Follow-up van behandeling biologicals
Uitgangsvraag
Op welke wijze dient men bij IMID patiënten de behandeling met biologicals te monitoren en evalueren?
Aanbeveling
Bij IMID patiënten behandeld met biologicals dient men de behandeling te monitoren door na 12 en 24 weken de repons vast te stellen aan de hand van de ziekteactiviteit.
Specifiek bij IBD wordt de effectiviteit van infliximab direct na de inductietherapie (3 infusen) geëvalueerd.
Er dient tijdens de behandeling met biologicals rekening gehouden te worden met een verhoogde kans op opportunistische infecties, met een ernstiger beloop van banale infecties en met het opflakkeren van latente tuberculose.
Bij intercurrente ernstige infecties dient het gebruik van biologicals tijdelijk te worden onderbroken. De behandeling van deze infecties is niet anders dan gebruikelijk.
IMID patiënten dienen adequaat te worden geïnstrueerd hoe zij het risico op infecties kunnen verkleinen en hoe zij dienen te handelen bij tekenen van een mogelijke infectie (zoals koorts en malaise).
Het is gewenst om bij IMID patiënten die bekend zijn met een maligniteit in de voorgeschiedenis alert te zijn op de ontwikkeling van tumoren.
Er wordt geadviseerd om IMID patiënten die behandeld worden met biologicals te informeren over het mogelijk risico op de ontwikkeling van huidtumoren en het belang van zelfinspectie van de huid van armen/benen/hoofd/hals te benadrukken.
Wanneer bij een IMID patiënt een plaveiselcelcarcinoom van de huid wordt vastgesteld dient in overleg met de dermatoloog risico analyse plaats te vinden naar de kans op nieuwe tumoren. Indien verhoogd, dan dient overwogen te worden de behandeling met biologicals te staken.
Bij onvoldoende klinische respons kan overwogen worden te ‘switchen’ van een eerste TNFα-blokker naar een tweede TNFα-blokker, dan wel naar een ander klasse van biologicals.
Het is raadzaam om een wash-out periode te hanteren alvorens te starten met een andere biological. De volgende wash-out periode wordt geadviseerd, voor:
- etanercept 1 week;
- adalimumab 2 weken;
- infliximab 6 tot 8 weken;
- abatacept 3 maanden;
- rituximab 6 maanden.
Overwegingen
Monitoren en evalueren van effectiviteit
Vrijwel alle literatuur over dit onderwerp heeft betrekking op TNFα-blokkerende biologicals. Van de andere klassen van biologicals zijn alleen data uit registratietrials voorhanden. Bovendien heeft de schaarse literatuur over het monitoren van effectiviteit voornamelijk betrekking op de behandeling van RA en AS.
De onderliggende vraag is met welke frequentie patiënten die worden behandeld met biologicals dienen te worden gecontroleerd om vast te stellen of de biological effectief is, of deze veilig kan worden gecontinueerd en welke vorm en inhoud de controles dienen te hebben.
Géén van deze vragen is onderwerp van studie geweest in RCT’s. Diverse richtlijncommissies hebben zich gebogen over het vraagstuk monitoring, al dan niet in de context van het gebruik van biologicals. Van belang hierbij is de vraag hoe lang moet worden behandeld voordat kan worden gesproken van non-respons. De ASAS groep heeft in haar consensus statement over het gebruik van TNFα-blokkerende middelen bij AS opgenomen dat 6 tot 12 weken moet worden behandeld voordat kan worden beoordeeld of patiënten non-responder zijn (Braun et al, 2006). Zeer recentelijk is op basis van gegevens uit klinische trials besloten dat deze tijdsduur tenminste 12 weken moet zijn.
De Nederlandse, Britse en EULAR richtlijnen voor het gebruik van biologicals bij RA schrijven voor dat indien na drie maanden géén klinische respons is opgetreden, gemeten met de DAS28 (≥ 1.2 units daling), het gebruik van de biological dient te worden gestaakt respectievelijk te worden heroverwogen. Pocock et al hebben recentelijk gekeken naar 40 van de 189 patiënten bij wie na 3 maanden evaluatie géén DAS respons kon worden vastgesteld. Na zes maanden behandeling bleek bij 27 van de 40 patiënten alsnog een DAS-respons te kunnen worden vastgesteld (Pocock et al, 2008). Resultaten van deze strekking werden ook gerapporteerd door Kievit et al, die vonden dat 37% van de non-responders op 3 maanden alsnog responder werden na 6 maanden behandeling (Kievit et al, 2009). Het National Institute for Health and Clinical Excellence (NICE) stelt in haar richtlijn voor de behandeling van RA voor om 6 maanden te behandelen voordat wordt besloten dat een patiënt non-responder is.
