follow-up of 29 days in the current publication. The following relevant outcome measures were included; mortality, duration of hospitalization, respiratory support. The primary outcome was survival without IMV from day 3 to day 29. This was achieved in 198/223 (88.8%) patients in the intervention group, compared with 191/223 (85.6%) patients in the placebo group. This resulted in a difference of 3.1 % (95%CI -3.1 to 9.3).
Cremer (2021) (Three C study) described a double-blind, multicenter randomized controlled trial, which was conducted in 5 hospitals in Cleveland (the US). They evaluated the efficacy and safety of canakinumab in patients hospitalized with COVID-19. In total 45 patients were included as they met the inclusion criteria (e.g., hospitalized due to COVID-19 infection, documented SARS-CoV2 acute myocardial injury: Defined as upper respiratory tract specimen positive for COVID-19 and troponin greater than 99th percentile upper reference range without signs or symptoms of acute myocardial ischemia, NT-proBNP or BNP (brain natriuretic peptide) higher than upper reference limit, receiving current standard therapy, C-reactive protein (CRP) > 50 mg/L). These patients were randomized to receive canakinumab (600 mg [i.e., I1] or 300 mg [i.e., I2]) or placebo. Fifteen patients were included in the I1, 14 patients in I2, and 16 patients in the placebo group. The median (IQR) age was 68.8 (56.1-74.3) years, and 33/45 (73.3%) were males. Patients in the intervention group received a single dose of canakinumab (see Table 1), and patients in the placebo group received placebo. In addition, all patients continued to receive standard care treatment for COVID-19 per local practice. Medication which was initiated was antibiotics related to non-COVID-19 infections, antivirals related to COVID-19, corticosteroids, use of convalescent plasma, and other immunosuppressive agents. The length of the follow-up was 28 days. The following relevant outcome measures were included; mortality, respiratory support, and time to clinical improvement. The primary outcome was time to clinical improvement up to Day 14, defined as the time in days from randomization to either an improvement of two points on seven category ordinal scale or discharge from the hospital, whichever occurred first. This was achieved in 9/15 (60.0%) of the patients in I1, 7/14 (58.1%) patients in I2, and 9/16 (58.3%) in the control group. This resulted in a relative risk of 1.15 (95% CI 0.46 to 2.91) for I1 vs. control, and 0.61 (95% CI 0.23 to 1.64) for I2 vs. control.
Note: The authors published the study design and baseline characteristics of the first 20 patients earlier (Sheng, 2020). In the current summary of literature, the study of Cremer (2021) was used, which reported results of the primary and secondary endpoints of 30 patients.
Table 1. Overview of RCTs comparing IL-1 inhibitors with standard care (or placebo) in hospitalized COVID-19 patients.
|
Author |
Disease severity, based on need for respiratory support* |
Sample size |
Dosage |
|
Anakinra |
|||
|
Mixed: The majority of the patients needed administration of oxygen at baseline. Intervention: oxygen; N=50 - on non-invasive ventilation or high flow oxygen devices; N=44 - on invasive mechanical ventilation; N=17
Control: oxygen; N=119 - on non-invasive ventilation or high flow oxygen devices; N=84 - on invasive mechanical ventilation; N=22 |
I: N=112 C: N=230 Total: N=342 |
Patients allocated to the active drug were daily injected with 100 mg of anakinra in addition to standard care until discharge or a maximum of 28 days.
For the second randomization, a subgroup received siltuximab (11 mg/kg i.v. [single injection]) OR tocilizumab (8 mg/kg i.v. [not exceeding 800 mg; single injection]) |
|
|
Mixed: Moderate I; 3/15 (20%) Severe: I; 12/15 (80%) |
I: N=15 C: N=15 Total: N=30 |
Patients allocated to the active drug were daily injected with 100mg of anakinra in addition to standard care until discharge or a maximum of 14 days. |
|
|
Mixed: The majority of the patients needed administration of oxygen at baseline. Although patients were excluded if ratio or partial oxygen pressure to fraction of inspired oxygen less than 150 mmHg or need for NIV (CPAP or BPAP) or mechanical ventilation. |
I: N=405 C: N=189 Total: N=594 |
Patients allocated to the active drug were daily injected with 100 mg of anakinra for 7 to 10 days. |
|
|
Mariette (2021) |
Moderate: Mild-to-moderate COVID-19 pneumonia with a WHO-CPS score of 5 points, receiving at least 3 L/min of oxygen but without ventilation assistance (e.g., high-flow oxygen, non-invasive ventilation, or mechanical ventilation). |
I: 59 C: 55 Total: 114 |
Patients allocated to the active drug received anakinra (200 mg twice a day on days 1–3, 100 mg twice on day 4, 100 mg once on day 5) |
|
Canakinumab |
|||
|
Caricchio (2021) |
Mixed: The majority of the patients needed administration of oxygen at baseline. Regarding the WHO ordinal SCALE I: Intervention: - non-invasive ventilation or high-flow oxygen; N=52
Control: - non-invasive ventilation or high-flow oxygen; N=52 |
I: N=227 C: N=227 Total: N=454 |
Patients allocated to the active drug received a single dose of canakinumab (450 mg for body weight of 40-<60 kg, 600 mg for 60-80 kg, and 750 mg for >80 kg) |
|
Cremer (2021) |
Mixed: the majority needed supplementary oxygen at baseline: - requiring invasive mechanical ventilation: 22.2% - requiring nasal high-flow oxygen and/or non-invasive ventilation: 24.4% - requiring supplemental oxygen: 44.4% - not requiring oxygen: 8.9% |
I1: N=15 I2: N=14 C: N=16 Total: N= 45 |
Patients allocated to the active drug received a single dose of canakinumab. Intervention group 1 received 600 mg, and intervention group 2 received 300 mg. |
*Disease severity categories:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen [high flow nasal cannula (HFNC)/Optiflow], continuous positive airway pressure [CPAP], non-invasive ventilation [NIV], mechanical ventilation, extracorporeal membrane oxygenation [ECMO or ECLS]).
N: Total sample size; I: Intervention; C: Control
Results – IL-1 inhibitors
Since the number of studies and the number of patients included was relatively limited, we report all outcomes for the different IL-1 inhibitors combined. Also conclusions were reported for all IL-1 inhibitors combined.
Caricchio (2021) and Cremer (2021) included patients treated with canakinumab, the other studies treated patients with anakinra. Importantly, Cremer (2021) studied two interventions, namely 600 mg canakinumab or 300 mg canakinumab. Outcomes were only reported separately. Therefore, as 300 mg is a reduced dose, the 600 mg canakinumab was used in the main analysis, and the results for the 300 mg intervention were reported as ‘additional analysis’.
Mortality (crucial)
Caricchio (2021), Cremer (2021), Kyriazopoulou (2021) and Mariette (2021) reported mortality at day 28 in hospitalized patients with COVID-19. The results are presented separately for a follow-up of 28-30 days. Declercq (2021) reported mortality during the study (i.e., follow-up of 10-20 weeks), and the estimated mortality at day 28 based on Kaplan-Meier. Kharazmi (2021) reported mortality at day 14. These results are described separately.
Caricchio (2021) reported the mortality for patients with a mild, moderate, or severe disease state. The mortality was 12/227 (5.3%) in the intervention group, compared with 16/227 (7.0%) in the control group. The RR was 0.75 (95%CI 0.36 to 1.55), and RD was -0.02 (95%CI -0.06 to 0.03). This is not considered clinically relevant.
Cremer (2021) reported the mortality for patients with a mild, moderate, or severe disease state. The incidence was 1/15 (6.7%) in the intervention group, compared with 3/16 (19.0%) in the control group. The RR was 0.36 (95%CI 0.04 to 3.05), and RD was -0.12 (95%CI -0.35 to 0.11). This is considered clinically relevant.
Additional analysis 300 mg canakinumab
The incidence of mortality was 3/14 (21.4%) in patients treated with canakinumab 300mg, compared with 3/16 (18.8%) in the control group. This resulted in a RR of 1.14 (95% CI 0.27 to 4.78) and a RD of -0.02 (95%CI -0.06 to 0.03).
Kyriazopoulou (2021) reported the mortality specifically for patients with a mild or moderate disease state. The incidence was 13/405 (3.2%) in the intervention group, compared with 13/89 (6.9%) in the control group. The RR was 0.47 (95%CI 0.22 to 0.99), and RD was -0.04 (95%CI -0.08 to 0.00). This is considered clinically relevant.
Mariette (2021) reported the mortality specifically for patients with a moderate disease state. The incidence was 13/59 (22.0%) in the intervention group, compared with 13/55 (23.6%) in the control group. The RR was 0.93 (95%CI 0.47 to 1.83), and RD was -0.02 (95%CI -0.17 to 0.14). This is not considered clinically relevant.
Figure 1: Mortality (28-30days) in hospitalized patients
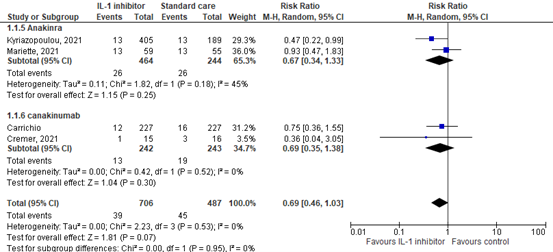
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Mortality, during the study
Declercq (2021) reported the number of deaths in the subgroups at a follow-up of 10-20 weeks. The incidence of mortality was 10/44 (22.7%) in patients treated with only additional anakinra, compared with 9/74 (12.2%) in the control group (i.e., receiving only usual care according to the randomizations). This resulted in a RR of 1.87 (95% CI 0.82 to 4.24) and a RD of 0.11 (95% CI -0.04 to 0.25). This is considered clinically relevant.
The incidence of mortality was 5/32 (15.6%) in patients treated with anakinra + tocilizumab, 6/36 (16.7%) in patients treated with anakinra + siltuximab, 10/81 (12.3%) in patients treated with usual care + tocilizumab, and 15/75 (20.0%) in patients treated with usual care + siltuximab.
In addition, the estimated mortality (i.e., based on Kaplan-Meier) at day 28 resulted in 16% (95% CI 8 to 31) in the intervention group, compared with 10% (95% CI 5 to 20) in the control group. This is considered clinically relevant, in favour of the control group.
Mortality, 14 days
Kharazmi (2021) reported the mortality at day 14 for patients with a moderate, or severe disease state. The incidence of mortality was 5/15 (33.3%) in the intervention group, compared with 7/15 (46.7%) in the control group. The RR was 0.71 (95%CI 0.29 to 1.75), and RD was -0.13 (95%CI -0.48 to 0.21). This is considered clinically relevant.
Note: It was not possible to perform stratified analyses per disease severity category as these results could not be obtained in the publications. Most of the studies included patients with mild, moderate or severe disease state at baseline, see Table 1.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 2 levels, because of inconsistency (1 level, due to considerable heterogeneity between the studies regarding medication and/or dose), and imprecision (1 level, 95% CI of the mean difference includes no effect [RD=0]). Therefore, level of evidence for the outcome ‘mortality’ is considered low.
