Diagnostische accuratesse ACTH-test
Uitgangsvraag
Welke plaats heeft de ACTH-test bij het testen van de bijnierfunctie van patiënten die langer dan 14 dagen glucocorticoïden hebben gebruikt?
Aanbeveling
Aanbeveling 1
Overweeg na een drie maanden glucocorticoïden vrij interval de bijnierfunctie te testen door middel van de low dose ACTH (1 mcg) test bij patiënten waarbij er was gekozen om een afbouwschema in te zetten (zie module ‘Risicofactoren voor een bijnierschorsinsufficiëntie en bijniercrisis’).
Aanbeveling 2
Overweeg om bij onvoldoende oploop van cortisol tijdens de low dose ACTH-test, deze test na 3 maanden te herhalen. Het gebruik van een stress-schema blijft gedurende deze 3 maanden noodzakelijk.
Aanbeveling 3
Overweeg om bij patiënten ≥6 jaar die glucocorticoïden hebben gebruikt en waarbij twee opeenvolgende low dose ACTH-testen onvoldoende oploop van cortisol lieten zien, om 3 tot 6 maanden later een metyrapon-test te (laten) doen in een gespecialiseerd centrum.
Overwegingen
De onderstaande overwegingen en aanbevelingen gelden voor het overgrote deel van de populatie waarop de uitgangsvraag betrekking heeft.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
De in de literatuuronderzoek gevonden studies hadden verschillende beperkingen: de gevonden studies waren over het algemeen klein, het bleef vaak onduidelijk hoe de studiepopulatie tot stand was gekomen, twee van de geïncludeerde studies hebben alleen kinderen met bijnierschorsinsufficiëntie geïncludeerd (waardoor de specificiteit niet bepaald kon worden) en soms was de timing van de index test ten opzichte van referentietest suboptimaal. Er is daarom sprake van een lage bewijskracht. Daarnaast is de patiëntpopulatie waarop we ons in deze richtlijn focussen (patiënten waarbij glucocorticoïden zijn afgebouwd) niet onderzocht in de geïncludeerde studies. Uit de literatuursamenvatting blijkt dat het mogelijk is dat de specificiteit van beide ACTH-testen voldoende is. Dit betekent dat wanneer een patiënt geen afwijkend resultaat heeft op deze test (afkapwaarde is afhankelijk van betreffend assay, >0,55 µmol/l bij de meeste assays of 0,43 µmol/L bij de nieuwste methode van Roche), er in dat geval (bij voldoende specificiteit) waarschijnlijk geen sprake is van bijnierschorsinsufficiëntie bij deze patiënt. Pas op bij patienten die orale anticonceptiva gebruiken (zie inleiding van de richtlijn).
Aangezien het gangbaar is in Nederland om de low dose ACTH-test te verrichten en er ten opzichte van de high dose ACTH-test geen grote nadelen zijn, raadt de werkgroep aan het gebruik van low dose ACTH-test te overwegen. Van deze low-dose test wordt gedacht dat het de gehele hypothalamus-hypofyse-bijnier (HPA-as) test, dit houdt in dat bij een adequate respons (geen afwijkend resultaat op de test, zie ook bovenstaande alinea), geen stress-schema meer nodig is (Plumpton, 1969; Ng, 2016; Chrousos, 2009).
Daar de low dose ACTH-test voor elke leeftijdscategorie geschikt is, er een kortere ziekenhuisopname nodig is (voor de metyrapon-test moeten kinderen een nacht in het ziekenhuis blijven) en de test minder belastend, minder risico’s en minder bijwerkingen kent, heeft deze test de voorkeur boven de metyrapon-test. Echter, wanneer de low dose ACTH-test twee keer een afwijkend beeld laat zien (twee keer een waarde < 0,55 µmol/L, afhankelijk van de assay), raadt de werkgroep aan om bij kinderen ≥6 jaar de metyrapon-test te doen in verband met een hogere sensitiviteit en specificiteit van de metyrapon-test. In Forest (2003) en in het farmacotherapeutisch kompas wordt aangegeven dat er weinig ervaring is met de metyrapon-test bij jonge kinderen. Het geven van metyrapon resulteert in een verminderde productie van cortisol en daarmee een risico op een bijniercrisis. Forest (2003) beschrijft ook het risico van neerslag in de witte stof van de hersenen van jonge kinderen. Het is niet duidelijk welke leeftijd bedoeld wordt met jonge kinderen. We hebben voor een veilige marge gekozen en daarom de grens bij 6 jaar gelegd. Bij kinderen <6 jaar wordt aangeraden om de ACTH test te herhalen.
Waarden en voorkeuren van patiënten (en eventueel hun verzorgers)
Ouders hebben over het algemeen geen bezwaar tegen het doen van een ACTH-test. Bij een normaal testresultaat hoeven zij immers geen stress-schema meer te hanteren, wat de belasting voor ouders, kind en omgeving sterk verlaagd. Een normale uitslag geeft een groot gevoel van zekerheid en voor het kind weer vertrouwen in het lichaam.
Kosten (middelenbeslag)
De kosten van de test komen neer op de kosten van een dagopname. De kosten van een eventuele bijniercrisis kunnen veel hoger liggen.
Aanvaardbaarheid voor de overige relevante stakeholders
Er zijn voor dit aspect geen noemenswaardige bezwaren vanuit het veld die van invloed zijn op de besluitvorming.
Haalbaarheid en implementatie
Het is belangrijk dat de verdunning van de tetracosactide altijd volgens het handboek parenteralia geschied en bij voorkeur door de apotheek wordt gedaan. Dit in verband met de nauwkeurigheid die vereist is om de ampul te verdunnen tot 1 mcg. Gezien het weinig gebruik en het risico op uitlokken van een bijniercrisis en het risico op hypoglycaemie bij de metyrapon-test heeft het de voorkeur dat deze test in een gespecialiseerd centrum wordt afgenomen. Er zijn voor dit aspect verder geen noemenswaardige bezwaren die van invloed zijn op de besluitvorming.
Rationale/balans tussen de argumenten voor en tegen de interventie
Aanbeveling 1
Een test om de bijnierfunctie te beoordelen wordt gedaan om het risico op een crisis zoveel mogelijk uit te kunnen sluiten. Het gebruiken van een stress-schema is een grote belasting voor de ouders, kind en omgeving. Daarnaast is het voor kinderen belangrijk dat zij weer op hun lichaam kunnen vertrouwen. De werkgroep raadt aan een dergelijke test te overwegen bij patiënten waarbij er was gekozen om een afbouwschema in te zetten (zie ook module ‘Risicofactoren voor een bijnierschorsinsufficiëntie en bijniercrisis’). Gezien de voordelen van de low dose ACTH-test boven de metyrapon-test en aangezien deze test in Nederland gangbaarder is dan de high dose ACTH-test, heeft de werkgroep een voorkeur om met de low dose ACTH-test te starten.
