Duur en continuïteit behandeling ADHD
Uitgangsvraag
Duur en continuïteit medicamenteuze behandeling van ADHD bij volwassenen.
Aanbeveling
In de medicamenteuze behandeling van ADHD bij volwassenen is een individuele benadering belangrijk; hulpverleners dienen op zijn minst jaarlijks de medicatie en de gebruikte doseringen te evalueren, evenals het effect van de behandeling op comorbide aandoeningen en stemmingswisselingen (PB).
In principe is de behandeling van volwassenen met ADHD een langetermijnbehandeling. Bij een adequate respons dient de medicamenteuze behandeling voortgezet te worden zolang deze klinisch effectief is. Dit moet men jaarlijks beoordelen. Een dergelijke beoordeling omvat een uitgebreide evaluatie van de klinische noodzaak, de voordelen en de bijwerkingen van de medicamenteuze behandeling. Hierbij wordt rekening gehouden met de visie van de patiënt zelf en waar mogelijk met die van een partner, ouder, vriend of betrokkene, met aandacht voor de verschillen tussen deze visies. Naast het evalueren van de gebruikte dosering moet men ook kijken naar het effect van gemiste doseringen, geplande dosisverlagingen en korte periodes zonder behandeling. Verder wordt de aanwezigheid van comorbide psychiatrische problematiek nagegaan. Ten slotte wordt de noodzaak van aanvullende psychologische, sociale en beroepsmatige ondersteuning van de patiënt geëvalueerd (PB).
Overwegingen
Er zijn bij deze uitgangsvraag geen overwegingen geformuleerd.
Onderbouwing
Vergeleken met kinderen is er bij volwassenen met ADHD nog relatief weinig onderzoek met ADHD-medicatie verricht, zoals uit het eerdere literatuuroverzicht is gebleken. Omdat ADHD bij kinderen en volwassenen echter dezelfde kenmerken, vergelijkbaar disfunctioneren en dezelfde neurobiologische achtergronden heeft, kunnen de resultaten van onderzoek bij kinderen ten dele geëxtrapoleerd worden naar volwassenen. Bij kinderen zijn de effectiviteit en de veiligheid van psychostimulantia over de langere termijn ook met onderzoek aangetoond (Hechtman & Greenfield, 2003). Dit sluit aan bij de positieve ervaringen in de klinische praktijk.
Bij volwassenen is onderzoek naar de effectiviteit van ADHD-medicatie over langere termijn nog beperkt (Biederman e.a., 2005). In de afgelopen 20 jaar is de klinische ervaring toegenomen, nu meer volwassenen met ADHD in behandeling gekomen zijn. De algemene trend hierbij is dat wanneer door middel van medicatie vermindering van de ADHD-symptomen is bereikt, de effectiviteit van de medicatie bij blijvend gebruik ook over langere termijn behouden blijft. De gebruikte medicijnen zijn ook op langere termijn veilig gebleken: het bijwerkingenprofiel verandert in de loop van de behandeling niet en toxische reacties op langere termijn zijn zeldzaam.
Omdat ADHD een chronische stoornis is, adviseert de werkgroep bij gebleken effectiviteit en geringe bijwerkingen, langdurig gebruik van deze medicatie. Wel blijven regele controles noodzakelijk om de effectiviteit en bijwerkingen van de medicatie te evalueren en de noodzaak van blijvend medicatiegebruik te beoordelen.
Zoekstrategie
Er werd een uitvoerig literatuuronderzoek uitgevoerd naar medicamenteuze interventies voor volwassenen met ADHD. Dit onderzoek resulteerde in 1192 referenties die potentieel relevant waren voor dit review. Van de 1192 referenties werd van 136 referenties in verband met mogelijke inclusie de volledige tekst opgevraagd. 1056 referenties werden geëxcludeerd, met als meest voorkomende redenen voor uitsluiting:
- het gebruikte medicijn viel niet onder dit overzicht;
- het onderwerp lag buiten het bestek van dit onderzoek;
- het artikel betrof geen patiëntgebonden onderzoek (bijv. een hoofdstuk uit een boek of commentaar, dan wel een secundaire analyse);
- de onderzochte populatie viel buiten het bestek van dit onderzoek (bijv. kinderen, of geen patiënten met ADHD).
Van de 116 referenties die mogelijk relevant waren, werden er 44 geïncludeerd en voldeden er 72 niet aan de inclusiecriteria, waardoor ze geëxcludeerd werden. Van de 44 geïncludeerde referenties die overbleven, betroffen 31 referenties oorspronkelijke onderzoeken (K=15 over methylfenidaat, K=4 over dexamfetamine, K=1 over bupropion, K=9 over atomoxetine, K=1 over MAOI’s (monoamine-oxidaseremmers) en K=1 anders). Verdere details over de geëxcludeerde onderzoeken zijn te vinden in de Appendix.
Daarnaast werden er 13 systematische reviews verkregen en werden de primaire gegevensbronnen van deze reviews beoordeeld op hun methodologische kwaliteit en op hun mogelijke inclusie in het review.
Hetzelfde literatuuronderzoek werd ook gebruikt om referenties te ziften voor de zoekvragen over therapietrouw en het risico op misbruik. Waar mogelijk werden er ook referenties verkregen door handmatig zoeken en werden referenties aangeleverd door de leden van de richtlijncommissie.
Op 5 maart 2013 vond een update van de literatuursearch plaats voor het hoofdstuk medicamenteuze behandeling. Deze update resulteerde in 403 referenties die potentieel relevant waren voor deze review. Van de 31 referenties die na de eerste zifting als mogelijk relevant werden beoordeeeld, werden 6 RCT’s en 1 systematic review geïncludeerd. 24 referenties voldeden niet aan de inclusiecriteria (vielen buiten de scope van de richtlijn, hadden geen placebogroep, waren open-labeltrials, waren post-hocanalyse van eerdere trials met uitkomstmaten buiten de scope van de richtlijn), waardoor ze geëxcludeerd werden.
