Medicamenteuze behandeling bij allergie van de bovenste luchtwegen
Uitgangsvraag
Met welk(e) middel(en) kunnen patiënten met allergie van de bovenste luchtwegen het beste worden behandeld?
Aanbeveling
Schrijf bij patiënten met allergie van de bovenste luchtwegen en neusverstoppingsklachten een nasaal corticosteroïd voor (eventueel in combinatie met een nasaal antihistaminicum).
Geef de patiënt voorlichting over het juiste gebruik en de veiligheid van nasale corticosteroïden.
Controleer bij voorkeur 2 weken (eventueel telefonisch) na de start van de behandeling of de klachten van patiënten met allergie van de bovenste luchtwegen voldoende onder controle zijn. Pas zo nodig de behandeling aan.
Overwegingen
Allergie van de bovenste luchtwegen kan worden behandeld met een combinatie van een nasaal corticosteroïd en een nasaal antihistaminicum (meest effectief), een nasaal corticosteroïd (minder effectief), een nasaal of oraal antihistaminicum (nog minder effectief) en tenslotte montelukast en chromoglycaat (zie zelfzorgmiddelen) (minst effectief).
Geef een patiënt met een allergie van de bovenste luchtwegen een nasaal corticosteroïd tenzij de klachten mild en intermitterend zijn. Geef altijd een nasaal corticosteroïd als neusverstopping één van de presenterende klachten is.
Step-up naar een combinatie van een nasaal corticosteroïd en een lokaal antihistaminicum als de klachten na twee weken gebruik van een nasaal corticosteroïd onvoldoende gecontroleerd zijn.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
De combinatie van azelastine/fluticasone resulteert waarschijnlijk in minder klachten ten gevolge van allergie van de bovenste luchtwegen dan fluticasone alleen bij volwassenen en adolescenten maar heeft waarschijnlijk geen aanvullend effect op de kwaliteit van leven.
Er is waarschijnlijk geen verschil in het optreden van bijwerkingen tussen de combinatie en alleen fluticasone.
Behandeling met nasale corticosteroïden resulteert waarschijnlijk in minder klachten en verbeterde kwaliteit van leven ten gevolge van allergie van de bovenste luchtwegen dan behandeling met orale antihistaminica bij volwassenen en kinderen zonder aantoonbaar verschil in bijwerkingen (lage Grade). Er is waarschijnlijk geen klinisch relevant verschil tussen behandeling met orale en nasale antihistaminica met betrekking tot klachten, kwaliteit van leven en bijwerkingen.
Alhoewel voor alle medicatie is aangetoond dat er een afname van symptomen is ten opzichte van placebo, zijn lang niet alle bovengenoemde (gecombineerde) opties onderling goed vergeleken. Ook lijken niet alle gevonden (significante) verschillen klinisch relevant te zijn. Met name bij kinderen is de bewijskracht over het algemeen laag.
Er zijn geen aanwijzingen dat nasale corticosteroïden een systemische corticosteroïdbelasting geven ook niet bij (jonge) patiënten die inhalatiecorticosteroïden gebruiken.
Placebo gecontroleerde studies naar antihistaminica, nasale corticosteroïden, combinaties van nasale corticosteroïden/antihistaminica en montelukast laten over het algemeen geen relevante bijwerkingen van deze producten zien. Als patiënten klagen over epistaxis/bloed in nasale mucus bij gebruik van een neusspray moet de spraytechniek gecontroleerd worden aangezien dit over het algemeen wijst op een onjuiste spray techniek waarbij direct tegen het neustussenschot aangespoten wordt. Aangeraden wordt de neusspray altijd kruislings te gebruiken: spuit met de rechterhand in het linker neusgat en met de linkerhand in het rechter neusgat, zodat er altijd naar de buitenkant van de neus wordt gericht (Sastre, 2012).
Sommige patiënten klagen over de onaangename smaak van Azelastine. Moderne antihistaminica geven, in tegenstelling tot de eerste generatie, niet significant vaker sufheid/slaperigheid dan placebo.
De impact van de klachten op het functioneren van de patiënt wordt door behandelaars vaak onderschat (Keith, 2012). Veel patiënten worden onvoldoende behandeld en vaak zijn hun klachten niet onder controle (Gani, 2018).
Kosten (middelenbeslag)
De prijs van alle genoemde medicatie is relatief laag (maximaal 20 euro per maand) waarbij veel patiënten medicatie slechts enkele maanden per jaar gebruiken. Deze kosten wegen niet op tegen mogelijke indirecte kosten, zoals niet kunnen werken of naar school kunnen gaan. Het doel van de behandeling zou dan ook moeten zijn om patiënten klachtenvrij te krijgen ook in situaties met matige allergeenexpositie.
Haalbaarheid en implementatie
Het gebruik van nasale corticosteroïden is al standaard zorg. De werkgroep voorziet daarom geen problemen op het gebied van de haalbaarheid en implementatie.
Rationale/ balans tussen de argumenten voor en tegen de interventie
|
Alleen nasale corticosteroïden hebben een klinisch relevant effect op neusverstoppingsklachten. Nasale corticosteroïden hebben niet meer bijwerkingen dan antihistaminica en geven, ook bij kinderen en langdurig gebruik, geen/minimale systemische belasting. |
|
Regelmatige controle van een behandelaar ten aanzien van de klachten lijkt geïndiceerd, omdat:
|
Onderbouwing
Tot enkele jaren geleden was de behandeling met nasale corticosteroïden in de meeste richtlijnen de eerste keus bij de behandeling van matig-ernstige allergie van de bovenste luchtwegen. Eerdere studies naar het toevoegen van orale antihistaminica hebben geen additief verschil aangetoond. Recent is de combinatie van fluticasone met azelastine op de markt gekomen. Azelastine is een nasaal antihistaminicum met andere “anti-inflammatoire” of corticosteroïd potentiërende effecten. De rol van Azelastine ten opzichte van orale antihistaminica en nasale corticosteroïden en de combinatie van deze middelen ten opzichte van nasale corticosteroïden is onduidelijk. Daarnaast zijn er vragen over de plaats van leukotrieën receptor antagonisten in de behandeling van allergie van de bovenste luchtwegen.
Azelastine fluticasone versus nasale corticosteroïden
Klachten ten gevolge van allergie van de bovenste luchtwegen
|
Redelijk GRADE |
De combinatie van azelastine/fluticasone resulteert waarschijnlijk in minder klachten ten gevolge van allergie van de bovenste luchtwegen dan fluticasone bij volwassenen en adolescenten.
Bronnen: Berger, 2014; Carr, 2012; Hampel 2010; Price 2013 |
Kwaliteit van leven
|
Redelijk GRADE |
Er is waarschijnlijk geen verschil in kwaliteit van leven tussen behandeling met azelastine/fluticasone ten opzichte van fluticasone alleen, bij patiënten met allergie van de bovenste luchtwegen.
Bronnen: Carr, 2012; Hampel 2010 |
Bijwerkingen (treatment related adverse events)
|
Redelijk GRADE |
Er is waarschijnlijk geen verschil in het optreden van bijwerkingen tussen azelastine/fluticasone versus fluticasone bij volwassen patiënten en adolescenten met allergie van de bovenste luchtwegen.
Bronnen: Berger, 2014; Carr, 2012; Hampel 2010 |
Bijwerkingen - kinderen
|
Laag GRADE |
Er is mogelijk geen verschil in het optreden van bijwerkingen tussen azelastine/fluticasone en fluticasone bij kinderen met allergie van de bovenste luchtwegen.
Bron: Berger, 2018 |
Orale antihistaminica versus nasale corticosteroïden
Klachten ten gevolge van allergie van de bovenste luchtwegen bij volwassenen
|
Hoog GRADE |
Behandeling met nasale corticosteroïden resulteert in minder klachten ten gevolge van allergie van de bovenste luchtwegen dan behandeling met orale antihistaminica bij volwassenen.
Bronnen: Condemi, 2000; Bernstein, 1994; Ford, 2015; Frølund, 1991; Gawchik, 1997; Gehanno, 1997; ; Kim, 2015; Ratner, 1998; Rinne, 2002Schoenwetter, 1995; Vervloet, 1997 |
Klachten ten gevolge van allergie van de bovenste luchtwegen bij kinderen
|
Laag GRADE |
Behandeling met nasale corticosteroïden resulteert mogelijk in minder klachten ten gevolge van allergie van de bovenste luchtwegen bij kinderen ten opzichte van behandeling met orale antihistaminica.
Bronnen: Fokkens, 2004; Malizia, 2018; Wartna, 2017 |
Kwaliteit van leven bij volwassenen
|
Hoog GRADE |
Behandeling met nasale corticosteroïden resulteert in een verbetering in kwaliteit van leven ten opzichte van behandeling met orale antihistaminica bij volwassenen met allergie van de bovenste luchtwegen, maar dit verschil is niet klinisch relevant.
Bronnen: Bhatia, 2005; Condemi, 2000; Ford, 2015; Kim, 2015; Ratner, 1998 |
Kwaliteit van leven bij kinderen
|
Laag GRADE |
Er is mogelijk een verbetering in kwaliteit van leven bij kinderen na behandeling met nasale corticosteroïden ten opzichte van behandeling met orale antihistaminica, maar dit verschil is niet klinisch relevant.
Bron: Malizia, 2018 |
Bijwerkingen
|
Redelijk GRADE |
Er is waarschijnlijk geen verschil in het optreden van bijwerkingen tussen behandeling met nasale corticosteroïden en orale antihistaminica bij volwassenen en kinderen met allergie van de bovenste luchtwegen.
Bronnen: Andrews, 2009; Bernstein, 2004; Condemi, 2000; D’Ambrosio, 1998; Gawchik, 1997; Kim, 2015 |
Orale versus nasale antihistaminica
Klachten ten gevolge van allergie
|
Redelijk GRADE |
Behandeling van allergie van de bovenste luchtwegen met nasale antihistaminica resulteert waarschijnlijk in een verbetering van klachten ten gevolge van allergie van de bovenste luchtwegen ten opzichte van orale antihistaminica, maar dit verschil is niet klinisch relevant.
Bronnen:(Berger, 2003; Berger, 2006; Charpin, 1995; Conde Hernández, 1995; Corren, 2005 |
Kwaliteit van leven
|
Redelijk GRADE |
Er is waarschijnlijk een verbetering in kwaliteit van leven bij behandeling van allergie van de bovenste luchtwegen met nasale antihistaminica ten opzichte van orale antihistaminica, maar dit verschil is niet klinisch relevant.
Bronnen: Berger, 2006; Corren, 2005 |
Bijwerkingen
|
Laag GRADE |
Er is mogelijk geen verschil in het optreden van bijwerkingen tussen behandeling van allergie van de bovenste luchtwegen met orale antihistaminica en nasale antihistaminica.
Bronnen: Charpin, 1995; Conde Hernández, 1995 |
Montelukast versus placebo
Klachten ten gevolge van allergie
|
Laag GRADE |
Montelukast resulteert mogelijk in een klinisch relevante vermindering van klachten ten gevolge van allergie van de bovenste luchtwegen dan placebo bij volwassenen en kinderen.
Bronnen: Chen, 2006; Ciebiada, 2006; Ciebiada, 2011; Hsieh, 2004; Kurowski, 2004; Philip, 2005; Okubo, 2008; Wei, 2016 |
Kwaliteit van leven
|
Laag GRADE |
Montelukast resulteert mogelijk in een niet-klinisch relevante verbetering van de kwaliteit van leven ten opzichte van placebo bij volwassenen en kinderen met allergie van de bovenste luchtwegen.
Montelukast resulteert mogelijk in verbetering in kwaliteit van leven ten opzichte van placebo bij kinderen met allergie van de bovenste luchtwegen.
Bronnen: Chen, 2006; Lombardo, 2006; Philip, 2005; Wei, 2016 |
Bijwerkingen
|
Laag GRADE |
Er is mogelijk geen verschil in het optreden van bijwerkingen tussen montelukast en placebo bij volwassenen en kinderen met allergie van de bovenste luchtwegen.
Bronnen: Bisgaard, 2009; Hsieh, 2004; Philip, 2005; Okubo, 2008 |
Montelukast versus antihistaminica
Klachten ten gevolge van allergie
|
Redelijk GRADE |
Er is waarschijnlijk geen verschil in effect op klachten ten gevolge van allergie van de bovenste luchtwegen tussen antihistaminica en montelukast bij volwassenen.
Bronnen: Andhale, 2016; Ciebiada, 2006; Kaur, 2017; Kurowski, 2004; Lombardo, 2006; Rajeshkar, 2018, Wei, 2016 |
Klachten ten gevolge van allergie bij kinderen
|
Laag GRADE |
Het antihistaminicum cetirizine resulteert mogelijk in een verbetering van klachten ten gevolge van allergie van de bovenste luchtwegen ten opzichte van montelukast bij kinderen.
Bronnen: Chen, 2006; Hsieh, 2004 |
Kwaliteit van leven
|
Laag GRADE |
Antihistaminica resulteren mogelijk in een niet-klinisch relevante verbetering in kwaliteit van leven ten opzichte van montelukast bij volwassenen met een allergie van de bovenste luchtwegen.
Er is mogelijk geen verschil in kwaliteit van leven bij behandeling van allergie van de bovenste luchtwegen bij kinderen tussen cetirizine en montelukast.
Bronnen: Chen, 2006; Lombardo, 2006; Wei, 2016; |
Bijwerkingen
|
Redelijk GRADE |
Er is waarschijnlijk geen verschil in het optreden van bijwerkingen tussen behandeling met montelukast of antihistaminica bij patiënten met allergie van de bovenste luchtwegen.
Bronnen: Lombardo, 2006; Wei, 2016 |
Bijwerkingen bij kinderen
|
Zeer laag GRADE |
Het is onduidelijk of er verschil is in bijwerkingen tussen montelukast en het antihistaminicum cetirizine bij behandeling van kinderen met allergie van de bovenste luchtwegen.
Bron: Hsieh, 2004 |
Beschrijving studies
Azelastine/fluticasone versus nasale corticosteroïden (PICO 1)
Berger (2014) en Price (2013) beschrijven een open-label RCT uitgevoerd in India in 2009; 388 patiënten kregen azelastine/fluticason en 199 kregen fluticason gedurende een jaar. In de azelastine/fluticasongroep was na een jaar nog 77% van de patiënten over en in de fluticasongroep nog 74% van de patiënten. Price (2013) rapporteerde de reflective nasal total symptom score (rTNSS) van deze patiënten. Berger (2014) rapporteerde adverse events. Kwaliteit van leven en ziekteverzuim werden niet gerapporteerd.
Carr (2012) poolde 3 RCT’s: MP 4002 (NCT00651118), MP4004 (NCT00740792) (Meltzer 2012) en MP4006 (NCT00883168) in een meta-analyse. Dit betroffen multicenter, gerandomiseerde, dubbelblinde, placebo-gecontroleerde studies. In verschillende seizoenen werden de patiënten gedurende 14 dagen behandeld. In totaal 848 patiënten kregen azelastine/fluticason toegediend en 846 patiënten kregen een corticosteroïden (fluticason propionate) toegediend. Gerapporteerde uitkomsten waren verandering ten opzichte van baseline van de som van in de ochtend en avond gescoorde rTNSS de maximum rTNSS of instantaneous nasale symptoom score was 24 (ie 4 symptomen x score van 3 x 2 voor ochtend en avond), kwaliteit van leven gemeten met de RQLQ (Rhinitis Quality of Life Questionnaire) en bijwerkingen. Ziekteverzuim werd niet gerapporteerd.
Hampel (2010) rapporteert een multicenter, gerandomiseerde, dubbelblinde, placebo-gecontroleerde RCT. De patiënten werden gedurende 14 dagen behandeld. In totaal 153 patiënten kregen azelastine/fluticason en 151 kregen fluticason. Gerapporteerde uitkomsten waren verandering ten opzichte van baseline van de som van in de ochtend en avond gescoorde rTNSS de maximum rTNSS of instantaneous nasale symptoom score was 24 (ie 4 symptomen x score van 3 x 2 voor ochtend en avond), kwaliteit van leven gemeten met de RQLQ en bijwerkingen. Ziekteverzuim werd niet gerapporteerd.
Kinderen
Berger (2018) beschrijft een open-label RCT uitgevoerd bij kinderen van 4 tot 11 jaar in de USA op 42 sites; 304 kinderen kregen azelastine/fluticason en 101 kinderen kregen fluticason gedurende een jaar. In de azelastine/fluticasongroep was na drie maanden nog 94% van de patiënten over en in de fluticasongroep nog 91% van de patiënten. Van deze studie werd als uitkomst alleen het aantal adverse events gerapporteerd.
Orale antihistaminica versus nasale corticosteroïden (PICO 2)
Een systematische review van 16 studies is opgenomen in de literatuuranalyse (Weiner, 1998). Zes van de studies beschreven in deze review voldeden aan de zoekcriteria en zijn geëvalueerd, de overige zijn buiten beschouwing gelaten. Daarnaast zijn 19 RCT’s opgenomen in de analyse. Als orale antihistaminicum behandeling werden astemizole, loratadine, desloratadine, cetirizine en levocetirizine bestudeerd, als corticosteroïden fluticasone propionate, triamcinolone acetonide, becalmethasone dipropionate, budenoside en ciclesonide. Het merendeel van de studies hanteerde een behandelingsduur van 2 tot 4 weken.
Drie studies bestudeerden specifiek een populatie jonger dan 18 jaar. Wartna (2017) onderzocht behandeling met levocetirizine en fluticasone propionate op indicatie bij SAR patiënten van 6 tot 18 jaar (n=150) gedurende 3 maanden. Malizia (2018) onderzocht cetirizine (10 mg) en beclomethasone dipropionate (tweemaal daags 100 μg per neusgat) in patiënten van 6 tot 14 jaar oud (n=68) gedurende 3 weken. Fokkens (2004) bestudeerde kinderen van 2 tot 4 jaar (n=26), en beschreef neussymptoomscores uitgesplitst in dag en nacht.
Orale versus nasale antihistaminica (PICO 3)
Geïncludeerde studies vergeleken orale antihistaminicum behandeling met azelastine nasale spray behandeling bij patiënten met seizoensgebonden allergie van de bovenste luchtwegen.
Ellis (2013) is een dubbel-blinde RCT en includeerde volwassenen gediagnosticeerd met seizoensgebonden allergische rhinitis (SAR). Patiënten (n=70) werden in een cross-over structuur gedurende 8 uur blootgesteld aan allergenen in een gecontroleerde omgeving en behandeld met orale loratadine, orale cetirizine, nasale azelastine of placebo. Gerapporteerde uitkomstmaten waren onset of action en nasale symptoomscore (totaal en individuele scores).
Berger (2006) is een dubbel-blinde multicenter RCT (ACT II) en includeerde patiënten van tenminste 12 jaar oud gediagnosticeerd met SAR. (n=360) Patiënten werden gedurende 2 weken behandeld met cetirizine (10 mg) of azelastine nasale spray (tweemaal daags 2 sprays per neusgat). Gerapporteerde uitkomstmaten waren een totale nasale symptoomscore, individuele symptoomscores en RQLQ.
Corren (2005) is een dubbelblinde multicenter RCT (ACT I) en includeerde patiënten van tenminste 12 jaar oud gediagnosticeerd met SAR. Patiënten (n=307) werden gedurende 2 weken behandeld met cetirizine (10 mg) of azelastine nasale spray (tweemaal daags 2 sprays per neusgat). Gerapporteerde uitkomstmaten waren een totale nasale symptoomscore en time of onset daarvan, individuele symptoomscores en RQLQ.
Berger (2003) is een dubbelblinde multicenter RCT en includeerde patiënten van tenminste 12 jaar oud gediagnosticeerd met SAR. Patiënten (n=440) werden gedurende 2 weken behandeld met desloratadine (5 mg), azelastine nasale spray (tweemaal daags 2 sprays per neusgat), een combinatie van beide behandelingen of placebo. Gerapporteerde uitkomstmaat was de verandering in totale nasale symptoomscore.
Charpin (2003) is een dubbelblinde multicenter RCT en includeerde patiënten van tenminste 12 jaar oud gediagnosticeerd met SAR. Patiënten (n=129) werden gedurende 2 weken behandeld met cetirizine (10 mg) of azelastine nasale spray (tweemaal daags 1 spray per neusgat). Gerapporteerde uitkomstmaten waren de door de onderzoeker vastgestelde symptoomscore (TSSI), verandering daarin, door de patiënt gerapporteerde VAS-score en tolerantie voor de medicatie.
Conde Hernández (1995) is een open-label RCT en includeerde patiënten gediagnosticeerd met SAR. Patiënten (n=63) werden gedurende 2 weken behandeld met ebastine (10 mg) of azelastine nasale spray (tweemaal daags 2 sprays per neusgat). Gerapporteerde uitkomstmaten waren de verandering in totale symptoomscore, afzonderlijke symptoomscores en actieve rhinomanometrie.
Gambardella (1993) is een dubbelblinde RCT en includeerde volwassen patiënten gediagnosticeerd met SAR. Patiënten (n=30) werden gedurende 6 weken behandeld met loratadine (10 mg) of azelastine nasale spray (tweemaal daags 1 spray per neusgat). Gerapporteerde uitkomstmaten waren de totale symptoomscore en de veranderingen daarin.
Kinderen
Er zijn geen studies gevonden binnen de gestelde criteria waarin het effect van de medicatie op kinderen afzonderlijk is beschreven.
Leukotrieën receptor antagonisten versus placebo of antihistaminica (PICO 4 en 5)
In de literatuuranalyse is de meta analyse van Wei (2106) opgenomen en ge-update met recente RCT’s (Rajashekar, 2018; Kaur, 2017). Daarnaast zijn er 10 RCT’s geïncludeerd gepubliceerd tussen 2000 en 2016 die niet in de meta-analyse van Wei (2016) zijn opgenomen. Hiervan zijn acht studies uitgevoerd bij volwassenen (Andhale, 2016; Ciebiada, 2011; Okubo, 2008; Ciebiada, 2006; Lombardo, 2006; Philip, 2004; Kurowski, 2004) en studies includeerden kinderen (Bisgaard, 2009; Hsieh, 2004; Chen, 2006)
De meta-analyse van Wei (2016) includeerde uitsluitend RCT’s (n=16). Alleen RCT’s die uitkomstmaten rapporteerden op basis van de meest gebruikte schalen voor symptoomscores werden hierbij meegenomen. De studiepopulatie betrof patiënten met seizoensgebonden en niet-seizoensgebonden allergie van de bovenste luchtwegen. Studies waarbij de populatie astma patiënten betrof, werden geëxcludeerd. Ook werden studies met een hoog risico op bias (geen blindering van patiënten en behandelaars) niet meegenomen in de meta-analyse. Geïncludeerde studies vergeleken behandeling met montelukast met een controle groep (placebo en/of antihistamine). Voor het beantwoorden van deze uitgangsvraag werden alleen de resultaten van de studies geëxtraheerd die montelukast vergeleken met een antihistaminicum en niet met combinatietherapie (n=8).
Rajeshekar (2018) is een dubbelblinde RCT en includeerde patiënten gediagnosticeerd met mild tot matige Persistent Allergische Rhinitis (PAR). Patiënten (n=70) werden gedurende drie weken behandeld met montelukast of ebastine. Gerapporteerde uitkomstmaat was de five nasal symptom scoring (T5NSS).
Kaur (2017) is een RCT en includeerde patiënten (15 tot 55 jaar) met AR (n=125). Patiënten werden gedurende zes weken behandeld met montelukast (10mg), levocetirizine (5mg), fexofenadine (180mg), desloratadine (5 mg) of chlorpheniramine (4 mg). Gerapporteerde uitkomstmaten waren een symptoomscore voor niezen en een symptoomscore voor nasal discharge/rhinorrhoe.
Andhale (2016) is een trial waarin patiënten met PAR (n=50) gedurende twee weken werden behandeld met montelukast (10 mg) of levocetirizine (5 mg). Gerapporteerde uitkomstmaten zijn gemeten op een VAS-schaal voor neus- en oogsymptomen gedurende de dag en de nacht.
Ciebiada (2011) is een RCT met een cross-over design en includeerde patiënten met PAR. Patiënten (n=20) werden gedurende 6 weken behandeld met montelukast (10 mg), levocetirizine (5 mg) of placebo met tussendoor ‘washout” periode van 2 weken. Gerapporteerde uitkomsten waren congestion symptoms geschat op basis van nasal symptoms score gerapporteerd in een patiënten dagboek.
Okubo (2008) is een dubbelblinde RCT bij patiënten met SAR met symptomen (n=945). Patiënten werden gedurende 2 weken behandeld met placebo, montelukast (5 mg of 10 mg). Gerapporteerde uitkomstmaten waren composite nasal symptom scores (CNSS), gedefinieerd als gemiddelde score van nasal symptom scores gedurende de dag en nacht, en bijwerkingen.
Ciebiada (2006) is een dubbelblinde RCT met cross-over design. Patiënten met PAR werden geïncludeerd en gedurende zes weken behandeld met montelukast, levocetirizine en placebo (n=20) met daartussen een ‘washout’ periode van twee weken. Gerapporteerde uitkomstmaten zijn total daytime nasal symptoms score en daytime eye symptom score.
Lombardo (2006) is een RCT en includeerde patiënten met SAR. Patiënten werden behandeld gedurende 4 weken met montelukast (10 mg), levocetirizine (5 mg) of placebo (n=254). Gerapporteerde uitkomstmaten zijn daytime nasal symptoms (DNSS), nighttime nasal symptoms (NSS), en daytime eye symptoms (DES) en RQLQ.
