Zwangerschap en biologicals
Uitgangsvraag
Welke werkwijze heeft bij de behandeling met biologicals de voorkeur bij zwangerschapswens (vrouw en man) tijdens de zwangerschap en tijdens borstvoeding?
Aanbeveling
Bij iedere vrouwelijke IMID patiënt met een zwangerschapswens dient in overleg met haar en haar partner een afweging te worden gemaakt omtrent het (dis)continueren van het biological. De volgende factoren zullen hierbij worden betrokken:
- fertiliteit respectievelijk fecundiciteit;
- potentieel risico van biological voor embryo en voor foetus;
- niveau/risico van ziekteactiviteit voor de moeder;
- risico van ziekteactiviteit voor embryo en voor foetus;
- behandelingsalternatieven.
Preconceptuele counseling en zwangerschapsbegeleiding door een gynaecoloog is bij een vrouwelijke IMID patiënt met een zwangerschapswens wenselijk.
TNFα-blokkers kunnen bij hoge ziekteactiviteit in het individuele geval (mede afwegende de ernst van onbehandelde ziekte) overwogen worden gedurende zwangerschap, maar ze kunnen momenteel niet als (geheel) veilig worden beschouwd.
Indien binnen de totale behandelingsplanning mogelijk, dient de behandeling met TNFα- blokkers 4 maal de halfwaarde tijd voor de geplande conceptie gestopt te worden.
De werkgroep is om pragmatische redenen van mening dat de behandeling van biologicals (Rituximab/Abatacept/Anakinra/Tocilizumab) 6 maanden vóór het staken van de anticonceptie gestopt dient te zijn.
Wegens gebrek aan bewijs geeft de werkgroep geen aanbeveling omtrent het (door)gebruiken van biologicals bij de man met een kinderwens.
Het wordt aanbevolen bij de vrouw die borstvoeding geeft geen biological te geven vanwege onvoldoende bewijs voor veiligheid. Bij doorgebruiken van een biological wordt daarom geadviseerd primair kunstvoeding te overwegen.
Overwegingen
Aanbevolen wordt om preconceptueel met beide partners medicatieregime, erfelijkheid van de betreffende IMID en ziektespecifieke zwangerschapsproblematiek te bespreken. Bij elke IMID-patiënt is informatie over zwangerschap(swens) bij diagnosestelling derhalve aan te raden. Ook eventuele invloed van ziekte(-activiteit) of medicatie op fertiliteit en fecundiciteit dienen vroegtijdig en in algemene zin besproken te worden.
Bij actieve IMID, zoals kan voorkomen bij RA en IBD, is er een verhoogd risico op gecompliceerd zwangerschapsverloop en/of vroeggeboorte. Daarom wordt in het algemeen aangeraden zwanger te worden in een rustige fase van de ziekte.
Zonder adequate controlegroep is veiligheid van biologicals lastig te bewijzen. Door spontane dataverzameling, en deze terug te calculeren naar de mogelijke populatie at risk, kan men toch enig inzicht in risicoverhoging verkrijgen. In de huidige registries zijn geen significante toename van congenitale malformaties gevonden, hetgeen geruststellend lijkt.
In de meest recente FDA-rapportage van spontane meldingen wordt wel een verhoogd risico gesuggereerd met betrekking tot het VATER-syndroom. Men dient dit licht verhoogde risico in de individuele situatie af te wegen tegen de risico’s van onbehandelbare, hoge ziekteactiviteit met verhoogd achtergrondrisico.
Bij een actieve inflammatoire (darm)ziekte dient men zich te realiseren dat de kans op zwangerschap verminderd is. Door adequate behandeling neemt de kans op zwangerschap toe. Dan rijst op enig moment de vraag of men de behandeling met biologicals moet stoppen wanneer de vrouw zwanger is geworden en het risico van opvlammen van de inflammatoire ziekte kan accepteren of niet. Immers, het is gezien bovenstaande gegevens niet uit te sluiten of behandeling met sommige biologicals niet extra risico’s voor de vrucht induceren. Zo is met name nog niets bekend over lange termijneffecten van intra-uteriene blootstelling op intellectuele ontwikkeling of maligniteitsrisico’s. Bij de informatie aan de vrouw en haar partner is het belangrijk de voor- en nadelen te bespreken, evenals het individuele risico op een opvlamming van de ziekte.
Voor- en nadelen van biologicals bij actieve inflammatoire ziekte zijn dermate complex dat volgens de werkgroep en de betreffende Wetenschappelijke Verenigingen begeleiding van een dergelijke zwangerschap vanuit de tweedelijn door ervaren experts aangewezen is. Bij aanvang van behandeling met biologicals is het wenselijk dat de arts een mogelijke zwangerschapswens bespreekt als onderdeel van de voorlichting. Bij behandeling met infliximab kan men overwegen door te gaan met behandeling (uiteraard zonder methotrexaat)
Het risico voor de vrucht bij bewust of onbewust doorgebruiken van biologicals is dermate laag ingeschat, dat bij accidentele zwangerschap weliswaar gynaecologische evaluatie sterk geadviseerd wordt, maar dat een eventuele abortus provocatus of zwangerschapsbeëindiging niet zonder meer geïndiceerd is.
Hoewel het niet is uitgesloten dat TNFα-blokkers de spermatogenese kunnen remmen, formuleert de werkgroep wegens gebrek aan bewijs geen aanbeveling omtrent het (door)gebruiken van biologicals bij de man met een kinderwens.
Over eventuele onveiligheid van borstvoeding van moeders die therapie met biologicals krijgen, is weinig bekend. In het algemeen is borstvoeding voor de pasgeborene gunstig voor de verdere ontwikkeling. Anderzijds is niet uitgesloten dat bij gebruik van biologicals enig effect via de borstvoeding op de pasgeborene kan plaatsvinden, bijvoorbeeld inductie van orale tolerantie. Veiligheidshalve is het te adviseren kunstvoeding te gebruiken.
Onderbouwing
Achtergrond
Veel typen van IMID komen vaker voor bij jonge vrouwen en mannen in de vruchtbare periode, al dan niet met kinderwens. Een richtlijn voor beleid rondom zwangerschapswens, gedurende zwangerschap en ten tijde van eventuele borstvoeding is derhalve van groot belang. Gecontroleerde studies zijn over dit onderwerp niet voorhanden Onze kennis hieromtrent is derhalve gebaseerd op dierexperimenteel onderzoek, op theoretische basisprincipes vanuit met name farmacologie en op ervaringen uit de alledaagse praktijk, dat wil zeggen casuïstiek en grote (inter)nationale sets met registratiegegevens over zwangerschap en geboortes bij IMID-patiënten en hun partners. Deze ervaringen brengen het gewicht van het bewijs volgens EBRO systematiek tot klasse 4 of hooguit klasse 3 bewijskracht.
