Antifosfolipiden syndroom (APS) en antistolling
Uitgangsvraag
Welk anticoagulans (Vitamine K-antagonisten of direct werkende orale anticoagulantia) heeft de voorkeur bij patiënten met veneuze trombose bij het antifosfolipiden syndroom?
Aanbeveling
Behandel de patiënten met een dubbel of drievoudig positief APS met een veneuze trombo-embolie en indicatie voor antistolling met een VKA (streef INR 2 tot 3).
Bespreek de optie van overzetten van DOAC naar VKA met patiënten met een veneuze trombo-embolie bij wie APS wordt vastgesteld.
Vanwege het gebrek aan bewijs kan er geen voorkeur uitgesproken worden voor DOACs of VKA bij patiënten met een enkelvoudig positief APS en een veneuze trombo-embolie.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Ten aanzien van de bepaling van de voorkeur voor type anticoagulans (DOAC versus VKA) bij patiënten met APS en trombose werden drie RCT’s geïncludeerd. RCTs starten op een hoog niveau van bewijskracht. De bewijskracht voor de cruciale uitkomstmaten (veneuze trombose; arteriële trombose; myocardinfarct; ischemische beroerte) varieerde van laag tot redelijk omdat één of twee niveaus werd afgewaardeerd voor imprecisie. Er was sprake van een laag aantal events en bij de uitkomstmaten veneuze trombose en myocardinfarct bevatte het 95% betrouwbaarheidsinterval zowel de bovenste als onderste grens van klinische relevantie (breed betrouwbaarheidsinterval). De overall bewijskracht voor de cruciale uitkomstmaten was laag.
De bewijskracht voor de belangrijke uitkomstmaten (majeure bloeding; klinisch relevante bloeding; mineure bloeding) was laag. Wederom werd afgewaardeerd voor imprecisie: het aantal events was laag en het 95% betrouwbaarheidsinterval bevatte zowel de bovenste als onderste grens van klinische relevantie (breed betrouwbaarheidsinterval).
Het diagnostisch proces voor APS is weergegeven in de studie van Limper (2019).
Verschillen in studiepopulatie
De drie geïncludeerde RCT’s verschillen in studiepopulatie. De RCT van Cohen (2016) includeerde APS-patiënten met enkel veneuze trombose, Ordi-Ros (2019) includeerde APS patiënten met veneuze of arteriële trombose. Bij Pengo (2018) was onderdeel van de inclusiecriteria de diagnose APS en veneuze, arteriële of PA bewezen micro-trombose.
Post-hoc analyses van Goldhaber (2016)
Goldhaber (2016) voerde post-hoc subgroep analyses uit met gepoolde data van de RE-COVER (II) trials en met data van de RE-MEDY trial, naar de effectiviteit van dabigatran etexilaat (DE) versus warfarine bij patiënten (≥ 18 jaar) met symptomatische proximale diepe veneuze trombose of longembolieën. In deze trials was het testen op APS niet verplicht, maar indien dergelijke testen werden uitgevoerd, werd dit vastgelegd. APS was gedefinieerd als tenminste één positieve test voor lupus anticoagulans en/of voor anticardiolipine antilichamen gecombineerd met symptomatisch, objectief vastgestelde veneuze trombo-embolieën. Het aandeel van patiënten met vastgestelde APS in de RE-COVER (II) en RE-MEDY trials was 2.2%. In totaal waren er 151 patiënten met vastgestelde APS, waarvan 71 patiënten tweemaal daags 150 mg dabigatran ontvingen en 80 patiënten warfarine (INR-range 2.0 tot 3.0). Patiënten werden 6 tot 36 maanden gevolgd. Een beperking van deze post-hoc subgroep analyse was dat de diagnose van APS niet werd gesteld volgens de internationale criteria (deze criteria veronderstellen onder meer twee testen met een tijdsinterval van tenminste 12 weken). Daarnaast werden niet alle patiënten getest op APS, waardoor de resultaten van de analyses niet het ware aantal patiënten met deze aandoening reflecteert.
Majeure bloeding werd gerapporteerd voor 1/70 (1,4%) APS-patiënten die dabigatran ontvingen in vergelijking met 2/77 (2,6%) patiënten die warfarine ontvingen (HR 0,46 (0,04 tot 5,43). De samengestelde uitkomstmaat majeure bloeding/klinisch relevante bloeding werd gerapporteerd voor 6/70 (8.6%) APS-patiënten die dabigatran ontvingen in vergelijking met 14/77 (18,2%) patiënten die warfarine ontvingen (HR 0,53 (0,20 tot 1,41).
Met de beperkte literatuur die er is lijkt het toch aannemelijk dat bij patiënten met APS en een trombose die daarmee een indicatie voor antistolling hebben, behandeling met Vitamine K-antagonisten een lagere kans op (recidief) arteriële trombotische complicaties geeft met een vergelijkbaar veneus trombose risico en een vergelijkbaar of lager bloedingsrisico in vergelijking met behandeling met een DOAC.
In alle 3 de studies moesten patiënten aan de internationale criteria voor APS voldoen, waarbij in de studie van Pengo (2018) alleen drievoudig positieve APS-patiënten met een trombose werden geïncludeerd. Voor deze groep lijkt het gunstige effect van VKA het meest waarschijnlijk, maar in de studie van Ordi-Ros (2019) werden ook patiënten met een enkele positieve APS-diagnostische test geïncludeerd en was het effect vergelijkbaar. Er was geen duidelijk, aantoonbaar verschil in bloedingsrisico tussen de behandeling met VKA of DOAC en als er al een verschil was, kwam dit ten gunste van de behandeling met VKA.
Er is een kennislacune in hoeverre het gunstig effect van VKA ten opzichte van DOAC geldt voor alle patiënten met APS en een trombose of alleen voor bepaalde APS patiënten, drievoudig positieve patiënten versus niet-drievoudig positieve patiënten of APS patiënten met een veneuze trombose of arteriële trombose. Er is geen literatuur over de behandeling met obstetrisch APS-syndroom en antistolling tijdens de zwangerschap of daarna.
Waarden en voorkeuren van patiënten (en eventueel hun verzorgers)
Voor een patiënt is een behandeling met VKA meer belastend dan behandeling met DOACs, door de frequente controles van de INR. Gezien de behandeling met VKA resulteert in een lagere kans op (recidief) arteriële trombotische complicaties geeft, is de werkgroep van mening dat dit de frequente controles van INR rechtvaardigt.