Er is weinig onderzoek gedaan naar welke variabelen zouden moeten worden gemonitord om vast te stellen of een biological effectief is. ASAS adviseert bij patiënten met AS het gebruik van de BASDAI om vast te stellen of er een respons is op TNF-blokkerende therapie: De BASDAI dient met tenminste 50% of met 2 units (op een schaal van 10) te zijn verminderd.
NICE adviseert bij patiënten met RA zowel een laboratoriummaat (CRP) als een samengestelde maat voor ziekteactiviteit (DAS28) te gebruiken voor dit doel: voorgesteld wordt om een daling van DAS28 ≥1.2 units als adequate respons te hanteren.
Bij IBD-patiënten wordt geadviseerd de kliniek te vervolgen, eventueel aangevuld met bepaling van activiteitsparameters zoals een CRP-concentratie, een fecale calprotectine, of – nog moderner – een vaststellen van herstel van het voorheen geülcereerde slijmvlies.
Een van de redenen voor verlies van effectiviteit van een TNFα-blokkerende biological is antistofvorming tegen het product; dit kan mogelijk relevant zijn bij secundaire ineffectiviteit en daarmee gevolgen hebben voor de kans dat een tweede anti-TNFα-therapie werkzaam zou kunnen zijn.
Monitoring van veiligheid
Een belangrijk facet van monitoring is veiligheid. Over het risico op infecties en het risico op maligniteiten tijdens behandeling met TNFα-blokkerende middelen is het meeste gerapporteerd buiten de context van (gesponsorde) klinische trials. De informatie over Rituximab, Anakinra, Abatacept en Tocilizumab is fragmentarisch en in het algemeen beperkt tot RCTs.
Ernstige infecties
In uitgangsvraag 3 is het risico op infecties uitgewerkt en hierin wordt geconcludeerd dat bij IMID patiënten, behandeld met TNFα-blokkers, het risico op zowel (myco)bacteriële als virale infecties (in het bijzonder herpes zoster) verhoogd is. Van de andere biologicals is dat niet duidelijk, waarbij moet worden aangetekend dat deze middelen vaak korter op de markt zijn, en dus de follow-up relatief beperkt is.
Voor de behandeling met biologicals dient te worden gescreend op (latente) tuberculose en, indien nodig, dient deze te worden behandeld.
Patiënten die worden behandeld met biologicals dienen zich bewust te zijn van het verhoogd risico op infecties, en bij tekenen van mogelijke infectie (zoals koorts en malaise) medisch advies in te winnen. Maar ook anderen, bijvoorbeeld huisartsen en andere specialisten die deze patiënten behandelen, dienen op de hoogte te zijn van dit verhoogde infectierisico.
Maligniteiten
In uitgangsvraag 6 is het risico op maligniteiten uitgewerkt en hierin wordt geadviseerd dat het niet geïndiceerd is om IMID patiënten, bij wie men een behandeling met biologicals wil starten, vooraf te screenen op maligniteiten. Maligniteit in de voorgeschiedenis bij IMID patiënten vormt geen absolute contra-indicatie om behandeld te worden met biologicals. Het is wenselijk om bij IMID patiënten die bekend zijn met een maligniteit in de voorgeschiedenis alert te zijn op de ontwikkeling van tumoren. Bij IMID patiënten die behandeld worden met biologicals wordt geadviseerd de huid van armen/benen/hoofd/hals jaarlijks te inspecteren op de ontwikkeling van huidtumoren, om wanneer een plaveiselcelcarcinoom wordt vastgesteld, de behandeling met biologicals te staken.
Switching
De vraag of het zinvol is om een tweede TNFα-blokkerende biological te starten bij patiënten met RA die hebben gefaald op de eerste TNFα-blokker, is onvolledig beantwoord. RCTs zijn niet voorhanden. Er is echter wel observationeel onderzoek verricht. In het Britse BSRBR werd gevonden dat RA-patiënten die hun eerste TNFα-blokker moesten staken in verband met ineffectiviteit een 2 tot 3 keer zo grote kans hadden om ook hun tweede TNFα-blokker om die reden te staken. Hetzelfde verband bestond voor staken in verband met bijwerkingen (Hyrich et al, 2007). In het Deense DANBIO register werd gevonden dat RA-patiënten die veranderden van TNFα-blokker in verband met ineffectiviteit een betere klinische respons lieten zien op de tweede dan op de eerste TNFα-blokker. Zo constateerden Wolbink et al (2009) dat switchers naar adalimumab een betere respons hadden bij antistoffen tegen infliximab dan zonder deze antistoffen. Patiënten die hun eerste TNFα-blokker staakten in verband met bijwerkingen hadden een zelfde klinische respons op de tweede TNFα-blokker.
