Omalizumab
Uitgangsvraag
Wat is de effectiviteit en veiligheid van omalizumab in de behandeling van chronische spontane urticaria?
Aanbeveling
Aanbeveling volgens de GRADE-methode
|
Sterk voor |
Er is een sterke aanbeveling voor behandeling van patiënten met chronische spontane urticaria met omalizumab als add-on therapie. |
Slotaanbevelingen
Op basis van de hoge kwaliteit van bewijs, het gunstige effect, in relatie tot de bijwerkingen en kosten past het gebruik van omalizumab bij patiënten met chronische spontane urticaria als derde stap in het stepped-care model.
Het wordt aanbevolen om niet eerder dan zes maanden na de eerste manifestatie van CSU te starten met omalizumab.
Het wordt aanbevolen te starten met omalizumab bij patiënten die onvoldoende reageren op opgedoseerde 2de generatie antihistaminica, met tenminste een matige ziekteactiviteit, gedefinieerd als UCT ≤ 12, UAS7 ≥16 en DLQI ≥ 6.
Bij een duidelijke discrepantie tussen de vragenlijsten kan door de behandelend arts worden afgeweken van deze criteria.
Bij gebruik van omalizumab beveelt de werkgroep aan ten minste de volgende parameters te monitoren op de baseline en daarna iedere 3-6 maanden te registreren: UAS7, UCT, DLQI en bijwerkingen.
Bij een (vrijwel) complete remissie wordt aanbevolen de therapie aan te passen. Bij gebrek aan bewijs in de literatuur wordt, op basis van expert opinion, aanbevolen het interval tussen de behandelingen met stappen van één week te verlengen tot 8 weken en vervolgens de therapie te staken.
Is er na drie maanden geen effect van de behandeling met omalizumab dan moet worden overwogen de behandeling aan te passen. De expert opinion van de werkgroep is om in dit geval de dosis te verhogen of het interval te verkorten.
Omalizumab is een add-on therapie. De verhoogde dosis antihistaminica is de basis van de behandeling en wordt tijdens de behandeling met omalizumab gecontinueerd, mits dit door de patiënt verdragen wordt.
Overwegingen
De algehele kwaliteit van bewijs omtrent het gebruik van omalizumab bij CSU is hoog. De geïncludeerde RCT’s zijn van hoge kwaliteit en kunnen met een zekere inschatting de voordelen en risico’s laten zien. De gunstige effecten overtreffen hierin de nadelige effecten / bijwerkingen aanzienlijk. Nadelige effecten die overwogen dienen te worden zijn de bijwerkingen zoals besproken en de ongemakken van subcutane toediening zoals reacties op de injectieplaats kunnen voorkomen zoals pijn, jeuk, erytheem en zwelling. Deze bijwerkingen lijken slechts kortdurend op te treden. Bijwerkingen voor omalizumab zijn voor CSU voor slechts zes maanden behandelduur beschreven, voor patiënten met astma zijn meer data beschikbaar. Uit de astmaliteratuur is bekend dat een anafylactische reactie op kan treden. De observatie omtrent behandeling met omalizumab is bij CSU vooralsnog niet anders dan wanneer er voor astma behandeld wordt. Ook is voorzichtigheid is geboden bij auto-immuunziekten, immuuncomplex-gemedieerde aandoeningen, reeds bestaande nier- of leverinsufficiëntie en bij patiënten met een groot risico van worminfecties, vooral wanneer zij naar gebieden reizen waar deze infecties endemisch zijn. Dit dient onder andere overwogen te worden bij patiënten met zowel CSU als astma. Een uitgebreid overzicht van alle contra-indicaties en interacties is tevens te vinden in het Farmacotherapeutisch Kompas of Informatorium Medicamentorum.
De werkgroep schat in dat vrijwel alle patiënten hetzelfde perspectief hanteren wat betreft de behandeling met omalizumab. De verwachting is dat een klein aantal patiënten gezien de bijwerkingen van de toediening d.m.v. injectie of het regelmatige ziekenhuisbezoek dat hiermee gepaard gaat, zal afzien van het geneesmiddel. Het is belangrijk om uit te leggen aan de patiënt dat omalizumab als add-on middel is geregistreerd. Soms is het mogelijk om andere add-on therapieën dan antihistaminica te staken.
De werkgroep heeft er vertrouwen in dat alle zorgverleners hetzelfde perspectief op de wenselijkheid van de aan te bieden interventie hanteren. Ook zijn er voldoende condities van zorg-organisatorische aard aanwezig om de interventie toe te passen. De zorg is vergelijkbaar met de zorg omtrent het toedienen van andere biologics, hoewel bij behandeling met omalizumab screening op nier- en leverfunctiestoornissen en latente TBC niet noodzakelijk is.
De gemiddelde dosering van omalizumab in de eerder genoemde gerandomiseerde gecontroleerde studies is 300mg per maand, toegediend als twee subcutane injecties van 150mg. Hierop is ook de GRADE analyse gebaseerd en in de dagelijkse praktijk is dit de meest gebruikte dosering. Het doseren van 600mg per maand is in één studie ook onderzocht, maar was in die studie op basis van meerdere uitkomstmaten niet effectiever dan placebo (Saini et al., 2011).
De mening van de werkgroep is dat in sommige gevallen het verhogen van de dosering of het verkorten van het interval tussen de injecties wel degelijk effect kan hebben.
De totale kosten van één behandeling met omalizumab (Xolair®) zijn €796,51 (2 injectiespuiten à 150mg) (medicijnkosten.nl, december 2014). Gezien de ernst van de ziekte en de gevolgen voor het functioneren van patiënten, de indirecte kosten en socio-economische impact is de werkgroep is van mening dat de netto-gunstige effecten de kosten / middelen waard zijn. Er is een kosteneffectiviteitsanalyse uitgevoerd door zowel de farbikant als door onafhankelijke medewerkers van het UMCU, zie rapport kosteneffectiviteit voor beide rapporten. Uit deze modelmatige analyse is omalizumab kosteneffectief gebleken ten opzichte van behandeling met de standaardzorg van antihistaminica.
De werkgroep vindt dat in het doorlopen van de stap van opgedoseerde tweede generatie antihistaminica naar omalizumab de ziekte-ernst in acht genomen moet worden (zie voor het stepped-care model de module stepped-care behandeling). De mogelijke grootte van de verbetering van kwaliteit van leven en ziektevermindering in ogenschouw nemende, stelt de werkgroep dat het pas nuttig lijkt met omalizumab te behandelen vanaf matige ziekte-ernst. In gerandomiseerde, gecontroleerde studies naar effectiviteit en veiligheid van omalizumab werd als inclusiecriterium een UAS7-score van ≥16 aangehouden (Kaplan et al., 2013; Maurer et al., 2013; Saini et al., 2014), waarvan men stelt dat dit overeenkomt met matige tot ernstige ziekteactiviteit. De werkgroep wil dit afkappunt graag overnemen. Ook stelt de werkgroep dat de UCT bij het onvoldoende ziektecontrole (UCT≤11) zou kunnen dienen als afkapwaarde voor het veranderen / opdoseren van therapie. De afkapwaarde van tenminste matige impact op kwaliteit van leven, een DLQI van zes of hoger, zou de werkgroep ook willen toevoegen aan de voorwaarden voor het starten van omalizumab.
Uitzondering hierop is zijn patiënten die reeds gebruik maken van systemische immunosuppressiva (zoals corticosteroiden of ciclosporine A) met onvoldoende effect.
Bij een duidelijke discprepantie tussen de drie scores kan de behandelend arts beslissen af te wijken van bovenstaande en toch te starten met omalizumab.
Omalizumab passeert de placenta. Er zijn geen gegevens bekend over teratogeniteit of over de overgang in moedermelk bij mensen. Bij dieren is dit wel aangetoond. Gebruik van omalizumab bij zwangerschap of lactatie wordt afgeraden. (Informatorium Medicamentorum en Farmacotherapeutisch kompas).
Op basis van expert opinion lijkt het de werkgroepleden zinvol, afhankelijk van de effectiviteit, elke 3-6 maanden de therapie te herevalueren.
Om extra data te genereren over de effectiviteit en veiligheid van omalizumab bij CSU patiënten wordt er in enkele centra in Nederland al gebruik gemaakt van (decentrale) registraties. Deze registraties bevatten tenminste:
- UAS7 (baseline en iedere 3 - 6 maanden tijdens de behandeling)
- UCT (baseline en iedere 3 - 6 maanden tijdens de behandeling)
- DLQI (baseline en iedere 3 - 6 maanden tijdens de behandeling)
- Bijwerkingen (iedere 3 - 6 maanden tijdens de behandeling)
- Lab (baseline).
