Hydroxychloroquine
Uitgangsvraag
Wat is de plaats van hydroxychloroquine bij de behandeling van COVID-19 patiënten?
Aanbeveling
Behandel patiënten met COVID-19 niet met hydroxychloroquine.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Er is literatuuronderzoek verricht naar de verschillen in klinische uitkomsten tussen behandeling met en zonder hydroxychloroquine bij patiënten met COVID-19. Tot 7 januari 2021 werden er 8 gerandomiseerde gecontroleerde studies (RCT’s) gevonden in patiënten die waren opgenomen in het ziekenhuis (n=3037 in de interventiegroep en n=4569 in de controlegroep), waaronder twee grote studies: de RECOVERY trial (Horby, 2020) en de SOLIDARITY trial (Pan, 2020). Drie studies onderzochten hydroxychloroquine in patiënten die niet waren opgenomen in het ziekenhuis (n=500 in de interventiegroep en n=520 in de controlegroep).
Omdat de geïncludeerde studies verschillende populaties onderzochten, zijn de resultaten opgesplitst in groepen zonder ziekenhuisopname (ambulant) en met ziekenhuisopname. De studies met opgenomen patiënten bevatten daarnaast heterogene patiëntgroepen. Waar mogelijk zijn deze studies opgesplitst voor patiënten met milde, matige en ernstige COVID-19 symptomen op basis van respiratoire ondersteuning bij inclusie. Er werden alleen RCT’s geïncludeerd in de analyse, waardoor de kwaliteit van bewijs initieel hoog was. Omdat er vijf open-label trials waren, met een mogelijk risico op vertekening van de studieresultaten (risk of bias) bij subjectieve uitkomstmaten, werd de kwaliteit van dit bewijs waar nodig naar beneden bijgesteld. Daarnaast waren er meerdere studies met een relatief kleine populatie en mede hierdoor een grote spreiding van de puntschatter van de uitkomstmaat (imprecision), waardoor de kwaliteit van dit bewijs ook naar beneden werd bijgesteld. Eventueel werd de kwaliteit van het bewijs naar beneden bijgesteld als er veel heterogeniteit tussen de studies was, bijvoorbeeld in rapportage van de uitkomstmaat. De verschillen in de geïncludeerde patiënten werden niet meegenomen in de gradering van de studies, maar zullen wel worden meegenomen in de overwegingen.
Studies in patiënten die waren opgenomen in het ziekenhuis
Op basis van de gevonden resultaten wordt er geconcludeerd dat er voor de cruciale uitkomstmaat ‘mortaliteit’ en ‘invasieve respiratoire ondersteuning’ geen voordeel is voor behandeling met hydroxychloroquine. De gevonden studies tonen ook geheel consistent géén positief klinisch effect van hydroxychloroquine op duur van ziekenhuisopname of andere uitkomstmaten. Behandeling met hydroxychloroquine resulteert mogelijk zelfs in een hogere mortaliteit (RR 1.09; 95% CI 0.99-1.19) en meer noodzaak voor mechanische ventilatie (RR 1.13; 95% CI 0.95-1.34). De bewijskracht van de cruciale uitkomstmaten binnen patiënten die opgenomen waren in het ziekenhuis komt uit op ‘redelijk’.
Studies in patiënten die niet waren opgenomen in het ziekenhuis
Op basis van de gevonden resultaten kan worden geconcludeerd dat er voor de cruciale uitkomstmaat mortaliteit geen klinisch relevant verschil is gevonden voor behandeling met hydroxychloroquine in vergelijking met behandeling zonder hydroxychloroquine bij patiënten met COVID-19 die niet zijn opgenomen in het ziekenhuis. Ook bij andere relevante uitkomstmaten was er geen overtuigend bewijs in het voordeel van hydroxychloroquine. Echter, de drie studies die hydroxychloroquine onderzochten bij ambulante patiënten waren klein en niet gepowerd om kleine verschillen in bijvoorbeeld mortaliteit vast te stellen. De bewijskracht van de deze uitkomstmaten komt daarom uit op ‘laag’ of ‘zeer laag’.
Aanvulling van literatuur tot 2-9-2021
In aanvulling op bovenstaande literatuur werd er een nieuwe zoekopdracht uitgevoerd op 2 september 2021. Deze zoekopdracht leverde 10 nieuwe RCT’s op. Geen van deze studies liet een voordeel van hydroxychloroquine zien ten opzichte van de standaardbehandeling. De grootste RCT bij ambulante (hoog risico) patiënten was de TOGETHER studie, bij opgenomen patiënten was de grootste studie de DisCoVeRy studie, waarin respectievelijk 214 en 145 patiënten werden gerandomiseerd naar hydroxychloroquine. Beide studies werden voortijdig gestaakt. De TOGETHER studie toonde geen voordeel van hydroxychloroquine in tijd totdat patiënten klachten vrij werden, de tijd tot opname in het ziekenhuis of opnameduur. Ook in de DisCoVeRy studie ontbrak een positief effect van hydroxychloroquine op de klinische status na 15 dagen (de primaire uitkomstmaat), gemeten op een 7-puntsschaal, bij een interim analyse (adjusted OR 0.93; 95% CI 0.62-1.41). De mortaliteit na 28 dagen was iets hoger in de hydroxychloroquine-groep: 6.5% versus 5.3%, maar dit was niet statistisch significant. Daarnaast is het artikel van Abd-Elsalam (2020), welke is opgenomen in de literatuuranalyse teruggetrokken door het tijdschrift (Abd-Elsalam: retraction note, 2022). Het weglaten van deze studie uit de gepoolde resultaten, resulteert niet in andere conclusies.
Concluderend, de aanbevelingen veranderen niet na een aanvullend literatuuronderzoek.
Overige overwegingen
Bijwerkingen
Ten gevolge van het gebruik van hydroxychloroquine kan cardiotoxiciteit optreden waardoor eerder ECG-monitoring van het QT-interval werd aanbevolen (Nord, 2004; Schrezenmeier, 2020). Er is tevens gepubliceerd dat er een verdubbeling van cardiovasculaire toxiciteit is gezien, wanneer hydroxychloroquine samen gegeven werd met azitromycyine (Lane, 2020). In een cohort uit New York werd bij 11% van de patiënten die hydroxychloroquine en azitromycine gelijktijdig toegediend kregen al op dag 3.6 + 1.6 (SD) na start therapie een QTc > 500 ms geregistreerd (Chorin, 2020). Dat er risico is op QT-tijdverlenging door hydroxychloroquine, waarbij dat risico nog verder toeneemt als er azitromycine aan toe werd gevoegd, bleek ook uit een andere Amerikaanse patiënten serie (Mercuro, 2020). Bij milde tot matig ernstige ziekte was het risico op QTc-tijd verlenging door hydroxychloroquine significant hoger in vergelijking met SOC (14.6 % vs. 1.7%), maar in deze studie verhoogde de combinatie met azitromycine het risico niet verder (14.7%; Cavalcanti, 2020).
Ook meerdere RCT’s die geëvalueerd werden in de huidige richtlijn beschrijven cardiale toxiciteit van opgenomen patiënten (Chen, 2020; RECOVERY trial (Horby, 2020); Self, 2020; Ulrich, 2020 (TEACH)). De RECOVERY trial (Horby, 2020) rapporteerde dat ritmestoornissen, onder andere supraventriculaire tachycardie, ventriculaire tachycardie of fibrillatie en atrioventriculaire blokkade die interventie behoefde, optraden in 60 van de 735 (8.2%) patiënten in de interventiegroep en in 90 van de 1420 (6.3%) patiënten in de controlegroep (RR 1.29; 95% CI 0.94 tot 1.76). Self (2020) rapporteerde een incidentie van ventriculaire tachycardie/fibrillatie van 5/242 (2%) in de interventiegroep, vergeleken met 6/237 (2.5%) in de controlegroep (RR 0.82, 95% CI 0.25 tot 2.64). De incidentie van cardiac arrest was in deze studie 10/242 (4.1%) in de interventiegroep en 4/237 (1.7%) in de controlegroep (RR 2.45; 95% CI 0.78 tot 7.70; adjusted OR 2.51; 95% CI 0.78 tot 8.12). In de studie van Chen en Ulrich werden geen ritmestoornissen gezien, maar bij deze laatste studie werd er wel frequenter een verlenging van het QT-interval >500 ms gezien (3/67 (4.5%) in de interventiegroep ten opzichte van 1/61 (1.6%) in de controlegroep (RR 2.73; 95% CI 0.29 tot 25.57)).
Al met al tonen meerdere studies een klinisch relevant percentage ernstige bijwerkingen, met name cardiale geleidingsstoornissen, die eventueel de sterfte kunnen verhogen. Ook Cochrane review toont dat behandeling met hydroxychloroquine negatief kan werken. Zij bekeken studies tot februari 2021 en concludeerden op basis van 6 trials dat er een 3 keer verhoogde kans was op bijwerkingen in de hydroxychloroquine groep (RR 2.90, 95% CI 1.49 tot 5.64), maar geen verschil in de kans op ernstige bijwerkingen.
Dosering
Farmacokinetische simulatiestudies voorspelden een adequate longconcentratie met een dosering van 2 x 400 mg hydroxychloroquine sulfaat op dag 1, gevolgd door 2 x 200 mg hydroxychloroquine sulfaat op dag 2-5. Dit is dezelfde dosering die in een van de eerste versies van de SWAB-leidraad medicamenteuze behandelopties van COVID-19 stond. Er werd gecalculeerd dat een oplaaddosis nodig is om in korte tijd een effectieve dosering te kunnen bereiken (Perinal, 2020). Het Engelse gerandomiseerde RECOVERY onderzoek gebruikte een veel hogere dosering gedurende 10 dagen: 2400 mg in de eerste 24 uur verdeeld over 4 giften en nog 9 dagen 2 dd 400 mg. Omdat er bij deze hogere dosering nog altijd geen effect werd gezien op klinische eindpunten als overlijden op dag 28, zijn er geen aanvullende positieve effecten van een andere dosering te verwachten.
Chloroquine
Er was één studie die de duur van ziekenhuisopname, opklaring van symptomen, bijwerkingen en virale klaring beschreef bij behandeling met en zonder chloroquine. Op basis van deze literatuur kan geen conclusie worden getrokken (overall bewijskracht zeer laag). Mortaliteit en respiratoire ondersteuning werden niet beschreven. Omdat er geen bewijs is voor klinische effectiviteit van hydroxychloroquine voor COVID-19, is er van het verwante molecuul chloroquine ook geen klinische effectiviteit te verwachten.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
In de verschillende studies werd er geen klinisch relevant effect gevonden bij patiënten die hydroxychloroquine kregen ten opzichte van de controle groepen. Wel werd er voor mortaliteit een klein (niet klinisch relevant) voordeel gevonden voor de controle groep binnen de patiëntengroep die was opgenomen in het ziekenhuis. Daarnaast kan het gebruik van hydroxychloroquine wel mogelijk gepaard gaan met ernstige bijwerkingen bij patiënten, zoals cardiotoxiciteit.
Kosten (middelenbeslag)
Gezien het gebrek aan effectiviteit wordt hydroxychloroquine niet (meer) voorgeschreven aan patiënten met COVID-19.
Aanvaardbaarheid, haalbaarheid en implementatie
Hydroxychloroquine wordt niet (meer) voorgeschreven bij de behandeling van COVID-19, dus de werkgroep voorziet geen problemen qua implementatie. Wel kan het zijn dat sommige patiënten nog denken dat hydroxychloroquine of chloroquine werkzaam is tegen COVID-19. Goede voorlichting is hierbij belangrijk om de aanvaardbaarheid bij deze patiënten te vergroten.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
In de verschillende studies werd er geen klinisch effect gevonden bij patiënten die hydroxychloroquine kregen ten opzichte van de controlegroepen. Daarentegen kan het gebruik van hydroxychloroquine wel mogelijk gepaard gaan met (klinisch relevantie) bijwerkingen en mogelijk een kleine toename van de mortaliteit. Op basis hiervan kan geconcludeerd worden dat hydroxychloroquine geen plaats heeft bij de behandeling van COVID-19.
Onderbouwing
Hydroxychloroquine wordt toegepast bij de behandeling van uiteenlopende ziektebeelden, waaronder reumatoïde artritis, Q-koorts en Systemische Lupus erythematodes (SLE). Hydroxychloroquine heeft een gelijkende molecuulstructuur als chloroquine: aan chloroquine werd een hydroxyethylgroep toegevoegd om de cumulatieve toxiciteit te reduceren. Al vroeg in de pandemie, werd er van zowel hydroxychloroquine als chloroquine in vitro een antiviraal effect aangetoond (Liu, 2020; Wang, 2020), al was dit middels niet klinisch gevalideerde testsystemen. In de beginfase van de COVID-19 pandemie werd hydroxychloroquine gegeven aan patiënten met COVID-19. In een redelijk vroege fase wezen de eerste redelijk uitgevoerde observationele vergelijkende studies al op de afwezigheid van een klinisch relevant effect.
Inmiddels is in diverse gerandomiseerde studies (RCT’s) de effectiviteit van hydroxychloroquine onderzocht om de plaats van hydroxychloroquine bij de behandeling van COVID-19 patiënten te bepalen.
Hydroxychloroquine in hospitalized COVID-19 patients
Mortality (crucial)
Extensive respiratory support (crucial)
|
Moderate GRADE |
Treatment with hydroxychloroquine probably results in little to no difference in need for extensive respiratory support compared with treatment without hydroxychloroquine in hospitalized patients with COVID-19.
Source: Abd-Elsalam, 2020; RECOVERY trial (Horby, 2020); SOLIDARITY trial (Pan, 2020) ; Ulrich, 2020 (TEACH). |
Duration of hospitalization (important)
|
Low GRADE |
Treatment with hydroxychloroquine may result in little to no difference in length of stay compared with treatment without hydroxychloroquine in hospitalized patients with COVID-19.
Source: Abd-Elsalam, 2020; RECOVERY trial (Horby, 2020); Pan, 2020; Self, 2020; Ulrich, 2020 (TEACH). |
Time to clinical improvement (important)
|
Very low GRADE |
The evidence is uncertain about the effect of treatment with hydroxychloroquine on time to clinical improvement compared to treatment without hydroxychloroquine in hospitalized patients with COVID-19.
Source: Abd-Elsalam, 2020; Chen, 2020; Lyngbakken, 2020; Self, 2020; Tang, 2002; Ulrich, 2020 (TEACH). |
Hydroxychloroquine in non-hospitalized COVID-19 patients
Mortality (crucial)
|
Low GRADE |
Treatment with hydroxychloroquine may result in little to no difference in mortality compared with treatment without hydroxychloroquine in non-hospitalized patients with COVID-19.
Sources: Mitjà, 2020; Omrani, 2020 (Q-PROTECT); Skipper, 2020 |
Hospitalization (important)
|
Very low GRADE |
The evidence is uncertain about the effect of treatment with hydroxychloroquine on risk of hospitalization compared to treatment without hydroxychloroquine in non-hospitalized patients with COVID-19.
Sources: Mitjà, 2020; Skipper, 2020 |
Time to clinical improvement (important)
|
Very low GRADE |
The evidence is uncertain about the effect of treatment with hydroxychloroquine on time to clinical improvement compared to treatment without hydroxychloroquine in non-hospitalized patients with COVID-19.
Sources: Mitjà, 2020; Omrani, 2020 (Q-PROTECT); Skipper, 2020 |
Hydroxychloroquine in hospitalized COVID-19 patients
Eight RCTs investigated hydroxychloroquine in hospitalized patients (Abd-Elsalam, 2020; Chen, 2020; RECOVERY trial (Horby, 2020); Lyngbakken, 2020; SOLIDARITY trial (Pan, 2020) ); Self, 2020; Tang, 2020; Ulrich, 2020 (TEACH)). An overview of the included RCTs is provided in Table 1.
Abd-Elsalam (2020) described a multicenter randomized controlled trial. Abd-Elsalam (2020) evaluated the efficacy and safety of hydroxychloroquine in addition to standard of care versus standard of care alone in patients with COVID-19. The study included hospitalized patients with suspected and confirmed COVID-19. Patients were stratified in mild, moderate, and severe disease according to the WHO interim guidelines. Mild cases represented patients with uncomplicated upper respiratory tract viral infection. Moderate cases represented patients with pneumonia but without need for supplemental oxygen. Severe cases represented cases with fever or suspected respiratory infection, respiratory rate >30 breath/min, sever respiratory distress, or SpO2 93% on room air. The intervention group (n=97) received 400 mg hydroxychloroquine twice per day (on day one) follow by 200 mg tablets twice per day added to the standard of care for 15 days. The control group (n=97) received standard of care only. Standard of care consisted of paracetamol, oxygen, fluids (according to assessment), empiric antibiotic (cephalosporins), oseltamivir if needed (75 mg/12 hours for 5 days), and invasive mechanical ventilation with hydrocortisone for severe cases if PaO2 < 60 mmHg, O2 saturation < 90% despite oxygen or non-invasive ventilation, progressive hypercapnia, respiratory acidosis (pH < 7.3), and progressive or refractory septic shock. The length of follow-up was four weeks. The following relevant outcome measures were included: mortality, respiratory support (mechanical ventilation/Optiflow), duration of hospitalization, time to clinical improvement. The primary outcomes were recovery within 28 days, need for mechanical ventilation, or death. There were no difference between the two groups regarding the primary outcomes.
Chen (2020) described an open-label randomized clinical trial. Chen (2020) evaluated the efficacy and tolerability of hydroxychloroquine in adult hospitalized patients with mild to moderate COVID-19 disease. The study included patients aged between 20 and 79 years and confirmed positive for SARS-CoV-2 infection by RT-PCR. Patients in the intervention group (n=21) received 400 mg hydroxychloroquine twice per day on day one and 200 mg twice per day for six days plus standard of care. The control group (n=12) received standard of care only. Standard of care consisted of supportive treatment without antibiotics for subjects with mild clinical COVID-19 symptoms and with antimicrobial therapy for subjects presenting with moderate clinical COVID-19 symptoms. The antimicrobial treatment consisted of: (1) ceftriaxone two gram daily for seven days plus azithromycin 500 mg on the first day and 250 mg on days two to five; or (2) levofloxacin 750 mg daily for five days; or (3) levofloxacin 500 mg daily; or (4) moxifloxacin 400 mg daily for seven to 14 days for subjects allergic to ceftriaxone or azithromycin or according to physician discretion. The length of follow-up was 14 days. The following relevant outcome measures were included: time to clinical improvement. The primary endpoint was to evaluate the time to negative rRT-PCR assessments from randomization, up to 14 days. The study showed that HCQ failed the primary endpoint of shortening the viral clearance interval.
Horby (2020) (RECOVERY trial) described a randomized controlled open-label trial conducted in 176 National Health Service (NHS) hospitals in the UK. The RECOVERY trial (Horby, 2020) reported the results of a comparison between hydroxychloroquine and usual care involving patients hospitalized with COVID-19. The study included patients who had clinically-suspected or laboratory confirmed SARS-CoV-2 infection and no medical history that might put patients at substantial risk if they were to participate in the trial. Patients in the intervention group (n=1561) received hydroxychloroquine sulfate (in the form of a 200 mg tablet containing a 155-mg base equivalent) in a loading dose of four tablets (total dose, 800 mg) at baseline and at six hours, which was followed by two tablets (total dose, 400 mg) starting at 12 hours after the initial dose and then every 12 hours for the next nine days or until discharge. The control group (n=3155) received standard of care only. The standard of care differed between hospitals but was not further described. At total of 288 (9.2%) patients received dexamethasone, 638 (20.3%) azithromycin or other macrolide, 84 (2.7%) tocilizumab or sarilumab, 6 (0.2%) Lopinavir-Ritonavir, 2 (0.1%) remdesivir, and 12 (0.4%) hydroxychloroquine. The length of follow-up was 28 days. The following relevant outcome measures were included: mortality, respiratory support (mechanical ventilation / optiflow), duration of hospitalization and cardiac-related events. The results . The primary outcome was 28-day mortality and occurred in 421 patients (27.0%) in the hydroxychloroquine group and in 790 (25.0%) in the usual-care group (rate ratio 1.09; 95% CI 0.97 to 1.23; P=0.15). These results suggested that patients in the hydroxychloroquine group were less likely to be discharged from the hospital alive within 28 days than those in the usual-care group
Lyngbakken (2020) described a single center two-arm open-label group-sequential pragmatic randomized controlled trial. Lyngbakken (2020) assessed the efficacy and safety of hydroxychloroquine therapy in addition to standard of care on SARS-CoV-2 oropharyngeal viral kinetics in hospitalized patients with moderately severe COVID-19. The study included patients. The intervention group (n=27) received 400 mg hydroxychloroquine sulfate twice per day for seven days in addition to standard of care. Standard care was similar for all
patients included in the study, and encompassed appropriate level and intensity of
medical treatment according to local and national guidelines, but was not further described. The pharmacological treatment of the included patients was not described. The control group (n=26) received standard of care only. The length of follow-up was 30 days. The following relevant outcome measures were included: mortality, time to clinical improvement, respiratory support (non-invasive ventilation). The primary outcome was rate of decline in SARS-CoV-2 viral load in the oropharynx from baseline through the first 96 h after randomization. The estimated mean difference between groups was 0.27 (95% CI −0.92 to 1.47) log10 RNA copies/mL at
randomization, 0.06 (95% CI −1.15 to 1.26) log10 RNA copies/mL at 48 h, and −0.16 (95% CI −1.67 to 1.36) log10 RNA copies/mL at 96 h. These results suggested that treatment with hydroxychloroquine did not result in a significantly greater rate of decline in SARS-CoV-2 oropharyngeal viral load compared to standard care alone during the first five days.
Pan (2020) (SOLIDARITY trial) described an open-label randomized controlled trial. This trial compared the efficacy of the addition of either hydroxychloroquine, remdesivir, lopinavir or interferon with or without lopinavir to standard of care in hospitalized patients with COVID-19. The study included patients who were aged 18 years or older, diagnosed with COVID-19, not known with receiving any trial drug, not expected to be transferred elsewhere within 72 hours, and hand no contraindication to any trial drug. Regarding hydroxychloroquine, the intervention group (n=947) orally received four tablets hydroxychloroquine at hour 0, four tablets at hour six, and, starting at hour 12, two tablets twice daily for ten days in addition to local standard of care. Each tablet contained 200 mg of hydroxychloroquine sulfate (155 mg of hydroxychloroquine base per tablet). The control group (n=906) received local standard of care at a time and place in which that drug was locally available. Pharmacological treatment was not further specified. The length of follow-up was 28 days or up to discharge. The following relevant outcome measures were included: mortality, respiratory support (mechanical ventilation/Optiflow) and duration of hospitalization. The primary outcome was in-hospital mortality and occurred in in 104 of 947 patients receiving hydroxychloroquine and in 84 of 906 receiving its control (rate ratio, 1.19; 95% CI, 0.89 to 1.59; P=0.23). Hydroxychloroquine had little to no effect on overall mortality.
Self (2020) described a double-blind randomized controlled trial. Self (2020) assessed whether hydroxychloroquine is an efficacious treatment for hospitalized adults with COVID-19. The study included adults aged 18 years or older who were hospitalized for less than 48 hours with laboratory-confirmed SARS-CoV-2 infection and symptoms of respiratory illness for less than ten days. The intervention group (n=242) received 400 mg of hydroxychloroquine sulfate in pill form twice per day for the first two doses and then 200 mg in pill form twice per day for the subsequent eight doses, for a total of ten doses over five days. The control group (n=237) received matching placebo in the same dosing frequency. Among the 479 patients in the trial, remdesivir, azithromycin, and corticosteroids were received by 104 (21.7%), 91 (19.0%), and 88 (18.4%) patients, respectively, during the same hospitalization in which they were enrolled in the trial. The length of follow-up was 28 days. The following relevant outcome measures were included: mortality, duration of hospitalization, time to clinical improvement, respiratory support (non-invasive ventilation). The primary outcome was clinical status 14 days after randomization assessed with a 7-category ordinal scale (the COVID Outcomes Scale) recommended by the World Health Organization. At day 14 the difference in COVID Outcomes Scale score between the hydroxychloroquine group (median (IQR) score, 6 (4-7)) and placebo group (median (IQR) score, 6 (4-7)) with an adjusted OR of 1.02 (95% CI, 0.73 to 1.42). Treatment with hydroxychloroquine did not significantly improve clinical status at day 14 compared with placebo.
Tang (2020) described a multicenter open-label randomized controlled trial. Tang (2020) assessed the efficacy and safety of hydroxychloroquine plus standard of care compared with standard of care alone in hospitalized adults with mild, moderate and severe COVID-19. The study included patients aged 18 years or older, ongoing SARS-CoV-2 infection confirmed in upper or lower respiratory tract specimens with RT-PCR, willingness to participate, and consent not to be enrolled in other clinical trials during the study period. Patients in the intervention group (n=75) received hydroxychloroquine in addition to standard of care within 24 hours after randomization, with a loading dose of 1200 mg daily for three days followed by a maintenance dose of 800 mg daily for the remaining days. The total treatment duration was two weeks for patients with mild to moderate disease and three weeks for those with severe disease. The control group (n=75) received standard of care only. Standard of care was aligned with the indications from the updated national clinical practice guidelines for COVID-19 in China. Minimum requirements for the standard of care included the provision of intravenous fluids, supplemental oxygen, regular laboratory testing, SARS-CoV-2 testing, hemodynamic monitoring, and intensive care, as well as the ability to deliver concomitant medications. The length of follow-up was 28 days. The following relevant outcome measures were included: mortality, time to clinical improvement. The primary outcomes for this trial were whether patients had negative conversion of SARS-CoV-2 by 28 days. The probability of negative conversion of SARS-CoV-2 among patients who were assigned to receive standard of care plus hydroxychloroquine by 28 days was 85.4% (95% CI: 73.8% to 93.8%) and 81.3% (71.2% to 89.6% in the standard care group. The difference in the probability of negative conversion between standard of care plus hydroxychloroquine and standard of care alone was 4.1% (95%CI: –10.3% to 18.5%).