Toxiciteit van de preparaten wordt geklassificeerd volgens systeem van FDA - tabel 1.
|
A adequate en goed-gecontroleerde studies bij zwangeren tonen aan dat er geen risico is voor de foetus in eerste trimester, noch later in zwangerschap |
|
B dierstudies tonen aan dat er geen risico is voor de foetus, en adequate, goed-gecontroleerde humane data zijn niet voorhanden C1 dierstudies hebben geen risico voor de foetus aangetoond maar er zijn geen adequate, goed-gecontroleerde studies bij zwangere vrouwen OF C2 dierstudies hebben nadelen/geringe risico’s voor de foetus aangetoond, maar adequate, goed-gecontroleerde studies bij zwangeren hebben dit niet bevestigd. D 1 dierstudies hebben nadelen/risico’s aangetoond, echter er zijn geen goed-gecontroleerde studies bij zwangere vrouwen, OF D2 er zijn geen dierstudies verricht, en er zijn geen adequate goed-gecontroleerde studies bij zwangeren |
|
X dierstudie of studie bij mensen tonen foetale afwijkingen aan: risico van gebruik duidelijk groter dan eventueel voordeel |
Er zijn verschillende medicamenten bij verschillende IMIDs die in het kader van veiligheid bij zwangerschapwens reeds geklassificeerd zijn. Methotrexaat en leflunomide zijn in dit kader onveilig c.q. geklassificeerd als klasse X. Sulfasalazine is veilig en beoordeeld als klasse B. Glucocorticoïden worden als veilig beschouwd gedurende de zwangerschap: ze zijn ingedeeld als FDA klasse C vanwege aangetoonde foetale veranderingen bij hoge doseringen. Infliximab staat te boek als klasse B door data bij patiënten met IBD en RA en dierexperimenteel onderzoek. Van sommige biologicals is nog geen internationale classificatie bekend.
Conclusies
|
Niveau 3 |
Zoals eigenlijk bij elke vorm van medicatie is ook de ervaring met biologicals vlak vóór en gedurende de zwangerschap te gering om te kunnen claimen dat (doorgaand) gebruik veilig is.
C Carter et al, 2006; Carter et al, 2009; Klink et al 2008; Ostensen&Förger 2009, Gisbert et al, 2009/2010 |
|
Niveau 3 |
Er zijn aanwijzingen dat TNFα-blokkers mogelijk geassocieerd zijn met het VATER-syndroom, met name bij hogere doseringen.
C Carter et al, 2006; Carter et al, 2009; |
|
Niveau 3 |
Het is onvoldoende bekend in welke mate TNFα-blokkers uitgescheiden worden in moedermelk, of dat ze relevante spiegels kunnen opbouwen via ingestie c.q. borstvoeding. Moeders die moedermelk willen geven, moeten geïnformeerd worden dat onvoldoende kennis voorhanden is voor een positief advies en dat er een goed alternatief voor borstvoeding is.
C Peltier et al, 2001; Ostensen et al, 2004 |
|
Niveau 3 |
Het is niet uitgesloten dat TNFα-blokkers de spermatogenese kunnen remmen.
C La Montagna et al, 2005; Mahadevan et al, 2005; Paschou et al, 2009 |
Samenvatting literatuur
TNF-α-blokkers
In dierstudies is geen risico voor de foetus aangetoond van TNFα-blokkers, net zo min als in series van zwangere patiënten die infliximab of adalimumab in de periode vlak voor zwangerschap, en gedeeltelijk of geheel tijdens de zwangerschap gebruikten. Het effect op de spermatogenese is onbekend maar vooralsnog zijn er geen aanwijzingen op ongunstige bijwerkingen. De TNFα-blokkers zijn derhalve geclassificeerd in groep B.
Een ‘task force panel’ vanuit de ACR heeft voor TNFα-blokkers geen specifieke aanbevelingen gedaan vanwege tegenstrijdige bewijsstukken in de literatuur (Saag 2008). In de Nederlandse richtlijn IBD, zowel als in internationale richtlijnen, heeft men uitgaande van de ongunstige effecten van actieve IBD op het beloop van de zwangerschap en de betrekkelijk grote kans op opvlamming van de ziekte na discontinueren van goed geïndiceerd gebruik van antiTNF-α medicatie, een genuanceerd positieve insteek voor het gebruik van een van de TNFα-blokkers (infliximab in het bijzonder) ten tijde van de zwangerschap aangegeven (richtlijn IBD 2009). Infliximab wordt door de FDA in klasse B geplaatst, hetgeen betekent dat voor de zwangerschap geen risico lijkt te bestaan. Er is beperkte informatie over het gebruik in de zwangerschap, deels afkomstig uit een register van de fabrikant (n=280, deels gepubliceerd (n=106), waarbij geen verhoogde kans op miskraam of perinatale complicaties vermeld werd (Caprilli et al., 2006; Katz et al., 2004; Mahadevan et al., 2005).
Fertiliteit
TNFα-blokker remt apoptose van zaadcellen, stimuleert androgeenreceptorexpressie op de cel van Sertoli, remt de steroidogenese op transcriptieniveau van de cel van Leydig. La Montagna (2005) beschrijft een studie van drie mannen met spondylitis ankylopoëtica behandeld met infliximab gedurende 8 tot 24 maanden en vindt asthenozoöspermie en teratozoöspermie, echter er zijn geen controlemonsters van voor de behandeling met infliximab. Het is dus niet duidelijk of de beschreven afwijkingen zijn veroorzaakt door blootstelling aan infliximab. Mahadevan et al. (2005) tonen door vergelijking van pre- en postinfusie metingen in semen aan dat na infliximab-infusie een groter volume ejaculaat, alsook een trend tot verminderde zaadcelmobiliteit is. Paschou et al (2009) vinden in een groep van 65 mannen met spondylitis ankylopoetica vier mannen die vier eerste (gezonde) kinderen kregen: dosering 5mg/kg infliximab, > 10 infusies; van hen hadden twee nog een tweede (gezond) kind verwekt na 14 en 23 infusies. Gezien deze resultaten concluderen de onderzoekers dat bij deze vier mannen geen sprake is van een fertiliteitsprobleem tijdens behandeling met infliximab. Fertiliteit en zaadkwaliteit zijn echter niet in statistisch adequaat onderbouwde studies bestudeerd. Over de invloed van TNFα blokkade op de vrouwelijke fertiliteit zijn geen data gepubliceerd.