Het is belangrijk om de voor- en nadelen van de behandeling met de patiënt te bespreken. Handreikingen hiervoor zijn te vinden in de module over communicatie met de patiënt (https://lta-antistollingszorg.nl/communicatie-met-patienten)
Aanvaardbaarheid, haalbaarheid en implementatie
Behandeling met VKA is al meer dan 75 jaar zorg die in Nederland door trombosediensten geleverd wordt. Nadeel is de frequente controle van de INR die noodzakelijk is. Zorg voor patiënten met DOAC wordt geleverd door medisch specialisten of huisartsen en is onderdeel van de routine. Er zijn geen belemmeringen.
Rationale van de aanbeveling: weging van argumenten voor en tegen de interventies
APS-patiënten met een trombose en indicatie voor antistolling hebben een lager risico op (recidief) arteriële trombotische complicaties bij behandeling met VKA ten opzichte van behandeling met een DOAC. De meest overtuigende studie is gedaan bij patiënten die drievoudig positief waren. Maar de grootte van het effect is ook aanwezig bij andere patiënten die volgens de internationale criteria APS hebben. Ten aanzien van bloedingen is er geen groot verschil tussen VKA en DOAC en als er al een verschil is, is dit ten gunste van de behandeling met VKA.
Onderbouwing
Het antifosfolipiden syndroom (APS) is gedefinieerd als het optreden van een veneuze of arteriële trombose en/of zwangerschap gerelateerde morbiditeit (> 3 onverklaarde opeenvolgende spontane abortus < 10 weken of ernstige (pre-)eclampsie < 34e week zwangerschap) in combinatie met bij herhaling aangetoonde aanwezigheid van antifosfolipiden antilichamen en/of een lupus anticoagulans. Een uitgebreide definitie is weergegeven in de studie van Limper (2019).
In deze module gaan we in op de vraag welke vorm van antistolling bij voorkeur gebruikt dient te worden bij patiënten met een veneuze trombose en de aanwezigheid van antifosfolipiden antistoffen en/of een lupus anticoagulans, zowel bij de initiële diagnose van een trombose als ter preventie van recidief trombose. APS wordt beschouwd als een primaire auto-immuun ziekte, maar kan ook secundair voorkomen bij bijvoorbeeld systemische lupus erythematodes (SLE) (30 tot 40% van de patiënten met SLE heeft APS) of ziektes als reumatoïde artritis, systemische sclerose en dermatomyositis. Bij APS is de kans op recidief trombose verhoogd, waardoor patiënten vaak langdurig met antistolling worden behandeld.
|
Laag GRADE |
Het gebruik van DOACs lijkt niet te resulteren in een hoger risico op veneuze trombose in vergelijking met het gebruik van VKA bij patiënten met APS en trombose.
Bronnen: (Cohen, 2016; Ordi-Ros, 2019; Pengo, 2018) |
|
Redelijk GRADE |
Het gebruik van DOACs resulteert waarschijnlijk in een hoger risico op arteriële trombose in vergelijking met het gebruik van VKA bij patiënten met APS en trombose.
Bronnen: (Cohen, 2016; Ordi-Ros, 2019; Pengo, 2018) |
|
Laag GRADE |
Het gebruik van DOACs zou kunnen resulteren in een hoger risico op myocardinfarct in vergelijking met het gebruik van VKA bij patiënten met APS en trombose.
Bronnen: (Pengo, 2018) |
|
Redelijk GRADE |
Het gebruik van DOACs resulteert waarschijnlijk in een hoger risico op ischemisch beroerte in vergelijking met het gebruik van VKA bij patiënten met APS en trombose.
Bronnen: (Ordi-Ros, 2019; Pengo, 2018) |
|
Laag GRADE |
Het gebruik van DOACs lijkt het risico op majeure bloedingen niet of nauwelijks te verhogen in vergelijking met het gebruik van VKA bij patiënten met APS en trombose.
Bronnen: (Cohen, 2016; Ordi-Ros, 2019; Pengo, 2018) |
|
Laag GRADE |
Het gebruik van DOACs zou kunnen resulteren in een hoger risico op klinisch relevante bloeding in vergelijking met het gebruik van VKA bij patiënten met APS en trombose.
Bronnen: (Cohen, 2016; Ordi-Ros, 2019) |
|
Laag GRADE |
Het gebruik van DOACs lijkt het risico op mineure bloedingen niet of nauwelijks te verhogen in vergelijking met het gebruik van VKA bij patiënten met APS en trombose.
Bronnen: (Cohen, 2016; Ordi-Ros, 2019) |
Beschrijving studies
Cohen (2016) (RAPS study) voerde een open label trial uit (randomised, controlled, open-label, phase 2/3, non-inferiority clinical trial) in het Verenigd Koninkrijk om de behandeling met rivaroxaban te vergelijken met warfarine bij trombotische APS-patiënten (≥ 18 jaar). Alle geïncludeerde patiënten voldeden aan de internationale consensus criteria voor APS. Verdere inclusiecriteria voor deze studie waren het doorgemaakt hebben van tenminste één veneuze trombo-embolie bij geen of subtherapeutisch gebruik van anticoagulantia en het gebruik van warfarine (standaard-intensiteit target INR 2 tot 5; range 2.0 tot 3,0) voor tenminste 3 maanden sinds de laatste veneuze trombo-embolie. Exclusiecriteria waren een arteriële trombo-embolie in de voorgeschiedenis ten gevolge van APS of terugkerende veneuze trombo-embolieën bij het gebruik van warfarine bij een therapeutische International Normalized Ratio (INR) van 2.0 tot 3.0. Patiënten in de interventiegroep (n=57) werden overgezet op rivaroxaban en ontvingen 20 mg orale rivaroxaban eenmaal daags gedurende 180 dagen. Patiënten in de controlegroep (n=59) bleven warfarine gebruiken met standaard intenstiteit (target INR 2,5; range 2.0 tot 3.0). In de interventiegroep was de dosis rivaroxaban eenmaal daags 15 mg in plaats van 20 mg bij patiënten met een creatineklaring van 30 tot 49 mL/min. SLE was een stratificatie-variabele in deze studie: in zowel de interventie- als de controlegroep was bij 19% van de patiënten sprake van SLE met APS. Patiënten werden gedurende 210 dagen gevolgd.
Ordi-Ros (2019) voerde een open label trial uit (open-label, fase 3, randomized non-inferiority trial) in 6 Spaanse universiteitsziekenhuizen waarin de behandeling met rivaroxaban werd vergeleken met VKA bij APS-patiënten met een arteriële - of veneuze trombose. Deelnemende patiënten moesten voldoen aan de internationale consensus criteria van APS en tussen de 18 en 75 jaar oud zijn. Er moest sprake zijn van twee positieve metingen op de aPL test (met een tijdsinterval van tenminste 3 maanden) en objectief vastgestelde arteriële of veneuze trombose. Patiënten in de interventiegroep (n=95) ontvingen 20 mg rivaroxaban eenmaal daags (of 15 mg eenmaal daags bij patiënten met een creatineklaring van 30 tot 49 mL/min/1.73m2), patiënten in de controlegroep bleven aangepaste doses VKA ontvangen (target INR, 2.0 tot 3.0, of 3.1 tot 4.0 voor degenen met een voorgeschiedenis van herhaalde trombose). Gestratificeerd werd op de aanwezigheid van SLE. Van secundaire APS (APS bij SLE) was sprake bij 31/95 (32.6%) patiënten in de interventiegroep en bij 27/95 (28.4%) van de patiënten in de controlegroep. De follow-up van patiënten bedroeg 36 maanden. Bij ongeveer 60% van de patienten in beide groepen was sprake van een drievoudig positieve APS (lupus anticoagulans, anticardiolipine en beta 2 microglobuline alle positief).
