Triple therapie in één versus in meerdere devices bij COPD
Uitgangsvraag
In hoeverre biedt het gebruik van triple (LABA/LAMA/ICS) therapie in één device voordelen voor patiënten met COPD in vergelijking met dezelfde therapie in twee of drie devices, als er een indicatie voor alle drie de bestandsdelen bestaat?
Aanbeveling
Schrijf bij het gebruik van inhalatiemedicatie bij COPD een inhalator of combinatie van inhalatoren passend bij de patiëntkarakteristieken, hulpmiddel gebonden factoren, en patiëntvoorkeuren voor.
Schrijf, indien er gekozen wordt voor meerdere inhalatoren, zoveel mogelijk hetzelfde type inhalator voor. Is hetzelfde type niet mogelijk, dan bestaat er een voorkeur voor combinaties van inhalatoren die eenzelfde type inhalatie behoeven.
Wissel het soort inhalator en/of de verpakking van de inhalator niet onnodig, maar alleen op medische indicatie of op voorkeur van de patiënt.
Overweeg triple therapie in 1 device uit gebruiksgemak en therapietrouw
Overweeg, indien flexibiliteit is gewenst, triple therapie voor te schrijven met meerdere, dezelfde type inhalatoren.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Gekeken naar de conclusies met betrekking tot de cruciale uitkomstmaten COPD-longaanvallen en mortaliteit is er, met betrekking tot het farmacologisch effect, geen sprake van een klinisch relevant verschil tussen triple therapie in 1 device vergeleken met triple therapie in meerdere devices. Slechts één studie vergeleek dezelfde moleculen in de triple versus duale bronchusverwijding. De bewijskracht van deze conclusies zijn redelijk tot laag vanwege beperkingen in studieopzet en brede betrouwbaarheidsintervallen. Daarnaast zijn er twijfels over de generaliseerbaarheid van de gerandomiseerde studies. Hierdoor is er met redelijke zekerheid te zeggen dat het gevonden effect daadwerkelijk niet klinisch relevant is.
Ook zijn er geen klinisch relevante verschillen gevonden in de belangrijke uitkomstmaten. Bij pneumonie werd na 12 weken een klinisch relevant effect gevonden in het voordeel van triple therapie in 1 device. Dit effect werd echter niet gezien in de studies die keken naar het optreden van een pneumonie na 24 of 52 weken. De bewijskracht van deze conclusie is laag vanwege studiebeperkingen en de brede betrouwbaarheidsintervallen.
Er is weinig voldoende kwalitatief onderzoek verricht naar de rol van één device in vergelijking met meerdere device. De onderzoeken die in deze richtlijn worden beschreven laten geen klinisch relevante verschillen zien. Real-world data verschijnt ook, maar de uitkomsten daarvan zijn zeer gevoelig voor bias en hebben een lage bewijskracht in vergelijking met de nu onderzochte studies.
Een argument vóór het gebruik van triple therapie in 1 device is het gegeven dat 1 op de 4 patiënten met COPD de medicatie anders gebruikt dan is voorgeschreven. Het is aannemelijk dat bij gebruik van meerdere devices de therapietrouw verder zal afnemen. Nader onderzoek zal dat moeten uitwijzen. In de publicatie van Bremner (2018) blijkt dat ‘time to first on-treatment moderate/severe exacerbation’ over de periode van 24 weken een hazard ratio toont van 0.87 (95% CI 0.68 to 1.12) voor de triple therapie in 1 device versus meerdere devices. Dit betekent dat het risico op een longaanval voor de patiënten in de triple therapiegroep in 1 device 13% lager was vergeleken met de triple therapiegroep in meerdere devices. Mogelijk speelt therapietrouw hier een rol. Ook is gevonden dat het gebruik van meerdere devices leidt tot het maken van meer fouten (Rootmensen, 2010; Sulku, 2021).
Indien de combinatie LAMA+LABA+ICS langdurig is geïndiceerd, zal een voorkeur kunnen worden uitgesproken voor een triple-medicijn ten opzichte van het gebruik van 2 of 3 devices. Dit zal leiden tot minder fouten bij gebruik en mogelijk tot een betere therapietrouw. Dit past ook bij de analyses en aanbevelingen zoals eerder gegeven zijn in de richtlijn polyfarmacie in de module bevordering therapietrouw uit 2020. Nadeel van een triple-medicijn is dat bij aanpassing van een geneesmiddel, hetzij stoppen van één van de bestanddelen dan wel veranderen van de dosering, dit te allen tijde zal leiden tot andere therapie en stoppen van het triple-medicijn.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Patiënten hebben bij monde van de patiëntenvereniging zowel in de invitational, als in diverse uitingen in tijdschriften en media laten weten een voorkeur te hebben voor een vast device. Zij ervaren veel hinder en gezondheidsschade door het regelmatig wisselen van devices en het preferentiebeleid, dat vaak geen rekening houdt met devices maar vooral met de werkzame stoffen. Het wisselen van device zonder medische noodzaak wordt dan door hen ook afgeraden. Recent is het rapport 'Wisselen van medicijnen en medische noodzaak' van Patiëntenfederatie Nederland verschenen waaruit blijkt dat veel patiënten hiermee te maken hebben. Over verantwoord wisselen van medicijnen zijn tussen diverse partijen afspraken gemaakt welke zijn vastgelegd in de ‘Leidraad verantwoord wisselen medicijnen’.
Kosten (middelenbeslag)
Er is geen analyse beschikbaar naar de kosten van triple therapie in 1 device versus triple therapie in meerdere devices. Bovendien is een vergelijking van de kosten met beide therapieën ingewikkeld, omdat niet altijd dezelfde werkzame stoffen in dezelfde devices vergeleken kunnen worden en tevens de hoeveelheid werkzame stof kan variëren. Daarbij komt dat door het preferentiebeleid de keuze van een triple therapie in 2 of 3 devices en daarmee de prijs kan variëren afhankelijk van waar de patiënt verzekerd is.
De enige werkelijke kostenvergelijking die je kunt maken is de volgende omdat je daarmee dezelfde doseringen in dezelfde toedieningsvorm vergelijkt: bron: www.medicijnkosten.nl
|
|
Kosten per dag, 1 device |
2 devices |
|
Fluticasonfuroaat/vilanterol/ umeclidinium |
€ 1,90 |
|
|
Fluticasonfuroaat/vilanterol |
|
€ 1,06 |
|
umeclidinium |
|
€ 1,06 |
|
Totaal |
|
€ 2,12 |
Dit zijn prijzen exclusief de afleverkosten van de apotheek (prijspeil 2023). Het meenemen van deze kosten zal het prijsverschil nog iets groter maken. Hier worden dezelfde werkzame stoffen in dezelfde sterke en in hetzelfde device vergeleken. In dit geval is de triple therapie in 1 device goedkoper dan dezelfde therapie in 2 devices.
Wordt er puur naar de kosten van de inhalers gekeken zou men kunnen concluderen dat een triple goedkoper is dan meerdere devices, echter bijvoorbeeld onduidelijk is of dit verschil overeind blijft in een officiële kostenanalyse waarbij bijvoorbeeld wel of niet vaker wisselen tussen types inhalatoren en benodigde aanpassingen bij dosis veranderingen en zo nodig extra medicatie in het kader van longaanval actieplannen wordt meegenomen.
Uiteindelijk is het effect van de therapie het belangrijkste, ook voor de kosten. Het juiste gebruik van een device, het langdurig gebruik van dezelfde device en een goede therapietrouw leidt tot een beter resultaat en daardoor minder consulten en opnames en uiteindelijke tot lagere kosten van de gezondheidszorg.
Aanvaardbaarheid, haalbaarheid en implementatie
Sinds enige jaren zijn de triple therapieën op de markt. Deze combinaties van LABA/LAMA/ICS zijn verkrijgbaar in verschillende devices. Fluticasonfuroaat/vilanterol/umeclidinium is verkrijgbaar in een Ellipta (een droogpoederinhalator). De combinaties beclomethason/formoterol/glycopyrronium in een aerosol en in een Nexthaler (een droogpoederinhalator), budesonide/formoterol/glycopyrronium in een Aerosphere ( een aerosol) en de combinatie mometason/indacaterol/glycopyrronium in een Breezhaler (een inhalatiecapsule).Dit laatste middel is geregistreerd voor de indicatie astma. Nog meer van belang dan de keuze van de werkzame stoffen is de keuze van het device. Het juiste gebruik van een device met een juiste inhalatietechniek is onontbeerlijk voor een goede therapie. In de praktijk blijkt dat de patiënt daarbij zijn/haar voorkeuren heeft en met het ene device een betere inhalatie bereikt dan met een ander device. Mede omdat elke device afhankelijk van de weerstand een andere inhalatietechniek vereist. Indien de keus is gemaakt voor een triple therapie zal daarmee rekening gehouden moeten worden. Op basis van prijs is er geen onderscheid elke vaste triple therapie heeft dezelfde dagprijs. Een kanttekening daarbij dient geplaatst te worden bij de aerosolen. Hierbij dient een voorzetkamer te worden gebruikt. Met deze toevoeging valt de prijs van deze middelen duurder uit.
In het geval voor een keuze van 2 of 3 devices als combinatie van LABA met LAMA en ICS is de uniformiteit van device van belang. Uit onderzoek is gebleken dat bij meerdere devices de kans op een foute inhalatietechniek toeneemt. Uit een recent onderzoek van de Stichting Farmaceutische Kengetallen is gebleken dat 41% van de inhalatorgebruikers in 2020 inhalatiemiddelen kreeg uit meerdere therapeutische groepen. Van deze groep ontving 31% zowel een dosisaerosol als een droogpoederinhalator Bij het maken van een keuze zou dan, op basis van een mogelijk betere therapietrouw, een voorkeur kunnen worden uitgesproken voor een triple therapie in 2 devices ten opzichte van 3 devices.
Vanuit patiëntperspectief zal de voorkeur zeer waarschijnlijk uitgaan naar langdurig dezelfde triple-therapie vanwege gebruiksgemak. Dit zal zeker gelden voor patiënten met geringe gezondheidsvaardigheden, lage sociale klassen, laag opleidingsniveau, laag inkomen, lage taalvaardigheden.
Op 4 maart 2021 heeft het College ter Beoordeling van Geneesmiddelen (CBG) in opdracht van het ministerie van Volksgezondheid, Welzijn en Sport (VWS) een overzicht vastgesteld van medicijnen waarbij (generiek) wisselen, in het belang van de patiënt, ongewenst is. Dit vanwege de kans op ernstige klinische gevolgen bij dubbele dosering of te late inname door verwarring, die mogelijk het gevolg is van de wisseling. Ook veel longmedicatie staat op deze lijst.
In het huidige zorgveld met preferentie en substitutie beleid zal deze richtlijn COPD geïmplementeerd worden. Er zijn in Nederland diverse initiatieven gaande op dit vlak en samenwerkingsverbanden. Het verdient aanbeveling om in de samenwerkingsverbanden die vaak bestaan tussen 1e en 2e lijn, apothekers en zorgverzekeraars afspraken te maken om de aanbevelingen uit de richtlijn en daarmee de kwaliteit van de zorg zoveel mogelijk te borgen.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
Op de binnen de PICO gedefinieerde wetenschappelijke eindpunten is geen voordeel aan te geven voor een van beide strategieën op het moment. Echter het gebruik van 1 geneesmiddel in de vorm van triple-therapie, mits geïndiceerd, geeft minder fouten in gebruik en mogelijk betere therapietrouw dan de verschillende medicamenten in verschillende soorten inhalatoren. In voorkomende gevallen is het wel mogelijk om duale therapie of monotherapie in 1 zelfde soort inhalator te geven. Hoewel literatuur hierover ontbreekt is het aannemelijk om te verwachten dat dan het voordeel voor de triple in 1 device wegvalt.