Reviewprotocol
Het reviewprotocol (hieronder in schema) beschrijft de criteria voor de onderzoekspopulaties, de interventies, de vergelijkingen en de uitkomstmaten, die bij dit literatuuronderzoek naar medicamenteuze interventies bij volwassenen met ADHD gebruikt zijn.
|
Component |
Description |
|
Objectives |
To evaluate the clinical effectiveness of pharmacological interventions for adults with ADHD. |
|
Population |
Adults and adolescents aged 18 years and older with suspected ADHD
Excluded groups include: Children (≤18 years of age)
For literature with a sample comprising of both children and adults, we should include when at least 50% of the sample population are adults.
For comorbidities, we should consider a sample to have comorbidities when at least 50% of the sample has a current comorbid diagnosis. |
|
Intervention(s) |
Pharmacotherapy |
|
Comparison |
Placebo, treatment as usual, control |
|
Critical outcomes |
|
|
Search Strategy |
Databases: MEDLINE, Embase, PsycINFO, Cochrane. Dates searched up until: March 5th 2013 |
|
Minimum sample size |
|
|
Study setting |
|
|
Study design |
|
|
Review strategy |
|
|
DSM=Diagnostic and Statistical Manual; ICD=International Classification of Diseases; RCT=Randomised Controlled Trial. ITT=Intention To Treat |
|
- Biederman, J., Spencer, T.J., Wilens, T.E., Weisler, R.H., Read S.C., & Tulloch S.J. (2005). Long-term safety and effectiveness of mixed amphetamine salts extended release in adults with ADHD. CNS Spectrums, 10(12,Suppl20), 16-25.
- Hechtman, L., & Greenfield, B. (2003). Long-term use of stimulants in children with attention deficit hyperactivity disorder: safety, efficacy, and long-term outcome. Peadiatric Drugs, 5(12), 787-794.
Beoordelingsdatum en geldigheid
Publicatiedatum : 08-03-2016
Beoordeeld op geldigheid : 08-07-2015
Uiterlijk in 2020 bepaalt het bestuur van de NVvP of deze richtlijn nog actueel is. Zo nodig wordt een nieuwe werkgroep geïnstalleerd om de richtlijn te herzien. De geldigheid van de richtlijn komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De NVvP is als houder van deze richtlijn de eerstverantwoordelijke voor de actualiteit van deze richtlijn. De andere aan deze richtlijn deelnemende wetenschappelijk verenigingen of gebruikers van de richtlijn delen de verantwoordelijkheid en informeren de eerstverantwoordelijke over relevante ontwikkelingen binnen hun vakgebied.
Algemene gegevens
In opdracht van de Nederlandse Vereniging voor Psychiatrie (NVvP) heeft de richtlijnwerkgroep ADHD bij volwassenen een monodisciplinaire richtlijn ontwikkeld over diagnostiek en medicamenteuze behandeling van ADHD bij volwassenen. De ontwikkeling van deze richtlijn werd gefinancierd vanuit het gealloceerde budget van de NVvP vanuit de Stichting Kwaliteitsgelden Medisch specialisten (SKMS) en methodologisch en organisatorisch ondersteund door het Trimbos-instituut.
De status van de richtlijn
De professionaliteit van hulpverleners in de gezondheidszorg brengt met zich mee dat zij (mede door het hanteren van een richtlijn) zo veel als mogelijk evidence-based handelen, volgens de laatste stand van de wetenschap. Wanneer richtlijnen door en binnen de beroepsgroep zijn opgesteld, normeren deze het medisch professioneel handelen en zijn ze een uitwerking van de medisch professionele standaard (Gevers & Aalst, 1998).
Richtlijnen zijn geen dwingende voorschriften, maar zo veel mogelijk op bewijs gebaseerde inzichten en aanbevelingen waaraan hulpverleners, organisaties, zorgverleners, beleidsmakers, inhoudelijk adviseurs en mensen met een psychiatrische diagnose kennis kunnen ontlenen om hoogwaardige zorg te verlenen, te waarborgen en te toetsen. De behandelaar kan, als hij of zij dat nodig acht, op basis van de eigen professionele autonomie afwijken van de richtlijn. Afwijken van richtlijnen is als de situatie dat vereist zelfs noodzakelijk, en dient schriftelijk te worden vastgelegd in het dossier (Wijmen e.a., 2004).
Uitgangsvragen
De richtlijn is ontwikkeld op geleide van uitgangsvragen, die gebaseerd zijn op knelpunten rondom diagnostiek en behandeling van volwassenen met ADHD, zoals tevoren aangegeven door patiëntenvereniging Impuls en leden van de NVvP middels een enquête. Deze uitgangsvragen zijn ook vastgesteld door NICE, op basis van een knelpuntenanalyse die is uitgevoerd in Engeland. De Nederlandse richtlijnwerkgroep heeft een deel van deze uitgangsvragen overgenomen voor de huidige richtlijn.
Een richtlijn is een document met praktische aanbevelingen. Dat betekent dat praktijkproblemen zo veel als mogelijk uitgangspunt zijn van de teksten in de richtlijn. De richtlijn is een document waarin staat hoe optimale diagnostiek en behandeling er inhoudelijk uitzien. In deze richtlijn worden de volgende ‘klinische uitgangsvragen’ behandeld in de verschillende modules.
Diagnostiek
Uitgangsvragen rond diagnostiek waren:
- Is er een gevalideerd instrument om de diagnose ADHD bij volwassenen betrouwbaar vast te stellen volgens de DSM-IV-criteria (Module CAADID)?
- Waar moet het afkappunt voor symptomen in de volwassenheid liggen om een betrouwbare diagnose ADHD bij volwassenen te kunnen stellen (Module DIS-L)?
