Biologicals bij CE
Uitgangsvraag
Wat is de effectiviteit en veiligheid van dupilumab bij constitutioneel eczeem (CE)?
Aanbeveling
Volwassen patiënten met ernstig constitutioneel eczeem komen in aanmerking voor behandeling met dupilumab bij ontoereikende respons op intensieve lokale therapie en het falen van minimaal één systemisch immunosuppressivum (met adequate dosering en duur).
De aanbevolen dosering van dupilumab voor de behandeling van volwassen patiënten met constitutioneel eczeem is een oplaaddosis van 600 mg s.c. gevolgd door 300 mg s.c. om de week.
Bij de start van een biological stelt de dermatoloog de huisarts zo spoedig mogelijk hiervan op de hoogte vanwege mogelijke bijwerkingen of interacties met andere medicatie.
Plaatsbepaling dupilumab
Om dupilumab doelmatig in te zetten bij de behandeling van ernstig, moeilijk behandelbaar CE, moet een goede afweging worden gemaakt welke patiënten met dupilumab behandeld zouden moeten worden. Het label schrijft voor dat dupilumab mag worden ingezet na het falen van lokale therapie. In tegenstelling tot de VS worden er in Europa veel vaker orale immunosuppressiva gebruikt voor de behandeling van ernstig CE. Daarom wordt in de meeste Europese richtlijnen geadviseerd eerst een oraal immunosuppressivum te gebruiken en bij falen hierop behandeling met dupilumab te overwegen.
Aangezien ciclosporine geregistreerd is voor volwassenen met ernstig CE, lijkt dit middel een logische eerste keuze bij indicatie systemische therapie. In de praktijk blijkt dat er in Nederland ook veel gebruik gemaakt wordt van de off-label orale immunosuppressiva. Om die reden stelt de richtlijn commissie voor de keuze van het immunosuppressivum voorafgaande aan eventuele behandeling met dupilumab aan de behandelaar over te laten. Wel is het van belang het middel voldoende lang en in adequate dosering voor te schrijven volgens het behandelalgoritme (zie de aanverwante producten). Na het falen van de behandeling met een oraal immunosuppressivum kan overwogen worden een ander oraal immunosuppressivum te starten of over te gaan op dupilumab.
Ter monitoring van het behandelresultaat van zowel orale immunosuppressiva als dupilumab dient gebruik te worden gemaakt van de Investigator Global Assessment (IGA) (zes-puntschaal: 0 = geen eczeem, 1 = bijna geen eczeem, 2 = mild eczeem, 3 = matig eczeem 4 = ernstig eczeem 5 = zeer ernstig eczeem) en Numeric Rating Scale (NRS) jeuk (de gemiddelde jeuk in de afgelopen zeven dagen op een schaal van 0-10) welke in het medisch dossier worden vastgelegd.
Overwegingen
De algehele zekerheid van bewijs omtrent het gebruik van dupilumab bij CE is hoog. De financiering van de studies werd verzorgd door de fabrikant van dupilumab. De werkgroep heeft besloten hier niet voor af te waarderen, omdat de studies goed waren opgezet met een duidelijk omschreven methodologie en een laag risico op bias (zie de evidence tabellen).
Dupilumab wordt tijdens de onderhoudsfase één keer per 2 weken subcutaan toegediend, waarmee de injectiefrequentie niet hoog en niet laag is. Patiënten kunnen bezwaren hebben tegen injecties (prikangst). Goed overleg met de patiënt en gedegen uitleg is cruciaal om deze eventuele bezwaren weg te nemen. Dupilumab moet gekoeld bewaard worden en is na opwarming op kamertemperatuur maximaal 14 dagen houdbaar, wat praktische bezwaren kan opleveren (o.a. bij reizen).
De toediening van 1 subcutane injectie van 300 mg dupilumab kost € 628,88 (inclusief BTW) per injectie van 300 mg. [Prestatie- en tariefbeschikking Add-on geneesmiddelen, maart 2020]. Er werd een doorrekening van de gewijzigde kosten ten gevolge van gebruik van dupilumab gemaakt in een zogenaamde Budget Impact Analyse (BIA). De volledige rapportage is opgenomen in de aanverwante producten.
Ofschoon er geen verschil is in het aantal bijwerkingen tussen de dupilumab- en placebo- groep, is er wel één bijwerking die bij het gebruik van dupilumab in het oog springt. Een veel voorkomende bijwerking is conjunctivitis. Het is belangrijk hier alert op te zijn en bij bestaande oogklachten oogheelkundig te vervolgen. Patiënten met conjunctivitis die niet goed reageren op behandeling met indifferente oogdruppels moeten verwezen worden naar de oogarts. [Wollenberg 2018] Geadviseerd wordt om een specifiek registratieformulier met betrekking tot specifieke oogheelkundige problematiek (met name Limbus) mee te geven aan patiënt (zie de aanverwante producten).
Het is op dit moment nog onduidelijk of er risico’s verbonden zijn aan langdurige behandeling met dupilumab: er lopen momenteel open-label extensiestudies en Nederlands (real world) registries in grote patiëntengroepen.
Gezien het soms snelle en sterke behandeleffect van dupilumab kan het voorkomen dat patiënten het gebruik van lokale corticosteroïden snel afbouwen of direct stoppen. Hierdoor kan een acute bijnierschorsinsufficiëntie ontstaan, zeker als de patiënt van te voren grote hoeveelheden lokale corticosteroïden gebruikt heeft. Dit probleem kan voorkomen worden door goede voorlichting m.b.t. afbouwen van lokale corticosteroïden, ondanks dat de huid rustig is, en meten van basaal cortisol ter controle. Als het basaal cortisol niet herstelt tijdens het afbouwen van de lokale corticosteroïden is verwijzing naar een endocrinoloog geïndiceerd. [Ariëns 2018]
Onderbouwing
Achtergrond
Dupilumab is een volledig humaan monoklonaal antilichaam wat specifiek bindt aan de interleukin-4 receptor alpha, resulterend in een remming van Il-4 en IL-13. Deze type 2-cytokines (inclusief Th2) spelen een belangrijke rol bij diverse allergische aandoeningen, zoals astma en CE. Dupilumab is het eerste biological voor patiënten met CE.
Label: behandeling van matig-ernstig tot ernstig CE bij volwassenen die in aanmerking komen voor een systemische behandeling.
Indicatiestelling: behandeling van ernstig, moeilijk behandelbaar CE bij volwassen patiënten, waarbij eerdere systemische immunosuppressieve behandeling heeft gefaald (zie ook het behandelalgoritme bij de aanverwante producten).
Registratie: FDA goedkeuring in maart 2017 in de Verenigde Staten en EU marktautorisatie in september 2017. De Add-on is toegekend per januari 2018 (tabel 19).
Tabel 19. registratie/dosering dupilumab
|
Registratiedatum voor CE |
September 2017 EMA, add-on januari 2018
|
|
Aanbevolen startdosering
|
Oplaaddosis: 600 mg s.c. |
|
Aanbevolen onderhoudsdosering
|
300 mg om de week s.c. |
|
Tijd waarin respons verwacht wordt |
16 weken |
Instructies voor gebruik
Monitoring
Hoewel het niet vermeld wordt in de bijsluiter en de SmPC-tekst van dupilumab adviseert de werkgroep laboratoriumonderzoek te verrichten voorafgaand en tijdens de behandeling met dupilumab. Dit wordt sterk aanbevolen bij patiënten met co-morbiditeit.
Het nut van laboratoriummonitoring zal over 3 jaar geëvalueerd worden. De werkgroep is van mening dat monitoring het volgende dient in te houden:
Voorafgaand aan de behandeling:
- Bij de start en bij ieder vervolgconsult worden de volgende meetinstrumenten sterk aanbevolen: Investigator Global Assessment, IGA (zespuntschaal: 0 = geen eczeem, 1 = bijna geen eczeem, 2 = mild eczeem, 3 = matig eczeem 4 = ernstig eczeem 5 = zeer ernstig eczeem) en de Numeric Rating Scale, NRS jeuk (gemiddelde jeuk in de afgelopen zeven dagen op een schaal van 0-10).
- Aanvullend kunnen de EASI, POEM en DLQI gebruikt worden.