Transplacentaire passage:
De moleculaire structuur van adalimumab, infliximab (respectievelijk humaan en chimeer IgG1 anti-TNFα antilichaam), en etanercept (dimeer met ligand bindingsplaats op de p75 receptor) staat toe dat enige placentaire passage mogelijk is gedurende het eerste trimester, en in toenemende mate in het tweede, en zeker in het derde trimester (Simister et al, 2003). Serumspiegels van infliximab waren bij vijf pasgeborenen niet verschillend van de spiegels bij hun moeders, en bleven aantoonbaar tot zelfs zes maanden post-partum: dit betekent wellicht een verlengde halfwaardetijd bij de neonaat (Vasiliauskas et al, 2005).
Zwangerschap bij patiënten met reumatische aandoeningen
De eerste ervaringen bij de mens zijn geruststellend. Cush (2005) beschrijft Noord-Amerikaanse real-life data: 378 normale bevallingen bij 454 RA-patiënten die zwanger werden tijdens gebruik van biologicals (142 patiënten gedurende gehele zwangerschapsperiode incl. conceptie), van wie negen prematuur kwamen, vijf-maal werd een abortus-lege-artis verricht en er waren 25 ‘spontane’ miskramen. Berthelot et al (2009) beschrijven de Franse register met 15-TNFα-blokkerblootstellingen: 3-maal vroegtijdig gestopt wegens elective abortus of miskraam en 12-maal gezonde babies. Het Britse “registry” (Hyrich 2006) beschrijft 35 zwangerschappen waarvan 32 met bekende afloop en 23 blootstellingen tijdens de conceptie waarbij uiteindelijk zes miskramen in het eerste trimester (3x tevens MTX-gebruik). Van de 32 patiënten verkoos 91% de zwangerschap te continueren, waarvan 24% eindigde in een miskraam gedurende het eerste trimester, en 76% in gezonde kinderen. Beoordeling wordt gecompliceerd doordat door sommigen ook MTX of leflunomide werd gebruikt. Bij alle gerapporteerde zwangerschappen onder gecontinueerde behandeling met infliximab is echter geen risicoverhoging aangetoond op een miskraam, noch op prematuriteit of vormafwijkingen bij de neonaat. Deze series zijn evenwel niet groot genoeg voor eenduidige uitspraken in deze.
De Organisation of Teratology Information Services (OTIS) heeft gegevens over 33 zwangerschappen bij RA-patiënten met blootstelling aan TNF-α-blokker gedurende eerste trimester (Chambers 2004; 2005). De meeste ervaringen zijn met etanercept: 29-maal blootstelling alleen in het eerste trimester, met soms blootstelling aan etanercept gedurende gehele zwangerschapsperiode (n=3) (Chambers 2004/2005). Uitkomsten: eenmaal trisomie 18 met spontane abortus, drie keer spontane abortus, eenmaal therapeutische zwangerschapsbeëindiging. Twee typen van misvorming zijn bij etanercept-gebruik gemeld: trisomie-18 (n=1) bij een miskraam en bij een 28-jarige patiënte met artritis psoriatica behandeld met etanercept (50 mg 2x per week) een VATER-syndroom (vertebrale afwijkingen, anaalatresie, tracheo-oesofageale fistel, oesofagusatresie en renale alsook radiusafwijkingen) (n=1) (Carter 2006). Het kind had ook een hypospadie en open foramen ovale. Het VATER-syndroom komt echter sporadisch (1,6 per 10.000 geboorten) ook voor, zodat een causale relatie tussen etanercept en het VATER-syndroom niet als bewezen kan worden beschouwd. Recent is er echter ook bij adalimumab-blootstelling een eerste kind beschreven met het VATER-syndroom (Carter 2009).
Een recent overzicht van 41 congenitale defecten ontstaan (post aut propter) gedurende intra-uteriene blootstelling aan de TNFα-blokkers etanercept en infliximab (en recent het eerste bij adalimumab) die spontaan gemeld zijn bij de FDA toont dat 59% een of meer afwijkingen heeft uit het zogenaamde spectrum van VATER-syndroom. Deze aantallen zijn teveel cq. boventallig (p<0,01) in vergelijking tot historische controles (Carter 2009). Complicerende factoren bij de interpretatie zijn dat 24 van de 41 moeders (59%) weliswaar geen andere medicatie gebruikten, maar 17 van de 41 moeders wel; en dat niet duidelijk is hoe groot de groep is die in totaal blootgesteld is geweest en feitelijk dit risico liepen. De begeleidende editorial besluit dat onbehandelde aandoeningen bij de moeder een forse risicoverhoging kunnen geven voor moeder alsook het ongeboren kind, en dat dit risico mee gewogen dient te worden in een risk/benefit ratio wanneer men zoekt naar het belang voor zowel moeder als kind (Koren 2009). Inmiddels zijn meer dan 200 vrouwen aan infliximab, meer dan 100 aan etanercept en ongeveer 50 aan adalimumab blootgesteld geweest tijdens de zwangerschap, meestal in het eerste trimester (Ostensen&Förger 2009). Negenentwintig van deze zwangerschappen zijn gedurende gehele zwangerschapsperiode aan TNFα-blokker blootgesteld geweest. Significante transplacentaire passage vindt pas plaats vanaf het tweede trimester.
Twee prospectieve studies door de OTIS-groep toonden vergelijkbare ratio’s miskramen en malformaties in blootgestelde en niet-blootgestelde zwangerschappen (Chambers 2004; Chambers 2006) zodat staken van TNFα-blokker bij patiënten vanaf gemiste menstruatie of eventueel staken op het moment van positieve zwangerschapstest, i.c. 4 tot 8 weken na conceptie, redelijk veilig lijkt (Ostensen&Förger 2009).
Zwangerschap bij IBD-patiënten
Bij een serie met 10 vrouwen die bewust zwanger raakten en vervolgens bevielen ten tijde van infliximabgebruik werden 10 normaalgevormde kinderen geboren (Mahadevan et al, 2005).
Verder is in de literatuur een review verschenen waarin de inmiddels 141 vervolgde IFX-zwangerschappen bij IBD-patiënten worden beschreven. In de samengestelde serie werden geen aanwijzingen gevonden voor een toegenomen percentage foetale afwijkingen of andere bedreigende gegevens (Gisbert et al, 2009). Vooralsnog zijn van slechts 3 vrouwen die IBD hadden en die adalimumab ten tijde van de zwangerschap gebruikten case reports verschenen.
Momenteel worden data verzameld in Amerikaanse en Franse IBD-cohorten van zwangeren die immunomodulerende medicatie gebruiken.
Borstvoeding
Er zijn verschillende case reports over onschadelijkheid van borstvoeding gedurende anti-TNFα therapie bij de moeder, waarbij het kind normale groeicurven en een normale ontwikkeling lijkt door te maken. Dit is in overeenstemming met het gegeven dat oraal toegediende TNFα blokkade niet werkzaam is.