De vraag of switching naar een andere klasse van biologicals zinvol is, is eveneens onvolledig beantwoord. Een recente analyse van het Zwitserse biologicals register suggereert dat patiënten die faalden op een TNFα-blokker vanwege ineffectiviteit een betere klinische respons mogen verwachten op rituximab dan op een tweede TNFα-blokker. Dit voordeel bestond niet indien patiënten faalden op hun eerste TNFα-blokker vanwege bijwerkingen (Finck et al, 2009).
Switching bij AS is slechts sporadisch onderzocht. In een analyse van Coates et al hadden 14 van de 15 patiënten die faalden op hun eerste TNFα-blokker een goede klinische respons (BASDAI verbetering >50%) op hun tweede TNFα blokker (Coates et al, 2008). Deze gegevens werden deels bevestigd door Pradeep et al (2008) die vonden dat vier van de vijf patiënten die veranderden van TNFα-blokker vanwege bijwerkingen, maar slechts 5 van de 12 patiënten die dit deden vanwege ineffectiviteit een significante klinische respons hadden.
Switching bij andere indicaties is niet of nauwelijks onderzocht.
Washout
In een recente RCT bij patiënten met RA werd aangetoond dat het geen verschil maakt met betrekking tot effectiviteit (kans op klinische respons) en veiligheid (bijwerkingen) of abatacept direct na het staken van de TNFα-blokker dan wel na een wash-out periode van tenminste 2 maanden wordt gestart (Schiff et al ARD 2008). In een aantal RCTs naar de werkzaamheid en veiligheid van biologicals (TNFα-blokkers en andere klassen) zijn patiënten ingesloten die voorheen behandeld waren met een TNFα-blokker. In deze onderzoeken werd in het algemeen een verplichte wash-out periode voor TNFα-blokkers gehanteerd, zodat het niet duidelijk is of direct kan worden overgeschakeld van de eerste TNFα-blokker naar tweede TNFα-blokker of biological van een andere klasse.
De vraag of direct na het staken van een biological kan worden gestart met een andere biological (zelfde of andere klasse) is slechts summier onderzocht. Bij gebrek aan betrouwbare data wordt bij het bepalen van een veilige washout periode momenteel vaak een marge van 5 keer de halfwaardetijd van het product aangehouden. Een dergelijke marge wordt in de klinische praktijk echter als uitermate onpraktisch ervaren. Min of meer gangbaar (maar niet geschraagd door enig wetenschappelijk bewijs) is dat infliximab 6 tot 8 weken moet worden gestaakt voordat een andere biological kan worden Voor etanercept is dit 1 week, voor adalimumab 2 weken, voor abatacept 3 maanden en voor rituximab 6 maanden.
Onderbouwing
Achtergrond
Bij wetenschappelijk onderzoek naar de effectiviteit en veiligheid van biologicals ligt de nadruk op het starten van een (voor de patiënt) nieuwe biological. In een RCT wordt onderzocht of de patiënt een vooraf bepaald doel wel of niet haalt dan wel of de patiënt de biological kan verdragen/continueren binnen de context (en tijdsduur) van de trial. Deze context verschilt fundamenteel van de dagelijkse klinische praktijk. Uitkomstmaten die gebruikt worden in RCTs zijn vaak ontwikkeld voor het vergelijken van groepen en zijn dientengevolge niet zomaar geschikt voor toepassing bij individuele patiënten in de klinische praktijk. De vraag hoe een individuele patiënt dient te worden gevolgd om vast te stellen of een biological effectief is, laat zich dan ook niet beantwoorden met gebruikmaking van de RCTs die de effectiviteit van een biological bewijzen. Dit geldt ook voor de vraag welke de beste monitoringstrategie is in de klinische praktijk en de vraag wanneer zonodig een volgend biological kan worden gestart.
Verantwoording
Autorisatiedatum en geldigheid
Laatst beoordeeld : 01-01-2011
Laatst geautoriseerd : 01-01-2011
Geplande herbeoordeling : 01-01-2014
De Nederlandse Vereniging voor Reumatologie is als houder van deze richtlijn de eerstverantwoordelijke voor de actualiteit van deze richtlijn. Uiterlijk in 2013 bepaalt de NVR of deze richtlijn nog actueel is. Zo nodig wordt een nieuwe werkgroep geïnstalleerd om de richtlijn te herzien. De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De andere aan deze richtlijn deelnemende beroepsverenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid ten aanzien van het bewaken van de actualiteit van de aanbevelingen in de richtlijn. Hen wordt verzocht relevante ontwikkelingen binnen hun vakgebied kenbaar te maken aan de eerstverantwoordelijke.
Algemene gegevens
Met ondersteuning van de afdeling Ondersteuning Professionele Kwaliteit van de Orde van Medisch Specialisten. De richtlijnontwikkeling werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (= SKMS).