- Laboratoriumonderzoek (zie tabel 20)
- Check contra-indicaties
- Bespreek voorafgaand aan de behandeling de kinderwens bij vrouwelijke patiënten. Zwangerschap (anamnestisch) uitsluiten (zie ook onder: ‘Kinderwens, zwangerschap en lactatie’).
Tabel 20. Monitoringsschema behandeling met dupilumab bij CE
|
Parameter |
Bij intake |
Vervolg consult |
Na 4 weken |
Tijdens onderhoudsdosering daarna elke 6 maanden) |
|
IGA en NRS jeuk* |
x |
x |
|
x |
|
Bloedonderzoek |
|
|
|
|
|
Hb, Leukocyten |
x |
|
x |
x |
|
Leukocyten differentiatie |
x |
|
x |
x |
|
Totaal eosinofielen** |
x |
|
x |
x** |
|
Levertesten: ALAT, Ɣ-GT*** |
x |
|
x |
x |
|
Serum creatinine*** |
x |
|
x |
x |
|
HIV |
x |
|
|
|
|
HBV/HCV |
x |
|
|
|
|
Faeces- of serumonderzoek op Parasieten**** |
x |
|
|
|
* Aanvullend kunnen de EASI, POEM of DLQI gebruikt worden.
** Eosinofielen: 1x per 3 maanden herhalen tot patiënten weer terug zijn op de waarde bij intake.
NB. Eosinofilie wordt regelmatig gezien tijdens de eerste fase van de behandeling met dupilumab. Het optreden van eosinofilie lijkt niet klinisch relevant, het is geen reden tot stoppen of een dosisreductie. Het hoort wel te normaliseren in het verloop van de behandeling.
*** Standaard bij intake, Verdere monitoring op indicatie (bijv. co-morbiditeit en afwijkende waarde bij start)
**** Op indicatie bij patiënten met darmklachten, eosinofilie of die in het afgelopen jaar in de tropen zijn geweest.
Contra-indicaties
- Overgevoeligheid voor de werkzame stof of één van de hulpstoffen (L-argininehydrochloride, l-histidine, polysorbaat 80, natriumacetaat, azijnzuur, sucrose)
- Intestinale worminfecties (eerst behandelen)
Bijwerkingen/veiligheid
Tabel 21. Overzicht bijwerkingen dupilumab
|
Zeer frequent |
Reacties op injectieplaats |
|
Frequent |
(Allergische) conjunctivitis, orale herpes, eosinofilie, hoofdpijn, oculaire pruritus, blefaritis |
|
Zeer zelden |
Serumziekte/ serumziekte-achtige reacties |
Bron: SmPC tekst Dupixent® 2017
Zie ook: https://www.farmacotherapeutischkompas.nl/bladeren/preparaatteksten/d/dupilumab
Overdosering
Er is geen specifieke behandeling voor een overdosering met dupilumab. In het geval van een overdosering moet de patiënt gecontroleerd worden op eventuele klachten en symptomen van bijwerkingen en moet er onmiddellijk een gepaste symptomatische behandeling gestart worden.
Interacties
De veiligheid en werkzaamheid van gelijktijdig gebruik van dupilumab en levende vaccins zijn niet onderzocht. Immuunreacties op vaccinatie werden beoordeeld in een onderzoek waarbij patiënten met CE werden behandeld met 300 mg dupilumab, eenmaal per week toegediend gedurende 16 weken. Na 12 weken behandeling met dupilumab kregen patiënten een Tdap-vaccin (T-celafhankelijk) en een meningokokken-polysacharidevaccin (T-celonafhankelijk) toegediend, waarna na 4 weken de immuunreacties werden beoordeeld. Reacties van antilichamen op zowel het Tdap-vaccin als het meningokokken-polysacharidevaccin waren vergelijkbaar bij patiënten die werden behandeld met dupilumab en met placebo. Tijdens het onderzoek werden geen negatieve interacties waargenomen tussen de niet-levende vaccins en dupilumab. Patiënten die met dupilumab worden behandeld, mogen daarom gelijktijdig vaccinatie met inactieve of niet-levende vaccins ondergaan.
In een klinisch onderzoek bij patiënten met CE zijn de effecten beoordeeld van dupilumab op de farmacokinetiek (FK) van CYP-substraten. De gegevens die in dit onderzoek werden verzameld, duidden niet op klinisch relevante effecten van dupilumab op de activiteit van CYP1A2, CYP3A4, CYP2C19, CYP2D6 of CYP2C9.
Kinderwens, zwangerschap en lactatie
Er zijn beperkte gegevens over het gebruik van dupilumab tijdens de zwangerschap. Dierstudies geven geen aanwijzingen voor schadelijke effecten wat betreft voortplantingstoxiciteit. Dupilumab dient tijdens de zwangerschap alleen te worden gebruik als het potentiele voordeel het potentiele risico voor de foetus rechtvaardigt. Het is niet bekend of dupilumab wordt uitgescheiden in de moedermelk of systemisch wordt opgenomen na inname. Er moet worden besloten of borstvoeding moet worden gestaakt od dat de behandeling met dupilumab moet worden gestaakt, waarbij het voordeel van borstvoeding van het kind en het voordeel van behandeling voor de vrouw in overweging moet worden genomen. Dieronderzoek toonde geen verminderde vruchtbaarheid. [SmPC 2017]
Door de werkgroep wordt het gebruik van dupilumab tijdens de zwangerschap of bij borstvoeding voorlopig afgeraden.
Conclusies
Dupilumab 300 mg om de week versus placebo
Effectiviteit
|
Hoog
|
Uitkomstmaat 1: verandering van EASI score t.o.v. baseline (cruciaal)
Dupilumab 300 mg om de week geeft een grote reductie van EASI score t.o.v. baseline vergeleken met placebo.
Thaçi 2016; Simpson 2016; Blauvelt 2017, de Bruin-Weller 2017 |
|
Hoog
|
Uitkomstmaat 2: proportie patiënten die IGA response (0= clear; 1= almost clear) behaalt (cruciaal)
Dupilumab 300 mg om de week geeft een grote proportie patiënten die een IGA 0/1 response behaalt in vergelijking met placebo.
Thaçi 2016; Simpson 2016; Blauvelt 2017, de Bruin-Weller 2017 |
|
Hoog
|
Uitkomstmaat 3: verandering in aangedane oppervlakte (gemeten met SCORAD) t.o.v. baseline (belangrijk)
Dupilumab 300 mg om de week geeft een grote reductie in aangedane oppervlakte (gemeten met SCORAD) t.o.v. baseline in vergelijking met placebo.
Thaçi 2016; Simpson, 2016; Blauvelt, 2017, de Bruin-Weller 2017 |
|
Hoog
|
Uitkomstmaat 4: verandering in peak pruritus NRS score t.o.v. baseline (belangrijk)
Dupilumab 300 mg om de week geeft een grote reductie in peak pruritus NRS score t.o.v. baseline in vergelijking met placebo. Simpson 2016; Blauvelt 2017, de Bruin-Weller 2017 |
|
Hoog
|
Uitkomstmaat 5: proportie patiënten die 75% verbetering van EASI score t.o.v. baseline behaalt (belangrijk) Dupilumab 300 mg om de week geeft een grote proportie patiënten die EASI-75 behaalt in vergelijking met placebo. Simpson 2016; Blauvelt 2017, de Bruin-Weller 2017 |
|
Hoog
|
Uitkomstmaat 6: verandering in DLQI score t.o.v. baseline (belangrijk) Dupilumab 300 mg om de week geeft een grote reductie in DLQI score t.o.v. baseline in vergelijking met placebo. Thaçi, 2016 Simpson 2016; Blauvelt 2017, de Bruin-Weller 2017 |
Veiligheid
|
Hoog
|
Uitkomstmaat 7: Proportie patiënten met tenminste één bijwerking (cruciaal)
Er is geen verschil in de proportie patiënten met tenminste één bijwerking tussen dupilumab 300 mg om de week en placebo.