Pengo (2018) (TRAPS study) voerde een open label trial uit (randomized open-label multicenter non-inferiority study) in 14 centra in Italië waarbij de effectiviteit en veiligheid van rivaroxaban met warfarine werd vergeleken in hoog-risico patiënten met APS en trombose. Patiënten tussen de 18 en 75 jaar met een bevestigde diagnose van APS en positief getest voor alle drie de aPL testen (‘triple positivity’), kwamen voor inclusie in aanmerking. Daarnaast moest er sprake zijn van een voorgeschiedenis van trombose (objectief bewezen arteriële, veneuze en/of PA bewezen microtrombose). Patiënten in de interventiegroep (n=59) ontvingen eenmaal daags 20 mg rivaroxaban, in het geval van een creatineklaring tussen 30-50 mL/min werd 15 mg rivaroxaban eenmaal daags voorgeschreven. Patiënten in de controlegroep (n=61) ontvingen warfarine, waarbij de doses aangepast werden om de INR binnen een range van 2.0 tot 3.0 te houden (target INR 2.5). De gemiddelde follow-up duur was 611 dagen voor het intention-to-treat cohort. De studie werd echter voortijdig stopgezet vanwege een overmaat aan trombo-embolische gebeurtenissen in de interventiegroep. Er was sprake van vier ischemische beroertes (4/59, 7%) en drie myocardinfarcten (3/59, 5%) in de interventiegroep, terwijl deze in de controlegroep niet voorkwamen.
Resultaten
Cohen (2016), Ordi-Ros (2019) en Pengo (2018) analyseerden en rapporteerden hun resultaten op verschillende wijze. Allen gingen uit van een non-inferiority design. Cohen (2016) gebruikte een gemodificeerde intention-to-treat (ITT) analyse om alle gerandomiseerde patiënten met beschikbare data in alle analyses op te nemen. Pengo rapporteerde (2018) zowel de resultaten van een ‘as treated’ analyse en een ITT-analyse. Ordi-Ros (2019) voerde voor een aantal uitkomstmaten eveneens beide analyses uit, maar voor de uitkomstmaat bloedingen werd de ‘as treated safety population’ gebruikt. Deze populatie werd door Ordi-Ros gedefinieerd als de populatie, die bestond uit alle patiënten die tenminste 1 dosis van studiemedicatie ontvingen. Door de studies werden hazard ratio’s en/of risk ratio’s gerapporteerd. Cohen (2016) rapporteerde als effectgrootte ‘treatment effect’ (BI 95%), welke verder niet werd gedefinieerd.
Veneuze trombose
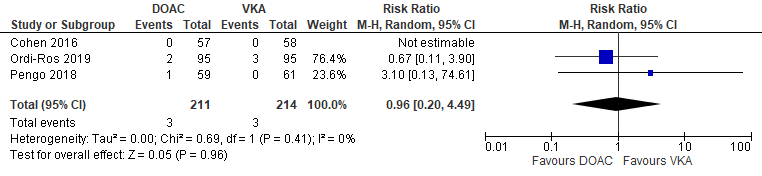
Drie RCTs (Cohen, 2016; Ordi-Ros, 2019; Pengo, 2018) vergeleken de behandeling met rivaroxaban versus VKA bij trombotische APS-patiënten op de uitkomstmaat veneuze trombose. Cohen rapporteerde diepe veneuze trombose en longembolie als separate uitkomstmaten, Ordi-Ros en Pengo rapporteerden veneuze trombo-embolieën in zijn totaliteit. Veneuze trombo-embolieën werden gevonden in 3/211 (1,4%) APS-patiënten die DOACs ontvingen in vergelijking met 3/214 (1,4%) APS patiënten die VKA ontvingen (RR 0,96 (95% BI 0,20 tot 4,49)) (Figuur 1).
Figuur 1 Veneuze trombose, vergelijking DOAC versus VKA
Arteriële trombose
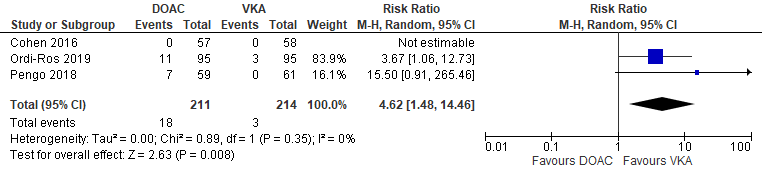
De uitkomstmaat arteriële trombose werd gerapporteerd door drie RCT’s (Cohen, 2016; Ordi-Ros, 2019; Pengo, 2018), welke de behandeling vergeleken met rivaroxaban versus VKA bij trombotische APS-patiënten. Arteriële trombo-embolieën werden gevonden in 18/211 (8,5%) APS-patiënten die DOACs ontvingen in vergelijking met 3/214 (1,4%) APS patiënten die VKA ontvingen (RR 4,62 (95% BI 1,48 tot 14,46)) (Figuur 2).
Figuur 2 Arteriële trombose, vergelijking DOAC versus VKA
Myocardinfarct
Eén RCT (Pengo, 2018) vergeleek de behandeling met rivaroxaban versus VKA bij trombotische APS-patiënten op de uitkomstmaat myocardinfarct. Myocardinfarct werd gerapporteerd voor 3/59 (5,1%) APS patiënten die DOACs ontvingen in vergelijking met 0/61 (0%) APS patiënten die VKA ontvingen (RR 7,23 (95% BI: 0,38 tot 137,08)).
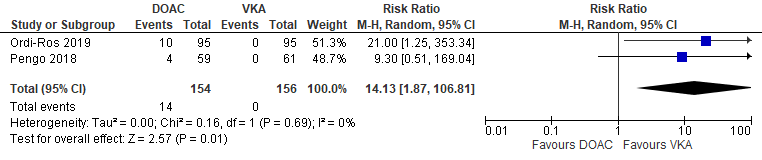
De uitkomstmaat ischemische beroerte werd gerapporteerd door twee RCT’s (Ordi-Ros, 2019; Pengo, 2018), die de behandeling vergeleken met rivaroxaban versus VKA bij trombotische APS-patiënten. Ischemische beroerte werd gevonden in 14/154 (9,1%) APS-patiënten die DOACs ontvingen in vergelijking met 0/156 (0%) patiënten die VKA ontvingen (RR 14,13 (95% BI: 1,87 tot 106,81)) (Figuur 3).