Doel en doelgroep
Deze richtlijn geeft een leidraad voor de dagelijkse praktijk van het gebruik van Biologicals. De richtlijn heeft een algemeen karakter, kan gebruikt worden om beroepsspecifieke richtlijnen te formuleren en biedt aanknopingspunten voor bijvoorbeeld lokale (instituuts- of regiogebonden) protocollen en/of zorgafspraken.
De richtlijn is primair geschreven voor medisch specialisten die patiënten met chronische ontstekingsziekten (IMID = Immune Mediated Inflammatory Disorders) behandelen met biologicals, alsmede voor deze patiënten zelf en hun overige behandelaars.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2009 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die met biologicals te maken hebben.
Bij het samenstellen van de werkgroep is rekening gehouden met geografische spreiding en evenredige vertegenwoordiging van verschillende verenigingen, ‘scholen’ en academische achtergrond. De werkgroepleden zijn door de wetenschappelijke verenigingen gemandateerd voor deelname en de samenstelling van de werkgroep is goedgekeurd door alle deelnemende wetenschappelijke verenigingen. De werkgroepleden zijn gezamenlijk verantwoordelijk voor de tekst.
- Dr. D.L. Baeten, reumatoloog, Academisch Medisch Centrum, Amsterdam
- Dr. M. Bijl, reumatoloog, Universitair Medisch Centrum Groningen
- Prof. dr. J.W.J. Bijlsma, reumatoloog, Universitair Medisch Centrum Utrecht (voorzitter)
- Dr. A.A. van Bodegraven, maag-, darm-, leverarts, VU Medisch Centrum, Amsterdam
- Dr. P.L.A. van Daele, internist-klinisch immunoloog, Erasmus Medisch Centrum, Rotterdam
- Prof. dr. M. Drent, longarts, Academisch Ziekenhuis Maastricht
- Mevr. drs. G.J. Geven, Reumapatiëntenbond, Amersfoort
- Mevr. drs. J.W. Hagemeijer, senior adviseur, Orde van medisch Specialisten, Utrecht
- Dr. T.L.Th.A. Jansen, reumatoloog, Medisch Centrum Leeuwarden
- Prof. dr. M.A.F.J. van de Laar, reumatoloog, Medisch Spectrum Twente, Enschede
- Prof. dr. R.B.M. Landewé, reumatoloog, Academisch Ziekenhuis Maastricht
- Prof. dr. W.F. Lems, reumatoloog, VU medisch centrum, Amsterdam
- Dr. M.T. Nurmohamed, reumatoloog, Jan van Breemen Instituut, Amsterdam
- Dr. E. Prens, dermatoloog, Erasmus Medisch Centrum, Rotterdam
- Mevr. drs. M.M.J.H. Scholte-Voshaar, Reumapatiëntenbond, Amersfoort
- Mevr. drs. M. Wessels, informatiespecialist, Orde van Medisch Specialisten, Utrecht
Belangenverklaringen
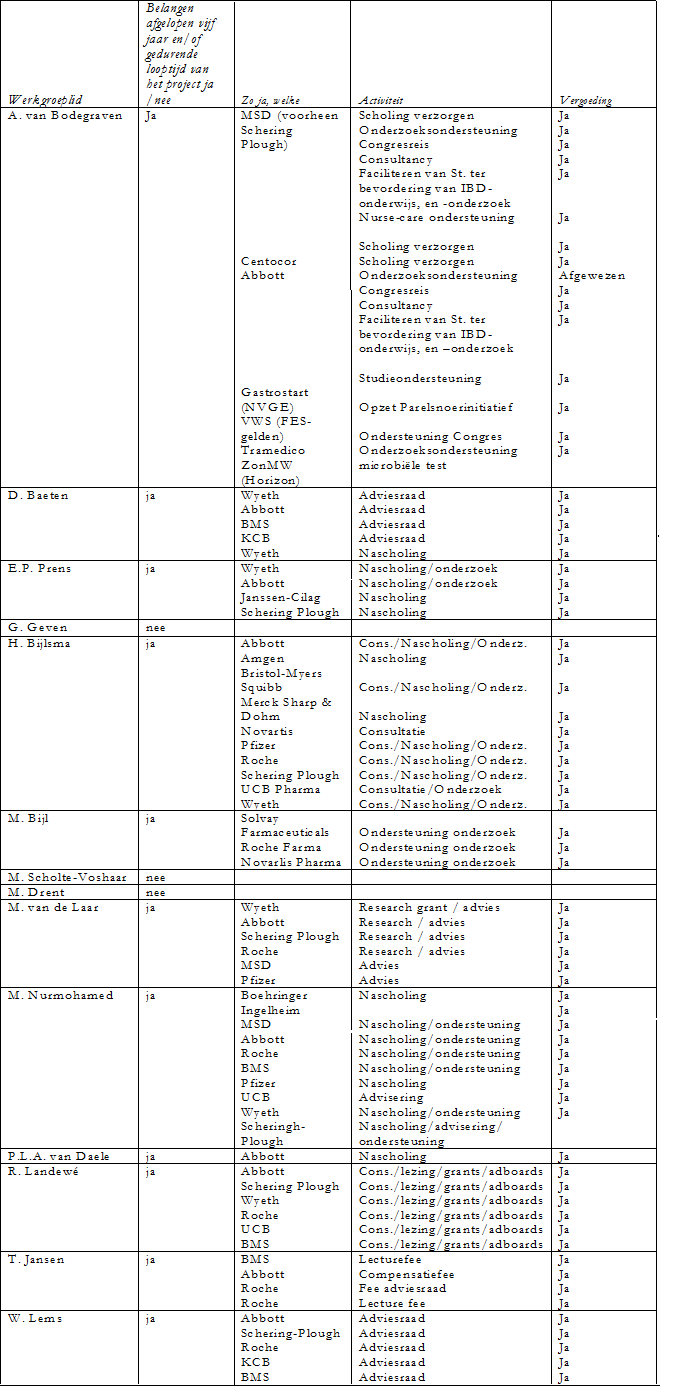
De werkgroepleden is gevraagd om aan te geven of er sprake is van een mogelijke belangenverstrengeling met commerciële bedrijven. Een overzicht hiervan is hieronder weergegeven. De verklaringen van werkgroepleden over mogelijke financiële belangenverstrengeling ligt ter inzage bij de afdeling Ondersteuning Professionele Kwaliteit van de Orde van Medisch Specialisten. Uit de ingevulde belangenverklaringen blijkt dat de werkgroepleden banden met de farmaceutische industrie hebben en dat deze banden gezien het onderwerp belangrijk zijn. Er wordt geconcludeerd dat deze banden geen invloed hebben gehad bij het totstandkomen van de richtlijn.