Bij het gebruik van 1 device als triple mis je de flexibiliteit van aanpassing van therapie die je wel hebt bij het gebruik van 2 of 3 middelen.
Tevens is aannemelijk gemaakt (richtlijn polyfarmacie bij ouderen) dat het wisselen tussen merken en verpakkingen van inhalatoren met als gevolg onder onder andere verschillen in gebruik, uiterlijk van de inhalatoren, vereiste inhalatietechniek, waarbij de indicatie en daarmee de werkzame stof gelijk blijft, de therapietrouw en daarmee de effectiviteit en de kosten negatief beïnvloedt.
Onderbouwing
Achtergrond
Sinds enige jaren zijn er triple therapieën (ICS/LAMA/LABA) verkrijgbaar naast de verschillende componenten als mono-ICS, mono-LAMA, mono-LABA en de duo-therapieën LAMA/LABA en LABA/ICS. Welk middel wanneer het beste gebruikt kan worden, wordt elders in de richtlijn besproken. Naast de werkzame stoffen is ook het kiezen van de optimale inhalatie device uitermate belangrijk voor de zorg van patiënten met COPD. Op dit vlak bestaat er veel praktijkvariatie. Door verschillende zorgverzekeraars, apothekers en patiënten wordt met enige regelmaat gevraagd van inhalatie device te wisselen, ook als een specifieke device is voorgeschreven door een arts en goed bevalt. Een van de discussiepunten in dit gebied betreft het gebruik van één inhalator per dag voor triple therapie in vergelijking met het gebruik van meerdere devices. Deze zijn voor 1 patiënt dan vaak van verschillende merken voor de drie verschillende groepen inhalatiemedicatie. Deze module poogt het beschikbare gerandomiseerde bewijs voor dit laatste te bundelen.
Conclusies / Summary of Findings
COPD exacerbations rate (crucial outcome measure)
|
Moderate GRADE |
The evidence suggests that triple therapy in one device likely results in little to no difference in COPD exacerbations when compared with triple therapy in two or three devices in patients with COPD. Sources: Bremner, 2018; Vestbo, 2017 |
Mortality (crucial outcome measure)
|
Low GRADE |
The evidence suggests that triple therapy in one device may result in little to no difference in mortality when compared with triple therapy in two devices in patients with COPD
Sources: Ferguson, 2020; Vestbo, 2017 |
Quality of life (important outcome measure)
|
Low GRADE |
The evidence suggests that triple therapy in one device may result in little to no difference in quality of life when compared with triple therapy in two devices in COPD patients.
Sources: Bremner, 2018; Ferguson, 2020; Vestbo, 2017 |
FEV1 (important outcome measure)
|
Low GRADE |
The evidence suggests that triple therapy in one device may result in little to no difference in FEV1 when compared with triple therapy in two devices in COPD patients.
Sources: Bremner, 2018; Ferguson, 2020; Vestbo, 2017 |
Serious adverse events (SAEs) (important outcome measure)
|
Low GRADE |
The evidence suggests that triple therapy in one device may result in little to no difference in pneumonia incidence when compared with triple therapy in two devices in COPD patients.
Sources: Bremner, 2018; Ferguson, 2020; Vestbo, 2017 |
|
Very Low GRADE |
Other SAEs The evidence is very uncertain about the effect of triple therapy in one device on other SAEs when compared with triple therapy in two devices in COPD patients.
Sources: Bremner, 2018; Ferguson, 2020; Vestbo, 2017 |
Samenvatting literatuur
Description of studies
Bremner (2018) performed a randomized, double-blind, parallel group non-inferiority study to evaluate the non-inferiority of single-inhaler fluticasone furoate/umeclidinium/vilanterol (FF/UMEC/VI) and placebo versus an alternative once-daily triple therapy regimen using two inhalers (FF/VI + UMEC) in the treatment of adults with COPD. After screening, eligible patients (n=1055) enrolled on combination maintenance therapy. After two weeks, they were randomly assigned to the intervention group, receiving FF/UMEC/VI 100 μg/62.5 μg/25 μg (n=527) and placebo; or the control group, receiving FF/VI 100 μg/25 μg + UMEC 62.5 μg (n=528). All patients were treated for 24 weeks, and treatments were delivered using the ELLIPTA inhaler once daily in the morning. After one week of follow-up, patients were evaluated on exacerbation frequency (moderate/severe), quality of life, FEV1 and serious adverse events.
Ferguson, (2020) presented results from two randomized, double-blind, triple-dummy non-inferiority trials comparing single- with multiple-inhaler inhaled corticosteroid/long-acting muscarinic antagonist/long-acting β2-agonist (ICS/LAMA/LABA) combination in COPD. Both trials were conducted from June 2018 to March 2019. After screening, eligible patients enrolled on maintenance therapy, in which patients continued existing COPD medications. After four weeks, all patients received BUD/FOR (400/12 μg) twice daily via metered-dose inhaler (MDI) plus TIO (18 μg) once daily via HandiHaler (Boehringer Ingelheim International GmbH) plus placebo once daily via Ellipta dry-powder inhaler (GlaxoSmithKline). Thereafter, patients were randomly assigned to the intervention group, receiving FF/UMEC/VI 100/62.5/25 μg once daily (via Ellipta plus two inhalations of placebo twice daily via MDI plus placebo once daily via Handihaler; or the control group, receiving two inhalations of BUD/FOR 200/6 μg twice daily via MDI plus TIO 18 μg once daily via HandiHaler (in the morning) plus placebo once daily (in the morning) via Ellipta. All patients were treated for 84 days. Directly after treatment and after one week of follow-up, patients were evaluated on FEV1, quality of life and serious adverse events. Subgroup analyses per performed by age (< 65, ≥ 65 years), percent predicted FEV1 at screening (≤ 30%, 30–< 50%, ≥ 50%), and baseline CAT score (< 20, ≥ 20).
Vestbo (2017) performed a double-blind, parallel group, double-dummy randomized controlled trial comparing treatment with extrafine beclomethasone dipropioate formoterol fumarate, and glycopyrronium bromide (BDP/FF/GB; fixed triple) with tiotropium, and BDP/FF plus tiotropium (open triple) in COPD patients. After screening, all patients (n=2580) received tiotropium 18 μg, one inhalation per day (in the morning) via single-dose dry-powder inhaler for two weeks. Thereafter, patients were randomly assigned to receive BDP/FF/GB and placebo (fixed triple, n=1032), tiotropium and placebo (n=1032) or tiotropium and BDP/FF (n=516). Patients were treated for 52 weeks in total, and were evaluated on exacerbation rate, quality of life, FEV1, and serious adverse events after 52 weeks. Outcome measures were presented per comparison: fixed triple versus tiotropium; fixed triple versus open triple; open triple versus tiotropium. To answer our clinical question, only the fixed triple group (intervention) and the open triple group (control) were compared.
Results
COPD exacerbations rate (crucial outcome measure)
Two studies (Bremner, 2018; Vestbo, 2017) reported on the rate of COPD exacerbations. Bremner (2018) assessed patient-reported exacerbations at week 24. Patients
identified exacerbation as worsening of COPD symptoms that were mild, moderate or severe. After 24 weeks 24% of the patients in the intervention group experienced moderate/severe exacerbations (129/527), compared to 27% of the patients in the control group (142/528) (RR 0.91 (95% CI: 0.74 to 1.12) in favour of the intervention group).
Vestbo (2017) assessed rate of severe and of moderate COPD exacerbations after 52 weeks of treatment, defined as a worsening of the patient’s respiratory symptoms that in the view of the patient’s healthcare provide required treatment with systemic corticosteroids, antibiotics, hospital admission or a combination. Events were classified as moderate or severe according to European Medicines Agency/Committee for Medicinal Products for Human Use guidelines. After 52 weeks of treatment, the intervention group (fixed triple treatment) showed an exacerbation rate of 33% (351/1077), compared to 31% in the control group (open triple therapy) (167/537) (RR 1.05 (95% CI 0.90 to 1.22) in favour of the control group).
Mortality in all patients (crucial outcome measure)
Two studies (Ferguson, 2020; Vestbo, 2017) reported on the number of deaths. Ferguson (2020) reported on the number of fatal serious adverse events, showing mortality of 0% in the intervention group receiving fixed triple therapy (0/556), compared to 0.1% in the control group receiving open triple therapy (1/549) (RR 0.33 (95% CI 0.01 to 8.06) in favour of the intervention group).
Vestbo (2017) reported on the adverse events leading to death. In the intervention group receiving fixed triple therapy mortality was 1.9% (20/1077), compared to 1.5% in the control group receiving open triple therapy (8/537) (RR 1.25 (95% CI 0.55 to 2.81) in favour of the control group).
Quality of life (important outcome measure)
Three studies (Bremner, 2018; Ferguson, 2020; Vestbo, 2017) reported on the SGRQ. Bremner (2018) assessed quality of life by the proportion of responders based on the SGRQ at Week 24. The proportion of responders was 50% in the intervention group receiving fixed triple therapy, compared to 51% in the control group receiving open triple therapy
Ferguson (2020) assessed the proportion of responders by the number of patients who experienced an improvement on the SGRQ that met or exceeded the minimum clinically important difference of at least 4 units after 12 weeks of treatment. The proportion of responders was 35% in the intervention group, compared to 34% in the control group
Vestbo (2017) assessed the proportion of responders after 26 weeks of treatment, defined as a SGRQ-score decrease of at least four units from baseline. After 26 weeks, the proportion of responders was 47% in the intervention group, compared to 51% in the control group
A pooled RR was calculated for the studies mentioned above 0.96 (95% CI 0.90 to 1.03) in favour of the intervention group) (Figure 1).
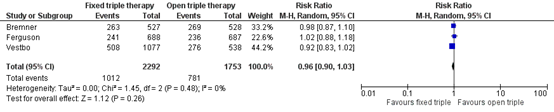
Figure 1 Rate of SGRQ improvement
FEV1 (important outcome measure)
Three studies (Bremner, 2018; Ferguson, 2020; Vestbo, 2017) reported on the FEV1. Bremner (2018) reported a between-treatment difference of 18 mL (95% CI -13 to 50) and Ferguson (2020) of 14 mL (95% CI -5 to 34) at 12 weeks, both in favour of the intervention. Vestbo (2017) reported the proportion of patients who showed a increase of at least 100 mL from baseline after 26 weeks of treatment. After 26 weeks, this proportion was 39% in the intervention group, compared to 38% in the control group (RR 1.03 (95% CI 0.90 to 1.18) in favour of the intervention. After 52 weeks, this proportion was 38% in the intervention group, compared to 39% in the control group (RR 0.95 (95% CI 0.77 to 1.18) in favour of the control group.
Serious adverse events (SAEs) (important outcome measure)
Pneumonia
Three studies (Bremner, 2018; Ferguson, 2020; Vestbo, 2017) reported on pneumonia incidence. Bremner assessed pneumonia after 24 weeks of treatment, resulting in
3% in the intervention group (14/527), compared to 4% in the control group (21/529) (RR 0.67 (95% CI 0.34 to 1.30) in favour of the intervention group.
Ferguson (2020) assessed pneumonia after 12 weeks of treatment, resulting in 1.0% in the intervention group (7/729) and 1.2% in the control group (9/731) (RR 0.78 (95% CI 0.29 to 2.08) in favour of the intervention group).
Vestbo (2017) assessed pneumonia after 52 weeks of treatment, resulting in 1.9% (21/1077) in the intervention group, compared to 1.7% in the control group (9/537) (RR 1.16 (95% CI 0.54 to 2.52) in favour of the control group). .