Aanbeveling 2
Indien deze test (low dose ACTH (1 mcg)) test onvoldoende oploop laat zien, is het advies om deze test na drie maanden te herhalen. In de tussentijd blijft het gebruik van het stress-schema noodzakelijk.
Aanbeveling 3
Bij kinderen ≥6 jaar waarbij de low dose ACTH-test twee keer afwijkend was, wordt geadviseerd om een metyrapon test te plannen. Deze test heeft een hogere sensitiviteit en specificiteit (Cegla, 2013).
Onderbouwing
Er zijn verschillende inzichten tussen de diverse kindergeneeskundige subspecialismen over de noodzaak en wijze van afbouwen na langdurig glucocorticoïdgebruik. Ook adviezen ten aanzien van de duur en de noodzaak van een stressdosis bij ziekte zijn wisselend. Het streven is naar een eenduidig beleid te komen in Nederland. Om aan te kunnen geven wanneer er geen stress-schema meer nodig is, dient er een adequate test voor handen te zijn. Momenteel wordt bij kinderen de low dose (1 mcg) ACTH-test gebruikt. Deze uitgangsvraag is gericht op het vaststellen van de diagnostische accuratesse van de ACTH-tests. Kunnen we met deze test een eventuele insufficiëntie van de HPA-as vaststellen dan wel uitsluiten?
|
Zeer laag GRADE |
Het is mogelijk dat de specificiteit van de low-dose en de high-dose ACTH-test voldoende is om bijnierschors(in)sufficiëntie aan te tonen bij patiënten <18 jaar.
Bronnen: O’Grady, 2003; Maghnie, 2005; Shankar, 1997; Kamrath, 2010; Weintrob, 1998 en Rose, 1999. |
Beschrijving studies
De systematische review van Ng (2016) is opgenomen in de literatuursamenvatting. In deze review werd de diagnostische waarde van standard short Synacthen-test (SSST) en low-dose short Synacthen-test (LDSST) met betrekking tot het vaststellen van insufficiëntie van de HPA-as in kinderen onderzocht. EMBASE, MEDLINE en The Cochrane Library werden tot 2016 doorzocht.
Er werden acht studies geïncludeerd (n=342), waarvan er zes in deze literatuursamenvatting werden geïncludeerd. Eén studie kwam niet overeen met de PICO door het gebruik van een andere referentietest (corticotrophin-releasing hormone test) en één studie werd na het raadplegen van de individuele full tekst artikelen geëxcludeerd wegens onvoldoende duidelijke weergave van de resultaten (zie ook de Zoeken en selecteren sectie).
Alle geïncludeerde studies betroffen cohortonderzoeken met kinderen met risico op het ontwikkelen van HPA-as insufficiëntie. Vijf studies (O’Grady, 2013; Weintrob, 1998; Maghnie, 2005; Shankar, 1997; Kamrath and Boehles, 2010) gebruikten de insuline tolerantie test (ITT) als referentietest en twee studies (Rose, 1999; Shankar, 1997) gebruikten (ook) de metyrapon-test. Vier studies (Rose, 1999; Weintrob, 1998; Maghnie, 2005; Shankar, 1997) onderzochten de diagnostische accuratesse van zowel de SSST en de LDSST en twee studies (O’Grady, 2013; Kamrath en Boehles, 2010) onderzochten enkel de LDSST. Van alle studies is de sensitiviteit gerapporteerd of berekend. Van twee studies (Kamrath and Boehles, 2010; O’Grady, 2012) kon de specificiteit niet bepaald worden, aangezien in deze studies alleen kinderen met bijnierschorsinsufficiëntie (bepaald met de referentietest) zijn geïncludeerd. In tabel 1 staat een overzicht van individuele studie karakteristieken samengevat. Gezien het beperkte aantal studies/patiënten, de heterogeniteit tussen de studies met betrekking tot de referentietest, doseringen en timing van de assessments, zijn de resultaten niet gepoold.
Resultaten
Een overzicht van de resultaten is weergegeven in tabel 1.
Tabel 1 Overzicht van de sensitiviteit en specificiteit van de LDSST en de SSST
|
Test |
Referentie test |
Studie |
Aantal patiënten leeftijd, range (jaren) |
Studiepopulatie/ indicatie |
Dosis tetracosactrin |
Timing van de meting (min), LDSST |
Cut-off score LDSST |
Sensitiviteit |
Specificiteit |
Risk of bias (RoB) en opmerkingen
|
|
LDSST |
Insulin Tolerance Test (ITT)
|
O’Grady, 2013
|
42/5,8-18 |
Patiënten met een abnormale cortisol respons tijdens een eerdere ITT. |
ITT: 0,1 U/kg |
- 30, 0, 15, 25, 35, 60, 90 |
≥500 nmol/L |
0,26 (0,14-0,42) |
Kan niet worden berekend, patiënten zonder insufficiëntie afwezig |
High RoB voor de patiëntselectie en de timing van de index test. Alleen inclusie kinderen met suboptimale (<500 nm) respons op ITT. LDSST binnen 3 maanden van ITT. |
|
LDSST : 1 mcg |
0, 30, 60 |
|||||||||
|
Weintrob, 1998 |
30/4,0-19 Controlegroep (n=13, met vroege /versnelde puberteit, zonder andere aanwijzingen pathologie) heeft geen ITT gekregen. |
Idiopathische multipele hypofysaire uitval, n=10 insufficiënt, n=20 sufficiënte respons op ITT. |
ITT: 0,1 U/kg |
0, 30, 60, 90, 120 |
≥520 nmol/L |
0,90 (0,55-1,00) |
0,90 [0,68-0,99] |
Unclear RoB voor Patiënt selectie. Heeft ook volwassenen geïncludeerd. Onduidelijk hoe sample tot stand is gekomen. |
||
|
LDSST: 1mcg/1,73 m2 |
0, 30, 60 |
|||||||||
|
Maghnie, 2005
|
16/4,2-31 (gemiddeld 9,6). N=24 inclusie maar=8 hadden contra-indicatie voor ITT |
Groeihormoon deficiëntie |
ITT: 0,1 U/kg |
ITT: 0, 30, 60, 90, 120 |
≥550 nmol/L |
0,45 (0,17-0,77) |
1.00 [0.48-1.00] |
RoB unclear voor patiëntenselectie. Heeft ook volwassenen geïncludeerd. |