De werkgroep pleit ervoor dat iedere dermatoloog een dergelijke registratie gaat bijhouden om de patiënt beter te kunnen volgen, en om eventueel in studieverband meer observationele data te genereren over de effectiviteit en veiligheid van omalizumab.
In de internationale en Amerikaanse richtlijn (Zuberbier et al., 2014; Bernstein et al., 2014) wordt niet gesproken over het eventuele moment van staken van omalizumab. Gezien de kosten en de kans een eventuele spontane remissie onder omalizumab heeft de werkgroep besloten hier wel enkele (expert opinion) aanbevelingen over te doen.
Bij aanvang van de therapie en vervolgens om de drie maanden dient de UAS7 afgenomen te worden.
Bij een (vrijwel) complete remissie wordt aanbevolen de therapie aan te passen. Bij gebrek aan bewijs in de literatuur beveelt de werkgroep aan, op basis van expert opinion, om het interval tussen de behandelingen met stappen van één week te verlengen tot 8 weken en vervolgens de therapie te staken.
Is er na drie maanden geen effect van de behandeling met omalizumab dan moet worden overwogen de behandeling aan te passen. De expert opinion van de werkgroep is om in dit geval de dosis te verhogen of het interval te verkorten. Blijft het effect onvoldoende dan kan men doorgaan naar stap vier in het stepped-care model.
Omalizumab is een add-on therapie. De verhoogde dosis antihistaminica is de basis van de behandeling en wordt tijdens de behandeling met omalizumab gecontinueerd, mits dit door de patiënt verdragen wordt.
Onderbouwing
Achtergrond
Omalizumab is een monoklonaal antilichaam dat selectief bindt aan humaan immunoglobuline E (IgE). Het complex dat zich vormt voorkomt binding IgE aan de hoog-affiene IgE receptor (FcɛRI) op het oppervlak van o.a. mestcellen en basofiele granulocyten. Hierdoor neemt de hoeveelheid vrije IgE die beschikbaar is af, en wordt tevens de receptorexpressie verminderd. Het precieze werkingsmechanisme is echter niet volledig opgehelderd. Omalizumab is sinds 2005 in Europa geregistreerd voor astma en sinds februari 2014 is dit middel ook geregistreerd als aanvullende therapie voor de behandeling van chronische spontane urticaria (CSU) bij volwassen en adolescente (12 jaar en ouder) patiënten die onvoldoende reageren op behandeling met H1-antihistaminica.
Conclusies / Summary of Findings
|
Hoog |
Uitkomstmaat: Verbetering ziekteactiviteit in vergelijking tot baseline (cruciaal)
Er is een hoge kwaliteit van bewijs dat de ziekteactiviteit, gemeten middels de UAS7, significant lager is na behandeling met omalizumab in vergelijking met placebo. De minimal important difference (MID) van de UAS7 ligt tussen de 9,5 en 10,5. Een verschil van -11,58 is hiermee dus een klinisch relevante verbetering
Saini et al., 2014; Kaplan et al., 2014; Maurer et al., 2013; Maurer et al,. 2011; Saini et al., 2011 |
|
Hoog |
Uitkomstmaat: Verbetering kwaliteit van leven (cruciaal)
Er is een hoge kwaliteit van bewijs dat de kwaliteit van leven gemeten middels de CU-Q2oL significant verbetert na behandeling met omalizumab in vergelijking met placebo. De klinische relevantie hiervan is echter nog niet duidelijk aangezien de ‘minimal important difference’ (MID) nog niet is vastgesteld.
Saini et al., 2014; Kaplan et al., 2014; Maurer et al., 2013; Maurer et al., 2011 |
|
Hoog |
Uitkomstmaat: Proportie patiënten met bijwerkingen (cruciaal)
Er is een hoge kwaliteit van bewijs dat de proportie patiënten met bijwerkingen na behandeling met omalizumab niet significant verschilt van behandeling met placebo.
Saini et al., 2014; Kaplan et al., 2014; Maurer et al., 2013; Maurer et al., 2011; Saini et al., 2011 |
|
Hoog |
Uitkomstmaat: Proportie patiënten met een partiële respons (belangrijk)
Er is een hoge kwaliteit van bewijs dat significant meer patiënten een partiële respons bereiken door de behandeling met omalizumab dan met de behandeling van placebo.
Saini et al., 2014; Kaplan et al., 2014; Maurer et al., 2013; Saini et al., 2011. |
|
Hoog |
Uitkomstmaat: Proportie patiënten met complete respons (belangrijk)
Er is een hoge kwaliteit van bewijs dat significant meer patiënten een complete respons (UAS7=0) bereiken door de behandeling met omalizumab versus placebo.
Saini et al., 2014; Kaplan et al., 2014; Maurer et al., 2013 |
|
Hoog |
Uitkomstmaat: Proportie angio-oedeem vrije dagen (belangrijk)
Er is een hoge kwaliteit van bewijs dat de proportie angio-oedeem vrije dagen significant hoger is na de behandeling met omalizumab versus placebo.
Saini et al., 2014; Kaplan et al., 2014; Maurer et al., 2013 |
|
- |
Uitkomstmaat: Proportie patiënten met remissie binnen 1 maand (onbelangrijk)
Geen van de geïncludeerde studies heeft deze uitkomstmaat bepaald. |
Samenvatting literatuur
Beschrijving studies
Vijf gerandomiseerde, gecontroleerde studies met in totaal 1116 patiënten werden geïncludeerd. Drie grote fase III studies werden verricht naar de effectiviteit en veiligheid van omalizumab als add-on bij CSU; ASTERIA I (Saini et al., 2014), ASTERIA II (Maurer et al., 2013), en GLACIAL (Kaplan et al., 2013). In ASTERIA I en II zijn de effectiviteit en veiligheid van 75mg, 150mg en 300mg omalizumab onderzocht in dubbelblinde, gerandomiseerde, placebogecontroleerde studies bij 319 en 323 patiënten respectievelijk met CSU die matige of ernstige klachten hadden ondanks behandeling met H1-antihstaminica. De medicatie werd elke vier weken subcutaan toegediend. Inclusiecriteria voor alle drie de studies waren een UAS7 van 16 of hoger (schaal 0-42) en een ISS over één week van 8 of hoger (schaal 0-21).De ASTERIA studies waren identiek in design, maar verschillen in behandelduur, met 24 weken behandeling in ASTERIA I en 12 weken in ASTERIA II. Beide studies hadden een follow-up duur van 16 weken. Beide studies hadden als primaire uitkomstmaat de verandering in wekelijkse jeukscore in vergelijking met de baseline en keken tevens naar de verandering in de UAS7, de kwaliteit van leven (DLQI en CU-Q2oL), het aantal patiënten met een complete en partiële respons en het aantal (ernstige) bijwerkingen.
De derde fase III studie, GLACIAL, was een dubbelblinde, placebogecontroleerde multicentrum RCT bij 335 patiënten met symptomatische CSU ondanks behandeling met tot maximaal vier maal daags gedoseerde antihistaminica gecombineerd met H2-antihistaminica en / of leukotriënenantagonisten (Kaplan et al., 2013). Behandeling met omalizumab 300mg elke vier weken werd vergeleken met placebo injecties gedurende 24 weken behandeling. De follow-up duur was 16 weken. Het primaire onderzoeksdoel was het bepalen van de veiligheid van omalizumab door middel van het registreren van (ernstige) bijwerkingen, veranderingen in vitale parameters en laboratoriumbepalingen. Daarnaast werden ook alle uitkomstmaten m.b.t. effectiviteit zoals in de ASTERIA I en II studies in deze studie onderzocht.
In zowel ASTERIA I als II (Saini et al., 2014; Maurer et al., 2013) mochten patiënten de H1-antihistaminica die ze gebruikten vóór de randomisatie door blijven gebruiken. Ook in de GLACIAL studie (Kaplan et al., 2013) mochten patiënten de medicatie (H1-antihistaminica met H2-antihistaminica en / of leukotriënenantagonisten) die ze vóór randomisatie gebruikten blijven gebruiken. In deze drie studies mocht diphenhydramine 25mg als noodmedicatie worden gebruikt (maximaal drie doseringen per 24 uur).