Other serious adverse events
Other SAEs were addressed by three studies (Bremner, 2018; Ferguson, 2020; Vestbo, 2017). Bremner assessed on-treatment non-fatal SAEs after 24 weeks of treatments but did not specify which events were included exactly. Data resulted a percentage of 9.8% in the intervention group (52/527) and 10.8% in the control group (57/528) (RR 0.91 (95% CI 0.64 to 1.30) in favour of the intervention.
Ferguson (2020) assessed the number of serious adverse events per 1000 patient-year but did not specify which events were included exactly. Data resulted in a percentage of 5.1% intervention group (37/729) and 4.2% in the control group (31/731) (RR 1.20 (95% CI 0.75 to 1.91)) in favour of the control group. .
Vestbo (2017) assessed serious adverse events, including COPD exacerbations, pneumonia, ischaemic heart disease and cardiac failure. Data resulted in a percentage of 13% in the intervention group (140/1077) and 12.7% in the control group (68/537) (RR 1.03 (95% CI 0.78 to 1.35) in favour of the control group.
Level of evidence of the literature
COPD exacerbations rate (crucial outcome measure)
The level of evidence regarding the outcome measure ‘COPD exacerbation rate’ started at high (RCT) and was downgraded by 1 level to moderate because of risk of bias (inadequate concealment allocation). Because funding did not influence results, the evidence was not downgraded for publication bias and since the confidence interval did not include the possibility of benefit or no harm, the evidence was not downgraded for imprecision.
Mortality (crucial outcome measure)
The level of evidence regarding the outcome measure ‘mortality’ for all participants started at high (RCT) and was downgraded by 2 levels to low because of risk of bias (inadequate concealment allocation) and imprecision (limited number of included patients). Since the confidence interval stayed in between the borders of clinical relevance, the evidence was not downgraded for imprecision due to wide confidence intervals. Furthermore, funding likely did not influence results, so the evidence was not downgraded for publication bias.
Quality of life (important outcome measure)
The level of evidence regarding the outcome measure ‘quality of life’ started at high (RCT) and was downgraded by 2 levels to low because of risk of bias (inadequate concealment allocation and lack of blinding).
FEV1 (important outcome measure)
The level of evidence regarding the outcome ‘FEV1’ started at high (RCT) and was downgraded by 2 level to low because of risk of bias (inadequate concealment allocation and publication bias). The evidence was not downgraded for imprecision.
Serious adverse events (SAEs) (important outcome measure)
Pneumonia
The level of evidence regarding the outcome ‘pneumonia’ started at high (RCT) and was downgraded by 2 levels to low due to risk of bias (inadequate concealment allocation) and imprecision (confidence interval include the possibility of benefit or no harm).
Other SAEs
The level of evidence regarding the outcome ‘other SAEs’ started at high (RCT) and was downgraded by three levels to very low because of risk of bias (inadequate concealment allocation) and inconsistency (clinical and statistical heterogeneity).
Zoeken en selecteren
A systematic review of the literature was performed to answer the following question: To what extent is triple therapy advantageous compared to triple therapy in two or three devices in patients with COPD?
P: patients COPD-patients, > 40 years old who use triple therapy (ICS/LAMA, LABA)
I: intervention triple therapy (any LABA/LAMA/ICS) in one device.
C: control triple therapy (any LABA/LAMA/ICS) in two or three devices.
O: outcome COPD exacerbations rate, mortality, quality of life, FEV1, adverse events
(including pneumonia and other adverse events)
Relevant outcome measures
The guideline development group considered COPD exacerbation rate and mortality as crucial outcome measures for decision making; all other outcome measures were considered important measures for decision making.
The working group defined a set of minimal clinically (patient) important differences (see introduction for further details). For this intervention, the following set was chosen:
• Exacerbation reduction: ≥20%
• Mortality: ≥10% difference in relative risk
• Pneumonia: ≥20% difference in relative risk
• CAT-score: >2 units
• CCQ-score: >0.4 units
• SGRQ-score: ≥4 units
• Hospital admissions: ≥20% difference in relative risk
• Adverse events: ≥25% difference in relative risk
• FEV1: >100 ml
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms from 2015 until 26-1-2021. The detailed search strategy is depicted under the tab Methods. The systematic literature search resulted in 203 hits. Studies were selected based on the following criteria: Systematic reviews, randomized controlled trials (RCTs) or observational studies. Initially 49 studies were selected based on title and abstract screening. After reading the full text, 46 studies were excluded (see the table with reasons for exclusion under the tab Methods), and three studies were included.
Results
Three studies were included in the analysis of the literature. Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
Referenties
- Bremner PR, Birk R, Brealey N, Ismaila AS, Zhu CQ, Lipson DA. Single-inhaler fluticasone furoate/umeclidinium/vilanterol versus fluticasone furoate/vilanterol plus umeclidinium using two inhalers for chronic obstructive pulmonary disease: a randomized non-inferiority study. Respir Res. 2018 Jan 25;19(1):19. doi: 10.1186/s12931-018-0724-0. PMID: 29370819; PMCID: PMC5785849.
- Ferguson GT, Brown N, Compton C, Corbridge TC, Dorais K, Fogarty C, Harvey C, Kaisermann MC, Lipson DA, Martin N, Sciurba F, Stiegler M, Zhu CQ, Bernstein D. Once-daily single-inhaler versus twice-daily multiple-inhaler triple therapy in patients with COPD: lung function and health status results from two replicate randomized controlled trials. Respir Res. 2020 May 29;21(1):131. doi: 10.1186/s12931-020-01360-w. PMID: 32471423; PMCID: PMC7257245.
- Rootmensen GN, van Keimpema AR, Jansen HM, de Haan RJ. Predictors of incorrect inhalation technique in patients with asthma or COPD: a study using a validated videotaped scoring method. J Aerosol Med Pulm Drug Deliv. 2010 Oct;23(5):323-8. doi: 10.1089/jamp.2009.0785. PMID: 20804428.
- Vestbo J, Papi A, Corradi M, Blazhko V, Montagna I, Francisco C, Cohuet G, Vezzoli S, Scuri M, Singh D. Single inhaler extrafine triple therapy versus long-acting muscarinic antagonist therapy for chronic obstructive pulmonary disease (TRINITY): a double-blind, parallel group, randomised controlled trial. Lancet. 2017 May 13;389(10082):1919-1929. doi: 10.1016/S0140-6736(17)30188-5. Epub 2017 Apr 3. PMID: 28385353.
Evidence tabellen
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3
|
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Bremner, 2018 |
Type of study: Study.
Setting and country: USA Funding and conflicts of interest: NCT02729051). ELLIPTA is owned by or licensed to the GSK group of companies. RB, NB, ASI, C-QZ, and DAL are employees of GSK and hold stocks/shares in the company. ASI is also an unpaid faculty member at McMaster University,
Canada. PRB has no conflicts of interest to disclose. |
Inclusion criteria: COPD Assessment Test™ (CAT) score ≥ 10 (13, 14); a post-albuterol/salbutamol FEV1/forced vital capacity ratio < 0.70; and a post-bronchodilator FEV1< 50% of predicted and ≥1 moderate/severe exacerbation in the previous 12 months or a post-bronchodilator FEV1≥ 50% to < 80% of predicted and ≥2 moderate exacerbations or ≥1 severe exacerbation requiring hospitalisation in the previous 12 months.
Exclusion criteria: a current diagnosis of asthma (patients with a prior history of asthma were eligible if they had a current diagnosis of COPD that was the primary cause of their respiratory symptoms); α1-antitrypsin deficiency; active tuberculosis; other respiratory disorders that were the primary cause of respiratory symptoms; lung resection surgery in the previous 12 months; risk factors for pneumonia (including immunosuppression and neurological disorders affecting control of the upper airway (e.g. Parkinson’s disease or myasthenia gravis); pneumonia and/or moderate/severe exacerbation that had not resolved at least 14 days prior to screening; respiratory infections; abnormal findings on chest X-ray; clinically significant comorbidities; unstable liver or cardiac disease; and cancer. Patients with a high risk for pneumonia (e.g. very low body mass index, severely malnourished, or very low FEV1) were only to be included at the discretion of the investigator.
N total at baseline: Intervention: 527 Control: 528
Important prognostic factors2: age ± SD: I: 66.7 ± 8.5 C: 65.9 ± 8.8
Sex: I: 74% M C: 75% M
Groups comparable at baseline? Yes
|
24 weeks of FF/UMEC/VI 100 μg/62.5 μg/25 μg in a single inhaler and placebo (second inhaler). all treatments/placebo were delivered using the ELLIPTA inhaler once daily in the morning.
|
24 weeks of FF/VI 100 μg/25 μg and UMEC 62.5 μg, in separate inhalers. all treatments/placebo were delivered using the ELLIPTA inhaler once daily in the morning.
|
Length of follow-up: One week after treatment.
Loss-to-follow-up: Intervention: 6% Reasons: n.r.
Control: 6% Reasons: n.r.
Incomplete outcome data: n.r.
|
COPD exacerbations rate
Effect measure: Risk ratio (95% CI):
Mortality
Quality of life Defined as the proportion of responders to the SGRQ.
Effect measure: Risk ratio (95% CI):
FEV1 Defined as the between treatment difference in FEV1
Pneumonia Defined as the number of participants with pneumonia.
Effect measure: Risk ratio (95% CI):
Serious adverse events Defined as the number of serious adverse events.
Effect measure: Risk ratio (95% Pneumonia: 0.67 (0.34 to 1.30) in favour of fixed triple therapy.
Other SAEs 0.91 (95% CI 0.64 to 1.30) in favour of fixed triple therapy. |
This study showed that single-inhaler FF/UMEC/VI 100 μg/62.5 μg/25 μg was non-inferior to FF/VI 100 μg/ 25 μg plus UMEC 62.5 μg based on change from baseline in trough FEV1 at Week 24 in patients with advanced COPD. Our findings confirm that single-inhaler triple therapy with FF/UMEC/VI offers similar efficacy, healthrelated quality of life, and safety benefits as the same triple therapy administered using two inhalers. |
|
Vestbo, 2017 |
Type of study: randomised controlled trial
Setting and country: UK
Funding and conflicts of interest: declare no competing interests. |
Inclusion criteria: Eligible patients were 40 years of age or older; current or ex-smokers; had a diagnosis of COPD, with postbronchodilator (salbutamol 400 μg) forced expiratory volume in 1 s (FEV1) of less than 50% and a ratio of FEV1 to forced vital capacity of less than 0·7; had at least one moderate or severe COPD exacerbation in the previous 12 months; and used an inhaled corticosteroid plus long-acting β₂-agonist (as an open or fixed combination), or inhaled corticosteroid plus long-acting muscarinic antagonist, or inhaled long-acting β₂-agonist plus long-acting muscarinic antagonist (as an open or fixed combination), or long-acting muscarinic antagonist monotherapy for at least 2 months before screening— patients receiving triple therapy of inhaled corticosteroid, long-acting β₂-agonist and long-acting muscarinic antagonist were not eligible. Additionally, eligible patients were symptomatic, with a COPD Assessment Test total score of at least 10.
Exclusion criteria: We excluded patients from the study if they had a diagnosis of asthma, or history of allergic rhinitis or atopy; COPD exacerbation in the 4 weeks before screening or during the run-in period; clinically significant cardiovascular conditions or laboratory abnormalities (including persistent, long-standing, or permanent atrial fibrillation); or unstable concurrent disease that could have influenced efficacy or safety (as judged by the investigator).