Inbreng patiëntenperspectief
Gedurende de ontwikkeling van de richtlijn is nadrukkelijk aandacht besteed aan het in kaart brengen van het patiëntenperspectief. In de werkgroep hebben twee patiëntenvertegenwoordigers zitting genomen en zij brachten het perspectief van de patiënten naar voren tijdens de bespreking van de teksten en de formulering van de aanbevelingen. Daarnaast is halverwege het traject een focusgroep georganiseerd waaraan 9 patiënten hebben deelgenomen. De uitgangsvragen zijn voorgelegd aan de leden van de focusgroep en hen is gevraagd naar hun ervaringen en overwegingen die zij van belang achten bij het formuleren van de aanbevelingen. Hiervan is een verslag gemaakt en aan de leden voorgelegd ter verifiëring en eventuele aanvulling. De leden van het schrijverscollectief hebben gebruikgemaakt van de inhoud van deze documentatie voor de formulering van overwegingen vanuit patiëntenperspectief. Een verslag hiervan is hieronder te vinden.
Patiëntenperspectief – verslag focusgroep
In totaal hebben 9 patiënten hun medewerking verleend aan het in kaart brengen van het patiëntenperspectief. Twee mannelijke patiënten met Sarcoidose met Remicade (via infuuskliniek (1 X p.m.) of Humira zelf injecteren (1X p.w.), één mannelijke patiënt met Psoriasis Enbrel zelf injecteren (1X p.w.), twee vrouwen met RA (Enbrel en Rituximab), één mannelijke patiënt met RA (Enbrel), twee vrouwelijke patiënten met Ziekte van Crohn (Humira en Remicade).
Drie personen waren aanwezig bij de focusgroepbijeenkomst die gehouden is op 12 november 2009. De overige 5 personen waren niet in staat om aanwezig te zijn en hebben op digitale wijze hun input geleverd door per uitgangsvraag hun ervaringen terug te koppelen.
Naast het bespreken van de ervaringen is ook gevraagd naar overwegingen die vanuit het perspectief van de patiënt van belang zijn en die naar hun idee meegewogen dienen te worden bij de formulering van de aanbevelingen.
Uit de besprekingen is naar voren gekomen dat de patiënten de geformuleerde uitgangsvragen complex vinden. De patiënten hebben aangegeven dat het maken van een patiëntenversie van de richtlijn belangrijk is omdat nog veel onduidelijk is over het gebruik van biologicals. Door het ontwikkelen van een patiëntenversie van de richtlijn ontvangen de patiënten een instrument waarmee zij ook kunnen bijdragen aan het goed en verantwoord gebruik van biologicals. Hierbij is als voorwaarde gesteld dat de patiëntenversie toegankelijk geschreven moet worden.