Figuur 3 Ischemische beroerte, vergelijking DOAC versus VKA
Majeure bloeding
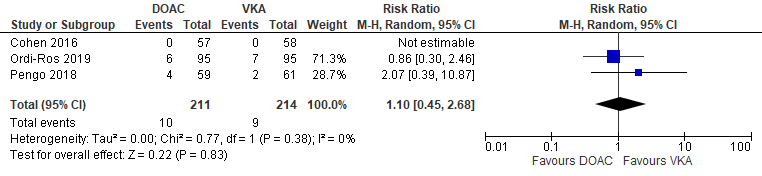
Drie RCTs (Cohen, 2016; Ordi-Ros, 2019; Pengo 2018) vergeleken de behandeling bestaande uit rivaroxaban versus VKA bij trombotische APS-patiënten op de uitkomstmaat majeure bloeding. Majeure bloeding werd gerapporteerd voor 10/211 (4,7%) APS-patiënten die DOACs ontvingen in vergelijking met 9/214 (4,2%) patiënten die VKA ontvingen (RR 1,10 (95% BI 0,45 tot 2,68)) (Figuur 4).
Figuur 4 Majeure bloeding, vergelijking DOAC versus VKA
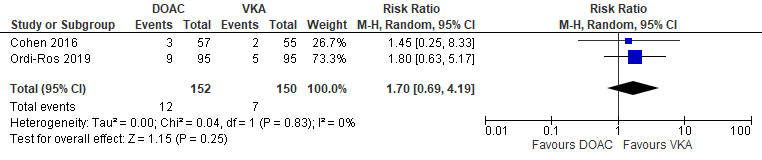
Klinisch relevante bloeding
De uitkomstmaat klinisch relevante bloeding werd gerapporteerd door twee RCT’s (Cohen, 2016; Ordi-Ros, 2019), die de behandeling vergeleken met rivaroxaban versus VKA bij trombotische APS-patiënten. Een klinische relevante bloeding werd gevonden bij 12/152 (7,9%) patiënten die DOACs ontvingen in vergelijking met 7/150 (4,7%) patiënten die VKA ontvingen (RR 1.70 (95% CI 0,69 tot 4,19)) (Figuur 5).
Figuur 5 Klinisch relevante bloeding, vergelijking DOAC versus VKA
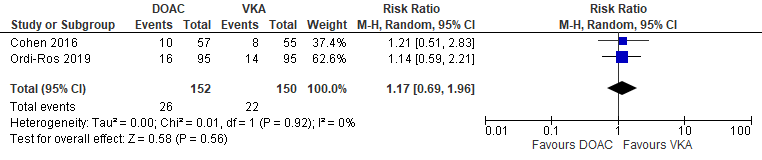
Mineure bloeding
Twee RCT’s (Cohen, 2016; Ordi-Ros, 2019) vergeleken de behandeling bestaande uit rivaroxaban versus VKA bij trombotische APS-patiënten op de uitkomstmaat mineure bloeding. Een mineure bloeding werd gerapporteerd voor 26/152 (17,1%) APS-patiënten die DOACs ontvingen in vergelijking met 22/150 (14,7%) APS patiënten die VKA ontvingen (RR 1.17 (95% BI 0.69, 1.96)) (Figuur 6).
Figuur 6 Mineure bloeding, vergelijking DOAC versus VKA
Bewijskracht van de literatuur
De bewijskracht voor RCTs start op hoog.
- De bewijskracht voor de uitkomstmaat veneuze trombose is met twee niveaus verlaagd naar een GRADE laag gezien de aanwezigheid van imprecisie (-2) (het aantal events was laag: 3/211 in de interventiegroep en 3/214 in de controlegroep. Het 95% betrouwbaarheidsinterval omvat zowel de bovenste als onderste grens van klinische relevantie).
- De bewijskracht voor de uitkomstmaat arteriële trombose is met één niveau verlaagd naar GRADE redelijk gezien de aanwezigheid van imprecisie (-1) (het aantal events was laag: 18/211 in de interventiegroep en 3/214 in de controlegroep).
- De bewijskracht voor de uitkomstmaat myocardinfarct is met twee niveaus verlaagd naar een GRADE laag gezien de aanwezigheid van imprecisie (-2) (één RCT en het aantal events was laag: 3/59 in de interventiegroep en 0/61 in de controlegroep. Het 95% betrouwbaarheidsinterval omvat zowel de bovenste als onderste grens van klinische relevantie).
- De bewijskracht voor de uitkomstmaat ischemische beroerte is met één niveau verlaagd naar een GRADE redelijk gezien de aanwezigheid van imprecisie (-1) (het aantal events was laag: 14/154 in de interventiegroep en 0/156 in de controlegroep).
- De bewijskracht voor de uitkomstmaat majeure bloeding is met twee niveaus verlaagd naar een GRADE laag gezien de aanwezigheid van imprecisie (-2) (het aantal events was laag: 10/211 in de interventie groep en 9/214 in de controlegroep. Het 95% betrouwbaarheidsinterval omvat zowel de bovenste als onderste grens van klinische relevantie).
- De bewijskracht voor de uitkomstmaat klinisch relevante bloeding is met twee niveaus verlaagd naar een GRADE laag gezien de aanwezigheid van imprecisie (-2) (het aantal events was laag: 12/152 in de interventie groep en 7/150 in de controlegroep. Het 95% betrouwbaarheidsinterval omvat zowel de bovenste als onderste grens van klinische relevantie).
- De bewijskracht voor de uitkomstmaat mineure bloeding is met twee niveaus verlaagd naar een GRADE laag gezien de aanwezigheid van imprecisie (-2) (het aantal events was laag: 26/152 in de interventie groep en 22/150 in de controlegroep. Het 95% betrouwbaarheidsinterval omvat zowel de bovenste als onderste grens van klinische relevantie).
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraagvraag:
Welk anticoagulans (Vitamine K-antagonisten of direct werkende orale anticoagulantia) heeft de grootste effectiviteit en laagste kans op complicaties bij patiënten met veneuze trombose bij het antifosfolipiden syndroom?
P: patiënten met trombose en antifosfolipiden antilichamen en/of een lupus anticoagulans;
I: direct werkende orale anticoagulantia (DOAC);
C: Vitamine K-antagonisten (VKA);
O: recidief trombose (arterieel en veneus), cardiovasculaire events, bloedingen.