N total at baseline: Intervention: 1077 Control: 537
Important prognostic factors2: age ± SD: I: 63.4 ± 8.7 C: 62.6 ± 8.9
Sex: I: 77% M C: 74% M
Groups comparable at baseline? Yes
|
Patients received extrafine 100 μg BDP/6 μg FF/12·5 μg GB, two actuations twice daily via pressurised metered-dose inhaler (pMDI; fixed triple group) + placebo
|
Patients received extrafine 100 μg BDP/6 μg FF, two actuations twice daily via pMDI plus tiotropium 18 μg, one inhalation per day via SDDPI (open triple group). |
Length of follow-up: 52 weeks.
Loss-to-follow-up: Intervention: N (%)
Control: N (%) 42 (7.8%)
Incomplete outcome data: n.r.
|
COPD exacerbations rate
Effect measure: Risk ratio (95% CI):
Mortality
Effect measure: Risk ratio (95% CI): 1.25 (95% CI 0.55 to 2.81) in favour of open triple therapy.
Quality of life Defined as the proportion of responders to the SGRQ.
Effect measure: Risk ratio (95% CI): After 24 weeks:
After 52 weeks:
FEV1 Defined as proportion of patients who improved at least with 100 mL from baseline.
After 24 weeks:
After 52 weeks: 0.97 (0.85 to 1.11) in favour of fixed triple therapy
Effect measure: Risk ratio (95% CI):
Serious adverse events Defined as the number of serious adverse events.
Effect measure: Risk ratio (95% CI): Pneumonia: After 52 weeks
Other SAEs After 52 weeks: |
In conclusion, in TRINITY treatment with extrafine fixed triple therapy had clinically beneficial effects compared with long-acting muscarinic antagonist monotherapy on different components of COPD, namely exacerbations, FEV1, hyperinflation, and health-related quality of life. This consistent improvement in different disease domains suggests that stepping up a patient from long-acting muscarinic antagonist to triple therapy will have a clinically meaningful impact. |
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors ((potential) confounders)
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Ferguson, 2020
|
SR of 2 RCTs
No literature search applicable.
A: Study 207608 B: Study 207609
Study design: A: replicate Phase IV, 12-week, randomized, double-blind, triple-dummy, parallel-group, multicenter, non-inferiority trials. B: replicate Phase IV, 12-week, randomized, double-blind, triple-dummy, parallel-group, multicenter, non-inferiority trials.
Setting and Country: A: 59 centres in 4 countries.
Source of funding and conflicts of interest: Funded by GSK. N Brown, C Compton, TC Corbridge, K Dorais, C Harvey, MC Kaisermann, DA Lipson, N Martin, M Stiegler, and C-Q Zhu are employees of GlaxoSmithKline (GSK) and are shareholders in GSK. GT Ferguson received grants, personal fees, and non-financial support from Boehringer Ingelheim, Novartis, AstraZeneca, Pearl Therapeutics, Sunovion, Theravance, and GSK, grants and personal fees from Verona, and Sanofi, and personal fees from Mylan, Innoviva, and Circassia, and grants from Altavant, unrelated to this work. C Fogarty has nothing to disclose. F Sciurba received research support from the COPD Foundation, Department of Defense Beta Blocker, Gala Therapeutics, Inc., GSK, NIH, Nuvaira, PCORI, PneumRX, Inc., PulmonX, and ResMed Corp, participated in advisory board for GSK, PneumRX, Inc., Theravance, and Verona and previously received research support from Astellas and AstraZeneca, unrelated to this work. D Bernstein has received grants and personal fees from GSK and ALK America, grants from Aimmune, Amgen, AstraZeneca, Genentech, Merck, Mylan, Novartis, Pearl, Shire, Teva, Adare, Knopp, Leo, Mandala, Gossamer, and Regeneron, and personal fees from Gerson Lehman, Guidepoint Global, and Covis, unrelated to this work.
|
Inclusion criteria SR: Informed consent, outpatients, 40 years and older, male and female, COPD diagnosis, smoking history, severity of COPD symptoms (10 or higher on the CAT), severity of FEV1 of <50% predictued to normal or post-brochodilatator FEV1 <80% of predicted normal, history of 2 or more moderate/severe exacerbations in previous 12 months, post albuterol/salbutamol FEV1/FVC ratio of <70, existing COPD maintenance treatment.
Exclusion criteria SR: Pregnancy, asthma, alpha1-antitrypsin deficiency, other respiratory disorders, lung resection, risk actors for pneumonia, pneumonia and/or moderate or sever COPD exacerbations, respiratory tract infection, abnormal chest x-ray, other diseases/abnormalities, unstable liver disease, unstable or life threatening cardiac disease, abnormal and clinically significant 12-lead ECG finding, contraindications, cancer, oxygen therapy, medication prior to spirometry, pulmonary rehabilitation, drug/alcohol abuse, non-complicance, questionable validity of consent, affiliation with investigator site, inability to read, medication prior to screening.
2 studies included
Important patient characteristics at baseline: N, mean age A: 728 patients, 65.1y B: 732 patients, 65.3y
Sex: A: 52.7% Male B: 51.0% Male
Groups comparable at baseline? Yes. |
Describe intervention:
A: single-inhaler triple therapy (FF/UMEC/VI) 100/62.5/25 μg once daily (in the morning) via Ellipta plus two inhalations of placebo twice daily via MDI (in the morning and evening) plus placebo once daily via HandiHaler (in the morning). B: single-inhaler triple therapy (FF/UMEC/VI) 100/62.5/25 μg once daily (in the morning) via Ellipta plus two inhalations of placebo twice daily via MDI (in the morning and evening) plus placebo once daily via HandiHaler (in the morning).
|
Describe control:
A: multiple-inhaler triple combination therapy (BUD/FOR+TIO) 200/6 μg twice daily via MDI (in the morning and evening) plus TIO 18 μg once daily via HandiHaler (in the morning) plus placebo once daily (in the morning) via Ellipta for 84 days B: multiple-inhaler triple combination therapy (BUD/FOR+TIO) 200/6 μg twice daily via MDI (in the morning and evening) plus TIO 18 μg once daily via HandiHaler (in the morning) plus placebo once daily (in the morning) via Ellipta for 84 days.
|
End-point of follow-up:
A: After four weeks of treatment, after 12 weeks of treatment, 7 days after treatment. B: After four weeks of treatment, after 12 weeks of treatment, 7 days after treatment.
For how many participants were no complete outcome data available? (intervention/control) A: 29/40 B: 35/26
|
COPD exacerbations rate
Mortality Effect measure: risk ratio (95% CI): Quality of life Defined as the proportion of responders to the SGRQ. (intervention/control) A: 117 (34%) / 117 (34%) Effect measure: risk ratio (95% CI): Pooled effect: 1.02 (95% CI 0.88 to 1.19) in favour of open triple therapy.
FEV1 n.r.
Serious adverse events Defined as the number of serious adverse events (intervention / control) Pneumonia A: 5 (1%) / 6 (2%)
Effect measure: Risk ratio (95% CI): Pooled effect: 0.78 (95% CI 0.29 to 2.08) in favour of fixed triple therapy.
Other serious adverse events
Effect measure: Risk ratio (95% CI): Pooled effect: 1.20 (95% CI 0.75 to 1.91) in favour of open triple therapy.
|
Author’s conclusion Once-daily single-inhaler triple therapy with FF/UMEC/VI provides similar overall improvements in weighted mean FEV1 and health status, and a similar safety profile, including a low incidence of pneumonia, as twice-daily multipleinhaler triple therapy with BUD/FOR+TIO. These results suggest that FF/UMEC/VI is a viable treatment option for patients who wish to simplify their treatment regimen from multiple- to single-inhaler triple therapy. The greater improvements with FF/UMEC/VI over BUD/FOR+TIO in trough FEV1 at 12 and 24 h further suggest that once-daily FF/UMEC/VI therapy may offer more consistent and sustained lung function benefits throughout the dosing interval compared with twice-daily BUD/FOR + once-daily TIO therapy. Personal remarks on study quality, conclusions, and other issues (potentially) relevant to the research question.
Risk of bias
|
Risk of bias tables
Risk of bias table for intervention studies (randomized controlled trials; based on Cochrane risk of bias tool and suggestions by the CLARITY Group at McMaster University)
Research question: 2 Triple therapie in 1 vs in meerdere devices bij COPD
|
Study reference
(first author, publication year) |
Was the allocation sequence adequately generated? a
Definitely yes Probably yes Probably no Definitely no |
Was the allocation adequately concealed?b
Definitely yes Probably yes Probably no Definitely no |
Blinding: Was knowledge of the allocated interventions adequately prevented?c
Were patients blinded?
Were healthcare providers blinded?
Were data collectors blinded?
Were outcome assessors blinded?
Were data analysts blinded?
Definitely yes Probably yes Probably no Definitely no |
Was loss to follow-up (missing outcome data) infrequent?d
Definitely yes Probably yes Probably no Definitely no |
Are reports of the study free of selective outcome reporting?e
Definitely yes Probably yes Probably no Definitely no |
Was the study apparently free of other problems that could put it at a risk of bias?f
Definitely yes Probably yes Probably no Definitely no |
Overall risk of bias If applicable/necessary, per outcome measureg
LOW Some concerns HIGH
|
|
Bremner, 2016 |
Definitely yes;
Reason: Eligible patients were randomized (1:1) to receive 24 weeks of FF/UMEC/VI 100 μg/62.5 μg/25 μg in a single inhaler and placebo (second inhaler) or FF/VI 100 μg/25 μg and UMEC 62.5 μg, in separate inhalers; all treatments/placebo were delivered using the ELLIPTA inhaler once daily in the morning. |
Definitely yes;
Reason: Randomization was stratified by the number of long-acting bronchodilators (0, 1, or 2) per day during the run-in. |
Definitely yes;
Reason: patients in both groups received their assigned study treatment using double-dummy blind inhalers, and so we were unable to directly assess the impact on adherence of a simplified single-inhaler triple therapy regimen versus the same treatments delivered using two inhalers.. |
Definitely yes;
Reason: Loss to follow-up was infrequent. |
Definitely yes;
Reason: Investigators who did the analyses were masked to group allocation. |
Definitely no;
Reason: Funding This study was funded by GSK (GSK study CTT200812; ClinicalTrials.gov identifier NCT02729051). ELLIPTA is owned by or licensed to the GSK group of companies. |
Some concerns
|
|
Ferguson, 2020 |
Definitely no;
Reason: Following run-in (Visit 2; Day 1 of study), an Interactive Web Response System was used to randomize patients 1:1 to receive FF/UMEC/VI or two inhalations of BUD/FOR plus TIO.
|
Definitely no;
Reason: Neither researchers nor staff involved was blinded to the study drug.
|
Definitely no;
Reason: Neither researchers nor staff involved was blinded to the study drug. However, dummy inhalers were used in these studies to ensure blinding and, as such, the studies were not designed to assess the impact of single or multiple inhalers nor once daily or twice-daily therapy on adherence. |
Probably yes
Reason: Loss to follow-up was infrequent. |
Definitely yes
Reason: All relevant outcomes were reported; |
Definitely no;
Reason: Funding The funders of the study had a role in the study design, data analysis, data interpretation, and writing of the report. ELLIPTA is owned by or licensed to the GSK Group of Companies. Patients at risk of non-compliance with study medication or attendance for scheduled visits, or unable to comply with the study procedures were excluded (subjective judgement). |
HIGH |
|
Vestbo, 2017 |
Definitely yes;
Reason: Patients were randomised to treatment by investigators contacting an interactive response technology (IRT) system, which used a randomisation list generated by the IRT provider. Randomisation was in the ratio 2:2:1 to the fixed triple group, tiotropium group, or open triple group. Randomisation was stratified by country and severity of airflow limitation (post-bronchodilator FEV1 <30% predicted, or 30–50% predicted, with a minimum of 20% of recruited patients to be <30% predicted). |
Definitely yes;
Reason: Patients were randomised to treatment by investigators contacting an interactive response technology (IRT) system, which used a randomisation list generated by the IRT provider. Randomisation was in the ratio 2:2:1 to the fixed triple group, tiotropium group, or open triple group. Randomisation was stratified by country and severity of airflow limitation (post-bronchodilator FEV1 <30% predicted, or 30–50% predicted, with a minimum of 20% of recruited patients to be <30% predicted). |
Definitely no;
Reason: Patients, investigators, site staff, and funder personnel were masked to treatment assignment for the duration of the study. To achieve this masking, a double-dummy approach was used, with all patients using a pMDI twice daily (containing BDP/FF/GB, BDP/FF, or placebo) and an SDDPI once daily (containing tiotropium or placebo). |
Probably yes
Reason: Loss to follow-up was infrequent. |
Definitely yes
Reason: All relevant outcomes were reported; |
Definitely no;
Reason: The funder was responsible for the design and analysis of the study, oversaw its conduct and was responsible for preparing the study report. All authors had full access to all of the data, with the lead author (JV) responsible for the decision to submit for publication. |
Some concerns |
- Randomization: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomization process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomization (performed at a site remote from trial location). Inadequate procedures are all procedures based on inadequate randomization procedures or open allocation schedules..