Alle deelnemers waren lid van een patiëntenvereniging en ervaren dit als zeer plezierig. Het geeft onder meer de mogelijkheid tot het uitwisselen van informatie met lotgenoten.
VOORAFGAANDE AAN DE BEHANDELING MET BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 1: - Patiënten geven aan dat de aanloop naar de behandeling met biologicals overwegend een langdurig proces is geweest. Er wordt overgegaan tot biologicals als alle andere behandelingsvormen niet blijken te helpen of te veel bijwerkingen veroorzaken. - Patiënten vinden het belangrijk dat alle aspecten die van belang zijn bij de behandeling van biologicals worden onderzocht, maar hebben geen zicht op welke aspecten dit dan moeten zijn. Zij geven aan dat het echter belangrijk is dat zij door de specialist of door de verpleegkundige deelgenoot worden gemaakt van het afwegingsproces door informatie te ontvangen zodat zij zelf actief kunnen meewerken aan het zorgproces. |
Overweging vanuit het patiëntenperspectief bij UV 2: - Patiënten vinden het belangrijk dat de arts door wie zij worden behandeld deskundig is, zodat zij erop kunnen vertrouwen in goede handen te zijn. - De (voorschrijvende) medisch specialist dient de regie in handen te houden en daarbij is het overleg met de andere betrokken professionals belangrijk. - Het is belangrijk dat de professionals communicatief zijn en duidelijk uitleg kunnen geven op niveau van de leek. Daarnaast is het wenselijk dat de medisch specialist toegankelijk en laagdrempelig is bij vragen; te weten via de telefoon of email. - Als het niet goed gaat met de patiёnt is het belangrijk dat de medisch specialist confronterend en direct is door aan te geven dat het helemaal niet goed gaat, dat de patiënt zich overvraagt of dat bijvoorbeeld gestopt moet worden met werken. Het is daarbij belangrijk dat de patiënt geen ruimte wordt gegeven om te marchanderen. - De partner is samen met de patiënt ziek en hiermee een essentiёle schakel die duidelijk betrokken behoort te worden in het zorgproces. |
BIJWERKINGEN VAN BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 3: - Patiënten vinden het belangrijk dat zij duidelijke en volledige (zowel schriftelijke als mondelinge) informatie ontvangen over de bijwerkingen die zich kunnen voordoen bij een behandeling met biologicals. - Patiënten vinden het belangrijk dat, als zij bijwerkingen ervaren, duidelijk is tot wie zij zich moeten richten, dus tot de huisarts of de medisch specialist. - Patiënten vinden het belangrijk dat, als zij bijwerkingen ervaren, hier serieus naar wordt gekeken en deskundig op wordt geacteerd. - Patiënten vinden het belangrijk dat zij instructies ontvangen wat zij zelf kunnen doen om bepaalde bijwerkingen te voorkomen. |
Overweging vanuit het patiëntenperspectief bij UV 4: - Patiënten vinden het belangrijk dat zij informatie ontvangen over de het risico op kanker bij biologicals. - Patiënten vinden het belangrijk dat zij actief worden gevolgd op het mogelijk ontstaan van kanker. |
ZWANGERSCHAP EN BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 5: Op basis van de focusgroep zijn geen overwegingen aan te dragen. |
BIOLOGICALS EN (OPERATIEVE) INGREPEN: |
Overweging vanuit het patiëntenperspectief bij UV 6: - Patiënten vinden het belangrijk dat de voorschrijvend specialist betrokken is bij het besluitvormingsproces van een operatie. Daarbij is het essentieel dat er direct contact is tussen de betrokken specialisten. - Patiënten vinden het belangrijk dat zij in bezit zijn van een ‘kaartje’ waarop staat dat zij biologicals gebruiken en dat met de specialist contact opgenomen moet worden in geval van tandheelkundige of operatieve ingrepen. - Patiënten vinden het belangrijk dat zij informatie ontvangen over de stappen die gezet moeten worden bij het staken of doorgaan van biologicals bij een operatie. Hierdoor zijn zij in staat zelf actief mee te werken en hebben meer het gevoel controle te hebben over hun behandeling met biologicals. |
FOLLOW-UP VAN DE BEHANDELING MET BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 7: - Patiënten vinden het belangrijk dat zij regelmatig (om de 3 tot 6 mnd) door de medisch specialist worden onderzocht waarbij de werking van de biologicals in kaart wordt gebracht. - Patiënten vinden het belangrijk dat zij worden geinformeerd over de uitslag van de periodieke controle zodat zij deelgenoot zijn van het zorgproces. - Patiënten geven aan geconfronteerd te worden met een veelheid aan informatiestromen die regelmatig tegenstrijdig zijn. Zij vinden het belangrijk dat hier meer regie over gevoerd gaat worden. - Patiënten vinden het belangrijk dat professionels aangeven wanneer men geen duidelijkheid ten aanzien van bepaalde vraagstukken kunnen geven omdat nog zoveel onbekend is bij de behandeling van biologicals. - Patiënten vinden het belangrijk regelmatig contact te hebben met de (specialistisch) verpleegkundige zodat zij eventuele vragen kan beantwoorden. |