||
|
LDSST: 1 mcg |
LDSST: 0, 10, 20, 30, 45 |
|||||||||
|
Shankar, 1997
|
22/ ? n=2 exclusie in verband met niet geldige ITT. |
Behandeld voor hersentumoren of schedel basis tumoren |
ITT: ? |
Niet gerapporteerd |
≥496 nmol/L |
0,40 (0,05-0,85) |
1.00 (0,78-1,00) |
RoB unclear voor meeste aspecten. Vijf testen in 2 dagen uitgevoerd. Dosis is mogelijk te hoog om als ‘lage dosis’ aangemerkt te worden. Dosis van ITT onbekend. |
||
|
LSDSST: 0,2 mcg/kg |
Niet gerapporteerd |
|||||||||
|
Kamrath and Boehles, 2010
|
11/5,5-14,5 |
Short stature en aangetoonde milde hypofyse-bijnieras insufficiëntie gebaseerd op een ITT. |
ITT: 0,05 IU/kg |
0, 30, 60 en eventueel aanvullend |
≥500 nmol/L |
0,09 (0,00-0,41) |
Kan niet worden berekend, patiënten zonder insufficiëntie afwezig |
High RoB voor de patiëntselectie en flow and timing. Index test heeft een mediaan 0,3 jaar na ITT plaatsgevonden |
||
|
0,5 mcg/m2 |
0, 20, 30 |
|||||||||
|
Metyrapon
|
Rose, 1999 |
120/1-18 N=19 exclusie in verband met inconclusieve resultaten op metyrapon-test |
At risk voor bijnierschors insufficiëntie |
Metyrapon: 1 g/m2 voor lichaamsgewicht <30 kg, en absolute dosis van 2 g voor 30-60 kg en 3 g voor >60 kg. |
8 uur later |
Serum 11-deoxy-cortisol: ≤200nmol/ |
1,00 (0,86-1,00) |
0,83 (0,73-0,91) |
High RoB voor patiëntenselectie. Drie categorieën aangehouden voor index-/referentietest. Hier de ‘Borderline ACTH’ resultaten meegenomen als positieve testen. |
|
|
LDSST: 1mcg/m2 |
0, 20 |
≥550 nmol/L |
||||||||
|
Shankar, 1997
|
22/? n=4 exclusie in verband met ongeldige metyrapon-test. |
Behandeld voor hersentumoren of hersenbasis tumoren |
Single dose Metyrapon: ? |
Niet gerapporteerd |
115 nmol/L |
0,40 (0,05-0,85) |
1,00 (0,75-1,00) |
RoB unclear voor meeste aspecten. Vijf testen in 2 dagen uitgevoerd. Dosis is mogelijk te hoog om als ‘lage dosis’ aangemerkt te worden. Dosis van metyrapon test onbekend. |
||
|
LDSST: 0,2 mcg/kg |
Niet gerapporteerd |
≥496 nmol/L |
||||||||
|
SSST |
ITT |
Weintrob, 1998 |
30/4.0-19 Controlegroep (n=13, met vroege /versnelde puberteit, zonder andere aanwijzingen voor pathologie) heeft geen ITT gekregen. |
Idiopathische multipele hypofysaire uitval, n=10 insufficiënt, n=20 sufficiënte respons op ITT. |
ITT: 0,1 U/kg |
0, 30, 60, 90, 120 |
≥520 nmol/L |
0,90 (0,55-1,00) |
1,00 (0,83-1,00) |
Unclear RoB voor Patiënt selectie. Heeft ook volwassenen geïncludeerd. Onduidelijk hoe sample tot stand is gekomen. |
|
SSST: 250mcg |
0, 30, 60 |
|||||||||
|
Maghnie, 2005 |
16 / 4,2-31 (gemiddeld 9,6) N=24 geïncludeerd, n=8 hadden contra-indicatie voor ITT |
Groeihormoon deficiëntie |
ITT: 0,1 U/kg |
ITT: 0, 30, 60, 90, 120 |
≥550 nmol/L |
0,27 (0,06-0,61) |
1,00 (0,48-1,00) |
RoB unclear voor patiëntenselectie. Heeft ook volwassenen geïncludeerd. |
||
|
SSST: 250mcg |
SSST: 0, 30, 60 |
|||||||||
|
Shankar, 1997
|
22/ ? n=2 exclusie i.v.m. niet geldige ITT. |
Behandeld voor hersentumoren of hersenbasis tumoren |
ITT: NR |
Niet gerapporteerd |
≥496 nmol/L |
0,40 (0,05-0,85) |
0,93 (0,68-1,0) |
RoB unclear voor meeste aspecten. Vijf testen in 2 dagen uitgevoerd. Dosis van ITT onbekend. |
||
|
SSST: 250mcg |
Niet gerapporteerd |
|||||||||
|
Metyrapon
|
Rose, 1999 |
38 / 1-18 N=3 exclusie in verband met inconclusieve resultaten op metyrapon test |
At risk voor bijnierschors insufficiëntie |
Metyrapon: 1 g/m2 voor lichaamsgewicht <30 kg, en absolute dosis van 2 g voor 30-60 kg en 3 g voor >60 kg. |
8 uur later
|
Serum 11-deoxy-cortisol:≤7 mcg/dL (≤200nmol/L) |
0,21 (0,05-0,51) |
0,95 (0,76-1,00) |
High RoB voor patiëntenselectie. drie categorieën aangehouden voor index-/referentietest. Hier de ‘Borderline ACTH’ resultaten meegenomen als positieve testen. |
|
|
SSST: 250 mcg |
0, 60 |
≥550 nmol/L |
||||||||
|
Shankar, 1997
|
22/2-18 N=4 exclusie in verband met ongeldige metyrapon-test |
Behandeld voor hersentumoren of hersenbasis tumoren |
metyrapon: NR |
Niet gerapporteerd |
≤115 nmol/L |
0,60 (0,15-0,95) |
1,0 (0,75-1,0) |
RoB unclear voor meeste aspecten. Vijf testen in 2 dagen uitgevoerd. Dosis van metyrapon-test onbekend |
||
|
SSST: 250 mcg |
Niet gerapporteerd |
≥496 nmol/L |
Sensitiviteit en specificiteit
LDSST versus ITT;
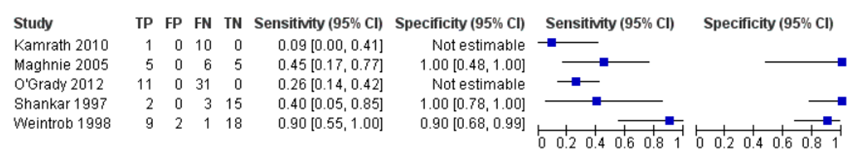
De gerapporteerde sensitiviteit van de LDSST met als referentietest de ITT besloeg een range van 9% (Kamrath, 2010) tot 90% (Weintrob, 1998). Van slechts drie studies kon de specificiteit van de LDSST berekend worden (Kamrath en Boehles, 2010 en O’Grady, 2013 hebben alleen kinderen met bijnierschorsinsufficiëntie (volgens referentietest) geïncludeerd). De gerapporteerde specificiteit varieerde van 90% (Weintrob, 1998) tot 100% (Maghnie, 2005; Shankar, 1997). Zie figuur 1.
Figuur 1: Sensitiviteit en specificiteit van de LDSST-test met ITT als referentietest
SSST versus ITT
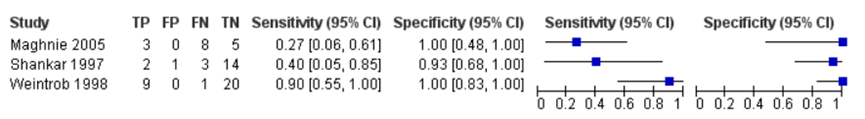
Slechts drie studies onderzochten de sensitiviteit van SSST met ITT als referentietest. Ook hier was er veel variatie in de gevonden waarden voor de sensitiviteit. Bij alle drie studies betrof de specificiteit >93%. Zie figuur 2.