Naast deze drie fase III studies waren ook twee fase II studies verricht. In de fase II studie (MYSTIQUE; Saini et al., 2011) met 90 patiënten met resistente CSU werd de eenmalige toediening van 75mg, 300mg en 600mg vergeleken met placebo in een dubbelblinde, placebogecontroleerde RCT met een follow-up duur van 12 weken. De studie was uitgevoerd in de VS en in Duitsland. In deze studie mochten patiënten als noodmedicatie diphenhydramine 25mg gebruiken. De primaire uitkomstmaat was de verandering in de ziekteactiviteit gemeten met behulp van de UAS7 (range 0-42) in vergelijking met de baseline.
In de fase II studie genaamd ‘X-QUISITE’ (Maurer et al. 2011) zijn 49 patiënten met resistente CSU en IgE-antilichamen tegen thyreoperoxidase (TPO) ondanks antihistaminica behandeld met omalizumab vs. placebo gedurende 24 weken. Patiënten kregen omalizumab 75-375mg iedere twee tot vier weken subcutaan toegediend volgens het schema dat werd gebruikt bij allergisch astma, waarbij de dosering omalizumab was gebaseerd op lichaamsgewicht en hoogte van de IgE-titer. Patiënten mochten als noodmedicatie loratadine 10mg of clemastine 1mg gebruiken. Ook in deze studie was de primaire uitkomstmaat de verandering in de UAS7 in vergelijking met de baseline.
Kwaliteit van bewijs
Alle studies zijn dubbelblinde, placebogecontroleerde multicenter RCT’s. De randomisatiemethode gebruikt in alle studies was adequaat beschreven. In de studies ASTERIA I en II, GLACIAL en MYSTIQUE was gerandomiseerd door middel van een ‘interactive voice and web response system’. Er was gebruik gemaakt van centrale randomisatie, waarmee de geblindeerde toewijzing (‘concealment of allocation’) gewaarborgd is (Kaplan et al., 2013; Maurer et al., 2013; Saini et al., 2011; Saini et al., 2014). In alle studies waren zowel de patiënten als de onderzoekers geblindeerd en de werkgroep heeft voldoende vertrouwen dat dit adequaat is gebeurd. De injecties in de studies hadden, behoudens het onderzoeksgeneesmiddel omalizumab, dezelfde ingrediënten en hadden een identiek uiterlijk. De studiemedicatie werd toegediend door een persoon die niet betrokken was bij het verzamelen van klinische gegevens van de patiënten. Ook het blinderen van de uitkomstmaten leek in alle onderzoeken adequaat te zijn gebeurd. De uitkomstmaten in deze studies waren zowel door patiënten als onderzoekers bepaald. De blindering van zowel patiënten als onderzoekers leek adequaat te zijn gebeurd. De statistici waren niet geblindeerd, zij waren echter niet in direct contact met de patiënten en onderzoekers, en werkten zij met reeds gerapporteerde en onwijzigbare data.
In de studies van ASTERIA I, II en GLACIAL (Kaplan et al., 2013; Maurer et al., 2013; Saini et al., 2014) is gebruik gemaakt van modified intention to treat (mITT) analyses, waarbij alle gerandomiseerde patiënten met ten minste één dosering in de analyse zijn meegenomen; bij deze studies was slechts één gerandomiseerde patiënt niet meegenomen in de analyse. Ondanks het gebruiken van een per-protocol analyse, heeft dit waarschijnlijk niet geleid tot bias. Het aantal patiënten dat tijdens de studie was uitgevallen was 10.2% tot 16.9%, de lost to follow up was bij alle studies <2% (laag), en beide waren redelijk tot goed gebalanceerd over de groepen. Dit is voor alle studies beoordeeld als een laag risico op (attrition) bias.
Alle studies hadden gebalanceerde studiegroepen tijdens de baseline. In de studies ASTERIA I, ASTERIA II en GLACIAL heeft meer dan 95% van de patiënten adequaat een symptoomdagboek ingevuld (Kaplan et al., 2013; Maurer et al., 2013; Saini et al., 2014). Alle studies leken vrij van selectieve rapportage. In alle studies werd gebruik gemaakt van gelden ontvangen uit de farmaceutische industrie. Voor het bepalen van de uitkomstmaten was gebruik gemaakt van gevalideerde vragenlijsten zoals de UAS7, de DLQI en de CU-Q2oL. Concluderend is de werkgroep van mening dat al deze studies een laag risico op bias hebben.
Effectiviteit omalizumab versus placebo
Voor de meta-analyses zijn de data van 300mg en 75-375mg omalizumab gepoold, derhalve zijn de data van in totaal 749 patiënten gepoold. Meer details van deze resultaten is weergegeven in de Evidence tabellen.
Verbetering ziekteactiviteit in vergelijking tot de baseline
Alle vijf studies met in totaal 749 patiënten (464 omalizumab, 285 placebo) hadden de ziekteactiviteit in vergelijking met de baseline onderzocht. De ziekteactiviteit was bepaald middels de UAS7. Gemiddeld was de UAS7 na behandeling 11,58 punten lager in de omalizumab 300mg groep dan in de placebo groep (mean difference -11,58; 95%CI -13,39 tot -9,77; P<0,00001). De minimal important difference (MID) van de UAS7 lag tussen de 9,5 en 10,5(Mathias et al., 2012). Een verschil van -11,58 is hiermee dus een klinisch relevante verbetering.
Verbetering kwaliteit van leven in vergelijking tot de baseline
Vier studies hassen de kwaliteit van leven in vergelijking met de baseline bepaald met in totaal 703 patiënten (439 omalizumab en 264 placebo) (Kaplan et al., 2013; Maurer et al., 2011; Maurer et al., 2013; Saini et al., 2014). De kwaliteit van leven was bepaald middels de CU-Q2oL (range 0-115). De CU-Q2oL was na de behandeling met omalizumab 300mg gemiddeld 13,12 punten lager dan na de behandeling met placebo (mean difference -13,12; 95%CI -16,3 tot -9,95; P<0,00001). De klinische relevantie van dit verschil is niet duidelijk omdat de MID nog niet bepaald is.
Proportie patiënten met complete respons
Complete respons was bij 749 patiënten bepaald in alle vijf studies (464 omalizumab, 285 placebo). Complete respons werd in 38,1% van de gevallen bereikt na behandeling met omalizumab 300mg, en in 5,6% van de gevallen na behandeling met placebo. In vier studies was de complete respons bepaald middels een UAS7 van 0 (Kaplan et al., 2013; Maurer et al., 2013; Saini et al., 2011; Saini et al., 2014), in één studie was dit bepaald middels een complete resolutie van urticaria symptomen (Maurer et al., 2011). Het relatieve risico van 6,44 (95%CI 3,93 tot 10,43; P<0,00001). Concreet betekent dit dat 305 keer vaker complete respons wordt bereikt per 1000 patiënten in vergelijking met placebo.
Proportie patiënten met een partiële respons
De uitkomstmaat partiële respons was in vier studies bepaald (700 patiënten waarvan 437 omalizumab en 263 placebo) (Kaplan et al., 2013; Maurer et al., 2013; Saini et al., 2011; Saini et al. 2014). Deze uitkomstmaat was bepaald middels de UAS7≤6, óf middels de proportie patiënten met 75% verbetering in UAS7 in vergelijking met de baseline. Het relatieve risico op een partiële respons was 4,08 (95%CI 2,98 tot 5,60; P<0,00001). Per 1000 patiënten wordt 422 keer vaker een partiële respons bereikt na de behandeling met omalizumab 300mg in vergelijking met placebo.
Proportie angio-oedeem vrije dagen
De proportie angio-oedeem vrije dagen (range 0-100%) was in drie studies onderzocht onder in totaal 576 patiënten (372 omalizumab, 204 placebo) (Kaplan et al., 2013; Maurer et al., 2013; Saini et al., 2014). Hierbij was het aantal dagen dat patiënten in hun studiedagboek hebben aangegeven dat zij geen angio-oedeem hadden gedeeld door het aantal dagen dat dit dagboek was ingevuld gedurende de periode 4-12 weken na de eerste gift. De proportie angio-oedeem vrije dagen was gemiddeld 5,66% hoger in vergelijking met placebo (mean difference 5,66%, 95%CI 2,55 tot 8,76); P=0,0004). Het percentage angio-oedeem vrije dagen in de placebogroep lag echter ook hoog (range 88,1%-89,2%). Alhoewel dit verschil statistisch significant is, is het de vraag of dit verschil klinisch relevant is.