Philip (2005) is een multicenter RCT en includeerde patiënten met SAR (ten minste milde symptomen) en actieve astma (n=831). Patiënten werden gedurende 6 weken behandeld met montelukast of placebo. Gerapporteerde uitkomstmaten waren een Daily Rhinitis Symptoms score, gedefinieerd als het gemiddelde van DNSS en NSS), RQLQ en bijwerkingen.
Kurowski (2004) is een dubbelblinde RCT en includeerde patiënten met SAR. Patiënten (n=41) werden gedurende 12 weken (6 weken pre-seizoen) behandeld met montelukast (10 mg), cetirizine (10 mg) of placebo. Gerapporteerde uitkomstmaat was een all-symptoms score (Rhinoconjunctivitis symptom score) gemeten op een 6-punts schaal.
Kinderen
Bisgaard (2009) is een studie waarin studieresultaten naar veiligheid van montelukast uit eerder gepubliceerde en niet gepubliceerde placebo gecontroleerde dubbelblinde en open-label pediatrische studies worden gerapporteerd. Alleen de resultaten van de dubbelblinde RCT (alleen gepubliceerd in Bisgaard, 2009) die kinderen (2 tot 14 jaar) met SAR (n=413) includeerde werden meegenomen in deze literatuursamenvatting. In deze studie werden alleen het aantal bijwerkingen gerapporteerd
Chen 2006 is een dubbelblinde RCT en includeerde kinderen (2 tot 6 jaar) met PAR (n=60). Patiënten werden gedurende 12 weken behandeld met cetirizine (5 mg), montelukast (4 mg) of placebo. Gerapporteerde uitkomstmaten waren een total symptoms score (TSS) en PRQLQ.
Hsieh (2004) is een dubbelblinde RCT die kinderen (6 tot 12 jaar) met PAR includeerde (n=65). Patiënten werden gedurende 12 weken behandeld met cetirizine (10 mg), montelukast (5 mg) of placebo. Gerapporteerde uitkomstmaten waren total symptoms score (TSS), PRQLQ en bijwerkingen.
Resultaten
Azelastine fluticasone versus nasale corticosteroïden (PICO 1)
Klachten ten gevolge van allergie van de bovenste luchtwegen (symptomen (rTNSS))
rTNSS werd gerapporteerd door Carr (2012), Price (2013), Hampel (2010) en Berger (2014). De resultaten van de rTNSS werden gecombineerd in een forest plot, zie figuur 1. Voor de studies met een follow-up van 14 dagen was het gepoolde effect -0,67 (95% BI -1,11 tot 0,23), dit effect is klinisch relevant aangezien het groter is dan de grens van 0,5 SD.
Bij de studie met een follow-up van 1 jaar werd een effect gezien van -0,27 (95% BI -0,76 tot -0,16) (Berger, 2014).
Figuur 1 Reflective total nasal symptom score voor de combinatie azelastine/fluticasone versus fluticasone
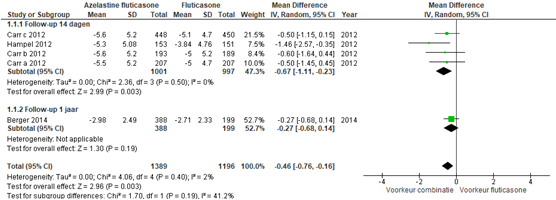
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval.
NB Hampel (2012) rapporteert geen SD van de eindscore, daarom is de baseline SD ingevuld
Kwaliteit van leven (RQLQ)
De RQLQ werd gerapporteerd door Carr (2012) en Hampel (2010). Carr (2012) vond een verschil op RQLQ van 1,6 (SD 1,3) voor azelastine/fluticasone en van 1,5 voor fluticasone; een verschil van -0,10 (95% BI -0,22 tot 0,00). Hampel vond een verschil op de RQLQ van 1,60 voor azelastine/fluticasone en 1,43 voor fluticasone; een verschil van -0,17 (95% BI -0,46 tot 0,12). Beide verschillen waren niet statistisch significant en niet klinisch relevant.
Kwaliteit van leven werd niet gerapporteerd voor kinderen.
Bijwerkingen
Een overzicht van bijwerkingen wordt gegeven in twee forest plots. Figuur 2 geeft een overzicht van alle bijwerkingen die gemeld werden tijdens de trial. Daarbij werd een risk ratio gevonden van 1,11 (95% BI 0,96 tot 1,28). Dit effect was niet statistisch significant en ook niet klinisch relevant. Figuur 3 geeft een overzicht van treatment related adverse events. Daarbij werd een risk ratio gevonden van 1,25 (95% BI 0,90 tot 1,75). Dit effect was ook niet statistisch significant noch klinisch relevant.
Berger (2014) geeft aan dat dysgeusia (bittere smaak) het meest frequent werd gerapporteerd (2,5%) net als hoofdpijn (4,3%). Hampel (2010) gaf geen totaal overzicht van hoeveel procent van de deelnemers last had van bijwerkingen maar rapporteerde hoe frequent een aantal bijwerkingen voorkwam.
Figuur 2 Treatment emergent adverse events van azelastine/fluticasone versus fluticasone
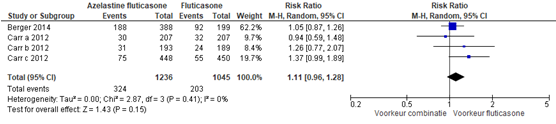
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Figuur 3 Treatment related adverse events van azelastine/fluticasone versus fluticasone
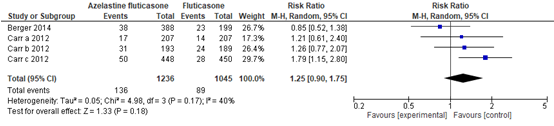
Z: p-waarde van het gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Kinderen
Er werd een vergelijkbaar percentage bijwerkingen (treatment emergent adverse events) gezien bij gebruik van azelastine/fluticasone 41% versus fluticasone (37%). Voor treatment related adverse events was dit azelastine/fluticasone 16% versus fluticasone (12%).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat klachten ten gevolge van allergie van de bovenste luchtwegen (symptomen (rTNSS)) is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en werd met één niveau verlaagd naar redelijk vanwege imprecisie (overlap met de grens voor klinische relevante).
De bewijskracht voor de uitkomstmaat klachten ten gevolge van allergie van de bovenste luchtwegen bij kinderen is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog. Er is 2 niveaus verlaagd gezien het zeer geringe aantal patiënten (imprecisie) en met 1 niveau wegens beperkingen in de onderzoeksopzet (risk of bias), waarmee de bewijskracht uitkomt op zeer laag.
De bewijskracht voor de uitkomstmaat kwaliteit van leven is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en werd met één niveau verlaagd vanwege imprecisie (slechts 2 studies rapporteren kwaliteit van leven).
De bewijskracht voor de uitkomstmaat treatment related adverse events is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en is met één niveau verlaagd naar redelijk vanwege het overschrijden van de grens van klinische relevantie (imprecisie).
De bewijskracht voor de uitkomstmaat treatment related adverse events bij kinderen is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en is met twee niveaus verlaagd naar laag gezien beperkingen in de onderzoeksopzet omdat het een open label studie betreft (risk of bias) en vanwege het geringe aantal patiënten (imprecisie).
Orale antihistaminica versus nasale corticosteroïden (PICO 2)
Klachten ten gevolge van allergie van de bovenste luchtwegen (symptoomscores)
In vier studies in volwassenen (d’Ambrosio, 1998; Kaszuba, 2001; Kulapaditharom, 2010; Bhatia, 2005) bij in totaal 286 patiënten werden de verbetering in totale symptoomscore vergeleken tussen orale antihistaminica en nasale corticosteroïden. De gecombineerde SMD was 0,32, met een 95% BI van -0,27 tot 0,90, zoals weergegeven in figuur 4. Frølund (1991) vond geen significant verschil in totale symptoomscore tussen loratadine en beclomethasone dipropionate behandeling gedurende 3 weken, maar verstrekte te weinig informatie om dit in de analyse mee te nemen. Charpin (1994) vond daarentegen een significant sterkere afname in symptoomscore bij behandeling met fluticasone propionate vergeleken met cetirizine, maar vermeldde ook niet voldoende gegevens om de data te kunnen analyseren.
Figuur 4 Verschil in Totale Symptom score (TSS) van orale antihistamine versus nasale corticosteroïd behandeling
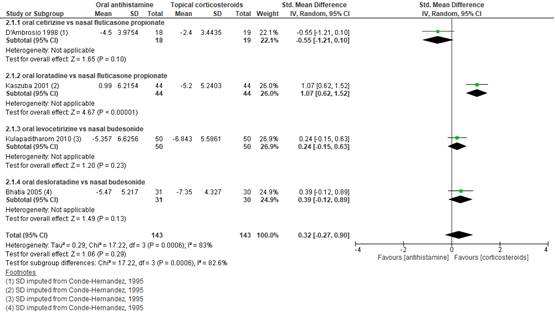
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Naast de totale symptoomscore werd ook de totale score van nasale symptomen beschreven. Uit de gepoolde data van 11 RCT s met in totaal 2510 patiënten werd consequent een sterkere symptoomscore verbetering gevonden bij corticosteroïden vergeleken met orale antihistaminicum, met een gestandaardiseerd gemiddeld verschil van 0,70 en een 95% BI van 0,45 tot 0,95 (figuur 5). Dit verschil is klinisch relevant. Van een aantal studies moesten de waarden omgerekend worden of moest de SD geïmputeerd worden op basis van vergelijkbare studies, omdat deze gegevens ontbraken.
Figuur 5 Verschil in Totale Nasale Symptoom Score (TNSS) van orale antihistamine versus nasale corticosteroïden behandeling
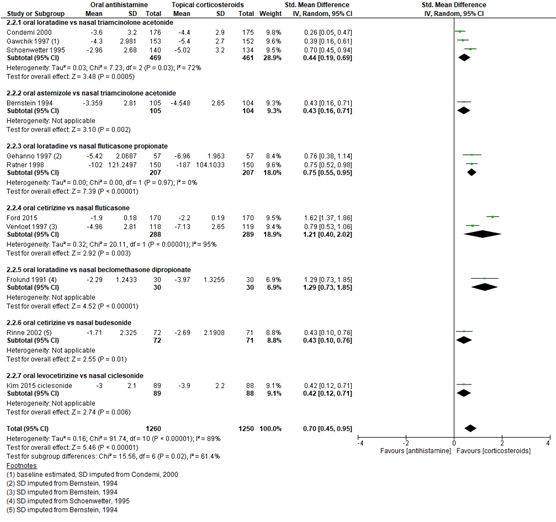
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Kinderen
Drie studies die bij kinderen behandeling met nasale corticosteroïden vergeleken met orale antihistaminica vonden een lagere symptoomscore bij gebruik van corticosteroïden. Bij kinderen van 6 tot 14 jaar oud (Malizia, 2018) werd een significant grotere afname in totale symptoomscore gevonden bij gebruik van nasale beclomethason (least square mean change -5,63) vergeleken met orale cetirizine (-3,54; P=0,008). Dagelijkse behandeling met fluticason resulteerde bij kinderen van 6 tot 18 jaar oud (Wartna, 2016) in een lagere dagelijkse symptoomscore (totaalscore: 3,90 ± 3,06; neussymptomen: 3,20± 1,79) dan levocetirizine gebruik op indicatie (totaalscore: 4,63 ± 2,82; neussymptomen: 4,14 ± 1,76), maar deze verschillen waren niet significant. Bij jonge kinderen (van 2 tot 4 jaar oud), werd een lagere gecombineerde symptoomscore gevonden na 4-6 weken gebruik van fluticason ten opzichte van ketotifen. Nachtsymptomen waren 4,2 ± 0,7 (standard error) bij gebruik van fluticason, versus 6,5 ± 0,7 (p=0,036) bij gebruik van ketotifen. Dagsymptomen waren bij fluticason 3,6 ± 0,6 versus 5,5 ± 0,6 (p=0,049) bij ketotifen (Fokkens, 2004). Door de verschillende manieren waarop de waarden zijn uitgedrukt en/of het ontbreken van baselinewaarden kunnen de gegevens van de drie studies niet gepoold worden weergegeven.
Kwaliteit van leven / Rhinoconjunctivitis quality of life questionnaire (RQLQ)
Vijf studies met in totaal 1229 patiënten rapporteerden de verbetering in RQLQ bij behandeling met verschillende medicatie. Nasale corticosteroïden gaven een sterkere verbetering dan orale antihistaminica, met een gemiddeld verschil van 0,46 (op een 7 puntsschaal) en een 95% BI van 0,28 tot 0,63. Dit verschil is niet klinisch relevant (figuur 6).
Kinderen
Malizia (2018) vond een vergelijkbaar effect bij kinderen, met een sterkere verbetering in kwaliteit van leven gemeten met de PRQLQ bij beclomethason vergeleken met cetirizine (-1,15 versus -0,69; P=0,031).
Figuur 6 Rhinitis Quality of Life Questionnaire (RQLQ) bij orale antihistamine versus nasale corticosteroïden behandeling
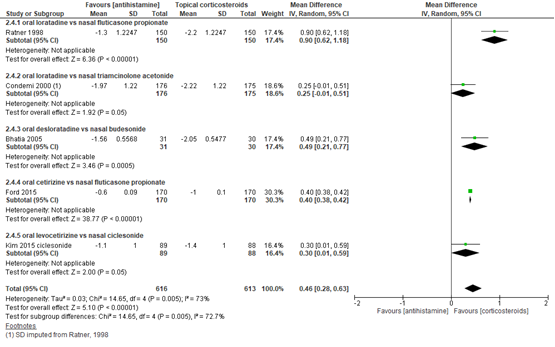
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Bijwerkingen
In 7 studies werd de incidentie van bijwerkingen kwantitatief beschreven. Bij in totaal 2403 patiënten was de het relatieve risico 1,03, met een 95% betrouwbaarheidsinterval van 0,95 tot 1,11 (figuur 7). Waarschijnlijk hebben de studies geen eenduidige criteria gehanteerd, aangezien de waarden binnen dezelfde type medicatie varieerden van 7% (Kim, 2015) tot 94% (Rinne, 2002).
Figuur 7 Bijwerkingen bij orale antihistamine versus nasale corticosteroïden behandeling
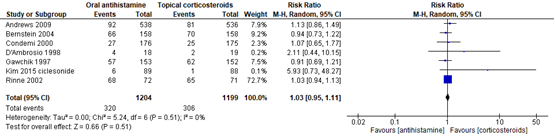
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Kinderen
Fokkens (2004) rapporteerde dat er geen ernstige bijwerkingen werden waargenomen (fluticason versus ketotifen).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaten is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog.
De bewijskracht van de uitkomstmaat klachten ten gevolge van allergie van de bovenste luchtwegen werd met twee niveaus verlaagd voor totale symptomen vanwege variatie in de resultaten (inconsistentie) en overschrijden van de grenzen van klinische relevantie (imprecisie) naar laag.
De bewijskracht voor de uitkomstmaat nasale symptomen is hoog en werd niet verlaagd.
De bewijskracht voor de uitkomstmaat klachten ten gevolge van allergie van de bovenste luchtwegen bij kinderen is met drie niveaus verlaagd naar laag gezien de indirectheid van de bepalingen en het beperkt aantal patiënten (imprecisie).
De bewijskracht voor de uitkomstmaat kwaliteit van leven is niet verlaagd en blijft hoog. De bewijskracht voor de uitkomstmaat kwaliteit van leven bij kinderen is verlaagd met 2 niveaus naar laag gezien het zeer beperkt aantal patiënten (imprecisie).
De bewijskracht voor de uitkomstmaat bijwerkingen is met één niveau verlaagd gezien de variatie in de definitie van bijwerkingen in de verschillende studies (inconsistentie) naar redelijk.
Orale versus nasale antihistaminica (PICO 3)
Klachten ten gevolge van allergie van de bovenste luchtwegen (symptoomscores)
De gecombineerde score van neus- oog- en keelsymptomen werd gerapporteerd door Gambardella (1993), Charpin (1995) en Conde Hernández (1995). Omdat het niet duidelijk was of dezelfde scores gehanteerd werden, zijn de resultaten weergegeven als gestandaardiseerd gemiddelde verschil (SMD). Het SMD bij in totaal 203 patiënten was -0,10 in het voordeel van orale medicatie, met een 95% BI van -0,37 tot 0,18. Dit is een klein, niet klinisch relevant verschil. De gecombineerde symptoomscore voor nasale symptomen na een behandeling van 2 weken werd in 5 studies beschreven. In een gecombineerde analyse (figuur 8) van Conde Hernández (1995), Charpin (1995), Berger (2003), Corren (2005) en Berger (2006) was het SMD 0,17 in het voordeel van nasale spray, met een 95% BI van 0,04 tot 0,30. Dit verschil is klein en niet klinisch relevant, zoals aangegeven in figuur 8. Ellis (2013) vond 6 uur na behandeling (orale loratadine of orale cetirizine versus nasale azelastine) een SMD van 0,19 met een 95% BI van -0,74 tot 1,12. Daarnaast beschreven Conde Hernández (1995) en Charpin (1995) een gecombineerde oogsymptoomscore, dat met een SMD van -0,32 en 95% BI van -0,62 tot -0,02 in het voordeel is van orale toediening, maar dit verschil is niet klinisch relevant.
Figuur 8 Gemiddeld verschil in TNSS bij orale versus nasale antihistamine behandeling
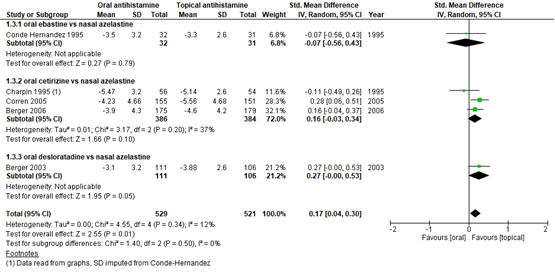
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Kwaliteit van leven/ Rhinoconjunctivitis quality of life questionnaire (RQLQ)
Twee studies rapporteerden de RQLQ (een gemiddelde score van 28 vragen op een zeven-puntsschaal). Corren (2005) en Berger (2006) vonden beide een sterkere verbetering in nasale dan in orale toediening, met een gemiddeld verschil van 0,36 en een 95% BI van 0,17 tot 0,54. Dit verschil is klinisch niet relevant.
Bijwerkingen
Het totale aantal bijwerkingen werd beschreven door Charpin (1995) en Conde Hernández (1995). Bij 22,7% van de 198 patiënten werden bijwerkingen gerapporteerd, met een relatief risico van 1,26 (95% BI 0,65 tot 2,43) in het voordeel van nasale toediening. Dit verschil is niet klinisch relevant. Diverse bijwerkingen werden gerapporteerd bij het gebruik van de medicatie. Bittere smaak en slaperigheid werden vaak gerapporteerd bij het gebruik van azelastine. Slaperigheid werd ook gerapporteerd bij cetririzine en ebastine. Verder kwamen diverse klachten zoals misselijkheid en hoofdpijn voor bij meerdere middelen.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaten klachten ten gevolge van allergie van de bovenste luchtwegen en kwaliteit van leven is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en is met één niveau verlaagd gezien het gering aantal patiënten (imprecisie) naar redelijk.
De bewijskracht voor de uitkomstmaat bijwerkingen is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en is met twee niveaus verlaagd gezien het beperkte aantal patiënten en de overlap van het betrouwbaarheidsinterval met 1 (imprecisie) naar laag.
Montelukast versus placebo (PICO 4)
Klachten ten gevolge van allergie
Symptomen werden gerapporteerd door 6 studies (Wei, 2016; Philip, 2005; Ciebiada, 2011; Okubo, 2008; Ciebiada, 2006; Kurowski, 2004), indien beschikbaar is de totaal score/ composite score gerapporteerd. Wei (2016) rapporteerde een composite symptoms score (CSS) op een 4-punts schaal (0-3). In de studies die werden geïncludeerd was de CSS gedefinieerd als de gemiddelde waarde van DNSS en NSS. Het gepoolde effect was -0,11 (95%BI -0.14 tot -0,07) in het voordeel van montelukast en wordt weergegeven in een forest plot (figuur 9).
Figuur 9 CSS montelukast vergeleken met placebo. Bron: Wei, 2016 aangevuld met studie Philip, 2005
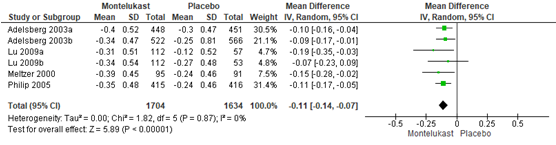
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Ciebiada (2011) rapporteerde neusverstopping (congestion symptoms). In de groep behandeld met montelukast werd op dag 42 een afname van 1,5 gerapporteerd en in de placebogroep een afname van 1,0 ten opzichte van baseline metingen (gemiddelde verschil -0,5; 95%BI -0,64 tot -0,37).
In de studie van Okubo (2008) was de gemiddelde afname in CNSS (gemiddelde DNSS en NSS) in de placebogroep -0,37 (SD en 95%BI niet gerapporteerd). Voor zowel de 5 mg als de 10 mg groep werd een significant grotere afname ten opzichte van de placebo groep van -0,47 gerapporteerd (SD en 95%BI niet gerapporteerd, p= 0,001). Ciebiada (2006) rapporteerde de DNSS en DES. Na 6 weken was er in de groep behandeld met montelukast een grotere afname in DNSS ten opzichte van baseline naar 3,44 (SD 2,5) ten opzichte van 4,99 (SD 3,4) in de placebogroep (p<0,001). Na 6 weken was er in de groep montelukast een grotere afname in DES ten opzichte van baseline naar 1,13 (SD 1,7) ten opzichte van 1,71 (SD 2,8) in de placebogroep (p<0.001).
Kurowski (2004) rapporteerde na 6 weken geen significante verschillen in de totale symptoms score tussen de groep behandeld met montelukast en de groep behandeld met placebo (zie resultaten uit figuur 10): symptoomscore van 1,0 in de placebogroep; symptoomscore van 1,2 in de groep montelukast.
Kinderen
Chen (2006) rapporteerde TSS gedefinieerd als de gemiddelde daily nasal en non-nasal symptoms over 7 dagen. In de groep behandeld met montelukast was de afname in TSS significant hoger met -0,43 (SD 0,23) ten opzichte van een afname in de placebogroep van -0,11 (SD 0,12) (p<0,001). Hsieh (2004) rapporteerde TSS gedefinieerd als somscore van nasal en non-nasal symptoms. In de groep behandeld met montelukast werd een klinisch relevante afname in symptomen gerapporteerd van 2,7 (baseline 8,9; 12 wk: 6,2) en in de groep behandeld met placebo een afname van 0,2 (baseline 8,5; 12 wk: 8,3, p<0,01).
Kwaliteit van leven (Rhinoconjunctivitis quality of life questionnaire (RQLQ))
Drie studies rapporteerden de RQLQ (Wei, 2016; Lombardo, 2006; Philip, 2005). Het gepoolde effect van de studies van Wei (2016) en Philip (2005) was -0,20 (95%BI -0,25 tot -0,14) in het voordeel van montelukast. Lombardo (2006) rapporteerde significant groter effect in de RQLQ score (7 puntschaal 0=best, 6=worst) in de groep montelukast van 0,6 ten opzichte van 0,4 in de placebo groep (p<0,01).
Figuur 10 RQLQ montelukast vergeleken met placebo. Bron: Wei, 2016 aangevuld met studie Philip, 2005
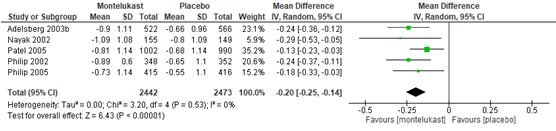
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Kinderen
Chen (2006) rapporteerde een afname in de PRQLQ in de groep behandeld met montelukast van -19,15 (SD 20,71) ten opzichte van een afname van -3,85 (SD 5,56) in de placebogroep (gemiddeld verschil: -15,3 95%BI -24,7 tot -5,7, p=0,028).
Bijwerkingen
Bijwerkingen werden in twee studies gerapporteerd (Okubo, 2008; Philip, 2005). Okubo (2008) rapporteerde geen verschillen in bijwerkingen; 4,1% in de placebogroep en respectievelijk 4,7% en 4,2% in de groep behandeld met montelukast 5mg en 10 mg. Hoofdpijn en sufheid kwamen in ongeveer 1% voor in alle groepen. Philip (2005) rapporteerde bijwerkingen bij respectievelijk 49 (11,8%) en 54 (13,0%) patiënten in de groep behandeld met montelukast en de placebogroep.
Kinderen
Bijwerkingen bij kinderen werden in twee studies gerapporteerd (Bisgaard, 2009, Hsieh, 2004). De studie in Bisgaard (2009) rapporteerde geen verschillen in tussen de groep behandeld met montelukast bijwerkingen (n=73, 26,1%) en de placebogroep (n=35, 26,3%). Hsieh (2004) rapporteerde geen significante verschillen in bijwerkingen tussen de studiearmen (hoofdpijn, sufheid en vermoeidheid). In de arm montelukast bijwerkingen bij één patiënt in de groep (hoofdpijn) en twee patiënten in de placebogroep (hoofdpijn en vermoeidheid).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaten klachten ten gevolge van allergie van de bovenste luchtwegen en kwaliteit van leven is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en werd met twee niveaus verlaagd vanwege beperkingen in de studieopzet (risk of bias) en overschrijden van de grenzen van klinische relevantie (imprecisie) naar laag.
De bewijskracht voor de uitkomstmaat bijwerkingen gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en is met twee niveaus verlaagd gezien het beperkt aantal patiënten (imprecisie) en beperkingen in de onderzoeksopzet (risk of bias) naar laag voor zowel volwassenen als voor kinderen.