Figuur 2: Sensitiviteit en specificiteit van de SSST test met ITT als referentietest
LDSST versus metyrapon
Slechts twee studies (Shankar, 1997 en Rose, 1999) onderzochten de sensitiviteit van LDSST met metyrapon als referentietest. Zie figuur 3.
Figuur 3: Sensitiviteit en specificiteit van de LDSST-test met metyrapon als referentietest

SSST versus metyrapon
Slechts twee studies (Shankar, 1997 en Rose, 1999) onderzochten de sensitiviteit van SSST met metyrapon als referentietest, zie figuur 4.
Figuur 4: Sensitiviteit en specificiteit van de SSST-test met metyrapon als referentietest

Positief en negatief voorspellende waarde van de LDSST en de SSST
De positief voorspellende waarde werd handmatig berekend door middel van: TP/(TP+FP), de negatief voorspellende waarde door TN/(FN+TN). De positief voorspellende waarde geeft aan welk percentage van patiënten met een positieve score op de LDSST of SSST, daadwerkelijk ook volgens de referentietest bijnierschorsinsufficiënt zijn. De negatieve voorspellende waarde geeft welk percentage van de patiënten die een negatief testresultaat hebben gekregen, daadwerkelijk geen bijnierschorsinsufficiëntie hebben. De resultaten zijn weergegeven in tabel 2.
Tabel 2: positief en negatief voorspellende waarde
|
Index test |
Referentietest |
Studie |
Positief voorspellende waarde |
Negatief voorspellende waarde |
|
LDSST |
ITT |
O’Grady, 2013 |
1,00 |
0 |
|
Weintrob, 1998 |
0,82 |
0,94 |
||
|
Maghnie, 2005 |
1,00 |
0,46 |
||
|
Shankar, 1997 |
1,00 |
0,83 |
||
|
Kamrath and Boehles, 2010 |
1,00 |
0 |
||
|
Metyrapon |
Rose, 1999 |
0,64 |
1,00 |
|
|
Shankar, 1997 |
1,00 |
0,81 |
||
|
SSST |
ITT |
Weintrob, 1998 |
1,00 |
0,95 |
|
Maghnie, 2005 |
1,00 |
0,38 |
||
|
Shankar, 1997 |
0,67 |
0,82 |
||
|
Metyrapon |
Rose, 1999 |
0,75 |
0,65 |
|
|
Shankar, 1997 |
1,00 |
1,00 |
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat diagnostische accuratesse is met 3 niveaus verlaagd gezien beperkingen in de onderzoeksopzet (risk of bias, zie ook de risk of bias-tabel), vaak is het niet duidelijk hoe de patiëntpopulatie is geworven, daarnaast is de timing van de testen in drie studies (Shankar, 1997; O’Grady, 2003; Kamrath en Boehles, 2010) ook suboptimaal (te lang of te kort na elkaar); de heterogeniteit in resultaten voor de sensitiviteit en het was niet mogelijk de resultaten te poolen, klein aantal cases, grens van de klinische relevantie bij specificiteit ligt bij de meeste studies binnen in het 95% betrouwbaarheidsinterval). Het uiteindelijke niveau van bewijskracht is ‘zeer laag’.
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat is de diagnostische accuratesse van de ACTH test?
P: patiënten (1 maand-18 jaar)
I: low/high dose ACTH test
C: insuline tolerance test of metyrapon test (referentie test)
O: diagnostische accuratesse met betrekking tot bijnierschorsinsufficiëntie
Relevante uitkomstmaten
De werkgroep achtte de diagnostische accuratesse (sensitiviteit en specificiteit, negatief voorspellende waarde, positief voorspellende waarde) voor de besluitvorming cruciale uitkomstmaten. De specificiteit is hierbij belangrijker dan sensitiviteit, aangezien we middels de ACTH voornamelijk zouden willen vaststellen of patiënten veilig kunnen stoppen met het stress-schema.
De werkgroep definieerde een specificiteit van 80% als de klinisch relevante grens; boven de 80% wordt de test als voldoende specifiek gezien.
Zoeken en selecteren (Methode)
Bij een eerste oriënterende search in Medline werd de systematische review en meta-analyse van Ng (2016) gevonden. Deze review beantwoordt onze zoekvraag geheel, en is zodoende als basis van deze literatuursamenvatting genomen. De search voor deze review is waarschijnlijk in november 2016 uitgevoerd (het artikel is hier niet helemaal duidelijk over). Om ook de meeste recente studies mee te kunnen nemen, is er door de werkgroep op 16 januari 2018 aanvullend in de databases Medline (via OVID) en Embase (via Elsevier) met relevante zoektermen gezocht naar studies die na 1 januari 2016 zijn verschenen en waarin bij kinderen de diagnostische accuratesse van de ACTH-test is bepaald. De zoekverantwoording van deze zoekactie is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 123 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria: diagnostische studie waarin de diagnostische accuratesse van de ACTH-test ten opzichte van de insuline tolerantie test en/of de metyrapon-test (referentietesten) is bepaald bij kinderen.
Op basis van titel en abstract werden in eerste instantie 12 (inclusief de review van Ng, 2016) van de 123 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens al deze studies geëxcludeerd (n=11); zie exclusietabel onder het tabblad Verantwoording), en werd hier alleen de reeds gevonden systematische review van Ng (2016) definitief geïncludeerd.
Eén systematische review is opgenomen in de literatuuranalyse. De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidence-tabellen. Bij de verwerking van de resultaten van deze studie bleek er echter informatie te missen, en/of bleken soms de resultaten in het artikel niet consequent. De individuele artikelen die voldeden aan de PICO (zeven van de acht door Ng (2006) geïncludeerde studies) zijn full-tekst geraadpleegd om de gegevens aan te vullen, te controleren en waar nodig aan te passen. Eén studie (Gonc, 2002) is na het raadplegen van het full tekst artikel alsnog geëxcludeerd, omdat de figuur waarin de benodigde data is gepresenteerd naar onze mening onvoldoende betrouwbaar kan worden afgelezen.
Voor de beoordeling van de individuele studieopzet (risk of bias) zijn de full tekst artikelen van de geïncludeerde studies bestudeerd, en is niet uitgegaan van de beoordeling door Ng (2016). De beoordeling is opgenomen in de risk of bias-tabellen.
- Cegla J, Jones B, Seyani L, et al. Comparison of the overnight metyrapone and glucagon stimulation tests in the assessment of secondary hypoadrenalism. Clin Endocrinol (Oxf). 2013;78(5):738-42. doi: 10.1111/cen.12043. PubMed PMID: 22998100.