BIOLOGICALS EN VACCINATIE: |
Overweging vanuit het patiëntenperspectief bij UV 8: - Patiënten vinden het belangrijk dat zij (tijdig) informatie ontvangen over welke vaccinaties zij toegediend kunnen krijgen. - Patiënten vinden het belangrijk dat zij informatie ontvangen bij welke verschijnselen, nadat zij een vaccinatie hebben ontvangen, zij contact op moeten nemen met de medisch speicalist. |
DE BEHANDELING MET BIOLOGICALS: |
Overweging vanuit het patiëntenperspectief bij UV 9: - Patiënten vinden het belangrijk dat de arts door wie zij worden behandeld deskundig is en over de meest recente inzichten beschikt zodat zij erop kunnen vertrouwen in goede handen te zijn. - De (voorschrijvende) medisch specialist dient de regie in handen te houden en daarbij is het overleg met de andere betrokken professionals belangrijk. - Het is belangrijk dat de professionals communicatief zijn en duidelijk uitleg kunnen geven op niveau van de leek. Daarnaast is het wenselijk dat de medisch specialist toegankelijk en laagdrempelig is bij vragen; te weten via de telefoon of email. - Als het niet goed gaat met de patiёnt is het belangrijk dat de medisch specialist confronterend en direct is door aan te geven dat het helemaal niet goed gaat, dat de patient zich overvraagt of dat bijvoorbeeld gestopt moet worden met werken. Het is daarbij belangrijk dat de patiënt geen ruimte wordt gegeven om te marchanderen. |
LANGDURIG VERBLIJF ELDERS: |
Overweging vanuit het patiëntenperspectief bij UV 10: - Patiënten vinden het belangrijk dat zij informatie ontvangen over: - hoe zij de biologicals het beste naar het buitenland kunnen vervoeren; - naar welke gebieden zij kunnen reizen; - de mogelijkheden om in het buitenland een infuus met biologicals te ontvangen.
|
Methode ontwikkeling
Evidence based
Implementatie
Tijdens de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn en de uitvoerbaarheid van de aanbevelingen. Daarbij is gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De richtlijn is verspreid onder alle relevante beroepsgroepen en instellingen. Een samenvatting van de richtlijn is gepubliceerd in het Nederlands Tijdschrift voor Geneeskunde en in tijdschriften van de deelnemende wetenschappelijke verenigingen.
Werkwijze
De werkgroep heeft een jaar aan de totstandkoming van de richtlijn gewerkt. Binnen de werkgroep was een schrijverscollectief ingesteld. De leden van het schrijverscollectief zochten systematisch naar literatuur en beoordeelden kwaliteit en inhoud ervan. Vervolgens schreven zij een concepttekst waarin de literatuur werd verwerkt. Tijdens vergaderingen lichtten zij hun teksten toe aan de overige leden van de werkgroep. De conceptrichtlijn is in februari 2010 schriftelijk aan alle betrokken wetenschappelijke verenigingen aangeboden en gevraagd de richtlijn aan hun leden voor te leggen. Daarnaast is de richtlijn ook naar wetenschappelijke verenigingen gestuurd die niet in de werkgroep hebben geparticipeerd, te weten oogartsen, neurologen, gynaecologen en tandartsen. De ontvangen commentaren zijn, waar relevant bevonden, verwerkt in de definitieve richtlijn.
Wetenschappelijke onderbouwing
De onderbouwing van de richtlijn is mede gebaseerd op bewijs uit gepubliceerd wetenschappelijk onderzoek. Relevante artikelen werden gezocht met systematische zoekacties. Er werd gezocht tussen 1998 en 2009 in Medline en Embase. Voor de oriënterende search werd ook gezocht in de Cochrane Library en werd specifiek gezocht naar al bestaande richtlijnen in online raadpleegbare (inter)nationale guideline clearinghouses.
Hierbij werd de taal gelimiteerd tot Nederlands, Engels, Duits en Frans. Daarnaast werden artikelen geëxtraheerd uit referentielijsten van opgevraagde literatuur. Dit leverde bij enkele uitgangsvragen nog aanvullende artikelen op.
Doordat de uitgangsvragen niet gericht waren op het beoordelen van de effectiviteit van de interventies maar veelal gingen over bijwerkingen, complicaties en diagnostiek bleek een beperking tot systematische reviews en RCTs vaak niet zinvol. De searches zijn verricht in mei en juni 2009. Voor alle uitgangsvragen is gebruik gemaakt van een uniforme formulering van de patiëntencategorie en de interventie.