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments, but this should not affect the risk of bias judgement. Blinding of those assessing and collecting outcomes prevents that the knowledge of patient assignment influences the process of outcome assessment or data collection (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is usually not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary. Finally, data analysts should be blinded to patient assignment to prevents that knowledge of patient assignment influences data analysis.
- If the percentage of patients lost to follow-up or the percentage of missing outcome data is large, or differs between treatment groups, or the reasons for loss to follow-up or missing outcome data differ between treatment groups, bias is likely unless the proportion of missing outcomes compared with observed event risk is not enough to have an important impact on the intervention effect estimate or appropriate imputation methods have been used.
- Results of all predefined outcome measures should be reported; if the protocol is available (in publication or trial registry), then outcomes in the protocol and published report can be compared; if not, outcomes listed in the methods section of an article can be compared with those whose results are reported.
- Problems may include: a potential source of bias related to the specific study design used (e.g. lead-time bias or survivor bias); trial stopped early due to some data-dependent process (including formal stopping rules); relevant baseline imbalance between intervention groups; claims of fraudulent behavior; deviations from intention-to-treat (ITT) analysis; (the role of the) funding body. Note: The principles of an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
- Overall judgement of risk of bias per study and per outcome measure, including predicted direction of bias (e.g. favors experimental, or favors comparator). Note: the decision to downgrade the certainty of the evidence for a particular outcome measure is taken based on the body of evidence, i.e. considering potential bias and its impact on the certainty of the evidence in all included studies reporting on the outcome.
Verantwoording
Beoordelingsdatum en geldigheid
Publicatiedatum : 15-09-2023
Beoordeeld op geldigheid : 31-08-2023
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen (zie hiervoor de Samenstelling van de werkgroep) die betrokken zijn bij de zorg voor patiënten met COPD.
Samenstelling van de werkgroep
Werkgroep
- Dr. F. (Folkert) Brijker, longarts, werkzaam in het Spaarne Gasthuis te Haarlem, NVALT (voorzitter, vanaf oktober 2022)
- Dr. J.S. (Jaring) van der Zee, longarts, NVALT (voorzitter, tot oktober 2022)
- Dr. W.H. (Wouter) van Geffen, longarts, werkzaam in het Medisch Centrum Leeuwarden te Leeuwarden, NVALT
- Drs. R. (Renée) van Snippenburg, werkzaam bij Ksyos en waarnemend longarts, NVALT
- Dr. J.C.C.M. (Hans) in ’t Veen, longarts, werkzaam in het Franciscus Gasthuis & Vlietland te Rotterdam, NVALT
- M. (Moniek) Wouters, longarts, werkzaam in het Ziekenhuis Gelderse Vallei te Arnhem, NVALT
- Prof. H.A.M. (Huib) Kerstjens, hoogleraar longziekten, longarts, werkzaam in het UMCG te Groningen, NVALT (vanaf oktober 2022)
- J. (Jeanine) Antons, longarts, werkzaam in het RadboudUMC te Nijmegen, NVALT (vanaf oktober 2022)
- Drs C.L.Y. (Chantal) Knoops, AIOS longgeneeskunde, werkzaam in het Catharina Ziekenhuis te Eindhoven (vanaf oktober 2022)
- Prof. J.W.M. (Jean) Muris, huisarts, werkzaam bij de Universiteit Maastricht, lid van de NHG-Expertgroep CAHAG, NHG
- Drs. E.R. (Erik) van der Meijs, apotheker, KNMP
- W.J.M. (Walter) van Litsenburg, verpleegkundig specialist longgeneeskunde, Catharina Ziekenhuis te Eindhoven, V&VN
- Dr. M.J.H. (Maurice) Sillen, fysiotherapeut, werkzaam bij CIRO, KNGF
- R.A. (Renée) Kool, projectleider, Longfonds
- R. (Ramona) Leysner, diëtiste, Nederlandse Vereniging van Diëtisten (NVD)
- Drs. M. (Menno) Wagenaar, patiëntvertegenwoordiger, Longfonds
- J. (Johan) Smit, patiëntvertegenwoordiger, Longfonds
Met ondersteuning van
- Dr. M. (Margriet) Moret, senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Drs. N. (Nicole) Verheijen, senior adviseur, Kennisinstituut van de Federatie Medisch Specialisten en longarts
- Dr. T. (Tim) Christen, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Inbreng patiëntenperspectief
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Brijker (voorzitter, vanaf oktober 2022) |
Longarts Spaarne Gasthuis |
Voorzitter sectie COPD NVALT. Dit is een onbetaalde functie binnen de longartsenvereniging NVALT. Lid regionale kwaliteitscommissie COPD/astma. Deze Commissie heeft 2x per jaar vergadering a 2 uur per keer in de avonduren en hiervoor ontvang ik onkostenvergoeding. Docent CASPIR cursussen. dit betreft scholing voor spirometrie voor huisartsen en POH-ers in de regio. Dit vindt een aantal keer per jaar plaats (<5 keer) in de avonduren en hiervoor ontvang ik een onkostenvergoeding |
Geen |
Geen |
|
*Van der Zee (voorzitter, tot oktober 2022) |
Longarts OLVG Amsterdam 0,2FTE tot 1-1-2020 Longarts Amsterdam UMC, locatie AMC, 0,2 FTE |
Lid MEC-U Locatie Nieuwegein, onkosten vergoeding Lid Gezondheidsraad Commissie Gespoten PUR, onkosten vergoeding Incidenteel medische expertises (o.a. DAS, ARAG, Triage, de Rechtspraak), betaald 2019 Speakers fee, Astra-Zeneca, Novartis, Chiesi 2019 1x Ad hoc Advies m.b.t. biologicals bij astma, GSK, betaald |
Geen |
Geen advieswerk tijdens het richtlijnontwikkeltraject |
|
Van Geffen |
Longarts Medisch Centrum Leeuwarden, maatschap Friese Longartsen |
Editorial board Cochrane Airways: Onbetaald Commisie Bronkhorst Nvalt: Onbetaald Richtlijn Commissie NVALT NSCLC: Onbetaald |
Deelname aan een investigator initiated onderzoek firma Novartis. financiering is overgemaakt aan UMCG (2017 beëindigd). Voor de bedrijven Chiesi, Roche, Boehringer en AstraZeneca deelname aan adviesraden betreffende oncologie. Deze gingen niet over COPD of biologicals. De hiervoor gebruikelijke CGR vergoeding werd geweigerd. Chiesi en Boehringer waren wel COPD, maar niet in de laatste 1.5 jaar. |
Geen advieswerk op gebied van COPD of biologicals tijdens het richtlijnontwikkeltraject |
|
In 't Veen |
Longarts bij In 't Veen Longarts BV. Verbonden aan de vakgroep longziekten en STZ expertisecentrum Astma, COPD & Respiratoire Allergie van het Franciscus Gasthuis en Vlietland, Rotterdam. |
Onbetaald: Opleider longziekten Franciscus Gasthuis en Vlietland Lid Concilium Opleiding NVALT Lid Vrij Ademen Akkoord namens NVALT Betrokken longarts bij Schone Lucht Akkoord Lid Move2Improve Lid werkgroep Ziektelastmeter; Generiek en COPD (afgerond) Voorzitter StichtingRoLeX (Rotterdam Leeuwarden eXpertise voor obstructieve longzieken), een stichting die nascholing voor longartsen (i.o.) verzorgd. Bestuurslid LAN Betaald: Longfunctiebeoordelaar Huisartsenlaboratorium STAR-SHL NHG richtlijn COPD namens NVALT Adviseurschap m.b.t. astma: Sanofi, GSK, Boehringer Ingelheim, Chiesi. (laatste 2 jaar (datum invullen 30-6-19) geen persoonlijke betrokkenhied als adviseur bij COPD gerelateerde issues, mede vanwege mijn betrokkenheid bij de NHG richtlijn). Speakers Bureau: Chiesie, Novartis, Boehringer Ingelheim, Inhalatie Technologie Werkgroep Health Agency Stichting RoLeX Sanofi |
Ik beoordeel longfuncties voor een huisarts laboratorium, en heb adviseurschap verricht voor diverse farmaceutische firma's. Er is nooit advies gegeven door mij over medicamenteuze COPD-behandeling, ook niet over biologicals. het genoemde adviseurschap is inmiddels meer dan 3-4 jaar geleden beëindigd. Zie eerder. Research faculty grants, (subsidiegevers Boehringer, Chiesi, Teva, Franciscus wetenschapsbureau) m.b.t. onderzoek bij astma en COPD, via ons expertisecentrum. Van belang hierbij is dat al het onderzoek niet medicatie-gerelateerd is. Zie boven bij onbetaald: Ik ben betrokken bij de bevordering van luchtkwaliteit en als zodanig word ik af en toe geconsulteerd met betrekking tot het schone Lucht Akkoord, een nationaal (door de overheid in gang gezet) platform dat maatregelen hierover in kaart brengt. Voorts ben ik betrokken bij het Vrij Ademen Akkoord, dat oa vanuit LAN, Longfonds, NRS en NVALT aandacht vraagt voor de (toekomstige) patient met een longziekte. |
Geen advieswerk over COPD of biologicals tijdens ontwikkeltraject van de richtlijn. Geen uitwerking van uitgangsvragen over longmedicatie of biologicals. |
|
Van Snippenburg |
Waarnemend Longarts; |
Secretaris Sectie COPD NVALT, onbetaald Werkgroep longen Huisartsen Utrecht Stad, betaald
|
Geen |
Geen |
|
Antons (vanaf oktober 2022) |
Longarts Radboudumc, Nijmegen |
Geen |
Geen |
Geen |
|
Kerstjens (vanaf oktober 2022) |
Hoogleraar longziekten UMCG, 1,0 FTE |
"Voorzitter Noordelijke CARA Stichting. Subsidiegevend orgaan. Onbetaald - Lid RvT bureau bijwerkingen geneesmiddelen LAREB. Betaald aan UMCG - Vz Stichting BEBO. Onafhankelijke METc. Betaald aan UMCG (per 1-1-2023 vz) - Vice-vz Netherlands Respiratory Society. Stichting ter bevordering van wetenschap en wetenschapsklimaat Longziekten NL. Onbetaald. (per 1-1-2023 vz).