- Chrousos GP, Kino T, Charmandari E. Evaluation of the hypothalamic-pituitary-adrenal axis function in childhood and adolescence. Neuroimmunomodulation. 2009;16(5):272-83. doi: 10.1159/000216185. Epub 2009 Jun 29. Review. PubMed PMID: 19571588; PubMed Central PMCID: PMC2790806.
- Forest MG: Adrenal Function tests; in Ranke MB (ed): Diagnostics of Endocrine function in children and adolescents, ed 3, Basel, Karger, 2003, pp 372-426
- Kamrath C, Boehles H. The low-dose ACTH test does not identify mild insufficiency of the hypothalamnic-pituitary-adrenal axis in children with inadequate stress response. J Pediatr Endocrinol Metab. 2010;23(11):1097-104. PubMed PMID: 21284322.
- Maghnie M, Uga E, Temporini F, et al. Evaluation of adrenal function in patients with growth hormone deficiency and hypothalamic-pituitary disorders: comparison between insulin-induced hypoglycemia, low-dose ACTH, standard ACTH and CRH stimulation tests. Eur J Endocrinol. 2005;152(5):735-41. PubMed PMID: 15879359.
- O'Grady MJ, Hensey C, Fallon M, et al. Requirement for age-specific peak cortisol responses to insulin-induced hypoglycaemia in children. Eur J Endocrinol. 2013;169(2):139-45. doi: 10.1530/EJE-13-0084. Print 2013 Aug. PubMed PMID: 23672955.
- Plumpton FS, Besser GM. The adrenocortical response to surgery and insulin-induced hypoglycaemia in corticosteroid-treated and normal subjects. Br J Surg. 1969;56(3):216-9. PubMed PMID: 5776687.
- Rose SR, Lustig RH, Burstein S, et al. Diagnosis of ACTH deficiency. Comparison of overnight metyrapone test to either low-dose or high-dose ACTH test. Horm Res. 1999;52(2):73-9. PubMed PMID: 10681636.
- Shankar RR, Jakacki RI, Haider A, et al. Testing the hypothalamic-pituitary-adrenal axis in survivors of childhood brain and skull-based tumors. J Clin Endocrinol Metab. 1997;82(6):1995-8. PubMed PMID: 9177419.
- Weintrob N, Sprecher E, Josefsberg Z, et al. Standard and low-dose short adrenocorticotropin test compared with insulin-induced hypoglycemia for assessment of the hypothalamic-pituitary-adrenal axis in children with idiopathic multiple pituitary hormone deficiencies. J Clin Endocrinol Metab. 1998;83(1):88-92.PubMed PMID: 9435421.
Evidence table for systematic reviews of diagnostic test accuracy studies
|
Study reference |
Study characteristics |
Patient characteristics
|
Index test (test of interest) |
Reference test
|
Follow-up |
Outcome measures and effect size |
Comments |
|
Ng, 2016
PS., the majority of the study characteristics and results are extracted from the SR, however the results and missing data was checked in the full text articles of the included studies. |
SR [and meta-analysis]
Literature search up to 2016.
A: O’Grady, 2013 B: Rose, 1999 C: Weintrob, 1998 D: Maghnie, 2005 E: Shankar, 1997 F: Kamrath and Boehles, 2010
Study design: A: cohort study B: non-RCT C: cohort study D: cohort study E: cohort study F: cohort study
Setting and Country: Not reported
Source of funding and conflicts of interest: SR: None declared Individual studies: A: No conflict of interest B Not reported C: Not reported D: Not reported E: non-commercial F: nothing to disclose
|
Inclusion criteria SR: * randomised, quasi-randomised trials, diagnostic or cohort studies * comparison between the use of the LDSST and/or SSST in the assessment of central adrenal insufficiency or HPA axis insufficiency in children (suspected or at risk of secondary adrenal insufficiency) compared with a reference standard (either ITT or metyrapone test or hCRH test).
Exclusion criteria SR: None reported
6 studies included. Ng, 2016 included 8 studies, but one (Maguire, 2008) used the human corticotrophin-releasing hormone (hCRH) as reference test and was excluded here. Furthermore, based on the full text article, Gonc, 2002 was excluded as the needed information was only presented in a figure from which it is difficult to extract the data.
Important patient characteristics:
N, age (range) years A: 42, 5.8-18 B: ?, 1-18 C: 30, 4.0-27 D: 16, 4.2-31 (mean age 9.6 years) E: 22 F: 11, 5.5-14.5
Indication A: suspected HPA axis insufficiency, suboptimal response on ITT (<500 nm) B: at risk of adrenal insufficiency C: idiopathic multiple pituitary hormone deficiencies. D: growth hormone deficiency E: under treatment for brain or skull-based tumors F: short stature and mild HPA insufficiency based on the ITT |
Synacthen test LDSST or SSST dose, sampling time
A: LDSST 1µg, 0, 30, 60 min B: n=38 SSST 250µg and n=120 LDSST 1µg/m2, 0,20 min C: SSST 250µg and LDSST 1µg/1.73m2,0, 30, 60 min D: SSST 250µg, 0, 30, 60 min and LDSST 1µg, 0, 10, 20, 30, 45 min E: SSST 250µg and LDSST 0.2 µg/kg F: LDSST 0.5µg/m2
|
Describe reference test and cut-off point(s): A: 500 nmol/L B: 550 nmol/L C: 520 nmol/L D: LDSST: 550 nmol/L E: >496 nmol/L F: 500 nmol/L
A: Insulin Tolerance Test (ITT) B: Metyrapone C: ITT D: ITT E: ITT/Metyrapone F: ITT
Prevalence (%) [based on reference test at specified cut-off point] A: 1.00 B: 0.36/.2 C: 0.33 D: 0.69 E: 0.20/0.28 (invalid tests excluded) F: 1.00
For how many participants were no complete outcome data available? N (%)
Reasons for incomplete outcome data described? A-G: Not reported |
Endpoint of follow-up: A-G: Not reported |
Door de mogelijke heterogeniteit van de referentietesten worden de uitkomsten gestratificeerd naar referentietest.
Outcome measures: Sensitivity
A: 0.26 [0.14-0.42] C: 0.90[0.55-1.00] D: 0.45[0.17-0.77] E: 0.40[0.05-0.85] F: 0.09[0.55-1.00]
SSST vs ITT C: 0.90 [0.55-01,00] D: 0.40[0.05-0,85] E: 0.40[0.05-0.85]
B: 1.00[0.86-1.00] E: 0.40 [0.05-0.85]
SSST vs Metyrapone B: 0.21[0.05-0.51] E: 0.60 [0.15-0.95]
Specificity
LDSST vs ITT A: not estimable C: LDSST: 0.90[0.68-0.99] D: LDSST: 1.00[0.48-1.00] E: LDSST: 1.00[0.78-1.00] F: not estimable
SSST vs ITT C: 1.00[0.83-1.00] D: 1.00[0.48-1.00] E: 0,93[0,68-1,00]
LDSST vs Metyrapone B: 0.83[0.73-0.91] E: 1.00 [0.75-1.00]
SSST vs Metyrapone B: 0,95[0.76-1.00] E: 1.00 [0.75-1.00] |
Place of the index test in the clinical pathway:
Brief description of author’s conclusion: The short Synacthen test (SSST) is more reliable in the exclusion of central adrenal insufficiency while low-dose Synacthen test (LDSST) is more reliable in the identification of patients with central adrenal insufficiency
Remarks:
|
Risk of bias assessment diagnostic accuracy studies (QUADAS II, 2011)
|
Study reference |
Patient selection
|
Index test |
Reference standard |
Flow and timing |
Comments with respect to applicability |
|
Maghnie, 2005 |
Was a consecutive or random sample of patients enrolled? Unclear
Was a case-control design avoided? Unclear
Did the study avoid inappropriate exclusions? Unclear, there were some exclusions because some patients had contraindications for the ITT test.