De bewijskracht voor de uitkomstmaten klachten ten gevolge van allergie van de bovenste luchtwegen en kwaliteit van leven bij kinderen is gebaseerd op gerandomiseerd onderzoek en start derhalve hoog en werd met twee niveaus verlaagd vanwege het zeer beperkte aantal patiënten (imprecisie) naar laag.
Montelukast versus antihistaminica (PICO 5)
Klachten ten gevolge van allergie van de bovenste luchtwegen
Klachten ten gevolge van allergie van de bovenste luchtwegen (symptomen) werden gerapporteerd door 7 studies (Andhale, 2016; Ciebiada, 2006; Kaur, 2017; Kurowski, 2004; Lombardo, 2006; Rajeshkar, 2018; Wei, 2016), indien beschikbaar is de totaal score/ composite score gerapporteerd.
Wei (2016) rapporteerde een composite symptoms score (CSS). Het gepoolde effect was
-0,03 (95% BI -0,01 tot 0,07) (figuur 11).
Figuur 11 CSS montelukast vergeleken met antihistaminica. Bron: Wei, 2016
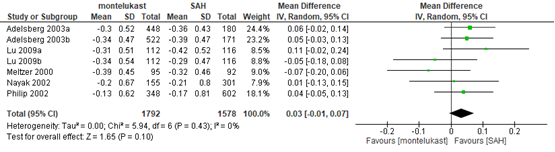
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Rajashekar (2018) rapporteerde een scoringssysteem gebaseerd op het totaal van vijf neussymptomen (T5NSS).
In de groep behandeld met montelukast werd een significante afname in de T5NSS gerapporteerd (baseline 161,72, SD 8,87; 4 weken 145,66, SD 7,7), ook in de groep behandeld met ebastine (baseline 157,89, SD 9,47; 4 weken 142,53, SD 9,5) werd een significante afname gerapporteerd. Na 4 weken was er echter geen significant verschil tussen de behandelgroepen (3,13 95%BI -0,91 tot 7,17).
Kaur (2017) rapporteerde symptoomscores voor niezen en neusuitvloed/rhinorrhoe na 2, 4 en 6 weken behandeling. Na 2 weken werd voor niezen een verschil gerapporteerd in het voordeel van levocetrizine ten opzichte van montelukast en andere SAHs. Na 4 en 6 weken werden er geen significante verschillen gerapporteerd tussen de behandelarmen op niezen en nasal discharge/rhinorrhoe.
Andhale (2016) rapporteerde in zowel de groep behandeld met montelukast als de groep behandeld met levocetirizine voor alle symptomen (zowel overdag en ’s nachts) significante afname na twee weken. De gemiddelde afname op de VAS-schaal (0-10) in de symptomen overdag was 3,2 voor de groep behandeld met montelukast en 2,8 voor de groep behandeld met levocetirizine. De gemiddelde afname op de VAS-schaal in de symptomen ’s nachts was 2,1 in de groep behandeld met montelukast en 2,3 in de groep behandeld met levocetirizine. Er werden geen verschillen in effectiviteit tussen de groepen gerapporteerd.
Ciebiada (2011) rapporteerde congestion symptoms. In de groep behandeld met montelukast werd op dag 42 een afname van 1,5 gerapporteerd en in de groep behandeld met levocetirizine een afname van 1,65 ten opzichte van baseline metingen (gemiddelde verschil 0,15 95%BI 0,02 tot 0,28).
Lombardo (2006) rapporteerde na 4 weken geen verschil in DNSS (0,09 95%BI -0,13 tot 0,31) tussen de groep behandeld met levocetirizine (gemiddeld effect 0,43, SD 0,74) en in de groep montelukast (gemiddeld effect 0,34, SD 0,82). Ook voor NSS (0,1 95%BI -0,10 tot 0,31) en DES (-0,06 95%BI -0,06 tot 0,38) werden geen verschil in effect tussen de groepen gerapporteerd.
Kurowski (2004) vond na 6 weken geen significante verschillen in de totale symptoms score tussen de groep behandeld met montelukast en de groep behandeld met cetirizine (resultaten herleidbaar uit figuur 12), symptoomscore groep montelukast 1,2; symptoomscore cetirizine 1,0)
Kinderen
Chen (2006) rapporteerde in de groep behandeld met montelukast een significant grotere afname in TSS met-0,60 (SD 0,25) in de groep behandeld met cetirizine ten opzichte van een afname in de groep behandeld met montelukast van -0,43 (SD 0,23) (p < 0,05).
Hsieh (2004) rapporteerde na 12 weken een significant grotere afname in TSS in de groep behandeld met cetirizine (afname van 8,8 op baseline naar 3,3 op 12 weken) vergeleken met de groep behandeld met montelukast (afname van 8,9 naar 6,2 op 12 weken) (P<0,001).
Kwaliteit van leven
Wei (2016) rapporteerde de RQLQ; het gepoolde effect was 0,09 (95%BI 0,02 tot 0,15) (figuur 12). Lombardo (2006) rapporteerde een verandering in de RQLQ score (schaal van 0-6) in de groep behandeld met montelukast van 0,64 ten opzichte van 0,78 in de groep behandeld met levocetirizine.
Figuur 12 RQLQ montelukast vergeleken met antihistamine. Bron: Wei, 2016
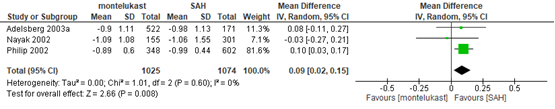
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval
Kinderen
De PRQLQ werd in twee studies gerapporteerd (Hsieh, 2004; Chen, 2006). Chen (2006) rapporteerde een verschil in PRQLQ in de groep behandeld met cetirizine van -31,15 (SD 23,36) en opzichte van -19,15 (SD 20,71) in de groep behandeld met montelukast (gemiddeld verschil: 12,0 95%BI -1,7 tot 25,7). De studie van Hsieh (2004) rapporteerde geen resultaten.
Bijwerkingen
Twee studies in Wei, 2016 (Meltzer 2000; Nayak, 2002) en Lombardo (2006) rapporteerden bijwerkingen van montelukast vergeleken met loratadine. Nayak (2002) rapporteerde in de groep behandeld met montelukast bij 5% bijwerkingen (vermoeidheid, hoofdpijn en anders) en in de groep behandeld met loratadine bij 6% bijwerkingen (vermoeidheid, hoofdpijn en anders). Meltzer (2000) rapporteerde hoofdpijn in de groep behandeld met montelukast bij 5,3% en in de groep behandeld met loratadine bij 8,7%. Lombardo (2006) rapporteerde in beide groepen geen bijwerkingen.
Kinderen
Hsieh (2004) rapporteerde geen significante verschillen in bijwerkingen tussen de studiearmen. In de groep behandeld met cetirizine werden bijwerkingen gerapporteerd bij één patiënt (sufheid) en bij één patiënt in de groep behandeld met montelukast (hoofdpijn).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat klachten ten gevolge van allergie van de bovenste luchtwegen is gebaseerd op gerandomiseerd onderzoek en start hoog en werd verlaagd met een niveau gezien de beperkingen in onderzoeksopzet (risk of bias) naar redelijk.
De bewijskracht voor de uitkomstmaat kwaliteit van leven is gebaseerd op gerandomiseerd onderzoek en start hoog en is met twee niveaus verlaagd naar laag gezien het overschrijden van de grenzen van klinische relevantie.
De bewijskracht voor de uitkomstmaat bijwerkingen is gebaseerd op gerandomiseerd onderzoek en start hoog en is met één niveau verlaagd naar redelijk gezien het beperkt aantal patiënten (imprecisie).
De bewijskracht voor de uitkomstmaten klachten ten gevolge van allergie van de bovenste luchtwegen en kwaliteit van leven bij kinderen is gebaseerd op gerandomiseerd onderzoek en start hoog en is met 2 niveaus verlaagd naar laag gezien het geringe aantal patiënten (imprecisie).
De bewijskracht voor de uitkomstmaat bijwerkingen bij kinderen is gebaseerd op gerandomiseerd onderzoek en start hoog en is met drie niveaus verlaagd naar zeer laag gezien het zeer beperkt aantal patiënten (slechts 1 studie en 2 patiënten met bijwerkingen, imprecisie).
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag (vragen):
Azelastine fluticasone versus nasale corticosteroïden
Wat is de effectiviteit van azelastine/fluticason (Dymista) bij patiënten met een allergie van de bovenste luchtwegen in vergelijking met nasale corticosteroïden (eventueel in combinatie met orale antihistaminica)?
P: kinderen, volwassenen en zwangere vrouwen met allergie van de bovenste luchtwegen;
I: nasale azelastine/fluticason (Dymista);
C: nasale corticosteroïden (evt. In combinatie met orale antihistaminica);
O: klachten ten gevolge van allergie van de bovenste luchtwegen, kwaliteit van leven, bijwerkingen.
Orale antihistaminica versus nasale corticosteroïden
Wat is de effectiviteit van orale antihistaminica bij patiënten met een allergie van de bovenste luchtwegen in vergelijking met nasale corticosteroïden?
P: kinderen, volwassenen en zwangere vrouwen met allergie van de bovenste luchtwegen;
I: orale antihistaminica (H1 antagonisten);
C: nasale corticosteroïden;
O: klachten ten gevolge van allergie van de bovenste luchtwegen, kwaliteit van leven, bijwerkingen.
Orale antihistaminica versus nasale antihistaminica
Wat is de effectiviteit van orale antihistaminica bij patiënten met een allergie van de bovenste luchtwegen in vergelijking met nasale antihistaminica?
P: kinderen, volwassenen en zwangere vrouwen met allergie van de bovenste luchtwegen;
I: orale antihistaminica (H1 antagonisten);
C: nasale antihistaminica;
O: klachten ten gevolge van allergie van de bovenste luchtwegen, kwaliteit van leven, bijwerkingen.
Leukotrieën receptor antagonisten versus placebo
Wat is de effectiviteit van leukotrieën receptor antagonisten bij patiënten met een allergie van de bovenste luchtwegen in vergelijking met placebo?
P: kinderen, volwassenen en zwangere vrouwen met allergie van de bovenste luchtwegen;
I: leukotrieën receptor antagonisten;
C: placebo;
O: klachten ten gevolge van allergie van de bovenste luchtwegen, kwaliteit van leven, bijwerkingen.
Leukotrieën receptor antagonisten versus antihistaminica
Wat is de effectiviteit van leukotrieën receptor antagonisten bij patiënten met een allergie van de bovenste luchtwegen in vergelijking met orale en nasale antihistaminica?
P: kinderen, volwassenen en zwangere vrouwen met allergie van de bovenste luchtwegen;
I: leukotrieën receptor antagonisten;
C: antihistaminica;
O: klachten ten gevolge van allergie van de bovenste luchtwegen, kwaliteit van leven, bijwerkingen.
Relevante uitkomstmaten
De werkgroep achtte klachten ten gevolge van allergie van de bovenste luchtwegen (meestal beschreven door middel van de total symptom score) en kwaliteit van leven voor de besluitvorming cruciale uitkomstmaten; en adverse events en ziekteverzuim voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde niet á priori de genoemde uitkomstmaten, maar hanteerde de in de studies gebruikte definities.
De werkgroep definieerde voor geen van de uitkomstmaten klinische (patiënt) relevante verschillen, maar sloot aan bij de door GRADE aangegeven default grenzen van 0,5 SD voor continue uitkomstmaten, RR < 0,75 of > 1,25) (GRADE recommendation) of Standardized mean difference (SMD=0,2 (klein); SMD=0,5 (matig); SMD=0,8 (groot).
Zoeken en selecteren (Methode)
Azelastine fluticasone versus nasale corticosteroïden (PICO 1)
In de databases Medline (via OVID), Embase (via Embase.com) is op 4 november 2018 met relevante zoektermen gezocht naar relevante literatuur. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 301 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: betrof het de juiste medicatie, originele publicaties en de geselecteerde uitkomstmaten. Provocatiestudies of studies met een medicijn die niet beschikbaar is in Nederland werden geëxcludeerd. Op basis van titel en abstract werden in eerste instantie acht studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens drie studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), en vijf publicaties die vier studies beschrijven definitief geselecteerd.
Orale antihistaminica versus nasale antihistaminica of nasale corticosteroïden (PICO 2 en 3)
Er is een overkoepelende search verricht voor PICO 2 en 3. In de databases Medline (via OVID), Embase (via Embase.com) is op 7 november 2018 met relevante zoektermen gezocht naar relevante literatuur. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 468 treffers op waarvan 376 RCT’s en 92 SR. Studies werden geselecteerd op grond van de volgende selectiecriteria: betrof de juiste medicatie, originele publicaties en de geselecteerde uitkomstmaten. Provocatiestudies of studies met een medicijn die niet beschikbaar is in Nederland werden geëxcludeerd. Op basis van titel en abstract werden in eerste instantie 83 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 56 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), en 27 studies definitief geselecteerd.
Leukotrieën receptor antagonisten versus placebo of antihistaminica (PICO 4 en 5)
Er is een overkoepelende search verricht voor PICO 4 en 5. In de databases Medline (via OVID), Embase (via Embase.com) is op 4 november 2018 met relevante zoektermen gezocht naar relevante literatuur. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 319 treffers op waarvan 256 RCT’s en 63 SR. Studies werden geselecteerd op grond van de volgende selectiecriteria: betrof het de juiste medicatie, originele publicaties en de geselecteerde uitkomstmaten. Provocatiestudies of studies met een medicijn die niet beschikbaar is in Nederland werden geëxcludeerd. Op basis van titel en abstract werden in eerste instantie 97 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 84 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording), en 13 studies definitief geselecteerd.
Resultaten
Azelastine fluticasone versus nasale corticosteroïden (PICO 1)
Vijf publicaties die vier RCT’s beschrijven zijn opgenomen in de literatuuranalyse.
Orale antihistaminica versus nasale corticosteroïden (PICO 2)
Een systematische review met 16 geïncludeerde studies is opgenomen in de literatuuranalyse (Weiner, 1998). Zes van de studies beschreven in deze review voldeden aan de zoekcriteria en zijn geïncludeerd in de literatuursamenvatting, de overige studies zijn buiten beschouwing gelaten. Daarnaast zijn 18 RCT’s opgenomen in de analyse. Als orale antihistaminicum behandeling werden astemizole, loratadine, desloratadine, cetirizine en levocetirizine bestudeerd, als corticosteroïden fluticasone propionate, triamcinolone acetonide, becalmethasone dipropionate, budenoside en ciclesonide. Het merendeel van de studies hanteerde een behandelingsduur van 2 tot 4 weken.
Orale versus nasale antihistaminica (PICO 3)
Zeven studies zijn opgenomen in de literatuuranalyse. De geïncludeerde studies vergeleken orale antihistaminicum behandeling met azelastine nasale spray behandeling bij patiënten met seizoensgebonden allergie van de bovenste luchtwegen.
Leukotrieën receptor antagonisten versus placebo of antihistaminica (PICO 4 en 5)
Dertien studies zijn opgenomen in de literatuuranalyse. De meerderheid van de studies had meerdere behandelarmen met zowel leukotrieën receptor antagonisten, placebo en antihistaminica.
De onderstaande beschrijving van de studies van PICO 2 en 3 zijn daarom gecombineerd, maar de resultaten zijn apart weergegeven.
De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk-of-biastabellen.
- Andhale S, Goel HC, Nayak S. Comparison of Effect of Levocetirizine or Montelukast Alone and in Combination on Symptoms of Allergic Rhinitis. Indian J Chest Dis Allied Sci. 2016 Apr-Jun;58(2):103-5. PubMed PMID: 30182671.
- Andrews CP, Martin BG, Jacobs RL, Mohar DE, Diaz JD, Amar NJ, Kaiser HB, Vandewalker ML, Bernstein J, Toler WT, Prillaman BA, Dalal AA, Lee LA, Philpot EE. Fluticasone furoate nasal spray is more effective than fexofenadine for nighttime symptoms of seasonal allergy. Allergy Asthma Proc. 2009 Mar-Apr;30(2):128-38. doi: 10.2500/aap.2009.30.3204. PubMed PMID: 19463203.
- Berger W, Sher E, Gawchik S, Fineman S. Safety of a novel intranasal formulation of azelastine hydrochloride and fluticasone propionate in children: A randomized clinical trial. Allergy Asthma Proc. 2018; 39(2):110-116. PubMed PMID: 29490769.
- Berger WE, Shah S, Lieberman P, Hadley J, Price D, Munzel U, Bhatia S. Long-term, randomized safety study of MP29-02 (a novel intranasal formulation of azelastine hydrochloride and fluticasone propionate in an advanced delivery system) in subjects with chronic rhinitis. J Allergy Clin Immunol Pract. 2014; 2(2):179-85. PubMed PMID: 24607046.
- Berger W, Hampel F Jr, Bernstein J, Shah S, Sacks H, Meltzer EO. Impact of azelastine nasal spray on symptoms and quality of life compared with cetirizine oral tablets in patients with seasonal allergic rhinitis. Ann Allergy Asthma Immunol. 2006 Sep;97(3):375-81. PubMed PMID: 17042145.
- Berger WE, White MV; Rhinitis Study Group. Efficacy of azelastine nasal spray in patients with an unsatisfactory response to loratadine. Ann Allergy Asthma Immunol. 2003 Aug;91(2):205-11. PubMed PMID: 12952117.
- Bernstein DI, Levy AL, Hampel FC, Baidoo CA, Cook CK, Philpot EE, Rickard KA. Treatment with intranasal fluticasone propionate significantly improves ocular symptoms in patients with seasonal allergic rhinitis. Clin Exp Allergy. 2004 Jun;34(6):952-7. PubMed PMID: 15196285.
- Bhatia S, Baroody FM, deTineo M, Naclerio RM. Increased nasal airflow with budesonide compared with desloratadine during the allergy season. Arch Otolaryngol Head Neck Surg. 2005 Mar;131(3):223-8. PubMed PMID: 15781762.
- Bisgaard H, Skoner D, Boza ML, Tozzi CA, Newcomb K, Reiss TF, Knorr B, Noonan G. Safety and tolerability of montelukast in placebo-controlled pediatric studies and their open-label extensions. Pediatr Pulmonol. 2009 Jun;44(6):568-79. doi:10.1002/ppul.21018. Review. PubMed PMID: 19449366.
- Carr W, Bernstein J, Lieberman P, Meltzer E, Bachert C, Price D, Munzel U, Bousquet J. A novel intranasal therapy of azelastine with fluticasone for the treatment of allergic rhinitis. J Allergy Clin Immunol. 2012; 129(5):1282-1289.e10. PubMed PMID: 22418065.
- Charpin D, Godard P, Garay RP, Baehre M, Herman D, Michel FB. A multicentre clinical study of the efficacy and tolerability of azelastine nasal spray in the treatment of seasonal allergic rhinitis: a comparison with oral cetirizine. Eur Arch Otorhinolaryngol. 1995;252(8):455-8. PubMed PMID: 8719584.
- Charpin D, Vervloet D. Treating seasonal rhinitis: antihistamines or intranasal corticosteroids? European Respiratory Review, 1994, 256-256.
- Chen ST, Lu KH, Sun HL, Chang WT, Lue KH, Chou MC. Randomized placebo-controlled trial comparing montelukast and cetirizine for treating perennial allergic rhinitis in children aged 2-6 yr. Pediatr Allergy Immunol. 2006 Feb;17(1):49-54. PubMed PMID: 16426255.
- Ciebiada M, Górska-Ciebiada M, DuBuske LM, Górski P. Montelukast with desloratadine or levocetirizine for the treatment of persistent allergic rhinitis. Ann Allergy Asthma Immunol. 2006 Nov;97(5):664-71. PubMed PMID:17165277.
- Ciebiada M, Gorska-Ciebiada M, Barylski M, Kmiecik T, Gorski P. Use of montelukast alone or in combination with desloratadine or levocetirizine in patients with persistent allergic rhinitis. Am J Rhinol Allergy. 2011 Jan-Feb;25(1):e1-6. doi: 10.2500/ajra.2011.25.3540. PubMed PMID: 21711959.
- Conde Hernández DJ, Palma Aqilar JL, Delgado Romero J. Comparison of azelastine nasal spray and oral ebastine in treating seasonal allergic rhinitis. Curr Med Res Opin. 1995;13(6):299-304. PubMed PMID: 8829888.
- Condemi J, Schulz R, Lim J. Triamcinolone acetonide aqueous nasal spray versus loratadine in seasonal allergic rhinitis: efficacy and quality of life. Ann Allergy Asthma Immunol. 2000 May;84(5):533-8. PubMed PMID: 10831008.
- Corren J, Storms W, Bernstein J, Berger W, Nayak A, Sacks H; Azelastine Cetirizine Trial No. 1 (ACT 1) Study Group. Effectiveness of azelastine nasal spray compared with oral cetirizine in patients with seasonal allergic rhinitis. Clin Ther. 2005 May;27(5):543-53. PubMed PMID: 15978303.
- D'Ambrosio FP, Gangemi S, Merendino RA, Arena A, Ricciardi L, Bagnato GF. Comparative study between fluticasone propionate and cetirizine in the treatment of allergic rhinitis. Allergol Immunopathol (Madr). 1998 Nov-Dec;26(6):277-82.Erratum in: Allergol Immunopathol (Madr) 1999 May-Jun;27(3):173. PubMed PMID:9934406.
- Ellis AK, Zhu Y, Steacy LM, Walker T, Day JH. A four-way, double-blind, randomized, placebo controlled study to determine the efficacy and speed of azelastine nasal spray, versus loratadine, and cetirizine in adult subjects with allergen-induced seasonal allergic rhinitis. Allergy Asthma Clin Immunol. 2013 May 1;9(1):16. doi: 10.1186/1710-1492-9-16. eCollection 2013. PubMed PMID:23635091; PubMed Central PMCID: PMC3655060.
- Fokkens WJ, Scadding GK. Perennial rhinitis in the under 4s: a difficult problem to treat safely and effectively? A comparison of intranasal fluticasone propionate and ketotifen in the treatment of 2-4-year-old children with perennial rhinitis. Pediatr Allergy Immunol. 2004 Jun;15(3):261-6. PubMed PMID: 15209960.
- Ford LB, Matz J, Hankinson T, Prillaman B, Georges G. A comparison of fluticasone propionate nasal spray and cetirizine in ragweed fall seasonal allergic rhinitis. Allergy Asthma Proc. 2015 Jul-Aug;36(4):313-9. doi:10.2500/aap.2015.36.3860. PubMed PMID: 26108088.
- Frølund L. Efficacy of an oral antihistamine, loratadine, as compared with a nasal steroid spray, beclomethasone dipropionate, in seasonal allergic rhinitis. Clin Otolaryngol Allied Sci. 1991 Dec;16(6):527-31. PubMed PMID: 1685945.
- Gambardella R. A comparison of the efficacy of azelastine nasal spray and loratidine tablets in the treatment of seasonal allergic rhinitis. J Int Med Res.1993 Sep-Oct;21(5):268-75. PubMed PMID: 7906659.
- Gani F, Lombardi C, Barrocu L, Landi M, Ridolo E, Bugiani M, Rolla G, Senna G,Passalacqua G. The control of allergic rhinitis in real life: a multicentre cross-sectional Italian study. Clin Mol Allergy. 2018 Feb 2;16:4. doi:10.1186/s12948-018-0082-y. eCollection 2018. PubMed PMID: 29434524; PubMed Central PMCID: PMC5797368.
- Gawchik SM, Lim J. Comparison of intranasal triamcinolone acetonide with oral loratadine in the treatment of seasonal ragweed-induced allergic rhinitis. Am J Manag Care. 1997 Jul;3(7):1052-8. PubMed PMID: 10173369.
- Hampel FC, Ratner PH, Van Bavel J, Amar NJ, Daftary P, Wheeler W, Sacks H. Double-blind, placebo-controlled study of azelastine and fluticasone in a single nasal spray delivery device. Ann Allergy Asthma Immunol. 2010 Aug;105(2):168-73. PubMed PMID: 20674829.
- Hsieh JC, Lue KH, Lai DS, Sun HL, Lin YH. A comparison of cetirizine and montelukast for treating childhood perennial allergic rhinitis. Pediatric Asthma, Allergy & Immunology, 2004; 17(1), 59-69.
- Jordana G, Dolovich J, Briscoe MP, Day JH, Drouin MA, Gold M, Robson R, Stepner N, Yang W. Intranasal fluticasone propionate versus loratadine in the treatment of adolescent patients with seasonal allergic rhinitis. J Allergy Clin Immunol. 1996 Feb;97(2):588-95. PubMed PMID: 8621843.
- Kaszuba SM, Baroody FM, deTineo M, Haney L, Blair C, Naclerio RM. Superiority of an intranasal corticosteroïd compared with an oral antihistamine in the as-needed treatment of seasonal allergic rhinitis. Arch Intern Med. 2001 Nov 26;161(21):2581-7. PubMed PMID: 11718589.
- Kaur, Gurpreet, Rachna Dhingra, and Manjinder Singh. "RESEARCH ARTICLE Assessment of Clinical Effectiveness of Various Drugs in the Patients of Allergic Rhinitis visiting Tertiary Care Hospital of Punjab, India."
- Keith PK, Desrosiers M, Laister T, Schellenberg RR, Waserman S. The burden of allergic rhinitis (AR) in Canada: perspectives of physicians and patients.Allergy Asthma Clin Immunol. 2012 Jun 1;8(1):7. doi: 10.1186/1710-1492-8-7. PubMed PMID: 22656186; PubMed Central PMCID: PMC3490734.