Extensive respiratory support (crucial)
Initiation of mechanical respiratory support in hospitalized patients with COVID-19 was reported in six studies (Caricchio, 2021; Cremer, 2021; Declercq, 2021; Kharazmi, 2021; Kyriazopoulou, 2021; Mariette, 2021). In the study of Caricchio (2021) this was defined as (1) intubation and mechanical ventilation and (2) ventilation plus additional organ support (vasopressors, kidney replacement therapy [KRT], ECMO). Cremer (2021) defined this outcome as requiring high flow nasal cannula (HFNC)/Optiflow, continuous positive airway pressure (CPAP), non-invasive ventilation (NIV), mechanical ventilation, extracorporeal membrane oxygenation (ECMO or ECLS). Declercq (2021) defined this outcome as number of invasive ventilation days. Kharazmi (2021) defined this as number of patients requiring (1) invasive mechanical ventilation or extracorporeal membrane oxygenation, or (2) non-invasive -ventilation or high flow oxygen, at day 7 and 14. This was defined as (1) the need for high-flow oxygen or non-invasive ventilation, (2) initiation of mechanical ventilation with P/F >150 mmHg, (3) initiation of mechanical ventilation with P/F <150 mmHg or vasopressors, (4) initiation of mechanical ventilation with P/F < 150 mmHg and vasopressors or haemodialysis or ECMO in the study of Kyriazopoulou (2021). In the study of Mariette (2021) this outcome was defined as the proportion of patients who had died or needed non-invasive or mechanical ventilation by day 14 (i.e., a score of >5 points on the WHO-CPS) at day 14.
Caricchio (2021) reported initiation of respiratory support in 2 categories. In the intervention group 5/227 (2.2%) needed intubation and mechanical ventilation, compared with 2/227 (0.8%) in the control group. This resulted in a RR of 2.50 (95% CI 0.49 to 12.75) and a RD of 0.01 (95%CI -0.01 to 0.04). This is not considered clinically relevant.
In addition, 2/227 (0.8%) patients in the intervention group needed ventilation plus additional organ support (pressors, KRT, ECMO), compared with 8/227 (3.5%) in the control group. This resulted in a RR of 0.25 (95% CI 0.05 to 1.16) and a RD of -0.03 (95% CI -0.05 to 0.01). This is not considered clinically relevant (<-0.03, due to rounding displayed as -0.03).
Cremer (2021) reported the number of patients requiring invasive respiratory support at day 14 and day 28. At day 14, 5/16 (31.1%) patients in the intervention group required invasive respiratory support, compared with 2/14 (14.3%) in the control group. This resulted in a RR of 2.19 (95%CI 0.50 to 9.56) and a RD of 0.17 (95% CI -0.12 to 0.46). However, two patients in the control group died before day 14. At day 28, 1/15 (6.3%) patients in the intervention group required invasive respiratory support, compared with 1/13 (7.7%) in the control group. This resulted in a RR of 0.87 (95% CI 0.06 to 12.52) and a RD -0.01 (95%CI -0.20 to 0.18). However, one patient died in the intervention group, and three patients in the control group.
Additional analysis 300 mg canakinumab
At day 14, 1/11 (9.1%) patients in the intervention group required invasive respiratory support, compared with 2/14 (14.3%) in the control group. This resulted in a RR of 0.64 (95% CI 0.07 to 6.14) and a RD of -0.05 (95% CI -0.30 to 0.20). However, three patients died in the intervention group, and two patients in the control group.
At day 28, 1/11 (9.1%) patients in the intervention group required invasive respiratory support, compared with 1/13 (7.7%) in the control group. This resulted in a RR of 1.18 (95% CI 0.08 to 16.78) and a RD 0.01 (95% CI -0.21 to 0.24). Three patients died in the intervention group, and three patients in the control group.
Declercq (2021) reported the mean number of invasive ventilator days, which was 5 (95%CI 3 to 9) in the intervention group (i.e., IL-1 blockade group), compared with 5 (95%CI 3 to 7) days in the control group (i.e., no IL-1 blockade group). This resulted in a mean difference of 0 days. This is not considered clinically relevant.
Kharazmi (2021) reported initiation of respiratory support at 2 different time points.
In the intervention group 2/15 (13.3%) patients were on invasive mechanical ventilation or extracorporeal membrane oxygenation or on non-invasive ventilation or high flow oxygen, compared with 5/15 (33.3%) in the control group at day 7. This resulted in a RR of 0.40 (95% CI 0.092 to 1.749) and a RD of -0.20 (95% CI -0.49 to 0.09). This is considered clinically relevant.
At day 14, the incidence was 0/15 in the intervention group, compared with 4/15 (26.7%) in the control group. This resulted in a RR of 0.11 (95% CI 0.01 to 1.90) and a RD of -0.27 (95% CI -0.50 to -0.03). This is considered clinically relevant.
Kyriazopoulou (2021) reported initiation of respiratory support in 4 categories. Results are provided in Table 2. The results are not considered clinically relevant.
Table 2. Results for the outcome respiratory support per category – mild and moderate disease.
|
Category |
Intervention (n/N) |
Control (n/N) |
RR (95% CI) |
RD (95% CI) |
|
the need for high-flow oxygen or non-invasive ventilation |
1/405 |
1/189 |
0.47 (0.03 to 7.42) |
-0.01 |
|
initiation of mechanical ventilation with P/F >150 mmHg |
1/405 |
1/189 |
0.47 (0.03 to 7.42) |
-0.01 |
|
initiation of mechanical ventilation with P/F <150 mmHg or vasopressors |
5/405 |
4/189 |
0.58 (0.16 to 2.15) |
-0.01 |
|
initiation of mechanical ventilation with P/F < 150 mmHg and vaso-pressors or haemodialysis or ECMO |
6/405 |
6/189 |
0.47 (0.15 to 1.43) |
-0.02 |
Mariette (2021) reported number of patients needing non-invasive ventilation, mechanical ventilation or died up to day 14. In the intervention group 28/59 (47.5%) achieved this outcome, compared with 28/55 (50.9%) in the control group. This resulted in a hazard ratio (HR; adjusted for age and centre) of 0.97 (95% CI 0.62 to 1.51).
By excluding the patients who died up to day 14, 19/50 (38.0%) patients in the intervention group, compared with 15/42 (35.7%) in the control group needed respiratory support. This resulted in a RR of 1.06 (95% CI 0.62 to 1.82) and a RD of 0.02 (95% CI -0.18 to 0.22). This is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 3 levels because of inconsistency (1 level, due to heterogeneity between the studies regarding medication and/or dose) and imprecision (2 levels, 95%CI of the mean difference includes no effect (RD=0), crosses both thresholds of clinical relevance, and low number of cases). The level of evidence for the outcome ‘respiratory support’ is considered very low.
Duration of hospitalization (important)
Duration of hospitalization in hospitalized patients with COVID-19 was reported in three studies (Caricchio, 2021 Kyriazopoulou, 2021; Mariette, 2021). Caricchio (2021) and Mariette (2021) defined this outcome as the number of patients which discharged at day 29 and 28, respectively. Kyriazopoulou (2021) defined this outcome as time to hospital discharge in days. In addition, time of ICU stay was reported in this study. Declercq (2021) defined this outcome as median number of days in the hospital and ICU, respectively. Kharazmi (2021) defined this outcome as length of hospital stay and ICU length of stay.
Caricchio (2021) reported that 199/211 (94.3%) patients in the intervention group discharged at day 29, compared with 187/206 (90.8%) in the control group. This resulted in a RR of 1.04 (95% 0.98 to 1.10) and a RD of 0.04 (95%CI -0.02 to 0.09). This is not clinically relevant.
Declercq (2021) reported that the mean number of days in hospital was 19 (95% CI 17 to 22) in the intervention group (i.e., IL-1 blockade group), compared with 19 (95% CI 17 to 21) days in the control group (i.e., no IL-1 blockade group). This resulted in a mean difference of 0 days, which is not considered clinically relevant. Regarding the number of days at ICU, this was 11 (95% CI 8 to 15) in the intervention group, compared with 10 (95% CI 8 to 13) in the control group. This resulted in a mean difference of -1 day, which is not considered clinically relevant.
Kharazmi (2021) reported that the mean (±SD) days of hospital stay was 9.5 (±4.5) in the intervention group (n=15), compared with 19 (±12) in the control group (n=15). This resulted in a mean difference of -9.5 days (95% CI -16 to -3). This is considered clinically relevant.
Regarding the length of stay at ICU, the mean (±SD) in days was 5.5 (±2) in the intervention group, compared with 16.5 (±9) in the control group. This resulted in a mean difference of -11 days (95%CI -15.5 to -6.5). This difference is considered clinically relevant.
Kyriazopoulou (2021) reported the median (IQR) time in days, which was 11 (7.8) days, compared with 12 (8.5) days in the control group. The difference is 1 day, this is not considered clinically relevant. The median (IQR) time of ICU stay was 10 (21) days in the intervention group, and 14 (22) days in the control groups. The difference is 4 days, this is considered clinically relevant.
Mariette (2021) reported that 34/59 (57.6%) patients in the intervention group discharged at day 28, compared with 34/55 (61.8%) in the control group. This resulted in a RR of 0.93 (95% CI 0.69 to 1.26) and a RD of -0.04 (95% CI -0.22 to 0.14). This is not clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 2 levels because of inconsistency (1 level, due to heterogeneity between the studies regarding medication and dose), and imprecision (1 level, 95%CI of the mean difference includes no effect [RD=0]). The level of evidence for the outcome ‘duration of hospitalization’ is considered low.
Time to clinical improvement (important)
Time to clinical improvement in hospitalized patients with COVID-19 was reported in two studies (Cremer, 2021; Declercq, 2021).
Cremer (2021) reported the number of patients who achieved clinical improvement or discharge. This was defined as the time in days from randomization to either an improvement of two points on seven category ordinal scale or discharge from the hospital, whichever occurred first, at day 28. This was achieved in 14/15 (93.5%) of the patients in the intervention group, compared with 11/16 (68.8%) of the patients in the control group. This resulted in a RR of 1.36 (95% CI 0.95 to 1.94), and a RD of 0.25 (95% CI -0.01 to 0.51). This is considered clinically relevant.
Additional analysis 300 mg canakinumab
This was achieved in 11/14 (78.6%) of the patients in the intervention group, compared with 11/16 (68.8%) of the patients in the control group. This resulted in a RR of 1.14 (95% CI 0.74 to 1.75), and a RD of 0.10 (95% CI -0.21 to 0.41). This is considered clinically relevant.
Declercq (2021) reported the median (95% CI) time to clinical improvement in days. This was 12 (10 to 16) days in the intervention group (i.e., IL-1 blockade group), compared to 12 (10 to 15) days in the control group. This resulted in a HR of 0.94 (95% CI 0.73 to 1.21). The absolute difference of 0 days is not considered clinically relevant.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by 3 levels because of risk of bias (1 level, selection bias might be present), and imprecision (2 levels, 95% CI of the mean difference includes no effect [HR=0, RD=0], crosses both thresholds of clinically relevance, and/or not meeting the optimal information size). The level of evidence for the outcome ‘time to clinical improvement’ is considered very low.
A systematic review of the literature was performed to answer the following question:
What is the effectivity of treatment with an interleukin (IL-)1 inhibitor compared to treatment without an interleukin (IL-)1 inhibitor in patients with COVID-19?
PICO
P: hospitalized with COVID-19 (subgroups mild, moderate, severe)
I: IL-1 inhibitor (i.e., anakinra or canakinumab) + standard care
C: standard care only / placebo treatment + standard care
O: 28-30 day mortality (if not available, any other reports of mortality), extensive respiratory support, duration of hospitalization, time to clinical improvement
Relevant outcome measures
For hospitalized COVID-19 patients, mortality and need for extensive respiratory support were considered as crucial outcome measures for decision making. Duration of hospitalization, and time to clinical improvement were considered as important outcome measures for decision making.
Extensive respiratory support was defined as high flow nasal cannula (HFNC)/Optiflow, continuous positive airway pressure (CPAP), non-invasive ventilation (NIV), mechanical ventilation or extracorporeal membrane oxygenation (ECMO or ECLS).
The working group defined 3% points absolute difference as a minimal clinically important difference for mortality (resulting in a NNT of 33), 3 days difference for duration of hospitalization and time to clinical improvement, 5% points absolute difference need for respiratory support and ICU admission (resulting in a NNT of 20).