Administration of hydroxychloroquine did not result in a significantly higher probability of negative conversion than standard of care alone in patients admitted to hospital.
Ulrich (2020) described a multicenter double-blind randomized controlled trial. Ullrich (2020) evaluated the efficacy and safety of hydroxychloroquine in hospitalized patients with COVID-19. The study included hospitalized patients who had a positive SARS-CoV-2 RT-PCR within 72 hours of enrolment, at least one COVID-19 symptom, and written informed consent. The intervention group (n=67) orally received 400 mg (two tablets of 200 mg) of hydroxychloroquine sulfate twice per day on day one and 200 mg by mouth two times per day on days two to five. The control group (n=61) received a placebo tablet of 400 mg calcium citrate in the same regimen as the intervention group. Possible concomitant medications were antibacterial agents (23.4% azithromycin and 24.2% ceftriaxone), anticoagulation (53.9% VTE prophylaxis, 35.9% therapeutic anticoagulation and 29.7% antiplatelet agents), zinc (14.1%), corticosteroids (10.2%), tocilizumab (3.9%), lopinavir-ritonavir (0.8%), remdesivir (0.8%), convalescent plasma (13.3%), and clazakizumab (3.1%). The length of follow-up was 30 days. The following relevant outcome measures were included: mortality, respiratory support (mechanical ventilation / Optiflow), duration of hospitalization, time to clinical improvement, respiratory support (non-invasive ventilation). The primary outcome was a composite end point (death, intensive care unit admission, mechanical ventilation, extracorporeal membrane oxygenation, and/or vasopressor use) at day 14. At day 14, 11 (16.4%) subjects assigned to HCQ and 6 (9.8%, p=0.35) subjects assigned to placebo met the severe disease progression end point.
Treatment with hydroxychloroquine did not significantly improve clinical status at day 14 compared with placebo.
Table 1 Overview of RCTs comparing hydroxychloroquine with standard care in hospitalized COVID-19 patients.
|
Author |
Disease severity, based on need for respiratory support* |
Sample size |
Dosage |
|
Abd-Elsalam (2020) |
Mild disease
|
I: N=97 C: N=97 Total: N=194 |
- 400 mg hydroxychloroquine twice per day (on day 1). - 200 mg tablets twice per day added to the standard of care for 15 days |
|
Chen (2020)
|
Mild disease
|
I: N=21 C: N=12 Total: N=33 |
- 400 mg hydroxychloroquine twice per day on day 1. - 200 mg twice per day for six days. |
|
Horby (2020) RECOVERY trial |
Mixed: mild, moderate and severe disease If possible, sub group results are presented |
I: N=1561 C: N=3155 Total: N=4716 |
- Four tablets of 200 mg hydroxychloroquine at baseline and at 6 hours. - Two tablets of 200 mg starting at 12 hours after initial dose and every 12 hours for the next nine days or until discharge. |
|
Lyngbakken (2020) |
Mixed: mild and moderate disease |
I: N=27 C: N=26 Total: N=53 |
400 mg hydroxychloroquine sulfate 2 times daily for 7 days. |
|
Pan (2020) SOLIDARITY trial |
Mixed: mild, moderate and severe disease If possible, sub group results are presented |
I: N=947 C: N=906 Total: N=1853 |
- 4 tablets of 200 mg hydroxychloroquine sulfate (155 mg of hydroxychloroquine base per tablet) at hour 0, 6 and 12. - 2 tablets twice daily for 10 days. |
|
Self (2020) |
Mixed: mild, moderate and severe disease
|
I: N=242 C: N=237 Total: N=479 |
- First 2 doses: 400 mg hydroxychloroquine in pill form 2 times daily. - 200 mg in pill form 2 times daily for the subsequent 8 doses, for a total of 10 doses over 5 days. |
|
Tang (2020) |
Mild disease
|
I: N=75 C: N=75 Total: N=150 |
1200 mg daily for three days followed by a maintenance dose of 800 mg daily for the remaining 11 days (mild to moderate patients) and 18 days (severe patients). |
|
Ulrich (2020) TEACH trial |
Mixed: mostly mild and moderate disease; 1.6% severe disease |
I: N=67 C: N=61 Total: N=128 |
2 tablets of 200 mg hydroxychloroquine |
*Disease severity categories:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen, CPAP, NIV, mechanical ventilation, ECMO).
N: Total sample size; I: Intervention; C: Control
Results
Mortality (crucial)
Mortality, 28-30 days
The pooled incidence of mortality in hospitalized patients in the intervention group was 564/3016 (18.7%), compared to 910/4557 (20.0%) in the control group. Pooled risk ratio (RR) was 1.09 (95% CI 0.99 to 1.19; figure 1). The pooled risk difference (RD) was in favour of the control group (1.1%; 95% CI -0.3% to 2.5%). This is not a clinically relevant difference. The minor discrepancy between the crude and pooled numbers arises due to the weighing in the random-effects model. The RECOVERY trial (Horby, 2020) has the heaviest weight (83.7%) and is in favour of the control group (27.0% in the intervention group versus 25.0% in the control group).
Figure 1: Mortality (28-30 days) in hospitalized patients
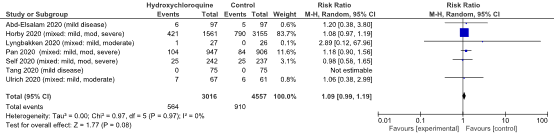
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Mild disease
Abd-Elsalam (2020) and Tang (2020) reported data of patients with mild disease. The mortality rate was 6 of 172 (%) in the intervention group and 5 of 172 (%) in the control group, with a risk ratio of 1.20 (95% CI 0.38 to 3.80). As no events occurred in the study of Tang (2020), this risk ratio is based on the study results of Abd-Elsalam (2020).
Figure 2: Mortality (28-30 days) in sub-groups of hospitalized patients
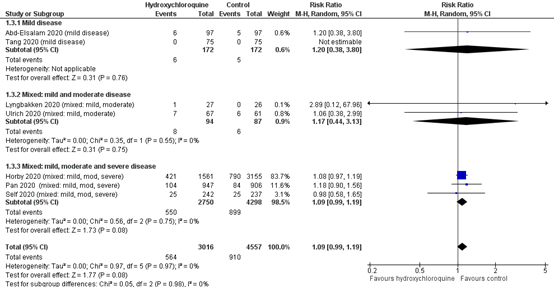
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Mild disease (not separately shown in meta-analysis)
In addition to the pooled results of Abd-Elsalam (2020) and Tang (2020) (figure 1), the RECOVERY trial (Horby, 2020) reported sub-group data of the patients that did not receive oxygen at baseline. The mortality rate was 58 of 362 (16.0%) in the intervention group and 99 of 750 (13.2%) in the control group.
Severe disease (not separately shown in meta-analysis)
The RECOVERY trial (Horby, 2020) reported subgroup data of the patients that received invasive mechanical ventilation at baseline. The mortality rate was 110 of 261 (42.1%) in the intervention group and 216 of 532 (40.6%) in the control group. The RR was 1.03 (95% CI 0.81 to 1.30).
The SOLIDARITY trial (Pan, 2020) reported subgroup data of the patients that received invasive mechanical ventilation at baseline. The HR for mortality was 1.26 (95% CI 0.76 to 2.10), in favour of the control group.
Mortality, other follow-up
Chen (2020) reported that no deaths took place in the intervention group (0/21) or control group (0/12) during the 14-day follow up.
Level of evidence of the literature
The level of evidence for hospitalized patients with COVID-19 started as high, because the studies were RCTs. The level of evidence was downgraded by one level because the confidence interval crosses both the threshold of no effect and the threshold of clinical relevance (imprecision, -1). The level of evidence for the outcome mortality in hospitalized patients is considered moderate.
Extensive respiratory support (crucial)
Extensive respiratory support in hospitalized patients with COVID-19 was reported in six studies (Abd-Elsalam, 2020; RECOVERY trial (Horby, 2020); Lyngbakken, 2020; SOLIDARITY trial (Pan, 2020) ; Self, 2020; Ulrich, 2020 (TEACH)).
The results were pooled in a meta-analysis (figure 3). The pooled incidence of mechanical ventilation in hospitalized patients in the intervention group was 212/2326 (9.1%) and 299/3605 (8.3%). The pooled RR was 1.12 (95% CI 0.95 to 1.32) and the pooled RD was 0.01 (95% CI -0.01 to 0.02) favouring the control group. This is not a clinically relevant difference.
Figure 3: Extensive respiratory support (28-30days) in hospitalized patients
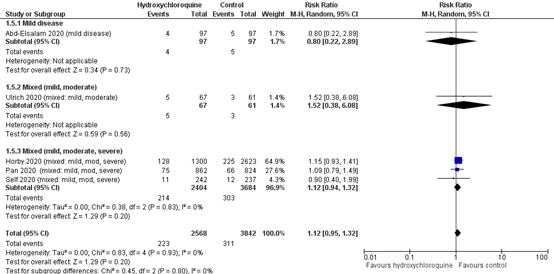
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Mixed – mild & moderate disease
Lyngbakken (2020) reported by clinical status at day 14 that the none of patients requiring invasive mechanical ventilation or ECMO in the intervention group and 1 patient (4.0%) in the control group. One patient (3.8%) in de intervention group required non-invasive ventilation or high flow oxygen devices respectively versus 0 patients in the control group.
In addition, Ulrich (2020) reported by clinical status at day 14 that the percentage of patients requiring ventilator or ECMO was 3.0% (n=2) in the intervention group and 0% in the control group; requiring non-invasive ventilation or high flow oxygen devices respectively 10.4% (n=7) and 3.3% (n=2).
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by one level because of crossing the threshold for clinical relevance (imprecision, -1). The level of evidence for the outcome ‘need for extensive respiratory support’, is considered moderate.
Duration of hospitalization (important)
Duration of hospitalization in hospitalized patients with COVID-19 was reported in five studies (Abd-Elsalam, 2020; RECOVERY trial (Horby, 2020); SOLIDARITY trial (Pan, 2020); Self, 2020; Ulrich, 2020 (TEACH)). Due to differences in reporting (mean, median or percentage), data were not pooled.
Mild disease
Abd-Elsalam (2020) reported that the mean (SD) duration of hospitalization was 11.04 (2.71) days in the intervention group (n=97), compared to 11.27 (2.19) days in the control group (n=97; MD -0.23 days; 95% CI -0.92 to 0.46)).
Mixed – mild & moderate disease
Ulrich (2020) found that the mean (SD) duration of hospitalization was 9.75 (10.3) days in the intervention group (n=67), compared to 6.80 (5.92) days in the control group (n=61). MD 2.95 days (95% CI 0.07 to 5.83), which was not considered as clinically relevant difference favouring the control group.
Mixed – mild, moderate & severe disease
The RECOVERY trial (Horby, 2020) reported that patients in the hydroxychloroquine group (n=1561) had a longer duration of hospitalization than those in the usual-care group (n=3155; median 16 days versus 13 days). In addition, the discharge rate at 28 days of follow-up was reported. In the intervention group, 931 of the 1561 (59.6%) patients were discharged, compared to 1983 of the 3155 (62.9%) patients in the control group (RR 0.90; 95% CI 0.83 to 0.98). This difference is considered as clinically relevant.
The SOLIDARITY trial (Pan, 2020) reported that the percentage of patients discharged from hospital at day 21 was 11.0% in the intervention group (n=947), compared to 10.0% in the control group (n=906), favouring the intervention group (RR 1.09; 95% CI 0.84 to 1.43). This difference is not considered as clinically relevant.
Self (2020) reported that the median number of hospital-free days through 28 days was 21 (IQR 11 to 24) in the intervention group and 20 (10 to 24) in the control group. The difference was 1 day (−1 to 3), which was not considered as clinically relevant difference.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because of study limitations (lack of blinding, -1) and heterogeneity in reporting ‘duration of hospitalization’ and variance of point estimates across studies (inconsistency, -1). The level of evidence for the outcome ‘duration of hospitalization’ is considered low.
Time to clinical improvement (important)
Symptom resolution was reported in six studies for hospitalized patients with COVID-19 (Abd-Elsalam, 2020; Chen, 2020; Lyngbakken, 2020; Self, 2020; Tang, 2020; Ulrich, 2020 (TEACH)).
Mild disease
Abd-Elsalam (2020): The mean (SD) duration to clinical improvement (definition not described) was 9.43 (1.87) days in the intervention group compared to 9.52 (2.94) days in the control group (MD -0.09 days; 95% CI -0.78 to 0.60).
Chen (2020) reported that the incidence of resolution of symptoms* by day 14 was 28.6% (6/21) in the intervention group and 41.7% (5/12) in the control group. The RR was 0.69 (0.26 to 1.78), which is a clinically relevant difference favouring the control group.
Tang (2020) reported the time to alleviation of symptoms**. The median time was 19 days in the intervention group and 21 days in the control group (HR 1.01; 95% CI 0.59 to 1.74).
Mixed – mild & moderate disease
Lyngbakken (2020) reported the change in NEWS2-score from randomization to 96 hours.
(higher score indicates worse condition). The mean change in the intervention group was 0.47
(95% CI −0.58 to 1.53) compared to 0.29 (95% CI −0.88 to 1.46) in the control group (MD in change score 0.18 (95% CI −1.40 to 1.76)).
Ulrich (2020) reported the number of fever-free days during the follow-up period of 30 days. The mean number of fever-free days was 6.40 (SD 0.94) in the intervention group and 6.31 (1.33) in the control group (MD 0.09).
Mixed – mild, moderate & severe disease
Self (2020) reported the time to recovery, defined as time to reach a clinical status of COVID Outcome Scale category 5, 6, or 7***. The median time to recovery was 5 (IQR 1 to 14) days in the intervention group and 6 (1 to 15) days in the control group.
* clinical recovery was defined as clinical recovery as the 1st time of three consecutive PCR tests and major symptoms showed negative results.
**: defined as resolving from fever to an axillary temperature of 36.6°C or below, normalisation of SpO2 (>94% on room air), and disappearance of respiratory symptoms including nasal congestion, cough, sore throat, sputum production, and shortness of breath.
*** 5, hospitalized, not receiving supplemental oxygen; 6, not hospitalized and unable to perform normal activities; and 7, not hospitalized and able to perform normal activities)
The definition of clinical improvement varied, and results were inconsistent between studies.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by three levels because of study limitations (lack of blinding, -1), heterogeneity in reporting ‘clinical improvement’ and variance of point estimates across studies (inconsistency, -1) and crossing thresholds for clinical relevance (imprecision, -1). The level of evidence for the outcome ‘clinical improvement’ is considered very low.
Description of studies: treatment with hydroxychloroquine in non-hospitalized COVID-19 patients
Three RCTs investigated hydroxychloroquine in non-hospitalized patients (Mitjà, 2020; Omrani, 2020 (Q-PROTECT); Skipper, 2020). An overview of the included RCTs is provided in Table 2.
Mitjà (2020) described an open-label randomized controlled trial. Mitjà (2020) aimed to determine whether early treatment with hydroxychloroquine would be more efficacious than no treatment for outpatients with mild COVID-19. The study included non-hospitalized patients aged 18 years or older with mild symptoms of COVID-19 for less than five days before enrollment, and with a positive PCR test for SARS-CoV-2. The intervention group (n=136) received 800 mg hydroxychloroquine – Dolquine on day one, followed by 400 mg once daily for six days. The control group (n=157) received no treatment aside from usual care. The length of follow-up was 28 days. The following relevant outcome measures were included: mortality, duration of hospitalization, time to clinical improvement. The primary outcome was the reduction of viral RNA load in nasopharyngeal swabs at days 3, and 7 after treatment start. A mean reduction of viral load (-1.41 vs -1.41 log10 copies/mL in the control and intervention arm, respectively) was observed at day 3, which further declined at day 7 (-3.37 vs -3.44). No significant differences were found in the mean reduction of viral load (-0.07 (95%CI: -0.44 to 0.29)).
Treatment with hydroxychloroquine did not result in a significantly greater rate of decline in SARS-CoV-2 oropharyngeal viral load compared to standard care alone during the first five days.
Omrani (2020) described a parallel randomized controlled trial. Omrani (2020) determined whether use of hydroxychloroquine or hydroxychloroquine plus azithromycin was associated with higher rates of viral clearance compared to placebo. The study included non-hospitalized patients who had a positive SARS-CoV-2 PCR-test with mild or COVID-19 symptoms, respiration >29/minute, or pulse oximetry on room air <93%. The intervention group (hydroxychloroquine only, n=152) received 200 mg of hydroxychloroquine three times a day during seven days. In addition, two placebo tablets for azithromycin will be taken on day one and one on days two to five. The control group (n=152) received placebo tablets for hydroxychloroquine and placebo tablets for azithromycin and followed the same regimen as the intervention groups. The length of follow-up was 21 days. The following relevant outcome measures were included: mortality, time to clinical improvement. The study’s primary outcome was achievement of virologic cure (PCR-negative status) as assessed on day six. At day six, 16 (10.5%) subjects assigned to the hydroxychloroquine + azithromycin group, 19 (12.8%) subjects assigned to the hydroxychloroquine group achieved virological cure and 18 (12.2%) in the placebo group achieved virological cure. No difference was found in groups’ proportions achieving virologic cure at day six (p=0.821). Treatment with hydroxychloroquine (with or without azithromycin) did not result in a greater rate of virological cure after six days of treatment.
Skipper (2020) described a randomized double-blind placebo controlled trial. Skipper (2020) investigated whether hydroxychloroquine could reduce COVID-19 severity in adult outpatients. The study included non-hospitalized adults who were required to have four or fewer days of symptoms and either PCR-confirmed SARS-CoV-2 infection or compatible symptoms after a high-risk exposure to a person with PCR-confirmed COVID-19 within the past 14 days. The intervention group (n=212) received 800 mg (four tablets) once, then 600 mg (three tablets) six to eight hours later, then 600 mg (three tablets) once daily for four more days (five days in total). The control group (n=211) received placebo tablets of 400 mcg folic acid as an identical regimen as the intervention group. The length of follow-up was 14 days. The following relevant outcome measures were included: mortality, duration of hospitalization, time to clinical improvement. The primary outcome was change in overall symptom severity over 14 days.
Change in symptom severity over 14 days did not differ between the hydroxychloroquine and placebo groups (difference in symptom severity: relative, 12%; absolute, -0.27 point (95% CI, -0.61 to 0.07 point); P=0.117). At 14 days, 49 subjects (24%) receiving hydroxychloroquine had ongoing symptoms compared with 59 (30%) receiving placebo (P=0.21). Treatment with hydroxychloroquine did not substantially reduce symptom severity.
Table 2. Overview of RCTs comparing hydroxychloroquine with standard care in non-hospitalized COVID-19 patients.
|
Author |
Disease severity |
Sample size |
Dosage |
|
Mitjà (2020) |
Mild |
I: N=136 C: N=157 Total: N=293 |
800 mg hydroxychloroquine – Dolquine on day one. - 400 mg once daily for 6 days. |
|
Omrani (2020) |
Mild or no symptoms included |
I: N=152 C: N=152 Total: N= 304 |
200 mg of hydroxychloroquine 3 times per day for 7 days. |
|
Skipper (2020) |
Mild |
I: N=212 C: N=211 Total: 423 |
- 800 mg (four tablets) once. - 600 mg (three tablets) 6 to 8 hours later. - 600 mg (three tablets) once daily for 4 more days (5 days in total). |
Results
Mortality (crucial)
Mortality was reported in three studies (Mitjà, 2020; Omrani, 2020 (Q-PROTECT); Skipper, 2020).
Mortality, 28-30 days
Mitjà (2020) reported no deaths among all participants up to 28-days of follow up.
Other
Omrani (2020) reported no deaths among all study participants up to 21 days of follow-up.
Skipper (2020) reported 1 death in the intervention group and 1 death in the control group after 14 days.
Level of evidence of the literature
The level of evidence for non-hospitalized patients with COVID-19 started as high, because the studies were RCTs. The level of evidence was downgraded by two levels because of the small number of participants and the small number of events in the studies (imprecision, -2). The level of evidence for the outcome mortality in non-hospitalized patients is considered low.
Hospitalization (important)
Two studies reported the incidence of hospitalization. Mitja (2020) reported that 8 of the 136 (5.9%) patients in the intervention group needed hospitalization in the 28-day follow-up period, compared to 11 of the 157 (7.0%) patients in the control group (RR 0.84; 95% CI 0.35 to 2.03).
Skipper reported hospitalization in the 18-day follow-up period for 4 of the 212 (1.9%) patients in the intervention group and 10 of the 211 (4.7%) patients in the control group. Two of the hospitalizations in the control group were reported not being related to the study (RR 0.40; 95% CI 0.13 to 1.25).
Level of evidence
The level of evidence for non-hospitalized patients with COVID-19 started as high, because the studies were RCTs. The level of evidence was downgraded by three levels because of study limitations (risk of bias, -1), the small number of participants and the small number of events in the studies and crossing both thresholds for clinical relevance (imprecision, -2). The level of evidence for the outcome mortality in non-hospitalized patients is considered very low.
Time to clinical improvement (important)
Clinical improvement was reported in three studies (Mitjà, 2020; Omrani, 2020 (Q-PROTECT); Skipper, 2020).
Mitjà (2020) reported a median (IQR) time to complete symptom resolution* of 10.0 (4 to 18) days in the intervention group and 12.0 (6 to 21) days in the control group. The difference was two days, not a clinically relevant difference in favour of the intervention group.
Omrani (2020) reported the number of patients that were symptomatic at day one and asymptomatic at day 7, 14 and 21. Of patients in the first intervention group, respectively 80.0%, 95.7% and 97.1% was asymptomatic at those follow-up moments, compared to 79.9%, 92.8% and 98.6% in the second intervention group and 88.1%, 96.7% and 93.3% in the control group.
Skipper (2020) reported change in symptom severity score based on the Visual Analog Scale (VAS)** from baseline to 14 days. The change in symptom severity score in the intervention group was -2.60 points (SE 0.12), compared to -2.33 (SE 0.12) in the control group. Absolute difference -0.27 (95% CI -0.61 to 0.07), which is not a clinically relevant difference.
*Time to complete resolution of symptoms was defined as median time from randomization to the resolution of COVID-19 symptoms. Resolution of symptoms was assessed sequentially using a symptoms questionnaire designed to gather information on the type of symptom and last day experienced; complete resolution was considered when no COVID-19-related symptoms were reported at all.
**Overall symptom severity was collected via a digital slider bar, which was marked with “0=no symptoms”; 5 (placed in the middle); and “10=severe symptoms”.
Level of evidence of the literature
The level of evidence started as high, because the studies were RCTs. The level of evidence was downgraded by three levels because of study limitations (risk of bias, -1), heterogeneity in reporting ‘clinical improvement’ (inconsistency, -1) and small number of participants (imprecision, -1). The level of evidence for the outcome ‘time to clinical improvement’ is considered very low.
Non-invasive respiratory support (important)
Respiratory support was not reported for non-hospitalized patients with COVID-19.
Level of evidence of the literature
-
A systematic review of the literature was performed to answer the following question:
What is the effectivity of treatment with hydroxychloroquine compared to treatment without hydroxychloroquine in patients with COVID-19?
PICO 1
P: hospitalized with COVID-19 (subgroups mild, moderate, severe)
I: hydroxychloroquine + standard care
C: standard care only / placebo treatment + standard care
O: 28-30 day mortality (if not available, any other reports of mortality), extensive respiratory support, duration of hospitalization, time to clinical improvement
PICO 2
P: non-hospitalized patients with COVID-19
I: hydroxychloroquine + standard care
C: standard care only / placebo treatment + standard care
O: 28-30 day mortality (if not available, any other reports of mortality), respiratory support, hospitalization, time to clinical improvement
Relevant outcome measures
PICO 1: For hospitalized COVID-19 patients, mortality and need for extensive respiratory support were considered as crucial outcome measures for decision making. Duration of hospitalization, and time to clinical improvement were considered as important outcome measures for decision making.
PICO 2: For non-hospitalized COVID-19 patients, mortality was considered as a critical outcome measure for decision making. Hospitalization, respiratory support and time to clinical improvement were considered as important outcome measures for decision making.
Extensive respiratory support was defined as high flow nasal cannula (HFNC)/Optiflow, continuous positive airway pressure (CPAP), non-invasive ventilation (NIV), mechanical ventilation or extracorporeal membrane oxygenation (ECMO or ECLS). Non-invasive respiratory support was defined as supplemental oxygen low flow oxygen or non-rebreathing mask.
The working group defined 3% points absolute difference as a minimal clinically important difference for mortality (resulting in a NNT of 33), 3 days for duration of hospitalization and time to clinical improvement, 5% points absolute difference need for respiratory support and hospital admission (resulting in a NNT of 20).