Begin van remissie binnen 1 maand
Deze uitkomstmaat is in geen van de studies onderzocht.
Veiligheid
Proportie patiënten met bijwerkingen
Het aantal bijwerkingen tijdens de behandeling was in alle studies beschreven bij in totaal 749 patiënten (464 omalizumab, 285 placebo). Deze uitkomstmaat was bepaald aan de hand van de proportie patiënten met bijwerkingen tijdens de behandeling. Bijwerkingen kwamen bij 64,2% voor in de placebo groep en bij 73,7% in de omalizumab 300mg groep. Het relatieve risico (RR) was 1,05, (95%CI 0,96 tot 1,16). Dit risico op bijwerkingen was niet significant verschillend in vergelijking met placebo (P=0,29). Concreet betekent dit dat 32 keer vaker een bijwerking werd gerapporteerd per 1000 patiënten in vergelijking met placebo.
De meest voorkomende bijwerkingen (1-10%) bij gebruik van omalizumab zijn hoofdpijn en lokale reacties op de injectieplaats zoals jeuk, roodheid en pijn.
Allergische reacties (anafylaxie, angio-oedeem, lanrynxoedeem) kunnen optreden (zelden: 0,01 – 0,1%), veelal binnen 2 uur na toediening, maar soms ook 2-24 uur na toediening (Informatorium Medicamentorum, kennisbank KNMP).
Het aantal ernstige bijwerkingen (gedefinieerd als een bijwerking die levensbedreigend is of die resulteert in (verlenging van) opname, aangeboren aandoeningen of overlijden, of wanneer een interventie nodig is om bovenstaande te voorkomen) tijdens de behandeling in de vijf geïncludeerde studies varieerde van 0%-6,9% in de placebogroep, en van 0%-7,1% in de omalizumab groepen.
In de productinformatie van Xolair® staan de bijwerkingen genoteerd die voorkomen bij ≥1% van de patiënten en bijwerkingen die ≥2% vaker voorkomen in de behandelgroep met omalizumab dan met placebo (na medische beoordeling) die gerapporteerd zijn in de drie fase III studies bij 300mg omalizumab (European Medicines Agency, 2013). Tabel 1 toont de gepoolde data van Maurer et al. (2013), Saini et al. (2014) en Kaplan et al. (2013).
Tabel 1. Bijwerkingen afkomstig van de gepoolde CSU veiligheidsdatabase (dag 1 tot week 24) bij 300mg omalizumab (vertaald van European Medicines Agency, 2013 en Novartis Europharm, 2014)
|
12 Weken Omalizumab studies 1, 2 en 3 Gepoold |
Frequentiecategorie |
||
|
Placebo N=242 |
300mg N=412 |
|
|
|
Infecties en parasitaire aandoeningen |
|
||
|
Sinusitis |
5 (2,1%) |
20 (4,9%) |
Vaak** |
|
Zenuwstelselaandoeningen |
|
||
|
Hoofdpijn |
7 (2,9%) |
25 (6,1%) |
Vaak |
|
Skeletspierstelsel- en bindweefselaandoeningen |
|
||
|
Artralgie |
1 (0,4%) |
12 (2,9%) |
Vaak |
|
Algemene aandoeningen en toedieningsplaatsstoornissen |
|
||
|
Reactie op de injectieplaats* |
2 (0,8%) |
11 (2,7%) |
Vaak |
|
24 Weken Omalizumab studies 1 en 3 Gepoold |
Frequentiecategorie |
||
|
Placebo N=163 |
300mg N=333 |
|
|
|
Infecties en parasitaire aandoeningen |
|
||
|
Bovenste luchtweginfectie |
5 (3,1%) |
19 (5,7%) |
Vaak |
* Alhoewel er geen verschil van 2% met placebo was aangetoond, zijn reacties op de injectieplaats wel opgenomen, aangezien alle gevallen zijn beoordeeld als causaal gerelateerd aan de studiebehandeling.
**vaak (≥1 / 100, <1 / 10)
Zoeken en selecteren
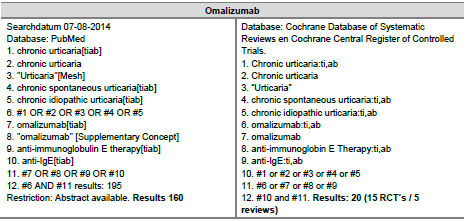
Voor deze uitgangsvraag is een systematische search verricht in de databases PubMed, the Cochrane Database of Systematic Reviews en Cochrane Central Register of Controlled Trials. De search leverde 160 artikelen op uit PubMed en 20 uit Cochrane.
Studies die voldeden aan de inclusiecriteria werden meegenomen in de beoordeling. Alleen studies over de behandeling van patiënten met chronische spontane of idiopathische urticaria werden geïncludeerd. Van de bruikbare literatuur is vervolgens een tweede selectie gemaakt op basis van de ‘piramide van evidence’. Hierbij werd eerst gekeken naar systematische reviews en meta-analyses, vervolgens naar gerandomiseerde, gecontroleerde trials (RCT’s) en vervolgens naar observationele onderzoeken. Op basis van de beschikbare literatuur is ervoor gekozen alleen de gerandomiseerde, gecontroleerde studies te includeren. Hierdoor zijn vijf RCT’s geïncludeerd die de veiligheid en effectiviteit van omalizumab bij patiënten met CSU hebben onderzocht (Kaplan et al., 2013; Maurer et al., 2011; Maurer et al., 2013; Saini et al., 2011; Saini et al., 2014). Meer details over de geïncludeerde studies zijn terug te vinden in de Evidencetabellen. In de literatuur is tevens een kleine RCT gevonden (20 patiënten) waar enkel een abstract met gelimiteerde data van is gepubliceerd, dit abstract is dan ook niet geïncludeerd (Gober et al., 2008).
Deze uitgangsvraag is uitgewerkt aan de hand van de GRADE-methode. De volgende voorafgaand bepaalde uitkomstmaten zijn gebruikt:
- verbetering ziekteactiviteit in vergelijking tot baseline (cruciaal)
- verbetering kwaliteit van leven in vergelijking tot baseline (cruciaal)
- proportie patiënten met bijwerkingen (cruciaal)
- proportie patiënten met complete respons (belangrijk)
- proportie patiënten met een partiële respons (belangrijk)
- proportie angio-oedeem vrije dagen (belangrijk)
- proportie patiënten met remissie binnen 1 maand (van beperkt belang).
Referenties
- Informatorium Medicamentorum, via kennisbank KNMP dd 7-7-2015.
- Gober LM, Sterba PM, Eckman JA, Saini SS. Effect of Anti-IgE (Omalizumab) in chronic idiopathic urticaria (CIU) patients. J Allergy Clin Immunol 2008;121:S147.
- Kaplan A, Ledford D, Ashby M, Canvin J, Zazzali JL, Conner E, Veith J, Kamath N, Staubach P, Jakob T, Stirling RG, Kuna P, Berger W, Maurer M, and Rosen K. 2013. Omalizumab in patients with symptomatic chronic idiopathic / spontaneous urticaria despite standard combination therapy. J. Allergy Clin. Immunol. 132 (1): 101-109.
- Mathias SD, Crosby RD, Zazzali JL, Maurer M, and Saini SS. 2012. Evaluating the minimally important difference of the urticaria activity score and other measures of disease activity in patients with chronic idiopathic urticaria. Ann. Allergy Asthma Immunol. 108 (1): 20-24.
- Maurer M, Altrichter S, Bieber T, Biedermann T, Brautigam M, Seyfried S, Brehler R, Grabbe J, Hunzelmann N, Jakob T, Jung A, Kleine-Tebbe J, Mempel M, Meurer M, Reich K, Rueff F, Schakel K, Sengupta K, Sieder C, Simon JC, Wedi B, Zuberbier T, Mahler V, and Staubach P. 2011. Efficacy and safety of omalizumab in patients with chronic urticaria who exhibit IgE against thyroperoxidase. J. Allergy Clin. Immunol. 128 (1): 202-209.
- Maurer M, Rosen K, Hsieh HJ, Saini S, Grattan C, Gimenez-Arnau A, Agarwal S, Doyle R, Canvin J, Kaplan A, and Casale T. 2013. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. N. Engl. J. Med. 368 (10): 924-935.