Op afroep (geen vaste contracten of afspraken) deelname aan adviesraden van farmaceutische industrieën, en betaling voor lezingen: AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Novartis. Alles betaald aan UMCG." |
"Geen persoonlijk financieel belang; alles wat er door mij binnenkomt wordt betaald aan UMCG. En krijg ik ook in tweede instantie nooit wat van. 2. Geen dienstverband 3. Betaald adviseurschappen zie bij overige item over nevenwerkzaamheden voor AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Novartis. 4. Geen directe fianicee belangen of via aandelen of opties. 5. Geen patenten"
"Veel gesponsord onderzoek, o.a. ZonMW VWS Innovatiefonds verzekeraars Industrie: AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Novartis." |
restricties ten aanzien van besluitvorming met betrekking tot modules over medicatie |
|
Knoops (vanaf oktober 2022) |
AIOS longziekten, Catharina Ziekenhuis Eindhoven |
Geen |
Geen |
Geen |
|
Tazmi (tot oktober 2020) |
Verpleegkundig specialist - werkzaam bij Laurens locatie Intermezzo |
Geen |
Geen |
Geen |
|
Van Jaarsveld |
Adviseur Zorg bij Longfonds |
Geen |
Geen |
Geen |
|
Muris |
Hoogleraar huisartsgeneeskunde, Universiteit Maastricht |
Vervangende werkzaamheden huisartspraktijk Geulle (betaald) |
Geen |
Geen |
|
Van der Meijs |
Apotheker, sinds 1 februari niet meer praktiserend lid namens de KNMP |
SIG-long (SIG = specialist interest group) van KNMP – vacatiegeld |
Geen |
Geen |
|
Van Litsenburg (vanaf oktober 2020) |
Verpleegkundig specialist astma en COPD 36 uur per week 24-uurs thuiszorgverpleegkundige 24 uur per week |
Bestuurslid IMIS (inhalatiemedicatie instructie school) 2u per week Coördinator IMIS Zuid Nederland IMIS trainer Docent Hogeschool Arnhem en Nijmegen Kernteam Picasso voor COPD (momenteel niet actief) Werkgroeplid palliatieve richtlijn COPD |
Kernteamlid Picasso (niet actief) |
Geen |
|
Leysner |
dietist Merem medische revalidatie in Hilversum" |
Incidenteel scholing geven aan studenten hogeschool Holland; betaald |
Geen |
Geen |
|
Wouters |
Bij aanvang AIOS longziekten, Rijnstate Ziekenhuis en thans longarts Ziekenhuis Gelderse Vallei Ede |
Geen |
Geen |
Geen |
|
Wagenaar |
Longervaringsdeskundige bij het Longfonds Geen betaalde functies |
Geen |
Geen |
Geen |
|
Kool |
Projectleider Zorgveld, Longfonds |
Geen |
Geen |
Geen |
|
Sillen |
Resultaatverantwoordelijk fysiotherapeut CIRO+, expertisecentrum voor chronisch orgaanfalen Horn |
Bestuurslid Vereniging voor Hart-, Vaat- en Longfysiotherapie (vacatievergoeding) Extern adviseur Fontys Hogeschool Eindhoven (betaald) Gastdocent Saxion Hogeschool, Enschede en Hogeschool van Amsterdam (betaald) |
Geen |
Geen |
|
Smit |
Longervaringsdeskundige Longfonds (vrijwilligerswerk) |
Geen |
Geen |
Geen |
Implementatie
Er werd aandacht besteed aan het patiëntenperspectief doordat een afgevaardigde patiëntenvereniging zitting nam in de werkgroep. De inbreng van de afgevaardigden is tevens verwerkt in de verslagen hiervan. De verkregen input is meegenomen bij het opstellen van de uitgangsvragen, de keuze voor de uitkomstmaten en bij het opstellen van de overwegingen. De conceptrichtlijn is tevens voor commentaar voorgelegd aan het Longfonds en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de voorbereidende fase inventariseerden de werkgroep de knelpunten in de zorg voor patiënten met COPD. De werkgroep beoordeelde de aanbevelingen uit de eerdere richtlijn (NVALT, 2010) op noodzaak tot revisie. Tevens zijn er knelpunten aangedragen door de NVALT, V&VN Longverpleegkundigen, het Longfonds, NHG, NAPA en KNMP via een invitational conference. Een verslag hiervan is opgenomen onder aanverwante producten.
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
De werkgroep heeft uitgebreid over de belangrijkste uitkomstmaten voor de te bespreken interventies gediscussieerd. Het werd wenselijk geacht de set uitkomsten over de verschillende modules waar mogelijk hetzelfde te houden, en zeker met de zelfde minimale klinisch relevante verschillen. Bij specifieke interventies werden soms specifieke relevante uitkomsten toegevoegd. Voor sommige parameters kon geen referentie gevonden worden en werd als onderstaand beschreven een consensus standpunt ingenomen. De werkgroep onderkent dat de absolute risico reductie klinisch beter interpretabel is dan de relatieve, maar kon voor onvoldoende studies die getallen er bij vinden en hield dus vast aan de relatieve risico’s, met toelichting waar mogelijk.
• Exacerbation reduction: ≥20% (Jones, 2014; Chapman 2013)
• Pneumonia: ≥20% difference in relative risk
• Hospital admissions: ≥20% difference in relative risk
• Mortality: ≥10% difference in relative risk
• SGRQ-score: ≥4 units (Jones, 2014)
• CAT-score: >2 units (Kon 2014)
• CCQ-score: >0.4 units
• FEV1: >100 ml (Donohue 2005; Jones, 2014)
• Adverse events: ≥25% difference in relative risk
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/) De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
Definitie |
|
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE-methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Kwalitatieve raming van mogelijke financiële gevolgen in het kader van de Wkkgz
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie het stroomschema op de Richtlijnendatabase).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
|
Module |
Uitkomst kwalitatieve raming |
Toelichting |
|
Module 1.1 Gebruik van inhalatiecorticosteroïden (ICS) bij COPD |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar is (>40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 1.2 Triple therapie in 1 vs in meerdere devices bij COPD |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar is (>40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 1.3 Onderhoudsantibiotica bij COPD
|
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 1.4 Poliklinische behandeling van een COPD-longaanval |
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar is (>40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 1.5 Biologicals bij COPD |
Geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbeveling(en) niet breed toepasbaar zijn (<5.000 patiënten) en zal daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven
|
|
Module 2.1 Fysiotherapie in de eerste lijn
|
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 2.2 tweede en derdelijnsrevalidatie bij COPD
|
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (5.000-40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 2.3 Voeding bij COPD
|
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar is (>40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 3.1 Zuurstoftherapie bij COPD
|
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar is (>40.000 patiënten), volgt ook uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 3.2 Longvolumereductie |
geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbeveling(en) niet breed toepasbaar zijn (<5.000 patiënten) en zal daarom naar verwachting geen substantiële financiële gevolgen hebben voor de collectieve uitgaven.
|
|
Module 3.3 Chronische beademing bij COPD
|
n.v.t. |
Verwijzing naar andere richtlijn |
|
Module 4 COPD zelfmanagement
|
Geen substantiële financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (>40.000 patiënten), volgt ook uit de toetsing het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. Er worden daarom geen substantiële financiële gevolgen verwacht.
|
|
Module 5 E-health
|
Geen substantiële financiële gevolgen |
Uit de toetsing volgt dat de aanbeveling(en) breed toepasbaar zijn (>40.000 patiënten) en dat het een nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft. Het inzetten van digitale zorgtoepassingen zal zijn georganiseerd op lokaal/regionaal niveau en daarom worden op landelijk niveau geen substantiële financiële gevolgen verwacht.
|
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Chapman KR, Bergeron C, Bhutani M, et al. Do we know the minimal clinically important
difference (MCID) for COPD exacerbations? COPD 2013;10(2):243-9.
DOI:10.3109/15412555.2012.733463.
Donohue JF. Minimal clinically important differences in COPD lung function. COPD
2005;2(1):111-124. (http://www.ncbi.nlm.nih.gov/pubmed/17136971).
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Jones PW, Beeh KM, Chapman KR, Decramer M, Mahler DA, Wedzicha JA. Minimal clinically
important differences in pharmacological trials. Am J Respir Crit Care Med
2014;189(3):250-5. DOI: 10.1164/rccm.201310-1863PP.
Kon SS, Canavan JL, Jones SE, et al. Minimum clinically important difference for the COPD
Assessment Test: a prospective analysis. Lancet Respir Med 2014;2(3):195-203. DOI:
10.1016/S2213-2600(14)70001-3.
Kocks JW, Tuinenga MG, Uil SM, van den Berg JW, Stahl E, van der Molen T. Health status
measurement in COPD: the minimal clinically important difference of the Clinical
COPD Questionnaire. Respir Res 2006;7(1):62.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. http://richtlijnendatabase.nl/over_deze_site/over_richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, Glasziou P, Jaeschke R, Vist GE, Williams JW Jr, Kunz R, Craig J, Montori VM, Bossuyt P, Guyatt GH; GRADE Working Group. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008 May 17;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008 May 24;336(7654). doi: 10.1136/bmj.a139.
Schünemann, A Holger J (corrected to Schünemann, Holger J). PubMed PMID: 18483053; PubMed Central PMCID: PMC2386626.
Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
Zoekverantwoording
Table of excluded studies
|
Author and year |
Reason for exclusion |
|
Baldi, 2020 |
does not fit PICO |
|
Bogart, 2020 |
poster discussie |
|
Canonica, 2019 |
does not fit PICO |
|
Compton, 2020 |
congress abstract |
|
Ferguson, 2020 |
poster discussie |
|
Ferguson, 2020 |
poster discussie |
|
Fergusion, 2020 |
Poster discussie |
|
Gessner, 2020 |
narrative review |
|
Goodall, 2018 |
voor/na vergelijking, geen controlegroep |
|
Halpin, 2020 |
poster discussie |
|
Hanania, 2020 |
poster discussie |
|
Harada, 2020 |
poster discussie |
|
Hoevelmann, 2020 |
poster discussie |
|
Scuri, 2017 |
poster discussie |
|
Scuri, 2017 |
poster discussie |
|
Shah, 2018 |
abstract ERS congress |
|
Singh, 2017 |
late breaking abstract |
|
Worsley, 2019 |
zelfde? |
|
Aziz, 2018 |
does not fit PICO |
|
Balmaceda, 2020 |
poster discussie |
|
Beeh, 2021 |
does not fit PICO |
|
Calzetta, 2020 |
congress abstract |
|
Georges, 2019 |
poster discussie |
|
Hövelmann, 2018 |
poster discussie |
|
Montuschi, 2016 |
narrative review |
|
Pelaia, 2020 |
voor/na vergelijking, geen controlegroep |
|
Rothnie, 2020 |
poster discussie |
|
Schroeder, 2019 |
poster discussie |
|
Schroeder, 2018 |
poster discussie |
|
Scuri, 2018 |
poster discussie |
|
Scuri, 2017 |
poster discussie |
|
Singh, 2020 |
poster discussie |
|
Singh, 2017 |
abstract ERS congress |
|
Singh, 2020 |
late breaking abstract |
|
Singh, 2017 |
zelfde? |
|
Singh, 2018 |
does not fit PICO |
|
Singh, 2018 |
narrative review |
|
Singh, 2019 |
Review, but describes all already included RCTs. |
|
Tan, 2018 |
poster discussie |
|
Topole, 2019 |
late breaking abstract |
|
Vanfleteren, 2018 |
narrative review |
|
Halpin, 2019 |
poster discussie |
|
Hovelmann, 2019 |
poster discussie |
|
Ismalia, 2017 |
past niet in PICO (geen vergelijking met kosten van triple therapie in verschillende devices) |
|
Ismalia, 2019 |
past niet in PICO (geen vergelijking met kosten van triple therapie in verschillende devices) |
|
Papi, 2019 |
poster discussie |
Literature search strategy
Zoekverantwoording
Algemene informatie
|
Richtlijn: NVALT Herziening COPD |
|
|
Uitgangsvraag: UV2 triple therapy one device |
|
|
Database(s): Embase, Ovid/Medline |
Datum: 26-1-2021 |
|
Periode: 2015- |
Talen: nvt |
|
Literatuurspecialist: Ingeborg van Dusseldorp |
|
|
BMI zoekblokken: voor verschillende opdrachten wordt (deels) gebruik gemaakt van de zoekblokken van BMI-Online https://blocks.bmi-online.nl/ Bij gebruikmaking van een volledig zoekblok zal naar de betreffende link op de website worden verwezen. |
|
|
Toelichting en opmerkingen:
Voor deze vraag is met de volgende elementen gezocht:
(Beclomathson AND formetorol AND glycopyroonium) OR (fluticason AND vilanterol AND umeclidinium) OR (Mometason AND indacaterol AND glypyrronium) OR (trimbow OR trelegy ellipta OR enerzair)
Alle sleutelartikelen worden gevonden. Normaliter worden conference abstracts achterwege gelaten. In deze zoekopdracht zijn ze meegenomen en worden ze apart getoond. Niet zozeer ten behoeve van de selectie maar meer vanwege de recente ontwikkelingen.