The results of studies in non-hospitalized and hospitalized patients are summarized separately. Studies of hospitalized patients were categorized based on the respiratory support that was needed at baseline (preferably based on patient inclusion/exclusion criteria; otherwise on baseline characteristics). The following categories were used:
- mild disease (no supplemental oxygen);
- moderate disease (supplemental oxygen: low flow oxygen, non-rebreathing mask);
- severe disease (supplemental oxygen: high flow oxygen [high flow nasal cannula (HFNC)/Optiflow], continuous positive airway pressure [CPAP], non-invasive ventilation [NIV], mechanical ventilation, extracorporeal membrane oxygenation [ECMO or ECLS]).
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until 7 January 2021. The detailed search strategy is outlined under Methods. Studies were selected based on the following criteria: randomized controlled trial, peer reviewed and published in an indexed journal, comparing treatment with hydroxychloroquine and standard care to standard care alone or treatment hydroxychloroquine and standard care to placebo and standard care in patients with COVID-19.
The systematic literature search resulted in 33807 hits. Studies were selected based on the following criteria: systematic review or randomized controlled trials. Eventually, eleven studies were included.
An additional search was performed until 2 September 2021 resulting in 77600 hits. Important study characteristics and results were summarized in the evidence tables. Ten additional studies were included. The assessment of the risk of bias is summarized in the additional risk of bias tables. None of the additional RCTs found a clinical benefit in favour of hydroxychloroquine. Since the new available studies did not change the recommendations, the literature summary was not updated.
Statistical analyses were conducted using Review Manager (RevMan) software 5.4. For dichotomous outcomes, Mantel Haenszel random‐effects risk ratios (RRs) and risk differences (RDs) were calculated. For continuous outcomes, a random‐effects mean difference (MD) weighted by the inverse variance was calculated. The random-effects model estimates the mean of a distribution of effects.
Results
In total, eleven RCTs were included in the analysis of the literature. Eight studies investigated hydroxychloroquine in hospitalized patients (see Table 1). Three studies investigated hydroxychloroquine in non-hospitalized patients (see Table 2). Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
- Abd-Elsalam S, Esmail ES, Khalaf M, Abdo EF, Medhat MA, Abd El Ghafar MS, Ahmed OA, Soliman S, Serangawy GN, Alboraie M. Hydroxychloroquine in the Treatment of COVID-19: A Multicenter Randomized Controlled Study. Am J Trop Med Hyg. 2020 Oct;103(4):1635-1639. doi: 10.4269/ajtmh.20-0873. PMID: 32828135; PMCID: PMC7543820.
- Abd-Elsalam S. Retraction Notice. Am J Trop Med Hyg. 2022 Sep 2;107(3):1. doi: 10.4269/ajtmh.1073ret. Epub ahead of print. PMID: 36099166; PMCID: PMC9490653.
- Ader, F., Peiffer-Smadja, N., Poissy, J., Bouscambert-Duchamp, M., Belhadi, D., Diallo, A., Delmas, C., Saillard, J., Dechanet, A., Mercier, N., Dupont, A., Alfaiate, T., Lescure, F. X., Raffi, F., Goehringer, F., Kimmoun, A., Jaureguiberry, S., Reignier, J., Nseir, S., Danion, F., … DisCoVeRy study group (2021). An open-label randomized controlled trial of the effect of lopinavir/ritonavir, lopinavir/ritonavir plus IFN-β-1a and hydroxychloroquine in hospitalized patients with COVID-19. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases, 27(12), 1826–1837. https://doi-org.saz.idm.oclc.org/10.1016/j.cmi.2021.05.020
- Arabi, Y. M., Gordon, A. C., Derde, L., Nichol, A. D., Murthy, S., Beidh, F. A., Annane, D., Swaidan, L. A., Beane, A., Beasley, R., Berry, L. R., Bhimani, Z., Bonten, M., Bradbury, C. A., Brunkhorst, F. M., Buxton, M., Buzgau, A., Cheng, A., De Jong, M., Detry, M. A., … REMAP-CAP Investigators (2021). Lopinavir-ritonavir and hydroxychloroquine for critically ill patients with COVID-19: REMAP-CAP randomized controlled trial. Intensive care medicine, 47(8), 867–886. https://doi-org.saz.idm.oclc.org/10.1007/s00134-021-06448-5
- Barratt-Due, A., Olsen, I. C., Nezvalova-Henriksen, K., Kåsine, T., Lund-Johansen, F., Hoel, H., Holten, A. R., Tveita, A., Mathiessen, A., Haugli, M., Eiken, R., Kildal, A. B., Berg, Å., Johannessen, A., Heggelund, L., Dahl, T. B., Skåra, K. H., Mielnik, P., Le, L., Thoresen, L., … NOR-Solidarity trial (2021). Evaluation of the Effects of Remdesivir and Hydroxychloroquine on Viral Clearance in COVID-19 : A Randomized Trial. Annals of internal medicine, 174(9), 1261–1269. https://doi-org.saz.idm.oclc.org/10.7326/M21-0653
- Brown, S. M., Peltan, I. D., Webb, B., Kumar, N., Starr, N., Grissom, C., Buckel, W. R., Srivastava, R., Harris, E. S., Leither, L. M., Johnson, S. A., Paine, R., 3rd, & Greene, T. (2020). Hydroxychloroquine versus Azithromycin for Hospitalized Patients with Suspected or Confirmed COVID-19 (HAHPS). Protocol for a Pragmatic, Open-Label, Active Comparator Trial. Annals of the American Thoracic Society, 17(8), 1008–1015. https://doi-org.saz.idm.oclc.org/10.1513/AnnalsATS.202004-309SD
- Cavalcanti, A. B., Zampieri, F. G., Rosa, R. G., Azevedo, L., Veiga, V. C., Avezum, A., Damiani, L. P., Marcadenti, A., Kawano-Dourado, L., Lisboa, T., Junqueira, D., de Barros E Silva, P., Tramujas, L., Abreu-Silva, E. O., Laranjeira, L. N., Soares, A. T., Echenique, L. S., Pereira, A. J., Freitas, F., Gebara, O., … Coalition Covid-19 Brazil I Investigators (2020). Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. The New England journal of medicine, 383(21), 2041–2052. https://doi-org.saz.idm.oclc.org/10.1056/NEJMoa2019014
- Chen (2020) CP, Lin YC, Chen TC, Tseng TY, Wong HL, Kuo CY, Lin WP, Huang SR, Wang WY, Liao JH, Liao CS, Hung YP, Lin TH, Chang TY, Hsiao CF, Huang YW, Chung WS, Cheng CY, Cheng SH; Taiwan HCQ Study Group. A multicenter, randomized, open-label, controlled trial to evaluate the efficacy and tolerability of hydroxychloroquine and a retrospective study in adult patients with mild to moderate coronavirus disease 2019 (COVID-19). PLoS One. 2020 Dec 2;15(12):e0242763. doi: 10.1371/journal.pone.0242763. PMID: 33264337; PMCID: PMC7710068.
- Chorin, E., Dai, M., Shulman, E., Wadhwani, L., Bar-Cohen, R., Barbhaiya, C., Aizer, A., Holmes, D., Bernstein, S., Spinelli, M., Park, D. S., Chinitz, L. A., & Jankelson, L. (2020). The QT interval in patients with COVID-19 treated with hydroxychloroquine and azithromycin. Nature medicine, 26(6), 808–809. https://doi.org/10.1038/s41591-020-0888-2
- Dubée, V., Roy, P. M., Vielle, B., Parot-Schinkel, E., Blanchet, O., Darsonval, A., Lefeuvre, C., Abbara, C., Boucher, S., Devaud, E., Robineau, O., Rispal, P., Guimard, T., d'Anglejean, E., Diamantis, S., Custaud, M. A., Pellier, I., Mercat, A., HYCOVID study group, HYCOVID investigators, … Study management: Data management (2021). Hydroxychloroquine in mild-to-moderate coronavirus disease 2019: a placebo-controlled double blind trial. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases, 27(8), 1124–1130. https://doi-org.saz.idm.oclc.org/10.1016/j.cmi.2021.03.005
- Gupta, S., Dixit, P. K., Ghana, P., Abhisheka, K., Khurana, H., Jha, V. K., Mahapatra, D., Goel, J., Ahmed, S., & Varadaraj, G. (2021). Open-label randomized control trial of hydroxychloroquine in patients with moderate to severe coronavirus disease 2019 infection. Medical journal, Armed Forces India, 77(Suppl 2), S305–S311. https://doi-org.saz.idm.oclc.org/10.1016/j.mjafi.2021.02.007
- Huang M, Tang T, Pang P, Li M, Ma R, Lu J, Shu J, You Y, Chen B, Liang J, Hong Z, Chen H, Kong L, Qin D, Pei D, Xia J, Jiang S, Shan H. Treating COVID-19 with Chloroquine. J Mol Cell Biol. 2020 May 18;12(4):322-325. doi: 10.1093/jmcb/mjaa014. PMID: 32236562; PMCID: PMC7232130.
- Lane, J. C. E., Weaver, J., Kostka, K., Duarte-Salles, T., Abrahao, M. T. F., Alghoul, H., Alser, O., Alshammari, T. M., Biedermann, P., Banda, J. M., Burn, E., Casajust, P., Conover, M. M., Culhane, A. C., Davydov, A., DuVall, S. L., Dymshyts, D., Fernandez-Bertolin, S., Fišter, K., . . . Prieto-Alhambra, D. (2020). Risk of hydroxychloroquine alone and in combination with azithromycin in the treatment of rheumatoid arthritis: a multinational, retrospective study. The Lancet Rheumatology, 2(11), e698–e711. https://doi.org/10.1016/s2665-9913(20)30276-9
- Liu J., Cao R., Xu M., Wang X., Zhang H., Hu H. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6:16.
- Lyngbakken MN, Berdal JE, Eskesen A, Kvale D, Olsen IC, Rueegg CS, Rangberg A, Jonassen CM, Omland T, Røsjø H, Dalgard O. A pragmatic randomized controlled trial reports lack of efficacy of hydroxychloroquine on coronavirus disease 2019 viral kinetics. Nat Commun. 2020 Oct 20;11(1):5284. doi: 10.1038/s41467-020-19056-6. PMID: 33082342; PMCID: PMC7576792.
- Mercuro, N. J., Yen, C. F., Shim, D. J., Maher, T. R., McCoy, C. M., Zimetbaum, P. J., & Gold, H. S. (2020). Risk of QT Interval Prolongation Associated With Use of Hydroxychloroquine With or Without Concomitant Azithromycin Among Hospitalized Patients Testing Positive for Coronavirus Disease 2019 (COVID-19). JAMA cardiology, 5(9), 1036–1041. https://doi.org/10.1001/jamacardio.2020.1834
- Mitjà O, Corbacho-Monné M, Ubals M, Tebe C, Peñafiel J, Tobias A, Ballana E, Alemany A, Riera-Martí N, Pérez CA, Suñer C, Laporte P, Admella P, Mitjà J, Clua M, Bertran L, Sarquella M, Gavilán S, Ara J, Argimon JM, Casabona J, Cuatrecasas G, Cañadas P, Elizalde-Torrent A, Fabregat R, Farré M, Forcada A, Flores-Mateo G, Muntada E, Nadal N, Narejos S, Gil-Ortega AN, Prat N, Puig J, Quiñones C, Reyes-Ureña J, Ramírez-Viaplana F, Ruiz L, Riveira-Muñoz E, Sierra A, Velasco C, Vivanco-Hidalgo RM, Sentís A, G-Beiras C, Clotet B, Vall-Mayans M; BCN PEP-CoV-2 RESEARCH GROUP. Hydroxychloroquine for Early Treatment of Adults with Mild Covid-19: A Randomized-Controlled Trial. Clin Infect Dis. 2020 Jul 16:ciaa1009. doi: 10.1093/cid/ciaa1009. Epub ahead of print. PMID: 32674126; PMCID: PMC7454406.
- Nord, J. E., Shah, P. K., Rinaldi, R. Z., & Weisman, M. H. (2004). Hydroxychloroquine cardiotoxicity in systemic lupus erythematosus: a report of 2 cases and review of the literature. Seminars in arthritis and rheumatism, 33(5), 336–351. https://doi.org/10.1016/j.semarthrit.2003.09.012
- Omrani AS, Pathan SA, Thomas SA, Harris TRE, Coyle PV, Thomas CE, Qureshi I, Bhutta ZA, Mawlawi NA, Kahlout RA, Elmalik A, Azad AM, Daghfal J, Mustafa M, Jeremijenko A, Soub HA, Khattab MA, Maslamani MA, Thomas SH. Randomized double-blinded placebo-controlled trial of hydroxychloroquine with or without azithromycin for virologic cure of non-severe Covid-19. EClinicalMedicine. 2020 Dec;29:100645. doi: 10.1016/j.eclinm.2020.100645. Epub 2020 Nov 20. PMID: 33251500; PMCID: PMC7678437.
- Perinel, S., Launay, M., Botelho-Nevers, É., Diconne, É., Louf-Durier, A., Lachand, R., Murgier, M., Page, D., Vermesch, R., Thierry, G., & Delavenne, X. (2020). Towards Optimization of Hydroxychloroquine Dosing in Intensive Care Unit COVID-19 Patients. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America, 71(16), 2227–2229. https://doi.org/10.1093/cid/ciaa394
- Réa-Neto, Á., Bernardelli, R. S., Câmara, B., Reese, F. B., Queiroga, M., & Oliveira, M. C. (2021). An open-label randomized controlled trial evaluating the efficacy of chloroquine/hydroxychloroquine in severe COVID-19 patients. Scientific reports, 11(1), 9023. https://doi-org.saz.idm.oclc.org/10.1038/s41598-021-88509-9
- RECOVERY Collaborative Group, Horby P, Mafham M, Linsell L, Bell JL, Staplin N, Emberson JR, Wiselka M, Ustianowski A, Elmahi E, Prudon B, Whitehouse T, Felton T, Williams J, Faccenda J, Underwood J, Baillie JK, Chappell LC, Faust SN, Jaki T, Jeffery K, Lim WS, Montgomery A, Rowan K, Tarning J, Watson JA, White NJ, Juszczak E, Haynes R, Landray MJ. Effect of Hydroxychloroquine in Hospitalized Patients with Covid-19. N Engl J Med. 2020 Nov 19;383(21):2030-2040. doi: 10.1056/NEJMoa2022926. Epub 2020 Oct 8. PMID: 33031652; PMCID: PMC7556338.
- Reis, G., Moreira Silva, E., Medeiros Silva, D. C., Thabane, L., Singh, G., Park, J., Forrest, J. I., Harari, O., Quirino Dos Santos, C. V., Guimarães de Almeida, A., Figueiredo Neto, A. D., Savassi, L., Milagres, A. C., Teixeira, M. M., Simplicio, M., Ribeiro, L. B., Oliveira, R., Mills, E. J., & TOGETHER Investigators (2021). Effect of Early Treatment With Hydroxychloroquine or Lopinavir and Ritonavir on Risk of Hospitalization Among Patients With COVID-19: The TOGETHER Randomized Clinical Trial. JAMA network open, 4(4), e216468. https://doi-org.saz.idm.oclc.org/10.1001/jamanetworkopen.2021.6468
- Schrezenmeier, E., & Dörner, T. (2020). Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nature reviews. Rheumatology, 16(3), 155–166. https://doi.org/10.1038/s41584-020-0372-x
- Schwartz, I., Boesen, M. E., Cerchiaro, G., Doram, C., Edwards, B. D., Ganesh, A., Greenfield, J., Jamieson, S., Karnik, V., Kenney, C., Lim, R., Menon, B. K., Mponponsuo, K., Rathwell, S., Ryckborst, K. J., Stewart, B., Yaskina, M., Metz, L., Richer, L., Hill, M. D., … ALBERTA HOPE COVID-19 Collaborators (2021). Assessing the efficacy and safety of hydroxychloroquine as outpatient treatment of COVID-19: a randomized controlled trial. CMAJ open, 9(2), E693–E702. https://doi-org.saz.idm.oclc.org/10.9778/cmajo.20210069
- Self WH, Semler MW, Leither LM, Casey JD, Angus DC, Brower RG, Chang SY, Collins SP, Eppensteiner JC, Filbin MR, Files DC, Gibbs KW, Ginde AA, Gong MN, Harrell FE Jr, Hayden DL, Hough CL, Johnson NJ, Khan A, Lindsell CJ, Matthay MA, Moss M, Park PK, Rice TW, Robinson BRH, Schoenfeld DA, Shapiro NI, Steingrub JS, Ulysse CA, Weissman A, Yealy DM, Thompson BT, Brown SM; National Heart, Lung, and Blood Institute PETAL Clinical Trials Network, Steingrub J, Smithline H, Tiru B, Tidswell M, Kozikowski L, Thornton-Thompson S, De Souza L, Hou P, Baron R, Massaro A, Aisiku I, Fredenburgh L, Seethala R, Johnsky L, Riker R, Seder D, May T, Baumann M, Eldridge A, Lord C, Shapiro N, Talmor D, O’Mara T, Kirk C, Harrison K, Kurt L, Schermerhorn M, Banner-Goodspeed V, Boyle K, Dubosh N, Filbin M, Hibbert K, Parry B, Lavin-Parsons K, Pulido N, Lilley B, Lodenstein C, Margolin J, Brait K, Jones A, Galbraith J, Peacock R, Nandi U, Wachs T, Matthay M, Liu K, Kangelaris K, Wang R, Calfee C, Yee K, Hendey G, Chang S, Lim G, Qadir N, Tam A, Beutler R, Levitt J, Wilson J, Rogers A, Vojnik R, Roque J, Albertson T, Chenoweth J, Adams J, Pearson S, Juarez M, Almasri E, Fayed M, Hughes A, Hillard S, Huebinger R, Wang H, Vidales E, Patel B, Ginde A, Moss M, Baduashvili A, McKeehan J, Finck L, Higgins C, Howell M, Douglas I, Haukoos J, Hiller T, Lyle C, Cupelo A, Caruso E, Camacho C, Gravitz S, Finigan J, Griesmer C, Park P, Hyzy R, Nelson K, McDonough K, Olbrich N, Williams M, Kapoor R, Nash J, Willig M, Ford H, Gardner-Gray J, Ramesh M, Moses M, Ng Gong M, Aboodi M, Asghar A, Amosu O, Torres M, Kaur S, Chen JT, Hope A, Lopez B, Rosales K, Young You J, Mosier J, Hypes C, Natt B, Borg B, Salvagio Campbell E, Hite RD, Hudock K, Cresie A, Alhasan F, Gomez-Arroyo J, Duggal A, Mehkri O, Hastings A, Sahoo D, Abi Fadel F, Gole S, Shaner V, Wimer A, Meli Y, King A, Terndrup T, Exline M, Pannu S, Robart E, Karow S, Hough C, Robinson B, Johnson N, Henning D, Campo M, Gundel S, Seghal S, Katsandres S, Dean S, Khan A, Krol O, Jouzestani M, Huynh P, Weissman A, Yealy D, Scholl D, Adams P, McVerry B, Huang D, Angus D, Schooler J, Moore S, Files C, Miller C, Gibbs K, LaRose M, Flores L, Koehler L, Morse C, Sanders J, Langford C, Nanney K, MdalaGausi M, Yeboah P, Morris P, Sturgill J, Seif S, Cassity E, Dhar S, de Wit M, Mason J, Goodwin A, Hall G, Grady A, Chamberlain A, Brown S, Bledsoe J, Leither L, Peltan I, Starr N, Fergus M, Aston V, Montgomery Q, Smith R, Merrill M, Brown K, Armbruster B, Harris E, Middleton E, Paine R, Johnson S, Barrios M, Eppensteiner J, Limkakeng A, McGowan L, Porter T, Bouffler A, Leahy JC, deBoisblanc B, Lammi M, Happel K, Lauto P, Self W, Casey J, Semler M, Collins S, Harrell F, Lindsell C, Rice T, Stubblefield W, Gray C, Johnson J, Roth M, Hays M, Torr D, Zakaria A, Schoenfeld D, Thompson T, Hayden D, Ringwood N, Oldmixon C, Ulysse C, Morse R, Muzikansky A, Fitzgerald L, Whitaker S, Lagakos A, Brower R, Reineck L, Aggarwal N, Bienstock K, Freemer M, Maclawiw M, Weinmann G, Morrison L, Gillespie M, Kryscio R, Brodie D, Zareba W, Rompalo A, Boeckh M, Parsons P, Christie J, Hall J, Horton N, Zoloth L, Dickert N, Diercks D. Effect of Hydroxychloroquine on Clinical Status at 14 Days in Hospitalized Patients With COVID-19: A Randomized Clinical Trial. JAMA. 2020 Dec 1;324(21):2165-2176. doi: 10.1001/jama.2020.22240. PMID: 33165621; PMCID: PMC7653542.
- Skipper CP, Pastick KA, Engen NW, Bangdiwala AS, Abassi M, Lofgren SM, Williams DA, Okafor EC, Pullen MF, Nicol MR, Nascene AA, Hullsiek KH, Cheng MP, Luke D, Lother SA, MacKenzie LJ, Drobot G, Kelly LE, Schwartz IS, Zarychanski R, McDonald EG, Lee TC, Rajasingham R, Boulware DR. Hydroxychloroquine in Nonhospitalized Adults With Early COVID-19 : A Randomized Trial. Ann Intern Med. 2020 Oct 20;173(8):623-631. doi: 10.7326/M20-4207. Epub 2020 Jul 16. PMID: 32673060; PMCID: PMC7384270.
- Tang W, Cao Z, Han M, Wang Z, Chen J, Sun W, Wu Y, Xiao W, Liu S, Chen E, Chen W, Wang X, Yang J, Lin J, Zhao Q, Yan Y, Xie Z, Li D, Yang Y, Liu L, Qu J, Ning G, Shi G, Xie Q. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. BMJ. 2020 May 14;369:m1849. doi: 10.1136/bmj.m1849. PMID: 32409561; PMCID: PMC7221473.
- Ulrich RJ, Troxel AB, Carmody E, Eapen J, Bäcker M, DeHovitz JA, Prasad PJ, Li Y, Delgado C, Jrada M, Robbins GA, Henderson B, Hrycko A, Delpachitra D, Raabe V, Austrian JS, Dubrovskaya Y, Mulligan MJ. Treating COVID-19 With Hydroxychloroquine (TEACH): A Multicenter, Double-Blind Randomized Controlled Trial in Hospitalized Patients. Open Forum Infect Dis. 2020 Sep 23;7(10):ofaa446. doi: 10.1093/ofid/ofaa446. PMID: 33134417; PMCID: PMC7543602.
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020 Mar;30(3):269–271.
- WHO Solidarity Trial Consortium, Pan H, Peto R, Henao-Restrepo AM, Preziosi MP, Sathiyamoorthy V, Abdool Karim Q, Alejandria MM, Hernández García C, Kieny MP, Malekzadeh R, Murthy S, Reddy KS, Roses Periago M, Abi Hanna P, Ader F, Al-Bader AM, Alhasawi A, Allum E, Alotaibi A, Alvarez-Moreno CA, Appadoo S, Asiri A, Aukrust P, Barratt-Due A, Bellani S, Branca M, Cappel-Porter HBC, Cerrato N, Chow TS, Como N, Eustace J, García PJ, Godbole S, Gotuzzo E, Griskevicius L, Hamra R, Hassan M, Hassany M, Hutton D, Irmansyah I, Jancoriene L, Kirwan J, Kumar S, Lennon P, Lopardo G, Lydon P, Magrini N, Maguire T, Manevska S, Manuel O, McGinty S, Medina MT, Mesa Rubio ML, Miranda-Montoya MC, Nel J, Nunes EP, Perola M, Portolés A, Rasmin MR, Raza A, Rees H, Reges PPS, Rogers CA, Salami K, Salvadori MI, Sinani N, Sterne JAC, Stevanovikj M, Tacconelli E, Tikkinen KAO, Trelle S, Zaid H, Røttingen JA, Swaminathan S. Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N Engl J Med. 2021 Feb 11;384(6):497-511. doi: 10.1056/NEJMoa2023184. Epub 2020 Dec 2. PMID: 33264556; PMCID: PMC7727327.
|
Hydroxychloroquine - hospitalized |
|||||||
|
Abd-Elsalam, 2020 |
Type of study: A multicenter randomized controlled study
Setting: Tertiary referral centers
Country: Egypt
Source of funding: None.
|
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 194 Intervention: 97 Control: 97
Important characteristics: Age, mean (SD): I: 40.35y (18.65) C: 41.09y (20.07) P=0.80
Sex, n/N (%) male: I: 56/97 male (57.7%) C: 58/97 male (59.8%) P=0.77
|
Hydroxychloroquine
Patients received HCQ 400 mg twice daily (in day 1) followed by 200 mg tablets twice daily added to the standard of care treatment adopted by the Egyptian Ministry of health for 15 days.
|
Standard of care
Patients received only the standard of care treatment adopted by the national Ministry of Health for 15 days. |
Length of follow up 28 days |
Mortality, 28d Death I: 6/97 (6.1%) C: 5/97 (5.1%) P=0.76 OR 0.824 (95% CI 0.243 to 2.797), p=0.757
Hospitalization Duration to hospital discharge; Mean (SD) I: 11.04 (2.71) C: 11.27 (2.19) P=0.52 Need for transfer to ICU I: n=11/97 (11.3%) C: n=13/97 (13.4%) P=0.83
Respiratory support Need for mechanical ventilation I: 4/97 (4.1%) C: 5/97 (5.2%) P = 0.75
Time to symptom resolution Duration to clinical improvement; Mean (SD) I: 9.43 (1.87) C: 9.52 (2.94) P=0.80
Disease severity (after 28 days), n (%) Recovered I: n=52/97 (53.6%) C: n=33/97 (34.0%) Mild I: n=23 (23.7%) C: n=39 (40.2%) Moderate I: n=8 (8.2%) C: n=11 (11.3%) Severe I: n=8 (8.2%) Death I: n=6 (6.1%) C: 5 (5.1%)
Safety Not reported
Viral clearance Time to negative PCR; Mean (SD) I: 17.01 (2.98) C: 17.64 (2.45) P= 0.11
|
Remarks: -definition of ‘clinical improvement’ not described
Authors conclusion: In conclusion, our trial adds extra evidence from Egypt that HCQ may not be beneficial as a treatment for COVID-19.
|
|
Chen, 2020
|
Type of study: RCT; open-label
Setting: 11 public hospitals between April 1 and May 31.