|
Were the index test results interpreted without knowledge of the results of the reference standard? Unclear
If a threshold was used, was it pre-specified? Yes
|
Is the reference standard likely to correctly classify the target condition? Yes
Were the reference standard results interpreted without knowledge of the results of the index test? Unclear
|
Was there an appropriate interval between index test(s) and reference standard? Yes
Did all patients receive a reference standard? Yes (results presented for the patient in which the reference standard could be performed)
Did patients receive the same reference standard? Yes
Were all patients included in the analysis? Yes |
Are there concerns that the included patients do not match the review question? Yes, 8 adults included
Are there concerns that the index test, its conduct, or interpretation differ from the review question? No
Are there concerns that the target condition as defined by the reference standard does not match the review question? No
|
|
CONCLUSION: Could the selection of patients have introduced bias?
RISK: UNCLEAR |
CONCLUSION: Could the conduct or interpretation of the index test have introduced bias?
RISK: likely LOW |
CONCLUSION: Could the reference standard, its conduct, or its interpretation have introduced bias?
RISK: likely LOW |
CONCLUSION Could the patient flow have introduced bias?
RISK: LOW |
|
|
|
Weintrob, 1998 |
Was a consecutive or random sample of patients enrolled? Unclear
Was a case-control design avoided? Unclear. The groups were made based on the results of the reference test.
Did the study avoid inappropriate exclusions? No
|
Were the index test results interpreted without knowledge of the results of the reference standard? Unclear
If a threshold was used, was it pre-specified? Yes
|
Is the reference standard likely to correctly classify the target condition? Yes
Were the reference standard results interpreted without knowledge of the results of the index test? Unclear, likely yes
|
Was there an appropriate interval between index test(s) and reference standard? YES
Did all patients receive a reference standard? Yes (control patients not included in own analyses)
Did patients receive the same reference standard? Yes
Were all patients included in the analysis? Yes |
Are there concerns that the included patients do not match the review question? Yes, some adults included
Are there concerns that the index test, its conduct, or interpretation differ from the review question? No
Are there concerns that the target condition as defined by the reference standard does not match the review question? no
|
|
|
CONCLUSION: Could the selection of patients have introduced bias?
RISK: UNCLEAR |
CONCLUSION: Could the conduct or interpretation of the index test have introduced bias?
RISK: Likely Low |
CONCLUSION: Could the reference standard, its conduct, or its interpretation have introduced bias?
RISK: LOW |
CONCLUSION Could the patient flow have introduced bias?
RISK: LOW |
|
|
Shankar, 19997 |
Was a consecutive or random sample of patients enrolled? Unclear
Was a case-control design avoided? Unclear
Did the study avoid inappropriate exclusions? yes
|
Were the index test results interpreted without knowledge of the results of the reference standard? Unclear, exact order remained unclear
If a threshold was used, was it pre-specified? Yes
|
Is the reference standard likely to correctly classify the target condition? Yes, but the used dosage is not reported
Were the reference standard results interpreted without knowledge of the results of the index test? Unclear
|
Was there an appropriate interval between index test(s) and reference standard? Unclear, five tests on two days might have caused stress, and the effects on the results of the tests are unknown.
Did all patients receive a reference standard? Yes, although some tests were invalid (these patients were excluded from the analyses)
Did patients receive the same reference standard? Yes
Were all patients included in the analysis? Yes, all patients with a valid test result |
Are there concerns that the included patients do not match the review question? Unclear, not described
Are there concerns that the index test, its conduct, or interpretation differ from the review question? The dose given during the low dose test might exceed the range for a low dose test.
Are there concerns that the target condition as defined by the reference standard does not match the review question? Yes, the used dosage is not reported.
|
|
|
CONCLUSION: Could the selection of patients have introduced bias?
RISK: UNCLEAR |
CONCLUSION: Could the conduct or interpretation of the index test have introduced bias?
RISK: Likely LOW |
CONCLUSION: Could the reference standard, its conduct, or its interpretation have introduced bias?
RISK: UNCLEAR |
CONCLUSION Could the patient flow have introduced bias?
RISK: UNCLEAR |
|
|
Rose, 1999 |
Was a consecutive or random sample of patients enrolled? Unclear
Was a case-control design avoided? Unclear
Did the study avoid inappropriate exclusions? yes, patients with inconclusive results on reference test excluded.
|
Were the index test results interpreted without knowledge of the results of the reference standard? Yes
If a threshold was used, was it pre-specified? Yes
|
Is the reference standard likely to correctly classify the target condition? Yes
Were the reference standard results interpreted without knowledge of the results of the index test? No
|
Was there an appropriate interval between index test(s) and reference standard Yes
Did all patients receive a reference standard? Yes
Did patients receive the same reference standard? Yes
Were all patients included in the analysis? Yes |
Are there concerns that the included patients do not match the review question? No
Are there concerns that the index test, its conduct, or interpretation differ from the review question? No
Are there concerns that the target condition as defined by the reference standard does not match the review question? No |
|
|
CONCLUSION: Could the selection of patients have introduced bias?
RISK: HIGH |
CONCLUSION: Could the conduct or interpretation of the index test have introduced bias?
RISK: LOW |
CONCLUSION: Could the reference standard, its conduct, or its interpretation have introduced bias?
RISK: LOW |
CONCLUSION Could the patient flow have introduced bias?
RISK: LOW |
|
|
O’Grady |
Was a consecutive or random sample of patients enrolled? yes
Was a case-control design avoided? NA
Did the study avoid inappropriate exclusions? Yes, only patients that ‘failed’ the ITT were included
|
Were the index test results interpreted without knowledge of the results of the reference standard? No
If a threshold was used, was it pre-specified? Yes
|
Is the reference standard likely to correctly classify the target condition? Yes
Were the reference standard results interpreted without knowledge of the results of the index test? Yes
|
Was there an appropriate interval between index test(s) and reference standard? No (max 0.3 months is quite long, recovery can occur).
Did all patients receive a reference standard? Yes
Did patients receive the same reference standard? Yes
Were all patients included in the analysis? Yes |
Are there concerns that the included patients do not match the review question? No
Are there concerns that the index test, its conduct, or interpretation differ from the review question? No
Are there concerns that the target condition as defined by the reference standard does not match the review question? No
|
|
|
CONCLUSION: Could the selection of patients have introduced bias?