Studies of hospitalized patients were categorized based on the respiratory support that was needed at baseline (preferably based on patient inclusion/exclusion criteria; otherwise on baseline characteristics). The following categories were used:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen [high flow nasal cannula (HFNC)/Optiflow], continuous positive airway pressure [CPAP], non-invasive ventilation [NIV], mechanical ventilation, extracorporeal membrane oxygenation [ECMO or ECLS]).
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until 20 January 2022. The detailed search strategy is outlined under the tab Methods. Studies were selected based on the following criteria: randomized controlled trial, peer reviewed and published in indexed journal, comparing treatment with an IL-1 inhibitor and standard care to standard care alone or treatment with an IL-1 inhibitor and standard care to placebo and standard care in patients with COVID-19. Studies with n<10 were excluded.
The systematic literature search resulted in 80.255 hits. Studies were further selected based on the following criteria: only systematic review or randomized controlled trials were included. Eventually, 5 studies were included. One additional study, which was not found by the literature search as the journal was not included in the databases, was included on request of the working group.
Statistical methods
Statistical analyses were conducted using Review Manager (RevMan) software 5.4. For dichotomous outcomes, Mantel Haenszel random‐effects risk ratios (RRs) and risk differences (RDs) were calculated. For continuous outcomes, a random‐effects mean difference (MD) weighted by the inverse variance was calculated. The random-effects model estimates the mean of a distribution of effects.
Results
In total, six studies were included in the analysis of the literature. Important study characteristics and results are summarized in the evidence tables. Studies are presented in alphabetical order, only results of the primary outcome is reported in the summary of literature. The assessment of the risk of bias is summarized in the risk of bias tables.
- Caricchio R, Abbate A, Gordeev I, Meng J, Hsue PY, Neogi T, Arduino R, Fomina D, Bogdanov R, Stepanenko T, Ruiz-Seco P, Gónzalez-García A, Chen Y, Li Y, Whelan S, Noviello S; CAN-COVID Investigators. Effect of Canakinumab vs Placebo on Survival Without Invasive Mechanical Ventilation in Patients Hospitalized With Severe COVID-19: A Randomized Clinical Trial. JAMA. 2021 Jul 20;326(3):230-239. doi: 10.1001/jama.2021.9508. PMID: 34283183; PMCID: PMC8293025.
- Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, Oltolini C, Castiglioni B, Tassan Din C, Boffini N, Tomelleri A, Farina N, Ruggeri A, Rovere-Querini P, Di Lucca G, Martinenghi S, Scotti R, Tresoldi M, Ciceri F, Landoni G, Zangrillo A, Scarpellini P, Dagna L. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020 Jun;2(6):e325-e331. doi: 10.1016/S2665-9913(20)30127-2. Epub 2020 May 7. PMID: 32501454; PMCID: PMC7252085.
- Cremer PC, Sheng CC, Sahoo D, Dugar S, Aguillon Prada R, Kai Ming Wang T, Abou Hassan OK, Hernandez-Montfort J, Wolinsky DA, Culver DA, Rajendram P, Duggal A, Brennan DM, Wolski KE, Lincoff AM, Nissen SE, Menon V, on behalf of the Three C study group, Double-blind randomized proof-of-concept trial of canakinumab in patients with COVID-19 associated cardiac injury and heightened inflammation, European Heart Journal Open, Volume 1, Issue 1, August 2021, oeab002
- CORIMUNO-19 Collaborative group (Mariette, 2021). Effect of anakinra versus usual care in adults in hospital with COVID-19 and mild-to-moderate pneumonia (CORIMUNO-ANA-1): a randomised controlled trial. Lancet Respir Med. 2021 Mar;9(3):295-304. doi: 10.1016/S2213-2600(20)30556-7. Epub 2021 Jan 22. PMID: 33493450; PMCID: PMC7825875.
- Davidson M, Menon S, Chaimani A, Evrenoglou T, Ghosn L, Graña C, Henschke N, Cogo E, Villanueva G, Ferrand G, Riveros C, Bonnet H, Kapp P, Moran C, Devane D, Meerpohl JJ, Rada G, Hróbjartsson A, Grasselli G, Tovey D, Ravaud P, Boutron I. Interleukin-1 blocking agents for treating COVID-19. Cochrane Database Syst Rev. 2022 Jan 26;1(1):CD015308. doi: 10.1002/14651858.CD015308. PMID: 35080773; PMCID: PMC8791232.
- Declercq J, Van Damme KFA, De Leeuw E, Maes B, Bosteels C, Tavernier SJ, De Buyser S, Colman R, Hites M, Verschelden G, Fivez T, Moerman F, Demedts IK, Dauby N, De Schryver N, Govaerts E, Vandecasteele SJ, Van Laethem J, Anguille S, van der Hilst J, Misset B, Slabbynck H, Wittebole X, Liénart F, Legrand C, Buyse M, Stevens D, Bauters F, Seys LJM, Aegerter H, Smole U, Bosteels V, Hoste L, Naesens L, Haerynck F, Vandekerckhove L, Depuydt P, van Braeckel E, Rottey S, Peene I, Van Der Straeten C, Hulstaert F, Lambrecht BN. Effect of anti-interleukin drugs in patients with COVID-19 and signs of cytokine release syndrome (COV-AID): a factorial, randomised, controlled trial. Lancet Respir Med. 2021 Dec;9(12):1427-1438. doi: 10.1016/S2213-2600(21)00377-5. Epub 2021 Oct 29. PMID: 34756178; PMCID: PMC8555973.
- Derde L, & REMAP-CAP Investigators. (2021). Effectiveness of tocilizumab, sarilumab, and anakinra for critically ill patients with COVID-19 the REMAP-CAP COVID-19 immune modulation therapy domain randomized clinical trial. medRxiv.
- Kharazmi AB, Moradi O, Haghighi M, Kouchek M, Manafi-Rasi A, Raoufi M, Shoaei SD, Hadavand F, Nabavi M, Miri MM, Salarian S, Shojaei S, Khalili S, Sistanizad M, Sadeghi S, Karagah A, Asgari S, Jaffaraghaei M, Araghi S. A randomized controlled clinical trial on efficacy and safety of anakinra in patients with severe COVID-19. Immun Inflamm Dis. 2021 Nov 11. doi: 10.1002/iid3.563. Epub ahead of print. PMID: 34762351.
- Kharazmi AB, Moradi O, Haghighi M, Kouchek M, Manafi-Rasi A, Raoufi M, Shoaei SD, Hadavand F, Nabavi M, Miri MM, Salarian S, Shojaei S, Khalili S, Sistanizad M, Sadeghi S, Karagah A, Asgari S, Jaffaraghaei M, Araghi S. A randomized controlled clinical trial on efficacy and safety of anakinra in patients with severe COVID-19. Immun Inflamm Dis. 2021 Nov 11. doi: 10.1002/iid3.563. Epub ahead of print. PMID: 34762351.
- Kyriazopoulou E, Poulakou G, Milionis H, Metallidis S, Adamis G, Tsiakos K, Fragkou A, Rapti A, Damoulari C, Fantoni M, Kalomenidis I, Chrysos G, Angheben A, Kainis I, Alexiou Z, Castelli F, Serino FS, Tsilika M, Bakakos P, Nicastri E, Tzavara V, Kostis E, Dagna L, Koufargyris P, Dimakou K, Savvanis S, Tzatzagou G, Chini M, Cavalli G, Bassetti M, Katrini K, Kotsis V, Tsoukalas G, Selmi C, Bliziotis I, Samarkos M, Doumas M, Ktena S, Masgala A, Papanikolaou I, Kosmidou M, Myrodia DM, Argyraki A, Cardellino CS, Koliakou K, Katsigianni EI, Rapti V, Giannitsioti E, Cingolani A, Micha S, Akinosoglou K, Liatsis-Douvitsas O, Symbardi S, Gatselis N, Mouktaroudi M, Ippolito G, Florou E, Kotsaki A, Netea MG, Eugen-Olsen J, Kyprianou M, Panagopoulos P, Dalekos GN, Giamarellos-Bourboulis EJ. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med. 2021 Oct;27(10):1752-1760. doi: 10.1038/s41591-021-01499-z. Epub 2021 Sep 3. Erratum in: Nat Med. 2021 Oct 8;: PMID: 34480127; PMCID: PMC8516650.
- Mertens M, Singh JA. Anakinra for rheumatoid arthritis. Cochrane Database Syst Rev. 2009 Jan 21;(1):CD005121. doi: 10.1002/14651858.CD005121.pub3. PMID: 19160248.
- Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, Sadoff JC, Slotman GJ, Levy H, Balk RA, Shelly MP, Pribble JP, LaBrecque JF, Lookabaugh J, Donovan H, Dubin H, Baughman R, Norman J, DeMaria E, Matzel K, Abraham E, Seneff M. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997 Jul;25(7):1115-24. doi: 10.1097/00003246-199707000-00010. PMID: 9233735.
- Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, Cron RQ, Opal SM. Interleukin-1 Receptor Blockade Is Associated With Reduced Mortality in Sepsis Patients With Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit Care Med. 2016 Feb;44(2):275-81. doi: 10.1097/CCM.0000000000001402. PMID: 26584195; PMCID: PMC5378312.
- Sheng CC, Sahoo D, Dugar S, Prada RA, Wang TKM, Abou Hassan OK, Brennan D, Culver DA, Rajendram P, Duggal A, Lincoff AM, Nissen SE, Menon V, Cremer PC. Canakinumab to reduce deterioration of cardiac and respiratory function in SARS-CoV-2 associated myocardial injury with heightened inflammation (canakinumab in Covid-19 cardiac injury: The three C study). Clin Cardiol. 2020 Oct;43(10):1055-1063. doi: 10.1002/clc.23451. Epub 2020 Aug 24. PMID: 32830894; PMCID: PMC7461303.
- van de Veerdonk FL, Giamarellos-Bourboulis E, Pickkers P, Derde L, Leavis H, van Crevel R, Engel JJ, Wiersinga WJ, Vlaar APJ, Shankar-Hari M, van der Poll T, Bonten M, Angus DC, van der Meer JWM, Netea MG. A guide to immunotherapy for COVID-19. Nat Med. 2022 Jan;28(1):39-50. doi: 10.1038/s41591-021-01643-9. Epub 2022 Jan 21. PMID: 35064248.
PICO: What is the effectivity of treatment with an IL-1 inhibitor compared to treatment without an IL-1 inhibitor in patients with COVID-19?
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Anakinra |
|||||||
|
Kyriazopoulou, 2021 |
Type of study: Prospective, doubleblind randomized controlled trial.
Setting: 37 study sites
Country: 29 sites in Greece and eight in Italy.
Source of funding: Not reported.