Country: Taiwan (northern, central, and southern).
Source of funding: Research grant from the Hospital and Social Welfare Organizations Administration Commission, Ministry of Health and Welfare. Taiwan Biotech Co. Ltd.: donation of investigational products.
The authors declare no conflicts of interests.
|
Non-hospitalized COVID-19 patients
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 33 Intervention: 21 Control: 12
Important characteristics: Age, mean (SD): I: 33.0 (12.0) C: 32.8 (8.3) Sex, n/N (%) male: I: 11/21 (52.4) C: 8/12 (66.7)
Groups comparable at baseline.
|
Hydroxychloroquine + standard of care (SOC):
HCQ 400 mg twice for 1 d and HCQ 200 mg twice daily for 6 days +SOC Enrollees were randomized within 4 days of diagnosis.
|
SOC
Stratified by mild or moderate illnesses within 4 days of diagnosis.
*mild clinical COVID-19 symptoms: supportive treatment
*moderate clinical COVID19 symptoms: antimicrobial therapy
(1) ceftriaxone 2 g daily for 7 days ± azithromycin 500 mg on day 1 and 250 mg on days 2–5; or (2) levofloxacin 750 mg daily for 5 d; or
(3) levofloxacin 500 mg daily; or (4) moxifloxacin 400 mg daily for 7–14 days for subjects allergic to ceftriaxone or azithromycin or according to physician discretion. Oseltamivir 75 mg b.i.d. will be administered for 5 days to subjects presenting with concomitant influenza A or B infection.
|
Follow-up From randomization up to 14 days.
Loss to follow-up 2 in the HCQ group and 1 in the SOC group had withdrawn consents before the first dose was administered.
|
Mortality, 14d I: 0/21 C: 0/12
Duration of hospitalization Discharge rate by day 14: announced but not reported
Respiratory support Not reported
Time to symptom resolution Clinical recovery (resolution of symptoms, not further specified) by day 14 I: 6/21, 28.6% C: 5/12, 41.7% p-value: 0.51
Safety – adverse events Serious adverse events: No severe adverse events were reported Severe QTc prolongation: No severe QTc prolongation was noted.
Viral clearance Time to negative rRT-PCR assessments from randomization to hospital day 14 (median) I: 5 days (95% CI 1-9 days) C: 10 days (95% CI 2-12 days) p-value: 0.40
Proportion of negative viral rRT-PCR on hospital day 14 (%) I: 17/21 (81.0) C: 9/12 (75.0) p-value: 0.36
Off-quarantined by day 14 I: 19.0% C: 16.7% p-value: not reported
|
Remarks: One (4.8%) in the HCQ group and two (16.7%) in the SOC group were concomitantly administered azithromycin.
Authors conclusion: HCQ failed the primary endpoint of shortening the viral clearance interval.
A retrospective observational study was performed as well by the authors: review of medical registers (n=37). The retrospective study also demonstrated that HCQ conferred no therapeutic benefit to the COVID-19 cases investigated. |
|
Horby, 2020 (RECOVERY trial) |
Type of study: Randomized, open label, controlled trial (RECOVERY trial)
Setting: Multi-centre, 176 Hospitals
Country: United Kingdom
Source of funding: NIHR grant, NIHR Oxford Biomedical Research Centre, Wellcome, the Bill and Melinda Gates Foundation, the Department for International Development, Health Data Research UK, the Medical Research Council Population Health Research Unit, the NIHR Health Protection Unit in Emerging and Zoonotic Infections, and NIHR Clinical Trials Unit Support Funding. Authors received funding from several parties.
|
Inclusion criteria: Clinically suspected or laboratory confirmed COVID-19 infection and no medical history that might, in the opinion of the attending clinician, put the patient at significant risk by participation in the trial. Initially, recruitment was limited to patients ≥ 18 years, but this limit was removed.
Exclusion criteria: Patients with known prolonged electrocardiograph QTc interval. (Co-administration with medications that prolong the QT interval was not a contraindication but it was advised to check QT interval by performing an ECG).
N total at baseline: N = 4716 Intervention: 1561 Control: 3155
Important characteristics: Age, mean (SD): I: 65.2 (15.2) C: 65.4 (15.4) P= not reported Sex, n/N (%) male: I: 960/1561 (61.5) C: 1974/3155 (62.6) P= not reported
Respiratory support — no. (%) No oxygen received I: 362/1561 (23.2) C: 750/3155 (23.8) Oxygen only I: 938/1561 (60.1) C: 1873/3155 (59.4) Invasive mechanical ventilation I: 261/1561 (16.7) C: 532/3155 (16.9)
Groups comparable at baseline.
|
Hydroxychloroquine + usual care
200mg tablet containing 155mg base equivalent. Patients received a loading dose of 4 tablets (800 mg) at zero and 6 hours, followed by 2 tablets (400 mg) starting at 12 hours after the initial dose and then every 12 hours for the next 9 days or until discharge (whichever occurred earlier).
|
Usual care alone |
A online follow-up form was to be completed when participants were discharged, had died or at 28 days after randomization (whichever occurred earlier). Further analyses at 6 months.
Follow-up information was complete for 4619 (98%) of the randomized patients.
|
Mortality, 28 days (n/N/%) I: 421/1561(27.0) C: 790/3155 (25.0) Rate Ratio (95%CI): 1.09 (0.97 to 1.23) P=0.15 Stratified for baseline respiratory support No oxygen received I: 58/362 (16.0) C: 99/750 (13.2) RR 1.24 (0.89−1.73) Oxygen only I: 253/938 (27.0) C: 475/1873 (25.4) RR 1.08 (0.93−1.26) Invasive mechanical ventilation 110/261 (42.1) RR 1.03 (0.81−1.30)
Duration of hospitalization Discharge within 28 days (n/N/%) I: 931/1561 (59.6) C:1983/3155 (62.9) Rate Ratio (95%CI): 0.90 (0.83 to 0.98) P= not reported
Time to symptom resolution Not reported
Respiratory support only reported in composite with death Invasive mechanical ventilation I: 128/1300 (9.8) C: 225/2623 (8.6) RR 1.15 (0.93–1.41)
Safety Cardiac-related events: Any major cardiac arrhythmia, including : Supraventricular tachycardia, Ventricular tachycardia or fibrillation and Atrioventricular block requiring intervention, info on those with complete FU forms: C: 90/1420 (6.3%)
|
Remarks: RECOVERY is a large, pragmatic, randomized, controlled platform trial designed to provide rapid and robust assessment of the impact of readily available potential treatments for COVID-19 on 28-day mortality. This paper published the preliminary results on hydroxychloroquine.
From 12 May 2020, extra information was recorded on the occurrence of new major cardiac arrhythmia.
Since preliminary data showed no beneficial effect of hydroxychloroquine, enrolment of participants was closed on 5 June and the preliminary result for the primary outcome was made public. Investigators were advised that any patients currently taking hydroxychloroquine as part of the study should discontinue the treatment.
Authors conclusion: The findings indicate that hydroxychloroquine is not an effective treatment for hospitalized patients with COVID-19 but do not address its use as prophylaxis or in patients with less severe SARS-CoV-2 infection managed in the community. |
|
Lyngbakken, 2020 |
Type of study: single center, two-arm, open label, group-sequential, pragmatic randomized controlled trial
Setting: Single-center
Country: Norway
Source of funding: The authors declare no competing interests.
|
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 53 Intervention: N = 27 Control: N = 26
Important characteristics: Age, median (IQR): I: 56 (41 to 72) C: 69 (51 to 74) P= not reported
Sex, n/N (%) male: I: 19/27 (70.4%) C: 16/26 (61.5%)
BMI in kg/m2 (range): I: 25.6 (23.9 to 29.4) C: 27.6 (24.2 to 33.0)
Groups comparable at baseline? Yes |
hydroxychloroquine sulfate (at a dose of 400 mg twice daily for 7 days) + standard care.
|
Standard care alone. |
30 days
|
Mortality, 28-30 day Mortality, 30d, n/N (%) I: 1/27 (3.9%). C: 0/26 (0%)
Duration of hospitalization Not reported
Time to symptoms resolution Change in degree of illness as quantified; change in NEWS2 (National Early Warning Score 2)-score from randomization to 96 h (higher score indicates worse condition): I: marginal mean change 0.47 [95% CI −0.58 to 1.53] C: 0.29 [95% CI −0.88 to 1.46] Diff between groups 0.18 [95% CI −1.40 to 1.76]
Respiratory support Clinical status at 7-point ordinal scale* at 14 days I: 1: 1 (3.8%) 2: 0 3: 1 (3.8%) 4: 0 5: 1 (3.8%) 6: 3 (11.5%) 7: 20 (76.9%) C: 1: 1 (4.0%) 2: 1 (4.0%) 3: 0 4: 2 (8.0%) 5: 0 6: 2 (8.0%) 7: 19 (76.0%) Cumulative odds ratio 1.11 [95% CI 0.31 to 4.01]
Safety Adverse events; included visual disturbances, gastrointestinal discomfort, diarrhea, headache, nausea, or dizziness I: 125 C: 112 Serious adverse events, n (%)b I: 5 (18.5%) C: 6 (23.1%) No patient had >1 SAE. SAEs included acute respiratory distress syndrome (n = 1), pneumonia (n = 2), respiratory failure (n = 7), and urinary tract infection (n = 1).
Viral clearance Rate of reduction in SARS-CoV-2 viral load; log10 RNA copies/mL/24 h I: 0.24 (95% CI 0.03 to 0.46) C: 0.14 (95% CI -0.10 to 0.37) MD= 0.11 (95% CI= -0.21 to 0.43). |
Definitions Clinical status at 7-point ordinal scale: 1. dead, 2. hospitalized, on invasive mechanical ventilation or extracorporeal membrane oxygenation, 3. hospitalized, on noninvasive ventilation or high flow oxygen devices, 4. hospitalized, requiring supplemental oxygen, 5. hospitalized, not requiring supplemental oxygen, 6. not hospitalized, but unable to resume normal activities, 7. not hospitalized, with resumption of normal activities
Remarks: Because of rapidly decreasing incidence of COVID-19 in Norway, the trial was prematurely stopped by the trial sponsor on May 25, 2020.
Authors conclusion: In conclusion, therapy with hydroxychloroquine did not impact SARS-CoV-2 viral kinetics in patients admitted to hospital with moderately severe COVID-19. Our results suggest no important antiviral effect of hydroxychloroquine in humans infected with SARS-CoV-2.
|
|
Pan, 2020 (WHO SOLIDARITY trial) |
Type of study: RCT (open-label, non-blinded)
Setting & country: 405 hospitals in 30 countries; WHO Solidarity Trial
Source of funding: Funded by the World Health Organization;
ISRCTN Registry nr, ISRCTN83971151; ClinicalTrials.gov nr, NCT04315948.)
|
N total at baseline: N = 11,330
Hydroxychloroquine arm I: 947 C: 906
Important characteristics: Age, n/N (%): I: <50y: 335/947(35.4%) 50-69y: 410/947(43.3%) ≥70y: 202/947 (21.3%) C: <50y: 317/906 (35.0%) 50-69y: 396/906 (43.7%) ≥70y: 193/906 (21.3%) Sex, n/N (%) male: I: 574/947 (60.6%) C: 535/906 (59.1%) Respiratory support I: No suppl. Oxygen at entry: 345/947(36.4%) Suppl. Oxygen at entry 517/947 (54.6%) Already receiving ventilation 85/947 (9.0%) C: No suppl. Oxygen at entry: 341/906 (37.6%) Suppl. Oxygen at entry 483/906 (53.3%) Already receiving ventilation 82/906 (9.2%) Previous days in hospital I: 0 days: 296/947 (32.2%) 1 day: 317/947 (33.5%) ≥2 days: 334/947 (35.3%) C: 0 days: 281/906 (31.0%) 1 day: 312/906 (34.4%) ≥2 days: 313/906 (34.5%) |
Hydroxychloroquine
Oral; 4 tablets at hour 0, 4 tablets at hour 6, and, starting at hour 12, two tablets twice daily for 10 days. Each tablet contained 200 mg of hydroxychloroquine sulfate (155 mg of hydroxychloroquine base per tablet; a little-used alternative involved 155 mg of chloroquine base per tablet). Discontinued for futility on June 19, 2020
Taking trial drug midway through scheduled duration*: I: 95% C: 6%
Use of non-study drug, n/N (%)s: Corticosteroids I: 140 (14.8%) C: 140 (15.5% Convalescent plasma I: 7 (0.7%) C: 3 (0.3%) Anti-IL-6 drug I: 21 (2.2%) C: 18 (2.0%) Non-trial interferon I: 2 (0.2%) C: 1 (0.1%) Non-trial antiviral I: 62 (6.6%) C: 54 (6.0%) |
Standard of care
|
Length of follow up: 28 days, or up to discharge
Loss to follow-up: I: 7/954 (0.7%) Reasons: no or unknown consent C: 3/909 (0.3%) Reasons: no or unknown consent
|
Mortality, 28-30 day All-cause in-hospital mortality, regardless of whether death occurred before or after day 28: I: 104/947 (10.2%) C: 84/906 (8.9%) RR=1.19 (0.89-1.59), Adjusted** HR=1.14 (0.89-1.46),
All-cause in-hospital mortality, stratified by ventilation at randomization: Ventilated: HR 1.26 (95% CI 0.76-2.10) Not ventilated: HR 1.16 (95% CI 0.82-1.65)
Duration of hospitalization Hospitalized, not discharged: Percentage of patients (rather than number of patients) ever reported as discharged who were still in the hospital: Day 7, % I: 64% C: 54% Day 14 I: 23% C: 20% Day 21 I: 11% C: 10%
Time to symptoms resolution Not reported
Respiratory support Initiation of mechanical ventilation, in those not receiving ventilation at baseline: I: 75/862 (8.7%) C: 66/824 (8.0%) RR 1.09 (95% CI 0.79 to 1.49)
Safety – adverse events Not reported
Viral clearance Not reported
Also reported: Composite death or initiation ventilation: I: 150/947 (15.5%) C: 131/906 (14.3%) RR 1.10 (95% CI 0.88 to 1.36) Publication: RR 1.11 [0.87-1.42] |
Definitions/information:
Taking trial drug midway through scheduled duration, %, calculated only among patients who died or were discharged alive, % patients who were taking the trial drug midway through its scheduled duration (or midway through the time from entry to death or discharge, if this was shorter). *Adjusted model all-cause mortality: some overlap between the 4 control groups; an exploratory sensitivity analysis used multivariate Cox regression to fit all 4 treatment effects simultaneously; adjusted for several prognostic factors (age, sex, diabetes, bilateral lung lesions at entry (no, yes, not imaged at entry), and respiratory support at entry (no oxygen, oxygen but no ventilation, ventilation).
Authors conclusion: These remdesivir, hydroxychloroquine, lopinavir, and interferon regimens had little or no effect on hospitalized patients with Covid-19, as indicated by overall mortality, initiation of ventilation, and duration of hospital stay. |
|
Self, 2020 |
Type of study: Double blind RCT
Setting: Multicenter, April 2, 2020, and June 19, 2020, at 34 hospitals in the US within the Prevention and Early Treatment of Acute Lung Injury (PETAL) Clinical Trials Network
Country: United States of America
Source of funding: National Heart, Lung, and Blood Institute
|
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 479 Intervention: 242 Control: 237
Important characteristics: Age, median (IQR): I: 58 y (45-69) C: 57 y (43-68) Sex, n (%) male: I: 135 (55.8%) C: 132 (55.7%) COVID Outcomes Scale, mean (SD): Defined by patient’s clinical status (1-7), mild to severe. 2: Hospitalized, receiving ECMO or invasive mechanical ventilation I: 13 (5.4) C: 19 (8.0) 3: Hospitalized, receiving noninvasive ventilation or nasal high-flow oxygen I: 28 (11.6) C: 27 (11.4) 4: Hospitalized, receiving supplemental oxygen without positive pressure or high flow I: 116 (47.9) C: 108 (45.6) 5: Hospitalized, not receiving supplemental oxygen I: 85 (35.1) C: 83 (35.0) Groups comparable at baseline? Yes |
400 mg of hydroxychloroquine sulfate in pill form twice a day for the first 2 doses and then 200 mg in pill form twice a day for the subsequent 8 doses, for a total of 10 doses over 5 days
|
Matching placebo in the same dosing frequency |
Length of follow up: 28 days
Loss to follow-up: I: 0 (0%) C: 0 (0%)
|
Mortality, 28-30 day All-cause mortality, 28 days, n (%): I: 25/242 (10.4%) C: 25/237 (10.6%) Absolute difference: - 0.2% (95% CI -5.7 to 5.3%); aOR: 1.07 (95% CI 0.54 to 2.09) 14 days I: 18 (7.5) C: 14 (5.9) Diff 1.5 (−2.9 to 6.0)
Duration of hospitalization Hospital-free days through 28 days, median (IQR) I: 21 (11 to 24) C: 20 (10 to 24) Diff 1 (−1 to 3) aOR 1.17 (0.85 to 1.61)
Time to symptoms resolution Time to recovery, defined as time to reach COVID Outcome Scale category 5, 6, or 7; median (IQR) I: 5 (1 to 14) C: 6 (1 to 15)
Respiratory support Clinical status after randomization Assessed; 7-point WHO scale*, median (IQR) 14 days I: 6 (4 to 7); C: 6 (4 to 7) Diff 0 aOR 1.02 (0.73 to 1.42) 28 days I: 6 (6 to 7); C: 6 (6 to 7) Diff 0 (−1 to 1) aOR 0.97 (0.69 to 1.38)
Clinical status at day 28 Hospitalized with oxygen I: 9 (3.7); C: 10 (4.2) 3: Noninvasive ventilation or high-fow nasal cannula I: 0 C: 0 Invasive mechanical ventilation or ECMO I: 11 (4.5); C: 12 (5.1)
Oxygen-free days, through 28 days, median (IQR) I: 21 (0 to 27) C: 20 (0 to 27) Ventilator-free days, through 28 days, median (IQR) I: 28 (28 to 28) C: 28 (28 to 28)
Safety – adverse events Systematically collected safety events, n (%): Cytopenia I: 92 (38); C: 87 (36.7) AST or ALT ≥2 times upper limit of normal I: 50 (20.7); C:65 (27.4) Cardiac arrest treated with CPR I: 10 (4.1); C: 4 (1.7) Symptomatic hypoglycemia I: 10 (4.1); C: 8 (3.4) Ventricular tachyarrhythmia I: 5 (2.1); C: 6 (2.5) Seizure I: 1 (0.4); C: 0 SAE I: 18 in 14 (5.8%) patients C: 12 in 11 (4.6%) patients Diff 1.1 (−3.0 to 5.2)
Viral clearance Not reported |
Definitions 7-point WHO scale for clinical status: 1, death; 2, hospitalized, receiving extracorporeal membrane oxygenation (ECMO) or invasive mechanical ventilation; 3, hospitalized, receiving noninvasive mechanical ventilation or nasal high-flow oxygen therapy; 4, hospitalized, receiving supplemental oxygen without positive pressure or high flow; 5, hospitalized, not receiving supplemental oxygen; 6, not hospitalized and unable to perform normal activities; and 7, not hospitalized and able to perform normal activities. Adjusted OR: Used covariables: age, sex, baseline COVID Outcome Scale category, baseline Sequential Organ Failure Assessment score, and duration of acute respiratory infection symptoms prior to randomization
Remarks:
Authors conclusion: Among adults hospitalized with respiratory illness from COVID-19, treatment with hydroxychloroquine, compared with placebo, did not significantly improve clinical status at day 14. These findings do not support the use of hydroxychloroquine for treatment of COVID-19 among hospitalized adults.
|
|
Tang, 2020 |
Type of study: multicentre, randomised, parallel, open label trial
Setting: 16 government designated covid-19 treatment centres in three provinces (Hubei, Henan, and Anhui); 11 to 29 February 2020
Country: China
Source of funding: Multiple funding.
|
Inclusion criteria:
Not mandatory: pneumonia on chest CT
Exclusion criteria:
kidney disease or poorly controlled metabolic diseases
continuous renal replacement therapy, haemodialysis, or peritoneal dialysis.
N total at baseline: 150 Intervention: 75 Control: 75
Important characteristics: Age, mean±SD: I: 48.0 ± 14.1 C: 44.1 ± 15.0 Sex, n/N (%) male: I: 42/75 (56%) C: 40/75 (53%) Days from disease onset to randomisation, mean±SD: C: 17.1±11.1
Groups comparable at baseline. Also described: exposure history, treatment before randomisation, vital signs, comorbidities, laboratory parameters |
Standard care + hydroxychloroquine (HCQ)
HCQ: start within 24 hours after randomisation, Day 1-3: loading dose 1200 mg daily for three days From day 4 on: maintenance dose 800 mg daily
Treatment duration: 2 weeks in mild to moderate disease, 3 weeks in severe disease
In case of adverse events related to HCQ as adjusted by investigators, the dose of HCQ was adjusted.
|
Standard care |
Follow-up duration: 28 days
Loss to follow-up: none
|
Mortality, 28-30 day I: 0; C: 0 “… and no patients died during follow-up.”
Duration of hospitalization Not reported
Time to symptoms resolution Alleviation of symptoms*; probability alleviation day 28 days I: 59.9%, (95% CI 45.0 - 75.3) C:66.6% (95% CI 39.5 - 90.9) Diff –6.6% (95% CI –41.3 - 28.0) Time to alleviation, days, Median: I: 19; C: 21 HR 1.01, 0.59 to 1.74 P=0.97
“As the trial was stopped early and only two patients with severe disease were enrolled, results on clinical improvement are not presented.”
Respiratory support Not reported
Safety – adverse events Safety (based on actual exposure) Duration HCQ treatment, days, median (range): I: 14 (1-22) C: n.a.
Adverse events, n/N (%): I: 21/70 (30%) C: 7/80 (9%) Serious adverse events, n/N (%): I: 2 /70 (3%) (1xupper respiratory tract infection, 1x disease progression C: 0
Viral clearance SARSCoV-2 negative conversion at 28 days: I: 85.4% (95% CI 73.8 - 93.8) C: 81.3% (95% CI 71.2 - 89.6) Mean diff: 4.1% (95% CI -10.3% to 18.5%). Median time to negative conversion, days: I: 8 (95% CI 5 – 10) C: 7 (95% CI 75 – 8) HR: 0.85, (95% CI 0.58 to 1.23) P=0.34
|
Definitions: Alleviation of symptoms* was defined as resolving from fever to an axillary temperature of 36.6°C or below, normalisation of SpO2 (>94% on room air), and disappearance of respiratory symptoms including nasal congestion, cough, sore throat, sputum production, and shortness of breath
Remarks:
Authors conclusion: Administration of hydroxychloroquine did not result in a significantly higher probability of negative conversion than standard of care alone in patients admitted to hospital with mainly persistent mild to moderate covid-19. Adverse events were higher in hydroxychloroquine recipients than in non-recipients.
|
|
Ulrich, 2020 (TEACH) |
Type of study: Multicenter, double-blind, randomized clinical trial
Setting: NYU Langone Health (Tisch Hospital and Kimmel Pavilion, NYU Langone—Brooklyn Hospital, and NYU Winthrop Hospital), NYC Health and Hospitals / Bellevue Hospital Center (BHC), and State University of New York (SUNY) Downstate Medical Center.
Country: USA
Source of funding: This work was supported by the New York University Grossman School of Medicine. R.J.U. is supported in part by the NYU CTSA grant (TL1 TR001445) from the National Center for Advancing Translational Sciences (NCATS) and the New York State Empire Clinical Research Investigator Program (ECRIP). M.J.M. and V.R. are supported by the National Institute of Allergy and Infectious Diseases (NIAID) at the National Institutes of Health (NIH) grant (UM1 AI148574). J.A.D. is supported by National Institutes of Health Fogarty grants (D43 TW010046, D43 TW010562, and D43 TW011532). This research was supported in part by an NYU CTSA grant (UL1 TR001445) from the National Center for Advancing Translational Sciences, National Institutes of Health. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest The authors have no relevant financial disclosures. All authors: no reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
|
Hospitalized patients with COVID-19
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 128 Intervention: 67 Control: 61
Important characteristics:
Age, mean (SD): I: 66.5 (16.4) years C: 65.8 (16.0) years P=0.804
Sex, n/N (%) male: I: 45/67 (67.2%) C: 31/61 (50.8%) P=0.089
Disease severity, median (IQR) Defined by COVID-19 severity score
3: Hospitalized, on non-invasive ventilation or high-flow nasal cannula I: 14/67 C: 7/61 Total: 21/128
4: Hospitalized, on supplemental oxygen I: 26/67 (38.8%) C: 36 (59.0%) Total: 62/128)
5: Hospitalized, not on O2, requiring ongoing medical care I: 26/67 C: 17/61 Total: 43/128
6: Hospitalized, not on O2, not requiring ongoing care I: 1/67 (1.5%) C: 1/61 (1.6%) Total: 2/128 (1.6%)
P=0.777 Symptom duration (days since symptom onset) I: 6.50 (6.00) C: 7.00 (10.0) P=0.091
Groups comparable at baseline. |
Hydroxychloroquine sulfate 400 mg by mouth 2 times per day (day 1) and 200 mg by mouth 2 times per day (days 2-5). The 5-day course was based on in vitro projections to optimize HCQ tissue levels against SARS-CoV-2.
|
Placebo (calcium citrate 400 mg by mouth 2 times per day (day 1) and 200 mg by mouth 2 times per day (days 2-5). The 5-day course was based on in vitro projections to optimize HCQ tissue levels against SARS-CoV-2. |
Length of follow up: 30 days.