RISK: HIGH |
CONCLUSION: Could the conduct or interpretation of the index test have introduced bias?
RISK: Likely LOW |
CONCLUSION: Could the reference standard, its conduct, or its interpretation have introduced bias?
RISK: LOW |
CONCLUSION Could the patient flow have introduced bias?
RISK: HIGH |
|
|
Kamrath and Boehles, 2010 |
Was a consecutive or random sample of patients enrolled? Unclear
Was a case-control design avoided? NA
Did the study avoid inappropriate exclusions? Yes, only patients that ‘failed’ the ITT were included
|
Were the index test results interpreted without knowledge of the results of the reference standard? No
If a threshold was used, was it pre-specified? Yes
|
Is the reference standard likely to correctly classify the target condition? Yes
Were the reference standard results interpreted without knowledge of the results of the index test? Yes
|
Was there an appropriate interval between index test(s) and reference standard? No, index test median 0.3 year after reference test.
Did all patients receive a reference standard? Yes
Did patients receive the same reference standard? Yes
Were all patients included in the analysis? Yes |
Are there concerns that the included patients do not match the review question? No
Are there concerns that the index test, its conduct, or interpretation differ from the review question? No
Are there concerns that the target condition as defined by the reference standard does not match the review question? No
|
|
|
CONCLUSION: Could the selection of patients have introduced bias?
RISK: HIGH |
CONCLUSION: Could the conduct or interpretation of the index test have introduced bias?
RISK: Likely LOW |
CONCLUSION: Could the reference standard, its conduct, or its interpretation have introduced bias?
RISK: LOW |
CONCLUSION Could the patient flow have introduced bias?
RISK: HIGH |
|
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Baek, 2016 |
Andere focus “therefore, the present study aimed to determine the recovery rate of adrenal function in patients with secondary AI within 1 to 2 years, and to assess the factors predictive of adrenal function recovery during the follow-up”. |
|
Bornstein, 2016 |
Primaire AO, geen resultaten van individuele studies. |
|
D’Aurizio, 2016 |
Italiaans. |
|
Dušková, 2016 |
Populatie bestaat uit gezonde volwassenen. |
|
Kline, 2017a |
kijkt naar de arrays waarmee cortisol niveau wordt gemeten, geen vergelijking met gouden standaard. |
|
Kline, 2017b |
Geen vergelijkende studie. |
|
Ospina, 2016 |
Search tot februari 2014. Review was breder ingestoken dan onze zoekvraag. Ook inclusie van volwassenen. |
|
Park, 2016 |
Review, niet systematsich. |
|
Reznik, 2018 |
Overzicht aanbevelingen van de French endocrinology society en de French pediatric endocrinology society. |
|
Schoelwer, 2016 |
Letter to the editor. |
|
Tan, 2018 |
geen vergelijking met Metarypon test of ITT test. |
|
Yanase, 2016 |
Geen origineel onderzoek, overzicht vanaanbevelingen in Japanse richtlijn. |
|
Younes, 2017 |
Review, niet systematisch. |
Beoordelingsdatum en geldigheid
Publicatiedatum : 02-05-2019
Beoordeeld op geldigheid : 15-05-2019
|
Module |
Regiehouder(s) |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn |
Frequentie van beoordeling op actualiteit |
Wie houdt er toezicht op actualiteit |
Relevante factoren voor wijzigingen in aanbeveling |
|
Diagnostische accuratesse ACTH test |
NVK |
2018 |
2023 |
Elke 5 jaar |
NVK |
Er worden momenteel geen studies verwacht die de conclusies sterk zullen veranderen. |
Uiterlijk in 2023 bepaalt het bestuur van de Nederlandse Vereniging voor Kindergeneeskunde of de modules van deze richtlijn nog actueel zijn. Op modulair niveau is een onderhoudsplan beschreven. Bij het opstellen van de richtlijn heeft de werkgroep per module een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De Nederlandse Vereniging voor Kindergeneeskunde is regiehouder van deze richtlijn en eerstverantwoordelijke op het gebied van de actualiteitsbeoordeling van de richtlijn.
Algemene gegevens
De richtlijnontwikkeling werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten (https://www.demedischspecialist.nl/kennisinstituut) en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijn.
Doel en doelgroep
Doel
In deze richtlijn worden aanbevelingen gedaan voor het afbouwen van glucocorticoïden bij kinderen (>1 maand & <18 jaar) die langer dan 14 dagen zijn behandeld met een glucocorticoïd. Hiermee wordt een bijdrage geleverd aan het verminderen van het risico op een bijniercrisis bij deze patiënten en verbetering van de kwaliteit van leven.
Doelgroep
Deze richtlijn is geschreven voor patiënten en alle leden van de beroepsgroepen die betrokken zijn bij de zorg voor kinderen (1 maand - 18 jaar) die langer dan 14 dagen zijn behandeld met glucocorticoïden.
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijn is in 2017 een werkgroep ingesteld, bestaande uit gemandateerde vertegenwoordigers van de NVK die betrokken zijn bij de zorg voor kinderen die worden behandeld met glucocorticoïden. De werkgroep is verantwoordelijk voor de integrale tekst van deze richtlijn.
Werkgroep:
- Dr. H.J. van der Kamp, kinderarts – endocrinoloog, werkzaam in het Wilhelmina Kinderziekenhuis, Universitair Medisch Centrum te Utrecht, NVK, voorzitter
- Dr. S.S.M. Kamphuis, kinderarts – reumatoloog/immunoloog, werkzaam in het Sophia Kinderziekenhuis, Erasmus Universitair Medisch Centrum te Rotterdam, NVK
- Dr. P.J.F.M. Merkus, kinderarts – pulmonoloog, werkzaam in het Radboud Amalia Kinderziekenhuis te Nijmegen, NVK
- Dr. E.J. Schroor, kinderarts, werkzaam in Isala te Zwolle, NVK
Met ondersteuning van:
- Dr. S. Persoon, adviseur, Kennisinstituut van de Federatie Medisch Specialisten te Utrecht
- Ing. L.H.M. Niesink-Boerboom MSc, literatuurspecialist, Kennisinstituut van de Federatie Medisch Specialisten
- A. Gallmann, projectsecretaresse, Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepsleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
|
Van der Kamp |
Kinderarts - endocrinoloog |
Regelmatig betrokken bij Bijniernet en NVACP |
- |
Geen actie nodig |
|
Schroor |
Kinderarts |
- |
- |
Geen actie nodig |
|
Merkus |
Kinderarts-pulmonoloog |
Voorzitter NVK-SKL sectiehoofd kindergeneeskunde lid WAC Longfonds lid LAN Lid Werkgroep Nosmokind
Betaald: Associate Editor ERJ |
- |
Geen actie nodig |
|
Kamphuis |
Kinderarts- reumatoloog/immunoloog |
Lid medische adviesraad patiëntenvereniging NVLE. Lid namens sectie kinderreumatologie in pCSO. Voorzitter Europese Lupus Working Party. |
- |
Geen actie |
|
Persoon |
Adviseur Kennisinstituut |
Gastvrijheidsaanstelling afdeling Revalidatie Academisch Medisch Centrum, Amsterdam, in verband met promotietraject. Project: Physical fitness to improve fitness and combat fatigue in patients with multiple myeloma or lymphoma treated with high dose chemotherapy. April-augustus 2018: Docent Team Technologie, Fontys Paramedische Hogeschool. Begeleiden van studenten bij afstudeerstages. Max 1 dag in de week, betaald. |
Geen, Promotieonderzoek werd gefinancierd door KWF, financier had geen invloed op uitkomsten onderzoek of op huidige werkzaamheden |
Geen actie nodig |
|
Niesink-Boerboom |
Informatiespecialist |
- |
- |
Geen actie nodig |
Inbreng patiëntenperspectief
Tijdens alle stappen van het ontwikkelproces is rekening gehouden met het patiëntperspectief. Met de Stichting Kind en Ziekenhuis en de Patiëntenfederatie heeft voorgaand aan het ontwikkelproces contact plaatsgevonden over het patiëntperspectief en de conceptrichtlijn is voor commentaar aan Stichting Kind en Ziekenhuis, de Patiëntenfederatie en Diabetesvereniging Nederland voorgelegd.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van de richtlijnontwikkeling is rekening gehouden met de implementatie van de richtlijnmodules en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de richtlijn in de praktijk kunnen bevorderen of belemmeren. De implementatietabellen zijn te vinden in de bijlagen van de afzonderlijke modules. De werkgroep heeft besloten geen indicatoren te ontwikkelen bij de huidige richtlijn, omdat er geen substantiële barrières konden worden geïdentificeerd die de implementatie van de aanbeveling zouden kunnen bemoeilijken.