- Saini S, Rosen KE, Hsieh HJ, Wong DA, Conner E, Kaplan A, Spector S, and Maurer M. 2011. A randomized, placebo-controlled, dose-ranging study of single-dose omalizumab in patients with H1-antihistamine-refractory chronic idiopathic urticaria. J. Allergy Clin. Immunol. 128 (3): 567-573.
- Saini SS, Bindslev-Jensen C, Maurer M, Grob JJ, Bulbul BE, Bradley MS, Canvin J, Rahmaoui A, Georgiou P, Alpan O, Spector S, and Rosen K. 2014. Efficacy and Safety of Omalizumab in Patients with Chronic Idiopathic / Spontaneous Urticaria Who Remain Symptomatic on H Antihistamines: A Randomized, Placebo-Controlled Study. J. Invest Dermatol.
Evidence tabellen
Omalizumab Evidence tabel
|
Auteur, jaartal |
Studie type |
N (M/ V) |
Patiënten populatie In/exclusie |
Studieopzet / Studiemedicatie / Dosering / Comedicatie |
Behandeld uur |
Followup duur (mndn) |
Leeftijd
|
Uitkomstmaten |
Overige opmerkingen |
|
Saini et al. 2014
ASTERI A I, fase III |
RCT DB PC MC
|
319
6580% vrouw
|
Inclusie: - ptn met resistente CsU/CIU >8 wkn ondanks H1-AH. - UAS ≥16, ISS ≥8 - Leeftijd 12-75 jaar (Duitsland > 18)
Exclusie: Fysische urticaria Aanwezigheid ziekte met symptomen van urticaria en/of angio-oedeem Gebruik orale corticosteroïden, hydrochloroquine, MTX, CsA, LTRA, H1- of H2antihistaminica voorafgaand aan randomisatie.
|
Omalizumab 75mg (n=78) vs. 150mg (n=80) vs. 300mg (n=81) vs. Placebo (n=80) s.c. iedere 4 wkn. (1:1:1:1)
Comedicatie: Week 1-12 pre-randomisatie medicatie toegestaan Week 13-24,1 extra H1-AH toegestaan
Noodmedicatie: Diphenhydramine 25mg, max 3dd / 24 uur |
24 wkn
Primaire uitkomstm aat na 12 wkn
|
16 wkn |
75mg: 40,7 (15,2)
150mg: 41,1 (14,0)
300mg: 42.4 (13,2)
Placebo: 40,4 (15,6) |
- Primaire uitkomst was de verandering in ISS in vergelijking met de baseline na 12 wkn. Secondaire uitkomstmaten o.a.: - Verandering in UAS7 in vergelijking met de baseline - Proportie ptn met een complete respons (UAS7=0) - Proportie of ptn met een partiële respons (UAS7 <6) - Proportie angioedeem vrije dagen gedurende week 4 tot 12. - Verandering in kwaliteit van leven (CU-Q2oL) in vergelijking met de baseline
|
Uitgevoerd in 53 centra in de VS, Denemarken, Frankrijk, Duitsland, Italië, Polen, Spanje en Turkije. |
|
Auteur, jaartal |
Studie type |
N (M/ V) |
Patiënten populatie In/exclusie |
Studieopzet / Studiemedicatie / Dosering / Comedicatie |
Behandeld uur |
Followup duur (mndn) |
Leeftijd
|
Uitkomstmaten |
Overige opmerkingen |
|
Maurer et al. 2011
X- QUISIT E, fase II |
RCT DB PC MC |
49
(11 M / 38 V) |
Inclusie: - ptn met rCsU + IgE-anti-TPO - Leeftijd > 18 jaar - CU > 6 wkn ondanks antihistaminica. - Serum IgE Spiegel tussen 30IU/mL en 700 IU/mL - Serum specifiek IgE- anti TPO Spiegel ≥5.0 IU/mL of groter - UAS7>10
Exclusie: Acute urticaria, chronische diarree, nier dysfunctie, verhoogd IgE door andere reden dan allergie. Gebruik corticosteroïden, MTX, CsA of andere immunosuppressiva voor randomisatie. |
Omalizumab 75-375mg (n=27) vs. placebo (n=22)(1:1) s.c. iedere 2-4 wkn (astma schema)
Noodmedicatie: loratadine 10mg/dag / clemastine 1mg/dag |
24 wkn
Primaire uitkomstm aat na 24 wkn |
- |
Omalizumab : 39,1 ± 9,0
Placebo: 42,3 ± 15,0 |
- Primaire uitkomstmaat verandering in UAS7 in vergelijking met de baseline. Secundaire uitkomstmaat o.a.: - Verandering in kwaliteit van leven (DLQI / Skindex-29 / CU-Q2oL) - (ernstige) bijwerkingen
|
Alleen ptn met IgE-anti-TPO. Uitgevoerd in 16 centra in Duitsland. |
|
Saini et al. 2011
MYSTI QUE, fase II |
RCT DB PC MC
|
90
68% vrouw
|
Inclusie: - ptn met rCsU -Leeftijd: 12-75 (Duitsland > 18)jaar -UAS7 ≥12
|
Omalizumab 600 (n=21) vs. 300 (n=25)vs. 75mg (n=23) vs. placebo (n=21) (1:1:1:1) s.c. eenmalig
Noodmedicatie: Diphenhydramine 25mg, max 3dd/24 uur
|
4 wkn
Primaire uitkomstm aat na 4 wkn |
12 wkn |
40,8 |
- Primaire uitkomstmaat was de verandering in UAS7 in vergelijking met de baseline. - Secondaire uitkomstmaat o.a. de verandering in ISS in vergelijking met de baseline. - (ernstige) bijwerkingen |
Effect enkele dosis.
Uitgevoerd in VS en Duitsland |
|
Auteur, jaartal |
Studie type |
N (M/ V) |
Patiënten populatie In/exclusie |
Studieopzet / Studiemedicatie / Dosering / Comedicatie |
Behandeld uur |
Followup duur (mndn) |
Leeftijd
|
Uitkomstmaten |
Overige opmerkingen |
|
Maurer et al. 2013
ASTERI A II, fase III |
RCT DB PC MC
|
323
76% vrouw en |
Inclusie: - ptn met rCsU (ondanks H1-AH) - 12-75 jaar (Duitsland > 18) - UAS7 ≥16, ISS ≥8
Exclusie: Fysische urticaria Gebruik orale corticosteroïden, hydrochloroquine, MTX, CsA, LTRA, H1- of H2-antihistaminica voorafgaand aan randomisatie.
|
Omalizumab 300mg (n=79) vs. 150mg (n=82) vs. 75mg (n=82) vs. placebo (n=79) (1:1:1:1) s.c. iedere 4 wkn
Comedicatie: ptn bleven hun prerandomisatie H1-AH gebruiken.
Noodmedicatie: Diphenhydramine 25mg, max 3dd / 24 uur
|
12 wkn
Primaire uitkomstm aat na 12 wkn |
16 wkn |
|
- Primaire uitkomstmaat was de verandering in ISS in vergelijking met de baseline. Secondaire uitkomstmaten o.a. - Verandering in UAS7 in vergelijking met de baseline - Proportie ptn met een complete respons (UAS7=0) - Proportie ptn met een partiële respons (UAS7 <6) - Proportie angioedeem vrije dagen in week 4 tot 12. - Verandering in kwaliteit van leven (DLQI / Skindex-29 / CU-Q2oL) |
Niet alle resultaten uitkomstmaten weergegeven in artikel, maar in supplementary appendix.
Uitgevoerd in de VS, DenemarkenFrankrijk, Duitsland, Italië, Polen, Spanje en Turkije. |
|
Auteur, jaartal |
Studie type |
N (M/ V) |
Patiënten populatie In/exclusie |
Studieopzet / Studiemedicatie / Dosering / Comedicatie |
Behandeld uur |
Followup duur (mndn) |
Leeftijd
|
Uitkomstmaten |
Overige opmerkingen |
|
Kaplan et al. 2013
GLACI AL, fase III |
RCT BD PC MC
|
335
71,9% vrouw |
Inclusie: - ptn met rCsU (ondanks tot 4x dosering H1,H2 of LTRA) - CIU/CSU 6 mnd of langer; - Leeftijd: Range 12-75 jaar(Duitsland > 18 years) - jeuk en/of kwaddels > 6 wkn ondanks behandeling met H1antihistaminica, plus H2antihistaminica, LTRAs of een combinatie hiervan. - UAS7 ≥16 - ISS≥8 Exclusie: Fysische urticaria Gebruik orale corticosteroïden, hydrochloroquine, MTX, CsA, LTRA, H1- of H2-antihistaminica voorafgaand aan randomisatie.
|
Omalizumab 300mg (n=252) vs. placebo (n=83) (3:1) s.c. Iedere 4 wkn
Comedicatie: ptn bleven hun prerandomization H1-AH, H2-AH, LTRAs of een combinatie hiervan gebruiken.