- Kim CH, Kim JK, Kim HJ, Cho JH, Kim JS, Kim YD, Lee HM, Kim SW, Cho KS, Lee SH, Rhee CS, Dhong HJ, Rha KS, Yoon JH. Comparison of intranasal ciclesonide,oral levocetirizine, and combination treatment for allergic rhinitis. Allergy Asthma Immunol Res. 2015 Mar;7(2):158-66. doi: 10.4168/aair.2015.7.2.158. Epub 2014 Dec 18. PubMed PMID: 25729623; PubMed Central PMCID: PMC4341337.
- Kulapaditharom B, Pornprasertsuk K, Boonkitticharoen V. Clinical assessment of levocetirizine and budesonide in treatment of persistent allergic rhinitis regarding to symptom severity. J Med Assoc Thai. 2010 Feb;93(2):215-23. PubMed PMID: 20302004.
- Kurowski M, Kuna P, Górski P. Montelukast plus cetirizine in the prophylactic treatment of seasonal allergic rhinitis: influence on clinical symptoms and nasal allergic inflammation. Allergy. 2004 Mar;59(3):280-8. PubMed PMID: 14982509.
- Lombardo G, Quattrocchi P, Lombardo GR, Galati P, Giannetto L, Barresi L. Concomitant levocetirizine and montelukast in the treatment of seasonal allergic rhinitis: Influence on clinical symptoms. Italian Journal of Allergy and Clinical Immunology 2006;16:63-68
- Malizia V, Fasola S, Ferrante G, Cilluffo G, Gagliardo R, Landi M, Montalbano L, Marchese D, La Grutta S. Comparative Effect of Beclomethasone Dipropionate and Cetirizine on Acoustic Rhinometry Parameters in Children With Perennial Allergic Rhinitis: A Randomized Controlled Trial. J Investig Allergol Clin Immunol. 2018 Dec;28(6):392-400. doi: 10.18176/jiaci.0263. Epub 2018 Apr 24. PubMed PMID:29688172.
- Meltzer EO, Philip G, Weinstein SF, LaForce CF, Malice MP, Dass SB, Santanello NC, Reiss TF. Montelukast effectively treats the nighttime impact of seasonal allergic rhinitis. Am J Rhinol. 2005 Nov-Dec;19(6):591-8. PubMed PMID: 16402647.
- Okubo K, Baba K. Therapeutic effect of montelukast, a cysteinyl leukotriene receptor 1 antagonist, on Japanese patients with seasonal allergic rhinitis. Allergol Int. 2008 Sep;57(3):247-55. doi: 10.2332/allergolint.O-07-515. Epub 2008 Jul 1. PubMed PMID: 18566548.
- Patel P, Philip G, Yang W, Call R, Horak F, LaForce C, Gilles L, Garrett GC, Dass SB, Knorr BA, Reiss TF. Randomized, double-blind, placebo-controlled study of montelukast for treating perennial allergic rhinitis. Ann Allergy Asthma Immunol. 2005 Dec;95(6):551-7. PubMed PMID: 16400895.
- Philip G, Nayak AS, Berger WE, Leynadier F, Vrijens F, Dass SB, Reiss TF. The effect of montelukast on rhinitis symptoms in patients with asthma and seasonal allergic rhinitis. Allergologie 2005; 28(9):343-354
- Price D, Shah S, Bhatia S, Bachert C, Berger W, Bousquet J, Carr W, Hellings P, Munzel U, Scadding G, Lieberman P. A new therapy (MP29-02) is effective for the long-term treatment of chronic rhinitis. J Investig Allergol Clin Immunol. 2013;23(7):495-503. PubMed PMID: 24654314.
- Rajashekar, Y. R., and S. N. Shobha. "Randomized prospective double-blind comparative clinical study of ebastine and its combined preparation of montelukast in persistent allergic rhinitis." National Journal of Physiology, Pharmacy and Pharmacology 2018: 8.3: 319-324.
- Ratner PH, van Bavel JH, Martin BG, Hampel FC Jr, Howland WC 3rd, Rogenes PR, Westlund RE, Bowers BW, Cook CK. A comparison of the efficacy of fluticasone propionate aqueous nasal spray and loratadine, alone and in combination, for the treatment of seasonal allergic rhinitis. J Fam Pract. 1998 Aug;47(2):118-25.PubMed PMID: 9722799.
- Rinne J, Simola M, Malmberg H, Haahtela T. Early treatment of perennial rhinitis with budesonide or cetirizine and its effect on long-term outcome. J Allergy Clin Immunol. 2002 Mar;109(3):426-32. PubMed PMID: 11897986.
- Sastre J, Mosges R. Local and systemic safety of intranasal corticosteroids. J Investig Allergol Clin Immunol. 2012;22(1):1-12. Review. PubMed PMID: 22448448.
- Spangler DL, Abelson MB, Ober A, Gotnes PJ. Randomized, double-masked comparison of olopatadine ophthalmic solution, mometasone furoate monohydrate nasal spray, and fexofenadine hydrochloride tablets using the conjunctival and nasal allergen challenge models. Clin Ther. 2003 Aug;25(8):2245-67. PubMed PMID: 14512132.
- Wartna JB, Bohnen AM, Elshout G, Pijnenburg MW, Pols DH, Gerth van Wijk RR, Bindels PJ. Symptomatic treatment of pollen-related allergic rhinoconjunctivitis in children: randomized controlled trial. Allergy. 2017 Apr;72(4):636-644. doi:10.1111/all.13056. Epub 2016 Oct 28. PubMed PMID: 27696447.
- Wei C. The efficacy and safety of H1-antihistamine versus Montelukast for allergic rhinitis: A systematic review and meta-analysis. Biomed Pharmacother.2016 Oct;83:989-997. doi: 10.1016/j.biopha.2016.08.003. Epub 2016 Aug 11. Review.PubMed PMID: 27522261.
- Weiner JM, Abramson MJ, Puy RM. Intranasal corticosteroids versus oral H1 receptor antagonists in allergic rhinitis: systematic review of randomised controlled trials. BMJ. 1998 Dec 12;317(7173):1624-9. PubMed PMID: 9848901; PubMed Central PMCID: PMC28740.
PICO 1 Azelastine fluticasone versus nasale corticosteroïden
Evidence table for intervention studies
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3
|
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Carrr, 2012
|
SR and meta-analysis of RCTs (MP4002 (NCT00651118), MP4004 (NCT00740792), and MP4006 (NCT00883168))
Literature search up to (month/year)
A: Hampel, 2010 B: Meltzer, 2012 C: Meltzer, 2013
Study design: RCTs parallel
Setting and Country: Multicentre, outpatient study in USA, Germany, Belgium and UK
Source of funding and conflicts of interest: funded by Meda Pharmaceuticals, Inc |
Inclusion criteria SR: Subjects (>12 years old) with a minimum 2-year history of SAR, significant current clinical rhinitis symptomatology, and a positive skin prick test response to relevant pollen were randomized. All subjects had moderate-to-severe SARdefined by a reflective total nasal symptom score (rTNSS) of at least 8 of 12, with a congestion score of 2 or 3 during screening. Inclusion criteria for the duration of symptoms for the 3 studies were slightly different.
Exclusion criteria SR: erosion, ulceration, or septal perforation or another disease (eg, sinusitis, rhinitis medicamentosa, polyposis, respiratory tract infection (within 14 days of screening), asthma (except intermittent asthma)), significant pulmonary disease, or symptomatic cardiac conditions or were taking concomitant medication that could interfere with the interpretation of study results
Important patient characteristics at baseline: N total at baseline: Intervention: 207+193+448=848 Control: 208+194+445=847
Sex: I: 36% M C: 36% M
Age ± SD: I: 36.7 (14.3) C: 35.9 (14.1)
Groups comparable at baseline? yes |
Describe intervention:
MP29-02 nasal spray (novel formulation of 137 mg of azelastine/50 mg of FP
|
Describe control:
corticosteroid (FP) Placebo spray comprised exactly the same vehicle/ formulation as the active treatments without any active agent
|
End-point of follow-up:
A: 14 days B: 14 days C: 14 days
For how many participants were no complete outcome data available? Loss-to follow-up: (intervention/control) A: Intervention Control B: Intervention Control C: Intervention Control
|
Outcome measures and effect size (include 95%CI and p-value if available):
Total symptom score (reduction from baseline): Baseline Change A I: 18.3 (3.0) -5.5 (5.2) n=207 C: 18.2 (3.2) -5.0 (4.7) n=207
B I: 18.2 (3.3) -5.6 (5.2) N=193 C: 18.2 (3.1) -5.0 (5.2) N=189
C I: 19.4 (2.4) -5.6 (5.2) N=448 C: 19.5 (2.4) -5.1 (4.7) N=450
Pooled I: -5.7 (SD 5.3) C: -4.4 (SD 4.8)
Quality of life: A: I: C:
B: I: C:
C: I: C:
Baseline I: -1.6 (SD 1.3) C: -1.4 (SD 1.3)
A I: 4 (2 %) C: 1 (0.5 %) B I: 3 (1.5 %) C: 1 (0.5 %) C I: 3(0.7 %) C: 4 (0.9 %)
Sick leave: Not reported
|
MP29-02 nasal spray (novel formulation of 137 mg of azelastine/50 mg of FP
MP 4002 MP 4004 beschreven in Meltzer, 2012 MP 4006
Facultative:
Brief description of author’s conclusion
Personal remarks on study quality, conclusions, and other issues (potentially) relevant to the research question
Level of evidence: GRADE (per comparison and outcome measure) including reasons for down/upgrading
Sensitivity analyses (excluding small studies; excluding studies with short follow-up; excluding low quality studies; relevant subgroup-analyses); mention only analyses which are of potential importance to the research question
Heterogeneity: clinical and statistical heterogeneity; explained versus unexplained (subgroup analysis) |
|
Berger, 2014 EudraCT no. 2011-001368-23
Price, 2013 |
Type of study: open-label RCT
Setting and country: India
Funding and conflicts of interest: W. E. Berger has received honoraria, consulted for, and received research support from MEDA and Alcon; has consulted for and received speaker fees from Sunovian and TEVA; has received speaker fees from Astra- Zeneca; and has received speaker fees and served as an adviser for Allergan. S. Shah has consulted and received research grants from MEDA and given lectures for TEVA, Alcon, and AstraZeneca. P. Lieberman is an adviser for the Allergy Foundation of America and Baxter and has given lectures for MEDA, Genentech, Ista, and TEVA. J. Hadley has consulted and served as an adviser for MEDA, Merck, and TEVA. D. Price has received consultancy and speaker fees from Merck, Mundipharma, Novartis, Medapharma, Kyorin, and TEVA; has received consultancy fees from GlaxoSmithKline, Almirall, and Chiese; has received consultancy fees and grants from Pfizer and AstraZeneca; has received consultancy fees and grants from Boehringer Ingelheim; has received speaker fees and grants from Aerocrine; has received grants from the UK National Health Service, Nycomed, and Medapharma; is director of research in Real Life LTD; is a guideline group member for Allergic Rhinitis and its Impact on Asthma and European Position Paper on Rhinosinusitis and Nasal Polyps; and has shares in AKL Ltd. U. Munzel is an employee of Medapharma. S. Bhatia has received research grants and consulted for MED |
Inclusion criteria: Male and female subjects 12 to 80 years of age with an established history (>=1 year) of chronic rhinitis due to perennial allergies or nonallergic triggers were eligible for the study. Subjects with a seasonal allergic component were included, provided that they had significant symptoms outside the allergy seasons. The diagnosis of rhinitis, whether allergic or nonallergic, was made on the basis of a thorough evaluation. This evaluation included medical history, physical examination, rhinitis symptoms, and skin testing, or validated in vitro tests for allergen-specific Ig E. All the subjects were symptomatic at study entry, with nasal symptoms of rhinitis for at least 2 days during the screening period. Subjects who received subcutaneous immunotherapy (antigen desensitization) were on a stable maintenance regimen for at least 30 days before the first study visit.
Exclusion criteria: If on nasal examination, they had any nasal ulceration (grade 3) or nasal septal perforation (grade 4); recent nasal or sinus surgery; chronic sinusitis; recent acute sinusitis; nasal disease(s), such as rhinitis medicamentosa, clinically significant nasal polyposis, or nasal structural abnormalities. Women who were pregnant or nursing and women of childbearing potential who were not using an acceptable method of contraception were not permitted to enroll. Subjects also were excluded if they had a history of asthma (with the exception of mild intermittent asthma), significant pulmonary disease, posterior subcapsular cataracts or glaucoma, clinically significant arrhythmia or prolonged corrected QT interval, a history of drug or alcohol abuse within the past 2 years, or a hypersensitivity to AZ or FP. Recent use of any investigational drug, systemic corticosteroids or omalizumab, inhaled corticosteroids or combination inhaled corticosteroids and/or long-acting b-agonists (within 30 days), or sublingual immunotherapy (within 6 months) was not permitted.
N total at baseline: Intervention: 388 Control: 199
Important prognostic factors2: Age ± SD: I: 32.8 ± 11.5 C: 35.3 ± 11.5
Sex: I: 59% M C: 52% M
Symptom score: I: 3.81 ± 2.49 C: 3.90 ± 2.32
Groups comparable at baseline? yes |
Describe intervention (treatment/procedure/test):
MP29-02 a Novel Intranasal Formulation of Azelastine Hydrochloride and Fluticasone Propionate in an Advanced Delivery System
administered at a dosage of 1 spray per nostril twice daily (AM and PM); each metered spray delivered 137 mcg of AZ and 50 mcg of FP per actuation, for a total daily dose of 548 mcg and 200 mcg for AZ and FP, respectively.
|
Describe control (treatment/procedure/test):
Fluticasone Propionate FP nasal spray (commercially available generic fluticasone; Boehringer Ingelheim/Roxane Laboratories, Columbus, Ohio) was administered at a dosage of 2 sprays per nostril once daily (AM), each metered spray 50 mcg, for a total daily dose of 200 mcg. |
Length of follow-up: Baseline evaluations were performed, with additional outpatient evaluations at months 1, 3, 6, 9, and 12.
Loss-to-follow-up: Intervention: N (%) 88 (23%) Reasons (describe): Adverse Event n = 11 Abnormal Test Result n = 2 Treatment Failure n = 0 Noncompliance n = 5 Withdrew Consent n = 12 Lost to follow-up n = 38 Administrative problems n = 15 Protocol Violation n = 3 Other n = 2
Control: N (%) 51 (26%) Reasons (describe): Adverse Event n = 6 Abnormal Test Result n = 0 Treatment Failure n = 2 Noncompliance n = 3 Withdrew Consent n = 8 Lost to follow-up n = 20 Administrative problems n = 6 Protocol Violation n = 3 Other n = 3
|
Outcome measures and effect size (include 95%CI and p-value if available):
Total symptom score (rTNSS) (after 52 weeks) I: -2.98 C: -2.71 Diff: -0.27; 95% CI; -0.56, 0.02; P=.0642
Quality of life: Not reported
Quality of life: Not reported
I: 9.4% C: 11.1% The most common TRAEs were dysgeusia (2.5%) in the MP29-02 group and headache (4.3%) in the FP group
Sick leave: Not reported
|
Two publications Price 2013 symptoms; adverse events Berger 2014
Study conducted at 37 investigational sites in India during 2008 and 2009 |
|
Hampel, 2010 NCT00660517 |
Type of study: randomized, double-blind, placebo-controlled, parallelgroup study conducted at 6 investigational sites
Setting and country: Texas
Funding and conflicts of interest: Funding for this study was provided by MedPointe Pharmaceuticals, Somerset, New Jersey. Dr Wheeler is the director of medical communications at Meda Pharmaceuticals. Dr Sacks is the chief medical officer of Meda Pharmaceuticals. |
Inclusion criteria: a minimum 2-year history of allergy to Texas mountain cedar pollen (J ashei), as confirmed by a positive prick-puncture skin test result to mountain cedar pollen within the past 12 months
At screening, eligible patients had to have a 12-hour reflective total nasal symptom score (TNSS) of at least 8 of a possible 12 and a congestion score of 2 or 3; Patients were also required to be in general ood health and free of any disease or concomitant treatment that could interfere with the interpretation of the study results as determined by the study investigator.
Exclusion criteria: (1) the presence of any nasal mucosal erosion, nasal ulceration, or nasal septal perforation (grade 1b-4) at either screening or randomization; (2) other nasal disease(s) likely to affect deposition of intranasal medication, such as sinusitis, rhinitis medicamentosa, clinically significant polyposis, or nasal structural abnormalities; (3) nasal surgery or sinus surgery within the previous year; or (4) more than 3 episodes per year of chronic sinusitis
N total at baseline: Intervention: 153 Control: 151
Important prognostic factors2: age ± SD: I: 39.5 (12-73) C:38.1 (12-74)
Sex: I: 36,6% M C: 33,8% M
Groups comparable at baseline? yes
|
Describe intervention (treatment/procedure/test):
Azelastine/fluticasone
Combination azelastine 0.1% and fluticasone, 1 spray per nostril twice daily (each metered spray of azelastine-fluticasone delivers 137microgram of azelastine hydrochloride and 50 [1]g of fluticasone propionate; therefore, a dosage of 1 spray per nostril twice daily delivers 548 microgram of azelastine hydrochloride and 200 microgram of fluticasone propionate)
|
Describe control (treatment/procedure/test):
1 spray per nostril twice daily (each metered spray of Flonase delivers 50 microgram of fluticasone propionate; therefore, a dosage of 1 spray per nostril twice daily delivers 200 [1]g of fluticasone); |
Length of follow-up: 14 days
Loss-to-follow-up: 33 (5%) discontinued participation in the study. Completion rates were similar in all treatment groups. One patient in the combination group, 3 in the azelastine group, 1 in the fluticasone group, and 1 in the placebo group discontinued participation in the study because of an adverse event. The most common reason for study withdrawal was noncompliance, followed by “other,” withdrawal of consent, and adverse events.
|
Outcome measures and effect size (include 95%CI and p-value if available):
Total symptom score TNSS baseline I: 18,8 (9-24) (SD 61.3) C: 18,3 (8-24) (SD 28.6)
TNSS end I: -5.31 (5.08) C:-3.84 (4.76)
Quality of life (RQLQ): I: 1.60 C: 1.43
I: Dysgeusia (bitter taste) 11 (7.2) Epistaxis 6 (3.9) Headache 4 (2.6) Pharyngolaryngeal pain 2 (1.3) Nasal discomfort 2 (1.3) Nausea 1 (0.7) Mucosal erosion 1 (0.7) Somnolence 1 (0.7)
C:
Dysgeusia (bitter taste) 3 (2.0) Epistaxis 4 (2.6) Headache 2 (1.3) Pharyngolaryngeal pain 1 (0.7) Nasal discomfort 0 (0.0) Nausea 2 (1.3) Mucosal erosion 1 (0.7) Somnolence 0 (0%)
Sick Leave: Not reported
|
|
|
Berger, 2018
NCT01794741 |
Type of study: parallel-group, open-label design
Setting and country: outpatient study, California USA
Funding and conflicts of interest: Research and medical writing support was funded by Mylan, Inc. (Canonsburg, PA) E. Sher is a national speaker for Mylan, Inc. and ALK Pharmaceuticals. S. Gawchik has served on the Advisory Board for Meda. The remaining authors have no conflicts of interest pertaining to this article |
Inclusion criteria: Male and female children 4–11 years old with a history of AR (SAR or PAR) who, in the opinion of the study center investigators, could benefit from treatment with MP-AzeFlu were eligible for inclusion. Children were required to be in generally good health, without concomitant disease or medical treatment that could interfere with interpretation of study results
Exclusion criteria: se of concomitant medication was discouraged; however, medications considered necessary and that did not interfere with the evaluation of study medications were allowed at investigator discretion. (See the Online Supplement Text for a list of prohibited medications.)
N total at baseline: Intervention: 304 Control: 100
Important prognostic factors2: age ± SD: I: 8.1 ± 2.1 C: 8.1 ± 1.9
Sex: I: 60% M C: 51% M
Groups comparable at baseline? yes
|
Describe intervention (treatment/procedure/test):
MP-AzeFlu (137microgram AZE and 50 microgram FP per spray)
Treatments were administered as one spray per nostril twice daily, morning and evening, which provided total daily doses of 548 microgram for MP-AzeFlu and 200microgram for FP |
Describe control (treatment/procedure/test):
FP nasal spray (50 g per spray).
Treatments were administered as one spray per nostril twice daily, morning and evening, which provided total daily doses of 548microgram for MP-AzeFlu and 200 microgram for FP |
Length of follow-up: 3 months
Loss-to-follow-up: Intervention: N (%) 19 (6%):
Reasons (describe) adverse events (6), treatment failure (1), protocol violation (1), noncompliance (2), withdrawal by subject (4), lost to follow-up (3), other (2)
Control: 9 (9%) N (%) Reasons (describe): adverse events (4), protocol violation (1), withdrawal by subject (1), lost to follow-up (2) other (1)
Incomplete outcome data: Not reported
|
Outcome measures and effect size (include 95%CI and p-value if available):
Total symptom score Not reported
Quality of life: Not reported
Treatment emergent adverse events I: 124 (41%) C: 37 (37)
Treatment related adverse events: I: 49 (16%) C: 12 (12%)
Sick leave: Not reported
|
children |
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation |
Bias due to inadequate concealment of allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?
(unlikely/likely/unclear) |
Bias due to loss to follow-up?
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?
(unlikely/likely/unclear) |
|
Berger, 2014 |
An interactive voice response system was used to sequentially assign the next available randomization number from a central randomization schedule. |
unlikely |
not blinded, unclear |
not blinded, unclear |
not blinded, unclear |
unclear |
unclear |
unlikely |
|
Berger, 2018 |
centralized, age-group stratified, block randomization scheme using Proc Plan (SAS, Cary, NC) was used to assign subjects to treatment (3:1 ratio). An interactive Web response system ensured proper stratification |
unlikely |
not blinded, unclear |
not blinded, unclear |
not blinded, unclear |
unclear |
unclear |
unlikely |
|
Carr, 2012 |
Patients were randomized and balanced by study site in blocks of 4. Eligible subjects received the study site’s next available randomization number in sequence |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
|
Hampel, 2010 |
randomized by a computer-generated randomization schedule |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
PICO 2 Orale antihistaminica versus nasale corticosteroïden
Evidence table for systematic review of RCTs and observational studies (intervention studies)
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Weiner, 1998 |
SR and meta-analysis of (RCTs / cohort / case-control studies)
Literature search up to 1997
A: Bunnag, 1992 B: Schoenwetter, 1995 C: Bernstein, 1995 D: Brooks, 1996 E: Gehanno, 1997 F: Vervloet, 1997
Study design: RCTs
Setting and Country: Department of Medicine, Monash University, Melbourne, Victoria, 3181, Australia
Source of funding and conflicts of interest: Funding: Astra Pharmaceuticals (Australia) provided library and research assistance and support for presentation at a scientific meeting. Astra did not provide any other direct financial support. Conflict of interest: Each of the authors is involved in clinical practice and prescribes both intranasal corticosteroids and oral antihistamines for patients. While the support of Astra Pharmaceuticals (manufacturers of budesonide brand nasal spray) is acknowledged, the authors produced this review independently and it was not subject to any editorial review or changes by Astra. The authors believe that no conflict of interest arose during the production of this paper. |
Inclusion criteria SR: randomised controlled trials of topical corticosteroids and rhinitis published between 1966 and 1997. At least one of the following clinical outcomes had to be reported: nasal symptoms (including total nasal symptom scores), eye symptoms, global symptoms, drug requirements for treating the rhinitis, nasal function (including measurements of nasal resistance), and assessment of quality of life.
Exclusion criteria SR: studies that reported only nasal challenge with specific allergens or nonclinical outcomes, such as in vitro results of inflammatory mediators.
16 studies included
Important patient characteristics at baseline:
N, mean age A: 69, 30yrs B: 298, 40 C: 239, 36 D: 60, not reported E: 114, 39 F: 238, 29
|
Describe intervention:
A: Astemizole 10 mg B: Loratadine 10 mg C: Astemizole 10 mg D: Loratadine 10 mg E: Loratadine 10 mg F: Cetirizine 10 mg
|
Describe control:
A: Budesonide, 200 µg B: Triamcinolone 220 µg C: Triamcinolone 220 µg D: Beclamethasone 336 µg E: Fluticasone 200 µg F: Fluticasone 200 µg
|
End-point of follow-up:
Not specified in SR
For how many participants were no complete outcome data available? (intervention/control)
Not specified in SR
|
Outcome measure-1 Total nasal symptom score
SMD (95% CI): B: -0.606 (-0.848 to -0.364) C: -0.427 (-0.701 to -0.152) E: -0.677 (-1.055 to -0.299) F: -0.062 (-0.317 to 0.193)
Pooled effect (fixed effects model): -0.423 (-0.531 to -0.315) favoring steroid treatment
χ2=26.82, df=8, P<0.001
Outcome measure-2 Ocular symptom score
SMD (95% CI): A: -0.216 (-0.696 to 0.265) B: -0.149 (-0.386 to 0.088) C: 0.000 (-0.271 to 0.271) D: -0.382 (-1.008 to 0.244)
Pooled effect (fixed effects model): -0.043 (-0.157 to 0.072) favoring steroid treatment
χ2=32.4, df=10, P<0.0005
|
|
Table of quality assessment for systematic reviews of RCTs and observational studies
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Weiner, 1998 |
Yes |
Yes |
Yes |
Yes |
Not applicable, only RCTs |
Yes |
Yes |
No |
Yes |
Evidence table for intervention studies
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Type of study: RCT, randomized, double-blind, double-dummy comparative study
Setting and country: Single-center study in Copenhagen, Denmark.
Funding and conflicts of interest: Not reported. |
Inclusion criteria: Unequivocal history of seasonal allergic rhinitis for the previous two seasons. The diagnosis was confirmed by a positive skin prick test.