Thaçi 2016; Simpson 2016; Blauvelt 2017, de Bruin-Weller 2017 |
Samenvatting literatuur
Beck et al. publiceerden de eerste gerandomiseerde, dubbelblinde, placebo-gecontroleerde trial bij volwassenen met matig-tot-ernstig CE. [Beck 2014] Dupilumab werd geëvalueerd als monotherapie met wekelijkse subcutane injecties in 2 vier-weken-durende trials en een 12-weken-durende trial en daarnaast in een andere trial van 4 weken waarbij lokale corticosteroïden gebruikt mochten worden. In de vier-weken-durende monotherapie studies resulteerde dupilumab in een snelle en dosisafhankelijke verbetering van klinische scores, biomarker levels en het transcriptoom. De resultaten van de 12-weken durende studie toonde tevens een 50% SCORAD reductie in 85% van de patiënten in de dupilumab groep en 35% in de placebogroep (p<0.001). Ook jeukscores en de investigator’s global assessment score verbeterden meer in de dupilumab groep (beide p<0.001). In de studie waarbij topicale corticosteroïden gebruikt mochten worden werd bij 100% van de deelnemers een 50% SCORAD daling gezien. Bijwerkingen zoals huidinfecties werden in de placebo groep beschreven; nasofaringitis en hoofdpijn waren de meest genoemde bijwerkingen in de dupilumab groep.
Thaçi et al. gingen op zoek naar de effectiviteit en veiligheid van verschillende doseringen dupilumab. [Thaçi 2016] In hun gerandomiseerde, placebo-gecontroleerde, dubbelblinde studie werden 379 volwassen patiënten met CE (EASI bij baseline ≥ 16) gedurende 16 weken behandeld met dupilumab 300mg 1x/week, 300mg 1x/2 weken, 200mg 1x/2 weken, 300 mg 1x/4 weken, 100mg 1x/4 weken of placebo elke week. EASI scores verbeterden in elke groep significant meer dan in de placebo groep (p<0.001). Na 16 weken was er een reductie van de EASI van respectievelijk 74%, 68%, 65%, 64%, 45% en 18%. Een 50% daling van de EASI (EASI50) werd bereikt bij respectievelijk 83%, 78%, 62%, 71%, 45% en 30% van de deelnemers. Er was geen significant verschil tussen de laagste dosis groep (100 mg/4 weken) en placebo. Alle andere klinische uitkomstmaten (EASI75, EASI90, IGA 0-1, SCORAD, aangedaan oppervlak) verbeterden significant in alle dupilumab groepen vergeleken met de placebo groep. Ook was er een snelle en sterke reductie van de jeukscore in alle behandelgroepen zichtbaar. De scores voor kwaliteit van leven lieten een significante dosisafhankelijke verbetering zien in de behandelgroepen versus placebo, m.u.v. de laagste dosis groep. De bijwerkingen waren over het algemeen mild en vergelijkbaar tussen de dupilumab groepen en de placebo groepen: de meest voorkomende bijwerking was nasofaryngitis.
Simpson et al. voerden twee gerandomiseerde, placebo gecontroleerde fase 3 studies uit van identieke opzet met 671 en 708 patiënten met CE. [Simpson 2016] Patiënten kregen gedurende 16 weken dupilumab 300mg subcutaan elke week, placebo elke week of dupilumab 300mg om de week, afgewisseld met placebo. De primaire uitkomstmaat was het percentage patiënten met een IGA score van 0 of 1 (geen eczeem of bijna geen eczeem) en een reductie van 2 of meer punten in de IGA score op week 16. Dit werd bereikt bij 36-38% van de patiënten met dupilumab 300 mg 1x/2 weken en 36-37% van de patiënten met dupilumab 300 mg 1x/week (placebogroep 8-10%; p< 0.001). EASI50 werd bereikt bij 61-69 % patiënten in de dupilumab groepen (placebogroepen 22-25%); EASI75 werd bereikt in 44-52% in de dupilumab-groepen vergeleken met 12-15% in de placebo groepen. Er was geen significant verschil tussen de beide doseringen. Bijwerkingen waren over het algemeen mild en vergelijkbaar in dupilumab en placebo groepen. Alleen conjunctivitis en lokale reacties op injecties werden frequenter gezien in de dupilumab groepen vergeleken met de placebo groepen.
In een 1 jaar durende gerandomiseerde, dubbelblinde, placebo gecontroleerde fase 3 studie van Blauvelt et al. werden volwassenen met matig tot ernstig CE en een onvoldoende klinische respons op lokale corticosteroïden geïncludeerd in 161 ziekenhuizen in 14 landen in Europa, Azië/Pasific en Noord Amerika. [Blauvelt 2017] Patiënten werden gerandomiseerd (3:1:3) voor subcutaan dupilumab 300 mg eens per week (qw), dupilumab 300mg elke 2 weken (q2w) of placebo. Lokale corticosteroïden en calcineurine remmers waren toegestaan op geleide van de ziekteactiviteit. Eindpunten waren onder andere het percentage patiënten dat een IGA van 0 of 1 bereikte en een 2 punten IGA daling had vanaf baseline, en het percentage patiënten dat een EASI-75 bereikte in week 16. In totaal werden 740 patiënten geïncludeerd: 319 voor dupilumab qw, 106 voor dupilumab q2w en 315 voor placebo. De resultaten in week 52 waren beschikbaar voor 623 patiënten. In week 16 werd een IGA score van 0 of 1 significant vaker gehaald (p<0.0001) door patiënten die behandeld werden met dupilumab in vergelijking met placebo: 39% (125 patiënten) bij dupilumab qw, 39% (41 patiënten) bij dupilumab q2w en 12% (39 patiënten) bij placebo. Ook EASI-75 werd significant vaker behaald in de dupilumab groep (p<0.001): 64% (204 patiënten) in dupilumab qw, 69% (73 patiënten) bij dupilumab q2w en 23% (73 patiënten) bij placebo. De resultaten in week 52 waren hetzelfde. Ernstige bijwerkingen werden gezien bij 9 patiënten bij dupilumab qw (3%), bij 4 patiënten (4%) bij dupilumab q2w en bij 16 patiënten (5%) in de placebo groep. In de dupilumab groep werden vaker lokale reacties op de injectieplaats en vaker conjunctivitis gezien. De incidentie van herpesinfecties was niet verschillend tussen de dupilumab en de placebo groep.
In een 16-weekse fase 3 studie, verricht in 10 Europese landen, werd de effectiviteit van dupilumab in combinatie met lokale corticosteroïden onderzocht bij CE patiënten die eerder gefaald hadden op ciclosporine (CsA) of bij wie een contra-indicatie hiervoor bestond. [de Bruin-Weller 2017] 325 personen van 18 jaar en ouder werden gerandomiseerd (1:1:1) naar lokale corticosteroïdtherapie + dupilumab qw, lokale corticosteroïdtherapie + dupilumab q2w of lokale corticosteroïdtherapie + placebo. Inclusiecriterium was onvoldoende effect van lokale therapie én onvoldoende respons op ciclosporine en/of ernstige bijwerkingen door ciclosporine en/of een contra-indicatie voor ciclosporine. Van de 318 deelnemers die de studie afmaakten, bereikten de patiënten die dupilumab hadden gebruikt (q2w en qw) significant vaker de primaire uitkomstmaat van 75% reductie van de klachten gemeten met de Eczema Area and Severity Index (EASI75) dan patiënten in de placebo (resp. 59.1%/62.6% vs 29.6%; p<0.0001). Gemeten met Investigator Global Assessment (IGA) behaalde in de placebogroep 13,9% een IGA van 0 of 1 én een reductie van minimaal 2 punten, terwijl dat in de dupilumab-groepen 40,2% (qw) en 39,1% (q2w) was (p<0.0001). Ook andere uitkomstenmaten zoals jeuk, pijn, slaapstoornissen, angst en depressie, en kwaliteit van leven verbeterden significant meer in de dupilumab-groepen dan in de placebogroep. Vergeleken met de placebogroep werden er in de dupilumab-groepen 30% (q2w) tot 40% (qw) minder lokale corticosteroïden gebruikt met een veel beter resultaat. Het percentage van de deelnemers dat 1 of meer bijwerkingen kreeg was in alle groepen gelijk. Daarbij werd conjunctivitis vaker gezien in de dupilumab-groepen dan bij placebo, terwijl in de placebogroep vaker huidinfecties voorkwamen.