Relevante uitkomstmaten
De werkgroep achtte recidief trombose (arterieel en veneus), myocardinfarct, ischemische beroerte voor de besluitvorming cruciale uitkomstmaten; en majeure bloeding, klinisch relevante bloeding en mineure bloeding voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde niet a priori de genoemde uitkomstmaten, maar hanteerde de in de studies gebruikte definities.
De werkgroep definieerde een relatief risico ≤ 0,8 als een klinisch (patiënt) relevant verschil voor alle uitkomstmaten.
Zoeken en selecteren (Methode)
De databases Medline (via OVID) en Embase (via Embase.com) zijn doorzocht met relevante zoektermen van 2010 tot 1 mei 2020. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 413 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: 1) de studiepopulatie bestaat uit patiënten met APS en/of lupus anticoagulans en 2) de studie vergelijkt DOAC met een VKA. Op basis van titel en abstract werden in eerste instantie 9 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 6 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording) en 3 studies definitief geselecteerd.
Resultaten
Drie RCTs zijn opgenomen in de literatuuranalyse (Cohen 2016; Ordi-Ros, 2019; Pengo, 2018). De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk-of-biastabellen.
- Cohen H, Doré CJ, Clawson S, et al. Rivaroxaban in antiphospholipid syndrome (RAPS) protocol: a prospective, randomized controlled phase II/III clinical trial of rivaroxaban versus warfarin in patients with thrombotic antiphospholipid syndrome, with or without SLE. Lupus. 2015;24(10):1087-1094. doi:10.1177/0961203315581207.
- Goldhaber SZ, Eriksson H, Kakkar A, et al. Efficacy of dabigatran versus warfarin in patients with acute venous thromboembolism in the presence of thrombophilia: Findings from RE-COVER®, RE-COVER™ II, and RE-MEDY™. Vasc Med. 2016;21(6):506-514. doi:10.1177/1358863X16668588.
- Limper, M., De Leeuw, K., Lely, A. T., Westerink, J., Teng, Y. K. O., Eikenboom, J.,... & Kruyt10, N. D. (2019). Diagnosing and treating antiphospholipid syndrome: a consensus paper. Neth J Med, 77(3), 98-108.
- Ordi-Ros J, Sáez-Comet L, Pérez-Conesa M, et al. Rivaroxaban Versus Vitamin K Antagonist in Antiphospholipid Syndrome: A Randomized Noninferiority Trial. Ann Intern Med. 2019;171(10):685-694. doi:10.7326/M19-0291.
- Pengo V, Denas G, Zoppellaro G, et al. Rivaroxaban versus warfarin in high-risk patients with antiphospholipid syndrome. Blood. 2018;132(13):1365-1371. doi:10.1182/blood-2018-04-848333.
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors ((potential) confounders)
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
Risk of bias tabellen
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Cohen, 2016 |
Randomisation was performed by a web-based independent randomisation service (Sealed Envelope, London, UK) to ensure allocation concealment. The schedule was created using permuted blocks with arandom block length, stratified by centre and patient type (with versus without systemic lupus erythematosus). |
Unlikely
Randomisation was performed by a web-based independent randomisation service.
|
Unlikely
The trial was open label to ensure optimum warfarin dosing: anticoagulant monitoring is necessary. Additionally, the management of bleeding events differs between patients receiving warfarin and rivaroxaban.
However, it is not likely that this results in risk of bias for the outcome measures (i.e., thrombotic events, bleeding events). |
Unclear
The trial was open label. |
Unlikely
Masking of treatment allocation was also not possible in the RAPS central laboratory because different tests were needed for the two anticoagulants, and samples taken at baseline and day 42 were tested simultaneously to minimise variability between assays.
Reports of SAEs, serious adverse reactions, and suspected unexpected serious adverse reactions were reviewed by external, independent, medically qualified staff.
Clinically relevant and minor bleeding events across all sites were pseudoanonymised and reviewed by one investigator (DAI) to remove the potential bias of interoperator variation. The classification of bleeding events as clinically relevant or minor, as per the protocol, was checked and changed if appropriate. |
Unlikely
Outcomes in the protocol and published report are comparable. |
Unlikely
3/57 (5.3%) patients in the intervention group and 3/59 (5.1%) patients in the control group did not contribute data to the primary outcome.
|
Unclear
A modified intention-to-treat approach was used to include all randomised patients with assessable data in all analyses.
However, because this was a noninferiority trial, per protocol analysis might be preferable. |
|
Ordi-Ros, 2019 |
The randomized list, stratified by center and presence of systemic lupus erythematosus, was created at VHH by using computer-generated random-number sequences (C4-Study design pack software (GlaxoSmithKline)) in blocks of 10. Sequentially numbered, concealed envelopes containing group assignments were provided to the investigators. |
Unlikely
Computer-generated random-number sequences were used. |
Unlikely
The trial was open label to ensure optimum VKA dosing and monitoring, and because bleeding management differs between VKAs and rivaroxaban.
However, it is not likely that this results in risk of bias for the outcome measures (i.e., thrombotic events, bleeding events). |
Unclear
The trial was open label. |
Unlikely
An independent committee blinded to the clinical end points applied protocol definitions to adjudicate suspected cases of thrombosis, death, and bleeding events that contributed to the prespecified end points. |
Unlikely
Outcomes in the Methods’ section and Result’s section were comparable.
|
Unlikely
In each group, 6.3% of patients permanently stopped their assigned therapy before a thrombotic event and before the end date. |
Unlikely
Because this was a noninferiority study, the primary efficacy analysis was prespecified to be performed in the per protocol population. An intention-to treat analysis was also performed. |
|
Pengo, 2018 |
After signing an informed consent form, patients underwent Web-based randomization using random block sizes of 2, 4, and 6 and stratified based on sex and the presence or absence of an associated autoimmune disease. In this way, 4 strata were constructed: females with or without associated autoimmune disease and males with or without associated autoimmune disease. |
Unlikely
Web-based randomization. |
Unlikely
Open-label trial.
However, it is not likely that this results in risk of bias for the outcome measures (i.e., thrombotic events, bleeding events). |
Unclear
Open-label trial. |
Unclear
Outcome assessors not described. |
Unlikely
Outcomes in the study protocol and published report were compared. Primary outcomes were comparable. Minor bleeding was not reported (secondary outcome).
However, the trial was terminated prematurely because of an excess of events in the intervention group. Therefore, selective reporting was unlikely. |
Unclear
12 patients exited the study prematurely. 9/59 (15%) of the intervention group and 3/61 (5%) of the control group. Reasons were reported.