Etanercept is in significante concentraties in de borstvoeding aantoonbaar gebleken bij een 30-jarige moeder die etanercept 25 mg 2x per week gebruikte (Ostensen&Obrist-Eigenmann, 2004). Recent is in 1 casus (40-jarige vrouw met RA) een ongecompliceerde zwangerschap beschreven gedurende bewust doorgebruiken van etanercept (25mg 2x/wk) met maternale etanerceptconcentraties van > 2200 ng/ml in elk trimester. Daarnaast een etanerceptconcentratie van 80 ng/ml bij de pasgeborene die overigens elke week daalt tot 2 ng/ml in week 3 en tot een waarde onder de detectiegrens in week 12; dit ondanks gebruik van borstvoeding met een etanerceptconcentratie van 3,5 ng/ml (Murashima 2009). In deze casus is het niet mogelijk gebleken detecteerbare etanerceptspiegels bij de pasgeborene te bereiken gedurende borstvoeding ondanks doorgebruiken van tweewekelijks etanercept.
Een task force panel vanuit de ACR heeft voor biologicals bij borstvoeding geen specifieke aanbevelingen gedaan vanwege gebrek aan bewijs in de literatuur (Saag 2008), waarbij in de IBD-populatie ook geen eenduidig advies voor borstvoeding bestaat. Het risico voor het kind, hoe klein misschien ook, blijft vooralsnog onbekend.
Ervaringen met T-/B-celremmers
De T- en B-celremmers zijn geklassificeerd als klasse C medicamenten. Een task force panel vanuit de ACR heeft voor deze biologicals geen specifieke aanbevelingen gedaan vanwege gebrekkig bewijs in de literatuur (Saag 2008).
T-celremming
Abatacept kan de placentabarrière passeren. Dierstudies met blootstelling tot 29 maal hogere doses dan bij de mens (zwangere rat/konijn) resulteerde niet in vormafwijkingen bij de vrucht (Golding 2007). Bij doses 11 maal hoger dan gegeven bij de mens werden wel veranderingen in immuunfuncties bij de nakomelingen gevonden, zoals toegenomen T-celafhankelijke antilichaamrespons bij vrouwelijke nakomelingen (thyroiditis) en toegenomen T-cel-afhankelijke antilichaamrespons in puppies. De relevantie voor de langere termijn is niet bekend.
In de dubbelblinde en open-label extensieperioden van 5 onderzoeken zijn 8 zwangerschappen gerapporteerd bij vrouwen met blootstelling aan abatacept. Van hen kregen er 7 eveneens MTX en 1 leflunomide als comedicatie. Drie ondergingen een spontane abortus gedurende het eerste trimester (één had al eerder 2 spontane abortussen gehad, en 1 had een voorgeschiedenis met een eerdere onvoldragen zwangerschap, en 1 had geen eerdere zwangerschappen gehad). Twee vrouwen beëindigden hun zwangerschap door inductie van abortus. Wat betreft de mannelijke fertiliteit: 1 echtgenote van een patiënt werd zwanger en beviel van een normale, gezonde baby. In een fase II MS studie (IM101200) werden 3 vrouwen zwanger: 2 patiënten uit de studie, en 1 partner van een patiënt uit de studie. Een vrouw beviel van een gezonde baby 10 maanden na beëindiging van de studie; de ander had een electieve abortus na 4 weken zwangerschap en de partner had een spontane abortus (miskraam) 2 maanden na de conceptie.
Abatacept dient ontraden te worden bij zwangerschap en men dient adequate anticonceptie te regelen tot 10 weken na stoppen van blootstelling aan abatacept. Tot op heden zijn geen ervaringen met borstvoeding bij de mens gepubliceerd.
B-celremming
Recente dierstudies met rituximab bij makaken, in de periode van de organogenese toegediend, tonen in verschillende doseringen (20, 50 of 100 mg/kg) embryotoxische noch teratogene effecten in het nageslacht. Zowel macroscopisch als histologisch zijn geen effecten waarneembaar. De enige opvallende bevinding is B-celdepletie bij de nakomelingen (Mc Keever 2003). Rituximab kan bij de mens de placenta passeren vanaf week 16 na de conceptie, en bereikt dan foetale spiegels vergelijkbaar met de maternale spiegels. Klink (2008) geeft een overzicht van 7 en voegt 1 eigen ervaring aan toe: 8 zwangerschappen met rituximab-blootstelling bij vrouwen met een lymfoom (n=5, 4-maal ook behandeld met CHOP) en autoimmuun-hemolytische anemie (n=3). De behandeling met 375 mg rituximab/week (in totaal 4x) bleef beperkt tot het eerste trimester (n=2) en tweede (n=4) en derde trimester (n=4). Bij de 2 laatste kinderen werd spontaan herstel van B-celaantal gezien. Na blootstelling gedurende het eerste trimester, werd enige lymfopenie, maar geen B-celdepletie, gevonden (Ojeda-Uribe 2006). Bij alle kinderen waren IgG spiegels normaal en allen vertoonden een normale respons op vaccinatie. In een ongepubliceerde raportage over 10 zwangerschappen werd gemeld dat alle resulteerden in levendgeboorte zonder vormafwijkingen en zonder serieuze infecties. Wel hadden 2 kinderen een te laag aantal granulocyten postpartum en 1 had tevens een anemie en lymfopenie.
De excretie van rituximab in de borstvoeding en eventuele veiligheid hiervan is echter niet vastgesteld.
Samenvattend: intra-uteriene blootstelling aan rituximab lijkt niet schadelijk voor de neonaat, alhoewel data beperkt zijn tot een dertigtal ervaringen. Derhalve blijft omzichtigheid geboden. Rituximab bij vrouwen die zwanger willen worden is slechts bij uitzondering te overwegen, rekening houdend met voor- en nadelen van hoge ziekteactiviteit bij moeder en effecten hiervan op moeder en kind (Klink 2008; Ostensen&Förger 2009).
Ervaringen met interleukineblokkers (IL)
IL-1 receptor antagonist (IL-1 RA: Anakinra) is geclassificeerd als klasse B medicament. Anti-IL-6 receptor (anti-IL-6R: Tocilizumab) is nog niet geclassificeerd door de FDA.