Exclusion criteria: Any concurrent disease that would interfere with the study results or require treatment or if pregnant or lactating, nasal polyps, deviated septa or any structural defect which might cause nasal obstruction or interfere with clinical evaluation, pre-seasonal or co-seasonal immunotherapy with antigen extracts started within the 12 months prior to the study or any maintenance doses of these preparations during the 12 months before entering the study; therapy with loratadine within 3 months, systemic or topical corticosteroids, sodium cromoglycate within 2 weeks prior to the study, decongestants within 24 h, astemizole within 4 weeks, and antihistamines other than astemizole 4 days prior to the study.
N total at baseline: 60 Intervention: 30 Control: 30
18 to 65 years, 61.7% male. No difference was seen between random allocation of patients or treatments. |
Loratadine 10 mg once daily for 3 weeks |
beclomethasone dipropionate (BDP) nasal spray 100 µg into each nostril twice daily for 3 weeks |
Length of follow-up: Duration of the treatment
Loss-to-follow-up: 6 patients in the LOR group and 3 patients in the BDP group discontinued because of treatment failure. 6 patients in the LOR group discontinued due to other reasons not related to treatment failure.
Incomplete outcome data: N.A. |
Both treatments reduced total symptom score compared to baseline, with no differences between the groups. This was true for both the medical visits and self-reported scores. On day 5, self-reported nasal symptoms score more improved in the BDP group (change from baseline -3.97) compared with LOR (-2.29). On the other hand, during the first 6 days, self-reported eye symptoms score was significantly lower in the LOR group. Ocular score was similar after both treatment (-1.73 with BDP, -1.8 with LOR).
One patient in the LOR group compared with 4 patients in the BDP group had to have a prescription for rescue medication (antihistamine- vasoconstrictor eye drops), whereas 1 patient in the BDP group used α-adrenic nasal spray. The rescue medication was not used continuously at any time during the study. |
|
|
|
Charpin, 1994 |
Type of study: RCT, randomized, double-blind, double-dummy comparative trial.
Setting and country: Multicenter study performed in 13 centers in France.
Funding and conflicts of interest: Not reported. |
Inclusion criteria: Patients aged 12 years and older with a moderate to severe seasonal allergic rhinitis diagnosed by a positive (2+) skin test reaction to grass pollen.
Exclusion criteria: Not described.
N total at baseline: 237 Intervention: 118 Control: 119
Patient characteristics were not provided. Groups were comparable at baseline.
|
1. Cetirizine tablet, 10 mg once daily for 3 weeks.
(2. Loratadine tablet, 10 mg once daily for 4 weeks.
3. Terfenadine tablet, 60 mg twice daily for 2 weeks.
4. Astemizole tablet, 10 mg once daily for 2 weeks.) |
Fluticasone propionate (FP) aqueous nasal spray, 200 µg once daily. |
Length of follow-up: Duration of the treatment
Loss-to-follow-up: None
Incomplete outcome data: N.A. |
FP-treated patients showed a significantly greater decrease in mean total symptom score at the end of the treatment (p=0.001) and a higher number of symptom-free days for each nasal symptom (P<0.001) compared to the CTZ-treated group. No values were provided. % symptom-free days per symptom: Nasal obstruction night: FP 57% CTZ 30% Nasal obstruction day: FP 53% CTZ 31% Sneezing: FP 46% CTZ 32% Nasal itching: FP 58% CTZ 42% Nasal discharge: FP 50% CTZ 33%. (P<0.001 for all symptoms) Fewer events were reported in the FP group. In particular, no drowsiness was reported with FP treatment. No values reported. |
|
|
Jordana, 1996 |
Type of study: RCT, double-blind, randomized, parallel- group study.
Setting and country: Multicenter study performed in 5 allergy clinics in southern Ontario, Canada.
Funding and conflicts of interest: Supported by Glaxo Canada. Conflict of interest not declared. |
Inclusion criteria: History of moderate to severe ragweed-induced seasonal allergic rhinitis.
Exclusion criteria: Concurrent perennial rhinitis, use of long-acting histamine antagonists within the past 6 weeks; inhaled, intranasal, or systemic corticosteroids or inhaled sodium cromoglycate within the past 4 weeks; or loratadine or another over-the-counter antihistamine within the last week. Other rhinitis therapy or clinical evidence of infection of the paranasal sinuses and/or of the upper or lower respiratory tract. Nasal surgery within the past year, structural nasal abnormalities or concurrent disease that could interfere with the validity of the study results, nursing/pregnant/ possibly pregnant
N total at baseline: 257 Intent to treat: 240 Intervention: 119 Control: 121
12-17 years of age. 56% male. Groups were comparable at baseline. |
Loratadine tablets, 10 mg daily for 4 weeks |
Fluticasone propionate aqueous nasal spray, 50 µg per actuation, 200 µg daily for 4 weeks |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: Six patients were withdrawn from the study because of violation of inclusion/exclusion criteria, and nine were withdrawn before randomization. Two patients elected not to participate in the trial just before the study medications were issued. 4 subjects were withdrawn because of suspected adverse events: 3 from the LOR group (infectious mononucleosis, angioedema, sinus headache), 1 from the FP group, (asthma exacerbation). 3 subjects discontinued from the FP group (failure to return, broken spray bottle, violation of exclusion criteria). 4 subjects in the LOR group and 1 in the FP group elected to withdraw because of ineffectiveness of study medication.
Incomplete outcome data: All but 3 subjects recorded valid data for at least 28 days. |
FP improved symptom-free days significantly compared to LOR for blockage (day), blockage (waking), sneezing, and nasal itching, but not for runny nose and eye irritation. In accordance, mean symptom scores were significantly lower in FP compared to LOR for all symptoms except eye irritation. There were no statistically significant differences between the groups for percentage of rescue-free days or use of rescue eye drops/ bronchodilator. No SD provided.
The most common adverse events in both treatment groups were headache and pharyngitis (42% and 16%, respectively, in the FP group and 25% and 10% in the LOR group, respectively). Headaches occurred in 50 subjects in the FP group and 27 subjects in the LOR group. Headaches were classified in as severe in nine and six subjects in FP and LOR groups, respectively (not significant), moderate in 29 and 14 subjects (p = 0.002), and mild in 66 and 29 subjects (p < 0.001). The event most frequently reported by the investigator as "drug-related" was epistaxis (7% and 4%, respectively). |
|
|
Gawchik, 1997 |
Type of study: RCT, double-blind, randomized, placebo-controlled parallel- group study.
Setting and country: Multicenter study performed in 10(?) centers throughout the USA.
Funding and conflicts of interest: The study was sponsored and supported by Rhône-Poulenc Rorer Pharmaceuticals. |
Inclusion criteria: Medical history of at least 2 consecutive seasons of allergic rhinitis characterized by rhinorrhea, sneezing, nasal stuffiness, nasal itch, and postnasal drip, with or without ocular symptoms.
Exclusion criteria: Lactating, (possible) pregnancy, long-acting steroid use, recent immunotherapy, medication that could affect study outcome, habitual abuse of nasal decongestants, a history of hypersensitivity or nonresponse to topical steroids/ antihistaminics, sinusitis, concomitant illness.
N total at baseline: 305 Intervention: 153 Control: 152
Mean age 33.1 years (12-68), 43% male. |
Loratadine 10 mg, for 4 weeks |
Triamcinolone acetonide (TAA) nasal aerosole inhaler, 55 µg/spray, 2 sprays/nostril, once daily, for 4 weeks |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: 89% completed the study in the intervention group, 92% in the control group.
Incomplete outcome data: Outcome for 1 patient in the LORA group was missing. |
All rhinitis and ocular symptoms improved significantly in both study groups, with no differences between the groups at week 1. During week 2, TAA patients showed significantly greater improvement than LOR in total nasal score (46 versus 36%), nasal itch (51 versus 41%), nasal stuffiness (40 versus 29%) and sneezing (54 versus 41%). Differences between the groups in postnasal drip, rhinorrhea, and ocular symptoms were not significant. During week 3 and 4, TAA treatment resulted in significantly greater improvement in total nasal score, nasal itch, nasal stuffiness, rhinorrhea, sneezing and ocular symptoms compared to LOR treatment. 3 patients from the LOR group were discontinued for lack of effectiveness.
Patient scores for the physician global evaluation at week 4 indicate a significantly (P<0.002) greater proportion of patients in the TAA group (68.4%) experienced moderate to complete relief of rhinitis symptoms than did the LOR group (59.2%)
8 subjects in either group were withdrawn due to adverse effects. Adverse events were reported in 41% of patients in TAA group, versus 37% in the LOR group. Headache was the most commonly described adverse effect in both groups. The only other drug-related adverse effect in more than 2% was increased rhinitis in the LOR group. 2 patients in the TAA group discontinued treatment, 1 due to moderate nervousness and the other due to mild epistaxis. In the LOR group, 1 patient discontinued due to moderate conjunctivitis and a moderate lacrimation disorder plus increased rhinitis symptoms. |
|
|
D’Ambrosio, 1998 |
Type of study: Placebo-controlled comparative trial.
Setting and country: Single-center study in Messina, Italy.
Funding and conflicts of interest: Not described. |
Inclusion criteria: >14 years old, clinical history of allergic rhinitis or rhino-conjunctivitis, positive allergy test for Parietaria Judaica, total initial symptom score of 6 at the end of run-in.
Exclusion criteria: Pathologies that may alter the treatment results, use of drugs that may interfere with the treatment results.
N total at baseline: 60 patients in 3 groups. CTZ: 18 patients, age 28.1 ± 10.4 years, 50% male. FP: 19 patients, age 30.0 ± 9.4 years, 42% male.
No differences between groups in age, sex and initial symptoms. |
Cetirizine tablet, 10 mg once daily for 60 days |
Fluticasone propionate 100 mg (not µg?) per nostril once daily by aerosol for 60 days. |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: 6 patients interrupted the treatment for personal reasons.
Incomplete outcome data: N.A. |
CTZ significantly reduced rhinorrhea, sneezing and nasal itching symptom scores compared to baseline. FP only improved nasal obstruction. Total symptom score decreased from 7.4 to 2.9 (=4.5) in the CTZ group, and from 7.2 to 4.8 in the FP group (=2.4, P<0.05 between groups). Symptom scores between days 20, 40 and 60 were limited for all symptoms.
EPC in nasal secretion decreased significantly in both groups (CTZ from 258.9 ±192.8 ng/ml to 74.6 ± 49.8 ng/ml; FP from 248.8 ± 238.7 ng/ml to 68.9 ± 75.9 ng/ml), with no difference between the interventions.
4 patients in the CTZ group reported dizziness, 1 gastric disorders and 1 visual problems. 2 patients in the FP group experienced dryness of the nasal mucosa and 1 reported a burning sensation of the nasal-throat mucosa. All adverse effects were light and temporary. |
|
|
Ratner, 1998 |
Type of study: RCT, double-blind, randomized, placebo-controlled parallel- group comparative trial.
Setting and country: Multicenter study in 5 sites across Texas, USA
Funding and conflicts of interest: The study was supported by a grant from Glaxo welcome Inc. |
Inclusion criteria: Male/female >12 years old, positive skin test to Juniperus ashei, appearance of the nasal mucosa consistent with allergic rhinitis, a history of seasonal onset and offset of symptoms for at least 2 previous mountain ceder pollen seasons, moderate to severe symptoms of rhinitis.
Exclusion criteria: Recent treatment with loratadine, astemizole, cromolyn sodium, nasal decongestants or systemic corticosteroids; septal deviation, nasal polyp, a history of nasal surgery or nasal septal perforation; candical inflation, pregnancy, lactating, any impairment that might affect completion of the study.
N total at baseline: 300 Intervention: 150, 40.1 (15-70) years old, 46% male Control: 150, 40.7 (13-80) years old, 45% male.
No differences in demographic characteristics and compliance between groups |
Loratadine tablet, 10 mg once daily for 14 days |
Fluticasone propionate (FP) aqueous nasal spray, 2 50-µg sprays per nostril once daily for 14 days |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: 95% completed the study. In the LOR group, 5% of patients were withdrawn, 1% due to adverse events, 2% due to lack of efficacy and 2 for other reasons. In the FP group, also 5% was withdrawn, 2% due to adverse events, and 3% due to lack of efficacy.
Incomplete outcome data: N.A.
|
Both after 7 and 14 days, clinician-rated total nasal symptoms (expressed as change from baseline) were significantly lower in the FP (187 on a scale of 400) than in the LOR group (102). This was also true for all individual nasal symptoms, i.e. blockage, discharge, itching and sneezing.
Both clinicians’ and patients’ overall evaluation (% of clinicians/patients scoring improvement on a 6 point scale) were significantly higher in FP than in LOR.
Improvements in mean global and individual RQLQ scores at day 14 were significantly higher in FP (2.2) than in LOR (1.3).
No significant differences in adverse events between the groups. 5-8% of patients experienced an adverse event possibly related to the medication. The most frequently reported drug-related adverse events were blood in the nasal mucus (1-2%), epistaxis (≤1%) and xerostomia (≤2%) for all treatments. |
|
|
Condemi, 2000 |
Type of study: RCT, double-blind, dummy-controlled randomized, parallel group study.
Setting and country: Multi-center study at 11 sites throughout the USA.
Funding and conflicts of interest: This study was funded by Rhône-Poulenc Rorer Pharmaceuticals Inc. |
Inclusion criteria: A 2-year consecutive history of allergic rhinitis symptoms verified by a positive skin prick test to grass pollens indigenous to the area; a combined rhinitis symptom score of 24 points or greater on 4 of the 5 baseline days according to a 4-point scale for the five symptoms evaluated
Exclusion criteria: Clinically significant abnormalities on physical examination or in urinalysis, hematology, or serum chemistry test results; nasal candidiasis, acute or chronic sinusitis, significant nasal polyposis or septum deviation; or rhinitis medicamentosa. Women who were pregnant, lactating, or of childbearing potential and not practicing an approved method of contraception; Recent or regular use of any of the following medications: topical corticosteroids, intranasal cromolyn, topical decongestants, systemic steroids, long-acting antihistamines, or investigational drugs; a history of habitual abuse of nasal decongestants; used medication for another indication that might affect the symptoms of seasonal allergic rhinitis; or had a history of hypersensitivity or nonresponse to topical steroids or antihistamines.
N total at baseline: 351 Intervention: 176 Control: 175
Mean age 32 (12-69) years, 55% male, 90% caucasian.
The 2 treatment groups were comparable at baseline |
Loratadine (10 mg) once daily, given as a single capsule, for 4 weeks. |
Triamcinolone acetonide (TAA) aqueous nasal spray 220 µg once daily, given as 2 sprays (55 µg/spray in each nostril) , for 4 weeks. |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: Control group: 15 patients (9%) withdrew from study for protocol deviations (6); adverse clinical events (4); treatment failure (4); and lost to follow-up (1). Intervention: 19 patients withdrew from the study for protocol deviations (9), adverse clinical events (3), treatment failure (5), and lost to follow-up (2).
Incomplete outcome data: N.A. |
Mean change in total nasal score was significantly stronger in TAA (-4.4 ± 2.9) than in LORA (-3.6 ± 3.2). Individual symptoms in which the change was also significant were nasal congestion (TAA -1.1 ± 0.9 versus -0.8 ± 1.0), sneezing (TAA -1.2 ± 0.9 versus -0.9 ± 0.9), and nasal itch (-1.1 ± 0.8 versus -0.9 ± 0.9). No differences between groups for rhinorrhea and ocular symptoms. Both treatment groups had improvement in symptoms as early as day 1, but the greatest improvement occurred over the first 3 days of treatment.
Physician global evaluation showed high relief levels in both groups. A similar number of patients in each group (4 TAA and 5 LOR) withdrew from the study early due to lack of efficacy.
Overall RQLQ was better in the TAA group both at 2 (TAA 1.79 ± 1.27 versus 2.13 ± 1.18) and 4 weeks (TAA 1.48 ± 1.24 versus 1.82 ± 1.29). In addition, At week 2, TAA-treated patients had significantly better QoL than LOR-treated patients in the dimensions of activity, emotions, nasal symptoms, and practical problems. At week 4, patients in the TAA group had better QoL in the dimensions of activity, nasal symptoms, and practical problems,
Headache was the most frequently reported adverse event with 25 TAA patients (14.3%) and 27 LOR patients (15.3%). Seven patients discontinued the study due to adverse events. The 4 TAA patients who discontinued study reported 6 adverse events; three of these events (headache, rhinitis, and chest pain) were possibly/probably related to study drug and occurred in one patient. The 3 LOR patients reported one adverse event each, none of which were related to study drug. |
|
|
Kaszuba, 2001 |
Type of study: Randomized, open-label, parallel-group study
Setting and country: Single-center study in Chicago
Funding and conflicts of interest: This study was supported in part by a grant from Glaxo Wellcome Inc, Research Triangle Park, NC; and grant AI 45583 from the National Institutes of Health, Bethesda, Md. |
Inclusion criteria: A history of rhinitis during at least the last 2 ragweed seasons in Chicago, Ill, and a positive puncture skin test result to ragweed antigen extract.
Exclusion criteria: Symptoms or physical signs suggestive of renal, hepatic, or cardiovascular disease; nasal polyps; a displaced septum; or perennial rhinitis, recent use of topical or systemic corticosteroids, antihistamines, decongestants, cromolyn sodium; pregnant or lactating women
N total at baseline: 88 Intervention: 44, mean age 30.0 (19-44) years, 57% male Control: 44, mean age 27.5 (18-48) years, 48% male
The groups were matched for age, sex, race, and skin test sensitivity |
Loratadine tablet (10 mg/d) if needed, for 4 weeks |
Fluticasone propionate nasal spray (100 µg/d per nostril) if needed, for 4 weeks |
Length of follow-up: Duration of the treatment.
Loss of follow-up: Two patients from each treatment group dropped out before completing the protocol. One patient moved away and 3 were noncompliant with respect to returning for their appointments.
Incomplete outcome data: N.A. |
Compared to LOR, the fluticasone group had significantly better scores on overall RQLQ and in the activity, sleep, practical, and nasal domains after 2 and 4 weeks.
Mean symptom scores were significantly better in the FP (improvement from baseline 5.2) than in the LOR (worsening of 0.99) group throughout the study period, with 4.0 with FP versus 7.0 at day 28. No SD reported.
Both total eosinophils and ECP levels were markedly and significantly lower in FP compared to LOR, after 2 and 4 weeks. The ECP levels followed the same pattern, with significant decreases in the intranasal corticosteroid–treated group and significant increases in the H1 receptor antagonist–treated group. No values described, only graphs.
Adverse events were not described. |
|
|
Rinne, 2002 |
Type of study: Double-blind, double-dummy, randomized, parallel-group design
Setting and country: Single-center study at the Skin and Allergy Hospital of the University of Helsinki, Finland
Funding and conflicts of interest: Supported by AstraZeneca R&D, Lund, Sweden, and Helsinki University Central Hospital grant 9303 |
Inclusion criteria: 16 to 68 years of age; symptoms of perennial rhinitis for 1 to 3 years; Rhinitis symptoms were required to be present for at least 1 hour on most days. Patients had to have eosinophils present in nasal smear (score ≥1), a positive skin prick test response for at least one perennial allergen, or both.
Exclusion criteria: Severe seasonal rhinitis symptoms; upper respiratory infection during the 4 weeks before randomization; concomitant illness or medication that might interfere with the study; previous steroid treatment >3 months at any time or within 1 month before randomization; systemic steroid therapy within 3 months or cutaneous application of steroids; immunotherapy for seasonal or perennial rhinitis within the past 3 years; women who were pregnant or lactating. Antihistamines, if used, were discontinued 48 hours before randomization (2 months before randomization in the case of astemizole), and intranasal cromoglycate was discontinued 14 days before randomization. Women of childbearing age were required to use adequate contraception.
N total at baseline: 143 patients entered the study.
Budenoside 71 patients, CTZ 72 patients. |
Oral cetirizine (Zyrtec) 10 mg, once daily for 1 year |
Intranasal budesonide (Rhinocort Turbuhaler), 400 μg, (equivalent to a delivered dose of 280 μg) once daily for 1 year |
Length of follow-up: 2 years
Loss-to-follow-up: 88% continued until the end of the 1-year treatment.
Incomplete outcome data: N.A. |
Reductions in total and individual nasal symptom scores during the first year were significantly greater with budesonide than with CTZ, except for the score for blocked nose at 12 months. For total nasal symptoms, budesonide was significantly more effective than cetirizine. At 28 days, total nasal symptom score improvement was greater with budesonide (2.69) than with CTZ (1.71).
The percentage of rhinitis-free days was significantly higher in the budesonide group (45.1%) compared with that in the CTZ group (25.9%). There was no significant difference between the groups in eye symptoms. Patients in the CTZ group used significantly more phenylpropanolamine tablets as rescue medication than patients in the budesonide group (P=0.002). Budesonide was significantly more effective in controlling rhinitis symptoms than CTZ. Substantial or total control of symptoms was achieved in 74% of budesonide-treated patients compared with in 50% of those receiving cetirizine (P=0.0023). Nasal peak expiratory flow (nPEF) increased from a mean of 236 to 257 L/min (8.9%) in the budesonide group and from 231 to 241 L/min (4.3%) in the cetirizine (P<0.05). Nasal smear eosinophilia decreased to a greater extent in patients treated with budesonide (from 0.79 to 0.16) than in those treated with CTZ (from 0.85 to 0.54 (P<0.01)
Adverse events occurred in 65 budesonide-treated patients and 68 patients receiving CTZ. The most common adverse events in each group were viral upper respiratory tract infections, which occurred in 48 budesonide-treated patients and 52 CTZ treated patients, blood in nasal secretion (ranging from blood-tinged mucus to nasal bleeding; 19 and 9 patients, resp.), conjunctivitis (8 and 5 patients, respectively), and headache (6 and 1 patients, respectively). Sinusitis was observed in 2 budesonide- treated patients and in one CTZ-treated patient. Most adverse events were mild or moderate in intensity. 3 budesonide-treated patients withdrew from the study at 6 months because of crusting of the nasal mucosa, bleeding, or both, and one CTZ-treated patient withdrew because of drowsiness. |
|
|
Type of study: Randomized, double-blinded study
Setting and country: Single-center study
Funding and conflicts of interest: This study was supported by an unrestricted grant from Alcon Laboratories, Inc., Fort Worth, Texas. The authors have no proprietary interest in the products studied. |
Inclusion criteria: Positive history of allergic rhinoconjunctivitis, a positive skin test result within 2 years.
Exclusion criteria: Not specified
N total at baseline: 73 mean age 45.3 (21-73) years, 42.5% male.
Group size per treatment not specified. |
fexofenadine hydrochloride 180-mg tablets, once daily for 1 week |
Mometasone furoate monohydrate 50-µg nasal spray, once daily for 1 week |
Length of follow-up: Duration of the treatment.
Two subjects did not complete the study: 1 was lost to follow-up at visit 3 and 1 took a disallowed medication
Incomplete outcome data: N.A. |
Mean ocular itching score and mean total nasal score not different between treatments conjunctival allergen challenge. Mean total nasal symptom scores after nasal allergen challenge were lower in the mometasone group than in the fexofenadine group.
Focus on conjunctival versus nasal challenge, limited data on medication differences. |
|
|
|
Fokkens, 2004 |
Type of study: Double-blind, double-dummy placebo-controlled RCT.
Setting and country: Dual-center study at the UK and the Netherlands.
Funding and conflicts of interest: This study was funded by GlaxoSmithKline R&D, UK. Potential conflict of interest was not specified. |
Inclusion criteria: Perennial rhinitis for more than 2 weeks, age 2-4 years, parental consent.
Exclusion criteria: Requirements for inhaled or intranasal corticosteroids, sodium cromoglycate or antihistamines, use of systemic corticosteroids, presence of nasal polyps or other anatomical deformations, presence of other severe illness, e.g. cleft palate, concurrent nasal infections or contraindications to steroids.
N total at baseline: 26 Age 2-4 years, 62% male. |
Oral ketotifen 1 mg once daily plus intranasal placebo, for 6 weeks. |
Fluticasone propionate aqueous nasal spray 100 µg once daily plus oral placebo, for 6 weeks |
Length of follow-up: Duration of the treatment.
Incomplete outcome data: 3 subjects could not be evaluated. No reason was provided. |
Patients treated with FPANS had a significant reduction in the total night-time rhinitis symptom assessment for weeks 4–6 (FPANS 4.2, ketotifen 6.5, no SD reported), and a significantly reduced total daytime rhinitis symptom score over the same period (FPANS 3.6, ketotifen 5.5, no SD). The corresponding measurements over weeks 1–3 for both total daytime and total night-times symptom scores were reduced in the fluticasone group but this difference was not statistically significant |
|
|
FNM 30033 |
Type of study: Double-blind, double-dummy placebo-controlled RCT.
Setting and country: Multi-center study at 14 investigative sites in the US
Funding and conflicts of interest: This study was funded by GlaxoSmithKline Inc., Research Triangle Park, NC. |
Inclusion criteria: At least 12 years of age with a history of allergic rhinitis for ≥2 years and a positive skin test to at least one allergen relevant to the spring pollen season and geographic region
Exclusion criteria: Not described
N total at baseline: 471, 80% between 18 and 64 years old, 42-38% male in each group, 80-90% Caucasian.
FP: n=158 LOR: n=158 (Placebo: 155) |
Encapsulated loratadine 10mg, once daily for 4 weeks |
Fluticasone propionate aqueous nasal spray, 200 µg (2 sprays of 50 µg per nostril) once daily for 4 weeks
(Placebo group not considered) |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: 87% of the LOR group, and 94% of the FP group completed the 28-day treatment period
The most common reasons for premature discontinuation were adverse events (4% in the LOR group and 3% in the FP group) and lack of efficacy (4% in the LOR group and 0% in the FP group).