Meta-analyse
Voor de kwantitatieve meta-analyse is gekozen voor de studies waarbij de dosering volgens het label is, namelijk 300 mg s.c. q2w. Hierdoor viel de studie van Beck et al. af. De studies zijn beoordeeld op Risk of Bias volgens het Cochrane Handbook for Systematic Reviews of Interventions [Higgins 2011] en de zekerheid van het bewijs middels de Grading of Recommendations Assessment, Development and Evaluation methodiek. [Schünemann 2013] Minimaal 4 en maximaal 5 studies konden op basis van de uitkomstmaten worden gepooled voor een meta-analyse met ReviewManager (RevMan versie 5.3), op de volgende uitkomstmaten:
- Verandering in EASI score t.o.v. baseline (cruciaal)
- Proportie patiënten die IGA response (0=clear; 1=almost clear) behaalt (cruciaal)
- Verandering in aangedane oppervlakte (gemeten met SCORAD) score t.o.v. baseline (belangrijk)
- Verandering van peak pruritus numeric rating scale (pruritus NRS) scores t.o.v. baseline (belangrijk)
- Proportie patiënten die 75% verbetering van EASI t.o.v. baseline behaalt (EASI75) (belangrijk)
- Verandering in DLQI score t.o.v. baseline (belangrijk)
- Proportie patiënten met tenminste één bijwerking (cruciaal)
Voor continue uitkomstmaten is gekozen voor het gemiddelde verschil (mean difference) tussen de dupilumab-groep en de placebogroep; voor dichotome uitkomstmaten de risk ratio. In het geval van uitkomstmaten met verschillende meetmethoden is gekozen voor de standardized mean difference (SMD). In het geval van verhoogde heterogeniteit (I2 > 50%) is een sensitiviteitsanalyse uitgevoerd om te bezien welke studie dit mogelijk veroorzaakt. Daarbij is tevens getest op het verschil tussen de uitkomsten met een fixed-effects en random-effects model. Gezien de richting van het effect van de interventie, is de kwaliteit van het bewijs niet verlaagd op basis van een verhoogde heterogeniteit (>50%), hetgeen voor twee uitkomstmaten het geval was, ook omdat I2 kleiner was dan 60%. Zie de evidence tabellen voor de Summary of Findings-tabel en de meta-analyse (forest plots).
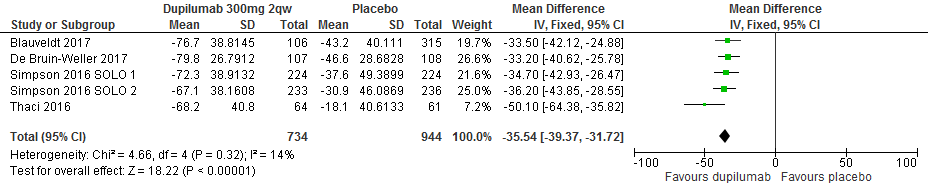
Percentuele verandering in EASI score t.o.v. baseline
De uitkomsten van 5 RCTs (1678 deelnemers) laat een gemiddeld verschil in percentuele reductie van EASI-scores zien van -35.54% (95%CI -39.37 tot -31.72) tussen dupilumab 300mg q2w en placebo (resp. 734 en 944 deelnemers). [Thaçi 2016, Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017]
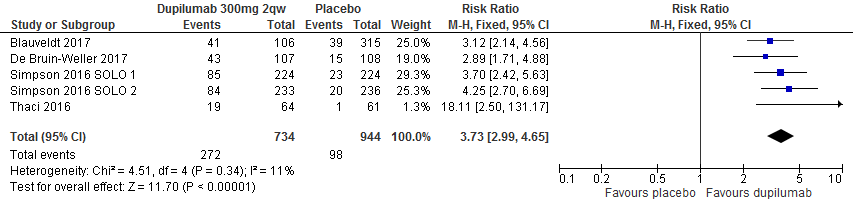
Proportie patiënten die IGA response (0=clear; 1=almost clear) behaalt
Op basis van 5 RCTs (1678 deelnemers) behaalde in de placebogroep 98/944 (10,4%) van de deelnemers een IGA score van 0 of 1, terwijl dat in de dupilumab-groep 272/734 (37,1%) was. Dat komt neer op een risk ratio van 3.73 (95%CI 2.99 - 4.65) ten voordele van dupilumab. [Thaçi 2016, Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017]
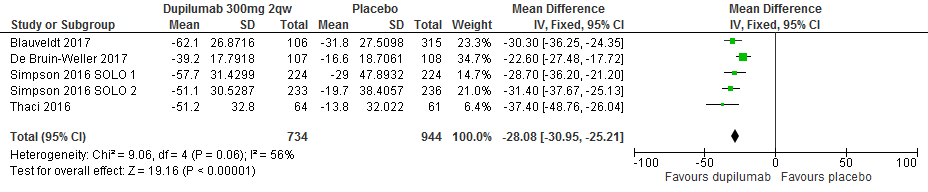
Verandering in aangedane oppervlakte (gemeten met SCORAD) score t.o.v. baseline
Het verschil tussen de dupilumab-groep (734) en de placebogroep (994) bedroeg op basis van vijf studies -28.08% (95%CI -30.95 tot -25.21) op de uitkomstmaat ‘Verandering in aangedane oppervlakte (gemeten met SCORAD) score t.o.v. baseline’ (1678 deelnemers), in het voordeel van dupilumab. [Thaçi 2016, Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017]
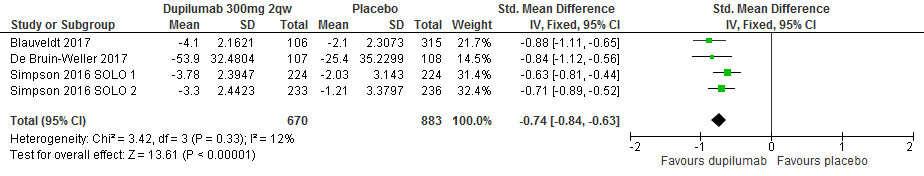
Verandering van peak pruritus numeric rating scale (pruritus NRS) scores t.o.v. baseline
Gezien de gerapporteerde uitkomstmaten konden hier vier studies worden gepooled, met in totaal 1553 deelnemers. Op de uitkomstmaat peak puritus (hoogste mate van jeuk in de afgelopen week, 0 is geen, 10 is maximale jeuk) was het verschil tussen de dupilumab-groep en de placebogroep een SMD van -0.74 (95%CI -0.84 tot -.63), wat geïnterpreteerd kan worden als een groot effect ten faveure van dupilumab. [Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017]
Proportie patiënten die 75% verbetering van EASI t.o.v. baseline behaalt (EASI75)
De uitkomstmaat EASI75, een 75% reductie in EASI- score t.o.v. baseline), kon gepooled worden in 4 studies (1553 deelnemers). 358/670 (53,4%) in de dupilumab-groep en 166/883 (18,8%) deelnemers in de placebogroep behaalde dit eindpunt. Dit betekent een risk ratio van 3.05 (95%CI 2.60 tot 3.58) in het voordeel van dupilumab. [Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017]
Verandering in DLQI score t.o.v. baseline
Voor de verandering in DLQI konden vijf studies worden gepooled, met in totaal 1678 deelnemers. Gezien de verschillen in de gerapporteerde mate van effect is gekozen voor een standardized mean difference. Het verschil tussen de dupilumab-groep en de placebogroep was een SMD van -0.78 (95%CI -0.89 tot -0.68) hetgeen een groot effect is in het voordeel van dupilumab. [Thaçi 2016, Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017]
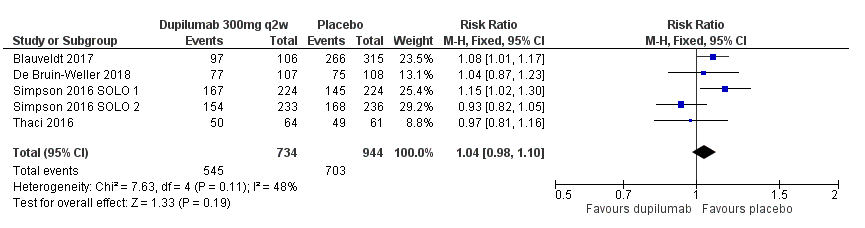
Proportie patiënten met tenminste één bijwerking
In vijf studies, met in totaal 1678 deelnemers, rapporteerde in placebogroep 703/944 (74,5%) van de deelnemers in de placebogroep minstens één bijwerking, in de dupilumab-groep was dat 545/734 (73,4%%). De risk ratio voor bijwerkingen tussen de groepen is 1.04 (95%CI 0.98 tot 1.10). [Thaçi 2016, Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017]
Zoeken en selecteren
De search naar de effectiviteit en veiligheid van dupilumab leverde 6 publicaties met 7 RCTs op, zie de zoekverantwoording voor de zoekstrategie. Deze uitgangsvraag werd middels de GRADE-methode uitgewerkt. Volgens deze methode werden relevante uitkomstmaten bepaald: verandering in EASI score t.o.v. baseline, proportie patiënten die IGA response (0=clear; 1=almost clear) behaalt, verandering in aangedane oppervlakte (gemeten met SCORAD) t.o.v. baseline, verandering van peak pruritus numeric rating scale (pruritus NRS) scores t.o.v. baseline, proportie patiënten die 75% verbetering van EASI t.o.v. baseline behaalt (EASI75), verandering in DLQI score t.o.v. baseline en proportie patiënten met tenminste één bijwerking. Afhankelijk van de gerapporteerde uitkomstmaten konden 4 of 5 (van de 6) studies worden gepoold voor een kwantitatieve meta-analyse.