The trial was terminated prematurely after the enrolment of 120 patients because of an excess of events among patients in the rivaroxaban arm. The prespecified sample size was 536 patients.
|
Unlikely
The primary outcome was analyzed according to “as treated” and intention to treat (ITT) principle. |
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Bala 2017 |
Verkeerde interventie (systematisch review over effectiviteit trombocytenaggregatieremmers of anticoagulantia, meestal vitamine-K antagonisten) |
|
Dufrost 2018 |
Verkeerd study design (systematisch review met bredere inclusiecritria dan RCT’s) |
|
Dufrost 2016 |
Verkeerd study design (systematisch review met bredere inclusiecritria dan RCT’s) |
|
Sanchez-Redondo 2019 |
Verkeerd study design (systematisch review met bredere inclusiecritria dan RCT’s) |
|
Tektonidou 2019 |
Voldoet niet aan PICO (brede focus op management APS) |
Beoordelingsdatum en geldigheid
Publicatiedatum : 24-11-2021
Beoordeeld op geldigheid : 15-11-2021
Belangrijkste wijzigingenten opzichte van de vorige versie:
De modules over beleid bij bloedingen bij VKA en TARS zijn gewijzigd, evenals de modules over oppervlakkige tromboflebitis en het continueren van antistolling na een acute veneuze tromboembolie. De module over antifosfolipiden syndroom en antistolling is een nieuwe module.
Algemene gegevens
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule. Patiëntenparticipatie bij deze richtlijn werd mede gefinancierd uit de Kwaliteitsgelden Patiënten Consumenten (SKPC) binnen het programma KIDZ.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodule is in 2018 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met stollingsproblemen.
Kerngroep
- Prof. dr. M.V. (Menno) Huisman, internist-vasculaire geneeskunde, LUMC, NIV (voorzitter)
- Dr. M.J.H.A. (Marieke) Kruip, internist-hematoloog, ErasmusMC, NIV, NVvH (Hematologie)
- Dr. F.A. (Erik) Klok, internist-vasculaire geneeskunde, LUMC, NIV
- Dr. M.A. (Marc) Brouwer, cardioloog, RadboudUMC, NVVC
- Dr. H.B. (Harmen) Ettema, orthopedisch chirurg, Isala, NOV
- Drs. B. (Banne) Nemeth, aios orthopedie, LUMC, NOV
- Dr. A.M. (Arno) Wiersema, vaatchirurg, NVVH
- Dr. M.E. (Maarten) Tushuizen, maag-darm-leverarts, LUMC, NVMDL
- Dr. J.M. (Jonathan) Coutinho, neuroloog, AMC-UVA, NVN
- Drs. A. (Andrew) Oostindjer, huisarts/Kaderhuisarts HVZ, NHG
Klankbordgroep
- Dr. J.J.C.M. (Sjef) van de Leur, arts klinische chemie, Isala, NVKC
- Dr. A.W.M.M. (Ankie) Koopman - van Gemert, anesthesioloog, ASZ, NVA
- Dr. M.G. (Mariëlle) van Pampus, gynaecoloog, OLVG, NVOG
- Drs. R.J. (Repke) Snijder, longarts, Antoniusziekenhuis, NVALT
- Drs. R.J. (Rutger) Lely, radioloog, VUMC, NVVR
- Dr. C. (Bibi) van Montfrans, dermatoloog, ErasmusMC, NVDV
- Dr. R.A. (Richard) Faaij, klinisch geriater, Diakonessenhuis, NVKG
- Dr. B. (Bauke) van Minnen, kaakchirurg, UMCG, NVMKA
- Drs. A (Annemarie) Auwerda, beleidsadviseur, Harteraad
- Prof. dr. S (Saskia) Middeldorp, internist vasculaire-geneeskunde, Radboudumc, NIV
- M.J. (Jacqueline) Krol- van Straaten, internist - nefroloog, Hagaziekenhuis, NIV
- Dr. L. Jakulj, internist-nefroloog, Amsterdam UMC, NIV
- Dr. M. (Marcel) Schouten, internist - nefroloog, Tergooi, NIV
- Dr. N. (Nakisa) Khorsand, ziekenhuisapotheker, OLVG, NVZA
- Dr. M.F. (Margreet) Warlé-van Herwaarden, openbaar apotheker, KNMP
Met ondersteuning van
- Dr. S.R. (Sabine) Zwakenberg, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- H. (Hanneke) Olthuis, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
Kerngroep
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
prof. dr. M.V. (Menno) Huisman |
Internist vasculaire geneeskunde Leids Universitair Medisch Centrum |
- Voorzitter commissie herziening Antitrombotisch Beleid, onbetaald |
- Adviseur farmaceutische bedrijven die (nieuwe) antistollingsmiddelen maken (gelden gaan naar afdeling Interne Geneeskunde LUMC) - ZONMW grant Dutch-AF - registry met onderzoek op het gebied van antistolling bij patiënten met atriumfibrilleren (gelden gaan naar afdeling Interne Geneeskunde LUMC) - Research grants van farmaceutische bedrijven die (nieuwe) antistollingsmiddelen make; (gelden gaan naar afdeling Interne Geneeskunde LUMC) |
Deelname in werkgroep als voorzitter in verband met expertise op dit gebied. In verband met adviseur farmaceutische bedrijven: participeert niet bij besluitvorming middelen |
|
dr. M.J.H.A. (Marieke) Kruip |
Internist-hematoloog Erasmus MC |
Medisch leider trombosedienst Star-SHL, gedetacheerd vanuit het Erasmus MC |
- Anti-thrombotic stewardship S-team introduction (2015-2016) Boehringer-Ingelheim - 2016: Symposium over Richtlijn antitrombotisch beleid, internistendagen en bijscholing / Bayer / € 1169,- + € 129,-, € 224,- ten gunste van de afdeling hematologie |
In verband met belangen geen participatie besluitvorming keuze middelen |
|
dr. F.A. (Erik) Klok |
Internist Vasculair geneeskundige, LUMC, Leiden |
Adjunct Professor Universiteit van Mainz, Duitsland (onbetaald) |
Dr. F.A. Klok heeft financiële steun ontvangen voor studies (unrestricted research grants) van Bayer, BMS/Pfizer, Boehringer Ingelheim, Daiichi Sankyo, MSD, Actelion, Trombose Stichting Nederland, de Hartstichting en ZonMW. Dit betroffen allen studies naar diagnostiek van longembolie/DVT of voorkomen van lange termijn effecten (pulmonale hypertensie, lagere kwaliteit van leven et cetera). |
Geen, gesponsorde studies gaan over diagnostiek. Diagnostiek komt vooralsnog niet aan bod in deze richtlijn. Mocht dit het geval zijn belangen opnieuw bespreken. Geen actie nodig |
|
dr. M.A. (Marc) Brouwer |