Werkwijze
AGREE
Deze richtlijn is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based richtlijn tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse, uitgangsvragen en uitkomstmaten
Tijdens de voorbereidende fase inventariseerden de voorzitter van de werkgroep en de adviseur de knelpunten. Op basis van de uitkomsten van deze inventarisatie zijn door de voorzitter en de adviseur concept uitgangsvragen opgesteld, die vervolgens met de werkgroep zijn besproken. Het conceptraamwerk is schriftelijk voorgelegd aan betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties. Door de Diabetesvereniging Nederland, Stichting Kind en Ziekenhuis, Lareb, Nederlandse Internisten Vereniging, Nederlandse Vereniging voor Dermatologie en Venereologie, Nederlandse Vereniging voor Kindergeneeskunde, Nederlandse Vereniging voor Neurologie, Nederlandse Vereniging van Ziekenhuisapothekers en de Vereniging Innovatieve Geneesmiddelen is inhoudelijke input op het conceptraamwerk geleverd. Deze input is met de werkgroep besproken, waarna de werkgroep de definitieve uitgangsvragen heeft vastgesteld. Een overzicht met antwoorden op de schriftelijk ingestuurd punten is in de richtlijn opgenomen onder aanverwante producten.
Vervolgens inventariseerde de werkgroep per uitgangsvraag welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. De werkgroep waardeerde deze uitkomstmaten op hun relatieve belang bij de besluitvorming rondom aanbevelingen, als 1) cruciaal, 2) belangrijk (maar niet cruciaal) en 3) onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Er werd voor de afzonderlijke uitgangsvragen aan de hand van specifieke zoektermen gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De databases waarin is gezocht, de zoekstrategie en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: AMSTAR – voor systematische reviews; ACROBAT-NRS – voor observationeel onderzoek; QUADAS II – voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidence-tabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur. Bij een voldoende aantal studies en overeenkomstigheid (homogeniteit) tussen de studies werden de gegevens ook kwantitatief samengevat (meta-analyse) met behulp van Review Manager 5.
Beoordelen van de kracht van het wetenschappelijke bewijs
A) Voor interventievragen (vragen over therapie of screening)
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor Grading Recommendations Assessment, Development and Evaluation (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
B) Voor vragen over diagnostische tests, schade of bijwerkingen, etiologie en prognose
De kracht van het wetenschappelijke bewijs werd eveneens bepaald volgens de GRADE-methode: GRADE-diagnostiek voor diagnostische vragen (Schünemann, 2008), en een generieke GRADE-methode voor vragen over schade of bijwerkingen, etiologie en prognose. In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje Overwegingen.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt (patient values and preferences), kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje Overwegingen.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
In de knelpuntenanalyse en bij de ontwikkeling van de richtlijn is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag.
Indicatorontwikkeling
Gelijktijdig met het ontwikkelen van de conceptrichtlijn heeft de werkgroep overwogen om interne kwaliteitsindicatoren te ontwikkelen om het toepassen van de richtlijn in de praktijk te volgen en te versterken. De werkgroep heeft besloten geen indicatoren te ontwikkelen bij de huidige richtlijn, omdat er geen substantiële barrières konden worden geïdentificeerd die de implementatie van de aanbeveling zouden kunnen bemoeilijken.
Kennislacunes
Tijdens de ontwikkeling van deze richtlijn is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvragen. Bij elke uitgangsvraag is door de werkgroep nagegaan of er (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Een overzicht van de onderwerpen waarvoor (aanvullend) wetenschappelijk van belang wordt geacht, is als aanbeveling in de Kennislacunes beschreven (onder aanverwante producten).
Commentaar- en autorisatiefase
De conceptrichtlijn werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijn aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijn werd aan Nederlandse Vereniging voor Kindergeneeskunde en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd.
Literatuur
Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub. 2010 Jul 5. Review. PubMed PMID: 20603348.
Medisch Specialistische Richtlijnen 2.0. Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html. 2012.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, et al. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008;336(7654). doi: 10.1136/bmj.a139. PubMed PMID: 18483053.
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van Medisch Specialisten.
Zoekverantwoording
|
Database |
Zoektermen |
Totaal |
|
Medline (OVID)
2016 – januari 2018
|
1 exp *Cosyntropin/ or exp *Adrenal Insufficiency/ or tetracosact*.ti. or synacthen*.ti. or acth.ti. or cosyntropin*.ti. or cortrosyn*.ti. or 'adrenal insufficien*'.ti. (23833) 2 test*.ab,ti. (2744287) 3 1 and 2 (3082) 4 diagnos*.ab,ti. (2102983) 5 3 and 4 (919) 6 limit 5 to yr="2016 -Current" (110) 7 6 not (Comment/ or Editorial/ or Letter/ or (animals/ not humans/)) (106)
= 106 |
123 |
|
Embase (Elsevier) |
(('tetracosactide'/exp/mj OR tetracosact*:ti OR synacthen*:ti OR acth:ti OR cosyntropin*:ti OR cortrosyn*:ti OR 'adrenal insufficiency'/exp/mj OR 'adrenal insufficien*':ti) AND test*:ab,ti) AND diagnos*:ab,ti AND [2016-2018]/py NOT 'conference abstract':it
= 90 (87 uniek) |