Loss to follow-up: I: n/N (%) Reasons: C: n/N (%) Reasons: |
Mortality, 28-30 day Mortality (day 14), n/N (%) I: 3/67 (4.5%) C: 5/61 (8.2%) Total: 8/128) P=0.350 Mortality (day 30), n/N (%) I: 7/67 (10.4%) Total: 13/128 (10.2) P=1.000
Duration of hospitalization Length of stay – admission to discharge, mean (SD) I: 9.75 (10.3) C: 6.80 (5.92) Total: 8.34 (8.59) P=0.053
Time to symptoms resolution (fever-free days), mean (SD) I: 6.40 (0.94) C: 6.31 (1.33) Total: 6.36 (1.13) P=0.631
Respiratory support Clinical improvement (COVID-severity score at day 14), n/N (%) 1: death I: 3/67 (4.5%) C: 5/61 (8.2%) Total: 8/128 (6.2%)
2: Ventilator or ECMO I: 2/67 (3.0%) C: 0/61 (0%) Total: 2/128 (1.6%)
3: Hospitalized, on NIV or high-flow nasal I: 7/67 (10.4%) C: 2/61 (3.3%) Total: 9/128 (7.0%)
4: Hospitalized, on supplemental oxygen I: 4/67 (6.0%) C: 1/61 (1.6%) Total: 5/128 (3.9%)
5: Hospitalized, not on O2, ongoing medical care I: 2/67 (3.0%) C: 0/61 (0%) Total: 2/128 (1.6%)
6: Hospitalized, not on O2, not requiring ongoing care I: 1/67 (1.5%) C: 2/61 (3.3%) Total: 3/128 (2.3%) 7: Outpatient, no limitation on activities or home O2 I: 13/67 (19.4%) C: 18/61 (29.5%) Total: 31/128 (24.2%)
8: Outpatient, no limitation on activities I: 28/67 (41.8%) C: 29/61 (47.5%) Total: 57 (44.5%)
Unknown: I: 7/67 (10.4%) C: 4/61 (6.6%) Total: 11/128 (8.6%)
O2-supplementation-free days, mean (SD) I: 4.63 (2.44) C: 4.43 (2.40) Total: 4.53 (2.41) P=640
Mechanical ventilation, n/N (%) day 14 I: 5/67 (7.5%) C: 4/61 (6.6%) Total: 9/128 (7/0%) P=1.000 day 30 I: 5/67 (7.5%) C: 3/61 (4.9%) Total: 8/128(6.2%) P=0.778
Safety – adverse events Cardiac-related events: (Arrhythmia, cardiac arrest) I: 0 C: 0 Total No. of patients with AE, n(%) I: 38/67 (56.7) C: 36/61 (59.0) P=.933 Of which possibly related to study treatment I: 7 (10.4) C: 4 (6.6) P=.639 AEs included GI symptoms, rash, headache Total No. of events I: 63 C: 59
Viral clearance Not reported |
Definitions: -
Remarks: -
Authors conclusion: Therapies against SARS-CoV-2 are urgently needed to improve COVID-19 morbidity and mortality. This double blind, placebo-controlled randomized trial did not suggest that HCQ is beneficial in preventing severe outcomes or improving clinical scores among non-ICU hospitalized patients with COVID19. Treatment with HCQ was associated with a slight QTc interval prolongation, increased D-dimer, and a trend toward increased length of stay. However, our findings are limited due to a relatively small sample size, and larger randomized trials are needed.
|
|
Hydroxychloroquine – non-hospitalized |
|||||||
|
Mitjà, 2020 |
Type of study: open-label, randomized, controlled trial
Setting: Multicenter, 3 health administrative regions in Catalonia
Country: Spain
Source of funding: This work was mainly supported by the crowdfunding campaign JoEmCorono (https://www.yomecorono.com/) with the contribution of over 72,000 citizens and corporations. The study also received financial support from Laboratorios Rubió, Laboratorios Gebro Pharma, Zurich Seguros, SYNLAB Barcelona, and Generalitat de Catalunya. Laboratorios Rubió also contributed to the study with the required doses of hydroxychloroquine (Dolquine®).
|
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 293 Intervention: 136 Control: 157
Important characteristics: Age, mean (SD): I: 41.6 (12.4) C: 41.7 (12.6) P=not reported
Sex, n/N (%) male: I: 98/136 (72.1%) C: 103/157 (65.6%) P=not reported
Groups comparable at baseline? Yes |
Hydroxychloroquine
HCQ - Dolquine®, 800 mg on day 1, followed by 400 mg once daily for six days) |
Usual care
No treatment aside from usual care |
28 days |
Mortality, 28d I: 0 C: 0 “No patients required mechanical ventilation or died during the study period.”
Hospitalization I: 5.9%, 8/136 C: 7.1%, 11/157 RR 0.75 [95% CI 0.32; 1.77]).
Time to symptom resolution, Complete resolution of symptoms* within 28 days, median I: 10.0, IQR 4–18 C: 12.0 days, IQR 6–21) log-rank-test for survival analysis p = 0.38
Respiratory support Mechanical ventilation I: 0 C: 0 “No patients required mechanical ventilation or died during the study period.”
Safety (in safety population) at least one AE during, 28d FU I: 121/169 (72.0%) C: 16/184 (8.7%) The most frequent treatment-related AEs among participants given HCQ were gastrointestinal (e.g., diarrhea, nausea, and abdominal pain) and nervous system disorders (e.g., drowsiness, headache, and metallic taste). SAE I: 8/169 (4.7%) C: 12/184 (6.5%) Of which none related to HCQ.
Viral clearance Viral RNA load in nasopharyngeal swab, reduction from baseline, mean difference: Day 3 I: 1.41 Log10 copies/mL C: -1.41 Log10 copies/mL (difference [d] 0.01 [95% CI –0.28; 0.29]) Day 7 I: –3.44 Log10 copies/mL C: 3.37 Log10 copies/mL (d –0.07 [–0.44; 0.29]). |
Definitions: *Resolution of symptoms was assessed sequentially using a symptoms questionnaire designed to gather information on the type of symptom and last day experienced; complete resolution was considered when no Covid-19-related symptoms were reported at all.
Remarks: - Initially, the protocol included the use of HCQ and cobicistat-boosted darunavir (DRVc) combined treatment, but it was adapted to HCQ alone after the recommendation of the pharmaceutical company not to use DRVc for the treatment of Covid-19 due to lack of activity in-vitro and the negative results in human clinical trials of closely related HIV protease inhibitors.
Authors conclusion: - Compared with usual care, early treatment with HCQ failed to reduce the RNA viral load in nasopharyngeal swabs after 3 and 7 days of treatment and shorten the time to complete resolution of symptoms in adults with PCR-confirmed mild Covid-19. - Our results indicate no impact on viral burden up to 7 days nor symptoms resolution or hospitalization rate up to 28 days following diagnosis. The added value of our study is the randomized-controlled design and the use of the agreed minimal outcome set for Covid-19 clinical trials, including RT-PCR to conclusively determine the viral burden. Our findings provide the scientific community and policymakers with essential insights on the inefficacy of HCQ as a therapeutic candidate for SARS-CoV-2, at least in similar settings and conditions to ours. |
|
Omrani, 2020 (Q-PROTECT) |
Type of study: RCT (parallel)
Setting: two units of Qatar’s national healthcare system, Hamad Medical Corporation (HMC). The first was the Emergency Department (ED) at HMC’s tertiary hospital, Doha’s Hamad General Hospital (HGH). The second unit was a 3500-bed quarantine facility 20 miles north of Doha, at Umm Qarn
Country: Qatar
Source of funding: The study was supported by internal institutional funds of the Hamad Medical Corporation (government health service of the State of Qatar).
Conflict of interest The authors have no financial or personal relationships with other people or organizations that could represent a conflict of interest.
|
SARS-CoV-2 PCR-positive males and females with mild or no symptoms.
Inclusion criteria:
Comorbidity (e.g. diabetes or hypertension)
Exclusion criteria:
N total at baseline: N = 456 Intervention-1: 152 Intervention-2: 152 Control: 152
Important characteristics: Age, median (IQR): I-1: 42 years (38-48) I-2: 40 years (31-47) C: 41 years (31-47) Sex, n/N (%) male: I-1: 150/152 (98.7%) I-2: 149/152 (98.0%) C: 150/152 (98.7%)
Disease severity, median (IQR): Symptoms at enrolment, Patient-reported fever I-1: 46 (30.3%) I-2: 51 (33.6%) C: 52 (34.2%) Respiratory symptoms I-1: 37 (24.3%) I-2: 36 (23.7%)
Groups comparable at baseline? Yes |
Hydroxychloroquine
Intervention-1: Oral hydroxychloroquine plus oral azithromycin (500 mg day one, 250 mg daily on days two through five)
Intervention-2: Oral hydroxychloroquine (600 mg daily for one week)
|
Placebo |
Length of follow up: 21 days
Loss to follow-up:
Reasons for loss to follow-up
Unavailability for contact. |
Mortality, 28d Not reported. Mortality other: Not reported
Duration of hospitalization Not reported.
Time to symptom resolution Symptomatic day one and: Asymptomatic day 7 I-1: 56 of 70 (80.0%, 68.7-88.6%) I-2: 55 of 69 (79.7%, 68.3-88.4%) C: 52 of 59 (88.1%, 77.1-95.1%) P=0.377 Asymptomatic day 14 I-1: 66 of 69 (95.7%, 87.8-99.1%) I-2: 64 of 69 (92.8%, 83.9-97.6%) C: 58 of 60 (96.7%, 88.5-99.6%) P=0.716 Asymptomatic day 21 I-1: 67 of 69 (97.1%, 89.9-99.6%) I-2: 68 of 69 (98.6%, 92.2-100.0%) C: 56 of 60 (93.3%, 83.8-98.2%)P=0.299
Respiratory support Not reported
Safety QT prolongation >30 msec I-1: 37 (24.3%; 17.8-32.0%) I-2: 31 (20.4%; 14.3-27.7%) C: 13 (8.8%; 4.8-14.6%) p= 0.001 >60 msec I-1: 5 (3.3%; 1.1-7.5%) I-2: 4 (2.6%; 0.76.6%) C: 2 (1.4%; 0.2-4.8%) p=0.641
Viral clearance Virologic cure, defined as (PCR-negative status), n/N (%) (95% CI) day six I-1: 16/152 (10.5%) (95% CI= 6.1 to 16.5%) I-2: 19/149 (12.8%) (95% CI= 7.9 to 19.2%) C: 18/147 (12.2%) (95% CI= 7.4 to 18.7%) P=0.821 day 14 I-1: 30/149 (20.1%) (95% CI= 14.0 to 27.5%) I-2 42/146 (28.8%) (95% CI= 21.6 to 36.8%) C: 45/143 (31.5%) (95% CI= 24.0 to 39.8%) P=0.072
Increase in cycle threshold from day one to day six, median (IQR) (95% CI) I-1: 7.2 (3.9 to 11.5) (95% CI= 6.1 to 8.8) I-2: 7.5 (3.4 to 11.5) (95% CI= 5.7 to 8.8) C: 8.0 (4.1 to 11.7) (95% CI= 7.3 to 9.0) P=0.634
|
Definitions: -
Remarks:
Authors conclusion: The lessons of Q-PROTECT must be considered in light of the trial strengths and weaknesses, the medication risks and benefits, and the existing evidence base. Taking all of these factors into account, the investigators conclude that HC±AZ shows no sign of usefulness in the population studied, and that there is low likelihood of undiscovered drug benefits outweighing therapeutic risks.
|
|
Skipper, 2020 |
Type of study: Randomized, double-blind, placebo-controlled trial
Setting: Internet-based trial across the United States and Canada (40 states and 3 provinces).
Country: USA and Canada
Source of funding: By Steve Kirsch, Jan and David Baszucki, the Minnesota Chinese Chamber of Commerce, the Alliance of Minnesota Chinese Organizations, and the University of Minnesota Foundation. Authors were supported by several funds. Apotex Canada and Rising Pharmaceuticals in the United States provided a donation of some of the hydroxychloroquine tablets used. |
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 423 Intervention: 212 Control: 211
Important characteristics: Age, median (IQR): I: 41 (33–49) C: 39 (31–50) P=not reported
Sex, n/N (%) male: I: 89/212 (42%) C: 96/211 (45%) P=not reported
Groups comparable at baseline? Yes |
Hydroxychloroquine
800 mg (4 tablets) once, then 600 mg (3 tablets) 6 to 8 hours later, then 600 mg (3 tablets) once daily for 4 more days (5 days in total).
Medication adherence I: 77% (157 of 203)
|
Placebo
Placebo tablets of folic acid, 400 mcg, were prescribed as an identical regimen for the control group. In Canada, the placebo tablets were lactose.
Medication adherence C: 86% (166 of 194)
|
Length of follow up 14 days
|
Mortality, 28d Not reported Mortality, other Death, 14d I: 1 (non-hospitalized) C: 1 (hospitalized)
Hospitalization Hospitalisation, 14d I: 4/212 C: 10/211 (of which 2 not related to study)
Total events hospitalization or death, 14 d I: 5 C: 10 P = 0.29
Respiratory support Not reported
Time to symptom resolution Change in symptom severity score over 14 days; on a 10-point VAS I: -2.6 points (SE 0.12) C: -2.33 points (SE 0.12) (absolute difference, -0.27 points [95% CI, -0.61 to 0.07 points]; P = 0.117)
Safety Adverse events I: 43% [92 of 212] C: 22% [46 of 211]; P < 0.001. Serious adverse events I: 0 C: 0
Viral clearance Not reported |
Remarks: -participants with no data were excluded from analyses (5.3%) -also probable COVID-19 patients were included. Only 58% of participants received SARS-CoV-2 testing because of severe U.S. testing shortages. -primary outcome was changes after interim analyses
Authors conclusion: - Hydroxychloroquine did not substantially reduce symptom severity or prevalence over time in nonhospitalized persons with early COVID-19. This trial may not inform whether an effect would be observed in populations at higher risk for severe COVID-19. Further randomized controlled clinical trials are needed in early COVID-19. - Hydroxychloroquine did not substantially reduce symptom severity in outpatients with early, mild COVID-19.
|
Risk of bias table for intervention studies (randomized controlled trials).
|
Hydroxychloroquine |
||||||||
|
|
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2 (unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3 (unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4 (unlikely/likely/unclear) |
Bias due to loss to follow-up?5 (unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6 (unlikely/likely/unclear) |
|
Hospitalized |
||||||||
|
Abd-Elsalam, 2020
|
Patients were randomized into two groups using a computerized random number generator using simple randomization with an equal allocation ratio. |
Unclear
Not described. |
Likely
Study reported ‘soft’ outcome measures.
Blinding not necessary because of ‘hard’ outcome measure (mortality). |
Likely
Study reported ‘soft’ outcome measures.
Blinding not necessary because of ‘hard’ outcome measure (mortality) |
Likely
Study reported ‘soft’ outcome measures.
Blinding not necessary because of ‘hard’ outcome measure (mortality) |
Unlikely
All outcome measures in the method section are reported in the results. |
Unlikely
No lost to follow-up. |
Unlikely
All included patients in the analysis are exactly the patients who were randomized into the trial. |
|
Chen, 2020
|
“Randomization was performed through a computer-generated list stratified by site” |
Unlikely |
Unclear
Blinding of participants was not described
|
Unclear
Blinding of care providers was not described |
Unclear
Blinding of assessors was not described |
Unclear
|
Unlikely
No loss to follow-up |
Unlikely
ITT analysis performed. |
|
Horby, 2020 (RECOVERY trial )
|
Eligible and consenting patients were assigned in a ratio of 2:1 to treatment group using web-based simple (unstratified) randomization with allocation concealment |
|
Likely
Patients were not blinded to the allocated treatment. However, primary outcome (mortality) is a hard outcome measure. It might have had an impact on other outcomes. |
Likely
Caregivers were not blinded to the allocated treatment. However, primary outcome (mortality) is a hard outcome measure. It might have had an impact on other outcomes. |
Likely
Local study staff was not blinded to the allocated treatment. However, primary outcome (mortality) is a hard outcome measure. It might have had an impact on other outcomes. |
Unlikely
Preliminary results are presented; not all data is reported yet.
|
Unlikely
Follow-up information was complete for 98% of the patients. |
Unlikely
All analyses were done according to the intention-to-treat principle. |
|
Lyngbakken, 2020 |
Computer randomization procedures
|
Unclear
Not described. |
Unlikely
Mortality and viral load not susceptible to patient knowledge on allocation. |
Unlikely
study personnel performing the RT-PCR and statistical analyses were blinded concerning group allocation |
Unlikely
study personnel performing the RT-PCR and statistical analyses were blinded concerning group allocation |
Unlikely
All predefined outcome measures were reported |
Unlikely
Only one patient was lost to follow-up in the control group. |
Unlikely
Intension-to-treat analysis were performed.
|
|
Pan, 2020
|
Computerized
Once a hospital has obtained approval, electronic entry of patients who have given informed consent takes only a few minutes. At the end of it, the randomly allocated treatment is displayed on the screen and confirmed by electronic messaging. |
Unlikely
Allocation displayed on screen after entry of patient in the system |
Likely
Unblinded trial (mortality: unlikely)
|
Likely
Unblinded trial (mortality: unlikely)
|
Likely
Unblinded trial (mortality: unlikely)
|
Unlikely
Protocol published
https://www.who.int/publications/m/item/an-international-randomised-trial-of-additional-treatments-for-covid-19-in-hospitalised-patients-who-are-all-receiving-the-local-standard-of-care |
Unlikely
Low rate of lost-to follow-up and reasons comparable between groups |
Unlikely
analyses performed according to intention-to-treat protocol |
|
Self, 2020
|
Using a centralized electronic system, we randomly assigned enrolled patients to hydroxychloroquine or placebo in a 1:1 ratio stratified by enrolling hospital using randomization block sizes of 2 and 4. |
Unclear
Allocation was concealed but was not further explained. |
Unlikely
Patients, treating clinicians, trial personnel, and outcome assessors were blinded to group assignment. |
Unlikely
Patients, treating clinicians, trial personnel, and outcome assessors were blinded to group assignment. |
Unlikely
Patients, treating clinicians, trial personnel, and outcome assessors were blinded to group assignment. |
Unlikely
All outcome measures were reported |
Unlikely
No loss to follow-up |
Unlikely
None of the participants switched therapies |
|
Tang, 2020
|
Computerized, envelopes opened at enrolment
According to disease severity, random 1:1 ratio.
“ LL designed the randomisation rules together with the principal investigators, and an independent statistician who was not involved in data analysis implemented them. Equal numbers of cards with each group assignment number randomly generated by computer were placed in sequentially numbered envelopes that were opened as the patients were enrolled.” |
Unclear
Unclear whether procedure is adequate as not all steps were computerized (envelopes) |
Unlikely
“Patients, investigators, and statisticians were not masked to treatment assignment.” |
Likely
“The dose of hydroxychloroquine was adjusted when adverse events were related to hydroxychloroquine, as judged by investigators.” / “Patients, investigators, and statisticians were not masked to treatment assignment.” |
Unlikely
“Laboratory technicians who did virological, chemical, and other routine measurements were unaware of treatment information.” |
Unlikely
Authors adequately explain why changes in enrolment (also severe disease patients) and outcomes (viral clearance at day 28 instead of day 10) took place. |
Unlikely
No loss to follow-up |
Unlikely
Intention-to-treat analysis used for effectivity analysis. Safety analyses, were based on the patients’ actual exposure. |
|
Ulrich, 2020 (TEACH) |
Enrolled subjects were randomized 1:1 to study drug or placebo and followed for 30 days.
|
Unclear
Randomization was stratified by age (>60 years old) and study site
|
Unlikely
Subjects and investigators were blinded to the treatment assignment, but in cases of rapid COVID-19 progression meeting our primary end point, or at the request of the treating physician, we allowed for subject unblinding. |
Unlikely
Subjects and investigators were blinded to the treatment assignment, but in cases of rapid COVID-19 progression meeting our primary end point, or at the request of the treating physician, we allowed for subject unblinding.
|
Unlikely
Subjects and investigators were blinded to the treatment assignment, but in cases of rapid COVID-19 progression meeting our primary end point, or at the request of the treating physician, we allowed for subject unblinding.
|
Unlikely
All predefined outcomes were reported |
Likely
N = 21 lost to follow-up in the intervention group versus N = 15 lost to follow-up in the control group. |
Unlikely
Primary analyses used the intention to-treat (ITT) paradigm in which participants are classified according to their randomized treatment assignment, regardless of treatment receipt or compliance. Secondary analyses assessed the safety population (those who received any dose of study medication) and the per-protocol population (those who received at least 80% of their assigned dose). |
|
Non-hospitalized |
||||||||
|
Mitjà, 2020
|
Computer-generated random-number list
Participants were randomized (1:1) using a computer-generated random-number list to either the control arm (no treatment aside from usual care) or the intervention arm (HCQ - Dolquine®, 800 mg on day 1, followed by 400 mg once daily for six days). |
Unlikely
Note: Random allocation was done remotely by a member of the study team not involved in participants’ enrollment |
Likely
Note: Masking was not possible because a placebo could not be prepared due to the emergency nature of the trial. |
Likely
Note: not clear whether care providers were blinded, and whether this have lead to potential bias. |
Unlikely
Note: Laboratory technicians were unaware of participants’ treatment allocation, treatment response, and previous PCR results at all time points |
Unlikely
outcomes well defined |
Unlikely
Note: loss to follow-up was small, and similar in both groups |
Unlikely
Note: All allocated patients were included in the intention-to-treat analysis. |
|
Omrani, 2020 (Q-PROTECT)
|
Randomization of the study’s projected n of 456 was executed by computer in a location (Imperial College London) remote from the study site. |
Unlikely
Central allocation concealment was used. The randomization scheme was transmitted to the study institution’s central pharmacy, where it was translated to identical-appearing sequentially numbered drug-bottle sets. Other routes of allocation concealment addressed three facets of appearances of bottles and study medications. |
Unlikely
Study participants were unaware of the specific contents of their medication bottles.
|
Unlikely
Study staff (physicians and nurses who enrolled participants, executed virologic sampling, and assessed and recorded participant’s clinical follow-up data) were unaware of study medication identity and did not see the contents of the study bottles.
|
Unlikely
Study staff (physicians and nurses who enrolled participants, executed virologic sampling, and assessed and recorded participant’s clinical follow-up data) were unaware of study medication identity and did not see the contents of the study bottles.
|
Unlikely
All primary and secondary outcomes are reported
|
Likely
|
Unlikely
Study planning dictated intent-to-treat (ITT) analysis. Per-protocol analysis was also executed, but only for exploratory assessments.
|
|
Skipper, 2020
|
Unclear method
Sequential randomization occurred at research pharmacies in Minneapolis, Minnesota, and Montreal, Canada.
|
Unlikely
The trial statistician generated a permuted block randomization, the research pharmacies held this list, and statisticians verified that the randomization sequence was followed. |
Unlikely
Participants were blinded. |
Unlikely
Care providers were blinded. |
Unclear
not reported whether outcome assessors were blinded |
Likely
primary outcome was changed after interim analyses, but reasons were not described |
Unlikelyonly patients with complete data were analysed. Drop-out rated were similar in both groups. |
Likelyit was reported that intention-to-treat analysis were performed, but only participants with data were included. |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Evidence tables of studies published between 7 January 2021 and 2 September 2021
|
Barratt-Due, 2021 |
Type of study: Independent, add-on RCT to WHO Solidarity trial, an open-label, adaptive RCT
Setting: 23 hospitals, between 28 March and 5 October 2020
Country: Norway
Source of funding: National Clinical Therapy Research, no role in RCT
Conflicts of interest: JTA reports grant from South-Eastern Norway Regional Health Authority. ABD reports paid lecture by Allergan Norden AB and participation in advisory board meeting SANOFI-AVENTIS Norwy. SGD reports grant from Research Council of Norway. FLJ reports funding from Oslo University Hospital and Helse SørØst. MT reports participation in European advisory board for Eli Lilly and coordinator of National reference group on COVID-19 treatment in Norway.