Medicamenteuze behandeling
Uitgangsvragen rond medicamenteuze behandeling waren:
- Leidt medicamenteuze behandeling van volwassenen met ADHD met (langwerkend) methylfenidaat, amfetamine (dexamfetamine en/of l-amfetamine, lisdexamfetamine), modafinil, atomoxetine of bupropion tot afname van ADHD-symptomen en tot een algehele klinische verbetering?
- Welke strategieën in de medicamenteuze behandeling van ADHD bij volwassenen verhogen de therapietrouw?
- Wat is het risico op misbruik (abuse) of verslaving (addiction) bij het gebruik van methylfenidaat en dexamfetamine in de behandeling van volwassenen met ADHD?
Doel en doelgroep
Doelstelling
De richtlijn ADHD bij volwassenen geeft aanbevelingen en handelingsinstructies voor de diagnostiek en medicamenteuze behandeling van volwassenen met ADHD. De richtlijn geeft aanbevelingen ter ondersteuning van de praktijkvoering van psychiaters die betrokken zijn bij de zorgverlening aan volwassenen met ADHD. Op basis van de resultaten van wetenschappelijk onderzoek en overige overwegingen geeft de richtlijn een overzicht van goed (‘optimaal’) handelen als waarborg voor kwalitatief hoogwaardige zorg.
De werkgroep moedigt het opstellen van lokale zorgprogramma’s en protocollen op basis van deze richtlijn aan, omdat dit bevorderlijk is voor de implementatie van de in de richtlijn beschreven optimale zorg.
Indien de aanbevelingen uit deze richtlijn in de concrete situatie niet aansluiten bij de wensen of behoeften van de volwassene met ADHD, dan kan beredeneerd worden afgeweken van de richtlijn, tenzij de wensen of behoeften van de persoon met ADHD hem of haar naar de mening van de behandelaar kunnen schaden dan wel geen nut hebben.
Doelgroep
De primaire doelgroep van deze richtlijn zijn volwassenen en adolescenten vanaf 18 jaar, bij wie sprake is van (een vermoeden van) ADHD. In de afbakening van de werkgroep valt ook de groep ‘ouderen’ binnen de groep volwassenen. Hoewel diagnostiek en behandeling van ouderen met ADHD relatief nieuwe fenomenen zijn, en RCT’s met medicatie bij deze groep nog ontbreken, is uit onderzoek gebleken dat ADHD ook bij ouderen voorkomt bij wie behandeling geïndiceerd kan zijn.
Richtlijngebruikers
De ontwikkeling van richtlijnen voor de GGZ geschiedt primair ter verbetering van de kwaliteit van de zorgverlening. De gebruikers van de richtlijn zijn allen professioneel betrokken bij de zorg voor volwassenen met (mogelijk) ADHD.
Afbakening
De richtlijnwerkgroep ADHD bij volwassenen heeft zich enerzijds gericht op het in kaart brengen van de knelpunten rondom diagnostiek en behandeling van deze aandoening en anderzijds op de beschikbare wetenschappelijk evidentie op dit gebied, waaronder de Engelse NICE-richtlijn voor ADHD bij volwassenen (2008). De richtlijnwerkgroep formuleerde uitgangsvragen voor het ontwikkelen van de monodisciplinaire richtlijn ADHD bij volwassenen voor de volgende vier onderwerpen: 1. Diagnostiek; 2. Comorbiditeit; 3. Medicamenteuze behandeling; 4. Niet-medicamenteuze behandeling. De onderwerpen diagnostiek en medicamenteuze behandeling worden behandeld in deze eerste fase van de richtlijn. De andere onderwerpen worden in het vervolgproject, de Zorgstandaard ADHD voor alle leeftijden, aan deze richtlijn toegevoegd (zie Voorwoord). Dit betekent dat de onderhavige eerste fase van de richtlijn slechts een deel van de volledige diagnostiek en behandeling van volwassenen met ADHD betreft.
Samenstelling werkgroep
De monodisciplinaire richtlijn ADHD bij volwassenen is ontwikkeld door de richtlijnwerkgroep ADHD bij volwassenen, in opdracht van de NVvP.
De richtlijnwerkgroep, onder voorzitterschap van dr. Sandra Kooij, bestond uit psychiaters, die door de beroepsvereniging werden afgevaardigd en een arts/psychotherapeut, tevens voorzitter van patiëntenvereniging Impuls. De richtlijnwerkgroep werd methodologisch en organisatorisch ondersteund door het technisch team van het Trimbos-instituut. Dit technisch team bestond uit projectleiding, een informatiespecialist, literatuurreviewers en projectassistentie. De volgende schema’s geven een overzicht van de samenstelling van de richtlijnwerkgroep en het ondersteunend technisch team.