Conflicts of interest: G.P. has received independent educational grants from Pfizer, MSD, Angelini and Bio -Rad. H.M. reports receiving honoraria, consulting fees and non -financial support from healthcare companies, including Amgen, Angelini, Bayer, Mylan, MSD, Pfizer and Servier. L.D. received consultation honoraria from Sobi. M.B. has received funds for research grants and/or advisor/consultant and/or speaker/chairman from Angelini, Astellas, Bayer, bioMérieux, Cidara, Cipla, Gilead, Menarini, MSD, Pfizer, Roche, Shionogi and Nabriva. M.G.N. is supported by an ERC Advanced Grant (no. 833247) and a Spinoza grant of the Netherlands Organization for Scientific Research. He has also received independent educational grants from TTxD, GSK and ViiV Healthcare. J.E. -O. is a co-founder, shareholder and CSO of ViroGates, Denmark, and is a named inventor on patients on suPAR owned by Copenhagen University Hospital Hvidovre, Denmark. P.P. has received honoraria from Gilead, Janssen and MSD. G.N.D. is an advisor or lecturer for Ipsen, Pfizer, Genkyotex, Novartis and Sobi, has received research grants from Abbvie and Gilead and has served as principal investigator in studies for Abbvie, Novartis, Gilead, Novo Nordisk, Genkyotex, Regulus Therapeutics, Tiziana Life Sciences, Bayer, Astellas, Pfizer, Amyndas Pharmaceuticals, CymaBay Therapeutics, Sobi and Intercept Pharmaceuticals. E.J.G.- B. has received honoraria from Abbott, bioMérieux, Brahms, GSK, InflaRx, Sobi and XBiotech; independent educational grants from Abbott, AxisShield, bioMérieux, InflaRx, Johnson & Johnson, MSD, Sobi and XBiotech; and funding from the Horizon 2020 Marie - Curie Project European Sepsis Academy (granted to the National and Kapodistrian University of Athens) and the Horizon 2020 European. Grants ImmunoSep and RISKinCOVID (granted to the Hellenic Institute for the Study of Sepsis). The other authors do not have any competing interests to declare.
|
Patients with COVID-19 at risk of progressing to respiratory failure
Inclusion criteria: • Adult patients of either sex; • For women, unwillingness of remain pregnant during the study period; • Confirmed infection by SARSCoV-2 by molecular test; • Findings in chest X-ray or chest computed tomography compatible with lower respiratory tract infection; • Need for hospitalization; • Plasma suPAR ≥6 ng ml-1.
Exclusion criteria: • Any stage IV malignancy; • Any do-not-resuscitate order; • Ratio or partial oxygen pressure to fraction of inspired oxygen less than 150 mmHg; • Need for NIV (CPAP or BPAP) or mechanical ventilation; • Any primary immunodeficiency; • Fewer than 1500 neutrophils per mm3; • Oral or intravenous intake of corticosteroids at a daily dose greater than or equal to 0.4 mg/kg-1 or prednisone for a period longer than the last 15 days; • Any anti-cytokine biological treatment including JAK inhibitors, during the last 1 month; • Severe hepatic failure; • End-stage renal failure necessitating hemofiltration or peritoneal hemodialysis; • Pregnancy of lactation.
N total at baseline: N = 594 Intervention: N = 405 Control: N = 189
Important characteristics: Age, mean (SD): I: 62.0 y (11.4) C: 61.5 y (11.3)
Sex, n/N (%)male:
Disease severity, mean (SD): Defined by WHO classification for COVID-19 at the time of screening (%)
Moderate pneumonia I: 39/405 (9.6%) C: 11/189 (5.8%)
Severe pneumonia I: 366/405 (90.4%) C: 178/189 (94.2%)
Groups comparable at baseline ? Yes. |
Anakinra
Patients allocated to the active drug were daily injected with 100mg of anakinra at a final volume of 0.67ml. |
Placebo
Patients allocated to placebo treatment were daily injected with 0.67ml of 0.9% sodium chloride. |
Length of follow up: Until day 28.
Loss to follow-up: Intervention: N = 1 Reasons: not reported.
Control: N = 0 Reasons: not reported. |
Clinical outcomes
Mortality day 28 I: 13/405 (3.2%) C: 13/189 (6.9%)
Duration of hospitalization Median (IQR) time to hospital discharge in days I: 11 (7.8) C: 12 (8.5) OR 1.22 (95% CI 1.02 to 1.47) P=0.033
Time to symptom resolution Not reported
Median (IQR) time of ICU stay in days I: 10 (21) C: 14 (22) OR 2.33 (95% CI 1.11 to 4.92) P=0.026
Respiratory support WHO-CPS by day 28 Fully recovered PCR -, n/N(%) I: 204/405 (50.4%) C: 50/189 (26.5%)
Asymptomatic PCR +, n/N(%) I: 40/405 (9.9%) C: 6/189 (3.2%)
Symptomatic independent, n/N (%) I: 93/405 (23.0%) C: 74/189 (39.2%)
Symptomatic assistance needed, n/N (%) I: 25/405 (6.2%) C: 21/189 (11.1%)
Hospitalized with no need for oxygen, n/N (%) I: 9/405 (2.2%) C: 3/189 (1.6%)
Hospitalized with nasal/mask oxygen, n/N (%) I: 8/405 (2.0%) C: 10/189 (5.3%)
Need for HFO or NIV, n/N (%) I: 1/405 (0.2%) C: 1/189 (0.5%)
Mechanical ventilation with P/F >150 mmHg, n/N (%) I: 1/405 (0.2%) C: 1/189 (0.5%)
MV with P/F <150 mmHg or vasopressors, n/N (%) I: 5/405 (1.2%) C: 4/189 (2.1%)
MV with P/F <150 mmHg and vasopressors or hemodialysis or ECMO, n/N (%) I: 6/405 (1.5%) C: 6/189 (3.2%)
Absolute decrease of WHO-CPS at day 28 from baseline day 1, median (IQR) I: 4 (2.0) C: 3 (2.5) OR 0.40 (95% CI 0.29 to 0.55) P<0.0001
Absolute decrease of WHO-CPS at day 14 from baseline day 1, median (IQR) I: 3 (2.0) C: 2 (3.0) OR 0.63 (95% CI 0.46 to 0.86) P=0.003
Absolute decrease of SOFA score at day 7 from baseline day 1, median (IQR) I: 1 (2) C: 0 (1) OR 0.64 (0.47 to 088) P=0.007
Safety Adverse events At least one serious adverse event , n/N (%) I: 65/405 (16.0%) C: 41/189 (21.7%) P=0.107
At least one non-serious adverse event, n/N (%) I: 335/405 (82.7%) C: 156/189 (82.5%) P=1.00
Virological outcomes Viral clearance Not reported. |
Definitions: WHO-CPS: World Health Organisation Clinical Progression Scale SOFA: Sequential Organ Failure Assessment Score suPAR: “denotes the presence of dangerassociated molecular patterns (DAMPs), namely calprotectin (S100A8/A9) and IL-1α3, 4, both of which contribute to pathogenic inflammation in COVID-19." Serious adverse events: infections and infestations, ventilator-associated pneumonia, septic shock and multiple organ dysfunction, bloodstream infection, probable hospital-acquired infection, hospital-acquired pneumonia, acute pyelonephritis and pulmonary embolism.
Adverse events: anemia, neutropenia, thrombocytopenia, rash at the infection site, constipation, diarrhea, increase of liver function tests, bradycardia, headache, anxiety, creatinine increase, hyperglycemia, hyponatremia, hypernatremia, hypokalemia, hyperkalemia and hypocalcemia.
Remarks: - Authors conclusion: In conclusion, the SAVE -MORE trial showed that early start of treatment with anakinra guided by suPAR levels in patients hospitalized with moderate and severe COVID -19 significantly reduced the risk of worse clinical outcome at day 28. |
|
Mariette, 2021 CORIMUNO-ANA-1 trial |
Type of study: RCT; open-label
Setting: 16 University hospitals; sub-study of CORIMUNO-trial
Country: France
Source of funding: The Ministry of Health, Programme Hospitalier de Recherche Clinique, Foundation for Medical Research, and AP-HP Foundation; The funders of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. |
Patients with mild-to-moderate, RT-PCR confirmed, COVID-19 Pneumonia
Inclusion criteria: CORIMUNO-trial overall: • confirmed SARS-CoV-2 infection (RT-PCR and/or chest CT) • mild-to-moderate, severe, or critical pneumonia (ie, receiving oxygen at a flow of >3 L/min via mask or nasal cannula and a score of ≥5 points on the WHO Clinical Progression Scale [WHO-CPS] 10-point ordinal scale, which is described in the appendix 2 [pp 12–13]).
CORIMUNO-ANA-1 trial: • C-reactive protein serum concentration > 25 mg/L not requiring admission to hospital ICU at time of admission • mild-to-moderate COVID-19 pneumonia with a WHO-CPS score of 5 points, receiving at least 3 L/min of oxygen but without ventilation assistance (eg, high-flow oxygen, noninvasive ventilation, or mechanical ventilation).
Exclusion criteria: • known hypersensitivity to anakinra or any of its excipients • pregnancy • current documented bacterial infection • absolute neutrophil count of 1·0 × 10⁹ per L or less • platelet concentration <50 G/L • serum aspartate aminotransferase or serum alanine aminotransferase > 5x upper limit of normal • severe renal insufficiency defined by an estimated glomerular filtration rate of < 30 mL/min. See full list of criteria in appendix 2
N total at baseline: N = 116 Intervention: 59 Control: 57 (55 included in baseline characteristics and analysis)
Important characteristics: Age, median (IQR): I: 67·0 (55·5–74·3) C: 64·9 (59·5–78·3) Sex, n/N (%) male: I: 43/59 (73%) C: 37/55 (67%)
Time from symptoms onset to randomisation, median (IQR) days I: 10·0 (8·0–13·0; n=59) C: 10·0 (7·0–13·0; n=54) Groups comparable at baseline. |
Anakinra + usual care Anakinra (Sobi, Puteaux, France); intravenously;
Day 1-3 after randomization: 200 mg twice a day (total 400 mg) Day 4: 100 mg twice a day (total 200 mg) Day 5: 100 mg once If no improvement was seen on the morning of day 4 (improvement was determined as a reduction in requirement of oxygen of more than 50%, but the decision was left to the treating physician), 3 supplementary days of treatment at 400 mg per day were done on days 4–6, followed by a decrease to 200 mg per day on day 7 and 100 mg per day on day 8, and no treatment thereafter.
Actual treatment: • All received 2–15 injections of anakinra (median 11 [IQR 9–15]). • Median dose of anakinra by • Perfusion: 180 mg (IQR 167–186) • 55 (93%) patients • received seven perfusions or more. • Median cumulative dose of anakinra was 1900 mg (1500–2700). |
Usual care
Usual care (antibiotic drugs, antiviral drugs, corticosteroids, vasopressor support, anticoagulants) was provided at the discretion of the site clinicians. |
Length of follow up: 90 days Loss to follow-up: I: 4/59 (6.8%) Reasons: lost contact C: 2/57 (3.5%) Reasons: withdrew consent |
Also reported: composite score for non-invasive ventilation, high-flow oxygen, mechanical ventilation, or death; mechanical ventilation of death at day 14;
Clinical outcomes Mortality Survival with no need for mechanical or non-invasive ventilation median posterior HR 0·97 (90% CrI 0·62 to 1·52)
Mortality, incidence, day 90 I: 16/59 patients C: 15/55 patients died
Overall survival; adjusted HR (95% CI) Day 14 0·56 (0·23–1·39) Day 28 0·77 (0·33–1·77) Day 90 overall survival 72% (95% CI 61 to 85) overall survival 72%, (95% CI 62 to 85) adjusted HR 0·97 (95% CI 0·46 to 2·04)
Duration of hospitalization Discharge from hospital, day 28 I: 34/59; 58% (95% CI 44– 69) C: 34/55; 62% (47–73) Adjusted HR 0·91 (95% CI 0·56 to 1·48)
Symptom resolution WHO-CPS score ≥ 5, day 4 I: 21/59 (36%) C: 21/55 (38%) Absolute risk diff –2·5% (90% CrI –17·1 to 12·0)
WHO-CPS score (10-point scale); median (IQR) and median posterior odds ratio adjusted for age and centre. Day 4 I: 5 (5 to 6) C: 5 (5 to 6) OR 0·80 (95% 0.38 to 1.68 Day 7 I: 5 (5 to 7) C: 5 (5 to 7) OR 0·69 (95% CrI 0·33 to 1·43) Day 14 I: 5 (2 to 8) C: 5 (3 to 8) OR 0·70 (95% CrI 0·35 to 1·38) Day 2 to 14 (longitudinal analysis) OR 0·92 (95% CrI 0·32 to 2·65)
Need for respiratory Support Oxygen independence, day 28 I: 63% (95% CI 49–74) C: 69% (55–80) adjusted HR 1·01 (95% CI 0·64 to 1·61)
Safety Adverse events (I: n=59; C: n=55) Patients with at least one AE I: 29 (49%) C: 23 (42%)
Patients with multiple AEs I: 19 (32%) C: 14 (25%) Total number of adverse events I: 113 C: 60 Serious adverse events Patients with at least one SAE I: 27 (46%) C: 21 (38%) Patients with multiple SAEs I: 8 (14%) C: 5 (9%) Total number of SAEs I: 42 C: 28
Virological outcomes Viral clearance not reported Also reported: C-reactive protein, neutrophil count, lymphocyte count, lymphocyte to neutrophil ratio |
Definitions: WHO-CPS score Uninfected • 0, Uninfected;
Ambulatory: Mild disease • 1, Asymptomatic; viral RNA detected; • 2, Symptomatic; independent; • 3, Symptomatic; assistance needed;
Hospitalised: moderate disease • 4, hospitalized, not requiring oxygen; • 5, hospitalized, requiring oxygen by mask or nasal prongs;
Hospitalised: severe disease • 6, hospitalized, requiring nasal highflow oxygen therapy, non-invasive mechanical ventilation, or both; • 7, hospitalized, requiring • intubation and mechanical ventilation, pO2/FIO2 ≥150 or SpO2/FIO2 ≥ 200; • 8, hospitalized, requiring • mechanical ventilation, (pO2/FIO2 <150 OR SpO2/FIO2 < 200) OR vasopressors (norepinephrine < 0.3 microg/kg/min); • 9, Mechanical ventilation, pO2/FIO2 <150 AND vasopressors (norepinephrine > 0.3 • microg/kg/min), OR dialysis OR ECMO
Dead • 10, Dead.