Noodmedicatie: diphenhydramine 25mg, max 3dd / 24 uur. |
24 wkn
Primaire uitkomstm aat na 24 wkn, veiligheid na 12 wkn |
16 wkn |
43,1 (14,1) |
Primaire uitkomstmaten: - (ernstige )bijwerkingen Secundaire uitkomstmaten o.a.: - Verandering in ISS in vergelijking met de baseline - Verandering in UAS7 in vergelijking met de baseline - Verandering in UAS7 in vergelijking met de baseline - Proportie ptn met een complete respons (UAS7=0) - Proportie ptn met een partiële respons (UAS7 <6) proportie angioedeem vrije dagen in week 4 tot 12. - Verandering in kwaliteit van leven (DLQI / CU-Q2oL) |
Uitgevoerd in Australie, Duitsland, Nieuw-Zeeland, Polen, Singapore, UK en VS. |
Omalizumab: Summary of Findings table
Date: 2014-10-27
Question: Should omalizumab 300mg be used in chronic spontaneous urticaria?1,2,3
Settings: (Global and national) multicentre hospital trials
|
Quality assessment |
No of patients |
Effect |
Quality |
Importan ce |
Comments |
||||||||
|
No of participants (studies) |
Design |
Risk of bias |
Inconsistency |
Indirectness |
Imprecision |
Other considerations |
Omalizumab 300mg |
Control |
Relative (95% CI) |
Absolute |
|||
|
Change in disease activity from baseline (treatment 4-24 weeks4; measured with: Change in UAS7 from baseline; range of scores: 0-42; Better indicated by lower values) |
|
||||||||||||
|
749 (5 studies5 ) |
randomised trials |
no serious risk of bias |
no serious inconsistency |
no serious indirectness |
no serious imprecision |
none |
464 |
285 |
- |
MD 11.58 lower (13.39 to 9.77 lower) |
ÅÅÅÅ HIGH |
CRITICA L |
The MID of UAS7 lies between 9.5 and 10.5, therefore this difference of 11.58 is clinically important |
|
Change in quality of life from baseline (treatment 12-24 weeks4; measured with: Change in CU-Q2oL from baseline; range of scores: 0-115; Better indicated by lower values) |
|
||||||||||||
|
703 (4 studies6) |
randomised trials |
no serious risk of bias |
no serious inconsistency |
no serious indirectness |
no serious imprecision |
none |
439 |
264 |
- |
MD 13.12 lower (16.3 to 9.95 lower) |
ÅÅÅÅ HIGH |
CRITICA L |
|
|
Adverse events (treatment 4-24 weeks4; assessed with: Any adverse events during treatment) |
|
||||||||||||
|
749 (5 studies5) |
randomised trials |
no serious risk of bias |
no serious inconsistency |
no serious indirectness |
no serious imprecision |
none |
342/464 (73.7%) |
183/285 (64.2%) |
RR 1.05 (0.96 to 1.16) |
32 more per 1000 (from 26 fewer to 102 more) 24 more per 1000 (from 19 fewer to 77 more) 43 more per 1000 (from 34 fewer to 138 more) |
ÅÅÅÅ HIGH |
CRITICA L |
|
|
|
48% |
||||||||||||
|
|
86% |
||||||||||||
|
Complete response (treatment 4-24 weeks4; assessed with: UAS7=0 or complete resolution of urticarial symptoms8) |
|
||||||||||||
|
749 (5 studies5) |
randomised trials |
no serious risk of bias |
no serious inconsistency |
no serious indirectness |
no serious imprecision |
none |
177/464 (38.1%) |
16/285 (5.6%) |
RR 6.44 (3.95 to 10.49) |
305 more per 1000 (from 166 more to 533 more)
272 more per 1000 (from 148 more to 475 more) |
ÅÅÅÅ HIGH |
IMPORT ANT |
|
|
|
5% |
||||||||||||
|
Quality assessment |
No of patients |
Effect |
Quality |
Importance |
Comments |
||||||||
|
No of participants (studies) |
Design |
Risk of bias |
Inconsistency |
Indirectness |
Imprecision |
Other considerations |
Omalizumab 300mg |
Control |
Relative (95% CI) |
Absolute |
|||
|
Partial response (treatment 4-24 weeks4; assessed with: UAS7<6 or proportion of patients with 75% improvement in UAS7) |
|
||||||||||||
|
700 (4 studies9) |
randomised trials |
no serious risk of bias |
no serious inconsistency |
no serious indirectness |
no serious imprecision |
none |
241/437 (55.1%) |
36/263 (13.7%) |
RR 4.08 (2.98 to 5.6) |
422 more per 1000 (from 271 more to 630 more) |
ÅÅÅÅ HIGH |
IMPORTA NT |
|
|
|
9% |
277 more per 1000 (from 178 more to 414 more) |
|||||||||||
|
|
20% |
616 more per 1000 (from 396 more to 920 more) |
|||||||||||
|
Angioedema free days (treatment 12-24 weeks7; measured with: Proportion of angioedema free days; range of scores: 0-100; Better indicated by higher values) |
|
||||||||||||
|
576 (3 studies10) |
randomised trials |
no serious risk of bias |
no serious inconsistency |
no serious indirectness |
no serious imprecision |
none |
372 |
204 |
- |
MD 5.66 higher (2.55 to 8.76 higher) |
ÅÅÅÅ HIGH |
NOT IMPORTA NT |
|
|
Begin of remission within 1 month - not measured |
|
||||||||||||
|
- (0 studies) |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
|
NOT IMPORTA NT |
|
1. Study Maurer 2011 used an 'asthma scheme' of omalizumab 75-375mg
2. All patients were patients with CSU who remained symptomatic despite treatment,
3. Study Maurer 2011 comprised patients with CSU and IgE autoantibodies against TPO with persistent symptoms despite standard antihistamine therapy
4. Subcutaneous injections of omalizumab or placebo every 2-4 weeks
5. Studies Saini 2014; Saini 2011; Kaplan 2013; Maurer 2011; Maurer 2013
6. Studies Saini 2014; Maurer 2011; Maurer 2013; Kaplan 2013
7. Subcutaneous injections of omalizumab 300mg or placebo every 4 weeks
8. Study Maurer 2011 assessed complete resolution of urticarial symptoms using investigator global assessment of symptoms
9. Studies Saini 2014; Saini 2011; Maurer 2013; Kaplan 2013
10. Studies Kaplan 2013; Maurer 2013; Saini 2014
Verantwoording
Beoordelingsdatum en geldigheid
Publicatiedatum : 21-12-2015
Beoordeeld op geldigheid : 03-12-2015
Algemene gegevens
Deelnemende verenigingen/instanties:
Nederlandse Vereniging voor Allergologie (NVvA)
Nederlands Huisartsen Genootschap (NHG)
Nederlandse Vereniging voor Kindergeneeskunde (NVK) - Huidpatiënten Nederland (HPN).
Financiering
Deze richtlijn is tot stand gekomen met financiële steun vanuit het SKMS-programma
Deze richtlijn is geautoriseerd door (beoogd):
Nederlandse Vereniging voor Allergologie (NVvA)
Nederlandse Vereniging voor Kindergeneeskunde (NVK)
Huidpatiënten Nederland (HPN)
Zorgverzekeraars Nederland (ZN)
Nefarma.
Aanleiding
Urticaria wordt gekenmerkt door het plotseling optreden van urticae (kwaddels), angio-oedeem of beide. Bij continue of terugkerende klachten van urticae en / of angio-oedeem gedurende meer dan zes weken wordt er gesproken van chronische urticaria. Chronische urticaria kan een zeer belastende aandoening zijn. Het is een huidziekte die resulteert in rode jeukende, soms verheven kwaddels (urticae) op de huid van verschillende omvang en vorm, meestal gepaard gaand met heftige jeuk. Ook kan er sprake zijn van diepe zwellingen (angio-oedeem). Patiënten met chronische urticaria kunnen vele kwaddels ontwikkelen en elke dag klachten hebben. Het kan vele jaren duren en kan grote invloed hebben op de kwaliteit van leven, zowel lichamelijk als emotioneel, alsook voor diens omgeving. De ziektelast van deze aandoening kan aanzienlijk zijn. Deze richtlijn bespreekt de huidige stand van zaken omtrent de nomenclatuur, bepaling van de ziektelast van chronische urticaria en de behandeling van chronische spontane urticaria.