Leden werkgroep
|
|
Naam |
Organisatie |
Beroep |
|
1. |
Sandra Kooij, voorzitter, MD PhD |
PsyQ Den Haag |
Psychiater |
|
2. |
Marita Braam-van Neer, MD |
ZGP |
Psychiater |
|
3. |
Nannet Buitelaar, MD |
De Waag, onderdeel van de Forensische Zorgspecialisten |
Psychiater |
|
4. |
Pieter-Jan Carpentier, MD PhD |
Reinier van Arkel Groep |
Psychiater |
|
5. |
Lex Pull, MD |
Brijder Verslavingszorg (tot 1 mei 2012) |
Arts / Psychotherapeut/ Voorzitter patiëntenvereniging Impuls |
|
6. |
Jeannette Rutgers, MD |
Pro Persona |
Psychiater |
|
7. |
Laetitia Smarius, MD |
De Bascule |
Psychiater |
Methodologische ondersteuning
|
|
Naam |
Ondersteuning |
|
1. |
Hedda van ’t Land, MsC PhD |
Programmahoofd, Programma Zorginnovatie, Trimbos-instituut |
|
2. |
Danielle van Duin, MsC |
Projectleider / richtlijnadviseur, Trimbosinstituut |
|
3. |
Jolanda Meeuwissen, MsC |
Projectleider / richtlijnadviseur, Trimbosinstituut |
|
4. |
Marleen Hermens, MsC PhD |
Reviewer, Trimbos-instituut |
|
5. |
Matthijs Oud, MsC |
Reviewer, Trimbos-instituut |
|
6. |
Angita Peterse |
Informatiespecialist, Trimbos-instituut |
|
7. |
Laura Shields, MsC |
Reviewer, Trimbos-instituut / NICE |
|
8. |
Nelleke van Zon |
Projectassistent, Trimbos-instituut |
In totaal kwam de richtlijnwerkgroep voorafgaand aan de commentaarfase acht keer bijeen in een periode van 31 maanden (september 2010-april 2013). In deze periode werden de stappen van de methodiek voor evidence-based richtlijnontwikkeling (EBRO) doorlopen. Voorafgaand aan dit project verrichtte de informatiespecialist in overleg met de werkgroepleden op systematische wijze literatuuronderzoek en maakte een selectie in de gevonden onderzoeken (zie voor informatie over de zoekstrategie en de selectiecriteria het reviewprotocol). De reviewers van het Trimbos-instituut beoordeelden de kwaliteit en inhoud van de aldus verkregen literatuur en verwerkten deze in evidencetabellen, beschrijvingen van de wetenschappelijke onderbouwing en wetenschappelijke (gewogen) conclusies. Leden van de richtlijnwerkgroep gingen op basis van de gevonden literatuur met elkaar in discussie over overige overwegingen en aanbevelingen. De werkgroepleden schreven samen met het technisch team van het Trimbos-instituut de conceptrichtlijntekst, die ter becommentariëring openbaar werd gemaakt. De ontvangen commentaren van leden van de NVvP, en een klankbordgroep bestaande uit leden van patiëntenvereniging Impuls, hoogleraren psychiatrie en andere betrokkenen werden vervolgens verwerkt in een commentaartabel, die tijdens een werkgroep bijeenkomst werd besproken. Na het doorvoeren van de op deze bijeenkomst voorgestelde wijzigingen werd de definitieve richtlijn aan de opdrachtgever aangeboden. Hierop volgden autorisatie door de beroepsvereniging, druk en verspreiding.
Belangenverklaringen
De leden van de werkgroep hebben schriftelijk verklaard of ze gedurende de totstandkoming van de richtlijn een (financieel ondersteunde) betrekking onderhielden met commerciële bedrijven, organisaties of instellingen die in verband staan met het onderwerp van de richtlijn. Daarin zijn door de opdrachtgever, het uitvoeringsorgaan en de werkgroepleden van de richtlijn geen strijdigheden bevonden met de te verrichten taken voor de richtlijn. Deze procedure is vastgelegd conform de ´Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling´, opgesteld door de Koninklijke Nederlandse Academie van Wetenschappen (KNAW), Koninklijke Nederlandsche Maatschappij tot bevordering der Geneeskunst (KNMG), Gezondheidsraad (GR), Centraal BegeleidingsOrgaan (CBO), Nederlands Huisartsen Genootschap (NHG) en Orde van Medisch Specialisten (OMS) in 2012. Een overzicht van deze verklaringen ligt ter inzage bij de Nederlandse Vereniging voor Psychiatrie.
Methode ontwikkeling
Evidence based
Implementatie
Recent onderzoek laat zien dat er bij richtlijntrajecten voornamelijk aandacht is voor de ontwikkeling van de richtlijn. Planmatig opgezette invoering komt maar heel beperkt van de grond. Hierop dient gericht te worden aangestuurd door financiers, zodat hier al bij de start geld voor beschikbaar is. De Regieraad voor richtlijntrajecten onderschrijft dan ook een programmatische aanpak van de ontwikkeling, invoering én evaluatie van richtlijnen inclusief (financiële en organisatorische) middelen, om het gebruik van de richtlijnen te stimuleren, monitoren en evalueren. TNO, CBO en het Trimbos-instituut besloten deze opdracht samen uit te voeren, met als doel de complementaire inzichten en expertise te vertalen in een gemeenschappelijke visie op richtlijnen. Dit resulteerde in een praktisch instrument voor een analyse van richtlijntrajecten: Kwaliteit Richtlijnontwikkeling, Invoering en Evaluatie (KRIE). Voor informatie over implementatie en evaluatie van de richtlijn wordt verwezen naar de module Implementatie.
Werkwijze
De richtlijn is ontwikkeld volgens de methodiek van de evidence-based richtlijnontwikkeling (EBRO).
Zoekstrategie
Om de klinische uitgangsvragen te beantwoorden is door de informatiespecialist, in 15 overleg met de werkgroepleden, op systematische wijze literatuuronderzoek verricht en is een selectie gemaakt binnen de gevonden onderzoeken volgens vooraf vastgestelde selectiecriteria. Er is gezocht naar bestaande (buitenlandse) evidence-based richtlijnen voor de zorg aan volwassenen met ADHD, systematische reviews en oorspronkelijke onderzoeken. In de literatuursearches is gezocht naar literatuur in de Engelse, 20 Nederlandse, Franse, en Duitse taalgebieden. Voor het zoeken naar publicaties is gebruikgemaakt van de volgende informatiebronnen:
|
|
|
|
|
|
Selectiestrategie
Bij de selectie van artikelen zijn de volgende criteria gehanteerd:
- Geeft het onderwerp van het gevonden onderzoek voldoende antwoord op de uitgangsvraag (worden bijvoorbeeld de vastgestelde kritische en belangrijke uitkomstmaten in het onderzoek geëvalueerd)?