|
|
Declercq, 2021 |
Type of study: Multicentre, open-label, 2x2 factorial, randomised, controlled phase 3 trial
Setting: Hospital-based, between April 4 and December 6, 2020
Country: 16 hospitals in Belgium
Source of funding: Belgian Health Care Knowledge Centre and VIB Grand Challenges program. The funder of the study (Belgian Health Care Knowledge Centre) was involved in purchasing study medication and study design, but was not involved in data collection, data analysis, data interpretation, writing of the manuscript, or the decision to submit. The funder (VIB Grand Challenges) was involved in purchasing reagents for measuring biomarkers.
Conflicts of interest: Conflicts of interest were transparently and extensively reported. |
Hospitalised patients with COVID-19, hypoxia, and signs of a cytokine release syndrome
Inclusion criteria:
Exclusion criteria:
N total at baseline: Randomized: N = 342 ITT population: N = 342
Intervention 1:: N = 112 Control 1: N = 230 Intervention 2a: N = 113 Intervention 2b: N = 114 Control 2: N = 115
Important characteristics: Age, median (IQR): I1: 67 y (56-74) C1: 64 y (54-72)
I2: 59 y (52-74) C2: 62 y (55-71)
Sex, n/N (%) male: I1: 87/112 (78%) C1: 178/230 (77%)
I2: 175/227 (77%) C2: 90/115 (78%)
Mechanical ventilation at day of randomisation Invasive I1: 17/112 (15%) C1: 22/230 (10%)
I2: 22/227 (10%) C2: 17/115 (15%)
Non-invasive or high flow oxygen device I1: 44/112 (39%) C1: 84/230 (37%)
I2: 89/227 (39%) C2: 39/115 (34%)
Groups were comparable at baseline. |
Intervention 1: anakinra 100 mg 1 dd s.c. for 28 days or until hospital discharge + standard care
Intervention 2: siltuximab 11 mg/kg i.v. (single injection) (= intervention 2a)
OR
tocilizumab 8 mg/kg i.v. (not exceeding 800 mg; single injection) (= intervention 2b) |
Control 1: standard care
Control 2: standard care |
Length of follow-up: 28 days
Loss-to-follow-up: I1: 0/112 (0%) Reasons: -
C1: 6/230 (3%) Reasons:
I2a: 2/113 (2%) Reasons:
I2b: 2/114 (2%) Reasons:
C2: 2/115 (2%) Reasons: withdrew consent (n = 2) |
Clinical outcomes Mortality Number of deaths I1: 10/44 (23%) I1+I2a: 5/32 (16%) I1+I2b: 6/36 (17%) I2a: 10/81 (12%) I2b: 15/75 (20%) C: 9/74 (12%)
Estimated mortality at day 28 I1: 16% (95%CI: 8-31) I1+I2a: 13% (95%CI: 5-30) I1+I2b: 17% (95%CI: 8-33) I2a: 11% (95%CI: 6-20) I2b: 13% (95%CI: 7-23) C: 10% (95%CI: 5-20)
Estimated mortality at day 90 I1: 23% (95%CI: 13-38) I1+I2a: 16% (95%CI: 7-34) I1+I2b: 17% (95%CI: 8-33) I2a: 12% (95%CI: 7-22) I2b: 19% (95%CI: 12-30) C: 13% (95%CI: 7-23)
Duration of hospitalization Time until discharge Days, median I1: 14 (95%CI: 11-19) C1: 12 (95%CI: 11-18) HR 0.90 (95%CI: 0.70-1.16)
I2: 12 (95%-CI: 11-18) C2: 13 (95%-CI: 11-19) HR 1.02 (95%CI: 0.80-1.31)
Number of days in hospital Mean I1: 19 (95%CI: 17-22) C1: 19 (95%CI: 17-21) expected count ratio 1.01 (95%CI: 0.85-1.21)
I2: 20 (95%-CI: 18-22) C2: 19 (95%-CI: 16-22) expected count ratio 1.03 (95%CI: 0.86-1.22)
Number of days in ICU Mean I1: 11 (95%CI: 8-15) C1: 10 (95%CI: 8-13) expected count ratio 1.05 (95%CI: 0.69-1.59)
I2: 11 (95%-CI: 8-14) C2: 10 (95%-CI: 7-15) expected count ratio 1.03 (95%CI: 0.68-1.56)
Number of days in ICU in patients ventilated at day of randomisation Mean I1: 20 (95%CI: 15-27) C1: 22 (95%CI: 17-29) expected count ratio 0.89 (95%CI: 0.60-1.32)
I2: 20 (95%-CI: 16-27) C2: 22 (95%-CI: 16-29) expected count ratio 0.94 (95%CI: 0.64-1.40)
Number of days in ICU relative to the number of days alive the first 28 days after randomisation Mean I1: 42% (95%CI: 31-56) C1: 36% (95%CI: 29-46) expected count ratio 1.14 (95%CI: 0.79-1.66)
I2: 38% (95%-CI: 30-48) C2: 40% (95%-CI: 29-54) expected count ratio 0.96 (95%CI: 0.66-1.39)
Time to symptom resolution Time to clinical improvement (primary outcome)† Days, median I1: 12 (95%CI: 10-16) C1: 12 (95%CI: 10-15) HR 0.94 (95%CI: 0.73-1.21)
I2: 11 (95%-CI: 10-16) C2: 12 (95%-CI: 11-16) HR 1.00 (95%CI: 0.78-1.29)
Estimated probability of having experienced clinical improvement at day 28 I1: 75% (95%CI: 67-83) C1: 73% (95%CI: 67-79)
I2: 74% (95%-CI: 68-79) C2: 74% (95%-CI: 66-82)
Respiratory support Median time until independence from supplemental O2 or discharge Days, median I1: 12 (95%CI: 10-20) C1: 12 (95%CI: 10-15) HR 0.91 (95%CI: 0.71-1.17)
I2: 11 (95%-CI: 10-15) C2: 12 (95%-CI: 10-15) HR 1.00 (95%CI: 0.78-1.28)
Median time until independence from invasive ventilation Days, median I1: 21 (95%CI: 8-not estimable) C1: 27 (95%CI: 9-not estimable) HR 1.21 (95%CI: 0.54-2.71)
I2: 23 (95%-CI: 8-not estimable) C2: 54 (95%-CI: 9-not estimable) HR 1.45 (95%CI: 0.63-3.33)
Median time until first use of high-flow oxygen device, ventilation, or death Days, median I1: < 50% reached event C1: < 50% reached event HR 0.97 (95%CI: 0.52-1.82)
I2: < 50% reached event C2: < 50% reached event HR 0.85 (95%CI: 0.47-1.55)
Number of days without supplemental O2 use up to 28 days after randomisation Mean I1: 9 (95%CI: 7-12) C1: 9 (95%CI: 7-12) expected count ratio 0.97 (95%CI: 0.68-1.38)
I2: 10 (95%-CI: 8-12) C2: 8 (95%-CI: 6-11) expected count ratio 1.17 (95%CI: 0.82-1.68)
Number of invasive ventilator days Mean I1: 5 (95%CI: 3-9) C1: 5 (95%CI: 3-7) expected count ratio 1.05 (95%CI: 0.54-2.03)
I2: 5 (95%-CI: 3-7) C2: 5 (95%-CI: 3-9) expected count ratio 0.89 (95%CI: 0.46-1.72)
Number of invasive ventilator days in patients ventilated at day of randomisation Mean I1: 15 (95%CI: 11-20) C1: 16 (95%CI: 13-21) expected count ratio 0.93 (95%CI: 0.63-1.37)
I2: 15 (95%-CI: 12-20) C2: 16 (95%-CI: 12-22) expected count ratio 0.96 (95%CI: 0.65-1.42)
Number of invasive ventilator days relative to the number of days alive the first 28 days after randomisation Mean I1: 23% (95%CI: 14-38) C1: 21% (95%CI: 14-31) expected count ratio 1.08 (95%CI: 0.57-2.05)
I2: 21% (95%-CI: 14-30) C2: 23% (95%-CI: 14-38) expected count ratio 0.89 (95%CI: 0.47-1.70)
Number of invasive ventilator-free days up to 28 days after randomisation Mean I1: 18 (95%CI: 15-21) C1: 18 (95%CI: 16-20) expected count ratio 1.00 (95%CI: 0.84-1.19)
I2: 18 (95%-CI: 17-20) C2: 17 (95%-CI: 15-20) expected count ratio 1.07 (95%CI: 0.90-1.27)
Number of invasive ventilator-free days up to 28 days after randomisation in patients ventilated at day of randomisation Mean I1: 6 (95%CI: 3-14) C1: 6 (95%CI: 3-13) expected count ratio 1.01 (95%CI: 0.33-3.07)
I2: 7 (95%-CI: 4-15) C2: 5 (95%-CI: 2-12) expected count ratio 1.39 (95%CI: 0.46-4.20)
Safety Serious infections Sepsis I1: 5/44 (11%) I1+I2a: 2/32 (6%) I1+I2b: 3/36 (8%) I2a: 4/81 (5%) I2b: 11/75 (15%) C: 6/74 (8%)
Septic shock I1: 5/44 (11%) I1+I2a: 1/32 (3%) I1+I2b: 3/36 (8%) I2a: 3/81 (4%) I2b: 6/75 (8%) C: 3/74 (4%)
Serious adverse events not leading to mortality Infectious disorder (not COVID-19) I1: 1/44 (2%) I1+I2a: 2/32 (6%) I1+I2b: 1/36 (3%) I2a: - I2b: 4/75 (5%) C: 1/74 (1%)
Bleeding I1: 2/44 (5%) I1+I2a: 1/32 (3%) I1+I2b: - I2a: 1/81 (1%) I2b: - C: 1/74 (1%)
Thrombosis I1: 1/44 (2%) I1+I2a: - I1+I2b: 1/36 (3%) I2a: - I2b: 1/75 (1%) C: 1/74 (1%)
Acute kidney injury I1: 1/44 (2%) I1+I2a: - I1+I2b: - I2a: - I2b: 1/75 (1%) C: 1/74 (1%)
Cardiac disorder I1: - I1+I2a: - I1+I2b: - I2a: 1/81 (1%) I2b: 1/75 (1%) C: 1/74 (1%)
Other I1: - I1+I2a: 2/32 (6%) I1+I2b: 2/36 (6%) I2a: 3/81 (4%) I2b: 1/75 (1%) C: 1/74 (1%)
Virological outcomes Viral clearance Not reported.