Cardioloog, Radboudumc |
- Afgevaardigde van de NVVO bij LSKA (onbetaald) |
De researchafdeling cardiologie heeft voor verschillende projecten financiële ondersteuning ontvangen van: Astra Zeneca, BMS-Pfizer, Boehringer Ingelheim, Beyer, Daiichi Sankyo - Dutch AF registry |
Geen, ander onderwerp |
|
dr. H.B. (Harmen) Ettema |
Orthopedisch chirurg Isala Klinieken, Zwolle |
|
In verleden deelname verschillende RCT waarvan de laatste > 3 jaar gelden |
RCT's >3 jaar geleden, geen actie |
|
drs. B. (Banne) Nemeth |
AIOS Orthopedie LUMC Leiden PhD Klinische Epidemiologie en Orthopedie, LUMC Leiden |
|
|
geen |
|
dr. A.M. (Arno) Wiersema |
Vaatchirurg in het Westfriesgasthuis te Hoorn en AUMC, locatie Vumc |
Grant onderzoek ACT en heparine arteriële vaatingrepen Medtronic, geen onderwerp van deze richtlijn |
Grant onderzoek ACT en heparine arteriële vaatingrepen Medtronic, geen onderwerp van deze richtlijn |
geen |
|
dr. M.E. (Maarten) Tushuizen |
MDL-arts, staflid Leids Universitair Medisch Centrum, Leiden |
MDL-arts; DC-kliniek Almere, Almere, verrichten van endoscopie (betaald) |
|
geen |
|
dr. J.M. (Jonathan) Coutinho |
Neuroloog Amsterdam UMC |
|
RESPECT-CVT studie. Gefinancierd door Boehringer. Ondergetekende zit in het steering Committee van deze studie |
geen actie, studie gaat over andere patiëntpopulatie |
|
A. (Andrew) Oostindjer |
Huisarts - eigen praktijk in Oldenzaal. |
Als Kaderhuisarts HVZ: Consulent voor Pfizer - beoordelen van informatie materiaal bedoeld voor patiënten en medici. |
Momenteel wel lid van de antistollingscommissie van het Medisch Spectrum Twente (onbetaald) |
geen |
|
Klankbordgroep |
||||
|
dr. J.J.C.M. (Sjef) van de Leur |
Arts klinische chemie, Isala |
Bestuurslid FNT, onbetaald |
|
Geen |
|
dr. A.W.M.M. (Ankie) Koopman - van Gemert |
Anesthesioloog, opleider Albert Schweitzer ziekenhuis (pensioen 1-7-2019) 1-11-2019 – 1-2-2021 MM IC en VZ intensivisten ZGT Almelo |
- participatie richtlijnen: neuraxisblokkade, bloedtransfusie, ESA (vacatiegelden) tot 1-7-2019 - bestuur TRIP en vz hemovigilantiekamer (onbetaald) tot oktober - gebruikersgroep sanquin (onbetaald) – idem - VZ examencie NVA en ESA tot 1-1-2020
|
|
Geen |
|
dr. M.G. (Mariëlle) van Pampus |
Gynaecoloog OLVG |
|
|
Geen |
|
drs. R.J. (Repke) Snijder |
Longarts st. Antonius Ziekenhuis Nieuwegein |
|
|
Geen |
|
drs. R.J. (Rutger) Lely |
(Interventie)radioloog AUMC, locatie Vumc |
|
|
Geen |
|
dr. C. (Bibi) van Montfrans |
Dermatoloog Erasmus MC |
- Lid board European academy of dermatology and venereology (EADV) (onbetaald) |
|
Geen |
|
dr. R.A. (Richard) Faaij |
Klinisch geriater Diakonessenhuis Utrecht-Zeist-Doorn |
|
|
Geen |
|
dr. B. (Bauke) van Minnen |
Kaakchirurg UMCG |
|
|
Geen |
|
mw. A (Annemarie) Auwerda |
Beleidsadviseur, Harteraad |
|
|
Geen |
|
prof. dr. S (Saskia) Middeldorp |
Internist-vasculair geneeskundige, Radboudumc |
Onbetaald: NVTH-voorzitter, Stichting Haemophilia-bestuurslid, Stichting Amstol - bestuurslid Betaald: Diverse honoraria naar AMR Medical Research B.V. Bedrijven: Aspen - Bayer - BMS/Pfizer - Boehringer Ingelhei - Daiichi Sankyo - GKS - Portola - Sanquin -Sanofi. Voor lezingen/onderwijs |
Research support, wordt betaald aan AMC Medical Research B.V. Bedrijven: Aspen - Bayer - BMS/Pfizer - Boehringer lngelheim - Daiichi Sankyo - GSK - Portola - Sanquin - Highlow studie, GSK, overgenomen door Aspen, financiering voor investigator initiated trial, wordt gestort op AMC Medical Research B.V - Sanquin, financiering voor vrijwilligersstudie naar reversal van NOACs in gezonde vrijwilligers, wordt gestort op AMR Medical Research BV - BMS/Pfizer, financiering voor vrijwilligersstudie naar reversal van NOACs in gezonde vrijwillers, wrodt gestort op AMR Medical Research BV - Daiichi Sankyo, financiering voor investigator initiated study naar lange termijn gevolgen van VTE |
Participeert niet bij besluitvorming omtrent middelen. In rol van klankbordgroeplid is hier echter ook geen sprake van. Participatie wegens expertise. Openheid over deze belangen is voldoende. |
|
M.J. (Jacqueline) Krol- van Straaten |
Internist - Nefroloog Haga ziekenhuis, Den Haag |
Lid werkgroep KNMP Geneesmiddelen en Dialyse. Reis- en onkosten vergoeding (1-4x per jaar) Richtlijncommissie Nederlandse Federatie voor Nefrologie - betaald (vacatiegelden) |
|
Geen |
|
dr. M. (Marcel) Schouten |
Internist-nefroloog Tergooi |
Richtlijncommissie Nederlandse Federatie voor Nefrologie - betaald (vacatiegelden) |
|
Geen |
|
dr. N. (Nakisa) Khorsand |
Ziekenhuisapotheker, OLVG, Amsterdam |
Voorzitter special interest group hematologie van NVZA (onbetaald) |
|
Geen |
|
dr. M.F. (Margreet) Warlé- van Herwaarden |
Apotheker, Apotheek Groesbeek |
participeert af en toe in onderzoek (onbetaald) |
|
Geen |
|
Dr. L. Jakulj |
Internist-nefroloog Stichting Dianet Internist-nefroloog Amsterdam UMC locatie AMC” |
Lid richtlijnencommissie Nederlandse Federatie voor Nefrologie (onbetaald) |
|
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde patiëntenvereniging in de klankbordgroep. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de Harteraad en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Methode ontwikkeling
Evidence based
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de werkgroep de knelpunten middels een enquête. Naast de in de kern- en klankbordgroep vertegenwoordigde partijen werden de FNT, IGJ, KiMO, NHG, NFU, NVZ, STZ, V&VN, Verenso, VIG, VWS, ZiNL en ZN uitgenodigd deze enquête in te vullen.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nul effect) liggen dan de MCID (Hultcrantz, 2017).