Anakinra en Tocilizumab zijn immuunglobulines die de placenta kunnen passeren vanaf week 6 na de conceptie. Zes maanden na stopzetten van de IL-blokkerende therapie kan de zwangerschapswens worden gerealiseerd, althans zo is door de fabrikant geadviseerd. Dierstudies hebben geen risico op misvorming aangetoond. Over schadelijkheid bij de mens is weinig bekend. Recent is 1 casus beschreven: Anakinra (bewust) gegeven aan een 33-jarige vrouw met een therapie-refractaire ziekte van Still (Berger 2009). Ondanks dat Anakinra werd doorgebruikt, werd na een normaal zwangerschapsbeloop een kind van 2700 gram geboren. Manuele placentaverwijdering was nodig maar het kind vertoonde een normale psychomotore ontwikkeling. Over zwangerschap en lactatie bij Anakinra zijn verder geen gegevens gepubliceerd.
Het effect van tocilizumab op de embryonale ontwikkeling is bestudeerd in een embryofoetale toxiciteitstudie in de cynomolgus aap in doseringen tot wel 50 mg/kg/dag. Er is geen abnormaal fenotype gerapporteerd en evenmin effect op de reproductiemogelijkheden als gevolg van IL-6 depletie. Niet-klinische data suggereren dat IL-6 niet beschouwd kan worden als groeifactor van enig belang voor de ontwikkeling van het musculoskeletale systeem noch als groeifactor van belang voor de ontwikkeling van andere orgaansystemen. Er werd echter een hogere incidentie gezien van abortus of embryofoetale dood: in de 50 mg/kg/dag doseringsgroep met een systemische blootstelling >100x boven de humaan effectieve plasmaconcentratie. Ook in de 10 mg/kg/dag groep werden meer abortus gezien dan in de controlegroep. Foetale blootstelling aan tocilizumab rondom de tijd dat abortus plaatsvond wordt beschouwd als nagenoeg afwezig. Dit suggereert meer een onderliggende maternale dan een direct foetotoxisch effect. De bevindingen kunnen niet goed verklaard worden door een tocilizumab-gerelateerd werkingsmechanisme. Zowel in-vitro als in-vivo studies tonen consistent dat IL-6 geen rol speelt in de zwangerschapsfysiologie. Er is ook geen preklinisch bewijs dat IL-6 betrokken is in reproductieprocessen. Noch apen met blootstelling aan tocilizumab gedurende meer dan 6 maanden, noch IL-6 k.o. muizen tonen morfologische veranderingen in weefsels van het immuunsysteem of in enig ander orgaan.
Verantwoording
Autorisatiedatum en geldigheid
Laatst beoordeeld : 01-01-2011
Laatst geautoriseerd : 01-01-2011
Geplande herbeoordeling : 01-01-2014
De Nederlandse Vereniging voor Reumatologie is als houder van deze richtlijn de eerstverantwoordelijke voor de actualiteit van deze richtlijn. Uiterlijk in 2013 bepaalt de NVR of deze richtlijn nog actueel is. Zo nodig wordt een nieuwe werkgroep geïnstalleerd om de richtlijn te herzien. De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De andere aan deze richtlijn deelnemende beroepsverenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid ten aanzien van het bewaken van de actualiteit van de aanbevelingen in de richtlijn. Hen wordt verzocht relevante ontwikkelingen binnen hun vakgebied kenbaar te maken aan de eerstverantwoordelijke.
Algemene gegevens
Met ondersteuning van de afdeling Ondersteuning Professionele Kwaliteit van de Orde van Medisch Specialisten. De richtlijnontwikkeling werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (= SKMS).
Doel en doelgroep
Deze richtlijn geeft een leidraad voor de dagelijkse praktijk van het gebruik van Biologicals. De richtlijn heeft een algemeen karakter, kan gebruikt worden om beroepsspecifieke richtlijnen te formuleren en biedt aanknopingspunten voor bijvoorbeeld lokale (instituuts- of regiogebonden) protocollen en/of zorgafspraken.
De richtlijn is primair geschreven voor medisch specialisten die patiënten met chronische ontstekingsziekten (IMID = Immune Mediated Inflammatory Disorders) behandelen met biologicals, alsmede voor deze patiënten zelf en hun overige behandelaars.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2009 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die met biologicals te maken hebben.
Bij het samenstellen van de werkgroep is rekening gehouden met geografische spreiding en evenredige vertegenwoordiging van verschillende verenigingen, ‘scholen’ en academische achtergrond. De werkgroepleden zijn door de wetenschappelijke verenigingen gemandateerd voor deelname en de samenstelling van de werkgroep is goedgekeurd door alle deelnemende wetenschappelijke verenigingen. De werkgroepleden zijn gezamenlijk verantwoordelijk voor de tekst.
- Dr. D.L. Baeten, reumatoloog, Academisch Medisch Centrum, Amsterdam