Several subgroups were prespecified on the basis of allocated treatment for the other randomisation, invasive ventilation and serum concentrations of CRP, IL-1β, and IL-6 on the day of randomisation. Post-hoc subgroup analyses included serum concentrations of IL-1RA, admission status to ICU or concomitant glucocorticoid use at randomisation. |
Definitions: * Patients needed to have either a single ferritin concentration measurement of more than 2000 μg/L at inclusion when they immediately required high flow oxygen or mechanical ventilation, or a ferritin concentration of more than 1000 μg/L, which had been increasing over the previous 24 h, or lymphopenia below 800/mL with two of the following criteria: an increasing ferritin concentration of more than 700 μg/L; an increasing lactate dehydrogenase concentration of more than 300 international units (IU)/L; an increasing CRP concentration of more than 70 mg/L; or an increasing D-dimers concentration of more than 1000 ng/mL. If the patient had three of the previous criteria at hospital admission with lymphopenia of less than 800/μL, there was no need to document an increase over 24 h.
† Defined as the time in days from randomisation until either an increase of at least two points on a 6-category ordinal scale (compared with the worst status at day of randomisation) or to discharge from the hospital alive, whichever occurred first. The 6-category ordinal scale was defined as 1=death; 2=hospitalised, on invasive mechanical ventilation or extracorporeal membrane oxygenation; 3=hospitalised, on non-invasive ventilation or high-flow oxygen devices; 4=hospitalised, requiring supplemental oxygen; 5=hospitalised, not requiring supplemental oxygen; 6=not hospitalised.
Remarks: -
Authors conclusion: Drugs targeting IL-1 or IL-6 did not shorten the time to clinical improvement in this sample of patients with COVID-19, hypoxic respiratory failure, low SOFA score, and low baseline mortality risk. |
|
Kharazmi, 2021 |
Type of study: Open-label, randomized controlled trial.
Setting: Imam Hossein Medical Centre affiliated with Shahid Beheshti University of Medical Sciences
Country: Iran
Source of funding: No information.
Conflicts of interest: The authors declare that there are no conflict of interests.
|
Patients with severe COVID-19
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 30 Intervention: N= 15 Control: N = 15
Important characteristics: Age, mean (SD): I: 49.25 y (19.12) C: 59.00 y (1.79) P=0.424
Sex, n/N (%) male: I: 8/15 (53.3%) C: 11/15 (73.3%)
Disease severity ordinal scale, n/N (%) 1. death I: 0/0 (0%) C: 0/0 (0%)
2. Hospitalized, on invasive mechanical ventilation or extracorporeal membrane oxygenation. I: 2/15 (13.3%) C: 3/15 (20.0%)
3. Hospitalized, on noninvasive ventilation or high flow oxygen I: 10/15 (66.7%) C: 6/15 (40%)
4. Hospitalized, requiring low flow supplemental oxygen I: 3/15 (20%) C: 6/15 (40%)
5. Hospitalized, not requiring supplemental oxygen—requiring ongoing medical care (COVID‐19 related or otherwise). I: 0/15 (0%) C: 0/15 (0%)
6. Hospitalized, not requiring supplemental oxygen—no longer required ongoing medical care. I: 0/15 (0%) C: 0/15 (0%)
7. Not hospitalized I: 0/15 (0%) C: 0/15 (0%)
Groups comparable at baseline? Yes.
|
Anakinra group
Patients in the intervention group received 100 mg anakinra as an intravenous (IV) infusion once daily in addition to the standard protocol for COVID‐19 based on the sixth and seventh national COVID‐19 committee guideline, until discharge or a maximum of 14 days.
|
Control group
In the control group, patients received medication based on the sixth and seventh national protocol released on April 29, 2020 and June 27, 2020, respectively.
|
Length of follow-up: 14 days.
Loss-to-follow-up: None.
Incomplete outcome data: None.
|
Clinical outcomes Mortality (28-30 day) Not reported.
Length of hospital stay, mean (SD) I: 9.50 (4.45) C: 19.00 (12.04)
ICU length of stay, mean (SD) I: 5.43 (1.72) C: 16.60 (9.04)
Disease severity day 7, n/N (%) 1. death : 4/15 (26.7%) C: 5/15 (33.3%)
2. Hospitalized, on invasive mechanical ventilation or extracorporeal membrane oxygenation. I: 1/15 (6.7%) C: 5/15 (33.3%)
3. Hospitalized, on noninvasive ventilation or high flow oxygen I: 1/15 (6.7%) C: 0/15 (0%)
4. Hospitalized, requiring low flow supplemental oxygen I: 4/15 (26.7%) C: 4/15 (26.7%)
5. Hospitalized, not requiring supplemental oxygen—requiring ongoing medical care (COVID‐19 related or otherwise). I: 1/15 (6.7%) C: 0/15 (0%)
6. Hospitalized, not requiring supplemental oxygen—no longer required ongoing medical care. I: 0/15 (0%) C: 0/15 (0%)
7. Not hospitalized I: 4/15 (26.7%) C: 1/15 (6.7%)
Disease severity day 14, n/N (%) 1. death : 5/15 (33.3%) C: 7/15 (46.7%)
2. Hospitalized, on invasive mechanical ventilation or extracorporeal membrane oxygenation. I: 0/15 (0%) C: 2/15 (13.3%)
3. Hospitalized, on noninvasive ventilation or high flow oxygen I: 0/15 (0%) C: 2/15 (13.3%)
4. Hospitalized, requiring low flow supplemental oxygen I: 0/15 (0%) C: 4/15 (26.7%)
5. Hospitalized, not requiring supplemental oxygen—requiring ongoing medical care (COVID‐19 related or otherwise). I: 0/15 (0%) C: 0/15 (0%)
6. Hospitalized, not requiring supplemental oxygen—no longer required ongoing medical care. I: 0/15 (0%) C: 0/15 (0%)
7. Not hospitalized I: 10/15 (66.7%) C: 5/15 (33.3%)
Time to symptom resolution Not reported.
Respiratory support Not reported.
Safety Adverse events I: 1/15 (6.7%) (sputum culture) C: 3/15 (20%) (positive microbiologic culture: one blood culture, two sputum cultures)
Virological outcomes Viral clearance Not reported.
|
Definitions: -
Remarks: There are some limitations to our study. First, this is a pilot study with a small sample size. Second, we did not control the trial with a placebo.
Authors conclusion: According to the study results, in general, anakinra is effective in improving the respiratory condition and significantly reduces the need for invasive mechanical ventilation in patients with severe COVID‐19. And also, the reduction was observed in hospitalization duration, which makes the medication an effective immunomodulatory agent to combat cytokine storm. As an important consideration, the reliable safety of anakinra in patients with critical conditions is one of this medication's most important properties
|
|
Canakinumab |
|||||||
|
Caricchio, 2021 |
Type of study: RCT (double blind)
Setting: 39 hospitals in Europe and the United States
Country: Europe and the United States
Source of funding: Novartis Pharma AG, Basel, Switzerland.
Conflicts of interest: All authors received funding from Novartis during the conduct of the study. Dr Caricchio reported receiving grants from Janssen and personal fees from Janssen, GlaxoSmithKline, Bristol Myers Squibb, Eli Lilly, and Siemens outside the submitted work. Dr Abbate reported receiving grants from Kiniksa, Janssen, Olatec, and Serpin Pharma; personal fees from Janssen, Kiniksa, Cromos, Olatec, Serpin Pharma, Eli Lilly, and Merck; and nonfinancial support from Swedish Orphan Biovitrum outside the submitted work. Dr Hsue reported receiving honoraria from Gilead and Merck outside the submitted work. Dr Neogi reported receiving personal fees from Novartis outside the submitted work. Drs Chen, Li, Whelan, and Noviello reported being employees of Novartis. Dr Whelan reported having a patent pending through Novartis. Dr Noviello reported being a former/employee/stockholder of Bristol Myers Squibb and stockholder of Johnson & Johnson; in addition, Dr Noviello reported having a patent pending through Novartis.
ClinicalTrials.gov:NCT04362813 |
Hospitalized patients with severe COVID-19.
Inclusion criteria: • at least 12 years old (United States) or 18 years old (Europe); • Had hypoxemia but did not require IMV; • diagnosis of infection with SARSCoV-2 within 7 days prior to randomization; • diagnosis of pneumonia with pulmonary infiltrates on chest x-ray or computed tomographic scan within 5 days prior to randomization; • peripheral capillary oxygen saturation of 93%or less on room air or arterial oxygen partial pressure/fraction of inspired oxygen less than 300mmHg; • blood levels of CRP of 20mg/L or greater or ferritin of 600 μg/L or greater
Exclusion criteria: • Had been treated with therapies targeting IL-1 or IL-6 • had a suspected or known untreated active infection due to another pathogen • or if progression to death was imminent within 24 hours according to the investigator.
N total at baseline: N = 454 Intervention:227 Control: 227
Important characteristics: Age, median (IQR): I: 59 (49-69) C: 57 (50-68)
Sex, n/N (%) male: I:135/227 (59%) C:132/227 (58%)
Disease severity, mean (SD): Defined by WHO ordinal scale I: 0: 0 1:0 2:0 3: 14 (6.2) 4: 161 (70.9) 5: 52 (22.9) 6: 0 7: 0 8: 0
C: 0: 0 1:0 2:0 3: 12 (5.4) 4: 160 (71.4) 5: 52 (23.2) 6: 0 7: 0 8: 0 Groups comparable at baseline? Yes |
Canakinumab single dose of canakinumab (450mg for body weight of 40-<60 kg, 600mg for 60-80 kg, and 750 mg for >80 kg) in 250 mL of 5% dextrose infused intravenously over 2 hours |
Placebo placebo in 250 mL of 5% dextrose infused intravenously over 2 hours |
Length of follow-up: 127 days; Interim analysis reported for 29 days follow up
Loss-to-follow-up: Intervention: 1 Control: 1 (2%)
Incomplete outcome data: Intervention: 13 Discontinued the study (12 Died; 1 Participant decision (hospitalized)) Control: 16 Discontinued the study (died) |
Clinical outcomes
Mortality (28-30 day) Day 29: No. (%) I: 12 (5.3) C: 16 (7.1) OR :0.67 (95% CI, 0.30-1.50).
Survived without requiring IMV (3-29 day) I:198/223 (88.8%) C: 191/223 (85.7%) OR: 1.39 (95% CI, 0.76-2.54; P =.29).
Duration of hospitalization Discharged at day 29 I: 199/211 C: 187/206
Time to symptom resolution Not reported
Respiratory support Intubation and mechanical ventilation I: 5 (2.2) C: 2 (0.9)
Ventilation plus additional organ support (pressors, KRT, ECMO): I: 2 (0.9) C: 8 (3.6)
Safety Adverse events Any AE: No. (%) I: 122 (54.2) C: 120 (53.8)
SAEs: No. (%) I: 36 (16.0) C: 46 (20.6)
Virological outcomes Viral load > day 22: ≥ 500 I: 1/15 (6.7) C: 5/17 (29.4)
<500 I: 14/ 15 (93.3) C: 12/ 17 (70.6) |
Definitions: World Health Organization’s 9-point ordinal scale: 0 No clinical or virological evidence of infection 1 No limitation of activities 2 Limitation of activities 3 Hospitalized, no oxygen therapy 4 Oxygen by mask or nasal prongs 5 Noninvasive ventilation or high-flow oxygen 6 Intubation and mechanical ventilation 7 Ventilation plus additional organ support (pressors, KRT, ECMO) 8 Death.