Referenties
- Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 2014;371:130-9.
- Schünemann H, Brożek J, Guyatt G, Oxman A. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. Available at http://www.gradeworkinggroup.org/.
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from http://training.cochrane.org/handbook
- Thaçi D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet 2016;387:40-52.
- Simpson EL, Bieber T, Guttman-Yassky E et al. Two phase 3 trials of Dupilumab versus placebo in atopic dermatitis. N Engl J Med 2016;375:2335-2348.
- SmPC teksten: www.geneesmiddeleninformatiebank.nl
- Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, pracebo-controlled, phase 3 trial. Lancet 2017; 389(10086):2287-2303.
- de Bruin-Weller M, Thaçi D, Smith CH et al. Dupilumab with concomitant topical corticosteroids in adult patients with atopic dermatitis who are not adequately controlled with or are intolerant to ciclosporin A, or when this treatment is medically inadvisable: a placebo-controlled, randomized phase 3 clinical trial (LIBERTY AD CAFÉ). Br J Dermatol. 2018 May;178(5):1083-1101.
- Ariëns LFM, van der Schaft J, Stades AME et al. Successful Treatment with Dupilumab in a Patient with Severe, Difficult to Treat Atopic Dermatitis: Beware of Symptomatic Adrenal Insufficiency due to Abrupt Discontinuation of Potent Topical Corticosteroids. Acta Derm Venereol. 2018 Jun 8;98(6):601-602.
- Wollenberg A, Ariens L, Thurau S, van Luijk C et al. Conjunctivitis occurring in atopic dermatitis patients treated with dupilumab-clinical characteristics and treatment. J Allergy Clin Immunol Pract. 2018; 6(5):1778-1780.e1.
Evidence tabellen
Summary of findings
Dupilumab 300 mg om de week versus placebo
|
Dupilumab 300mg q2w compared to placeboa for people with moderate-to-severe atopic dermatitis |
||||||
|
Patient or population: people with moderate-to-severe atopic dermatitis Setting: Multi-center, international phase-3 trials in dermatology clinics Intervention: Dupilumab 300mg q2w Comparison: placeboa |
||||||
|
Outcomes |
Anticipated absolute effects* (95% CI) |
Relative effect |
№ of participants |
Certainty of the evidence |
Comments |
|
|
Risk with placebo |
Risk with Dupilumab 300mg q2w |
|||||
|
Mean percentage change in EASI score from baseline |
The mean mean percentage change in EASI score from baseline was 0 |
MD 35,54 lower (39,37 lower to 31,72 lower) |
- |
1678 |
⨁⨁⨁⨁ |
Dupilumab 300mg q2w results in large reduction in mean percentage change in EASI score from baseline. |
|
Number of patients achieving IGA response 0/1 |
10 per 100 |
39 per 100 |
RR 3.73 |
1678 |
⨁⨁⨁⨁ |
Dupilumab 300mg q2w results in large reduction in number of patients achieving IGA response 0/1. |
|
(Mean) Percentage reduction in affected body surface area |
The mean (Mean) Percentage reduction in affected body surface area was 0 |
MD 28,08 lower (30,95 lower to 25,21 lower) |
- |
1678 |
⨁⨁⨁⨁ |
Dupilumab 300mg q2w results in large reduction in (Mean) Percentage reduction in affected body surface area. |
|
Mean change from baseline in peak pruritus NRS scores |
- |
SMD 0.74 SD lower |
- |
1553 |
⨁⨁⨁⨁ |
Dupilumab 300mg q2w results in large reduction in mean change from baseline in peak pruritus NRS scores. |
|
Proportion of patients achieving 75% reduction from baseline in EASI score |
19 per 100 |
57 per 100 |
RR 3.05 |
1553 |
⨁⨁⨁⨁ |
Dupilumab 300mg q2w results in large reduction in proportion of patients achieving 75% reduction from baseline in EASI score. |
|
Percentage/mean change from baseline in DLQI score |
- |
SMD 0.79 SD lower (0.89 to 0.68 lower} |
- |
1678 |
⨁⨁⨁⨁ |
Dupilumab 300mg q2w results in large reduction in percentage/mean change from baseline in DLQI score. |
|
Proportion of patients with at least one adverse event |
74 per 100 |
77 per 100 |
RR 1.04 |
1678 |
⨁⨁⨁⨁ |
Dupilumab 300mg q2w likely results in little to no difference in proportion of patients with at least one adverse event. |
|
*The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). |
||||||
|
GRADE Working Group grades of evidence |
||||||
Explanations
a. The placebo-arm can either be with with concomitant use of emollients only (Thaçi 2016, Simpson SOLO 1/2 2016) or emollients + topical active treatment (Blauveldt 2017, De Bruin-Weller 2017)
b. Thaçi 2016, Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017
c. All studies were funded by Sanofi/Regeneron, but we decided not to downgrade for this.
d. Simpson 2016 SOLO1/2, Blauvelt 2017, de Bruin-Weller 2017
e. Not downgraded for imprecision, because although the confidence interval includes both no effect and a positive effect, it is very narrow.
Forest plots meta-analysis
Dupilumab 300mg q2w-vs-placebo, outcome:
1 Mean percentage change in EASI score from baseline.
Dupilumab 300mg q2w-vs-placebo, outcome:
2 Number of patients achieving IGA response 0/1.
Dupilumab 300mg q2w-vs-placebo, outcome:
3 (Mean) Percentage reduction in affected body surface area.
Dupilumab 300mg q2w-vs-placebo, outcome:
4 Mean change from baseline in peak pruritus NRS scores.
Dupilumab 300mg q2w-vs- placebo, outcome:
5 Proportion of patients achieving 75% reduction from baseline in EASI score.
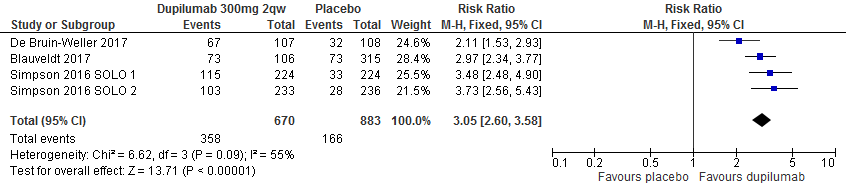
Dupilumab 300mg q2w-vs-placebo, outcome:
6 Percentage/mean change from baseline in DLQI score.
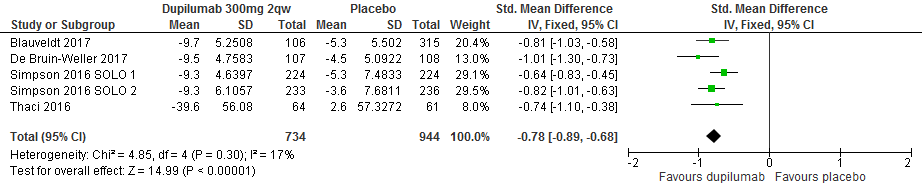
Dupilumab 300mg q2w-vs-placebo, outcome:
7 Proportion of patients with at least one adverse event.
Risk of Bias meta-analyse

Verantwoording
Autorisatiedatum en geldigheid
Laatst beoordeeld : 18-12-2019
Laatst geautoriseerd : 18-12-2019
Geplande herbeoordeling :
Voor het beoordelen van de actualiteit van deze richtlijn is de werkgroep uit 2014 gedeeltelijk in stand gehouden. Op modulair niveau is een onderhoudsplan beschreven. Bij het opstellen van de richtlijn heeft de werkgroep per module een inschatting gemaakt van de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update).