|
Hospitalized patients with confirmed SARS-CoV-2 infection
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 181 Remdesivir: n=42 Hydroxychloroquine: n=52 Control remdesivir: n=57 Control HSQ: n=54 (some controls were in both groups)
Important characteristics: Age, mean (SD): 59.8 (15.3) R: 59.7 y (16.5) CR: 58.1 y (15.7) H: 60.3 y (13.3) CH: 59.2 y (16.4)
Sex, n/N (%) male: R: 29/42 (69%) CR: 43/57 (75%) H: 31/52 (60%) CH: 34/54 (63%)
Disease severity, mean (SD): Defined by Viral load (log10 count/1000 cells) R: 1.6 (1.6) CR: 2.3 (1.8) H: 2.3 (1.5) CH: 2.0 (1.5)
Anti-SARS-CoV-2 antibodies, seroconverted (RDB ≥5) n/N (%) R: 14/42 (42.4%) CR: 18/57 (46.2%) H: 15/52 (42.9%) CH: 20/54 (54.1%)
Groups are not comparable on baseline regarding sex, comorbid conditions, ICU admission, P-F ratio less than 40 kPa, ACE and ARB medication, LDH level, D-dimer level, AST level, ALT, level. |
R: standard of care plus 200 mg intravenous remdesivir on day 1, 100 mg daily up to day 9
H: standard of care plus 800 mg oral hydroxychloroquine 2x day on day 1, 400 mg 2x day up to day 9
All study treatment were discontinued at discharge. |
Standard of care according to WHO guidelines recommending systemic steroids |
Length of follow-up: 3 months
Loss-to-follow-up: R: 9 (21%)
CR: 9 (16%)
H: 13 (24%)
CH: 8 (15%)
Incomplete outcome data: Missing data due to discharge or participant withdrawal were imputed with best outcome. Not reported how many.
|
Clinical outcomes Mortality (28 day) R: 2.4 (0.1; 10.1) CR: 5.2 (1.3-13.1) RD: -2.9 (-10.3; 4.5) H: 7.5 (2.4; 16.7) CH: 1.8 (0.1; 7.6) RD: 5.8 (-2.2; 13.7) Mortality (60 day) R: 7.1 (1.8; 17.5) CR: 5.3 (1.3; 13.1) RD: 1.9 (-7.8; 11.6) H: 7.5 (2.4; 16.7) CH: 1.8 (0.1; 7.6) RD: 5.8 (-2.2; 13.7) In-hospital mortality R: 7.1 (1.8; 17.5) CR: 7.0 (2.2; 15.6) RR: 1.0 (0.2; 4.6) H: 7.5 (2.4; 16.7) CH: 3.6 (0.6; 10.6) RR: 2.2 (0.4; 10.8)
Duration of hospitalization Not reported Admission to ICU during hospitalization R: 19.0 (9.2; 32.6) CR: 19.3 (10.5; 30.8) RD: -0.3 (-15.9; 15.4) H: 22.6 (12.8; 35.0) CH: 16.1 (8.1; 27.1) RD: 6.6 (-8.2; 21.4)
Time to symptom resolution Not reported
Respiratory support Mechanical ventilation R: 9.5 (3.1; 20.8) CR: 7.0 (2.2; 15.6) RD: 2.5 (-8.6; 13.6) H: 15.1 (7.2; 26.3) CH: 10.7 (4.4; 20.5) RD: 4.4 (-8.2; 17.0) Time to receipt mechanical ventilation RR R vs CR: 1.4 (0.4; 5.8) RR H vs CH: 2.1 (0.7; 6.2) Duration of mechanical ventilation Reported in appendix figure 1
Safety Adverse events n/N (%) R: 34/42 (81%) H: 26/52 (50%) C: 33/87 (n=38%) Serious adverse events n/N (%) R: 13/42 (31%) H: 12/52 (23%) C: 20/87 (n=23%)
Virological outcomes Viral clearance Not reported Viral load Reported in a figure 2 and appendix figure 2 Subgroup analysis based on symptom duration before hospitalization, the presence of ARS-CoV-2 antibodies, high or low viral load at hospital admission, degree of inflammation, and age were performed. |
Definitions: not applicable
Remarks:
Authors conclusion: The overall lack of effect of remdesivir and HCQ on the clinical course of patients hospitalized for COVID-19 was accompanied by a paucity of effect on SARS-CoV-2 viral clearance in the oropharynx. Our findings question the antiviral potential of these drugs in hospitalized patients with COVID-19.
|
|
Arabi, 2021 |
Type of study: Randomized, embedded, multifactorial adaptive platform trial for community-acquired pneumonia (REMAP-CAP).
Setting: 187 sites across 11 countries.
Country: United States of America, Canada, France, Germany, Ireland, Netherlands, Portugal, United Kingdom, Saudi Arabia, Australia, New Zealand.
Source of funding: Supported by the European Union; See the full-text publication of the study for the full description.
Conflicts of interest: See the full-text publication of the study for the full description.
NCT02735707
|
Severe COVID-19 ICU patients
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 694; analysed: N = 677 Intervention 1: N = 255; N = 249 Intervention 2: N = 50; N = 49 Intervention 3: N = 27 ; N = 26 Control: N = 362; N = 353
Important characteristics: Age, mean (SD): Intervention 1: 61 y (13) Intervention 2: 56.3 y (13) Intervention 3: 60.3 y (8.9) Control: 60.8 y (12.9)
Sex, n/N (%) male: Intervention 1: 182/254 (71.7%) Intervention 2: 35/50 (70%) Intervention 3: 19/27 (70.4%) Control: 252/362 (69.6%)
Disease severity, mean (SD): Defined by APACHE II score, median (IQR) Intervention 1: 13.0 (8 to 18) Intervention 2: 12.5 (7.8 to 20.2) Intervention 3: 14 (10.2 to 20.8) Control: 13 (8 to 19)
Groups comparable at baseline? Yes.
|
Lopinavir-ritonavir, hydroxychloroquine, combination therapy of lopinavir-ritonavir and hydroxychloroquine
Lopinavir-ritonavir was administered for 5 days minimum, up to a maximum of 14 days or until ICU discharge whichever occurred frst. Lopinavir–ritonavir was administered at a dose of 400 mg of lopinavir and 100 mg of ritonavir every 12 h. For patients with a gastric tube who were unable to swallow tablets, lopinavir-ritonavir (at the same dose) was administered as a 5-ml suspension every 12 h or alternatively as two dissolved tablets or four crushed tablets (double dose), noting that systemic absorption is reduced by approximately 50% for crushed tablets
Hydroxychloroquine
Hydroxychloroquine was administered as two loading doses of 800 mg, 6-h apart, followed 6 h later by 400 mg 12 hourly for 12 doses. Tis dose regimen was supported by pharmacokinetic modelling and by guidance regarding safety from clinicians with experience with the use of hydroxychloroquine for the treatment of severe malaria. If a patient was unable to swallow, crushed hydroxychloroquine tablets were administered via an enteral tube.
Combination therapy
Combination of Lopinavir-ritonavir and hydroxychloroquine. |
No antiviral agents against COVID-19 |
Length of follow-up: 90 days
Loss-to-follow-up: Intervention 1: N = 6 (%) Reasons: not reported.
Intervention 2: N = 1 (%) Reasons: not reported.
Intervention 3: N = 1 (%) Reasons: not reported.
Control: N = 9 (%) Reasons: not reported.
Incomplete outcome data: None.
|
Clinical outcomes In-hospital deaths, n/N (%) Intervention 1: 88/249 (35.3%) Intervention 2: 17/49 (34.7%) Intervention 3: 13/26 (50.0%) Control: 106/353 (30.0%)
90-day survival (time-to-event analysis), adjusted HR (95% CI) Intervention 1: aHR 0.83 (95% CI 0.65 to 1.07) Intervention 2: aHR 0.71 (95% CI 0.45 to 0.97) Intervention 3: aHR 0.58 (95% CI 0.36 to 0.92) Control: 1
Duration of hospitalization Time to hospital discharge, adjusted HR, median (95% CI) Intervention 1: aHR 0.83 (95% CI 0.68 to 0.99) Intervention 2: aHR 0.76 (95% CI 0.56 to 0.97) Intervention 3: aHR 0.63 (95% CI 0.42 to 0.89) Control: 1
Time to ICU discharge, adjusted HR, median (95% CI) Intervention 1: aHR 0.87 (95% CI 0.72 to 1.07) Intervention 2: aHR 0.74 (95% CI 0.52 to 0.94) Intervention 3: aHR 0.63 (95% CI 0.44 to 0.89) Control: 1
Time to symptom resolution WHO scale at day 14, adjusted HR, median (95% CI) Intervention 1: aHR 0.85 (95% CI 0.68 to 0.99) Intervention 2: aHR 0.76 (95% CI 0.56 to 0.97) Intervention 3: aHR 0.63 (95% CI 0.42 to 0.89) Control: 1
Respiratory support Progression to invasive mechanical ventilation, ECMO, or death, restricted to those not intubated at baseline, n/N (%); adjusted OR, median (IQR) Intervention 1: 89/176 (50.6%); aOR 0.75 Intervention 2: 17/24 (70.8%); aOR 0.58 (95% CI 0.24 to 1) Intervention 3: 11/14 (78.6%); aOR 0.42 (95% CI 0.16 to 0.95)
Cardiovascular support-free days, median (IQR); adjusted OR (95% CI) Intervention 1: 14 (-1 to 21); aOR 0.66 (95% CI 0.49 to 0.89) Intervention 2: 13 (-1 to 19); aOR 0.60 (95% CI 0.39 to 0.86) Intervention 3: -1 (-1 to 14); aOR 0.39 (95% CI 0.22 to 0.69) Control: 18 (-1 to 21); aOR 1.00.
Respiratory support-free days, median (IQR); adjusted OR (95% CI) Intervention 1: 3 (-1 to 15); aOR 0.75 (95% CI 0.56 to 0.99) Intervention 2: 0 (-1 to 9); aOR 0.64 (95% CI 0.4 to 0.92) Intervention 3: -1 (-1 to 7); aOR 0.47 (95% CI 0.27 to 0.83) Control: 5 (-1 to 16); aOR 1.00.
Safety Serious adverse events Patients with ≥1 serious adverse events, n/N (%); adjusted OR (95% CI) Intervention 1: 13/255 (5.1%); aOR 0.55 (95% CI 0.24 to 1.22) Intervention 2: 3/50 (6.0%); aOR 0.65 (95% CI 0 to 2.38) Intervention 3: 1/27 (3.7%); aOR 0.97 (95% CI 0.24 to 4.79) Control: 12/362 (3.3%)l aOR 1.00.
Serious ventricular arrhythmia or sudden unexpected death, n/N (%); adjusted OR (95% CI) Intervention 1: 6/239 (2.5%); aOR 1.30 (95% CI 0.56 to 3.28) Intervention 2: 2.49 (4.1%); aOR 0.88 (95% CI 0.27 to 3.55) Intervention 3: 2/26 (7.7%); aOR 0.62 (95% CI 0.18 to 2.60) Control: 10/345 (2.9%); aOR 1.00.
Virological outcomes Viral clearance Not reported. |
Definitions: - Organ support included the provision of invasive mechanical ventilation, non-invasive mechanical ventilation, high-flow nasal cannulae with a flow rate of at least 30 L per minute and a fractional inspired oxygen concentration of 0.4 or higher, or the infusion of vasopressor or inotropes for shock.
Remarks: -
Authors conclusion: In conclusion, among critically ill patients with COVID-19, treatment with lopinavir-ritonavir, hydroxychloroquine, or combination therapy resulted in worse outcomes compared to no COVID-19 antiviral therapy
|
|
Schwartz, 2021 |
Type of study: randomized, double-blind, placebo-controlled trial
Setting: community-based in Alberta, with enrolment beginning April 15, 2020 and paused on May 22, 2020
Country: Canada
Source of funding: Calgary Health Trust, the University of Calgary, Alberta Innovates Health Solutions, Alberta Health Services and the Alberta Government provided funding. Hydroxychloroquine and matching placebo were provided by Apotex. Funders had no role in trial design, interpretation or publication decisions.
Conflicts of interest: Conflicts of interest were transparently and extensively reported. Most importantly, the last author (MH) reports that Apotex Pharma provided the drug and placebo for the current trial as an in-kind contribution to the study. He was the main contact with Apotex Pharma and has no other relationship with the company; there were no obligations attached to this donation of drug and placebo. |
Community-dwelling individuals with confirmed COVID-19
Inclusion criteria:
Exclusion criteria:
N total at baseline: Randomized: N = 148 Intervention: N = 111 Control: N = 37
Important characteristics: Age, mean (SD): I: .46.7 y (11.5) C: .46.9 y (11.0)
Sex, n/N (%) male: I: 65/111 (58.6%) C: 17/37 (45.9%)
Participants in the intervention group less often had diabetes or asthma. Also, they less often had shortness of breath, malaise, myalgias, coryza, dysgeusia and diarrhea, but more often had nausea; unclear whether these differences are statistically significant. |
hydroxychloroquine 800 mg orally in divided doses on day 1 followed by 200 mg twice daily for 4 days (12 tablets over 5 days) |
placebo |
Length of follow-up: 30 days
Loss-to-follow-up: Based on the information presented in the flow chart (Fig. 2), no participants were lost to follow-up in the ITT population. However, the table of results (Tab. 2) shows that for most of the outcomes data was missing for ≥ 1 participants; the PP population consisted of 74/111 patients in the intervention group, and 31/37 patients in the control group.
Incomplete outcome data: Mortality within 30 days I: 0/111 (0%) C: 0/37 (0%)
Admission to ICU within 30 days I: 1/111 (0.9%) C: 0/37 (0%)
Hospitalization within 30 days I: 1/111 (0.9%) C: 0/37 (0%)
Time to symptom resolution Days, median I: 22/111 (19.8%) C: 2/37 (5.4%)
Development of severe disease, defined as the composite of hospitalization, invasive mechanical ventilation or death within 30 days I: 1/111 (0.9%) C: 0/37 (0%)
The safety population consisted of 91/111 patients in the intervention group and 33/37 patients in the placebo group who took at least 1 tablet of the study drug. |
Primary outcome was the development of severe disease, defined as the composite of hospitalization, invasive mechanical ventilation, or death within 30 days.
Clinical outcomes Mortality Mortality within 30 days I: 0/111 (0%) C: 0/37 (0%)
Duration of hospitalization Admission to ICU within 30 days I: 1/110 (0.9%) C: 0/37 (0%)
Hospitalization within 30 days I: 4/110 (3.6%) C: 0/37 (0%)
Time to symptom resolution Time to COVID-19 recovery Days, median I: 14 (95%CI: 10-20); data available for 89/111 participants C: 12 (95%CI: 7-18); data available for 35/37 participants p = 0.3
Respiratory support Not reported.
Other Development of severe disease, defined as the composite of hospitalization, invasive mechanical ventilation or death within 30 days I: 4/110 (3.6%) C: 0/37 (0%) p = 0.6
Disposition at day 30 Recovered vs. Ongoing symptoms, not hospitalized vs. Unknown, not hospitalized or deceased I: 67/110 (60.9%) vs. 23/110 (20.9%) vs. 20/11020/ (18.2%); missing for 1 patient C: 29/37 (78.4%) vs. 5/37 (16.2%) vs. 2/37 (5.4%)
Safety Serious adverse events within 30 days I: 3/91 (3.3%) C: 0/33 (0%) p = 0.6
Emesis within 30 days I: 5/91 (5.5%) C: 0/33 (0%) p = 0.3
Virological outcomes Viral clearance Not reported. |
Definitions: -
Remarks: The trial recruited only 10% of the target sample size, stopping early because of a report on hydroxychloroquine safety (that was subsequently retracted) and a rapid decline in disease prevalence coinciding with control of the first wave of the COVID-19 pandemic.
Authors’ conclusion: There was no evidence that hydroxychloroquine reduced symptom duration or prevented severe outcomes among outpatients with proven COVID-19, but the early termination of our study meant that it was underpowered. |
|
Ader, 2021 |
Type of study: RCT, phase 3 open-label (DisCoVeRY)
Setting: 30 sites in France and 2 in Luxembourg, between March 22nd and June 29th.
Country: France and Luxembourg
Source of funding: The study was founded by Programme Hospitalier de Recherche Clinique (PHRC-20-0351) (Ministry of Health), from the DIM One Health Île-de-France (R20117HD), and from REACTing, a French multi-disciplinary collaborative network working on emerging infectious diseases. The funding sources had no role in the analysis of the data nor in the decision of publication.
Conflicts of interest: F.R. reports personal fees from Gilead Sciences, personal fees from MSD, personal fees from Pfizer, personal fees from TheraTechnologies, personal fees from ViiV Healthcare, outside the submitted work. F.G. reports grants from BioMerieux, personal fees and non-financial support from Gilead, non-financial support from Corevio, outside the submitted work. G.P.reports grants and personal fees from Gilead Sciences, grants and personal fees from Merck, grants and personal fees from ViiV Healthcare, grants and personal fees from TheraTechnologies, outside the submitted work. K.L. reports personal fees and non-financial support from Gilead, personal fees and non-financial support from Janssen, personal fees and non-financial support from MSD, personal fees and non-financial support from ViiV Healthcare, personal fees and non-financial support from Abbvie, during the conduct of the study. Y.Y. has nothing to disclose. He has been a board member receiving consultancy fees from ABBVIE, BMS, Gilead, MSD, J&J, Pfizer, and ViiV Healthcare, however all these activities have been stopped in the 03 past years. F.L. reports personal fees from Gilead personal fees and non-financial support from MSD, non-financial support from Astellas, non financial support from Eulmedica, outside the submitted work. A.K. reports personal fees from Baxter, personal fees from Aspen, personal fees from Aguettant, outside the submitted work. S.N. reports personal fees from MSD, personal fees from Pfizer, personal fees from Gilead, personal fees from Biomérieux, personal fees from BioRad, outside the submitted work. F.D. reports personal fees from Gilead, outside the submitted work. J.N. reports non448 financial support from MSD France, non-financial support from GILEAD Sciences, personal fees from PASCALEO, outside the submitted work. J.M. reports non-financial support from GILEAD, outside the submitted work. A.M. reports personal fees from MSD, personal fees from GILEAD, personal fees from JANSSEN, personal fees from Viiv Healthcare, outside the submitted work. M.H. reports grants from Fonds Erasme- COVID-Université Libre de Bruxelles, grants from Belgian health Care Knowledge Center, during the conduct of the study; personal fees from Gilead advisory board on education on invasive fungal infections, personal fees from Pfizer: moderator for session on Isavuconazole, outside the submitted work. D.C. reports personal fees from Gilead, grants and personal fees from Janssen, outside the submitted work. C.B. reports personal fees from Da Volterra, personal fees from Mylan Pharmaceuticals, outside the submitted work. F.M. reports grants from Sanofi, grants and personal fees from Da Volterra, outside the submitted work. All other authors have nothing to disclose. |
Hospitalized patients, peripheral oxygen saturation ≤94% (moderate) or requiring supplemental oxygen (severe)
Inclusion criteria: adults (≥ 18-year-old) a PCR-proven (< 72 hours) SARS-CoV-2 infection pulmonary rales or crackles with a peripheral oxygen saturation ≤94% or requiring supplemental oxygen
Exclusion criteria: Enrolment in another investigative trial Use of other antivirals
N total at baseline: N = 583 Intervention: -L/r: 145 -L/r + IFN: 145 -HCQ: 145 Control: 148
Important characteristics: Age, median [IQR]: L/r: 63 [55-71] L/r + IFN: 64 [53-71] HCQ: 65 [55-71] C: 62 [52-71] Sex, n/N (%) male: L/r: 106/145 (73.1%) L/r + IFN: 103/145 (71.0%) HCQ: 104/145 (71.7%) C: 105/148 (70.9%) Baseline severity of COVID-19, n/N (%): Moderate disease (receiving low-flow supplemental oxygen or not requiring oxygen) L/r: 94/145 (64.8%) L/r + IFN: 91/145 (62.8%) HCQ: 93/145 (64.1%) C: 94/148 (63.5%)
Severe disease (requiring non-invasive ventilation or high-flow oxygen devices, invasive mechanical ventilation or ECMO) L/r: 51/145 (35.2%) L/r + IFN: 54/145 (37.2%) HCQ: 52/145 (35.9%) C: 54/148 (36.5%)
Groups comparable at baseline? Not reported. |
-lopinavir/ritonavir (L/r) (400 mg lopinavir and 100 mg ritonavir 234 orally twice on day for 14 days) + SoC
- lopinavir/ritonavir plus IFN-ß-1a (L/r + IFN) (44 µg of subcutaneous IFN-ß-1a on days 1, 3, and 6 +SoC
-hydroxychloroquine (HCQ) (400 mg orally, 236 twice on day 1 as a loading dose followed by 400 mg once daily for 9 days + SoC
Supportive treatments corticosteroids, anticoagulants or immunomodulatory agents were allowed |
Standard of care (Soc) Supportive treatments corticosteroids, anticoagulants or immunomodulatory agents were allowed. |
Length of follow up: 29±3 days
Loss to follow-up: L/r: 1/145 (0.7%) Reason: did not receive at least one dose of intervention. L/r + IFN: 1/145 (0.7%) Reason: did not receive at least one dose of intervention. HCQ: 2/145 (1.4%) Reason: did not receive at least one dose of intervention. C: 0/148 (0%)
|
All analyses were stratified by severity at randomization, and adjusted effect measures are reported.
Clinical outcomes Mortality (within 28 days), n (%) Moderate subgroup L/r: 4 (4.3%) L/r + IFN: 4 (4.4%) HCQ: 6 (6.5%) C: 5 (5.3%)
Severe subgroup L/r: 10 (19.6%) L/r + IFN: 13 (24.1%) HCQ: 5 (9.6%) C: 7 (13.0%)
L/r vs control: OR=1.24 (0.55 to 2.82) [P=0.60] L/r + IFN vs control: OR=1.51 (0.69 to 3.34) [P=0.30] HCQ vs control: OR=0.93 (0.40 to 2.20) [P=0.88]
Duration of hospitalization Time to hospital discharge within 29 days L/r vs control: HR=0.77 (0.58 to 1.02) [P=0.07] L/r + IFN vs control: HR=0.72 (0.54 to 0.96) [P=0.026] HCQ vs control: HR=0.83 (0.62 to 1.10) [P=0.20]
Time to symptom resolution Clinical status at 7-point ordinal scale* at day 15 L/r vs control: OR=0.83 (0.55 to 1.26) [P=0.39] L/r + IFN vs control: OR=0.69 (0.45 to 1.04) [P=0.08] HCQ vs control: OR=0.93 (0.62 to 1.41) [P=0.75]
Clinical status at 7-point ordinal scale* at day 29 L/r vs control: OR=0.93 (0.62 to 1.41) [P=0.74] L/r + IFN vs control: OR=0.76 (0.50 to 1.15) [P=0.19] HCQ vs control: OR=1.16 (0.77 to 1.75) [P=0.49]
Respiratory support Ventilator-free days until day 29 L/r vs control: LSMD=-0.98 (-2.96 to 1.00) [P=0.33] L/r + IFN vs control:LSMD=-2.01 (-4.03 to 0.00) [P=0.05] HCQ vs control: LSMD=0.09 (-1.93 to 2.10) [P=0.93]
Safety Adverse events, n/N (%) Any adverse events L/r: 119/144 (82.6%); p=0.02 L/r + IFN: 117/144 (81.3%); p=0.04 HCQ: 109/143 (76.2%); p=0.35 C: 105/148 (70.9%)
Any serious adverse events L/r: 76/144 (52.8%); p=0.02 L/r + IFN: 78/144 (54.2%); p=0.01 HCQ: 63/143 (44.1%); p=0.34 C: 57/148 (38.5%)
Virological outcomes Viral clearance The slope of the decrease of the viral loads in Nasopharyngeal swab (NPS)over time was not significantly affected by any of the investigational treatments. No significant difference in the proportion of participants with detectable viral loads at each sampling time was observed in the NPS nor in the lower respiratory tract (LRT) specimens.
Also available: Secondary efficacy outcome measures were the clinical status at day 29 and the time to an improvement of 2 categories as measured on the 7-point ordinal scale or hospital discharge until day 29, the time to National Early Warning Score 2 (NEWS2) ≤2 or hospital discharge until day 29, the time to hospital discharge until day 29, oxygenation- and ventilator-free days until day 29, 29-day mortality, and the SARS-CoV-2 detection and quantitative normalized viral loads. Trough plasma concentrations of lopinavir, ritonavir and hydroxychloroquine were measured at days 1 and 3. Secondary safety outcomes included the cumulative incidence of any grade 3 or 4 AE, or of any serious adverse event (SAE, according to the DAIDS Table 264 for Grading the Severity of Adult and Paediatric Adverse Events, v2.1, July 2017) and the proportion of patients with a premature suspension or discontinuation for any reason of the investigational treatments. |
Definitions: *7-point ordinal scale of the WHO Master Protocol (v3.0, March 3, 2020): 1. Not hospitalized, no limitation on activities; 2. Not hospitalized, limitation on activities; 3. Hospitalized, not requiring supplemental oxygen; 4. Hospitalized, requiring supplemental oxygen; 5. Hospitalized, on non-invasive ventilation or high flow oxygen devices; 6. Hospitalized, on invasive mechanical ventilation or ECMO; 7. Death.