Primary end point: the proportion of patients who survived without ever requiring IMV from day 3 to day 29 (inclusive).
The key secondary outcome: the proportion of patients who died of COVID-19 (causality assessed by investigators) between days 1 and 29.
Remarks: The sponsor, in consultation with investigators, designed and conducted the study. Collection of data and management of trial sites were conducted by the sponsor. An independent data monitoring committee reviewed trial safety data weekly. All authors contributed to the interpretation of the data, including sponsor co-authors. A preliminary draft of the manuscript was prepared by a writer contracted by Novartis.
Authors conclusion: Among patients hospitalized with severe COVID-19, treatment with canakinumab, compared with placebo, did not significantly increase the likelihood of survival without IMV at day 29.
|
|
Cremer, 2021 |
Type of study: RCT (double blind)
Setting: 5 hospitals in Cleveland Clinics
Country: the United States
Source of funding: This study was funded by an investigator-initiated grant fromNovartis.
Conflicts of interest: Paul Cremer has served on scientific advisory committees for Sobi and Kiniksa pharmaceuticals and has received investigator-initiated grants from Kiniksa andNovartis pharmaceuticals. |
Hospitalized patients with severe COVID-19.
Inclusion criteria: - Written informed consent must be obtained before any assessment is performed. - Hospitalized due to COVID-19 infection - Documented SARS-CoV2 acute myocardial injury: Defined as upper respiratory tract specimen positive for COVID-19 AND troponin greater than 99th percentile upper reference range without signs or symptoms of acute myocardial ischemia - NT-proBNP or BNP greater than upper reference limit - Receiving current standard therapy - C-reactive protein (CRP) > 50 mg/L
Exclusion criteria: - Alternative explanation for acute cardiac injury (Type I or Type II MI according to 4th Universal Definition of Myocardial Infarction, which in addition to a rise and fall of tropnonin above the 99th percentile upper reference limit, includes symptoms of acute myocardial ischemia, new ischemic ECG changes, development of pathologic Q waves, and imaging evidence of damage in a pattern consistent with an ischemic etiology) - Chronic Systolic Heart Failure with EF<35% - Age < 18 years-old - Uncontrolled systemic bacterial or fungal infection - Concomitant viral infection (e.g., Influenza or other respiratory virus) - Pregnant. Breast-feeding women are eligible with the decision to continue or discontinue breast-feeding during therapy taking into account the risk of infant exposure, the benefits of breast-feeding to the infant, and benefits of treatment to the mother. - On mechanical circulatory support - On mechanical ventilation for greater than 48 hours - Resuscitated cardiac arrest - Has a known hypersensitivity to canakinumab or any of its excipients - Neutrophil count <1000/mm3 - Has a history of myeloproliferative disorder or active malignancy receiving chemotherapy - Known active tuberculosis or history of incompletely treated tuberculosis - Current treatment with immunosuppressive agents - Chronic prednisone use >10 mg/daily (for more than 3 weeks prior to admission) - Has a history of solid-organ or bone marrow transplant - Severe pre-existing liver disease with clinically significant portal hypertension - End-stage renal disease on chronic renal replacement therapy - Enrollment in another investigational study using immunosuppressive therapy - In the opinion of the investigator and clinical team, should not participate in the study - If male and sexually active, must have documented vasectomy or must practice birth control and not donate sperm during the study and for 3 months after study drug administration. - Women of child-bearing potential, defined as all women physiologically capable of becoming pregnant, unless they are using highly effective methods of contraception during dosing of investigational drug.
N total at baseline: N = 45 Intervention1: 14 Intervention 2: 15 Control: 16
Important characteristics: Age, median (IQR): 68.8 (61.1-74.3)
Sex, n/N (%) male: 33/48 (73.3%)
|
a single infusion of canakinumab 600 mg intravenous (IV), canakinumab 300 mg IV
Intervention 1 = 600mg Intervention 2 = 300mg |
Equal volume (250mL) of 5% dextrose for placebo infusion |
Length of follow-up: 28 days Loss-to-follow-up: None.
Incomplete outcome data: None.
|
Clinical outcomes
Mortality (28-30 day) Day 28: No. (%) I-1: 1 (6.7) I-2: 3 (21.4) C: 3 (18.3)
HR-1: 0.20 (95%CI 0.02 to 1.98) HR-2: 1.21 (95%CI 0.23 to 5.59)
Respiratory support: Requiring invasive respiratory support (high flow nasal cannula (HFNC)/Optiflow, continuous positive airway pressure (CPAP), non-invasive ventilation (NIV), mechanical ventilation, extracorporeal membrane oxygenation (ECMO or ECLS)) At day 14: I1: 5/16 I2:1/11* C:2/14*
At day 28: I1: 1/18* I2:1/11* C:1/13*
Requiring non-invasive respiratory support (supplemental oxygen low flow oxygen, non-rebreathing mask) At day 14: I1: 1/16 I2: 3/11* C:4/14*
At day 28: I1: 0/15* I2:1/11* C:1/13*
duration of hospitalization: not reported.
time to symptom resolution:
clinical improvement or discharge at day 28, n (%)
Safety: |
Primary endpoint: Time to clinical improvement up to Day 14, defined as the time in days from randomization to either an improvement of two points on seven category ordinal scale or discharge from the hospital, whichever occurred first.
Remarks: The funder of the study provided the study drug and assisted with the study design. The funder was not involved in data collection or analysis. The corresponding author (P.C.C.) had full access to all of the data and the final responsibility to submit for publication.
Authors conclusion: There was no difference in time to clinical improvement at Day 14 in patients treated with canakinumab, and no safety concerns were identified. Future studies could focus on high dose canakinumab in the treatment arm and assess efficacy outcomes at Day 28. |
Risk of bias table for intervention studies (randomized controlled trials).
|
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2 (unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4 (unlikely/likely/unclear) |
Bias due to loss to follow-up?5 (unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6 (unlikely/likely/unclear) |
|
|
Anakinra |
||||||||
|
Kyriazopoulou, 2021 |
“Patients were electronically 1:2 randomized into treatment with placebo or anakinra using four randomization strata: classification into moderate or severe disease using the WHO definition based on the need for oxygen support (https://www.who.int/p ublications/i/item/ clinical-management-ofcovid-19); need for dexamethasone intake; body mass index (BMI) higher than 30kg m−2 ; and country.”) |
Unlikely
Patients were randomized using for randomization strata: • classification into moderate or severe disease using the WHO definition based on the need for oxygen support (https://www.who. int/publications/i/i tem/ clinicalmanagement-ofcovid-19); • need for Dexamethasone intake; • body mass index (BMI) higher than 30kg m−2; • country. |
Unlikely
Double-blind study. |
Unlikely
The study drug was prepared by an unblinded pharmacist with access to the electronic study system using a separate username and password. Administration was done by a blinded study nurse |
Unlikely
Data were captured after review of all medical and nursing charts by a physician team blinded to the allocation group |
Unlikely
All predefined outcome measures were reported. |
Unlikely
Only one patient was lost to follow-up in the study. The patient was initially allocated to the intervention group. |
Unlikely
Participants included in the analysis are exactly those who were randomized into the trial . |
|
Mariette, 2020 |
Computerized
randomization Participants were assigned by means of a centralized computer program to each intervention, starting with balanced assignment for tocilizumab, sarilumab, or control, with actual proportions dependent on the number of interventions available at each site |
Unclear
Centralized randomization was used that was remote from study sites. |
Likely
(mortality: unlikely) ‘Patients and investigators were not masked to treatment assignment due to the nature of the intervention.’ |
Likely
(mortality: unlikely) ‘Patients and investigators were not masked to treatment assignment due to the nature of the intervention.’ |
Likely
(mortality: unlikely) ‘Patients and investigators were not masked to treatment assignment due to the nature of the intervention.’
|
Unlikely
Not all outcomes that are reported in trial registration are reported in publication. Registered ClinicalTrials.gov: NCT04341584. |
Unlikely
4 patients (6.8%) in the intervention group en 2 patients (3.5%) in the control group were lost to FU or withdrew consent. This is not expected to induce a bias in the results. |
Mariette, 2020 |
|
Declercq, 2021 |
Computerised
Included patients were randomly assigned by means of permuted block randomization with varying block size and stratification by center. Patients were allocated in a 1:2 ratio to anakinra or no IL-1 blockade. Simultaneously, patients were randomly assigned in a 1:1:1 ratio to siltuximab, tocilizumab, or no IL-6 blockade. Randomization and subsequent data collection were done by means of the web based system REDCap. |
Unlikely
Randomization and subsequent data collection were done by means of the web based system REDCap. |
Likely
open-label trial |
Likely
open-label trial |
Unclear
not described |
Unlikely
All outcome measures described in the methods are reported in the results. |
Unlikely
The percentage of patients lost to follow-up is less than 10% and is similar between treatment groups. |
Unlikely
The primary and supportive efficacy endpoints were assessed in the intention-to-treat population. Safety was assessed in the safety population. |
|
Kharazmi, 2021
|
The sample size of 30 patients was divided into parallel groups of intervention and control considered.
|
Unlikely
Permuted block randomization with the sample size of four patients in each block which was stratified based on receiving invasive mechanical ventilation at baseline was performed |
Likely
Open-label study |
Likely
Open-label study |
Likely
Open-label study |
Unlikely
All predefined outcome measures were reported.
|
Unlikely
No loss to follow-up reported.
|
Unlikely
Participants included in the analysis are exactly those who were randomized into the trial.
|
|
Canakinumab |
||||||||
|
Caricchio, 2021 |
Block randomisation The randomization was stratified by country, with a block size of 4 within each stratum. It was implemented using an interactive response technology system in which a new patient meeting the inclusion and exclusion criteria was randomly assigned to a treatment group based on a random allocation sequence. This sequence was created by a separate randomization office of the sponsor and kept blinded to the study team until the scheduled unblinding for data analysis. |
Unlikely It was implemented using an interactive response technology system in which a new patient meeting the inclusion and exclusion criteria was randomly assigned to a treatment group based on a random allocation sequence. |
Unlikely It was implemented using an interactive response technology system in which a new patient meeting the inclusion and exclusion criteria was randomly assigned to a treatment group based on a random allocation sequence. |
Unlikely Double blinded |
Unlikely Double blinded |
Unlikely All outcomes mentioned in Methods were reported. ClinicalTrials.gov Identifier: NCT04362813 |
Unclear Reason for loss to follow up was not reported |
Unlikely Patients randomized in the study were the same patients who were analysed. |
|
Cremer, 2021 |
Computerised
Randomization was centralized through REDCap Cloud, |
Unlikely
Randomization will be centralized through REDcap Cloud. Randomization will be stratified on whether or not the patient is intubated at the time of enrollment (in addition to site). |
Unlikely
Randomization will be centralized through REDcap Cloud. Randomization will be stratified on whether or not the patient is intubated at the time of enrollment (in addition to site). |
Unlikely
Subjects, investigator staff, persons performing the assessments, and the clinical trial team will be blinded to treatment. Only the Research Pharmacist and members of the Data Safety Monitoring Board will have access to the treatment assignments. If the clinical team decides that unblinding the patient is essential for subsequent significant clinical management, then the patient may be unblinded after discussion between the clinical team and the Principal Investigator, Paul Cremer. |
Unlikely
Subjects, investigator staff, persons performing the assessments, and the clinical trial team will be blinded to treatment. Only the Research Pharmacist and members of the Data Safety Monitoring Board will have access to the treatment assignments. If the clinical team decides that unblinding the patient is essential for subsequent significant clinical management, then the patient may be unblinded after discussion between the clinical team and the Principal Investigator, Paul Cremer. |
Unlikely
Reported in study protocol. This is included in the supplementary file. |
Unlikely
None of the participants were lost to follow up |
Unlikely.
|
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Beoordelingsdatum en geldigheid
Publicatiedatum : 07-11-2022
Beoordeeld op geldigheid : 03-10-2022
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut). Deze ondersteuning werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De werkgroep werd gefinancierd uit een VWS subsidie.