De sleutelartikelen worden gevonden.
|
|
|
Te gebruiken voor richtlijnen tekst: In de databases Embase en Ovid/Medline is op 26 januari 2021 met relevante zoektermen gezocht naar SRs, RCTs en observationele studies over triple therapie in 1 inhaler. De literatuurzoekactie leverde193 unieke treffers op. |
|
Zoekopbrengst
|
|
EMBASE |
OVID/MEDLINE |
Ontdubbeld |
|
SRs |
31 |
6 |
35 |
|
RCTs |
110 |
81 |
129 |
|
Observationele studies |
30 |
6 |
29 |
|
Totaal |
|
|
193 |
|
Conference abstracts |
|
|
203 |
Zoekstrategie
Embase
|
No. |
Query |
Results |
|
#36 |
#30 AND #35 sleutelartikel |
1 |
|
#35 |
plus AND umeclidinium AND using AND two AND inhalers AND for AND chronic AND obstructive AND pulmonary AND disease AND bremner |
1 |
|
#34 |
#30 AND #33 sleutelartikel |
1 |
|
#33 |
'once daily' AND 'single inhaler' AND versus AND 'twice daily' AND 'multiple inhaler' AND triple AND therapy AND in AND patients AND with AND copd AND 2020 |
2 |
|
#31 |
#20 AND (1-1-2015)/sd NOT ('editorial'/it OR 'letter'/it OR 'note'/it) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) AND 'conference abstract'/it, Conference abstracts |
205 |
|
#30 |
#25 OR #26 OR #27 |
171 |
|
#29 |
#27 NOT #26 NOT #25 Observationee; |
30 |
|
#28 |
#26 NOT #25 RCT |
110 |
|
#27 |
#21 AND #24 |
98 |
|
#26 |
#21 AND #23 |
136 |
|
#25 |
#21 AND #22 SR |
31 |
|
#24 |
'major clinical study'/de OR 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR 'prospective study'/de OR 'comparative study'/de OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (('case control' NEAR/1 (study OR studies)):ab,ti) OR (('follow up' NEAR/1 (study OR studies)):ab,ti) OR (observational NEAR/1 (study OR studies)) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti) |
6318929 |
|
#23 |
('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it |
2500470 |
|
#22 |
('meta analysis'/exp OR 'meta analysis (topic)'/exp OR metaanaly*:ti,ab OR 'meta analy*':ti,ab OR metanaly*:ti,ab OR 'systematic review'/de OR 'cochrane database of systematic reviews'/jt OR prisma:ti,ab OR prospero:ti,ab OR (((systemati* OR scoping OR umbrella OR 'structured literature') NEAR/3 (review* OR overview*)):ti,ab) OR ((systemic* NEAR/1 review*):ti,ab) OR (((systemati* OR literature OR database* OR 'data base*') NEAR/10 search*):ti,ab) OR (((structured OR comprehensive* OR systemic*) NEAR/3 search*):ti,ab) OR (((literature NEAR/3 review*):ti,ab) AND (search*:ti,ab OR database*:ti,ab OR 'data base*':ti,ab)) OR (('data extraction':ti,ab OR 'data source*':ti,ab) AND 'study selection':ti,ab) OR ('search strategy':ti,ab AND 'selection criteria':ti,ab) OR ('data source*':ti,ab AND 'data synthesis':ti,ab) OR medline:ab OR pubmed:ab OR embase:ab OR cochrane:ab OR (((critical OR rapid) NEAR/2 (review* OR overview* OR synthes*)):ti) OR ((((critical* OR rapid*) NEAR/3 (review* OR overview* OR synthes*)):ab) AND (search*:ab OR database*:ab OR 'data base*':ab)) OR metasynthes*:ti,ab OR 'meta synthes*':ti,ab) NOT (('animal'/exp OR 'animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) NOT ('conference abstract'/it OR 'conference review'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) |
533576 |
|
#21 |
#20 AND (1-1-2015)/sd NOT ('conference abstract'/it OR 'editorial'/it OR 'letter'/it OR 'note'/it) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) |
246 |
|
#20 |
#4 OR #11 OR #18 OR #19 |
544 |
|
#19 |
'beclometasone dipropionate plus formoterol fumarate plus glycopyrronium bromide'/exp OR 'chf 5993':ti,ab,kw OR 'chf5993':ti,ab,kw OR 'riarify':ti,ab,kw OR 'trimbow':ti,ab,kw OR 'trydonis':ti,ab,kw OR 'fluticasone furoate plus umeclidinium plus vilanterol'/exp OR 'elebrato ellipta':ti,ab,kw OR 'temybric ellipta':ti,ab,kw OR 'trelegy ellipta':ti,ab,kw OR 'glycopyrronium bromide plus indacaterol plus mometasone furoate'/exp OR 'enerzair':ti,ab,kw OR 'qvm 149':ti,ab,kw OR 'qvm149':ti,ab,kw OR 'zimbus':ti,ab,kw |
244 |
|
#18 |
#12 AND #17 |
65 |
|
#17 |
#15 OR #16 |
950 |
|
#16 |
#13 AND #14 |
780 |
|
#15 |
'glycopyrronium bromide plus indacaterol'/exp OR 'qva 149':ti,ab,kw OR 'qva 149a':ti,ab,kw OR 'qva149':ti,ab,kw OR 'qva149a':ti,ab,kw OR 'ultibro':ti,ab,kw OR 'utibron':ti,ab,kw OR 'utibron neohaler':ti,ab,kw OR 'xoterna':ti,ab,kw |
520 |
|
#14 |
'glycopyrronium'/exp OR 'ad 237':ti,ab,kw,tn,dn OR 'ad237':ti,ab,kw,tn,dn OR 'ahr 504':ti,ab,kw,tn,dn OR 'ahr504':ti,ab,kw,tn,dn OR 'asecryl':ti,ab,kw,tn,dn OR 'cuvposa':ti,ab,kw,tn,dn OR 'drm 04':ti,ab,kw,tn,dn OR 'drm04':ti,ab,kw,tn,dn OR 'enurev':ti,ab,kw,tn,dn OR 'gastrodyn':ti,ab,kw,tn,dn OR 'glersa':ti,ab,kw,tn,dn OR 'glycopyrrolate':ti,ab,kw,tn,dn OR 'glycopyrronium':ti,ab,kw,tn,dn OR 'glyrx-pf':ti,ab,kw,tn,dn OR 'lonhala magnair':ti,ab,kw,tn,dn OR 'mobinul':ti,ab,kw,tn,dn OR 'nodapton':ti,ab,kw,tn,dn OR 'nva 237':ti,ab,kw,tn,dn OR 'nva237':ti,ab,kw,tn,dn OR 'robinal':ti,ab,kw,tn,dn OR 'robinol':ti,ab,kw,tn,dn OR 'robinul':ti,ab,kw,tn,dn OR 'seebri':ti,ab,kw,tn,dn OR 'sialanar':ti,ab,kw,tn,dn OR 'sroton':ti,ab,kw,tn,dn OR 'strodin':ti,ab,kw,tn,dn OR 'tarodyl':ti,ab,kw,tn,dn OR 'tarodyn':ti,ab,kw,tn,dn OR 'tovanor':ti,ab,kw,tn,dn |
7538 |
|
#13 |
'indacaterol'/exp OR 'arcapta':ti,ab,kw,tn,dn OR 'hirobriz breezhaler':ti,ab,kw,tn,dn OR 'indacaterol':ti,ab,kw,tn,dn OR 'onbrez':ti,ab,kw,tn,dn OR 'oslif breezhaler':ti,ab,kw,tn,dn OR 'qab 149':ti,ab,kw,tn,dn OR 'qab149':ti,ab,kw,tn,dn |
1714 |
|
#12 |
'mometasone furoate'/exp OR 'allermax aqueous':ti,ab,kw,tn,dn OR 'asmanex':ti,ab,kw,tn,dn OR 'belloseta':ti,ab,kw,tn,dn OR 'bloctimo':ti,ab,kw,tn,dn OR 'breso':ti,ab,kw,tn,dn OR 'brusonex':ti,ab,kw,tn,dn OR 'cutticom':ti,ab,kw,tn,dn OR 'dermotasone':ti,ab,kw,tn,dn OR 'dermovel':ti,ab,kw,tn,dn OR 'desdek':ti,ab,kw,tn,dn OR 'ecural':ti,ab,kw,tn,dn OR 'edelan':ti,ab,kw,tn,dn OR 'elica':ti,ab,kw,tn,dn OR 'elocom':ti,ab,kw,tn,dn OR 'elocon':ti,ab,kw,tn,dn OR 'elocyn':ti,ab,kw,tn,dn OR 'elomet':ti,ab,kw,tn,dn OR 'eloson':ti,ab,kw,tn,dn OR 'flumeta':ti,ab,kw,tn,dn OR 'frondava':ti,ab,kw,tn,dn OR 'ivoxel':ti,ab,kw,tn,dn OR 'kalmente':ti,ab,kw,tn,dn OR 'lorome':ti,ab,kw,tn,dn OR 'mefurosan':ti,ab,kw,tn,dn OR 'metaspray':ti,ab,kw,tn,dn OR 'metmin':ti,ab,kw,tn,dn OR 'mipasu':ti,ab,kw,tn,dn OR 'mofunder':ti,ab,kw,tn,dn OR 'momate':ti,ab,kw,tn,dn OR 'momeallerg':ti,ab,kw,tn,dn OR 'momecutan':ti,ab,kw,tn,dn OR 'momederm':ti,ab,kw,tn,dn OR 'momegalen':ti,ab,kw,tn,dn OR 'momekort':ti,ab,kw,tn,dn OR 'momesonex':ti,ab,kw,tn,dn OR 'momespir':ti,ab,kw,tn,dn OR 'momester':ti,ab,kw,tn,dn OR 'mometahexal':ti,ab,kw,tn,dn OR 'mometason':ti,ab,kw,tn,dn OR 'mometasone 17 furoate':ti,ab,kw,tn,dn OR 'mometasone furoate':ti,ab,kw,tn,dn OR 'mometop':ti,ab,kw,tn,dn OR 'mommox':ti,ab,kw,tn,dn OR 'monovel':ti,ab,kw,tn,dn OR 'monovo':ti,ab,kw,tn,dn OR 'morecort':ti,ab,kw,tn,dn OR 'motaderm':ti,ab,kw,tn,dn OR 'mundoson':ti,ab,kw,tn,dn OR 'muofuder':ti,ab,kw,tn,dn OR 'nasehaler':ti,ab,kw,tn,dn OR 'nasoaldo':ti,ab,kw,tn,dn OR 'nasomet':ti,ab,kw,tn,dn OR 'nasometin':ti,ab,kw,tn,dn OR 'nasonex':ti,ab,kw,tn,dn OR 'nasotasone':ti,ab,kw,tn,dn OR 'netonox':ti,ab,kw,tn,dn OR 'novasone cream':ti,ab,kw,tn,dn OR 'novasone lotion':ti,ab,kw,tn,dn OR 'novasone ointment':ti,ab,kw,tn,dn OR 'ovison':ti,ab,kw,tn,dn OR 'ovixan':ti,ab,kw,tn,dn OR 'pronasal':ti,ab,kw,tn,dn OR 'rinelon':ti,ab,kw,tn,dn OR 'rinometasone':ti,ab,kw,tn,dn OR 'rivelon':ti,ab,kw,tn,dn OR 'sch 32088':ti,ab,kw,tn,dn OR 'sch32088':ti,ab,kw,tn,dn OR 'sebanez':ti,ab,kw,tn,dn OR 'sinuva':ti,ab,kw,tn,dn OR 'sonalox':ti,ab,kw,tn,dn OR 'temon':ti,ab,kw,tn,dn OR 'uniclar':ti,ab,kw,tn,dn OR 'zhekort':ti,ab,kw,tn,dn |