- Sluit de doelgroep van het gevonden onderzoek voldoende aan bij de doelgroep van de richtlijn, te weten volwassenen en adolescenten ouder dan 18 jaar bij wie men vermoedt dat zij ADHD hebben? (literatuur gezocht vanaf > 15 jaar)
- Is de bestudeerde groep voldoende groot (minimaal tien deelnemers per arm)?
- Is er sprake van een geschikte onderzoeksopzet om de uitgangsvragen te beantwoorden? Gaat het om een gerandomiseerde gecontroleerde trial (RCT), cohortonderzoek, cross-sectioneel onderzoek, patiënt-controleonderzoek of goed kwalitatief onderzoek (waaronder goed observationeel onderzoek en case series)? Bij een longitudinaal onderzoek: is er een voldoende lange follow-upperiode?
Zie voor meer informatie over de Zoekverantwoording en Evidence tabellen.
Beoordeling van de kwaliteit van het bewijs
De methodologische kwaliteit van de gebruikte artikelen is beoordeeld met voor het betreffende onderzoekstype relevante formulieren ter beoordeling van de methodologische kwaliteit, afkomstig van het EBRO-platform (Handleiding voor werkgroepleden CBO, 2005). Voor het bewijs rondom interventies is daarna het bewijs van de onderzoeken per uitkomstmaat gegradeerd volgens het GRADE-systeem. (GRADE staat voor: Grading of Recommendations Assessment, Development and Evaluation.) De kwaliteit van het bewijs kent daarbij vier niveaus, te weten: zeer laag, laag, matig en hoog. De studie-opzet bepaalt de uitgangspositie van de kwaliteit van bewijs. RCT’s hebben over het algemeen meer bewijskracht dan observationele studies. Daarom is hun uitgangspositie hoog, terwijl de uitgangspositie van observationele studies laag is. De kwaliteit van het bewijs per uitkomstmaat wordt, behalve door de methodologische kwaliteit van de individuele onderzoeken, ook bepaald door andere factoren, zoals de mate van consistentie van de gevonden resultaten uit de verschillende onderzoeken en de precisie van de gevonden uitkomst (zie de onderstaande tabel). GRADE is niet toegepast bij vragen rondom ‘case identification’ en diagnostiek. De belangrijkste reden hiervoor is dat GRADE momenteel nog vooral geschikt is voor interventieonderzoeken.
Tabel 1. Grade: Factoren voor downgraden en upgraden
Het niveau van de kwaliteit van het bewijs (zeer laag, laag, matig en hoog) verwijst naar de mate van vertrouwen dat men heeft in de schatting van het effect van een behandeling.
|
We downgraden het niveau van de kwaliteit van bewijs van studies met een hoge uitgangspositie (RCT’s), bij: |
We upgraden het niveau van de kwaliteit van bewijs van observationele studies bij: |
|
1. Beperkingen in de onderzoeksopzet of uitvoering (study limitations): hierbij gaat het om de methodologische kwaliteit. Voorbeelden zijn dat de randomisatieprocedure niet optimaal was, dat beoordelaars van subjectieve uitkomsten niet geblindeerd waren, dat er selectief is gerapporteerd over de uitkomsten en dat er veel uitvallers waren. |
1. Een groot effect (large magnitude of effect): hiervan is sprake als er in de resultaten een groot effect of een sterk bewijs van associatie gevonden wordt. Dit kan tot uitdrukking komen in de hoogte van het relatieve risico (RR). |
|
1. Inconsistentie van de resultaten (inconsistency): hierbij gaat het om heterogeniteit van de resultaten van verschillende studies. Er kunnen beperkingen zijn als er een grote variatie is in de schattingen van het effect van een behandeling of als er nauwelijks overlap is tussen de 95%-betrouwbaarheidsintervallen (BI’s). |
2. Mogelijke confounders die het ‘ware’ effect verminderd hebben (plausible confounding): hiervan kan sprake zijn als er een achterliggende variabele is, zoals de ernst van de aandoening van de patiënten die met het onderzoek meedoen, die van invloed is op het effect van de interventie. |
|
2. Indirect bewijs (indirectness): er worden twee soorten indirect bewijs onderscheiden. Enerzijds gaat het om indirecte vergelijkingen, bijvoorbeeld wanneer er alleen interventies met een placebo worden vergeleken en geen interventies met elkaar worden vergeleken. Anderzijds gaat het om verschillen in patiëntenpopulatie, inhoud van de interventie of keuze van de uitkomstmaten tussen de beschikbare studies en de uitgangsvraag die in de richtlijn wordt gesteld. |
3. Bewijs van een verband tussen de dosering en de repons (dose-response gradient): hiervan kan sprake zijn als stijgende doseringen van een bepaald medicijn meer effect geven.
|
|
3. Onnauwkeurigheid van de resultaten (imprecision): hierbij gaat het om de onzekerheid van de uitkomst, bijvoorbeeld als de 95%-BI’s heel breed zijn vanwege kleine patiëntenaantallen. |
|
|
4. Kans op selectieve publicatie (publication bias) van onderzoeken of uitkomstmaten. Een voorbeeld van een beperking is wanneer niet alle studies gepubliceerd worden, bijvoorbeeld kleine studies die geen effecten ten gunste van de interventie konden aantoonden. |
|
Samenvatten van de resultaten
Van elk artikel is een samenvatting gemaakt in een zogenaamde ‘evidencetabel’, waarin de belangrijkste kenmerken van individuele onderzoeken zijn opgenomen (bij een RCT zijn dat bijvoorbeeld het doel van het onderzoek, het onderzoeksdesign, patiëntenkenmerken, interventies, uitkomstmaten en de resultaten). De resultaten van onderzoeken naar case identification en diagnostiek zijn op beschrijvende wijze samengevat (narratieve review). Bij de uitgangsvragen over interventies was het oorspronkelijk doel om voor elke uitkomstmaat een meta-analyse uit te voeren, om de omvang van het klinisch effect van de interventie samen te vatten. De data uit oorspronkelijke onderzoeken werden hiervoor verwerkt in een forest plot, die een grafische weergave van de meta-analyse geeft (zie de onderstaande tabel voor een voorbeeld van een forest plot).