- Dr. M. Bijl, reumatoloog, Universitair Medisch Centrum Groningen
- Prof. dr. J.W.J. Bijlsma, reumatoloog, Universitair Medisch Centrum Utrecht (voorzitter)
- Dr. A.A. van Bodegraven, maag-, darm-, leverarts, VU Medisch Centrum, Amsterdam
- Dr. P.L.A. van Daele, internist-klinisch immunoloog, Erasmus Medisch Centrum, Rotterdam
- Prof. dr. M. Drent, longarts, Academisch Ziekenhuis Maastricht
- Mevr. drs. G.J. Geven, Reumapatiëntenbond, Amersfoort
- Mevr. drs. J.W. Hagemeijer, senior adviseur, Orde van medisch Specialisten, Utrecht
- Dr. T.L.Th.A. Jansen, reumatoloog, Medisch Centrum Leeuwarden
- Prof. dr. M.A.F.J. van de Laar, reumatoloog, Medisch Spectrum Twente, Enschede
- Prof. dr. R.B.M. Landewé, reumatoloog, Academisch Ziekenhuis Maastricht
- Prof. dr. W.F. Lems, reumatoloog, VU medisch centrum, Amsterdam
- Dr. M.T. Nurmohamed, reumatoloog, Jan van Breemen Instituut, Amsterdam
- Dr. E. Prens, dermatoloog, Erasmus Medisch Centrum, Rotterdam
- Mevr. drs. M.M.J.H. Scholte-Voshaar, Reumapatiëntenbond, Amersfoort
- Mevr. drs. M. Wessels, informatiespecialist, Orde van Medisch Specialisten, Utrecht
Belangenverklaringen
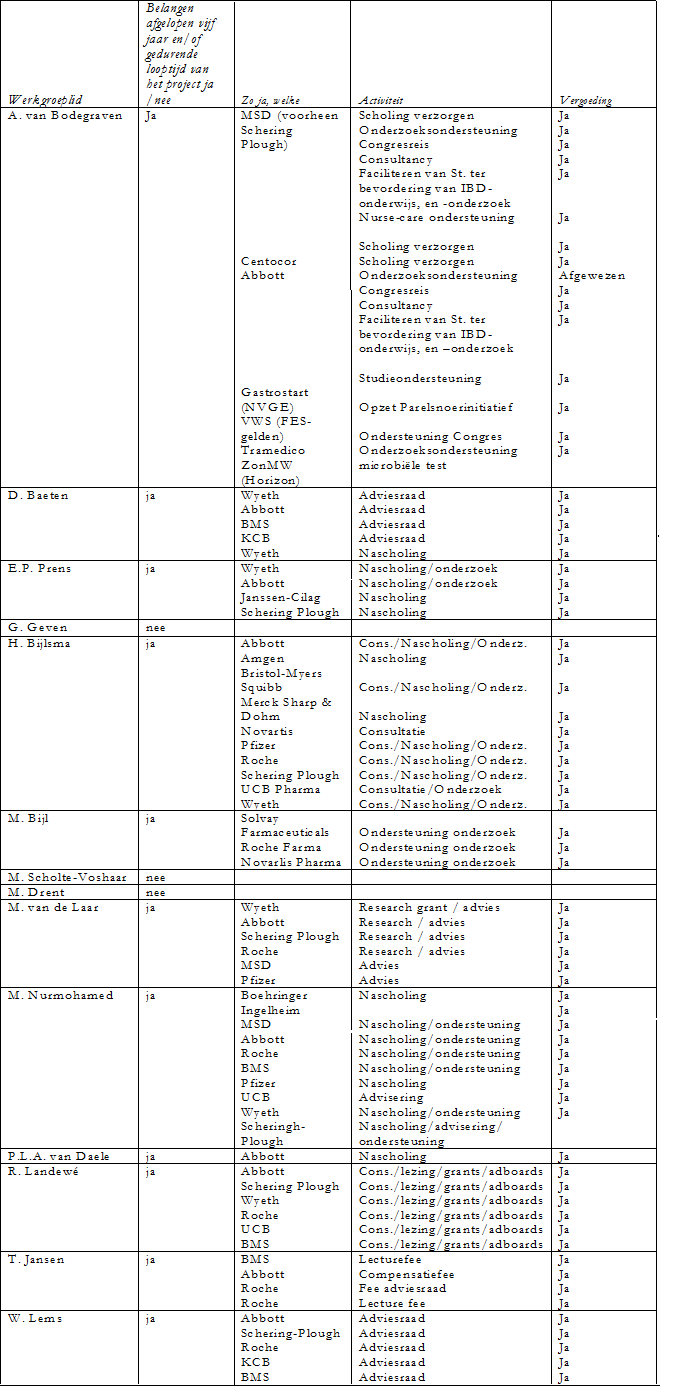
De werkgroepleden is gevraagd om aan te geven of er sprake is van een mogelijke belangenverstrengeling met commerciële bedrijven. Een overzicht hiervan is hieronder weergegeven. De verklaringen van werkgroepleden over mogelijke financiële belangenverstrengeling ligt ter inzage bij de afdeling Ondersteuning Professionele Kwaliteit van de Orde van Medisch Specialisten. Uit de ingevulde belangenverklaringen blijkt dat de werkgroepleden banden met de farmaceutische industrie hebben en dat deze banden gezien het onderwerp belangrijk zijn. Er wordt geconcludeerd dat deze banden geen invloed hebben gehad bij het totstandkomen van de richtlijn.
Inbreng patiëntenperspectief
Gedurende de ontwikkeling van de richtlijn is nadrukkelijk aandacht besteed aan het in kaart brengen van het patiëntenperspectief. In de werkgroep hebben twee patiëntenvertegenwoordigers zitting genomen en zij brachten het perspectief van de patiënten naar voren tijdens de bespreking van de teksten en de formulering van de aanbevelingen. Daarnaast is halverwege het traject een focusgroep georganiseerd waaraan 9 patiënten hebben deelgenomen. De uitgangsvragen zijn voorgelegd aan de leden van de focusgroep en hen is gevraagd naar hun ervaringen en overwegingen die zij van belang achten bij het formuleren van de aanbevelingen. Hiervan is een verslag gemaakt en aan de leden voorgelegd ter verifiëring en eventuele aanvulling. De leden van het schrijverscollectief hebben gebruikgemaakt van de inhoud van deze documentatie voor de formulering van overwegingen vanuit patiëntenperspectief. Een verslag hiervan is hieronder te vinden.
Patiëntenperspectief – verslag focusgroep
In totaal hebben 9 patiënten hun medewerking verleend aan het in kaart brengen van het patiëntenperspectief. Twee mannelijke patiënten met Sarcoidose met Remicade (via infuuskliniek (1 X p.m.) of Humira zelf injecteren (1X p.w.), één mannelijke patiënt met Psoriasis Enbrel zelf injecteren (1X p.w.), twee vrouwen met RA (Enbrel en Rituximab), één mannelijke patiënt met RA (Enbrel), twee vrouwelijke patiënten met Ziekte van Crohn (Humira en Remicade).
Drie personen waren aanwezig bij de focusgroepbijeenkomst die gehouden is op 12 november 2009. De overige 5 personen waren niet in staat om aanwezig te zijn en hebben op digitale wijze hun input geleverd door per uitgangsvraag hun ervaringen terug te koppelen.
Naast het bespreken van de ervaringen is ook gevraagd naar overwegingen die vanuit het perspectief van de patiënt van belang zijn en die naar hun idee meegewogen dienen te worden bij de formulering van de aanbevelingen.
Uit de besprekingen is naar voren gekomen dat de patiënten de geformuleerde uitgangsvragen complex vinden. De patiënten hebben aangegeven dat het maken van een patiëntenversie van de richtlijn belangrijk is omdat nog veel onduidelijk is over het gebruik van biologicals. Door het ontwikkelen van een patiëntenversie van de richtlijn ontvangen de patiënten een instrument waarmee zij ook kunnen bijdragen aan het goed en verantwoord gebruik van biologicals. Hierbij is als voorwaarde gesteld dat de patiëntenversie toegankelijk geschreven moet worden.
Alle deelnemers waren lid van een patiëntenvereniging en ervaren dit als zeer plezierig. Het geeft onder meer de mogelijkheid tot het uitwisselen van informatie met lotgenoten.