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwaliteit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html.
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, Glasziou P, Jaeschke R, Vist GE, Williams JW Jr, Kunz R, Craig J, Montori VM, Bossuyt P, Guyatt GH; GRADE Working Group. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008 May 17;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008 May 24;336(7654). doi: 10.1136/bmj.a139.
Schünemann, A Holger J (corrected to Schünemann, Holger J). PubMed PMID: 18483053; PubMed Central PMCID: PMC2386626.
Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
Zoekverantwoording
|
Uitgangsvraag: Welk anticoagulans (vitamine K-antagonisten of direct werkende orale anticoagulantia) heeft de grootste effectiviteit en laagste kans op complicaties bij patiënten met veneuze trombose en het antifosfolipiden syndroom? |
|
|
Database(s): Medline, Embase |
Datum search: 01-05-2020 |
|
Periode: ≥ 2010 |
Talen: Engels |
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
2010 – mei 2020
|
1 exp THROMBOEMBOLISM/ or exp Venous Thrombosis/ or (VTE or venous thrombosis or vein thrombosis or arterial thrombosis or DVT or thrombo-embolic or thromboembolic or thromboembolism* or thrombo-embolism* or pulmonary embolism* or lung embolism*).ti,ab,kw. or exp Pulmonary Embolism/ (186863) 2 exp Antiphospholipid Syndrome/ or antiphospholipid.ti,ab,kw. or aps.ti,ab,kw. or lupus anticoagulans.ti,ab,kw. (21924) 3 exp Anticoagulants/ or exp Rivaroxaban/ or exp Dabigatran/ or (anticoagul* or anti coagul* or antithrombotic or doac* or noac* or rivaroxaban or xarelto or dabigatran or apixaban or edoxaban or thrombin inhibitor* or betrixaban or melagatran or ximelagatran or xi-melagatran or exanta).ti,ab,kw. (271607) 4 1 and 2 and 3 (2105) 5 limit 4 to (english language and yr="2010 -Current") (697) 6 (meta-analysis/ or meta-analysis as topic/ or (meta adj analy$).tw. or ((systematic* or literature) adj2 review$1).tw. or (systematic adj overview$1).tw. or exp "Review Literature as Topic"/ or cochrane.ab. or cochrane.jw. or embase.ab. or medline.ab. or (psychlit or psyclit).ab. or (cinahl or cinhal).ab. or cancerlit.ab. or ((selection criteria or data extraction).ab. and "review"/)) not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (442815) 7 (exp clinical trial/ or randomized controlled trial/ or exp clinical trials as topic/ or randomized controlled trials as topic/ or Random Allocation/ or Double-Blind Method/ or Single-Blind Method/ or (clinical trial, phase i or clinical trial, phase ii or clinical trial, phase iii or clinical trial, phase iv or controlled clinical trial or randomized controlled trial or multicenter study or clinical trial).pt. or random*.ti,ab. or (clinic* adj trial*).tw. or ((singl* or doubl* or treb* or tripl*) adj (blind$3 or mask$3)).tw. or Placebos/ or placebo*.tw.) not (animals/ not humans/) (1974951) 8 Epidemiologic studies/ or case control studies/ or exp cohort studies/ or Controlled Before-After Studies/ or Case control.tw. or (cohort adj (study or studies)).tw. or Cohort analy$.tw. or (Follow up adj (study or studies)).tw. or (observational adj (study or studies)).tw. or Longitudinal.tw. or Retrospective*.tw. or prospective*.tw. or consecutive*.tw. or Cross sectional.tw. or Cross-sectional studies/ or historically controlled study/ or interrupted time series analysis/ (Onder exp cohort studies vallen ook longitudinale, prospectieve en retrospectieve studies) (3417878) 9 5 and 6 (58) 10 (5 and 7) not 9 (71) 11 (5 and 8) not 9 not 10 (166) 12 9 or 10 or 11 (295)
= 295 |
878 (135 SR’s, 279 RCT’s en 464 observationele studies) |
|
Embase (Elsevier) |
('thromboembolism'/exp OR vte:ti,ab OR 'venous thrombosis':ti,ab OR 'vein thrombosis':ti,ab OR 'arterial thrombosis':ti,ab OR dvt:ti,ab OR 'thrombo-embolic':ti,ab OR thromboembolic:ti,ab OR thromboembolism*:ti,ab OR 'thrombo-embolism*':ti,ab OR 'pulmonary embolism*':ti,ab OR 'lung embolism*':ti,ab) AND ('antiphospholipid syndrome'/exp OR antiphospholipid:ti,ab OR aps:ti,ab OR 'lupus anticoagulans':ti,ab) AND ('anticoagulant agent'/exp OR 'rivaroxaban'/exp OR 'dabigatran'/exp OR 'apixaban'/exp OR 'edoxaban'/exp OR 'thrombin inhibitor'/exp OR 'betrixaban'/exp OR 'melagatran'/exp OR 'ximelagatran'/exp OR anticoagul*:ti,ab OR 'anti coagul*':ti,ab OR antithrombotic:ti,ab OR doac*:ti,ab OR noac*:ti,ab OR rivaroxaban:ti,ab OR xarelto:ti,ab OR dabigatran:ti,ab OR apixaban:ti,ab OR edoxaban:ti,ab OR 'thrombin inhibitor*':ti,ab OR betrixaban:ti,ab OR melagatran:ti,ab OR ximelagatran:ti,ab OR 'xi-melagatran':ti,ab OR exanta:ti,ab OR 'direct oral anticoagulant'/exp OR 'direct oral anticoagulant agent'/exp) AND (english)/lim AND (2010-2020)/py NOT 'conference abstract':it
Sytematische reviews ('meta analysis'/de OR cochrane:ab OR embase:ab OR psycinfo:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT (('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) NOT 'human'/exp) = 110
RCT’s ('clinical trial'/exp OR 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti) NOT 'conference abstract':it = 260
Observationeel onderzoek 'major clinical study'/de OR 'clinical study'/de OR 'case control study'/de OR 'family study'/de OR 'longitudinal study'/de OR 'retrospective study'/de OR 'prospective study'/de OR 'cohort analysis'/de OR ((cohort NEAR/1 (study OR studies)):ab,ti) OR (('case control' NEAR/1 (study OR studies)):ab,ti) OR (('follow up' NEAR/1 (study OR studies)):ab,ti) OR (observational NEAR/1 (study OR studies)) OR ((epidemiologic NEAR/1 (study OR studies)):ab,ti) OR (('cross sectional' NEAR/1 (study OR studies)):ab,ti)
= 407 Totaal = 777 |