De Nederlandse Vereniging voor Dermatologie en Venereologie (NVDV) is regiehouder van deze richtlijn(modules) en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de richtlijn(modules). De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
Algemene gegevens
De richtlijnontwikkeling werd ondersteund door arts-onderzoekers van de NVDV en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijn.
Doel en doelgroep
Doel
Deze richtlijn over constitutioneel eczeem is een document met aanbevelingen ter ondersteuning van de dagelijkse praktijkvoering. De richtlijn berust op de resultaten van wetenschappelijk onderzoek en aansluitende meningsvorming gericht op het vaststellen van goed medisch handelen. De richtlijn geeft aanbevelingen over begeleiding en behandeling van patiënten met constitutioneel eczeem.
Doelgroep
De richtlijn is bestemd voor alle zorgverleners die betrokken zijn bij de zorg voor patiënten met constitutioneel eczeem. Zoals dermatologen, gespecialiseerd verpleegkundigen, huisartsen, bedrijfsartsen, jeugdgezondheidsartsen, allergologen, kinderartsen, klinische chemici, apothekers en psychologen. Voor patiënten is een afgeleide tekst van de richtlijn beschikbaar op de website van de NVDV (www.nvdv.nl) en op www.thuisarts.nl.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2007 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met constitutioneel eczeem en patiëntenvertegenwoordiger(s) vanuit de Huidpatiënten Nederland en Vereniging voor mensen met Constitutioneel Eczeem (VMCE) (zie voor het overzicht van de werkgroepleden de tabellen hieronder). Wetenschappelijke verenigingen en beroepsverenigingen zoals de NVK, NVZA, NVvA, V&VN, NHG, NVAB en NIP en stakeholders zoals VIG, ZN, NVZ, en NFU werden voor de knelpuntenanalyse en commentaarronde uitgenodigd. Voor de modulaire update in 2018 van de richtlijn werden bovenstaande partijen weer uitgenodigd.
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname.
De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Werkgroepleden – (modulaire) herziening 2019
|
Werkgroeplid |
Affiliatie en vereniging |
|
Dhr. B.W.M. Arents |
VMCE |
|
Mw. Dr. M.A. Breukels, kinderarts |
Elkerliek ziekenhuis, NVK |
|
Mw. Dr. M.S. de Bruin-Weller, dermatoloog (voorzitter) |
UMC Utrecht, NVDV |
|
Mw. Drs. Y.Y. Chung, arts-onderzoeker (secretaris) (tot december 2017) |
Bureau NVDV, Utrecht, NVDV (tot 2018) |
|
Dhr. Dr. J.J.E. van Everdingen, dermatoloog n.p. |
Directeur NVDV |
|
Mw. Dr. F.M. Garritsen, AIOS dermatologie |
UMC Utrecht |
|
Mw. Drs. M.F. Hofhuis, arts-onderzoeker (secretaris) (vanaf januari 2018) |
Bureau NVDV, Utrecht, NVDV (vanaf 2018) |
|
Dhr. Drs. W.N.M. Kouwenhoven |
VMCE |
|
Dhr. Dr. T. Rustemeyer, dermatoloog |
VUMC, NVDV |
|
Mw. Drs. A.A.J. van der Sande, arts-onderzoeker (secretaris) (tot december 2017) |
Bureau NVDV, Utrecht, NVDV (tot 2018) |
|
Mw. Dr. M.L.A. Schuttelaar, dermatoloog |
UMC Groningen, NVDV |
|
Mw. Drs. L. Teligui, arts-onderzoeker (secretaris) (januari tot december 2018) |
Bureau NVDV, Utrecht, NVDV (vanaf 2018) |
|
Dhr. Dr. R. Tupker, dermatoloog |
St. Antonius ziekenhuis Nieuwegein, NVDV |
|
Werkgroep vroege introductie voedingsallergenen |
Affiliatie en vereniging |
|
Mw. Dr. M. de Graaf dermatoloog |
UMC Utrecht, NVDV |
|
Mw. Dr. I. M. Haeck, dermatoloog |
Reinier de Graaf Groep, NVDV |
|
Dhr. Dr. A.C. Knulst, dermatoloog |
UMC Utrecht, NVDV |
|
Mw. Dr. T. M. Le, dermatoloog |
UMC Utrecht, NVDV |
|
Mw. Prof. dr. S.G.M.A. Pasmans, dermatoloog |
Erasmus MC, NVDV |
|
Mw. Drs. L.S. van der Schoot, arts-onderzoeker (secretaris), vanaf april 2019 |
Bureau NVDV, Utrecht, NVDV (vanaf 2019) |
Werkgroepleden – versie 2014
|
Werkgroeplid |
Affiliatie en vereniging |
|
Mw. Dr. M.A. Breukels, kinderarts |
NVK |
|
Mw. Prof. dr. C.A.F.M. Bruijnzeel-Koomen, dermatoloog (voorzitter) |
NVDV |
|
Mw. Dr. M.S. de Bruin-Weller, dermatoloog |
NVDV |
|
Mw. Drs. C.A.M. Eggen, arts-onderzoeker (secretaris) |
Bureau NVDV, Utrecht, NVDV (tot 2014) |
|
Dhr. Dr. J.J.E. van Everdingen, dermatoloog n.p. |
Directeur NVDV |
|
Dhr. Dr. F. Jungbauer, bedrijfsarts |
NVAB |
|
Dhr. Drs. W.N.M. Kouwenhoven |
VMCE |
|
Mw. Drs, R.A. Kuin, arts-onderzoeker (secretaris) |
Bureau NVDV, Utrecht, NVDV (vanaf 2014) |
|
Dhr. Prof. dr. A. Oranje, dermatoloog |
NVDV |
|
Mw. Dr. J.N.G. Oude Elberink, internist-allergoloog |
Vakgroep Allergologie (+NVvA) |
|
Mw. Dr. H. van Os-Medendorp |
V&VN |
|
Mw. Prof. dr. S. G.M.A. Pasmans, dermatoloog |
NVDV |
|
Dhr. Dr. T. Rustemeyer, dermatoloog |
NVDV |
|
Mw. Dr. M.L.A. Schuttelaar, dermatoloog |
NVDV |
|
Mw. Prof. dr. P.I. Spuls, dermatoloog |
NVDV |
|
Dhr. Dr. R.A. Tupker, dermatoloog |
NVDV |
|
Mw. Drs. C.J.H. de Vries, huisarts |
NHG (RL) |
|
Mw. Drs. W. Zijlstra |
NIP |
|
Ondersteuning werkgroep |
Affiliatie en vereniging |
|
Mw. Drs. K.B. Fieten |
NAD |
|
Mw. C. Frima |
student UMCU |
|
Mw. Drs. F.M. Garritsen |
AIOS UMCU |
|
Mw. Drs K. Hiemstra |
student UMCU |
|
Mw. Drs. E. Roekevisch |
AIOS AMC |
|
Mw. Drs. J.L. Thijs |
student UMCU |
|
Mw. Dr. S.G.A. van Velsen |
AIOS VUmc |
|
Dhr. Drs. G. Weststrate |
student UMCU |
|
Mw. Dr. M.J. Wiegman |
AIOS UMCG |
Werkgroepleden – versie 2007
|
Werkgroeplid (kernwerkgroep) |
Affiliatie en vereniging |
|
Mw. Prof. dr. C.A.F.M. Bruijnzeel-Koomen, dermatoloog (voorzitter) |
UMC Utrecht, NVDV |
|
Mw. drs. P.C.M. Eland-de Kok, verpleegkundig specialist |
UMC Utrecht |
|
Dhr. Dr. J.J.E. van Everdingen, dermatoloog |
CBO, secretaris vanaf 1 januari 2006 |
|
Mw. dr.ir. C.W.P.M. Hukkelhoven, epidemioloog, |
CBO, secretaris tot 1 januari 2006 |
|
Dr. J.P.C. Jaspers stafmedewerker vakinhoudelijke ontwikkeling |
UMC Groningen |
|
Dhr. Dr. J.H. Sillevis Smitt, dermatoloog |
AMC Amsterdam |
|
Werkgroep |
|
|
B.W.M. Arents |
VMCE (tot 27 november 2005) |
|
Mw. Drs. F.S. Boukes, huisarts |
NHG |
|
Mw. Dr. M.S. de Bruin-Weller, dermatoloog |
UMC Utrecht |
|
Dr. B.J.G. Daemen, apotheker |
Den Haag, KNMP/WINAP |
|
Mw. drs. P.C. Dirven-Meijer, huisarts |
Huisartsenpraktijk Renswoude |
|
Dr. M.O. Hoekstra, kinderarts |
UMC Utrecht |
|
Drs. E.J. Jansen, verpleegkundig consulent |
UMC Groningen |
|
Dr. E.J.M. van Leent, dermatoloog |
AMC Amsterdam |
|
Mw. Dr. J.N.G. Oude Elberink, internist-allergoloog |
UMC Groningen |
|
Drs. W.P. Piebenga, bedrijfsarts |
Velp |
|
Mw. drs. J.G.M. Rijntjes, kinderarts |
Emma Kinderziekenhuis, Amsterdam |
|
Dr. K.H. Tjiam, dermatoloog |
Renier de Graaf Groep, Delft |
|
Dr. A.W. van Toorenenbergen, klinisch chemicus |
Erasmus MC, Rotterdam |
|
Dr. R.A. Tupker, dermatoloog |
Sint Antonius Ziekenhuis, Nieuwegein |
|
Mw. Y. de Vries, hulplijn/columnist |
VMCE |
|
Mw. drs. C.L. Wensing-Souren, jeugdarts |
Huisartsenpraktijk Chaam |
Belangenverklaringen
Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen is opgenomen in bijlage A. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van de NVDV.