VOORAFGAANDE AAN DE BEHANDELING MET BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 1: - Patiënten geven aan dat de aanloop naar de behandeling met biologicals overwegend een langdurig proces is geweest. Er wordt overgegaan tot biologicals als alle andere behandelingsvormen niet blijken te helpen of te veel bijwerkingen veroorzaken. - Patiënten vinden het belangrijk dat alle aspecten die van belang zijn bij de behandeling van biologicals worden onderzocht, maar hebben geen zicht op welke aspecten dit dan moeten zijn. Zij geven aan dat het echter belangrijk is dat zij door de specialist of door de verpleegkundige deelgenoot worden gemaakt van het afwegingsproces door informatie te ontvangen zodat zij zelf actief kunnen meewerken aan het zorgproces. |
Overweging vanuit het patiëntenperspectief bij UV 2: - Patiënten vinden het belangrijk dat de arts door wie zij worden behandeld deskundig is, zodat zij erop kunnen vertrouwen in goede handen te zijn. - De (voorschrijvende) medisch specialist dient de regie in handen te houden en daarbij is het overleg met de andere betrokken professionals belangrijk. - Het is belangrijk dat de professionals communicatief zijn en duidelijk uitleg kunnen geven op niveau van de leek. Daarnaast is het wenselijk dat de medisch specialist toegankelijk en laagdrempelig is bij vragen; te weten via de telefoon of email. - Als het niet goed gaat met de patiёnt is het belangrijk dat de medisch specialist confronterend en direct is door aan te geven dat het helemaal niet goed gaat, dat de patiënt zich overvraagt of dat bijvoorbeeld gestopt moet worden met werken. Het is daarbij belangrijk dat de patiënt geen ruimte wordt gegeven om te marchanderen. - De partner is samen met de patiënt ziek en hiermee een essentiёle schakel die duidelijk betrokken behoort te worden in het zorgproces. |
BIJWERKINGEN VAN BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 3: - Patiënten vinden het belangrijk dat zij duidelijke en volledige (zowel schriftelijke als mondelinge) informatie ontvangen over de bijwerkingen die zich kunnen voordoen bij een behandeling met biologicals. - Patiënten vinden het belangrijk dat, als zij bijwerkingen ervaren, duidelijk is tot wie zij zich moeten richten, dus tot de huisarts of de medisch specialist. - Patiënten vinden het belangrijk dat, als zij bijwerkingen ervaren, hier serieus naar wordt gekeken en deskundig op wordt geacteerd. - Patiënten vinden het belangrijk dat zij instructies ontvangen wat zij zelf kunnen doen om bepaalde bijwerkingen te voorkomen. |
Overweging vanuit het patiëntenperspectief bij UV 4: - Patiënten vinden het belangrijk dat zij informatie ontvangen over de het risico op kanker bij biologicals. - Patiënten vinden het belangrijk dat zij actief worden gevolgd op het mogelijk ontstaan van kanker. |
ZWANGERSCHAP EN BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 5: Op basis van de focusgroep zijn geen overwegingen aan te dragen. |
BIOLOGICALS EN (OPERATIEVE) INGREPEN: |
Overweging vanuit het patiëntenperspectief bij UV 6: - Patiënten vinden het belangrijk dat de voorschrijvend specialist betrokken is bij het besluitvormingsproces van een operatie. Daarbij is het essentieel dat er direct contact is tussen de betrokken specialisten. - Patiënten vinden het belangrijk dat zij in bezit zijn van een ‘kaartje’ waarop staat dat zij biologicals gebruiken en dat met de specialist contact opgenomen moet worden in geval van tandheelkundige of operatieve ingrepen. - Patiënten vinden het belangrijk dat zij informatie ontvangen over de stappen die gezet moeten worden bij het staken of doorgaan van biologicals bij een operatie. Hierdoor zijn zij in staat zelf actief mee te werken en hebben meer het gevoel controle te hebben over hun behandeling met biologicals. |
FOLLOW-UP VAN DE BEHANDELING MET BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 7: - Patiënten vinden het belangrijk dat zij regelmatig (om de 3 tot 6 mnd) door de medisch specialist worden onderzocht waarbij de werking van de biologicals in kaart wordt gebracht. - Patiënten vinden het belangrijk dat zij worden geinformeerd over de uitslag van de periodieke controle zodat zij deelgenoot zijn van het zorgproces. - Patiënten geven aan geconfronteerd te worden met een veelheid aan informatiestromen die regelmatig tegenstrijdig zijn. Zij vinden het belangrijk dat hier meer regie over gevoerd gaat worden. - Patiënten vinden het belangrijk dat professionels aangeven wanneer men geen duidelijkheid ten aanzien van bepaalde vraagstukken kunnen geven omdat nog zoveel onbekend is bij de behandeling van biologicals. - Patiënten vinden het belangrijk regelmatig contact te hebben met de (specialistisch) verpleegkundige zodat zij eventuele vragen kan beantwoorden. |
BIOLOGICALS EN VACCINATIE: |
Overweging vanuit het patiëntenperspectief bij UV 8: - Patiënten vinden het belangrijk dat zij (tijdig) informatie ontvangen over welke vaccinaties zij toegediend kunnen krijgen. - Patiënten vinden het belangrijk dat zij informatie ontvangen bij welke verschijnselen, nadat zij een vaccinatie hebben ontvangen, zij contact op moeten nemen met de medisch speicalist. |
DE BEHANDELING MET BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 9: - Patiënten vinden het belangrijk dat de arts door wie zij worden behandeld deskundig is en over de meest recente inzichten beschikt zodat zij erop kunnen vertrouwen in goede handen te zijn. - De (voorschrijvende) medisch specialist dient de regie in handen te houden en daarbij is het overleg met de andere betrokken professionals belangrijk. - Het is belangrijk dat de professionals communicatief zijn en duidelijk uitleg kunnen geven op niveau van de leek. Daarnaast is het wenselijk dat de medisch specialist toegankelijk en laagdrempelig is bij vragen; te weten via de telefoon of email. - Als het niet goed gaat met de patiёnt is het belangrijk dat de medisch specialist confronterend en direct is door aan te geven dat het helemaal niet goed gaat, dat de patient zich overvraagt of dat bijvoorbeeld gestopt moet worden met werken. Het is daarbij belangrijk dat de patiënt geen ruimte wordt gegeven om te marchanderen. |
LANGDURIG VERBLIJF ELDERS: |
Overweging vanuit het patiëntenperspectief bij UV 10: - Patiënten vinden het belangrijk dat zij informatie ontvangen over: - hoe zij de biologicals het beste naar het buitenland kunnen vervoeren; - naar welke gebieden zij kunnen reizen; - de mogelijkheden om in het buitenland een infuus met biologicals te ontvangen.
|
Methode ontwikkeling
Evidence based
Implementatie
Tijdens de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn en de uitvoerbaarheid van de aanbevelingen. Daarbij is gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De richtlijn is verspreid onder alle relevante beroepsgroepen en instellingen. Een samenvatting van de richtlijn is gepubliceerd in het Nederlands Tijdschrift voor Geneeskunde en in tijdschriften van de deelnemende wetenschappelijke verenigingen.