Tabel 2. Voorbeeld van een forest plot met toelichting
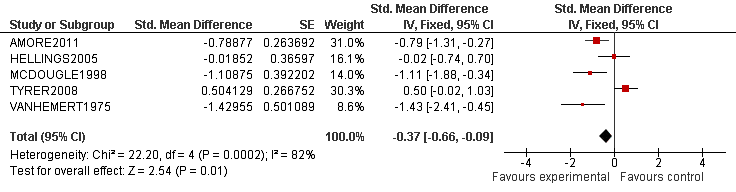
- De verticale lijn is de lijn van geen effect.
- Elke blokje geeft de puntschatting van het interventie-effect van een onderzoek aan.
- De grootte van het blokje correspondeert met het gewicht van het onderzoek binnen de meta-analyse.
- De horizontale lijn is een weergave van het betrouwbaarheidsinterval (95%-BI)
- De diamant is een weergave van het overalleffect (gemiddeld gewogen effect).
- De significantie van het overalleffect wordt onderaan de plot gegeven (z-score en p-waarde).
- De I2 geeft de mate van heterogeniteit van de studies aan (consistentie van de 10 resultaten).
Wanneer er onvoldoende data beschikbaar waren om een meta-analyse uit te voeren, stelden de reviewers een narratieve (beschrijvende) review van het beschikbare bewijs op. Bij de interventieonderzoeken is de waardering van de kwaliteit van het bewijs met behulp van de GRADE-methodiek in deze narratieve reviews opgenomen. De kwaliteitsbeoordeling en een samenvattende beschrijving van de verschillende onderzoeken worden voor elke uitgangsvraag gegeven onder het kopje ‘wetenschappelijke onderbouwing’.
Uitgangsvragen
De richtlijn is ontwikkeld op geleide van uitgangsvragen, die gebaseerd zijn op knelpunten rondom diagnostiek en behandeling van volwassenen met ADHD, zoals tevoren aangegeven door patiëntenvereniging Impuls en leden van de NVvP middels een enquête. Deze uitgangsvragen zijn ook vastgesteld door NICE, op basis van een knelpuntenanalyse die is uitgevoerd in Engeland. De Nederlandse richtlijnwerkgroep heeft een deel van deze uitgangsvragen overgenomen voor de huidige richtlijn.
Een richtlijn is een document met praktische aanbevelingen. Dat betekent dat praktijkproblemen zo veel als mogelijk uitgangspunt zijn van de teksten in de richtlijn. De richtlijn is een document waarin staat hoe optimale diagnostiek en behandeling er inhoudelijk uitzien. In deze richtlijn worden de volgende ‘klinische uitgangsvragen’ behandeld in de verschillende hoofdstukken.
Aanbevelingen
Aanbevelingen kunnen gegradeerd worden als sterk of zwak (voorwaardelijk). Wanneer de kwaliteit van het bewijs voor de positieve en negatieve effecten van een interventie hoog is, kan dit leiden tot een sterke aanbeveling, en omgekeerd, wanneer de bewijskracht laag tot zeer laag is, kan dit een zwakke aanbeveling opleveren. Een zwakke aanbeveling geeft meer ruimte om af te wijken en aandacht te schenken aan alternatieven die passen bij de behoeften van de patiënt, terwijl bij een sterke aanbeveling die ruimte beperkt is. De kracht van het wetenschappelijke bewijs is echter niet de enige factor die de sterkte van de aanbevelingen bepaalt. De aanbevelingen zijn gebaseerd op wetenschappelijk bewijs enerzijds en overige overwegingen anderzijds, zoals praktijkervaringen van de werkgroepleden, ervaringen en voorkeuren van mensen met ADHD en familie, kosten, beschikbaarheid (in verschillende echelons) en organisatorische aspecten. Deze laatste zijn opgenomen onder het kopje ‘Overige overwegingen’ (zie onderstaande tabel).