Doel en doelgroep
Doel
Deze richtlijn is een document met aanbevelingen ter ondersteuning van de dagelijkse praktijkvoering en dient als leidraad voor de dagelijkse praktijk om zo de kwaliteit van de zorg voor alle patiënten met chronische urticaria te bevorderen. De richtlijn berust op de resultaten van wetenschappelijk onderzoek (‘evidence- based’) en aansluitende meningsvorming gericht op het vaststellen van goed medisch handelen. Deze richtlijn en de daarvan afgeleide documenten geven aanbevelingen over de nomenclatuur, bepaling van de ziektelast en behandeling van chronische spontane urticaria. Het doel van deze richtlijn is meer uniformiteit wat betreft laatstgenoemde aspecten te creëren, waardoor een betere afstemming, begeleiding en follow-up van patiënten door zorgaanbieders wordt bereikt. De ontwikkeling van deze richtlijn zal gefaseerd gelopen; in eerste instantie zullen alleen de nomenclatuur, de vragenlijsten omtrent kwaliteit van leven en ziekteactiviteit en de behandeling van chronische spontane urticaria aan bod komen.
Doelgroep
De doelgroep wordt gevormd door alle zorgaanbieders die werkzaam zijn op het gebied van chronische urticaria of betrokken zijn bij de behandeling van patiënten met chronische spontane urticaria. Dit betreft zowel de medische, paramedische als verpleegkundige beroepsgroepen.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn werd in 2014 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die met patiënten met chronische urticaria te maken hebben. Deelnemende verenigingen in de werkgroep zijn de Nederlandse Vereniging voor Dermatologie en Venereologie (NVDV), de Nederlandse Vereniging voor Allergologie (NVvA), de Nederlandse Vereniging voor Kindergeneeskunde (NVK), het Nederlands Huisartsen Genootschap (NHG) en Huidpatiënten Nederland (HPN).
|
Werkgroepleden |
Vereniging |
|
Dr. A.C. Knulst (voorzitter) |
NVDV |
|
Dr. C. Nieuwhof |
NVvA |
|
Drs. M. Stadermann |
NVK |
|
Dr. R.A. Tupker |
NVDV |
|
Dr. M.B.A. van Doorn |
NVDV |
|
Drs. S.M. Franken |
NVDV |
|
Mevr. F. Das |
HPN |
|
Ds. W. Poldervaart |
HPN |
|
Drs. C. de Vries |
NHG |
|
Drs. M. van den Elzen |
Arts-onderzoeker UMCU |
|
Drs. E.J. van Zuuren |
NVDV |
|
Dr. J.J.E. van Everdingen |
Directeur NVDV |
|
Drs. M.C. Urgert (t / m half januari 2015) |
Arts-onderzoeker NVDV (secretariaat) |
|
Drs. W.R. Veldkamp (vanaf half januari 2015) |
Arts-onderzoeker NVDV (secretariaat) |
|
Drs. G.E. van der Kraaij (vanaf half maart 2015) |
Arts-onderzoeker NVDV (secretariaat) |
Inbreng patiëntenperspectief
Tijdens het vaststellen van de uitkomstmaten en het graderen hiervan volgens GRADE was er een patiënt aanwezig. Deze heeft ook actief bijgedragen tijdens de pressurecooker, met als doel het formuleren van de aanbevelingen.
Methode ontwikkeling
Evidence based
Implementatie
Bij het opstellen van deze richtlijn is veel aandacht besteed aan de implementatie en de praktische uitvoerbaarheid, effectiviteit, nut en noodzaak. De richtlijn wordt via het web verspreid onder alle relevante beroepsgroepen en ziekenhuizen en er zal in verschillende specifieke vaktijdschriften aandacht worden besteed aan de richtlijn. Tevens zal een samenvatting en patiëntenversie worden gemaakt. De voorlichtingsfolder van de NVDV zal worden afgestemd op de richtlijn.
Werkwijze
De werkgroep startte in 2014. In de voorbereidingsfase werd een ‘stakeholdersbijeenkomst’ georganiseerd. Hiervoor werden behalve de bovengenoemde partijen ook Zorgverzekeraars Nederland (ZN) en Nefarma uitgenodigd. Tijdens deze bijeenkomst werden de door de werkgroep opgestelde uitgangsvragen besproken en nader geëxpliceerd. Voor de ontwikkeling van deze richtlijn is gebruik gemaakt van zowel de EBRO-criteria als de GRADE-methode. Gekozen is om de therapeutische uitgangsvragen ciclosporine en omalizumab uit te werken volgens de GRADEmethode. Bij de GRADE-methode wordt wetenschappelijk bewijs beoordeeld aan de hand van uitkomstmaten. GRADE veronderstelt dat de werkgroep in het beginstadium van de richtlijnontwikkeling uitkomstmaten vaststelt. Een volledig uitleg over de GRADE-methode valt buiten het bestek van deze richtlijn, zie hiervoor het ‘GRADE handbook’ (Schünemann et al., 2013). Voor de twee uitgangsvragen die volgens GRADE zijn uitgewerkt, zijn tijdens de invitational conference patiënt-relevante uitkomstmaten bepaald en vervolgens ingedeeld in kritieke, belangrijke en minder belangrijke uitkomstmaten. De gekozen uitkomstmaten zijn als volgt:
- Verbetering ziekteactiviteit in vergelijking tot de baseline (cruciaal)
- Verbetering kwaliteit van leven in vergelijking tot de baseline (cruciaal)
- Proportie patiënten met bijwerkingen (cruciaal)
- Proportie patiënten met complete respons (belangrijk)
- Proportie patiënten met een partiële respons (belangrijk)
- Proportie angio-oedeem vrije dagen (belangrijk)
- Proportie patiënten met remissie binnen 1 maand (van beperkt belang).
Wetenschappelijke onderbouwing
Per uitgangsvraag werd een systematische search verricht in de databases PubMed, de Cochrane Database of Systematic Reviews en Cochrane Central Register of Controlled Trials), tenzij anders aangegeven. Ook werden relevante nationale en internationale richtlijnen aangaande chronische urticaria geraadpleegd, met name de recente internationale richtlijn ‘EAACI / GA2LEN / EDF / WAO guideline on urticaria, 2014’, ontwikkeld volgens AGREE methode, en de Amerikaanse richtlijn ‘The diagnosis and management of acute and chronic urticaria: 2014 update’ (Bernstein et al., 2014; Zuberbier et al., 2014). De zoekstrategie en resultaten van de searches zijn terug te vinden onder Zoekverantwoording en Evidencetabellen.
De volgende inclusie / exclusiecriteria zijn gebruikt, tenzij anders aangegeven:
|
Inclusiecriteria: |
Exclusiecriteria: |
|
Chronische spontane / idiopathische urticaria |
Acute urticaria |
|
Alle leeftijden |
Andere vormen van chronische urticaria |
|
Behandeling met het betreffende geneesmiddel |
Narrative reviews |
|
|
Case series / observationele studies met minder dan vijf patiënten |
|
|
Studies in andere taal dan Engels |
|
|
Studies met onvoldoende informatie over effectiviteit en / of veiligheid |
|
|
In vitro onderzoek |
|
|
Dubbele publicaties |
|
|
Onderzoek bij dieren. |
Na selectie van de literatuur bleven artikelen over die als onderbouwing bij de verschillende conclusies staan vermeld. De werkgroepleden beoordeelden de kwaliteit en inhoud ervan. Vervolgens schreven de werkgroepleden een paragraaf of module voor de conceptrichtlijn, waarin de beoordeelde literatuur werd verwerkt.
Bij de uitwerking van de uitgangsvragen volgens de GRADE-methode zijn de stappen beschreven in het ‘GRADE handbook’ nauwkeurig gevolgd (Schünemann et al. 2013). Tevens is gebruik gemaakt van ‘the Cochrane Handbook for Systematic Reviews of Intervention’ voor het uitwerken van de uitgangsvragen en voor het verrichten van de meta-analyses (Higgins and Green 2011).
Elke module van de richtlijn is volgens een vast stramien opgebouwd, dat onderstaand is weergegeven. Een van de doelen is om een richtlijn zo transparant mogelijk te laten zijn, zodat elke gebruiker kan zien op welke literatuur en overwegingen bepaalde aanbevelingen zijn gebaseerd.