Incomplete outcome data: N.A. |
Total ocular symptom score change was significantly greater in FP (-88.7 ± 5.3) than in LOR (-72.5 ± 5.4), as well as all separate symptoms itching, redness, and tearing. Nasal congestion improvement was also greater in FP (-35.3 ± 1.9) than in LOR (-25.0 ± 1.9).
Patients’ overall assessment of the effectiveness of the treatment on a seven-point categorical scale was significantly different between groups. 82% of patients in the FP group reported (significant/ moderate/mild) improvement against 64% in the LOR group.
The incidence of adverse events was similar among the treatment groups, with 42% of the LOR group, and 44% of the FP group reporting at least 1 adverse event during the study. The most common event was headache, 18% with LOR and 17% with FP. |
|
|
Andrews, 2009 |
Type of study: RCT, double-blind, randomized, placebo-controlled parallel- group study.
Setting and country:
Funding and conflicts of interest: GlaxoSmithKline funded the research (FFU109045 and FFU109047) described in this article; GlaxoSmithKline funded Dr. Saiers’ work. H Kaiser, D Mohar, R Jacobs, and B Martin are all research sponsors for GlaxoSmithKline; H Kaiser and M Vandewalker are speakers for GlaxoSmithKline; L Lee, W Toler, B Prillaman, E Philpot, and A Dalal are employees of GlaxoSmithKline |
Inclusion criteria: At least 12 years old, with a diagnosis of seasonal allergic rhinitis as defined by a clinical history of nasal allergy symptoms during each of the last two respective allergy seasons and a positive skin test to mountain cedar allergen (study 1) or ragweed allergen (study 2) within 12 months of screening
Exclusion criteria: Plans to travel outside the region for >48 hours of the study, (attempt at) pregnancy, severe physical obstruction of the nose (e.g., deviated septum or nasal polyp); recent nasal septal surgery or perforation; asthma, unless it was mild intermittent asthma; rhinitis medicamentosa; ocular or upper respiratory bacterial or viral infection; chronic use of medications that could affect allergic rhinitis or assessments of efficacy of study medication; current tobacco use: recent use of subcutaneous omalizumab, corticosteroids short-acting prescription or over-the-counter antihistamines, oral or intranasal decongestants, anticholinergics, long-acting β-agonists, or oral antileukotrienes; long-acting antihistamines and intranasal antihistamines or intranasal or ocular cromolyn.
N total at baseline: Study 1: 936 FFNS, n=312 mean age 37.8 years, 33% male. Fex, n=311, mean age 39.6 years, 36% male.
Study 2: 680 FFNS, n=224, mean age 34.0 years, 41% male. FEX, n=227, mean age 34.8 years, 39% male. |
Fexofenadine (FEX), 180 mg oral tablet once daily for 2 weeks |
Fluticasone furoate nasal spray (FFNS) 110 µg once daily for 2 weeks
(placebo group not considered) |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: In study 1, premature withdrawal was 4% in the fluticasone furoate group and 8% in the fexofenadine group. The most common reason for premature withdrawal was protocol violation. In study 2, the incidence of premature withdrawal was 7% in the fluticasone furoate group and 6% in the fexofenadine group.
Incomplete outcome data: N.A.
|
FFNS was significantly (p < 0.001) more effective than fexofenadine with respect to the mean change from baseline in the nighttime symptoms score in both studies. FFNS also showed greater improvement to all three individual components of the nighttime symptoms score—nasal congestion on awakening, nighttime awakenings due to nasal symptoms, and difficulty going to sleep because of nasal symptoms. FFNS was significantly (p < 0.001) more effective than FEX in both studies with respect to the mean change from baseline in the nighttime reflective TNSS. FFNS was significantly more effective than FEX in nighttime reflective total ocular symptom score in study 2, but not in study 1. FFNS was significantly (p < 0.001) more effective than fexofenadine and placebo with respect to the mean changes from baseline in morning peak nasal inspiratory flow. FFNS was significantly (p <0.001) more effective in both studies than FEX with respect to the mean changes from baseline in the daytime rTNSSs and evening peak nasal inspiratory flow measurements. FFNS was significantly more effective with respect to the mean change from baseline in the daytime reflective total ocular symptom score in study 2, but not in study 1.
FFNS was significantly (p < 0.001) more effective than FEX in both studies with respect to the mean change from baseline in the overall nocturnal RQLQ total score.
The percentage of patients with at least 1 adverse event was similar among groups in study 1 (16% in the FFNS group, 18% in the FEX group). The only adverse events reported in >1% of patients in a treatment group and reported more often with active treatment than placebo was pharyngo-laryngeal pain (2% FFNS, <1% FEX). The number of patients who prematurely withdrew from the study because of adverse events was 2 in the FFNS group (one each with sinusitis and influenza), 5 in the FEX group (one each with cholecystitis, hypersensitivity, cough, upper respiratory infection, and sinusitis). Three adverse events in the FEX group (cholecystitis, upper respiratory infection, and sinusitis), but none of the other adverse events leading to premature withdrawal, were designated by the investigator as having possibly been caused by study treatment. The percentage of patients in study 2 with at least 1 adverse event was also similar among groups (14% in the FFNS group, 16% in the FEX group). The only adverse event reported in >1% of patients in a treatment group and reported more often with active treatment than placebo was epistaxis (no reports with FFNS, 2% FEX). The number of patients who prematurely withdrew from the study because of adverse events was two in the FFNS group (upper respiratory infection and urticaria), one in the FEX group (lymph gland infection). The adverse event of urticaria in the FFNS group was the only event considered by the investigator as possibly caused by study treatment. |
|
|
Kulapaditha-rom, 2010 |
Type of study: RCT, randomized, parallel-group study.
Setting and country: Single-center study in Ramathibodi Hospital, Bangkok, Thailand
Funding and conflicts of interest: Faculty of Medicine, Ramathibodi Hospital, Mahidol University partially supported this study and played no role in study execution. |
Inclusion criteria: At least 18 years of age, a documented history of allergic rhinitis of more than 1 year.
Exclusion criteria: Allergic bronchial asthma, non-allergic rhinitis (infection or drug-induced, etc), structural abnormality of the nose (deviation of nasal septum, obstructive nasal polyps), known cardiac disease, renal or hepatic dysfunction and pregnant or breastfeeding women. Excluded medications were other antihistamines and corticosteroids, leukotriene inhibitors, decongestants and immunotherapies in the last 6 weeks
N total at baseline: 100 Levocetirizine 50, Budenoside 50
26% male, aged 35.72 ± 10.96, no significant differences between the two treatment groups. |
Levocetirizine 5 mg daily for 4 weeks |
Budesonide nasal spray 256 mg daily (two spays into each nostril, 64 mg per spray) for 4 weeks |
Length of follow-up: 29-56 days
Loss-to-follow-up: xx% completed the study in the intervention group, xx% in the control group.
Incomplete outcome data: Not described |
Fractional reduction of TSS compared to baseline: Budesonide 0.19, Levocetirizine 0.33. No SD reported for these values.
Highly indirect presentation of TSS scores and response time (time for the drop in TSS to a value of ≤4) by calculating area under curve for fractional change, and separating high TSS and low TSS at baseline.
Adverse events were not described. |
|
|
Bhatia, 2005 |
Type of study: RCT, double-blind, randomized, placebo-controlled parallel- group study.
Setting and country: Single-center study in Chicago, USA
Funding and conflicts of interest: This study was supported by a grant from the Investigator Sponsored Studies Program of AstraZeneca, Westborough, Mass. |
Inclusion criteria: Age 18 to 45 years, a clinical history of sensitivity to tree or grass pollens, positive skin test result and symptoms during the spring season for the past 2 years.
Exclusion criteria: Use of systemic corticosteroids in the previous 30 days, oral antihistamines or decongestants in the past 7 days, and topical antihistamines or decongestants in the past 24 hours, long-term antiasthma medications, immunotherapy in the previous 2 years; pregnancy or nursing
N total at baseline: 61 Desloratadine n=31, 39% male, mean age 26.4 ± 7.41; Budesonide n=30, 53% male, mean age 25.6 ± 5.97; No differences between groups. |
Desloratadine, 5 mg once daily for 2 weeks |
Budesonide, 32-μg per nostril once daily, for 2 weeks |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: xx% completed the study in the intervention group, xx% in the control group.
Incomplete outcome data: Not reported |
The primary outcome variable was nasal peak inspiratory flow (NPIF). Comparing the total NPIF between budesonide and desloratadine showed a significant difference on all of the treatment days except days 5 and 6, with budesonide providing more improvement in nasal airflow than desloratadine. The total change over the baseline showed a significantly greater improvement for budesonide (493.1, no SD reported) than for desloratadine (248.5, P=0.01).
RQLQ at the end of treatment was similar with Desloratadine 1.30 ± 0.17 versus Budesonide 1.08 ± 0.92., with no difference in improvement from baseline between the groups (DES 1.56, BUD 2.05)
Total symptom score (sneezing, runny nose, stuffy nose, itchy eyes/nose) improved with 7.35 points in the Budesonide group, and 5.47 in the Desloratadine group. No SD reported.
No adverse events reported.
|
|
|
Ford, 2015 |
Type of study: RCT, double-blind, randomized, placebo-controlled parallel- group study.
Setting and country: Multicenter at 32 investigative sites across the USA.
Funding and conflicts of interest: The study was funded by GSK (study 200165)
L.B. Ford has received support from GSK to conduct this study and her institution has received research grants from Teva, F. Hoffmann-La Roche, Novartis, AstraZeneca, Merck, Sanofi, Circassia, Forest, Johnson and Johnson, Amgen, Mylan, Aquinox, Perrigo, Oriel, Pfizer, Roxanne, and Biota. J. Matz has received support from GSK to conduct this study, has served as a consultant for Biota, has received payment for the development of educational programs from Novartis, and his institution has received research grants or has grants pending from Teva, Perrigo, Merck, Shionogi, and Greer. T. Hankinson, B. Prillaman, and G. Georges were employees of GSK and held stock in GSK at the time of study conduct and during the writing of this manuscript |
Inclusion criteria: Previous diagnosis with SAR or a positive skin-prick test result to a prevalent seasonal allergen; allergy treatable on an outpatient basis; at least 18 years of age.
Exclusion criteria: Significant medical conditions, use of study protocol–prohibited concurrent medications, or hypersensitivity to any excipient of the study treatments.
N total at baseline: 682 170 patients FP 170 patients CTZ And 2 placebo groups
The mean age was similar among the four treatment groups, with 98% to 99% of the subjects 18 to 64 years old. 36-44% male in all treatment groups. The majority of subjects in all groups (78–84%) reported having SAR for at least 10 years. |
Cetirizine 10 mg once daily for 2 weeks |
Fluticasone propionate (FP) nasal spray 200 µg once daily for 2 weeks |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: One subject in the FPNS group withdrew due to an adverse event. One patient in the CTZ group was withdrawn as a result of protocol deviation.
Incomplete outcome data: Not mentioned. |
The primary efficacy measurement defined for this study was the change in the rTNSS from baseline over the 2-week treatment period. FP -2.2 ± 0.19, CTZ -1.9 ± 0.18. Instantaneous ocular TSS change FP -1.9 ± 0.18, CTZ -1.3 ± 0.17. Change in nocturnal RQLQ for FP was -1.0 ± 0.10, versus -0.6 ± 0.09 with CTZ.
“All treatments (…) were well tolerated, with comparable rates of adverse events and no serious adverse events.” |
|
|
Kim, 2015
NCT 01430260 |
Type of study: RCT, open-label, 3-arm, parallel-group randomized study
Setting and country: Multicenter study at 13 sites
Funding and conflicts of interest: “There are no financial or other issues that might lead to conflict of interest.”
Funding was provided by Handok Inc. and Takeda Pharmaceutical Co. (Clinical trial registry URL and registration number: www.clinicaltrials.gov /ct2/show/ NCT01430260). |
Inclusion criteria: At least 18 years, signs and symptoms of AR for longer than 1 year. In accordance with ARIA guidelines, only subjects with moderate to severe SAR or PAR were included. All subjects tested were positive for skin allergy tests (skin prick test) or serological allergy tests, including for multiple allergen simultaneous tests (MAST), to 1 or more specified allergens less than 1 year prior to screening.
Exclusion criteria: active asthma requiring treatment with inhaled or systemic corticosteroids and/or routine use of beta-agonists or any other medications; nasal pathologies, including nasal polyps or bronchial anomalies; chronic obstructive pulmonary disease; clinically significant renal disease, liver disease within 2 months before the screening visit; intranasal biopsy revealing ulcers, trauma, surgery, atrophic rhinitis or drug-derived rhinitis within 2 months before screening; respiratory infections within 2 weeks before or during screening; usage of systemic steroids within 2 months of screening or local steroids (hydrocortisone >1%) within 4 weeks of screening or antibiotic treatment within 2 weeks of screening; a possibility of receiving medications like systemic corticosteroids or high-dose steroids, beta-agonists or other investigational drugs that were not permitted in the protocol during the study period; known or suspected hypersensitivity to ciclesonide or hydroxyzine; and genetic disorders such as galactose intolerance, Lapp lactase deficiency or glucose-galactose malabsorption.
N total at baseline: 260 Ciclesonide n=88, age 32.9 ± 11.3, 43.2% male Levocetirizine n=89, age 32.9 ± 10.2, 54.5% male |
levocetirizine 5 mg once daily for 2 weeks |
Ciclesonide nasal spray 200 μg once daily for 2 weeks |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: 1 patient from the levocetirizine group was unable to locate their patient diary.
Incomplete outcome data: Two subjects were excluded from efficacy analysis but included for safety analysis. levocetirizine group experienced a randomization error and was inadvertently dosed with ciclesonide. This subject’s data were analyzed as belonging to the levocetirizine group when assessing efficacy, but as the ciclesonide group when assessing safety. |
The primary endpoint was the change from baseline in the mean value of patient-reported rTNSS (morning and evening) averaged over the 2-week treatment period. rTNSS (rhinorrhea, nasal itching, nasal congestion, and sneezing) changed in Cicle group -3.9 ± 2.2, versus-3.0 ± 2.1 with Levo. rTOSS (itchy eyes, red eyes, and watery eyes) in cicle group -1.4 ± 1.6 versus -0.9 ± 1.4 with Levo. Physician-assessed overall nasal signs (discoloration, swelling, discharge, and postnasal drip) and symptoms (rhinorrhea, itching, nasal congestion, and sneezing) severity improvement was 1.1 ± 0.7 with Cicle versus 0.8 ± 0.6 in the Levo group. Mean RQLQ decreased with 1.4 ± 1.0 with Cicle and 1.1 ± 1.0 in the Levo group.
Adverse events: One (1.1%)patient with Cicle: nasal pain. Six (6.8%) patients with 8 symptoms in the Levo group: dry mouth, sleepiness, dizziness, dry nose, headache, epistaxis |
|
|
Wartna, 2017
Dutch Trial Register No. 3429 |
Type of study: Single-blinded RCT
Setting and country: Multicenter study in 61 GP practices and 2 paediatric outpatient clinics in the South-West of the Netherlands
Funding and conflicts of interest: Mrs Wartna, Dr. Elshout, Dr. Bohnen and Dr. Bindels report grants from Lung Foundation Netherlands and Stichting Astma Bestrijding , during the conduct of the study; Mr. Pols and Dr. Pijnenburg have nothing to disclose; Dr. Gerth van Wijk reports personal fees from MSD, HAL, Crucell, ALK-Abello, Novartis, and Emerade, grants from NWO, STW, Novartis, Biomay, and DBV, personal fees from Allergopharma, Thermo Fisher, Chiesi, de Tijdstroom, and Bohn, Stafleu, van Loghum, outside the submitted work.
Supported by The Lung Foundation Netherlands (Longfonds) (grant number 3.4.11.049); Stichting Astma Bestrijding (grant number 2013/036). Both funders had no influence in the study design, in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. |
Inclusion criteria: Aged between 6 and 18 years; recruitment by a GP on ICPC R97 or prescription of AR-related medication in the previous season; sensitization to grass pollen; a minimum retrospective (previous year) AR symptom score of 7/21, written informed consent
Exclusion criteria: Recent use of antihistamines or intranasal corticosteroids, being pregnant or breastfeeding, not being able to speak Dutch sufficiently, contraindications, not having internet access.
N total at baseline: 150 48 patients antihistamines on demand; 50 patients INCS daily; 52 INCS on demand
Mean age 11.6 years, 52% male. No differences between groups. |
Levocetirizine dihydrochloride tablet, 5 mg/day on demand for 3 months |
Fluticasone propionate nasal spray, for 3 months.
Dose: aged <12 years 100 µg/day, aged ≥12 years 200 µg/day
1) daily 2) on demand |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: 132 patients were suitable for the ITT analysis (45 patients in the INCS daily group, 46 in the INCS on demand group, and 41 in the antihistamine group
18 patients were excluded due to insufficient data: 7 patients dropped out (stopped using study medication and without any measurement) and 11 patients neglected to fill in their diary. 8 patients discontinued using their study medication; but continued to fill out the diary and questionnaires, and had an end visit |
The INCS on demand group had the highest percentage of symptom free days (30%), followed by the INCS daily group (22%) and the antihistamine group (15%) The clinical relevant difference in symptom free days was set at 10%-point. The difference between INCS on the demand and INCS daily use was 8% more symptom free days in favour of on demand use, with a confidence interval of -5 to +21%. There was no difference between INCS and antihistamines for symptom free days. Comparison between antihistamine on demand and INCS daily use showed that INCS daily use resulted in a higher percentage of nose symptom-free days than antihistamine use, with sneezing-free days showing a significant difference (57% versus 38%, p=0.017). Also, antihistamine use resulted in more eye symptom-free days than INCS daily use; this difference was significant for itching eye-free days (43 versus 52, p=0.022). Comparing antihistamine use with INCS daily use, antihistamine use had a higher symptom score (3.90 95% CI 3.03-4.76) than INCS (4.63, 95% CI 3.81-5.46) on the combined symptoms and, more specifically, on nose symptoms (CS 3.20, 95% CI 2.69-3.71; AH 4.14, CI 3.63-4.66), but none of the differences were significant. No change in symptom scores could be calculated without baseline data.
Patients in the antihistamine group used on average 43 tablets on average for 91 days. in the INCS daily group, on average 23.1 ml fluticasone was used (± 257 sprays), and the INCS on demand group used on average 9.1 ml fluticasone (± 102 sprays) (p<0.0001). On average, patients used rescue medication on 12% of the days; there was no significant difference between the three groups. |
|
|
Malizia, 2018 ID: NCT02646904 |
Type of study: Open-label RCT
Setting and country: Single-center study in Palermo, Italy
Funding and conflicts of interest: The study was supported by CHIESI Farmaceutici S.p.A., Parma, Italy (Unrestricted Grant N. 3774/2015).
The authors declare that they have no conflicts of interest. |
Inclusion criteria: Age 6-14 years; clinical history of perennial allergic rhinitis in the previous year, positive skin prick test response to Dermatophagoides pteronyssinus; Total 5-Symptom Score (T5SS) ≥5 (rhinorrhea, nasal obstruction, nasal itching, sneezing, eye itching)
Exclusion criteria: positive skin prick test result to seasonal allergens and other perennial allergens; medical diagnosis of nasal anatomic defects or nasal polyp disease; medical diagnosis of asthma; upper or lower respiratory tract infection in the previous 2 weeks; use of oral antihistamines, decongestants, leukotriene antagonists, systemic/topical antibiotics or corticosteroids in the previous 4 weeks; ongoing allergen immunotherapy; active smoking; adherence <80%.
N total at baseline: 68 Intervention: 34 children, mean age 9.47 ± 2.36, 68% male Control: 34 children, mean age 10.06 ± 2.35, 62% male
No differences in demographic characteristics between groups. |
Beclomethasone dipropionate nasal spray suspension (nBDP), 100 μg per nostril twice daily, for 21 days |
Length of follow-up: Duration of the treatment.
Loss-to-follow-up: 1 consent withdrawal in the CTZ group, 2 lost to follow up (change of residence). No dropouts in the nBDP group.
Incomplete outcome data: Not specified |
Nasal volume values increased significantly after 21 days in the nBDP group (P=.033), while the change was not significant in the CTZ group (P=.900). Minimal cross sectional area values increased significantly in the nBDP group (P=.045), while the change was not significant in the CTZ group (P=.840). After adjusting for confounders, nBDP was found to improve nasal volume more than CTZ (LSmc, 2.21 cm3 versus 0.20 cm3; P=.013). Similarly, nBDP improved MCA more than CTZ (LSmc, 0.63 cm2 versus 0.13 cm2; P=0.002). In the nBDP group, with respect to the CTZ group, a more pronounced improvement was observed in eosinophil classes (LSmc, –1.10 versus –0.40, P=0.031) and neutrophil classes (LSmc, –0.97 versus –0.17; P=.010), T5SS total score (LSmc, –5.63 versus –3.54; P=.008), PSQI total score (LSmc, –1.30 versus –0.19; P=.025), and PRQLQ total score (LSmc, –1.15 versus –0.69; P=.031).
No adverse events were observed. |
|
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation |
Bias due to inadequate concealment of allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?
(unlikely/likely/unclear) |
Bias due to loss to follow-up?
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?
(unlikely/likely/unclear) |
|
Frølund, 1991 |
Patients were assigned a treatment group according to a computer-generated randomization code.