Werkwijze
De werkgroep heeft een jaar aan de totstandkoming van de richtlijn gewerkt. Binnen de werkgroep was een schrijverscollectief ingesteld. De leden van het schrijverscollectief zochten systematisch naar literatuur en beoordeelden kwaliteit en inhoud ervan. Vervolgens schreven zij een concepttekst waarin de literatuur werd verwerkt. Tijdens vergaderingen lichtten zij hun teksten toe aan de overige leden van de werkgroep. De conceptrichtlijn is in februari 2010 schriftelijk aan alle betrokken wetenschappelijke verenigingen aangeboden en gevraagd de richtlijn aan hun leden voor te leggen. Daarnaast is de richtlijn ook naar wetenschappelijke verenigingen gestuurd die niet in de werkgroep hebben geparticipeerd, te weten oogartsen, neurologen, gynaecologen en tandartsen. De ontvangen commentaren zijn, waar relevant bevonden, verwerkt in de definitieve richtlijn.
Wetenschappelijke onderbouwing
De onderbouwing van de richtlijn is mede gebaseerd op bewijs uit gepubliceerd wetenschappelijk onderzoek. Relevante artikelen werden gezocht met systematische zoekacties. Er werd gezocht tussen 1998 en 2009 in Medline en Embase. Voor de oriënterende search werd ook gezocht in de Cochrane Library en werd specifiek gezocht naar al bestaande richtlijnen in online raadpleegbare (inter)nationale guideline clearinghouses.
Hierbij werd de taal gelimiteerd tot Nederlands, Engels, Duits en Frans. Daarnaast werden artikelen geëxtraheerd uit referentielijsten van opgevraagde literatuur. Dit leverde bij enkele uitgangsvragen nog aanvullende artikelen op.
Doordat de uitgangsvragen niet gericht waren op het beoordelen van de effectiviteit van de interventies maar veelal gingen over bijwerkingen, complicaties en diagnostiek bleek een beperking tot systematische reviews en RCTs vaak niet zinvol. De searches zijn verricht in mei en juni 2009. Voor alle uitgangsvragen is gebruik gemaakt van een uniforme formulering van de patiëntencategorie en de interventie.
Voor de gehanteerde zoektermen wordt verwezen naar Appendix 1. Op verzoek zijn de volledige zoekstrategieën beschikbaar. Daarnaast werden artikelen geëxtraheerd uit referentielijsten van opgevraagde literatuur en zijn enkele relevante publicaties tot 1 november 2009 meegenomen. Lopend onderzoek is buiten beschouwing gelaten. Abstracts van congressen van de afgelopen 2 jaar (november 2007 tot 1 november 2009) zijn meegenomen bij de selectie van de literatuur. Relevante informatie vanuit deze abstract wordt uitgewerkt bij de overige overwegingen. Onder samenvatting van de literatuur / conclusies worden alleen gepubliceerde onderzoeken / richtlijnen uitgewerkt.
De geselecteerde artikelen zijn beoordeeld op kwaliteit van het onderzoek en gegradeerd naar mate van bewijs. Hierbij is de standaardindeling gebruikt: zie tabel 1. Na selectie bleven de artikelen over die als onderbouwing bij de verschillende conclusies staan vermeld. De beoordeling van de verschillende artikelen is opgenomen onder het kopje ‘samenvatting van de literatuur’. Het wetenschappelijk bewijs is vervolgens kort samengevat in een ‘conclusie’. De belangrijkste literatuur waarop deze conclusie is gebaseerd staat bij de conclusie vermeld, inclusief de mate van bewijs (zie tabel 2).
Voor het formuleren van een aanbeveling zijn, naast het wetenschappelijk bewijs, vaak nog andere aspecten van belang, bijvoorbeeld patiëntenvoorkeuren, kosten, beschikbaarheid of organisatorische aspecten. Deze aspecten worden, voor zover niet wetenschappelijk onderzocht, vermeld onder het kopje ‘overwegingen’. In de overige overwegingen spelen de ervaring en de mening van de werkgroepleden een belangrijke rol. De ‘aanbeveling’ is het resultaat van de combinatie van het beschikbare bewijs en de overige overwegingen.
Voor een aantal uitgangsvragen zijn evidencetabellen gemaakt en deze zijn te raadplegen in Appendix 3.
Tabel 1: Indeling van methodologische kwaliteit van individuele studies
|
|
Interventie |
Diagnostische accuratesse onderzoek |
Schade of bijwerkingen, etiologie, prognose* |
|
A1 |
Systematische review van ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van A2-niveau |
||
|
A2 |
Gerandomiseerd dubbelblind vergelijkend klinisch onderzoek van goede kwaliteit van voldoende omvang |
Onderzoek ten opzichte van een referentietest (een ‘gouden standaard’) met tevoren gedefinieerde afkapwaarden en onafhankelijke beoordeling van de resultaten van test en gouden standaard, betreffende een voldoende grote serie van opeenvolgende patiënten die allen de index- en referentietest hebben gehad |
Prospectief cohortonderzoek van voldoende omvang en follow-up, waarbij adequaat gecontroleerd is voor ‘confounding’ en selectieve follow-up voldoende is uitgesloten |
|
B |
Vergelijkend onderzoek, maar niet met alle kenmerken als genoemd onder A2 (hieronder valt ook patiënt-controleonderzoek, cohort-onderzoek) |
Onderzoek ten opzichte van een referentietest, maar niet met alle kenmerken die onder A2 zijn genoemd |
Prospectief cohortonderzoek, maar niet met alle kenmerken als genoemd onder A2 of retrospectief cohortonderzoek of patiënt-controleonderzoek |
|
C |
Niet-vergelijkend onderzoek |
||
|
D |
Mening van deskundigen |
||
* Deze classificatie is alleen van toepassing in situaties waarin om ethische of andere redenen gecontroleerde trials niet mogelijk zijn. Zijn die wel mogelijk dan geldt de classificatie voor interventies.
Tabel 2: Niveau van bewijs van de conclusie
|
Conclusie gebaseerd op |
|
|
1 |
Onderzoek van niveau A1 of ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van niveau A2 |
|
2 |
Eén onderzoek van niveau A2 of ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van niveau B |
|
3 |
Eén onderzoek van niveau B of C |
|
4 |
Mening van deskundigen |
Deze (concept)richtlijn is opgesteld aan de hand van het Appraisal of Guidelines for Research & Evaluation (AGREE) instrument. Dit instrument is in een Europees verband opgesteld om de procedurele kwaliteit van richtlijnen te kunnen beoordelen. Door de aspecten van AGREE te verwerken in de inleiding van de richtlijn, wordt duidelijk aan welke kwaliteitseisen is voldaan.
Zoekverantwoording
Zoekacties zijn opvraagbaar. Neem hiervoor contact op met de Richtlijnendatabase.