Uitgangsvraag
Een uitgangsvraag is een klinisch relevante vraag waarop tijdens de richtlijnontwikkeling een antwoord wordt geformuleerd.
Inleiding
Een korte introductie op de achtergrond van de uitgangsvraag.
Wetenschappelijke onderbouwing
Per uitgangsvraag is beknopt de zoekstrategie en de uitkomst hiervan beschreven. Meer gedetailleerde informatie over de zoekstrategie is beschreven in de Zoekverantwoording.
Samenvatting van de literatuur
De antwoorden op de uitgangsvragen zijn voor zover mogelijk gebaseerd op gepubliceerd wetenschappelijk onderzoek. De geselecteerde artikelen zijn door de schrijvende werkgroepleden beoordeeld op kwaliteit van het onderzoek en gegradeerd naar mate van bewijs, waarbij gebruik gemaakt is van de GRADE-methode en van de EBRO-methode. Voor de indeling van methodologische kwaliteit van studies volgens EBRO en GRADE zie tabel 1 en 4. Beschrijving en beschouwing van de gepubliceerde artikelen zijn indien van toepassing te vinden onder het kopje ‘samenvatting van de literatuur’. Meer gedetailleerde informatie is beschreven in de Evidencetabellen.
Conclusie
Het wetenschappelijk materiaal is samengevat in een conclusie, waarbij het niveau van het meest relevante bewijs is weergegeven. Voor het niveau van conclusies volgens EBRO en GRADE zie tabel 2 en 5.
Overige overwegingen
Voor het komen tot een aanbeveling zijn er naast het wetenschappelijke bewijs vaak andere aspecten van belang, bijvoorbeeld: patiënten voorkeuren, beschikbaarheid van speciale technieken of expertise, organisatorische aspecten, maatschappelijke consequenties of kosten. Ook bijwerkingen werden hierin meegenomen, voor zover die niet reeds uit wetenschappelijke literatuur waren gedestilleerd en waarvoor dan wel andere bronnen beschikbaar waren. In de overige overwegingen worden de conclusies op basis van de literatuur geplaatst in de context van de dagelijkse praktijk en vindt een afweging plaats van de voor- en nadelen van de verschillende beleidsopties.
Aanbeveling
De uiteindelijk geformuleerde aanbeveling is het resultaat van het beschikbare bewijs in combinatie met deze overwegingen. Het volgen van deze procedure en het opstellen van de richtlijn in dit ‘format’ heeft als doel de transparantie van de richtlijn te verhogen. Het biedt ruimte voor een efficiënte discussie tijdens de werkgroep vergaderingen en vergroot bovendien de helderheid voor de gebruiker van de richtlijn. Voor de gebruikte niveaus van aanbevelingen volgens EBRO en GRADE zie tabel 3 en 6.
Uitwerking volgens de EBRO-methode
Tabel 1: Indeling van methodologische kwaliteit van individuele studies volgens EBRO
|
Kwaliteit |
Interventie |
Diagnostisch accuratesse onderzoek |
Schade / bijwerkingen*, etiologie, prognose |
|
A1 |
Systematische review van tenminste twee onafhankelijk van elkaar uitgevoerde onderzoeken van A2-niveau |
||
|
A2
|
Gerandomiseerd dubbelblind vergelijkend klinisch onderzoek van goede kwaliteit van voldoende omvang |
Onderzoek ten opzichte van een referentietest (een ‘gouden standaard’) met tevoren gedefinieerde afkapwaarden en onafhankelijke beoordeling van de resultaten van test en gouden standaard, betreffende een voldoende grote serie van opeenvolgende patiënten die allen de index- en referentietest hebben gehad |
Prospectief cohort onderzoek van voldoende omvang en follow-up, waarbij adequaat gecontroleerd is voor ‘confounding’ en selectieve follow-up voldoende is uitgesloten. |
|
B |
Vergelijkend onderzoek, maar niet met alle kenmerken als genoemd onder A2 (hieronder valt ook patiënt-controle onderzoek, cohortonderzoek) |
Onderzoek ten opzichte van een referentietest, maar niet met alle kenmerken die onder A2 zijn genoemd |
Prospectief cohort onderzoek, maar niet met alle kenmerken als genoemd onder A2 of retrospectief cohort onderzoek of patiënt-controle onderzoek |
|
C |
Niet-vergelijkend onderzoek |
||
|
D |
Mening van deskundigen |
||
* Deze classificatie is alleen van toepassing in situaties waarin om ethische of andere redenen gecontroleerde trials niet mogelijk zijn. Zijn die wel mogelijk dan geldt de classificatie voor interventies.
Tabel 2: Niveau van conclusies volgens EBRO
|
Niveau |
Conclusie gebaseerd op |
|
1 |
Onderzoek van niveau A1 of tenminste twee onafhankelijk van elkaar uitgevoerde onderzoeken van niveau A2 |
|
2 |
Eén onderzoek van niveau A2 of tenminste twee onafhankelijk van elkaar uitgevoerde onderzoeken van niveau B |
|
3 |
Eén onderzoek van niveau B of C |
|
4 |
Mening van deskundigen |
Tabel 3: Niveau van aanbeveling volgens EBRO
|
Aanbeveling |
Balans |
|
Sterk positief |
De interventie doet duideljk meer goed dan kwaad: Doe…. |
|
Zwak positief |
Het is onzeker of alle patienten gebaat zijn bij de interventie: doe bij voorkeur…. |
|
Zwak negatief |
Het is onzeker of alle patienten gebaat zijn bij de interventie: doe bij voorkeur niet…. |
|
Sterk negatief |
De interventie doet meer kwaad dan goed: doe niet…. |
Uitwerking volgens de GRADE-methode
Tabel 4: Indeling van methodologische kwaliteit van studies volgens GRADE
|
|
GRADE systeem |
|
Type bewijs |
Gerandomiseerd onderzoek = hoog Observationele studie = laag Elk ander bewijs = zeer laag |
|
Factoren die de kwaliteit van bewijs kunnen verlagen*: |
- Ernstige of zeer ernstige beperkingen in de kwaliteit van de studie - Indirectheid van het bewijs - Belangrijke inconsistentie tussen studies - Imprecisie - Grote kans op ‘publicatiebias’ |
|
Factoren die de kwaliteit van bewijs kunnen verhogen**: |
- Sterk bewijs voor een associatie—significant relatief risico van > 2 ( < 0,5) gebaseerd op consistent bewijs uit twee of meer observationele studies, zonder plausibele ‘confounders’ (+1) - Zeer sterk bewijs voor een associatie—significant relatief risico van > 5 ( < 0,2) gebaseerd op direct bewijs zonder belangrijke bedreigingen voor de validiteit (+2) - Bewijs voor een dosis respons gradiënt (+1) - Alle plausibele ‘confounders’ zouden het effect hebben verminderd (+1) |
*Elk criterium kan de kwaliteit verminderen met 1 stap of bij zeer ernstige beperkingen met twee stappen. ** Verhogen kan alleen indien er geen beperkingen zijn t.a.v. de studiekwaliteit, imprecisie, inconsistentie, indirectheid en publicatiebias.
Tabel 5: Niveau van conclusies volgens GRADE
|
Conclusie - Hoog = nader onderzoek zal zeer onwaarschijnlijk het vertrouwen in de inschatting van een effect veranderen - Middelmatig = nader onderzoek zal waarschijnlijk een belangrijke invloed hebben op het vertrouwen in de inschatting van een effect en kan de inschatting van een effect veranderen - Laag = nader onderzoek zal zeer waarschijnlijk een belangrijke invloed hebben op het vertrouwen in de inschatting van een effect en zal waarschijnlijk de inschatting van een effect veranderen - Zeer laag = elke inschatting van een effect is zeer onzeker |
Tabel 6: Niveau van aanbevelingen volgens GRADE
|
Klinische aanbeveling - Sterk voor/tegen = als clinici, gebaseerd op het beschikbare bewijs, zeer zeker zijn dat de voordelen de nadelen of risico’s overtreffen, of andersom, dan zal er een sterke aanbeveling worden gedaan - Zwak voor/tegen = als clinici, gebaseerd op het beschikbare bewijs, denken dat de voordelen en de nadelen of risico’s in balans zijn of als er een bepaalde onzekerheid bestaat over de grootte van de voordelen en risico’s, moeten ze een zwakke aanbeveling maken |
Zoekverantwoording