|
Unlikely, a double-blind, double-dummy technique was Used, in which test medication was provided as identical, opaque capsules, supplied in strips labelled with Patient No., Week No., and Prescribed Time of Administration, and as identically labelled pressurized canisters. |
Unlikely |
Unlikely |
Unlikely |
Unlikely, outcomes presented in Results are consistent with outcomes described in Methods. |
Likely, loss to follow-up is considerable and dissimilar between treatment groups. |
Unclear |
|
Charpin, 1994 |
Not described |
Unlikely, double-blind, double-dummy |
Unlikely |
Unlikely |
Unlikely |
Unclear |
Unlikely |
Unclear, not described |
|
Jordana, 1996 |
“Subjects were randomized as a cohort” |
Unlikely, double-blind, double-dummy |
Unlikely |
Unlikely |
Unlikely |
Unlikely, outcomes presented in Results are consistent with outcomes described in Methods. |
Unlikely |
Unlikely, intention to treat analysis was performed. |
|
Gawchik, 1997 |
Not described |
Unlikely, study was double blind and placebo-controlled. |
Unlikely |
Unlikely |
Unlikely |
Unlikely, all outcome measures described in the methods are quantified in the results. |
Unlikely, loss to follow up was limited and similar between groups. |
Unclear |
|
D’Ambrosio, 1998 |
Randomization not certain: “patients were divided into three homogeneous groups, according to age and symptom score” |
Unlikely, the study was placebo-controlled. |
Unlikely |
Unclear |
Unclear |
Unlikely |
Unclear |
Unclear |
|
Ratner, 1998 |
Not specified |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely, loss to follow up was limited |
Unclear |
|
Condemi, 2000 |
Not specified |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unclear |
Unlikely |
Unlikely, loss to follow up was limited and similar between groups. |
Unclear |
|
Kaszuba, 2001 |
subjects were randomized in balanced blocks of 4 |
Likely, patients were not blinded |
Likely |
Likely |
Unclear |
Unlikely |
Unlikely, loss to follow up was limited |
Unclear |
|
Rinne, 2002 |
Not described |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unclear |
Unlikely, data were analyzed on an intention-to-treat basis |
|
Spangler, 2003 |
Not described |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unclear |
|
Fokkens, 2004 |
Not described |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Likely, 3 out of 26 were lost, and reasons and distribution between the groups was not specified. |
Unlikely, data were analyzed on an intention-to-treat basis |
|
Bernstein, 2004 |
randomly assigned in a 1 : 1 : 1 ratio |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely, loss to follow up was limited |
Unclear |
|
Bhatia, 2005 |
randomization was assigned by a code in blocks of 4, |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Unclear |
Unclear |
Unclear |
|
Andrews, 2009 |
Patients were randomized 1:1:1 |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Unclear |
Unlikely, loss to follow up was limited |
Unlikely, ITT analysis was specified |
|
Ford, 2015 |
subjects were randomized with equal allocation to one of the treatment groups: |
Unlikely, the study was placebo-controlled |
Unlikely |
Unlikely |
Unlikely |
Likely, a seemingly subjective cut-off value was used to separate the group in high versus low allergy when describing results. |
Unclear |
Unlikely, ITT analysis was specified |
|
Kim, 2015 |
Block randomization at each study center (2 block sizes were combined for randomization), with random numbers generated using the SAS® Software Package |
Likely, patients were not blinded |
Likely |
Likely |
Unclear |
Unlikely |
Unlikely |
Unlikely |
|
Wartna, 2017 |
computerized block-randomization |
Likely, patients were not blinded |
Likely, patients were not blinded |
Likely, care providers were not blinded |
Unlikely, outcome assessors were blinded |
Unlikely |
Unclear |
Unlikely, ITT analysis was specified |
|
Malizia, 2018 |
computer-generated randomization sequence (1:1 allocation) unknown to the physicians |
Likely, patients were not blinded |
Likely |
Unlikely |
Unclear |
Unlikely |
Unlikely, loss to follow up was limited |
Unlikely, ITT analysis was specified |
PICO 3 Orale antihistaminica versus nasale antihistaminica
Evidence table for intervention studies
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Gambardella, 1993 |
Not described |
Unclear, not described |
Unlikely. |
Unlikely, the study is double-blind, double-dummy. |
Unlikely, the study is double-blind. |
Unlikely, all outcome measures described in the methods are reported in the results. However, it is unclear why from 1 patient the global assessments at 7 weeks were not reported. |
Unlikely, 1 patient in the intervention group withdrew from the study due to a congested nose and 2 in the control group due to side effects of the treatment. |
Unclear. |
|
Conde Hernández, 1995 |
“The patients were included in either group on the basis of the randomization scheme previously established.” |
Unclear, not described |
Likely, the study was not blinded and treatment allocation was obvious. |
Likely, the study was not blinded and treatment allocation was obvious. |
Likely, the study was not blinded and treatment allocation was obvious. |
Unlikely, all outcome measures described in the methods are quantified in the results. |
Unlikely, loss to follow up is limited and similar between groups. |
Unlikely. |
|
Charpin, 1995 |
Not described |
Unclear, not described |
Unlikely, the study is double-blind, double-dummy. |
Unlikely, the study is double-blind. |
Unlikely, the study is double-blind. |
Likely, some outcome measures presented in the results were not specified in the Methods. Extra analysis “for the population of patients included in the efficacy analysis” was not described. |
Likely, the number of analysed/excluded patients differs between outcome measures. |
Unclear. |
|
Berger, 2003 |
Not described |
Unlikely, the study was double-blind and placebo controlled |
Unlikely, the study was double-blind and placebo controlled |
Unlikely, the study was double-blind and placebo controlled |
Unlikely, the study was double-blind and placebo controlled |
Unlikely, all outcome measures described in the methods are quantified in the results. |
Unlikely, loss to follow up was limited and similar between groups. |
Unlikely, primary analysis was an intent-to-treat analysis. |
|
Corren, 2005 |
“A computer-generated randomization schedule was used to assign eligible patients to the 2 treatment groups in blocks of 4.” |
Unlikely, “blinding of the study was preserved at each study site until all patients had completed the study and the database was locked.” |
Unlikely, the study was double-blind and placebo controlled |
Unlikely, the study was double-blind and placebo controlled |
Unlikely, the study was double-blind and placebo controlled |
Unlikely, all outcome measures described in the methods are quantified in the results. |
Unlikely, loss to follow up was limited and similar between groups. |
Unlikely, “the primary analysis was an intent-to-treat analysis”. |
|
Berger 2006 |
“Patients who met the inclusion and exclusion criteria were randomized to treatment groups by means of a computergenerated randomization schedule” |
Unlikely, “access to the random code was confidential and accessible only to authorized persons who were not involved in the study. Blinding of the study was preserved at each study site until all the patients completed the study and the database was locked.” |
Unlikely |
Unlikely |
Unlikely |
Unlikely, all outcome measures described in the methods are quantified in the results. |
Unlikely, loss to follow up is limited and similar between groups. |
Unlikely, the authors clearly indicate the group was analysed with intention-to-treat. However, additional analysis was performed were statistical significance was not obtained with the ITT population. |
|
Ellis, 2013 |
“Randomization occurred in a 1:1:1:1 ratio” |
Unlikely, study was double blind and placebo-controlled. |
Unlikely |
Unlikely |
Unlikely |
Unlikely, all outcome measures described in the methods are quantified in the results. |
Unlikely |
Unlikely |
PICO 4 Leukotrieën receptor antagonisten versus placebo
PICO 5 Leukotrieën receptor antagonisten versus antihistaminica
Evidence table for systematic review of RCTs and observational studies (intervention studies)
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C) |
Follow-up |
Outcome measures and effect size |
Comments |
|
Wei, 2016
|
SR and meta-analysis of RCTs
Literature search up to March 2016
A: Meltzer, 2000 B: Philip, 2002 C: Adelsberg, 2003a D: Adelsberg, 2003b E: Lu, 2009a F: Lu, 2009b G: Nayak, 2002 H: van Cauwenberge, 2000 I: Philip, 2004 J: Holmberg, 2009 K: Bachert, 2009 L: Bousquet, 2010 M: Jauregui, 2011 N: Pradalier, 2007 O: Patel, 2005 P: Demoly: 2009
Study design: RCT
Setting and Country:
Source of funding and conflicts of interest: (commercial / non-commercial / industrial co-authorship)
|
Inclusion criteria SR: 1) randomised controlled trail (RCT) design; 2) patients with SAR or PAR. 3) with specific endpoint evaluation standards; 4) provision of efficacy or safety information; 5) comparing SAHs with Montelukast or placebo, or Montelukast with placebo or combination therapy; 6) data present as (mean +/- SD) or data to estimate them.
Exclusion criteria SR: 1) reviews, systematic reviews or Meta-analysis; 2) duplicate reports or reports on same subjects; 3) if providing insufficient data; 4) if focused on patient with asthma; 5) if combined therapy contained interventions other than SAHs and Montelukast.
8 studies included
Important patient characteristics at baseline:
N, mean age A: 278, 34 yrs B: 1312, 36 yrs C: 1079, 36 yrs D: 1295, 36 yrs E: 285, 35 yrs F: 281, 35 yrs G: 605, 37 yrs O: 1992, 36 yrs
Groups comparable at baseline? |
Describe intervention:
A: Montelukast 10 mg; B: Montelukast 10 mg; C: Montelukast 10 mg; D: Montelukast 10 mg; E: Montelukast 10 mg; F: Montelukast 10 mg; G: Montelukast 10 mg; O: Montelukast 10 mg; |
Describe control:
A: Loratadine 10 mg/placebo; B: Loratadine 10 mg/placebo; C: Loratadine 10 mg/placebo; D: Loratadine 10 mg/placebo; E: Loratadine 10 mg/placebo; F: Loratadine 10 mg/placebo; G: Loratadine 10 mg/placebo; O: Placebo |
End-point of follow-up:
A: 2 weeks B: 2 weeks C: 4 weeks D: 2 weeks E: 2 weeks F: 2 weeks G: 2 weeks H: 2 weeks I: 2 weeks J: 3 weeks K: 2 weeks L: 4 weeks M: 2 weeks N: 2 weeks O: 2 weeks P: 2 weeks
For how many participants were no complete outcome data available? (intervention/control) A: - B: - C: - D: - E: - F: - G: - O:-
|
1. Montelukast versus placebo
Outcome measure-1 CSS (composite symptoms score) Defined as mean DNSS and NNSS (4 point scale 0-3)
Pooled effect (fixed effects model): -0.11 (95% CI -0.15 to -0.06) favoring Montelukast Heterogeneity (I2): 0%
Outcome measure-2 RQLQ (rhino conjunctivitis quality of life questionnaire)
Pooled effect (fixed effects model): -0.20 (95% CI -0.26 to -0.13) favoring Montelukast Heterogeneity (I2): 5%
Outcome measure-1 CSS (composite symptoms score) Defined as mean DNSS and NNSS (4 point scale 0-3)
Pooled effect (random effects model / fixed effects model): -0.03 (95% CI -0.01 o 0.07) Heterogeneity (I2): 0|%
Outcome measure-2 RQLQ (rhino conjunctivitis quality of life questionnaire)
Pooled effect (fixed effects model): 0.09 (95% CI 0.02 to 0.15) Heterogeneity (I2):0%
|
Facultative:
Brief description of author’s conclusion
Personal remarks on study quality, conclusions, and other issues (potentially) relevant to the research question
Level of evidence: GRADE (per comparison and outcome measure) including reasons for down/upgrading
|
Table of quality assessment for systematic reviews of RCTs and observational studies
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/notapplicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Wei, 2016 |
Yes |
Yes |
Yes |
Yes |
NA |
Yes, however incomplete? |
Yes |
Yes |
No |
Evidence table for intervention studies
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Rajashekar, 2018 |
‘The enrolled subjects were randomly divided into three groups and were received following oral treatment for 3 weeks, once daily post cibum at 8:00 PM.’ |
Unclear |
Unlikely, Drug treatment was double-blinded for both investigators and patients. |
Unlikely |
Unlikely, Drug treatment was double-blinded for both investigators and patients. |
unclear |
Unclear |
unclear |
|
Kaur, 2017 |
‘All the patients were randomly divided into five groups of 25 each’ |
Unclear |
unclear |
unclear |
unclear |
unlikely |
unlikely |
unlikely |
|
Andhale, 2016 |
‘Randomly studied’ |
Unclear |
Likely |
Likely |
Likely |
Unlikely |
Unclear |
Unlikely |
|
Ciebada, 2011 |
‘Each patient received medications in crossover, blinded fashion.’ |
Unclear |
Unlikely |
Unlikely |
Unlikely |
Unclear |
Unclear |
Unlikely |
|
Bisgaard, 2009 |
unclear |
unclear |
unlikely |
unlikely |
unlikely |
Unclear |
unclear |
unclear |
|
Okubo, 2008 |
‘The patients were randomized in a 1 : 1 : 1 ratio to receive either montelukast 5 mg, 10 mg, or placebo groups.’ |
Unclear |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
Unclear |
Likely |
|
Chen, 2006 |
‘A randomized, double-blind, placebo-controlled, parallel-group study design was used. Patients were divided into three groups, 20 for each group, (…)‘ |
Unclear |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
Unlikely |
|
Ciebada, 2006 |
‘Eligible patients were randomly assigned to the group receiving montelukast alone, desloratadine alone, both agents, or placebo (20 patients) or to the group receiving montelukast alone, levocetirizine alone, both agents, or placebo (20 patients). The sequence of treatment was randomly assigned.’ |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
unlikely |
|
Lombardo, 2006 |
Not described. |
Unclear |
unlikely |
unlikely |
unlikely |
unlikely |
Unlikely |
unclear (Itt, however excluded patients from analysis: lack of efficacy and protocol deviation, small number of patients, all study arms) |
|
Patel, 2005
|
“After a single-blind, placebo run-in period of 5 to 7 days, patients were randomized (using a computer-generated schedule) |
Unlikely |
unlikely |
unlikely |
Unlikely |
Unlikely |
Unlikely |
Unlikely (stopped lack of efficacy in both groups) |
|
Philip, 2005 |
‘Following a singleblind placebo run-in period of 3 days–5 days, patients were randomized (using a computer-generated schedule) to oral montelukast 10 mg (n = 415) or placebo (n = 416) daily at bedtime during the 2-week active-treatment period.’ |
Unlikely |
Unlikely, double-blind, double-dummy study |
Unlikely, double-blind, double dummy study |
Unlikely, double blind, double dummy study |
Unlikely |
Unlikely |
Unclear (excluded patients from analysis: lack of efficacy and protocol deviation, however small number of patients, all study arms) |
|
Hsieh, 2004 |
‘By means of a computer-generated randomization code, survey-qualifying patients were assigned to one of the three treatment groups’ |
Unlikely |
Unlikely, double-blinded, parallel-group, placebo-controlled design |
Unlikely, double-blinded, parallel-group, placebo-controlled design |
Unlikely, double-blinded, parallel-group, placebo-controlled design |
Unlikely |
Unclear |
Unlikely |
|
Kurowski, 2004 |
‘Patients were randomly assigned to one of the following treatments:group A, placebo for both cetirizine and montelukast before the expected beginning of grass pollen season (i.e. before May 15) and active cetirizine 10mg/day plus active montelukast 10mg/day after May 15; group B, active montelukast 10mg/day plus placebo for cetirizine; group C, active cetirizine 10mg/day plus placebo for montelukast; and group D, active cetirizine 10mg/day plus active montelukast 10mg/day throughout the study period.’ |
unclear |
Unlikely, double blind, parallel-group, placebo-controlled study |
Unlikely, double blind, parallel-group, placebo-controlled study |
Unlikely, double blind, parallel-group, placebo-controlled study |
unlikely |
Likely, reasons for study discontinuation : exacerbation of symptoms, violation of protocol, Lost to follow-up |
Unlikely |
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Azelastine fluticasone versus nasale corticosteroïden (PICO 1) |
|
|
Bousquet 2018 |
No total symptom score provided, focuses on reactions after 5 minutes |
|
Meltzer 2012 |
Included in publication Carr, 2012 |
|
Meltzer 2013 |
Post hoc anaylsis Hampel. 2010; does not include prioritised outcomes |
|
Orale antihistaminica versus nasale corticosteroïden of nasale antihistaminica (PICO 2 en 3) |
|
|
Bousquet 2018 |
Voldoet niet aan PICO: andere interventies |
|
Dahl 2004 |
Fulltekst alleen in Duits beschikbaar |
|
Sheikh 2007 |
Fulltekst niet beschikbaar |
|
Benninger 2010 |
Review met te nauwe inclusiecriteria |
|
Zhonghua 2014 |
Fulltekst alleen in Chinees beschikbaar |
|
Parle-Pechera 2012 |
Geen review of origineel artikel |
|
Górska-Ciebiada 2004 |
Fulltekst alleen in Pools beschikbaar |
|
Korsgren 2007 |
Allergeen provocatie studie |
|
Takahashi 2012 |
Voldoet niet aan PICO: andere interventies |
|
Zieglmayer 2005 |
Voldoet niet aan PICO: andere interventies |
|
Howard 2001 |
Kosten-effectiviteitsanalyse |
|
Feng 2016 |
Voldoet niet aan PICO: andere interventies |
|
Juel-Berg 2017 |
Review met te nauwe inclusiecriteria, individuele studies uit review zijn geïncludeerd |
|
May 2017 |
Overview, geen systematisch review |
|
Seresirikachorn 2018 |
Voldoet niet aan PICO: andere interventies |
|
Yanai 2012 |
Overview, geen systematisch review |
|
Azevedo 1995 |
Geen systematisch review of originele studie |
|
Lange 2003 |
Fulltekst in Duits |
|
Noble 1995 |
Overview, geen systematisch review |
|
Sheikh 2009 |
Review met beperkte rapportage, , individuele studies uit review zijn geïncludeerd |
|
Storms 1995 |
Overview, geen systematisch review |
|
Waddell 2003 |
Overview, geen systematisch review |
|
Weinberger 2018 |
Overview, geen systematisch review |
|
Livostin Study Group 1993 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Bahmer 1994 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Bernstein 1996 |
Geïncludeerd in review Weiner |
|
Bronsky 1996 |
Geïncludeerd in review Weiner |
|
Brooks 1996 |
Geïncludeerd in review Weiner |
|
Bunnag 1992 |
Geïncludeerd in review Weiner |
|
Choo 1998 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Frantz 2002 |
Geen fulltekst beschikbaar |
|
Gehanno 1997 |
Geïncludeerd in review Weiner |
|
Gaspar 1994 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Greiff 1997 |
Provocatiestudie |
|
Hilberg 1995 |
Provocatiestudie |
|
Jacobi 1999 |
Provocatiestudie |
|
Juniper 1997 |
Voldoet niet aan PICO: andere interventies |
|
Karaayvaz 1995 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Kozma 1996 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Kremer 1996 |
Voldoet niet aan PICO: andere interventies |
|
LaForce 1996 |
Chlorpheniramine maleate wordt niet meer gebruikt in Nederland |
|
LaForce 2004 |
Voldoet niet aan PICO: andere interventies |
|
Lau 1990 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Li 2018 |
Voldoet niet aan PICO: andere interventies |
|
Meltzer 1994 |
Chlorpheniramine maleate wordt niet meer gebruikt in Nederland |
|
Mosges 1995 |
Fulltekst in Duits |
|
Odeback 1994 |
Voldoet niet aan PICO: andere interventies |
|
Osuna 1998 |
Fulltekst in Spaans |
|
Schoenwetter 1995 |
Geïncludeerd in review Weiner |
|
Simpson 1993 |
Geïncludeerd in review Weiner |
|
Sohoel 1993 |
Terfenadine is niet meer op de markt vanwege bijwerkingen |
|
Storms 1994 |
Chlorpheniramine maleate wordt niet meer gebruikt in Nederland |
|
Van Bavel 1994 |
Geïncludeerd in review Weiner |
|
Vervloet 1997 |
Geïncludeerd in review Weiner |
|
Wang 1996 |
Provocatie studie |
|
Weiler 1997 |
Voldoet niet aan PICO: andere interventies |
|
Leukotrieën receptor antagonisten versus antihistaminica of placebo (PICO 4 en 5) |
|
|
Erdogan 2014 |
Voldoet niet aan PICO: andere interventies |
|
Segundo 2009 |
Populatie per arm n<10 patiënten |
|
Tatar 2013 |
Voldoet niet aan PICO: andere interventies |
|
Sardana 2010 |
Voldoet niet aan PICO: alleen RSS score gerapporteerd voor azelastine versus budesonide en niet voor leukotrieën |
|
Barnes 2007 |
Voldoet niet aan PICO: andere interventies |
|
Benítez 2005 |
Voldoet niet aan PICO: andere interventies |
|
Dumitru 2011 |
Voldoet niet aan PICO: andere interventies |
|
Esteitie 2010 |
Voldoet niet aan PICO: andere interventies |
|
Gotoh 2012 |
Artificial exposure chamber |
|
Jindal 2016 |
Voldoet niet aan PICO: andere interventies |
|
Keskin 2006 |
Voldoet niet aan PICO: andere interventies |
|
Krug 2014 |
Voldoet niet aan PICO: andere interventies |
|
Li 2009 |
Voldoet niet aan PICO: andere interventies |
|
Phipatanakul 2000 |
Voldoet niet aan PICO: cat-induced astma |
|
Pullerits 1999 |
Zafirlukast is niet verkrijgbaar in Nederland |
|
Wakabayashi 2012 |
Klinische setting (exposure chamber) |
|
Wilson 2001a |
Voldoet niet aan PICO: andere interventies |
|
Wilson 2001b |
Voldoet niet aan PICO: andere interventies |
|
Chen 2018 |
Voldoet niet aan PICO: andere interventies |
|
Cingi 2010 |
Voldoet niet aan PICO: andere interventies |
|
Day 2009a |
Voldoet niet aan PICO: andere interventies |
|
Day 2009b |
Voldoet niet aan PICO: andere interventies |
|
Ho 2007 |
Zafirlukast is niet verkrijgbaar in Nederland |
|
Horak 2010 |
Voldoet niet aan PICO: andere interventies |
|
Jiang 2006 |
Zafirlukast is niet verkrijgbaar in Nederland |
|
Lu 2009 |
Geïncludeerd in review Wei 2016 |
|
Patel 2009 |
Geïncludeerd in review Wei 2016 |
|
Prenner 2009 |
Voldoet niet aan PICO: andere interventies |
|
Sasaki 2009 |
Pranlukast is niet beschikbaar in Nederland |
|
Watabasinsuru 2008 |
Voldoet niet aan PICO: andere interventies |
|
Yonekura 2009 |
Pranlukast is niet beschikbaar in Nederland |
|
Goh 2014 |
Voldoet niet aan PICO: andere interventies |
|
Ciebiada 2008 |
Voldoet niet aan PICO: andere interventies |
|
Ciebiada 2009 |
Voldoet niet aan PICO: andere interventies |
|
Dalgic 2017 |
Voldoet niet aan PICO: andere interventies |
|
Day 2008 |
Klinische setting (exposure chamber) |
|
Di Lorenzo 2004 |
Voldoet niet aan PICO: andere interventies |
|
Donnelly 1995 |
ICI 204,219 niet beschikbaar in Nederland |
|
Feng 2015 |
Voldoet niet aan PICO: andere interventies |
|
Erdogan 2014 |
Voldoet niet aan PICO: andere interventies |
|
Gonyeau 2003 |
Recenter review beschikbaar |
|
Grainger 2006 |
Recenter review beschikbaar |
|
Haberal 2003 |
Recenter review beschikbaar |
|
Ichimaru 2008 |
Populatie per arm n<10 patiënten |
|
Lal 2004 |
Geen originele studie of systematisch review |
|
Knapp 1990 |
Populatie per arm n<10 patiënten |
|
Liu 2018 |
Voldoet niet aan PICO: andere interventies |
|
Lu 2014 |
Chinees |
|
Lee 2004 |
Voldoet niet aan PICO: andere uitkomstmaat |
|
Lopez 2004 |
Recenter review beschikbaar |
|
Maynard 2001 |
Recenter review beschikbaar |
|
Martin 2006 |
Voldoet niet aan PICO: andere interventies |
|
Nathan 2003 |
Recenter review beschikbaar |
|
Meltzer 2000 |
Geïncludeerd in review Wei 2016 |
|
Nayak 2007 |
Recenter review beschikbaar |
|
Moinuddin 2004 |
Voldoet niet aan PICO: andere interventies |
|
Philip 2009 |
Voldoet niet aan PICO: populatie niet specifiek allergie patiënten |
|
Nathan 2007 |
Voldoet niet aan PICO: populatie niet specifiek allergie patiënten |
|
Nayak 2002 |
Geïncludeerd in review Wei 2016 |
|
Rachelefsky 2013 |
Recenter review beschikbaar |
|
Rodrigo 2006 |
Recenter review beschikbaar |
|
Okubo 2017 |
Pranlukast is niet beschikbaar in Nederland |
|
Philip 2002 |
Geïncludeerd in review Wei 2016 |
|
Sheikh 2009 |
Recenter review beschikbaar |
|
Sheikh 2007 |
Recenter review beschikbaar |
|
Prenner 2002 |
Voldoet niet aan PICO: andere interventies |
|
Ratner 2003 |
Voldoet niet aan PICO: andere interventies |
|
Saengpanich 2003 |
Voldoet niet aan PICO: andere interventies |
|
Saverno 2009 |
Economische evaluatie |
|
Wallace 2017 |
Richtlijn |
|
Wilson 2002 |
Voldoet niet aan PICO: andere interventies |
|
Xiao 2016 |
Review Wei 2016 bevat bredere PICO |
|
Xu 2014 |
Review Wei 2016 bevat bredere PICO |
|
Yonekura 2012 |
Pranlukast is niet beschikbaar in Nederland |
|
Van Adelsberg 2003a |
Geïncludeerd in review Wei 2016 |
|
Van Adelsberg 2003b |
Geïncludeerd in review Wei 2016 |
|
Wilson 2001 |
Voldoet niet aan PICO: andere interventies |
|
Yoshihara 2017 |
Voldoet niet aan PICO: andere interventies |
|
Nathan 2005 |
Letter to the editor |
|
Glacy 2013 |
Recenter review beschikbaar |
|
Kim 2018 |
Voldoet niet aan PICO: andere interventies |
|
Meltzer 2002 |
Recenter review beschikbaar |
|
Storms 2007 |
Geen originele studie of systematisch review |
|
Sander 2002 |
Editorial |
Beoordelingsdatum en geldigheid
Publicatiedatum : 18-12-2019
Beoordeeld op geldigheid : 12-02-2020
Uiterlijk in 2024 bepaalt het bestuur van de Nederlandse Vereniging voor Keel-Neus-Oorheelkunde en Heelkunde van het Hoofd-Halsgebied of de modules van deze richtlijn nog actueel zijn. Op modulair niveau is een onderhoudsplan beschreven. Bij het opstellen van de richtlijn heeft de werkgroep per module een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De Nederlandse Vereniging voor Keel-Neus-Oorheelkunde en Heelkunde van het Hoofd-Halsgebied is regiehouder van deze richtlijn en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied. Op modulair niveau is een onderhoudsplan beschreven.
|
Module |
Regiehouder(s) |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn |
Frequentie van beoordeling op actualiteit |
Wie houdt er toezicht op actualiteit |
Relevante factoren voor wijzigingen in aanbeveling |
|
Medicamenteuze behandeling |
NVKNO |
2019 |
2021 |
Twee jaarlijks |
NVKNO |
Beschikbaarheid nieuwe middelen |
Algemene gegevens
Deze richtlijn kreeg goedkeuring door de Vereniging van Allergie Patiënten (VAP) en de Patiëntenfederatie Nederland.
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijn.
Doel en doelgroep
Doel
Deze multidisciplinaire richtlijn beoogd de kwaliteit van zorg en het komen tot een goede klinische besluitvorming bij de behandeling van allergie van de bovenste luchtwegen op basis van de huidige wetenschappelijke inzichten te verhogen. Deze zorg behelst zowel de diagnostiek als behandeling van de allergie en aandacht voor de bijkomende co-morbiditeit. Daarnaast beoogt deze richtlijn een uniform beleid ten aanzien van het doorverwijzen en terugverwijzen van de patiënt te geven. Deze zorg wordt geleverd in de eerste lijn en in verschillende groepen binnen tweede en derde lijn, vandaar de samenwerking tussen huisartsen, KNO-artsen, allergologen, kinderartsen, oogartsen, longartsen en apothekers. Dit schept meer duidelijkheid voor patiënten, verwijzers en zorgverzekeraars.
Doelgroep
Deze richtlijn is geschreven voor alle leden van de beroepsgroepen die betrokken zijn bij de tweede en derde lijn zorg voor volwassenen en kinderen met allergische klachten van de bovenste luchtwegen. Voor de eerste lijn wordt verwezen naar de NHG-Standaard ‘Allergische en niet-allergische rhinitis’.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2018 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de zorg voor patiënten met allergie van de bovenste luchtwegen te maken hebben.