Remarks: Based on interim analyses (see Supplementary Appendix), enrolment in the hydroxychloroquine arm was prematurely stopped on June 17th , and enrolment in lopinavir containing arms was stopped on June 29th 2020 (futility and safety concerns). The trial was performed in the early phase of the COVID-19 pandemics and the SoC underwent substantial changes over time
Authors conclusion: In patients admitted to hospital with COVID-19, lopinavir/ritonavir, lopinavir/ritonavir plus IFN-β-1a and hydroxychloroquine were not associated with clinical improvement at day 15 and day 29, nor reduction in viral shedding, and generated significantly more SAEs in lopinavir/ritonavir-containing arms. These findings do not support the use of these investigational treatments for patients hospitalized with COVID-19.
|
|
Réa‑Neto, 2021 |
Type of study: RCT (open-label)
Setting: 6 hospitals in Curitiba, Brazil April 16 to August 06, 2020
Country: Brazil
Source of funding: Not reported.
Conflicts of interest: No competing interest.
clinicaltrials.gov nr, NCT04420247, “Retrospectively Registered”
|
Patients admitted to ICU
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 142 Intervention: 71 Control: 71 The modified intention-to-treat analysis: N=105 I: 53 C: 52
Important characteristics: Age, mean (SD): I: 54.7 y (12.1) C: 52.8 y (12.6) Sex, n/N (%) male: I: 36/53 (67.9%) C: 34/52 (65.4%)
Disease severity: according to ventilatory support Intensive mechanical ventilation at baseline, no. (%): I: 9/53 (17) C: 10/52 (19.2) Score on nine-point ordinal scale*, no. (%)d 3: I: 1 (1.9), C: 0 (0) 4: I: 43 (81.1), C:42 (80.8) 5: I: 0 (0), C: 0 (0) 6: I: 1 (1.9), C: 3 (5.8) 7: I: 8 (15.1), C: 7 (13.5)
Groups were comparable at baseline, with the exception of the number of patients with hypertension (I: 19 vs. C: 21), DM (I: 11 vs. C: 16) and Immunocompromised state (I: 2 vs. C: 4). No p values reported. |
Chloroquine or hydroxychloroquine for 5 days plus standard treatment
Chloroquine: 450 mg twice a day on day 1 and 450 mg once daily from days 2-5
Hydroxychloroquine: 400 mg twice a day on day 1 and 400 mg once daily from days 2-5 |
standard treatment only |
Length of follow up: 28 days
Loss to follow-up: I: 0/53 (0%) C: 1/52 (1.9%)
|
The modified intention-to-treat analysis was restricted to 105 patients. Results also reported for Clq and HClq separately (see supplement of article)
Clinical outcomes Mortality (28 day) I: 16 (30%) C: 10 (19%) (Difference (95% CI):1.57 (0.79 to 3.13), p= 0.236) RR 1.57 [95% CI 0.79 to 3.13], p=0.196
Duration of hospitalization ICU LOS (Length of stay) among survivors, median (IQR), days: I: 3.5 (1–12) C: 3 (0–7) (Difference (95% CI):1 (− 2.6 to 4.6), p= 0.368)
Hospital LOS among survivors, median (IQR), days: I: 7.5 (5–16) C: 7 (4–12) (Difference (95% CI): 1 (− 2.9 to 4.9), p= 0.257)
Time to symptom resolution Not reported
Respiratory support Mechanical ventilation -free days, median (IQR), days: I: 25 (3–28) C:28 (4–28) (Difference (95% CI): − 3 (− 11.7 to 5.7), p= 0.236)
Invasive mechanical ventilation incidence, no (%): I: 18 (41) C: 8 (19) (Difference (95% CI): 2.15 (1.05 to 4.40), p= 0.03)
WHO 9-point ordinal score*, n(%): day 14 Score 0 : I: 11 (20.8) C: 22 (42.3) Score 1: I: 5 (9.4) C: 6 (11,5) Score 2: I: 10 (18.9) C: 6 (11.5) Score 3: I: 0 (0) C: 0 (0) Score 4: I: 6 (11.3) C: 2 (3.8) Score 5: I: 2 (3.8) C: 3 (5.8) Score 6: I: 3 (5.7) C: 3 (5.8) Score 7: I: 6 (11.3) C: 3 (5.8) Score 8: I: 10 (18.9) C: 7 (13.5) OR (95% CI): 2.41 (1.17 to 4.93; odds for worse clinical condition) P=0.016 Day 28 OR 2.47 (1.15-5.30), p= 0.020
Safety Arrhythmias, no (%) I: 4/68 (5.9) C: 1/70 (1.9) (Difference (95% CI): 3.92 (0.45 to 33.9), p= 0.176) One patient in the Clq/HClq group stopped treatment on the third day owing to severe arrhythmia
Virological outcomes Not reported.
Also available: Coagulopathy incidence, Acute renal dysfunction incidence, clinical status on days 5, 7, 10 and 28.
|
Primary outcome: Clinical status measured on day 14 after randomization with a WHO 9-point ordinal scale.
Secondary outcomes: all-cause mortality; invasive MV use; the incidence of acute renal dysfunction in 28 days; clinical status of patients on days 5, 7, 10 and 28
Definitions: *WHO Score on nine-point ordinal scale: (0) non-hospitalized and no clinical or virological evidence of infection; (1) non-hospitalized and no limitation on activities; (2) non-hospitalized, but with limitation on activities; (3) hospitalized, but not requiring supplemental oxygen; (4) hospitalized and on oxygen via mask or nasal prongs; (5) hospitalized, on non IVM or high-flow oxygen or pressure support ventilation in weaning mode; (6) hospitalized, intubated and on MV; (7) hospitalized on MV and additional organ support (renal replacement therapy, vasoactive drugs or extracorporeal membrane oxygenation), and (8) dead
Remarks: Open-label trial; The trial was stopped before reaching the planned sample size due to harmful effects; Retrospectively Registered protocol.
Authors conclusion: In conclusion, the addition of Clq/HClq to standard care in patients admitted to the hospital with severe COVID-19 resulted in clinical worsening and higher incidences of IMV and renal dysfunction, even though there was no difference in mortality. According to these findings, the use of Clq/HClq in patients with more severe forms of COVID-19 pneumonia is strongly contraindicated, and these results can inform clinical practice and guidelines.
|
|
Reis, 2021 |
Type of study: RCT TOGETHER Trial
Setting: local public health authorities in Brazil, participants enrolled between June 2 and September 30, 2020.
Country: Brazil
Source of funding: The trial was supported by the Bill and Melinda Gates Foundation. The funder had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Conflicts of interest: None reported by the authors.
|
High-risk adult outpatients (see inclusion criteria)
Inclusion criteria:
aged 50 years or older; presence of pulmonary disease, specifically moderate or severe persistent asthma, chronic obstructive pulmonary disease, pulmonary hypertension, or emphysema; diabetes requiring oral medication or insulin; hypertension requiring treatment; known cardiovascular diseases (congestive heart failure of any etiology, documented coronary artery disease, clinically manifest miscellaneous heart disease); symptomatic lung disease on chronic treatment; history of transplantation; obesity (body mass index 30 [calculated as weight in kilograms divided by height in meters squared]); immunocompromised status due to disease (eg, those living with HIV with a CD4 T-cell count of <200 cells/mm3 , confirmed malignant neoplasm); immunocompromised status due to medication (eg, people taking 10 mg or more of prednisone equivalents a day); and patients with cancer
Exclusion criteria:
N total at baseline: N = 685 Intervention hydro: 214 Intervention lopi-rito: 244 Control: 227
Important characteristics: Age, median (IQR): I hydro: 53 y (18-84) I lopi-rito: 54 y (18-94) C: 53 y (18-80) Sex, n/N (%) male: I hydro: 92/214 (43.0 %) I lopi-rito: 110/244 (45.1%) C: 106/227 (46.7 %) Participants with multiple risk factors, n/N (%) I hydro: 121/214 (56.5%) I lopi-rito: 134/244 (54.9%) C: 134/227 (59.0%)
Groups comparable at baseline? With respect to covariates of age, body mass index, and comorbidities, the groups were generally well balanced. |
hydroxychloroquine group loading dose of 800 mg at the time of randomization and then 400 mg in daily doses at 8:00 AM for 9 days.
lopinavir-ritonavir group a loading dose of 800 mg of lopinavir and 200 mg of ritonavir at the first 2 intakes, followed by 400 mg of lopinavir and 100 mg of ritonavir every 12 hours for the next 9 days.
|
placebo group corresponding tablets of inert material (talc). Placebo bottles were matched for the same number of tablets as active hydroxychloroquine (placebo of hydroxychloroquine) and active lopinavir-ritonavir (placebo of lopinavir-ritonavir). |
Length of follow up: 90 days
Loss to follow-up: I hydro: 16/214 (7.5%) Reasons: -7 Did not receive assigned Treatment -9 Had adhered <80% to assigned treatment
I lopi-rito: 44/244 (18.0%) Reasons: -12 Did not receive assigned Treatment -32 Had adhered <80% to assigned treatment
C: 19/227 (8.4%) Reasons: -7 Did not receive assigned Treatment -12 Had adhered <80% to assigned treatment
At the end of the trial, 79 participants (11.5%) did not complete all phases of the study. The lopinavir-ritonavir intervention group had 44 participants (18%) who did not complete the study, which was more than either of the other 2 groups.
|
Clinical outcomes Mortality At the end of the trial, we recorded 3 fatalities, 1 in the placebo group and 2 in the lopinavir-ritonavir intervention group.
Duration of hospitalization COVID-19 hospitalization, n/N (%) I hydro: 8/214 (3.7) HR 0.76 (95% CI 0.30-1.88) I lopi-rito: 14/244 (5.7) HR 1.16 (955 CI 0.53-2.56) C: 11/227 (4.8) HR 1 [reference]
Time to hospitalization, median (IQR), d I hydro: 4.8 (1.40-6.12) I lopi-rito: 3.6 (2.50-4.76) C: 2.4 (0.76-3.20)
All-cause hospitalization, n/N (%) I hydro: 11/214 (5.1) HR 0.96 (95% CI 0.42-2.17) I lopi-rito: 16/244 (6.6) HR 1.22 (95% CI 0.58-2.57) C: 12/227 (5.3) HR 1 [reference]
Time to hospitalization, median (IQR), d I hydro: 3.8 (1.88-6.43) I lopi-rito: 3.1 (2.31-4.75) C: 2.2 (0.79-3.12)
Time to symptom resolution WURSS Scale (Wisconsin Upper Respiratory Symptom Survey) We found no difference in the resolution of combined symptoms using the WURSS scale between either hydroxychloroquine and placebo or lopinavir-ritonavir and placebo, or for individual symptoms.
Respiratory support Not reported.
Safety Adverse events Any Treatment-Emergent Adverse Events (TEAE), n/N (%) I hydro: 46/207 (22.2) I lopi-rito: 92/232 (39.7) C: 46/220 (20.9)
Serious TEAE, n/N (%) I hydro: 11 (5.3) I lopi-rito: 20 (8.6) C: 12 (5.5)
Virological outcomes Virological clearance Defined as 1 negative swab since Baseline.
In the mixed-effect logistic model, the clearance for the hydroxychloroquine (odds ratio [OR], 0.91; 95% CI, 0.82-1.02) and lopinavir-ritonavir (OR, 1.04; 95% CI, 0.94-1.16) groups did not differ in comparison with the control group.
Neither hydroxychloroquine nor lopinavir-ritonavir showed difference in viral clearance across all prespecified subgroups based on ITT analysis.
|
Remarks: The independent DSMB, based on interim analysis results, made the decision to stop enrollment to the hydroxychloroquine and lopinavir-ritonavir groups because of a low number of emerging events. This study reports on the final results inclusive of patients who had been randomized to hydroxychloroquine or lopinavir-ritonavir between the time of data-cut for interim analysis to the time of the DSMB meeting.
Authors conclusion: This randomized clinical trial found no clinical benefit to support the use of either hydroxychloroquine or lopinavir-ritonavir in an outpatient population. This adds to the growing evidence that these drugs should not be used for the treatment of COVID-19. While evidence emerges to evaluate these drugs as prophylaxis, as treatment for both outpatients and inpatients, hydroxychloroquine and lopinavir-ritonavir do not appear to confer any clinical benefit.
|
|
Gupta, 2021a |
Type of study: Open-label randomized controlled trial with unblinded assessment.
Setting: Tertiary care centre of Indian Armed Forces Medical Services located in a metropolitan city.
Country: India.
Source of funding: No information.
Conflicts of interest: All authors have none to declare.
|
Patients with moderate to severe COVID-19 infections who require hospitalized care.
Inclusion criteria:
Exclusion criteria:
N total at baseline: N = 110 Intervention: N=55 Control: N=55
Important characteristics: Age, mean (SD): I: 57.8 y (12.6) C: 57.3 y (14.1) P=0.72
Sex, n/N (%) male: I: 43/55 (76.8%) C: 37/55 (68.5%) P=0.20
Disease severity, mean (SD): Defined by SOFA score at admission (SD) I: 2.6 (1.7) C: 2.4 (1.6) P=0.56
Groups comparable at baseline? Yes. |
Hydroxychloroquine + standard of care
Patients in the HCQ arm received the drug as per the following schedule: 400 mg twice on day 1, followed by 400 mg once daily from day 2 today 5.
|
Standard of care
Standard of care included intravenous (IV) antibiotics to cover respiratory pathogens, IV dexamethasone at a dose of 4 mg every 8 h for 5 days and subcutaneous low-molecular-weight heparin(LMWH) enoxaparin in dose of 40 mg (0.4 ml) once a day for 5days.
|
Length of follow-up: Four days.
Loss-to-follow-up: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
Incomplete outcome data: Intervention: N (%) Reasons (describe)
Control: N (%) Reasons (describe)
|
Clinical outcomes Mortality (28-30 day), n/N (%) I: 10/55 (18.2%) C: 2/55 (3.6%) P=0.01
Duration of hospitalization Days of hospitalization, mean (SD) I: 13.89 (5.85) C: 13.67 (5.83) P=0.98
Time to symptom resolution Days to normalization of SaO2, mean (SD) I: 6.54 (4.48) C: 7.59 (5.06) P=0.26
Respiratory support Days on oxygen, mean (SD) I: 8.49 (6.38) C: 7.98 (5.45) P=0.26
Number of needing ventilator, n/N (%) I: 10/55 (18.2%) C: 4/55 (7.27%) P=0.09
Days from admission to ventilator, mean (SD) I: 4.90 (4.88) C: 1.5 (2.38) P=0.18
Days on ventilator, mean (SD) I: 8.33 (8.60) C: 8.75 (3.09) P=0.37
Safety Adverse events Not reported.
Virological outcomes Viral clearance Not reported. |
Definitions: SOC: Standard of care included intravenous (IV) antibiotics to cover respiratory pathogens, IV dexamethasone at a dose of 4 mg every 8 h for 5 days and subcutaneous low-molecular-weight heparin(LMWH) enoxaparin in dose of 40 mg (0.4 ml) once a day for 5days.
Remarks: -
Authors conclusion: In conclusion, in the rapidly changing world of COVID-19therapeutics, our open-label, parallel group, unblinded ran-domized control trial suggests that HCQ does not change outcomes in moderate to severe COVID-19 infection. It sup-ports some of the other observational studies and trials con-ducted in the last few months. We could not comment on thetoxicity of HCQ as trial was not designed to assess it. Thestrength of our trial is that it was randomized and therandomization was good as seen by the well matched baselinecharacteristics; we recruited more patients than our calcu-lated sample size and this helped us perform the adjustedanalysis without losing the strength of the study and ouroutcomes were both clinical and laboratory based. The chieflimitation of our trial is that there was no blinding atrandomization or at assessment. Our trial is relevant becauseit is one of the first few from India. It will help clinicians in notprescribing a drug which does not change outcomes in mod-erate to severe COVID-19 infection and may be potentiallytoxic. It will help policy makers in closing the chapter on arepurposed drug which had gained a lot of popularity and spotlight at the beginning of the pandemic.
|
|
Dubée, 2021
HYCOVID trial |
Type of study: RCT; double-blind, placebo-controlled
Setting: 48 hospitals in France and the Principality of Monaco; April 2 to May 21, 2020 Start: April 1, 2020 Suspended: May 26, 2020 Stopped: June 9, 2020
Country: France, Monaco
Source of funding: This work was supported by a grant from the French Ministry of Health through a national call for proposals for therapeutic trials on COVID-19. The trial also received an exceptional donation from the Pays de la Loire region and from the Angers Loire Métropole conurbation . None of the funding organizations played any role in the trial design, data collection, analysis of results, or writing of the manuscript.
Conflicts of interest: The authors have declared that there are no conflicts of interest in relation to this work.
|
Mild-to-moderate COVID-19 patients; almost all hospitalized (99%), with (60%) or without supplemental oxygen (severely ill patients requiring oxygen therapy needed > 3L / min or ICU admission excluded)
Inclusion criteria:
SARS-CoV-2 RT-PCR on a nasopharyngeal swab within 2 days or chest-CT
Exclusion criteria:
N total at baseline: Randomized: N = 250 Intervention: 125 Control: 125 In analysis: I: 123 C: 124
Important characteristics: Median age (IQR) – yr I: 76 (60-85) C: 78 (57-87) Male sex – no. (%) I: 65 (52.0) C: 56 (44.8) Median time (IQR) from onset of symptoms to inclusion – days I: 5 (3-9) C: 5 (3-8) Score on ordinal scale – no. (%) 1. Ambulatory, no limitation of activity I: 1 (0.8) C: 1 (0.8) 2. Ambulatory, limitation of activity I: 1 (0.8) C: 0 (0.0) 3. Hospitalized, no oxygen therapy I: 46 (36.8) C: 50 (40.0) 4. Hospitalized, oxygen therapy (≤3 L/min) I: 77 (61.6) C: 74 (59.2)
Groups comparable at baseline. |
Hydroxychloroquine + standard care
Hydroxychloroquine (200 mg tablets, orally) at a dose of two tablets twice daily on the first day followed by one tablet twice daily for 8 days (total hydroxychloroquine dose of 4 g) plus standard care |
Placebo + standard care
Matching placebo at a dose of two tablets twice daily on the first day followed by one tablet twice daily for 8 days plus standard care |
Length of follow up: 28 days days
Loss to follow-up: I: 2/125 (1.6%) Reason: withdrew consent C: 1/25 (0.8%) Reason: withdrew consent
Incomplete data: Clinical status data are missing at Day 14 and 28 for two patients in each group; not further described |
Clinical outcomes Mortality - day 28 I: 6 (4.8) C: 11 (8.9) RR 0.54 (0.21–1.42)
Mortality other: day 14 I: 6 (4.8) C: 6 (4.9) RR 0.99 (0.33–2.99)
Duration of hospitalization ‘not reported’
Time to symptom resolution Clinical evolution; WHO 9-point Ordinal Scale for Clinical Improvement for COVID-19*; RR (95% CI); definitions: absence of deterioration: stability or decrease of at least one point on the ordinal scale; clinical improvement: decrease of at least one point on the ordinal scale; recovery: score of 0, 1, or 2 Day 14 Absence of deterioration I: 112 (90.3) C: 111 (90.2) RR 1.01 (0.93–1.09) Clinical improvement I: 84 (67.7) C: 81 (65.9) RR 1.01 (0.86–1.18) Recovery I: 71 (57.3) C: 68 (55.3) RR 1.03 (0.83–1.27) Day 28 Absence of deterioration I: 115 (92.7) C: 112 (91.1) RR 1.02 (0.95–1.10) Clinical improvement I: 98 (79.0) C: 93 (75.6) RR 1.03 (0.90–1.16) Recovery I: 91 (73.4) C: 84 (68.3) RR 1.06 (0.91–1.24)
Respiratory support Use of intubation and mechanical ventilation Day 14 I: 3 (2.4) C: 3 (2.4) RR 0.99 (0.20–4.82) Day 28 I: 3 (2.4) C: 4 (3.3) RR 0.74 (0.17–3.26)
Safety Adverse events I: 124 C: 120 Any adverse event - no. of patients (%) I: 70 (56.5%) C: 61 (50.8) Adverse event leading to discontinuation of treatment - no. of patients (%) I: 4 (3.2) C: 2 (1.7) Of which: cardiac rhythm or conduction disorders: I: 4; C: 1 Skin rash: I: 0; C: 1 Serious adverse event - no. of patients (%) I: 3 (2.4) C: 4 (3.3) Of which: cardiac rhythm or conduction disorders: I: 3; C: 3 Rash: I: 0; C: 1
Virological outcomes Viral clearance Positive SARS-CoV-2 RT-PCR Day 5 I: 75/103 (72.8) C: 73/100 (73.0) RR 1.00 (0.84–1.18) Day 10 I: 52/91 (57.1) C: 47/83 (56.6) RR 1.01 (0.78–1.31) |
Definitions: WHO Ordinal Scale for Clinical Improvement: 0: patient uninfected, no clinical or virological signs of infection; 1: patient at home, without limitation of activities; 2: patient at home, with limitation of activities; 3: patient hospitalized without oxygen therapy; 4: patient with oxygen therapy by mask or nasal prongs; 5: patient under non-invasive ventilation or high-flow oxygen; 6: patient under invasive mechanical ventilation; 7; patient under invasive mechanical ventilation and additional organ support, including vasopressors, renal replacement therapy, and extracorporeal membrane oxygenation; 8: death.
Remarks: -trial prematurely stopped
Authors conclusion: In this underpowered trial involving mainly older patients with mild-to-moderate COVID-19, patients treated with hydroxychloroquine did not experience better clinical or virological outcomes than those receiving the placebo.
|
|
Brown, 2020 |
Type of study: Setting: Country: Source of funding:
|
Hospitalized COVID-19 patients with symptomatic laboratory-confirmed COVID-19, within 1-0 days of a positive test for COVID-19. Inclusion criteria: Exclusion criteria: Important characteristics: Sex, n/N (%) female: [Disease severity], median (IQR): Hospitalized, no oxygen (%) Mechanical ventilation & other organ support
|
Hydroxychloroquine sulfate was administered orally as a loading dose of 400mg twice on the first day followed by 200mg twice daily for the following 4 days (total dose 2.4gm) or until discharge or death
|
Azithromycin was administered orally as a loading dose of 500mg on the first day, followed by 250mg daily for the next 4 days (total dose 1.5gm) or until discharge or death.
|
Length of follow up: Loss to follow-up: |
Clinical outcomes *An OR >1 favors azithromycin over hydroxychloroquine for this comparison. 28-day mortality Day 7 COVID scale score Day 14 COVID scale score Day 28 COVID scale score Ventilator-free days at 28 days Hospital-free days at 28 days Median (IQR) ICU-free days at 28 daysI: 18 (8 to 22) Ventilator-free days at 28 days Days to a 1-point decrease in WHO COVID scale Subgroup analysis (Median; Mean; Mode; 95% CI) Duration symptoms <10 days (N=52) Duration symptoms >10 days (N=33) Not in ICU at enrolment (N=48) In ICU at enrolment (N=37) |
Definitions:
Authors conclusion:
|
|
Cavalcanti, 2020 |
Type of study: RCT
Setting: Multicenter, 55 hospitals in Brazil
Country: Brazil
Source of funding: The trial was funded by the hospitals and research institutes participating in Coalition Covid-19 Brazil. EMS Pharma provided additional funding and logistic support for the trial and also donated and supplied the trial drugs.
|
Inclusion criteria: Consecutive patients who were 18 years of age or older and who had been hospitalized with suspected or confirmed Covid-19 with 14 or fewer days since symptom onset.
Exclusion criteria: Among the reasons for exclusion from the trial were the use of supplemental oxygen at a rate of more than 4 liters per minute as administered by a nasal cannula or at a level of at least 40% as administered by a Venturi mask; the use of supplemental oxygen administered by a high-flow nasal cannula or invasive or noninvasive ventilation; previous use of chloroquine, hydroxychloroquine, azithromycin, or any other macrolide for more than 24 hours before enrollment (and since the onset of symptoms); and a history of severe ventricular tachycardia or electrocardiographic findings with a corrected QT interval (QTc) of at least 480 msec.
N total at baseline: N = 665 Total / with positive RT-PCR I (HCQ): 221 / 159 I (HCQ+AZI): 217 /172 Control: 227 / 173
Important characteristics: Age, mean (SD): I (HCQ): 51.3±14.5 I (HCQ+AZI): 49.6±14.2 C: 49.9±15.1 P=not reported
Sex, n/N (%) male: I (HCQ): 142/221 (64$) I (HCQ+AZI): 123/217 (57%) C: 123/227 (54%) P=not reported
Groups comparable at baseline? Not reported, but likely because of randomisation |
Hydroxychloroquine alone (HCQ) Standard care plus hydroxychloroquine at a dose of 400 mg twice daily for 7 days. The use of macrolides was not allowed in the hydroxychloroquine-alone group.
Hydroxychloroquine + azithromycin (HCQ+AZI) Standard care plus hydroxychloroquine at a dose of 400 mg twice daily plus azithromycin at a dose of 500 mg once a day for 7 days. |
Standard care The current standard care for Covid-19 was at the discretion of the treating physicians. The use of glucocorticoids, other immunomodulators, antibiotic agents, and antiviral agents was allowed. The administration of hydroxychloroquine or chloroquine and macrolides were not allowed in the control group. |
15 days |
Clinical status at 15days Seven-level ordinal scale. Effect estimate (95% CI) HCQ+AZI vs control: 0.99 (0.57 to 1.73) HCQ vs control: 1.21 (0.69 to 2.11) HCQ+AZI vs HCQ: 0.82 (0.47 to 1.43)
Clinical status at 7 days Six-level ordinal scale. Effect estimate (95% CI) HCQ+AZI vs control: 0.81 (0.54 to 1.22) HCQ vs control: 0.92 (0.61 to 1.38) HCQ+AZI vs HCQ: 0.89 (0.58 to 1.34)
Use of high-flow nasal cannula or noninvasive ventilation (n (%) HCQ+AZI: 16 (9.3) HCQ: 17 (10.7) Control: 16 (9.2)
Use of mechanical ventilation (n (%) HCQ+AZI: 19 (11.0) HCQ: 12 (7.5) Control: 12 (6.9)
Duration of hospital stay (n (%) HCQ+AZI: 10.3±8.4 HCQ: 9.6±6.5 Control: 9.5±7.2 Not significantly different.