De financiers hebben geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodules is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de behandeling van patiënten met COVID-19.
In 2020 is een multidisciplinair expertiseteam behandeling ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van het expertiseteam behandeling) die betrokken zijn bij de zorg voor patiënten met COVID-19. Dit expertiseteam fungeerde als stuurgroep, welke opdracht heeft gegeven tot het ontwikkelen van de module, alsmede fungeerde als klankbordgroep.
Werkgroep
- Dr. Marjolein Hensgens, internist-infectioloog, Afdeling Infectieziekten, UMC Utrecht en LUMC Leiden (Stichting Werkgroep Antibiotica Beleid)
- Drs. Emilie Gieling, apotheker, Afdeling Klinische Farmacie, UMC Utrecht.
- Prof. Dr. Dylan de Lange, intensivist, Afdeling Intensive Care, UMC Utrecht.
- Dr. Wim Boersma, longarts, Afdeling Longziekten, Noordwest Ziekenhuisgroep, Alkmaar.
- Dr. Paul van der Linden, apotheker, Afdeling Klinische Farmacie, Tergooi MC, Hilversum (Stichting Werkgroep Antibiotica Beleid).
- Prof. Dr. Bhanu Sinha, arts-microbioloog, Afdeling Medische Microbiologie & Infectiepreventie, UMCG, Groningen (Stichting Werkgroep Antibiotica Beleid).
- Dr. Mark de Boer, internist-infectioloog, Afdelingen Infectieziekten en Klinische Epidemiologie, LUMC, Leiden (Stichting Werkgroep Antibiotica Beleid).
- Tot 1-11-2021 tevens deel van de werkgroep: Dr. Albert Vollaard, internist-infectioloog, LCI, RIVM
Stuurgroep (expertiseteam Behandeling COVID-19)
- Dr. L.M. van den Toorn (voorzitter), longarts, Erasmus Medisch Centrum (Erasmus MC), NVALT
- Dr. M.G.J. de Boer, internist-infectioloog, Leids Universitair Medisch Centrum (LUMC), SWAB/NIV)
- Drs. A.J. Meinders, internist-intensivist, St. Antonius Ziekenhuis, NVIC
- Prof. dr. D.W. de Lange, intensivist-toxicoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), NVIC
- Dr. C.H.S.B. van den Berg, infectioloog-intensivist Universitair Medisch Centrum Groningen (UMCG), NVIC
- Dr. S.U.C. Sankatsing, internist-infectioloog, Diakonessenhuis, NIV
- Dr. E.J.G. Peters, internist-infectioloog, Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. M.S. Boddaert, arts palliatieve geneeskunde, Leids Universitair Medisch Centrum (LUMC), IKNL
- Dr. P.L.A. Fraaij, kinderarts-infectioloog, Erasmus Medisch Centrum (Erasmus MC), Sophia Kinderziekenhuis, NVK
- Dr. E. van Leeuwen, gynaecoloog, Amsterdam University Medical Centers (Amsterdam UMC), NVOG
- Dr. J.J. van Kampen, arts-microbioloog, Erasmus Medisch Centrum (Erasmus MC), NVMM
- Dr. M. Bulatović-Ćalasan, internist allergoloog-immunoloog en klinisch farmacoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. A.F.J. de Bruin, anesthesioloog-intensivist, St. Antonius Ziekenhuis, NVA
- Drs. A. Jacobs, klinisch geriater, Catharina Ziekenhuis, NVKG
- Drs. B. Hendriks, ziekenhuisapotheker, Leids Universitair Medisch Centrum (LUMC), NVZA
- Drs. M. Nijs, huisarts, NHG
- Dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
Meelezer
- Drs. K. (Klaartje) Spijkers, senior adviseur patiëntenbelang, Patiëntenfederatie Nederland, Utrecht
Met ondersteuning van:
- dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
- dr. L.M.P. Wesselman, adviseur, Kennisinstituut van Medisch Specialisten
- dr. D. Nieboer, adviseur, Kennisinstituut van Medisch Specialisten
- drs. A.L.J. (Andrea) Kortlever - van der Spek, adviseur, Kennisinstituut van Medisch Specialisten
-
M. Griekspoor MSc., junior adviseur, Kennisinstituut van Medisch Specialisten
- drs. I. van Dusseldorp, senior literatuurspecialist, Kennisinstituut van Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
De Lange |
1. Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht (0,6 fte) |
Secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE), onbezoldigd. |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, klinisch epidemioloog, senior medisch specialist, Leids Universitair Medisch Centrum, afdeling Infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Sinha |
Arts-microbioloog/hoogleraar, Universitair Medisch Centrum Groningen (voltijd) (zie ook https;//www.rug.nl/staff/b.sinha/) |
- SWAB-bestuur: secretaris [onbetaald; vacatiegeld voor instelling] |
- Projectsubsidie EU (Cofund): deelprojecten, cofinanciering
Mogelijk boedbeeldfunctie SWAB |
Geen actie nodig |
|
Van der Linden |
Ziekenhuisapotheker |
Penningmeester SWAB, vacatiegeld |
Geen |
Geen actie nodig |
|
Vollaard |
Internist-infectioloog, Landelijke Coordinatie Infectieziektebestrijding, RIVM |
Arts voor ongedocumenteerde migranten, Dokters van de Wereld, Amsterdam (onbetaald) |
Geen |
Geen actie nodig |
|
Gieling |
Ziekenhuisapotheker - Klinisch Farmacoloog, UMC Utrecht |
Lid OMT Nederlandse Vereniging voor Ziekenhuisapothekers (onbetaald) |
Geen |
Geen actie nodig |
|
Boersma |
Longarts Noordwest Ziekhuisgroep |
Lid sectie infectieziekten NVALT, onbetaald |
Eenmalige digitale deelname aan adviesraad MSD Pneumovax over Pneumococcal disease, betaald
|
Geen actie nodig |
|
Hensgens |
Internist-infectioloog, UMC Utrecht (0.8 aanstelling, waarvan nu 0.4 gedetacheerd naar LUMC) Internist-infectioloog, LUMC (via detachering, zie boven) |
Geen |
Geen |
Geen actie nodig |
Stuurgroep
|
Achternaam stuurgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Van den Toorn (voorzitter) |
Voorzitter NVALT |
Geen |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, senior medisch specialist, LUMC, afdeling infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Meinders |
Internist-intensivist, St.-Antonius ziekenhuis, Nieuwegein |
commissie werk |
Geen |
Geen actie nodig |
|
De Lange |
Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht |
secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE) (onbetaald) |
Geen |
Geen actie nodig |
|
Van den Berg |
Infectioloog-intensivist, UMCG |
Geen |
Geen |
Geen actie nodig |
|
Sankatsing |
Internist-infectioloog/internist-acute geneeskunde, Diakonessenhuis, Utrecht |
- Bestuurslid Nederlandse Vereniging van Internist-Infectiologen (NVII) (onbetaald). |
Geen |
Geen actie nodig |
|
Peters |
Internist - aandachtsgebieden infectieziekten en Acute Geneeskunde Amsterdam UMC, locatie Vumc |
Wetenschappelijk Secretaris International Working Group on the Diabetic Foot (onbetaald) |
Geen |
Geen actie nodig |
|
Boddaert |
Medisch adviseur bij Integraal Kankercentrum Nederland (IKNL) en Palliatieve Zorg Nederland (PZNL) Arts palliatieve geneeskunde in LUMC |
Geen |
Geen |
Geen actie nodig |
|
Fraaij |
Kinderarts infectioloog- immunoloog, Erasmus MC-Sophia, Rotterdam |
Bestuur Stichting Infecties bij Kinderen (onbetaald) |
deelname aan RECOVER, European Union's Horizon 2020 research |
Geen actie nodig |
|
Van Leeuwen |
Gyaecoloog Amsterdam Universitair Medisch Centra |
Geen |
Geen |
Geen actie nodig |
|
Van Kampen |
Arts-microbioloog, afdeling Viroscience, Erasmus MC |
- associate editor antimicrobial resistance & infection control (onbetaald) - lid antibioticacommissie Erasmus MC (onbetaald) |
1. Mede uitvinder patent: 1519780601-1408/3023503 2. R01AI147330 (NIAID/NH) (HN onderzoek (1+2 niet gerelateerd aan COVID-19)
|
Geen actie nodig |
|
Bulatovic |
Internist allergoloog-immunoloog en klinische farmacoloog, UMC Utrecht en Diakonessenhuis Utrecht |
Functie 1: arts |
Geen |
Geen actie nodig |
|
De Bruin |
Anesthesioloog - Intensivist St. Antonius ziekenhuis Nieuwegein en Utrecht |
Geen |
Geen |
Geen actie nodig |
|
Jacobs |
Klinisch geriater en klinisch farmacoloog |
Geen |
Geen |
Geen actie nodig |
|
Hendriks |
Ziekenhuisapotheker farmaceutische patiëntenzorg, afd. Kiinische Farmacie en Toxicoiogie, Leids Universitair Medisch Centrum |
Lid SWAB werkgroep surveillance antibioticagebruik, onbetaald Lid SWAB richtlijncommissie antibiotica allergie, onbetaald |
Geen |
Geen actie nodig |
|
Nijs |
Huisarts |
Geen |
Geen |
Geen actie nodig |
|
Hofstede |
Senior adviseur Kennisinstituut van Medisch Specialisten |
Geen |
Geen |
Geen actie nodig |
Meelezer
|
Achternaam |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Spijkers |
Senior adviseur patiëntenbelang |
Voorzitter Stichting Samen voor Duchenne |
Geen |
Geen actie nodig |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde patiëntenvereniging in de klankbordgroep. De verkregen input is meegenomen bij het opstellen van de module. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Werkwijze
Van leidraad naar richtlijnmodules
Bij aanvang van de pandemie in 2020 was het onduidelijk of bestaande of nieuwe medicijnen een relevante bijdrage konden leveren aan het herstel van patiënten geïnfecteerd met het SARS-CoV-2. Vandaar dat eind februari 2020 werd aangevangen met de eerste versie van de leidraad ‘Medicamenteuze behandeling voor patiënten met COVID-19 (infectie met SARS–CoV-2)’, welke begin maart 2020 online beschikbaar werd gesteld op de website van de SWAB (https://swab.nl/nl/covid-19). Sindsdien werd het adviesdocument op wekelijkse basis gereviseerd en indien nodig op basis van nieuwe publicaties van onderzoek aangepast. Het initiatief en de coördinatie hiertoe werden genomen door de SWAB Leidraadcommissie, ondersteund door het kennisinstituut van de Federatie Medisch Specialisten en een brede klankbordgroep waarbinnen de betrokken specialisten(verenigingen) zijn vertegenwoordigd. In september 2021 is gestart met het doorontwikkelen van de leidraad naar richtlijnmodules.
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de COVID-19 pandemie zijn knelpunten op verschillende manieren geïnventariseerd:
1. De expertiseteams benoemde de knelpunten in de zorg voor patiënten met COVID-19.
2. Er is een mailadres geopend (covid19@demedischspecialist.nl) waar verschillende partijen knelpunten konden aandragen, die vervolgens door de expertiseteams geprioriteerd werden.
3. Door de Federatie van Medisch Specialisten zijn webinars georganiseerd waarbij vragen konden worden ingestuurd. Deze vragen zijn na afloop van de webinars voorgelegd aan de expertiseteams en geprioriteerd.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. Wanneer mogelijk werd de data uit verschillende studies gepoold in een random-effects model. Review Manager 5.4 werd gebruikt voor de statistische analyses. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
Definitie |
|
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
Bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Zoekverantwoording
On request.