Tabel 3. Bepaling van de sterkte van aanbeveling
|
Sterkte van aanbeveling |
Kwaliteit wetenschappelijk bewijs: graad van evidentie |
Overige overwegingen: |
Implicaties |
|
1A = sterke aanbeveling |
Hoge graad van evidentie: RCT’s zonder beperkingen of sterk overtuigende evidentie van observationele onderzoeken |
Onder andere: voordelen overtreffen duidelijk de nadelen of risico’s |
sterke aanbeveling, kan worden toegepast bij de meeste mensen met een psychiatrische diagnose en in de meeste omstandigheden |
|
1B = sterke aanbeveling |
Matige graad van evidentie: RCT’s met beperkingen of sterke evidentie vanuit observationele onderzoeken |
Onder andere: voordelen overtreffen duidelijk de nadelen of de risico’s |
sterke aanbeveling, kan worden toegepast bij de meeste mensen met een psychiatrische diagnose en in de meeste omstandigheden |
|
1C = sterke aanbeveling |
Lage of zeer lage graad van evidentie: Uitkomsten van observationele onderzoeken of casestudies of RCT’s met veel beperkingen |
Onder andere: voordelen overtreffen duidelijk de nadelen of de risico’s |
sterke aanbeveling, maar dit kan veranderen als er hogere evidentie beschikbaar komt |
|
2A = zwakke aanbeveling |
Hoge graad van evidentie: RCT’s zonder beperkingen of sterk overtuigende evidentie van observationele onderzoeken |
Onder andere: evenwicht tussen voor- en nadelen of risico’s |
zwakke aanbeveling, de beste actie kan verschillen naar gelang de omstandigheden, mensen met een psychiatrische diagnose of maatschappelijke waarden |
|
2B = zwakke aanbeveling |
Matige graad van evidentie: RCT’s met beperkingen of sterke evidentie vanuit observationele onderzoeken |
Onder andere: evenwicht tussen voor- en nadelen of risico’s |
zwakke aanbeveling, de beste actie kan verschillen naargelang de omstandigheden, mensen met een psychiatrische diagnose of maatschappelijke waarden |
|
2C = zwakke aanbeveling |
Lage of zeer lage graad van evidentie: Uitkomsten van observationele onderzoeken of casestudies of RCT’s met veel beperkingen |
Onder andere: onzekerheid over voor- of nadelen - evenwicht tussen beide is mogelijk |
erg zwakke aanbeveling, alternatieven kunnen evengoed te verantwoorden zijn |
Tabel 4. Indeling van methodologische kwaliteit van individuele onderzoeken
|
|
Interventie |
Onderzoek naar diagnostische accuratesse |
Schade/bijwerkingen*, etiologie, prognose |
|
A1 |
Systematische review van ten minste twee onafhankelijk van elkaar uitgevoerde onderzoeken van A2-niveau |
||
|
A2 |
Gerandomiseerd dubbelblind vergelijkend klinisch onderzoek van goede kwaliteit van voldoende omvang |
Onderzoek ten opzichte van een referentietest (een ‘gouden standaard’) met tevoren gedefinieerde afkapwaarden en onafhankelijke beoordeling van de resultaten van test en gouden standaard, betreffende een voldoende grote serie van opeenvolgende patiënten die allen de index- en referentietest hebben gehad |
Prospectief cohortonderzoek van voldoende omvang en follow-up, waarbij adequaat gecontroleerd is voor ‘confounding’ en selectieve follow-up voldoende is uitgesloten. |
|
B |
Vergelijkend onderzoek, maar niet met alle kenmerken als genoemd onder A2 (hieronder valt ook patiënt-controleonderzoek, cohortonderzoek) |
Onderzoek ten opzichte van een referentietest, maar niet met alle kenmerken die onder A2 zijn genoemd |
Prospectief cohortonderzoek, maar niet met alle kenmerken als genoemd onder A2 of retrospectief cohortonderzoek of patiënt-controleonderzoek |
|
C |
Niet-vergelijkend onderzoek |
||
|
D |
Mening van deskundigen |
||
* Deze classificatie is alleen van toepassing in situaties waarin om ethische of andere redenen gecontroleerde trials niet mogelijk zijn. Zijn die wel mogelijk, dan geldt de classificatie voor interventies.
|
We downgraden het niveau van de kwaliteit van bewijs van studies met een hoge uitgangspositie (RCT’s), bij: |
We upgraden het niveau van de kwaliteit van bewijs van observationele studies bij: |
|
1. Beperkingen in de onderzoeksopzet of uitvoering (study limitations): hierbij gaat het om de methodologische kwaliteit. Voorbeelden zijn dat de randomisatieprocedure niet optimaal was, dat beoordelaars van subjectieve uitkomsten niet geblindeerd waren, dat er selectief is gerapporteerd over de uitkomsten en dat er veel uitvallers waren. |
1. Een groot effect (large magnitude of effect): hiervan is sprake als er in de resultaten een groot effect of een sterk bewijs van associatie gevonden wordt. Dit kan tot uitdrukking komen in de hoogte van het relatieve risico (RR). |
|
1. Inconsistentie van de resultaten (inconsistency): hierbij gaat het om heterogeniteit van de resultaten van verschillende studies. Er kunnen beperkingen zijn als er een grote variatie is in de schattingen van het effect van een behandeling of als er nauwelijks overlap is tussen de 95%-betrouwbaarheidsintervallen (BI’s). |
2. Mogelijke confounders die het ‘ware’ effect verminderd hebben (plausible confounding): hiervan kan sprake zijn als er een achterliggende variabele is, zoals de ernst van de aandoening van de patiënten die met het onderzoek meedoen, die van invloed is op het effect van de interventie. |
|
2. Indirect bewijs (indirectness): er worden twee soorten indirect bewijs onderscheiden. Enerzijds gaat het om indirecte vergelijkingen, bijvoorbeeld wanneer er alleen interventies met een placebo worden vergeleken en geen interventies met elkaar worden vergeleken. Anderzijds gaat het om verschillen in patiëntenpopulatie, inhoud van de interventie of keuze van de uitkomstmaten tussen de beschikbare studies en de uitgangsvraag die in de richtlijn wordt gesteld. |
3. Bewijs van een verband tussen de dosering en de repons (dose-response gradient): hiervan kan sprake zijn als stijgende doseringen van een bepaald medicijn meer effect geven.