5548 |
|
#11 |
#5 AND #10 |
305 |
|
#10 |
#8 OR #9 |
721 |
|
#9 |
#6 AND #7 |
588 |
|
#8 |
'umeclidinium plus vilanterol'/exp OR 'laventair':ti,ab,kw OR 'anoro':ti,ab,kw |
389 |
|
#7 |
'umeclidinium'/exp OR 'ellipta':ti,ab,kw OR 'gsk 573719':ti,ab,kw OR 'gsk 573719a':ti,ab,kw OR 'gsk-573719':ti,ab,kw OR 'gsk-573719a':ti,ab,kw OR 'gsk573719':ti,ab,kw OR 'gsk573719a':ti,ab,kw OR 'incruse':ti,ab,kw OR 'rolufta':ti,ab,kw OR 'umeclidinium':ti,ab,kw |
930 |
|
#6 |
'vilanterol'/exp OR 'gsk 642444':ti,ab,kw OR 'gsk642444':ti,ab,kw OR 'gw 642444':ti,ab,kw OR 'gw642444':ti,ab,kw OR 'vilanterol':ti,ab,kw |
1107 |
|
#5 |
'fluticasone'/exp OR 'fluticasone':ti,ab,kw |
13024 |
|
#4 |
#1 AND #2 AND #3 |
148 |
|
#3 |
'glycopyrronium'/exp OR 'ad 237':ti,ab,kw,tn,dn OR 'ad237':ti,ab,kw,tn,dn OR 'ahr 504':ti,ab,kw,tn,dn OR 'ahr504':ti,ab,kw,tn,dn OR 'asecryl':ti,ab,kw,tn,dn OR 'cuvposa':ti,ab,kw,tn,dn OR 'drm 04':ti,ab,kw,tn,dn OR 'drm04':ti,ab,kw,tn,dn OR 'enurev':ti,ab,kw,tn,dn OR 'gastrodyn':ti,ab,kw,tn,dn OR 'glersa':ti,ab,kw,tn,dn OR 'glycopyrrolate':ti,ab,kw,tn,dn OR 'glycopyrronium':ti,ab,kw,tn,dn OR 'glyrx-pf':ti,ab,kw,tn,dn OR 'lonhala magnair':ti,ab,kw,tn,dn OR 'mobinul':ti,ab,kw,tn,dn OR 'nodapton':ti,ab,kw,tn,dn OR 'nva 237':ti,ab,kw,tn,dn OR 'nva237':ti,ab,kw,tn,dn OR 'robinal':ti,ab,kw,tn,dn OR 'robinol':ti,ab,kw,tn,dn OR 'robinul':ti,ab,kw,tn,dn OR 'seebri':ti,ab,kw,tn,dn OR 'sialanar':ti,ab,kw,tn,dn OR 'sroton':ti,ab,kw,tn,dn OR 'strodin':ti,ab,kw,tn,dn OR 'tarodyl':ti,ab,kw,tn,dn OR 'tarodyn':ti,ab,kw,tn,dn OR 'tovanor':ti,ab,kw,tn,dn |
7538 |
|
#2 |
'formoterol'/exp OR 'eformoterol':ti,ab,kw OR 'formoterol':ti,ab,kw |
7795 |
|
#1 |
'beclometasone'/exp OR 'beclometasone':ti,ab,kw OR 'beclomethasone':ti,ab,kw |
10269 |
Ovid/Medline
|
# |
Searches |
Results |
|
24 |
22 not 21 not 20 OBS |
6 |
|
23 |
21 not 20 RCT |
81 |
|
22 |
17 and 19 |
18 |
|
21 |
16 and 19 |
86 |
|
20 |
15 and 19 SR |
6 |
|
19 |
18 not ((exp animals/ or exp models, animal/) not humans/) not (letter/ or comment/ or editorial/) |
128 |
|
18 |
limit 14 to dt="20150101-20220101" |
134 |
|
17 |
Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or cohort.tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ (Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies) |
3753572 |
|
16 |
(exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) |
2084298 |
|
15 |
(meta-analysis/ or meta-analysis as topic/ or (metaanaly* or meta-analy* or metanaly*).ti,ab,kf. or systematic review/ or cochrane.jw. or (prisma or prospero).ti,ab,kf. or ((systemati* or scoping or umbrella or "structured literature") adj3 (review* or overview*)).ti,ab,kf. or (systemic* adj1 review*).ti,ab,kf. or ((systemati* or literature or database* or data-base*) adj10 search*).ti,ab,kf. or ((structured or comprehensive* or systemic*) adj3 search*).ti,ab,kf. or ((literature adj3 review*) and (search* or database* or data-base*)).ti,ab,kf. or (("data extraction" or "data source*") and "study selection").ti,ab,kf. or ("search strategy" and "selection criteria").ti,ab,kf. or ("data source*" and "data synthesis").ti,ab,kf. or (medline or pubmed or embase or cochrane).ab. or ((critical or rapid) adj2 (review* or overview* or synthes*)).ti. or (((critical* or rapid*) adj3 (review* or overview* or synthes*)) and (search* or database* or data-base*)).ab. or (metasynthes* or meta-synthes*).ti,ab,kf.) not (comment/ or editorial/ or letter/ or ((exp animals/ or exp models, animal/) not humans/)) |
479432 |
|
14 |
4 or 8 or 12 or 13 |
149 |
|
13 |
trimbow.ti,ab,kf. or trelegy ellipta.ti,ab,kf. or enerzair.ti,ab,kf. |
12 |
|
12 |
9 and 10 and 11 |
9 |
|
11 |
Glycopyrrolate/ or ad 237.ti,ab,kf. or ad237.ti,ab,kf. or ahr 504.ti,ab,kf. or ahr504.ti,ab,kf. or asecryl.ti,ab,kf. or cuvposa.ti,ab,kf. or "drm 04".ti,ab,kf. or drm04.ti,ab,kf. or enurev.ti,ab,kf. or gastrodyn.ti,ab,kf. or glersa.ti,ab,kf. or glycopyrrolate.ti,ab,kf. or glycopyrronium.ti,ab,kf. or glyrx-pf.ti,ab,kf. or lonhala magnair.ti,ab,kf. or mobinul.ti,ab,kf. or nodapton.ti,ab,kf. or nva 237.ti,ab,kf. or nva237.ti,ab,kf. or robinal.ti,ab,kf. or robinol.ti,ab,kf. or robinul.ti,ab,kf. or seebri.ti,ab,kf. or sialanar.ti,ab,kf. or sroton.ti,ab,kf. or strodin.ti,ab,kf. or tarodyl.ti,ab,kf. or tarodyn.ti,ab,kf. or tovanor.ti,ab,kf. |
1696 |
|
10 |
(arcapta or hirobriz breezhaler).ti,ab,kf. or indacaterol.mp. or onbrez.ti,ab,kf. or oslif breezhaler.ti,ab,kf. or qab 149.ti,ab,kf. or qab149.ti,ab,kf. |
510 |
|
9 |
Mometasone Furoate/ or mometason.ti,ab,kf. or mometasone 17 furoate.ti,ab,kf. or mometasone furoate.ti,ab,kf. or mometop.ti,ab,kf. or mommox.ti,ab,kf. or monovel.ti,ab,kf. or monovo.ti,ab,kf. or morecort.ti,ab,kf. or motaderm.ti,ab,kf. or mundoson.ti,ab,kf. or muofuder.ti,ab,kf. or nasehaler.ti,ab,kf. or nasoaldo.ti,ab,kf. or nasomet.ti,ab,kf. or nasometin.ti,ab,kf. or nasonex.ti,ab,kf. or nasotasone.ti,ab,kf. or netonox.ti,ab,kf. or novasone cream.ti,ab,kf. or novasone lotion.ti,ab,kf. or novasone ointment.ti,ab,kf. or ovison.ti,ab,kf. or ovixan.ti,ab,kf. or pronasal.ti,ab,kf. or rinelon.ti,ab,kf. or rinometasone.ti,ab,kf. or rivelon.ti,ab,kf. or sch 32088.ti,ab,kf. or sch32088.ti,ab,kf. or sebanez.ti,ab,kf. or sinuva.ti,ab,kf. or sonalox.ti,ab,kf. or temon.ti,ab,kf. or uniclar.ti,ab,kf. or zhekort.ti,ab,kf. |
1062 |
|
8 |
5 and 6 and 7 |
114 |
|
7 |
(ellipta or gsk 573719 or gsk 573719a or gsk-573719 or gsk-573719a or gsk573719 or gsk573719a or incruse or rolufta or umeclidinium).ti,ab,kf. |
336 |
|
6 |
(gsk 642444 or gsk642444 or gw 642444 or gw642444 or vilanterol).ti,ab,kf. |
393 |
|
5 |
Fluticasone/ or fluticason*.ti,ab,kf. |
4662 |
|
4 |
1 and 2 and 3 |
37 |
|
3 |
eformoterol.ti,ab,kf. or formoterol.mp. |
2634 |
|
2 |
Glycopyrrolate/ or ad 237.ti,ab,kf. or ad237.ti,ab,kf. or ahr 504.ti,ab,kf. or ahr504.ti,ab,kf. or asecryl.ti,ab,kf. or cuvposa.ti,ab,kf. or "drm 04".ti,ab,kf. or drm04.ti,ab,kf. or enurev.ti,ab,kf. or gastrodyn.ti,ab,kf. or glersa.ti,ab,kf. or glycopyrrolate.ti,ab,kf. or glycopyrronium.ti,ab,kf. or glyrx-pf.ti,ab,kf. or lonhala magnair.ti,ab,kf. or mobinul.ti,ab,kf. or nodapton.ti,ab,kf. or nva 237.ti,ab,kf. or nva237.ti,ab,kf. or robinal.ti,ab,kf. or robinol.ti,ab,kf. or robinul.ti,ab,kf. or seebri.ti,ab,kf. or sialanar.ti,ab,kf. or sroton.ti,ab,kf. or strodin.ti,ab,kf. or tarodyl.ti,ab,kf. or tarodyn.ti,ab,kf. or tovanor.ti,ab,kf. |
1696 |
|
1 |
beclometasone/ or beclometason*.ti,ab,kf. or beclomethason*.ti,ab,kf. |
3941 |