Inbreng patiëntenperspectief
Er is aandacht besteed aan het patiëntenperspectief door deelname van vertegenwoordigers vanuit de patiëntenvereniging VMCE in de werkgroep (zie ook samenstelling van de werkgroep). De deelnemers zijn betrokken geweest bij het opstellen van de conceptteksten. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de VMCE.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn(module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren, zoals het niet meer vergoeden van bepaalde behandelingen (lichttherapie en (ureumhoudende) indifferente middelen) en de introductie van nieuwe systemische medicatie (dupilumab). Het implementatieplan is te vinden op de richtlijnen database (RLDB) onder aanverwante producten. Omdat de maatschappelijke impact van het gebruik van het nieuwe geneesmiddel dupilumab als groot werd ingeschat, is in 2019 een doorrekening van de gewijzigde kosten bij integrale implementatie van de richtlijn gerealiseerd in een zogenaamde Budget Impact Analyse (BIA). De volledige rapportage is opgenomen in de aanverwante producten.
Werkwijze
De werkgroep constitutioneel eczeem heeft de vraag- en doelstellingen van deze richtlijn met elkaar afgestemd en uitgewerkt. De eerste versie van de richtlijn stamt uit 2007. De richtlijn is in 2012-2014 herzien op een groot aantal onderdelen, de hoofdstukken ‘diagnostiek’, ‘lokale therapie’ en ‘overige systemische therapie’ gedeeltelijk herzien. Per hoofdstuk staat aangegeven in welk jaartal deze al dan niet herzien is.
De ontwikkeling van de modulaire herziening in 2019 is o.a. gebaseerd op de update van de systematische review van Roekevisch et al. over de effectiviteit en veiligheid van systemische immunomodulerende middelen bij patiënten met matig tot ernstig constitutioneel eczeem. [Roekevisch 2014] Ook is gebruik gemaakt van de ‘Guidelines of care for the management of atopic dermatitis, section 3. Management and treatment with phototherapy and systemic agents‘. [Sidbury 2014]
Hieronder wordt de werkwijze van de richtlijnontwikkeling toegelicht.
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. [Medisch Specialistische Richtlijnen] Dit rapport is gebaseerd op het AGREE II-instrument (Appraisal of Guidelines for Research & Evaluation II), dat een internationaal breed geaccepteerd instrument is. [Brouwers 2010] Voor een stap-voor-stapbeschrijving hoe een evidence-based richtlijn tot stand komt, wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van Medisch Specialisten.
Knelpuntenanalyse
In de eerste vergadering zijn knelpunten en wensen ten aanzien van de richtlijn geïnventariseerd door de werkgroepleden. De werkgroep heeft de aanbevelingen beoordeeld uit de eerdere richtlijn (NVDV, 2014) op noodzaak tot revisie. Tevens zijn er knelpunten aangedragen door de patiëntenvereniging. Tevens zijn de VIG, ZN, NVZ en NFU uitgenodigd om knelpunten aan te dragen tijdens de eerste bijeenkomst.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroep uitgangsvragen opgesteld. Per uitgangsvraag zijn klinisch relevante uitkomstmaten opgesteld, waarbij zowel naar gewenste als ongewenste effecten is gekeken. De werkgroep heeft deze uitkomstmaten gewaardeerd volgens hun relatieve klinisch belang bij de besluitvorming rondom aanbevelingen. Specifieke uitkomstmaten per uitkomstvraag worden in de betreffende paragrafen uitgewerkt. Klinische uitkomstmaten zoals het proportie patiënten die IGA response (clear/almost clear) behaalt en de reductie in ernst van het constitutioneel eczeem (EASI50 en SCORAD50). Maar ook patiënt gerapporteerde uitkomsten zoals afname en/of controle van symptomen (NRS-jeuk), verandering in DLQI score. Met betrekking tot daily practice research is drug survival de meest gebruikte uitkomstmaat. Deze werd meegenomen voor de lange-termijnveiligheid van systemische therapie.
Strategie voor zoeken en selecteren van literatuur
Voor de afzonderlijke uitgangsvragen is aan de hand van specifieke zoektermen een systematische zoekstrategie uitgevoerd in (verschillende) elektronische databases Embase, MEDLINE en Cochrane. In eerste instantie is gezocht naar studies met de hoogste mate van bewijs. De gevonden studies zijn steeds door twee arts-onderzoekers van de NVDV dan wel werkgroepleden onafhankelijk van elkaar geselecteerd op basis van titel en abstract en vooraf opgestelde selectiecriteria. De beoordeling en uiteindelijke selectie op basis van volledige tekst is gedaan door arts-onderzoekers van de NVDV en werkgroepleden. De geselecteerde studies zijn gebruikt om de uitgangsvraag te beantwoorden. De zoekstrategie is te vinden in bijlage B.
Kwaliteitsbeoordeling wetenschappelijk bewijs
De beoordeling van de kwaliteit van het wetenschappelijk bewijs en onderzoeksgegevens is in de (modulaire herziening van de) richtlijn voor het grootste deel tot stand gekomen met de EBRO-methode. De paragraaf ‘Dupilumab’ uit het hoofdstuk ‘systemische immunosuppressieve therapie’ is met de GRADE-methode uitgewerkt net als de paragraaf ‘effectiviteit van ureum bij patiënten met constitutioneel eczeem’ uit het addendum Ureum.
Kwaliteitsbeoordeling wetenschappelijk bewijs middels GRADE
Bij de GRADE-methode (Grading Recommendations Assessment, Development and Evaluation) worden individuele studies systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. [Schünemann 2013]
Tabel 1 geeft een kort overzicht van de indeling van methodologische kwaliteit van het wetenschappelijk bewijs volgens GRADE. De beoordelingen van de methodologische kwaliteit kunt u vinden in de Risk of Bias (RoB)-tabellen, deze zijn op te vragen via de NVDV en/of terug te vinden in bijlage C. Hiervoor is gebruikgemaakt van de Cochrane risk of bias tool. [Higgins 2011] GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag (tabel 1). Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie. [Schünemann, 2013] De kwaliteit van het bewijs per interventie per uitkomstmaat is te vinden in de tabellen met de Summary of Findings (zie bijlage C).