In-hospital death (n (%) HCQ+AZI: 5 (2.9) HCQ: 7 (4.4) Control: 6 (3.5) Not significantly different.
Thromboembolic complications (n (%) HCQ+AZI: 2 (1.2) HCQ: 3 (1.9) Control: 2 (1.2) Not significantly different.
Acute kidney injury (n (%) HCQ+AZI: 6 (3.5) HCQ: 4 (2.5) Control: 5 (2.9) Not significantly different.
Number of days alive and free from respiratory support up to 15 days (mean (sd)) HCQ+AZI: 11.1±4.9 HCQ: 11.2±4.9 Control: 11.1±4.9 Not significantly different.
Safety outcomes More adverse events were reported in patients who received hydroxychloroquine plus azithromycin (39.3%) or hydroxychloroquine alone (33.7%) than in those who received azithromycin alone (18.0%) or none of the trial drugs (22.6%). Serious adverse events occurred in nine patients. Prolongation of the QTc interval was more common in patients receiving hydroxychloroquine plus azithromycin or hydroxychloroquine alone than in patients in the control group; however, fewer patients in the control group had serial electrocardiographic studies performed during follow-up than did patients in the other two groups. Elevation in liver-enzyme levels was more common in patients receiving hydroxychloroquine plus azithromycin than in the control group. |
Remarks: - Seven-level ordinal scale of primary outcome: score of 1 indicated not hospitalized with no limitations on activities; 2, not hospitalized but with limitations on activities; 3, hospitalized and not receiving supplemental oxygen; 4, hospitalized and receiving supplemental oxygen; 5, hospitalized and receiving oxygen supplementation administered by a high-flow nasal cannula or noninvasive ventilation; 6, hospitalized and receiving mechanical ventilation; and 7, death. - 24% tested negative on RT-PCR or test results were unavailable - analyses were performed among those with positive RT-PCR - despite intense efforts to maintain adherence to the assigned treatments, a lack of medications that were perceived as beneficial by clinicians and patients led to some protocol deviations. - the use of hydroxychloroquine plus azithromycin was widespread among patients hospitalized with Covid-19 in participating hospitals. The enrollment of patients with no previous use of these medications was challenging, so we decided to enroll patients provided that their previous use since the onset of symptoms was limited to 24 hours.
Authors conclusion: - In this trial involving hospitalized patients with mild-to-moderate Covid-19, we did not find a significant difference in a 15-day ordinal clinical-status outcome among groups that received standard care, hydroxychloroquine alone,or hydroxychloroquine plus azithromycin. Patients who received hydroxychloroquine, either with azithromycin or alone, had more frequent events of QTc interval prolongation and elevation of liver-enzyme levels than patients who did not receive either agent.
|
Risk of bias table for intervention studies (randomized controlled trials)
|
|
Describe method of randomisation |
Bias due to inadequate concealment of allocation? (unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation? (unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation? (unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation? (unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results? (unlikely/likely/unclear) |
Bias due to loss to follow-up? (unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis? (unlikely/likely/unclear) |
|
Barratt-Due, 2021
|
Computerized.
Eligible patients were allocated in an equal ratio using computer randomization procedures. There were 2 separate allocation lists. The first was the global list, in which the allocation sequence was prepared by an independent statistician appointed by the international trial steering group. A secondary national list was additionally prepared as a backup if allocation according to the global list was not available. The randomization procedure ensured that a patient could be allocated only to an available treatment. The randomization lists were not stratified or blocked; thus, the randomization can be regarded as simple. |
Unlikely
The first was the global list, in which the allocation sequence was prepared by an independent statistician appointed by the international trial steering group. A secondary national list was additionally prepared as a backup if allocation according to the global list was not available. |
Likely
Open label |
Likely
Open label |
Unclear
Despite being a randomized controlled trial with blinded analyses of all relevant data, it did not include a placebo group. |
Unlikely
All outcomes were reported in main article of appendix
ClinicalTrials.gov: NCT04321616 |
Likely
15 to 24% of participants in study groups were lost to follow-up. Missing data on outcomes due to discharge or participant withdrawal were imputed with best outcome. |
Unlikely
Each pairwise intention-to-treat analysis was between the remdesivir or HCQ group and its respective SoC. Some participants receiving SoC act as controls for both active treatment groups, whereas some act in one or the other, giving a partial overlap of the 2 control groups. |
|
Arabi, 2021
|
Online randomization system
Using a concealed online randomization system, patients were randomized to receive lopinavir-ritonavir, hydroxychloroquine, combination therapy of lopinavir-ritonavir and hydroxychloroquine or control (no antiviral agents against COVID-19). Based on local equipoise and drug availability, investigators at each participating site selected a priori two or more interventions, one of which had to be control, to which patients could be randomized. Te REMAP-CAP platform uses response-adaptive randomization; however, the allocation in the COVID-19 Antiviral Terapy Domain did not deviate from the starting equal ratio before enrollment was halted. Although the interventions were given as open-label drugs, neither the clinical staf nor the ITSC were provided any information about aggregate patient outcomes.
|
Unlikely
Using a concealed online randomization system, patients were randomized to receive lopinavir-ritonavir, hydroxychloroquine, combination therapy of lopinavir-ritonavir and hydroxychloroquine or control (no antiviral agents against COVID-19).
|
Likely
Unblinded cohort: Restricted to patients randomized to an intervention in domains that have been unblinded including the COVID-19 Antiviral Therapy Domain and domains that have ceased recruitment
|
Unlikely
Although the interventions were given as open-label drugs, neither the clinical staff nor the ITSC were provided any information about aggregate patient outcomes.
|
Unlikely
Although the interventions were given as open-label drugs, neither the clinical staff nor the ITSC were provided any information about aggregate patient outcomes.
|
Unlikely
All predefined outcome measures were reported.
NCT02735707 |
Unlikely
Lost to follow-up differences between the groups were small.
|
Unlikely
Participants included in the analysis are exactly those who were randomized into the trial.
|
|
Schwartz, 2021 |
Computerized
Randomization was conducted using a custom-developed online tool to allow for dynamic randomization and allocation concealment. We used a minimal sufficient balance randomization tool to ensure balance on age, sex, risk status, days since symptom onset and provincial health zone. Participants were randomly assigned in a stochastically governed (not blocked) 2:1 ratio. |
Unlikely
Masking to allocation sequence was complete because randomization assignment was determined dynamically at randomization. |
Unlikely
All participants were blinded. |
Unlikely
The research team was blinded, except for the research pharmacist and randomization website programmer. |
Unlikely
The research team was blinded, except for the research pharmacist and randomization website programmer. |
Unlikely
All outcome measures described in the methods are reported in the results. |
Unclear
Based on the information presented in the flow chart (Fig. 2), no participants were lost to follow-up in the ITT population. However, the table of results (Tab. 2) shows that for most of the outcomes data was missing for ≥ 1 participants, for which the reasons are not specified per treatment arm in the case of time to COVID-19 recovery. |
Likely
For most of the outcomes data was missing for ≥ 1 participants. As a result, not all randomized participants were included in the ITT analyses, which is in contrary to information presented in the flow chart (Fig. 2). |
|
Ader, 2021
|
Computerized randomisation
Participants were randomly assigned to treatment arms in a 1:1:1:1 ratio, through computer227 generated blocks of various sizes and stratification by administrative region and severity of disease at enrolment |
Unlikely
Randomization was implemented in the electronic Case Report Form to ensure appropriate allocation concealment |
Likely
Open-label study |
Likely
Open-label study |
Likely
Open-label study |
Unlikely
Outcomes mentioned in the Methods section were reported in the Results section. |
Unlikely
Loss to follow-up was limited : L/r: 1/145 (0.7%) L/r + IFN: 1/145 (0.7%) HCQ: 2/145 (1.4%) C: 0/148 (0%)
|
Unlikely
The intention-to-treat population included all randomized participants for whom a valid consent form was obtained. |
|
Réa-Neto, 2021 |
Web-bases in blocks of variable size (2, 4 and 6)
Patients were randomized (1:1) within 48 h of admission to take Clq or HClq for 5 days plus standard treatment or control (standard treatment only). Randomization was performed in blocks of variable size (2, 4 and 6) in a centralized web-based automated system and stratified by site and whether the patient was on IMV. |
Unlikely
The allocated group was disclosed to the investigator only after all information regarding patient enrollment had been recorded in the web system. |
Likely
open-label trial |
Likely
open-label trial |
Likely
open-label trial |
Likely
Trial registered (NCT04333589); Retrospectively Registered protocol. “Although the outcomes presented in the latest version were updated late on ClinicalTrials.org, on October 23, 2020, these outcomes were already present in the trial protocol approved by the Brazilian National Commission for Ethics in Research on April 8, 2020 (approval number: 3960331) and amending the protocol, approved by the same National Commission on May 25, 2020 (approval number: 4044848)”
|
Unclear
Loss to follow-up: I: 0/53 (0%) C: 1/52 (1.9%)
The reason is not reported. |
Likely
33 patients from ITT population (n=138) received the intervention (Clq/HClq) until the test result was returned. |
|
Reis, 2021
|
Randomization schedule
We randomized patients to the hydroxychloroquine, lopinavir-ritonavir, and placebo groups at 1:1:1.Randomization was stratified by site, age (aged 50 years or older vs less than 50 years), and time of onset of flulike symptoms (at least 5 days vs less than 5 days). Patients, investigators, health care practitioners, and sponsors were masked to the study drug assignment.
|
Unlikely
The randomization schedule was prepared by a masked statistician and provided to site-level pharmacists. |
Unlikely
Patients, investigators, health care practitioners, and sponsors were masked to the study drug assignment. |
Unlikely
Patients, investigators, health care practitioners, and sponsors were masked to the study drug assignment. |
Unlikely
Patients, investigators, health care practitioners, and sponsors were masked to the study drug assignment.
|
Unlikely
Outcomes mentioned in the Methods section were similar as the outcomes reported in the Results section.
|
Likely
At the end of the trial, 79 participants (11.5%) did not complete all phases of the study. The lopinavir-ritonavir intervention group had 44 participants (18%) who did not complete the study, which was more than either of the other 2 groups.
In addition, the independent DSMB, based on interim analysis results, made the decision to stop enrollment to the hydroxychloroquine and lopinavir-ritonavir groups because of a low number of emerging events. |
Unlikely
The Cox proportional hazards model was used for the analysis of time-to-event outcomes of COVID-19–associated and all-cause hospitalizations for both intention-to-treat (ITT) and per-protocol (PP) analyses. |
|
Gupta, 2021
|
Randomization was carried out by simple alternate allocation of patients in a1:1 fashion by the first author to either the control or HCQ arm. |
Likely
The allocation was not concealed either from the patient or the physicians and other medical staff.
|
Likely
“This open-label randomized control trial with unblinded assessment…”
“The chief limitation of our trial is that there was no blinding at randomization or at assessment…”
|
Likely
“This open-label randomized control trial with unblinded assessment…”
“The chief limitation of our trial is that there was no blinding at randomization or at assessment…”
|
Likely
“This open-label randomized control trial with unblinded assessment…”
“The chief limitation of our trial is that there was no blinding at randomization or at assessment…”
|
Unlikely
All predefined outcome measures were reported.
|
Unlikely
No loss to follow-up reported.
|
Unlikely
Participants included in the analysis are exactly those who were randomized into the trial.
|
|
Dubée, 2021
|
Computerized
“Patients were randomized immediately after their inclusion into the study using an online application on the study website” |
Unclear
Unclear at what point in time allocation concealment was available.
|
Unlikely
“The allocation arm was concealed for the patient and for all medical and paramedical staff.”
|
Unlikely
“The allocation arm was concealed for the patient and for all medical and paramedical staff.”
|
Unlikely
“The allocation arm was concealed for the patient and for all medical and paramedical staff.”
|
Likely
No trial register available or link to study protocol; Prematurely stopped after the inclusion of 19% of the planned number of patients; possible bias for over- of underestimating results and missing data |
Unlikely
Both intention-to-treat and per-procotol analyses conducted; only small percentage of lost to follo-up; no bias expected
|
Unlikely
Both intention-to-treat and per-procotol analyses conducted; only small percentage of lost to follo-up; no bias expected
|
|
Brown, 2020
|
Permuted blocks with concealed allocation |
Unlikely Eligible patients were randomly assigned (permuted blocks with concealed allocation) in a 1:1 ratio to hydroxychloroquine or azithromycin. Randomization was stratified by study site.
|
Likely Remdesivir was differentially prescribed to patients in the hydroxychloroquine arm. The differential use of remdesivir among hydroxychloroquine patients, which has expected efficacy in this patient population, likely biases our estimates in favor of hydroxychloroquine. |
Unclear |
Unlikely The statistical analysis plan was finalized by investigators/statisticians blinded to trial data
|
Unlikely
|
Unlikely |
Unlikely Analyses were performed according to the intention to treat principle.
|
|
Cavalcanti, 2020 |
Electronic case-report form system
Randomization was performed in blocks of six and was stratified according to the use or nonuse of supplemental oxygen at the time of randomization. Randomization was performed centrally by means of an electronic case-report form system (RedCap) as described in the Supplementary Appendix |
Unclear |
Likely |
Likely
Note: care providers were not blinded |
Likely
|
Unlikely
Note: outcomes well defined |
Unclear Note: loss to follow-up was not described |
Unclear Note only patients with confirmed diagnosis were analysed. |
Table of excluded studies
On request.
Beoordelingsdatum en geldigheid
Publicatiedatum : 07-11-2022
Beoordeeld op geldigheid : 03-10-2022
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut). Deze ondersteuning werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De werkgroep werd gefinancierd uit een VWS subsidie.
De financiers hebben geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodules is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de behandeling van patiënten met COVID-19.
In 2020 is een multidisciplinair expertiseteam behandeling ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van het expertiseteam behandeling) die betrokken zijn bij de zorg voor patiënten met COVID-19. Dit expertiseteam fungeerde als stuurgroep, welke opdracht heeft gegeven tot het ontwikkelen van de module, alsmede fungeerde als klankbordgroep.
Werkgroep
- Dr. Marjolein Hensgens, internist-infectioloog, Afdeling Infectieziekten, UMC Utrecht en LUMC Leiden (Stichting Werkgroep Antibiotica Beleid)
- Drs. Emilie Gieling, apotheker, Afdeling Klinische Farmacie, UMC Utrecht.
- Prof. Dr. Dylan de Lange, intensivist, Afdeling Intensive Care, UMC Utrecht.
- Dr. Wim Boersma, longarts, Afdeling Longziekten, Noordwest Ziekenhuisgroep, Alkmaar.
- Dr. Paul van der Linden, apotheker, Afdeling Klinische Farmacie, Tergooi MC, Hilversum (Stichting Werkgroep Antibiotica Beleid).
- Prof. Dr. Bhanu Sinha, arts-microbioloog, Afdeling Medische Microbiologie & Infectiepreventie, UMCG, Groningen (Stichting Werkgroep Antibiotica Beleid).
- Dr. Mark de Boer, internist-infectioloog, Afdelingen Infectieziekten en Klinische Epidemiologie, LUMC, Leiden (Stichting Werkgroep Antibiotica Beleid).
- Tot 1-11-2021 tevens deel van de werkgroep: Dr. Albert Vollaard, internist-infectioloog, LCI, RIVM
Stuurgroep (expertiseteam Behandeling COVID-19)
- Dr. L.M. van den Toorn (voorzitter), longarts, Erasmus Medisch Centrum (Erasmus MC), NVALT
- Dr. M.G.J. de Boer, internist-infectioloog, Leids Universitair Medisch Centrum (LUMC), SWAB/NIV)
- Drs. A.J. Meinders, internist-intensivist, St. Antonius Ziekenhuis, NVIC
- Prof. dr. D.W. de Lange, intensivist-toxicoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), NVIC
- Dr. C.H.S.B. van den Berg, infectioloog-intensivist Universitair Medisch Centrum Groningen (UMCG), NVIC
- Dr. S.U.C. Sankatsing, internist-infectioloog, Diakonessenhuis, NIV
- Dr. E.J.G. Peters, internist-infectioloog, Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. M.S. Boddaert, arts palliatieve geneeskunde, Leids Universitair Medisch Centrum (LUMC), IKNL
- Dr. P.L.A. Fraaij, kinderarts-infectioloog, Erasmus Medisch Centrum (Erasmus MC), Sophia Kinderziekenhuis, NVK
- Dr. E. van Leeuwen, gynaecoloog, Amsterdam University Medical Centers (Amsterdam UMC), NVOG
- Dr. J.J. van Kampen, arts-microbioloog, Erasmus Medisch Centrum (Erasmus MC), NVMM
- Dr. M. Bulatović-Ćalasan, internist allergoloog-immunoloog en klinisch farmacoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. A.F.J. de Bruin, anesthesioloog-intensivist, St. Antonius Ziekenhuis, NVA
- Drs. A. Jacobs, klinisch geriater, Catharina Ziekenhuis, NVKG
- Drs. B. Hendriks, ziekenhuisapotheker, Leids Universitair Medisch Centrum (LUMC), NVZA
- Drs. M. Nijs, huisarts, NHG
- Dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
Meelezer
- Drs. K. (Klaartje) Spijkers, senior adviseur patiëntenbelang, Patiëntenfederatie Nederland, Utrecht
Met ondersteuning van:
- dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
- dr. L.M.P. Wesselman, adviseur, Kennisinstituut van Medisch Specialisten
- dr. D. Nieboer, adviseur, Kennisinstituut van Medisch Specialisten
- drs. A.L.J. (Andrea) Kortlever - van der Spek, adviseur, Kennisinstituut van Medisch Specialisten
-
M. Griekspoor MSc., junior adviseur, Kennisinstituut van Medisch Specialisten
- drs. I. van Dusseldorp, senior literatuurspecialist, Kennisinstituut van Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
De Lange |
1. Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht (0,6 fte) |
Secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE), onbezoldigd. |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, klinisch epidemioloog, senior medisch specialist, Leids Universitair Medisch Centrum, afdeling Infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Sinha |
Arts-microbioloog/hoogleraar, Universitair Medisch Centrum Groningen (voltijd) (zie ook https;//www.rug.nl/staff/b.sinha/) |
- SWAB-bestuur: secretaris [onbetaald; vacatiegeld voor instelling] |
- Projectsubsidie EU (Cofund): deelprojecten, cofinanciering
Mogelijk boedbeeldfunctie SWAB |
Geen actie nodig |
|
Van der Linden |
Ziekenhuisapotheker |
Penningmeester SWAB, vacatiegeld |
Geen |
Geen actie nodig |
|
Vollaard |
Internist-infectioloog, Landelijke Coordinatie Infectieziektebestrijding, RIVM |
Arts voor ongedocumenteerde migranten, Dokters van de Wereld, Amsterdam (onbetaald) |
Geen |
Geen actie nodig |
|
Gieling |
Ziekenhuisapotheker - Klinisch Farmacoloog, UMC Utrecht |
Lid OMT Nederlandse Vereniging voor Ziekenhuisapothekers (onbetaald) |
Geen |
Geen actie nodig |
|
Boersma |
Longarts Noordwest Ziekhuisgroep |
Lid sectie infectieziekten NVALT, onbetaald |
Eenmalige digitale deelname aan adviesraad MSD Pneumovax over Pneumococcal disease, betaald
|
Geen actie nodig |
|
Hensgens |
Internist-infectioloog, UMC Utrecht (0.8 aanstelling, waarvan nu 0.4 gedetacheerd naar LUMC) Internist-infectioloog, LUMC (via detachering, zie boven) |
Geen |
Geen |
Geen actie nodig |
Stuurgroep
|
Achternaam stuurgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Van den Toorn (voorzitter) |
Voorzitter NVALT |
Geen |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, senior medisch specialist, LUMC, afdeling infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Meinders |
Internist-intensivist, St.-Antonius ziekenhuis, Nieuwegein |
commissie werk |
Geen |
Geen actie nodig |
|
De Lange |
Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht |
secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE) (onbetaald) |
Geen |
Geen actie nodig |
|
Van den Berg |
Infectioloog-intensivist, UMCG |
Geen |
Geen |
Geen actie nodig |
|
Sankatsing |
Internist-infectioloog/internist-acute geneeskunde, Diakonessenhuis, Utrecht |
- Bestuurslid Nederlandse Vereniging van Internist-Infectiologen (NVII) (onbetaald). |
Geen |
Geen actie nodig |
|
Peters |
Internist - aandachtsgebieden infectieziekten en Acute Geneeskunde Amsterdam UMC, locatie Vumc |
Wetenschappelijk Secretaris International Working Group on the Diabetic Foot (onbetaald) |
Geen |
Geen actie nodig |
|
Boddaert |
Medisch adviseur bij Integraal Kankercentrum Nederland (IKNL) en Palliatieve Zorg Nederland (PZNL) Arts palliatieve geneeskunde in LUMC |
Geen |
Geen |
Geen actie nodig |
|
Fraaij |
Kinderarts infectioloog- immunoloog, Erasmus MC-Sophia, Rotterdam |
Bestuur Stichting Infecties bij Kinderen (onbetaald) |
deelname aan RECOVER, European Union's Horizon 2020 research |
Geen actie nodig |
|
Van Leeuwen |
Gyaecoloog Amsterdam Universitair Medisch Centra |
Geen |
Geen |
Geen actie nodig |
|
Van Kampen |
Arts-microbioloog, afdeling Viroscience, Erasmus MC |
- associate editor antimicrobial resistance & infection control (onbetaald) - lid antibioticacommissie Erasmus MC (onbetaald) |
1. Mede uitvinder patent: 1519780601-1408/3023503 2. R01AI147330 (NIAID/NH) (HN onderzoek (1+2 niet gerelateerd aan COVID-19)
|
Geen actie nodig |
|
Bulatovic |
Internist allergoloog-immunoloog en klinische farmacoloog, UMC Utrecht en Diakonessenhuis Utrecht |
Functie 1: arts |
Geen |
Geen actie nodig |
|
De Bruin |
Anesthesioloog - Intensivist St. Antonius ziekenhuis Nieuwegein en Utrecht |
Geen |
Geen |
Geen actie nodig |
|
Jacobs |
Klinisch geriater en klinisch farmacoloog |
Geen |
Geen |
Geen actie nodig |
|
Hendriks |
Ziekenhuisapotheker farmaceutische patiëntenzorg, afd. Kiinische Farmacie en Toxicoiogie, Leids Universitair Medisch Centrum |
Lid SWAB werkgroep surveillance antibioticagebruik, onbetaald Lid SWAB richtlijncommissie antibiotica allergie, onbetaald |
Geen |
Geen actie nodig |
|
Nijs |
Huisarts |
Geen |
Geen |
Geen actie nodig |
|
Hofstede |
Senior adviseur Kennisinstituut van Medisch Specialisten |
Geen |
Geen |
Geen actie nodig |
Meelezer
|
Achternaam |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Spijkers |
Senior adviseur patiëntenbelang |
Voorzitter Stichting Samen voor Duchenne |
Geen |
Geen actie nodig |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde patiëntenvereniging in de klankbordgroep. De verkregen input is meegenomen bij het opstellen van de module. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Werkwijze
Van leidraad naar richtlijnmodules
Bij aanvang van de pandemie in 2020 was het onduidelijk of bestaande of nieuwe medicijnen een relevante bijdrage konden leveren aan het herstel van patiënten geïnfecteerd met het SARS-CoV-2. Vandaar dat eind februari 2020 werd aangevangen met de eerste versie van de leidraad ‘Medicamenteuze behandeling voor patiënten met COVID-19 (infectie met SARS–CoV-2)’, welke begin maart 2020 online beschikbaar werd gesteld op de website van de SWAB (https://swab.nl/nl/covid-19). Sindsdien werd het adviesdocument op wekelijkse basis gereviseerd en indien nodig op basis van nieuwe publicaties van onderzoek aangepast. Het initiatief en de coördinatie hiertoe werden genomen door de SWAB Leidraadcommissie, ondersteund door het kennisinstituut van de Federatie Medisch Specialisten en een brede klankbordgroep waarbinnen de betrokken specialisten(verenigingen) zijn vertegenwoordigd. In september 2021 is gestart met het doorontwikkelen van de leidraad naar richtlijnmodules.
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de COVID-19 pandemie zijn knelpunten op verschillende manieren geïnventariseerd:
1. De expertiseteams benoemde de knelpunten in de zorg voor patiënten met COVID-19.
2. Er is een mailadres geopend (covid19@demedischspecialist.nl) waar verschillende partijen knelpunten konden aandragen, die vervolgens door de expertiseteams geprioriteerd werden.
3. Door de Federatie van Medisch Specialisten zijn webinars georganiseerd waarbij vragen konden worden ingestuurd. Deze vragen zijn na afloop van de webinars voorgelegd aan de expertiseteams en geprioriteerd.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. Wanneer mogelijk werd de data uit verschillende studies gepoold in een random-effects model. Review Manager 5.4 werd gebruikt voor de statistische analyses. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
Definitie |
|
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
Bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Zoekverantwoording
On request.