Voor de gehanteerde zoektermen wordt verwezen naar Appendix 1. Op verzoek zijn de volledige zoekstrategieën beschikbaar. Daarnaast werden artikelen geëxtraheerd uit referentielijsten van opgevraagde literatuur en zijn enkele relevante publicaties tot 1 november 2009 meegenomen. Lopend onderzoek is buiten beschouwing gelaten. Abstracts van congressen van de afgelopen 2 jaar (november 2007 tot 1 november 2009) zijn meegenomen bij de selectie van de literatuur. Relevante informatie vanuit deze abstract wordt uitgewerkt bij de overige overwegingen. Onder samenvatting van de literatuur / conclusies worden alleen gepubliceerde onderzoeken / richtlijnen uitgewerkt.
De geselecteerde artikelen zijn beoordeeld op kwaliteit van het onderzoek en gegradeerd naar mate van bewijs. Hierbij is de standaardindeling gebruikt: zie tabel 1. Na selectie bleven de artikelen over die als onderbouwing bij de verschillende conclusies staan vermeld. De beoordeling van de verschillende artikelen is opgenomen onder het kopje ‘samenvatting van de literatuur’. Het wetenschappelijk bewijs is vervolgens kort samengevat in een ‘conclusie’. De belangrijkste literatuur waarop deze conclusie is gebaseerd staat bij de conclusie vermeld, inclusief de mate van bewijs (zie tabel 2).
Voor het formuleren van een aanbeveling zijn, naast het wetenschappelijk bewijs, vaak nog andere aspecten van belang, bijvoorbeeld patiëntenvoorkeuren, kosten, beschikbaarheid of organisatorische aspecten. Deze aspecten worden, voor zover niet wetenschappelijk onderzocht, vermeld onder het kopje ‘overwegingen’. In de overige overwegingen spelen de ervaring en de mening van de werkgroepleden een belangrijke rol. De ‘aanbeveling’ is het resultaat van de combinatie van het beschikbare bewijs en de overige overwegingen.
Voor een aantal uitgangsvragen zijn evidencetabellen gemaakt en deze zijn te raadplegen in Appendix 3.
Tabel 1: Indeling van methodologische kwaliteit van individuele studies
|
|
Interventie |
Diagnostische accuratesse onderzoek |
Schade of bijwerkingen, etiologie, prognose* |
|
A1 |
Systematische review van ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van A2-niveau |
||
|
A2 |
Gerandomiseerd dubbelblind vergelijkend klinisch onderzoek van goede kwaliteit van voldoende omvang |
Onderzoek ten opzichte van een referentietest (een ‘gouden standaard’) met tevoren gedefinieerde afkapwaarden en onafhankelijke beoordeling van de resultaten van test en gouden standaard, betreffende een voldoende grote serie van opeenvolgende patiënten die allen de index- en referentietest hebben gehad |
Prospectief cohortonderzoek van voldoende omvang en follow-up, waarbij adequaat gecontroleerd is voor ‘confounding’ en selectieve follow-up voldoende is uitgesloten |
|
B |
Vergelijkend onderzoek, maar niet met alle kenmerken als genoemd onder A2 (hieronder valt ook patiënt-controleonderzoek, cohort-onderzoek) |
Onderzoek ten opzichte van een referentietest, maar niet met alle kenmerken die onder A2 zijn genoemd |
Prospectief cohortonderzoek, maar niet met alle kenmerken als genoemd onder A2 of retrospectief cohortonderzoek of patiënt-controleonderzoek |
|
C |
Niet-vergelijkend onderzoek |
||
|
D |
Mening van deskundigen |
||
* Deze classificatie is alleen van toepassing in situaties waarin om ethische of andere redenen gecontroleerde trials niet mogelijk zijn. Zijn die wel mogelijk dan geldt de classificatie voor interventies.
Tabel 2: Niveau van bewijs van de conclusie
|
Conclusie gebaseerd op |
|
|
1 |
Onderzoek van niveau A1 of ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van niveau A2 |
|
2 |
Eén onderzoek van niveau A2 of ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van niveau B |
|
3 |
Eén onderzoek van niveau B of C |
|
4 |
Mening van deskundigen |
Deze (concept)richtlijn is opgesteld aan de hand van het Appraisal of Guidelines for Research & Evaluation (AGREE) instrument. Dit instrument is in een Europees verband opgesteld om de procedurele kwaliteit van richtlijnen te kunnen beoordelen. Door de aspecten van AGREE te verwerken in de inleiding van de richtlijn, wordt duidelijk aan welke kwaliteitseisen is voldaan.
Zoekverantwoording
Zoekacties zijn opvraagbaar. Neem hiervoor contact op met de Richtlijnendatabase.