Een volledige uitleg over de GRADE-methode valt buiten het bestek van deze richtlijn, zie hiervoor het ‘GRADE handbook’. [Schünemann 2013, www.gradeworkinggroup.org]
Tabel 1. Indeling van kwaliteit van wetenschappelijk bewijs volgens GRADE
|
GRADE-systeem |
||
|
Kwaliteitsindeling bewijs |
Hoog |
|
|
Redelijk |
|
|
|
Laag |
|
|
|
Zeer laag |
|
|
|
Startkwalificatie |
- Gerandomiseerd onderzoek = hoog - Observationele studie = laag |
|
|
Factoren die de kwaliteit van bewijs kunnen verlagen* |
- Ernstige of zeer ernstige beperkingen in de kwaliteit van de studie - Indirectheid van het bewijs - Belangrijke inconsistentie tussen studies - Imprecisie - Grote kans op ‘publicatiebias’ |
|
|
Factoren die de kwaliteit van bewijs kunnen verhogen** |
- Sterk bewijs voor een associatie – significant relatief risico van > 2 (< 0,5) gebaseerd op consistent bewijs uit twee of meer observationele studies, zonder plausibele ‘confounders’ (+1) - Zeer sterk bewijs voor een associatie – significant relatief risico van > 5 (< 0,2) gebaseerd op direct bewijs zonder belangrijke bedreigingen voor de validiteit (+2) - Bewijs voor een dosis respons gradiënt (+1) - Alle plausibele ‘confounders’ zonder het effect te hebben verminderd (+1) |
|
* Elk criterium kan de kwaliteit verminderen met 1 stap of bij zeer ernstige beperkingen met 2 stappen.
** Verhogen kan alleen indien er geen beperkingen zijn t.a.v. de studiekwaliteit, imprecisie, inconsistentie, indirectheid en publicatiebias
Beoordelen van het niveau van het wetenschappelijke bewijs middels EBRO
Bij de EBRO-methode (Evidence Based RichtlijnOntwikkeling) wordt een andere classificatie voor de beoordeling van de kwaliteit van studies aangehouden (zie tabel 2). [van Everdingen 2004] Hierbij ligt de belangrijkheid van de uitkomstmaten niet van tevoren vast en is er geen vastgelegde procedure voor upgraden en downgraden van bewijs, zoals die bij GRADE geldt.
Tabel 2. Indeling van methodologische kwaliteit van individuele studies volgens EBRO
|
Kwaliteit |
Interventie |
Diagnostisch accuratesse- onderzoek |
Schade/ bijwerkingen*, etiologie, prognose |
|
A1 |
Systematische review van ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van A2-niveau |
||
|
A2
|
Gerandomiseerd dubbelblind vergelijkend klinisch onderzoek van goede kwaliteit van voldoende omvang |
Onderzoek ten opzichte van een referentietest (een ‘gouden standaard’) met tevoren gedefinieerde afkapwaarden en onafhankelijke beoordeling van de resultaten van test en gouden standaard, betreffende een voldoende grote serie van opeenvolgende patiënten die allen de index- en referentietest hebben gehad |
Prospectief cohortonderzoek van voldoende omvang en follow-up, waarbij adequaat gecontroleerd is voor ‘confounding’ en selectieve follow-up voldoende is uitgesloten. |
|
B |
Vergelijkend onderzoek, maar niet met alle kenmerken als genoemd onder A2 (hieronder valt ook patiënt-controleonderzoek, cohortonderzoek) |
Onderzoek ten opzichte van een referentietest, maar niet met alle kenmerken die onder A2 zijn genoemd |
Prospectief cohortonderzoek, maar niet met alle kenmerken als genoemd onder A2 of retrospectief cohortonderzoek of patiënt-controleonderzoek |
|
C |
Niet-vergelijkend onderzoek |
||
|
D |
Mening van deskundigen |
||
*Deze classificatie is alleen van toepassing in situaties waarin om ethische of andere redenen gecontroleerde trials niet mogelijk zijn. Zijn die wel mogelijk dan geldt de classificatie voor interventies.
Bij het werken volgens de EBRO-methode zijn op basis van de beschikbare literatuur een of meerdere conclusies geformuleerd. Afhankelijk van het aantal onderzoeken en de mate van bewijs is een niveau van bewijskracht toegekend aan de conclusie (zie tabel 3). [van Everdingen 2004]
Tabel 3. Niveau van conclusies volgens EBRO
|
Niveau |
Conclusie gebaseerd op |
|
1 |
Onderzoek van niveau A1 of ten minste 2 onafhankelijk van elkaar uitgevoerde onderzoeken van niveau A2 |
|
2 |
1 onderzoek van niveau A2 of ten minste 2 onafhankelijk van elkaar uitgevoerde onderzoeken van niveau B |
|
3 |
1 onderzoek van niveau B of C |
|
4 |
Mening van deskundigen |
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde studies zijn overzichtelijk weergegeven als ‘karakteristieken en resultaten van geïncludeerde studies’ zie bijlage C. De belangrijkste bevindingen uit de literatuur met betrekking op de vooraf opgestelde uitkomstmaten zijn beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies zijn de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Formuleren van conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de EBRO- of GRADE-methode. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten werden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overige overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn voor zowel de GRADE- en EBRO-methodiek gebaseerd op het beschikbare wetenschappelijke bewijs, de belangrijkste overige overwegingen en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht of het niveau van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling.
Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht (zie bijlage G).
Commentaar- en autorisatiefase
De conceptrichtlijn is aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren zijn verzameld in een commentaarformulier/tabel en besproken met de werkgroep. Naar aanleiding van de commentaren is de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn is aan de betrokken (wetenschappelijke) verenigingen en (patiënt)organisaties voorgelegd ter autorisatie en door hen geautoriseerd dan wel geaccordeerd. Zie daarvoor paragraaf ‘Autorisatie’.
Autorisatie
In 2014 werd de gehele richtlijn geautoriseerd door: Nederlandse Vereniging voor Dermatologie en Venereologie (NVDV), Nederlandse Vereniging voor Kindergeneeskunde (NVK), Nederlandse Vereniging voor Allergologie (NVvA), Nederlands Huisartsen Genootschap (NHG), Nederlands Instituut van Psychologen (NIP), Nederlandse Vereniging voor Arbeids- en Bedrijfsgeneeskunde (NVAB), Verpleegkundigen & Verzorgenden Nederland (V&VN) en de Vereniging voor Mensen met Constitutioneel Eczeem (VMCE)
De in 2019 herziene modules in deze richtlijn werden geautoriseerd door: Nederlandse Vereniging voor Dermatologie en Venereologie (NVDV), Nederlandse Vereniging voor Allergologie (NVvA) en de Vereniging voor Mensen met Constitutioneel Eczeem (VMCE) op 15 november 2019. De V&VN en HPN onderschrijven de richtlijn Het Nederlands Huisartsen Genootschap (NHG) gaf een verklaring van geen bezwaar af. De Nederlandse Vereniging voor Kindergeneeskunde (NVK) heeft de richtlijn geautoriseerd onder voorwaarde dat de module over monitoring en dosering van systemische en immunosuppressieve therapie bij kinderen herzien wordt conform de aanbevelingen van het Kinderformularium én de NVK richtlijn Medicamenteuze behandeling van kinderen met juveniele idiopathische artritis.
Literatuur
- Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348.
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
- Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwaliteit. Online beschikbaar op http://richtlijnendatabase.nl/
- van Everdingen JJE, Burgers JS, Assendelft WJJ, et al. Evidence-based richtlijnontwikkeling. Bohn Stafleu Van Loghum 2004.
- Roekevisch E, Spuls PI, Kuester D, et al. Efficacy and safety of systemic treatments for moderate-to-severe atopic dermatitis: A systematic review. J Allergy Clin Immunol 2014;133(2):429-438
- Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
- Sidbury R, Davis DM, Cohen DE et al. American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014 Aug;71(2):327-49.