De werkgroepleden zijn door hun beroepsverenigingen gemandateerd voor deelname. De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Werkgroep
- Dr. R.J.H. (Robbert) Ensink, KNO-arts, Gelre ziekenhuizen, Zutphen, Nederlandse Vereniging voor Keel-Neus-Oorheelkunde en Heelkunde van het Hoofd-Halsgebied (voorzitter)
- Prof. Dr. W.J. (Wytske) Fokkens, KNO-arts, Academisch Medisch Centrum Amsterdam, Nederlandse Vereniging voor Keel-Neus-Oorheelkunde en Heelkunde van het Hoofd-Halsgebied
- Dr. C.H.M. (Cor) Stengs, KNO-arts, Rijnstate Ziekenhuizen, Arnhem, Nederlandse Vereniging voor Keel-Neus-Oorheelkunde en Heelkunde van het Hoofd-Halsgebied
- Dr. H. (Hans) de Groot, allergoloog, Reinier de Graaf Groep, Delft, Nederlandse Vereniging voor Allergologie en Klinische Immunologie en Nederlandse Vereniging voor Kindergeneeskunde
- Dr. J.A. (Joost) Aalberse, huisarts, Huisartsenpraktijk Postjesweg, Amsterdam, Nederlands Huisartsen Genootschap
- Dr. Ir. J. (Jasper) Kappen, longarts, Franciscus Gasthuis & Vlietland, Rotterdam, Vereniging van Artsen voor Longziekten en Tuberculose
- Dr. K. (Khaled) Mansour, oogarts, Tjongerschans, Heerenveen, Nederlands Oogheelkundig Gezelschap
- Dr. O. (Olivia) Liem, kinderarts-allergoloog i.o., Sophia Kinderziekenhuis Erasmus MC, Rotterdam, Nederlandse Vereniging voor Kindergeneeskunde
- Y. (Yola) de Vries, Secretaris bij de Vereniging van Allergie Patiënten (VAP), Wijk aan Zee, Vereniging van Allergie Patiënten
Meelezer
- Drs. M.A.G.E. (Michiel) Bannier (meelezer), kinderarts-pulmonoloog, Maastricht UMC, Maastricht, Nederlandse Vereniging voor Kindergeneeskunde
Met ondersteuning van
- D. (Dieuwke) Leereveld, MSc., adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- Dr. S.N. (Stefanie) Hofstede, adviseur, Kennisinstituut van de Federatie Medisch Specialisten
- I. (Ingeborg) van Dusseldorp, literatuurspecialist, Van Dusseldorp, Delvaux & Ket
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Ensink (voorzitter) |
KNO-arts in Gelre ziekenhuizen, locatie Zutphen |
Lid Adviescommissie Richtlijn Kennisinstituut Medisch Specialisten Lid Richtlijn commissie NVKNO |
Geen |
Geen |
|
Fokkens |
Hoogleraar Keel-neus-Oorheelkunde Academisch Medisch Centrum Amsterdam |
Secretaris generaal ERS: onbetaald Editor in Chief Rhinology: onbetaald Editor in Chief Rhinology online: onbetaald Accociate Editor Allergy: betaald |
Werkgroep ARIA EUFOREA
ZonMw MEDA EU Studies 1 en 2 zijn in patiënten met rhinitis en kunnen m.i. gezien worden als COI. Studies 3-5 zijn studies naar patiënten met neuspoliepen. 1. BM4SIT: Novel concept for Birch Pollen Allergy Treatment (immunotherapie hooikoorts, voornamelijk berkenpollen: EU, AMC (Prof. R. van Ree) is coördinerend centrum, geïnitieerd vanuit de industrie (Biomay AG) ondersteund door EU 2. Control in patients with allergic rhinitis (hooikoorts): MEDA (principal investigator) 3. Clinical Benefit and cost effectiveness of endoscopic sinus surgery (ESS) in adult patients with chronic rhinosinusitis with nasal polyps (CRSwNP): ZonMw (principal investigator) 4. Een gerandomiseerd, dubbelblind, placebo gecontroleerd, fase II onderzoek om de behandeling met meerdere doses AK001 bij patiënten met matige tot ernstige polyposis nasi te beoordelen: Allakos (deelnemend centrum) 5. A randomized, 24-week treatment, double-blind, placebo-controlled efficacy and safety study of dupilumab 300 mg every other week, in patients with bilateral nasal polyposis on a background therapy with intranasal corticosteroids: Sanofi (deelnemend centrum) |
De belangen zijn besproken. Betrokkene participeert vanwege specifieke expertise op het gebied van allergie in de keel, neus en oorheelkunde |
|
Stengs |
Lid vakgroep KNO en HHO chirurgie Rijnstate Ziekenhuizen als KNO arts/ aangezichtschirurg |
Voorzitter vakgroep: onbetaald lid allergie werkgroep: onbetaald lid aangezichtswerkgroep: onbetaald Docent masterclasses allergische rhinitis met vergoeding volgens vacatieregeling (visitatie) commentaar ronde NHG standaard allergische rhinitis: onbetaald |
Masterclasses Allergie opgestart om te komen tot een richtlijn voor de allergie van de bovenste luchtwegen en een uniform behandelplan voor deze aandoening voor de eerste en tweede lijn. |
Geen |
|
De Groot |
Allergoloog Reinier de Graaf Groep, Delft |
Jaarlijkse organisatie en geven van een masterclass voor KNO-artsen, deels gefinancierd door ALK Abello en MILAN |
Deelname aan de RELIEF studie, een postmarketing onderzoek met de huisstofmijt tablet (Sublinguale immunotherapie), gefinancierd door ALK Abello |
Geen actie. Er komt een aparte richtlijn voor immunotherapie, waardoor dit onderwerp buiten de afbakening van de richtlijn valt. |
|
Aalberse |
Huisarts Huisartsenpraktijk Postjesweg Amsterdam |
NHG werkgroep, ZZP werkzaam tegen betaling en arts allergoloog bij DC klinieken. Maar sinds uitbreiding werkzaamheden als huisarts sinds 2018 niet meer daar gewerkt. |
Geen |
Geen |
|
Kappen |
Longarts Franciscus |
Honorary Staff Member lmmunotollerance group Imperia! college Londen (onbetaald) Lid: EAACI Task Force Biomarkers in Immunotherapie (onbetaald) Lid: Commissie kwaliteit NVAL T (onbetaald) Lid: Sectie Astma NVAL T (onbetaald) |
2017 (eenmalig): ALK adviesraad biomarkers in immunotherapie |
Geen actie. Lid adviesraad is twee jaar geleden gestopt. |
|
Mansour |
Vertegenwoordiging van het NOG: Nederlands Oogheelkunde Gezelschap. Werkzaam in Tjongerschans ziekenhuis Heerenveen Nijsmellinghe Drachten |
Voorzitter OOG (wetenschappelijke vereniging), lid Fischerstichting, lid Catharina heerd stichting, advieslid Sjogren’s patiëntenvereniging, alle nevenwerkzaamheden zijn onbetaald |
Geen |
Geen |
|
Liem |
Kinderarts-allergoloog i.o., Sophia Kinderziekenhuis Erasmus MC |
Geen |
Geen |
Geen |
|
De Vries |
Secretaris bij de Vereniging van Allergie Patiënten |
Secretaris is vrijwilligerswerk, onbetaald. |
Geen |
Geen |
|
Bannier (meelezer) |
Kinderarts-pulmonoloog Maastricht UMC+ |
Geen |
Geen |
Geen |
|
Leereveld (Kennisinstituut) |
Adviseur Kennisinstituut van de Federatie Medisch Specialisten |
Geen |
Geen |
Geen |
|
Hofstede (Kennisinstituut) |
Adviseur Kennisinstituut van de Federatie Medisch Specialisten |
Geen |
Geen |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde van de Vereniging van Allergie Patiënten (VAP) in de werkgroep. De VAP heeft input gegeven tijdens de knelpuntenanalyses en op de teksten, waaronder de overwegingen. Daarnaast is er een module opgenomen over zelfmedicatie. De conceptrichtlijn is tevens voor commentaar voorgelegd aan de VAP.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijn (module) en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. Het implementatieplan is te vinden bij de aanverwante producten. De werkgroep heeft besloten geen indicatoren te ontwikkelen bij de huidige richtlijn, omdat er of geen substantiële barrières konden worden geïdentificeerd die implementatie van de aanbeveling zouden kunnen bemoeilijken.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. Tevens zijn er knelpunten aangedragen door Nederlandse Vereniging voor Keel-Neus-Oorheelkunde en Heelkunde van het Hoofd-Halsgebied (NVKNO), het Nederlands Huisartsen Genootschap (NHG), Verpleegkundigen & Verzorgenden Nederland (V&VN), Nederlands Oogheelkundig Gezelschap (NOG), College van Medisch Immunologen (CMI), Zorginstituut Nederland (ZiNL), Artsenfederatie (KNMG), de Koninklijke Nederlandse Maatschappij ter bevordering der Pharmacie (KNMP) en Nederlandse Vereniging voor Klinische Chemie en Laboratoriumgeneeskunde (NVKC) via een invitational conference. Een verslag hiervan is opgenomen onder aanverwante producten.
Uitgangsvragen en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de voorzitter en de adviseur concept-uitgangsvragen opgesteld. Deze zijn met de werkgroep besproken waarna de werkgroep de definitieve uitgangsvragen heeft vastgesteld. Vervolgens inventariseerde de werkgroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Er werd eerst oriënterend gezocht naar bestaande buitenlandse richtlijnen en systematische reviews in Medline en Embase. Vervolgens werd voor de afzonderlijke uitgangsvragen aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie voor de oriënterende zoekactie en patiëntenperspectief zijn opgenomen onder aanverwante producten.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR – voor systematische reviews en Cochrane – voor gerandomiseerd gecontroleerd onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A) Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk* |
|
|
Laag |
|
|
Zeer laag |
|
*in 2017 heeft het Dutch GRADE Network bepaalt dat de voorkeursformulering voor de op een na hoogste gradering ‘redelijk’ is in plaat van ‘matig’
B) Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008), en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de cruciale uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk om mee te wegen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag. Meer algemene, overkoepelende, of bijkomende aspecten van de organisatie van zorg worden behandeld in de module Organisatie van zorg.
Indicatorontwikkeling
Gelijktijdig met het ontwikkelen van de conceptrichtlijn heeft de werkgroep overwogen om interne kwaliteitsindicatoren te ontwikkelen om het toepassen van de richtlijn in de praktijk te volgen en te versterken. Meer informatie over de methode van indicatorontwikkeling is op te vragen bij het Kennisinstituut van de Federatie Medisch Specialisten. De werkgroep heeft besloten geen indicatoren te ontwikkelen bij de huidige richtlijn, omdat er of geen substantiële barrières konden worden geïdentificeerd die implementatie van de aanbeveling zouden kunnen bemoeilijken.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Brouwers, M. C., Kho, M. E., Browman, G. P., Burgers, J. S., Cluzeau, F., Feder, G., ... & Littlejohns, P. (2010). AGREE II: advancing guideline development, reporting and evaluation in health care. Canadian Medical Association Journal, 182(18), E839-E842.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwaliteit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van Medisch Specialisten.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann, H. J., Oxman, A. D., Brozek, J., Glasziou, P., Jaeschke, R., Vist, G. E., ... & Bossuyt, P. (2008). Rating Quality of Evidence and Strength of Recommendations: GRADE: Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ: British Medical Journal, 336(7653), 1106.
Wessels, M., Hielkema, L., & van der Weijden, T. (2016). How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. Journal of the Medical Library Association: JMLA, 104(4), 320.
Zoekverantwoording
PICO 1: Azelastine fluticasone versus nasale corticosteroïden
|
Database |
Zoektermen |
Totaal |
||||||||||||||||||||||||||||||||||||||||||
|
Seach 2010 - 3 november 2018 |
||||||||||||||||||||||||||||||||||||||||||||
|
Embase |
|
|
||||||||||||||||||||||||||||||||||||||||||
|
#8 |
#6 AND 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti |
301 |
||||||||||||||||||||||||||||||||||||||||||
|
#7 |
#6 AND ('meta analysis'/de OR cochrane:ab OR embase:ab OR psychlit:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT ('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) |
|
||||||||||||||||||||||||||||||||||||||||||
|
62#6 |
#5 NOT ‘conference abstract’/it |
646 |
||||||||||||||||||||||||||||||||||||||||||
|
#5 |
#4 AND (1-1-2010)/sd |
1050 |
||||||||||||||||||||||||||||||||||||||||||
|
#4 |
#1 AND #2 AND #3 |
2185 |
||||||||||||||||||||||||||||||||||||||||||
|
#3 |
(‘steroid’/exp OR steroid:ti,ab ORcorticosteroïd*:ti,ab OR glucocorticoid*:ti,ab OR beclomethason*:ti,ab OR fluticason*:ti,ab OR triamcinolon*:ti,ab OR budesonid*:ti,ab OR mometason*:ti,ab OR dexamethason*:ti,ab OR flunisolide:ti,ab OR ciclesonide:ti,ab OR 'adrenal cortex hormone':ti,ab OR 'adrenal cortex hormones':ti,ab OR 'corticosteroïd'/exp) AND ('intranasal drug administration'/exp OR intranasal:ti,ab OR endonasal*:ti,ab OR nose:ti,ab OR nasal*:ti,ab) |
18328 |
||||||||||||||||||||||||||||||||||||||||||
|
#2 |
'azelastine'/exp OR '4 (4 chlorobenzyl) 2 (hexahydro 1 methyl 1h azepin 4 yl) 1 (2h) phthalazinone':ti,ab OR 'a 05610':ti,ab OR 'a 5610':ti,ab OR 'a05610':ti,ab OR 'a5610':ti,ab OR 'afluon':ti,ab OR 'alastine':ti,ab OR 'alerdual':ti,ab OR 'alergodil':ti,ab OR 'allergodil':ti,ab OR 'allergodrop':ti,ab OR 'allergospray':ti,ab OR 'allespray':ti,ab OR 'amelor':ti,ab OR 'astelin':ti,ab OR 'astepro':ti,ab OR 'azela vision':ti,ab OR 'azelastin*':ti,ab OR 'azelavision':ti,ab OR 'azella':ti,ab OR 'azep':ti,ab OR 'carelastin':ti,ab OR 'corifina':ti,ab OR 'e 0659':ti,ab OR 'e0659':ti,ab OR 'lasticom':ti,ab OR 'lastin':ti,ab OR 'loxin':ti,ab OR 'oculastin':ti,ab OR 'optilast':ti,ab OR 'optivar':ti,ab OR 'proallergodil':ti,ab OR 'radethacin':ti,ab OR 'radethazin':ti,ab OR 'rhinolast':ti,ab OR 'rinelaz':ti,ab OR 'tebarat':ti,ab OR 'mp azeflu':ti,ab OR 'dymista':ti,ab OR 'fluticasone'/exp OR 'fluticasone':ti,ab OR 'fluticasone propionate'/exp OR 'aerosona':ti,ab OR 'armonair respiclick':ti,ab OR 'arquist':ti,ab OR 'asmatil':ti,ab OR 'asmo-lavi':ti,ab OR 'asmo-lavi diskus':ti,ab OR 'atemur':ti,ab OR 'axotide':ti,ab OR 'beconase allergy 24 hour':ti,ab OR 'brisovent':ti,ab OR 'casoflune':ti,ab OR 'cci 18781':ti,ab OR 'cci18781':ti,ab OR 'cortifil':ti,ab OR 'cutivat':ti,ab OR 'cutivate':ti,ab OR 'eustidil (fluticasone propionate)':ti,ab OR 'fanipos':ti,ab OR 'flixoderm':ti,ab OR 'flixonase':ti,ab OR 'flixotaide':ti,ab OR 'flixotide':ti,ab OR 'flixovate':ti,ab OR 'floebb inhaler':ti,ab OR 'flonase':ti,ab OR 'flovent':ti,ab OR 'flugenix':ti,ab OR 'flunase':ti,ab OR 'flunutrac':ti,ab OR 'flusonal':ti,ab OR 'fluspiral':ti,ab OR 'flutaide':ti,ab OR 'flutica-teva':ti,ab OR 'fluticrem':ti,ab OR 'flutide':ti,ab OR 'flutinase':ti,ab OR 'flutirin':ti,ab OR 'flutivate':ti,ab OR 'fluxonal':ti,ab OR 'gr 18781':ti,ab OR 'gr18781':ti,ab OR 'inalacor':ti,ab OR 'nasofan':ti,ab OR 'otri allergie (fluticasone propionate)':ti,ab OR 'otri-allergie (fluticasone propionate)':ti,ab OR 'pavetod':ti,ab OR 'pirinase hayfever relief':ti,ab OR 'prutica':ti,ab OR 'reviflut':ti,ab OR 'sonera':ti,ab OR 'trialona':ti,ab OR 'truflo':ti,ab OR 'ubizol':ti,ab OR 'xhance':ti,ab OR 'zoflut':ti,ab |
17126 |
||||||||||||||||||||||||||||||||||||||||||
|
#1 |
'allergic rhinitis'/exp OR 'allergic rhinitis':ti,ab OR 'allergic rhinopathy':ti,ab OR 'eosinophil rhinitis':ti,ab OR 'eosinophile rhinitis':ti,ab OR 'eosinophilic rhinitis':ti,ab OR 'eosinophilous rhinitis':ti,ab OR 'rhinitis allergica':ti,ab OR 'rhinitis eosinophila':ti,ab OR 'rhinitis, allergic':ti,ab OR 'perennial rhinitis`:ti,ab or `pollen allergy`:ti,ab or' OR 'hayfever:ti,ab or `hay fever`:ti,ab or':ti,ab OR 'pollinosis:ti,ab or pollenosis:ti,ab' OR (('hypersensitivity'/exp OR hypersensitiv*:ti,ab OR 'hyperreactivity'/exp OR hyperreactiv*:ti,ab OR allerg*:ti,ab OR rhinit*:ti,ab) AND cat:ti,ab OR cats:ti,ab OR dander:ti,ab OR mite*:ti,ab OR dog*:ti,ab OR ragweed:ti,ab OR pollen:ti,ab OR grass*:ti,ab OR cedar:ti,ab OR alder:ti,ab OR willow:ti,ab OR birch:ti,ab OR mugwort:ti,ab OR tree*:ti,ab OR weed*:ti,ab OR perennial*:ti,ab OR season*:ti,ab OR spring:ti,ab OR summer:ti,ab)) OR (nose:ti,ab OR nasal:ti,ab) AND (allerg*:ti,ab OR hypersensitiv*:ti,ab OR hyperreactiv*:ti,ab)
|
70307 |
||||||||||||||||||||||||||||||||||||||||||
|
PubMed |
|
|
||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
PICO 2 en 3: Montelukast versus placebo of antihistaminica
|
Database |
Zoektermen |
Totaal |
||||||||||||||||||||||||||||||||||||||||||
|
Periode 1998 – 7 november 2018 |
||||||||||||||||||||||||||||||||||||||||||||
|
Embase |
|
|
||||||||||||||||||||||||||||||||||||||||||
|
#8 |
#6 AND 'randomization'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp OR 'placebo'/exp OR 'prospective study'/exp OR rct:ab,ti OR random*:ab,ti OR 'single blind':ab,ti OR 'randomised controlled trial':ab,ti OR 'randomized controlled trial'/exp OR placebo*:ab,ti |
301 |
||||||||||||||||||||||||||||||||||||||||||
|
#7 |
#6 AND ('meta analysis'/de OR cochrane:ab OR embase:ab OR psychlit:ab OR cinahl:ab OR medline:ab OR ((systematic NEAR/1 (review OR overview)):ab,ti) OR ((meta NEAR/1 analy*):ab,ti) OR metaanalys*:ab,ti OR 'data extraction':ab OR cochrane:jt OR 'systematic review'/de) NOT ('animal experiment'/exp OR 'animal model'/exp OR 'nonhuman'/exp) |
|
||||||||||||||||||||||||||||||||||||||||||
|
62#6 |
#5 NOT ‘conference abstract’/it |
646 |
||||||||||||||||||||||||||||||||||||||||||
|
#5 |
#4 AND (1-1-2010)/sd |
1050 |
||||||||||||||||||||||||||||||||||||||||||
|
#4 |
#1 AND #2 AND #3 |
2185 |
||||||||||||||||||||||||||||||||||||||||||
|
#3 |
(‘steroid’/exp OR steroid:ti,ab ORcorticosteroïd*:ti,ab OR glucocorticoid*:ti,ab OR beclomethason*:ti,ab OR fluticason*:ti,ab OR triamcinolon*:ti,ab OR budesonid*:ti,ab OR mometason*:ti,ab OR dexamethason*:ti,ab OR flunisolide:ti,ab OR ciclesonide:ti,ab OR 'adrenal cortex hormone':ti,ab OR 'adrenal cortex hormones':ti,ab OR 'corticosteroïd'/exp) AND ('intranasal drug administration'/exp OR intranasal:ti,ab OR endonasal*:ti,ab OR nose:ti,ab OR nasal*:ti,ab) |
18328 |
||||||||||||||||||||||||||||||||||||||||||
|
#2 |
'azelastine'/exp OR '4 (4 chlorobenzyl) 2 (hexahydro 1 methyl 1h azepin 4 yl) 1 (2h) phthalazinone':ti,ab OR 'a 05610':ti,ab OR 'a 5610':ti,ab OR 'a05610':ti,ab OR 'a5610':ti,ab OR 'afluon':ti,ab OR 'alastine':ti,ab OR 'alerdual':ti,ab OR 'alergodil':ti,ab OR 'allergodil':ti,ab OR 'allergodrop':ti,ab OR 'allergospray':ti,ab OR 'allespray':ti,ab OR 'amelor':ti,ab OR 'astelin':ti,ab OR 'astepro':ti,ab OR 'azela vision':ti,ab OR 'azelastin*':ti,ab OR 'azelavision':ti,ab OR 'azella':ti,ab OR 'azep':ti,ab OR 'carelastin':ti,ab OR 'corifina':ti,ab OR 'e 0659':ti,ab OR 'e0659':ti,ab OR 'lasticom':ti,ab OR 'lastin':ti,ab OR 'loxin':ti,ab OR 'oculastin':ti,ab OR 'optilast':ti,ab OR 'optivar':ti,ab OR 'proallergodil':ti,ab OR 'radethacin':ti,ab OR 'radethazin':ti,ab OR 'rhinolast':ti,ab OR 'rinelaz':ti,ab OR 'tebarat':ti,ab OR 'mp azeflu':ti,ab OR 'dymista':ti,ab OR 'fluticasone'/exp OR 'fluticasone':ti,ab OR 'fluticasone propionate'/exp OR 'aerosona':ti,ab OR 'armonair respiclick':ti,ab OR 'arquist':ti,ab OR 'asmatil':ti,ab OR 'asmo-lavi':ti,ab OR 'asmo-lavi diskus':ti,ab OR 'atemur':ti,ab OR 'axotide':ti,ab OR 'beconase allergy 24 hour':ti,ab OR 'brisovent':ti,ab OR 'casoflune':ti,ab OR 'cci 18781':ti,ab OR 'cci18781':ti,ab OR 'cortifil':ti,ab OR 'cutivat':ti,ab OR 'cutivate':ti,ab OR 'eustidil (fluticasone propionate)':ti,ab OR 'fanipos':ti,ab OR 'flixoderm':ti,ab OR 'flixonase':ti,ab OR 'flixotaide':ti,ab OR 'flixotide':ti,ab OR 'flixovate':ti,ab OR 'floebb inhaler':ti,ab OR 'flonase':ti,ab OR 'flovent':ti,ab OR 'flugenix':ti,ab OR 'flunase':ti,ab OR 'flunutrac':ti,ab OR 'flusonal':ti,ab OR 'fluspiral':ti,ab OR 'flutaide':ti,ab OR 'flutica-teva':ti,ab OR 'fluticrem':ti,ab OR 'flutide':ti,ab OR 'flutinase':ti,ab OR 'flutirin':ti,ab OR 'flutivate':ti,ab OR 'fluxonal':ti,ab OR 'gr 18781':ti,ab OR 'gr18781':ti,ab OR 'inalacor':ti,ab OR 'nasofan':ti,ab OR 'otri allergie (fluticasone propionate)':ti,ab OR 'otri-allergie (fluticasone propionate)':ti,ab OR 'pavetod':ti,ab OR 'pirinase hayfever relief':ti,ab OR 'prutica':ti,ab OR 'reviflut':ti,ab OR 'sonera':ti,ab OR 'trialona':ti,ab OR 'truflo':ti,ab OR 'ubizol':ti,ab OR 'xhance':ti,ab OR 'zoflut':ti,ab |
17126 |
||||||||||||||||||||||||||||||||||||||||||
|
#1 |
'allergic rhinitis'/exp OR 'allergic rhinitis':ti,ab OR 'allergic rhinopathy':ti,ab OR 'eosinophil rhinitis':ti,ab OR 'eosinophile rhinitis':ti,ab OR 'eosinophilic rhinitis':ti,ab OR 'eosinophilous rhinitis':ti,ab OR 'rhinitis allergica':ti,ab OR 'rhinitis eosinophila':ti,ab OR 'rhinitis, allergic':ti,ab OR 'perennial rhinitis`:ti,ab or `pollen allergy`:ti,ab or' OR 'hayfever:ti,ab or `hay fever`:ti,ab or':ti,ab OR 'pollinosis:ti,ab or pollenosis:ti,ab' OR (('hypersensitivity'/exp OR hypersensitiv*:ti,ab OR 'hyperreactivity'/exp OR hyperreactiv*:ti,ab OR allerg*:ti,ab OR rhinit*:ti,ab) AND cat:ti,ab OR cats:ti,ab OR dander:ti,ab OR mite*:ti,ab OR dog*:ti,ab OR ragweed:ti,ab OR pollen:ti,ab OR grass*:ti,ab OR cedar:ti,ab OR alder:ti,ab OR willow:ti,ab OR birch:ti,ab OR mugwort:ti,ab OR tree*:ti,ab OR weed*:ti,ab OR perennial*:ti,ab OR season*:ti,ab OR spring:ti,ab OR summer:ti,ab)) OR (nose:ti,ab OR nasal:ti,ab) AND (allerg*:ti,ab OR hypersensitiv*:ti,ab OR hyperreactiv*:ti,ab)
|
70307 |
||||||||||||||||||||||||||||||||||||||||||
|
PubMed |
|
|
||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
PICO 4 en 5 Orale antihistaminica versus nasale antihistaminica of nasale corticosteroïden