|
|
3. Onnauwkeurigheid van de resultaten (imprecision): hierbij gaat het om de onzekerheid van de uitkomst, bijvoorbeeld als de 95%-BI’s heel breed zijn vanwege kleine patiëntenaantallen. |
|
|
4. Kans op selectieve publicatie (publication bias) van onderzoeken of uitkomstmaten. Een voorbeeld van een beperking is wanneer niet alle studies gepubliceerd worden, bijvoorbeeld kleine studies die geen effecten ten gunste van de interventie konden aantoonden. |
|
Zoekverantwoording
|
Verslag literatuursearch richtlijnontwikkeling ‘NVvP - ADHD bij volwassenen’ |
|
Vraag 1: |
|
Datum search |
Werkgroeplid aanwezig: |
Informatiespecialist: |
|
Gebruikte database: |
Interface: |
Vanaf publicatiejaar: |
|
Zoektermen en/of sleutelartikelen aangeleverd vóór de search: |
Ja, door Nannet Buitelaar |
|
|
Zoekstrategieën |
|
P patiëntenpopulatie: ADHD breed of smal en volwassenen |
|
|
P |
Zoektermen |
|
Breed |
(attenti$ or disrupt$ or implusiv$ or inattenti$).sh. OR ((attenti$ or disrupt$) adj3 (adult$ or behav$ or condition$ or difficult$ or disorder$ or learn$ or people or person$ or poor or problem$ or process$)).tw. OR disruptive$.tw. OR impulsiv$.tw. OR inattentiv$.tw. OR (adhd or addh or ad hd or ad??hd).tw. OR (attenti$ adj3 deficit$).tw. OR hyperactiv$.mp. OR (hyper adj1 activ$).tw. OR hyperkin$.mp. OR (hyper adj1 kin$).tw. OR hkd.tw. OR overactiv$.tw. not overactive bladder$.ti. OR (over adj1 activ$).tw. not overactive bladder$.ti. OR (minimal adj1 brain).tw. |
|
Smal |
*”attention deficit and disruptive behavior disorders”/ or attention deficit disorder with hyperactivity/ OR (adhd or attention deficit$).ti,ab. |
|
Volwassenen |
adult$.ti,ab. OR exp Adult/ |
|
I interventie: |
|
|
I |
Zoektermen |
|
Heteroanamnese |
e medical history taking/ or cornell medical index/ OR (((medical adj3 history) or anamnesis) adj8 (relative? or friend? or spouse? or family$)).ti,ab. OR (hetero-anamnesis or hetero?anamnesis or (hetero adj2 anamnesis)).ti,ab. OR ((report$ or assess$) adj5 (relative? or friend? or spouse? or family$)).ti,ab. |
|
O outcome: |
|
|
O |
Zoektermen |
|
Diagnose |
diagnos$.ti,ab,sh. |
|
Age of onset |
early diagnosis/ OR ((earl$ or initial or onset or preclinical or pre clinical) adj3 (detect$ or diagnos$ or distinguish$ or identif$ or intervention$ or recogn$ or therap$ or treat$)).ti,ab. |
|
Diagnosis |
Attention Deficit Disorder with Hyperactivity/di [Diagnosis] OR exp “Sensitivity and Specificity”/ OR likelihood functions/ OR exp Diagnostic Errors/ OR area under curve/ OR “reproducibility of results”/ OR Diagnosis, Differential/ OR (sensitivit$ or specificit$).ti,ab. OR predictive value$.ti,ab. OR likelihood ratio$.ti,ab. OR (false adj (negative$ or positive$)).ti,ab. OR (valid$ adj3 (adhd or attention deficit$ or hyperkin$ or diagnos$)).ti,ab. |
|
Predictive value |
“Predictive Value of Tests”/ OR ((predict$ or development$) adj3 (adhd or atttention deficit or hyperactiv$ or hyperkin$ or minimal brain)).ti,ab. OR (trajector$ adj2 (development$ or symptom$)).ti,ab. OR (“age of onset”/ AND di.fs.) |
|
Persistence |
(persist$ adj3 (adhd or atttentiondeficit$ or hyperactiv$ or hyperkin$ or minimal brain$ or age or aging or addulthood)).ti,ab. OR persistence.mp. and (age factors or age of onset or aging).sh |
|
Symptoms |
comorbid$.mp. OR ((dysfunction$ or function) adj2 (chang$ or executive$ or deficit$ or impair$)).tw. OR (neuropsychopatholog$ or psychopatholog$ or pathophysiolog$).mp. OR prevalen$.mp. and (diagnos$.mp. or di.fs.) OR exp Neuropsychological Tests/ OR exp Psychiatric Status Rating Scales/ OR exp Psychological Tests/ OR Psychometrics/ OR Mental Status Schedule/ OR (“diagnostic and statistical manual of mental disorders”/ OR “International Classification of Diseases”/ AND (diagnos$.mp. or di.fs.)) OR “diagnostic and statistical manual of mental disorders”/ OR behavioral symptoms/ or affective symptoms/ OR Attention Deficit Disorder with Hyperactivity/di [Diagnosis] OR ((adhd or attention deficit$ or hyperactiv$ or hyperkin$ or detect$ or diagnos$ or identif$ or pattern$ or recogni$ or warning$) adj2 (sign? or symptom?)).ti,ab. OR (clinical adj (feature$ or characteristic$) adj2 (adhd or attention deficit$ or hyperactiv$ or hyperkines$)).ti,ab. OR (symptom$ adj3 (impulsiv$ or inattenti$ or overactiv$)).ti,ab. |
|
Validiteit dsm4 |
“diagnostic and statistical manual of mental disorders”/ AND (“reproducibility of results”/ OR validation studies/ OR (valid$ or reproducib$).ti,ab.) OR (dsm-iv adj5 valid$).ti,ab. |
|
Diagnostiek dsm4 |
“diagnostic and statistical manual of mental disorders”/ OR dsm-iv$.ti,ab. |
|
Limiteringen |
|
|
Op jaartal |
1990 tot huidig |
|
Op taal |
Nederlandse, Duitse, Franse of Engelse taal |
|
Op species |
Zonder onderzoeken uitsluitend over dieren |
|
Resultaten van deze search: |
||
|
Database |
Bijgewerkt tot |
Bestandsnaam |
|
combinatie: P:smal AND P:volwassenen AND I:heteroanamnese |
||
|
Medline |
28-04-2010 |
med100429 P smal adult en heteroanamnese |
|
Embase |
week 17 2010 |
emb100503 P smal adult en heteroanamnese |
|
PsycINFO |
12-4-2010 |
psy100503 P smal adult en heteroanamnese |
|
Cochrane |
geraadpleegd 3-5-2010 |
coc100503 P smal adult en heteroanamnese clinical trials |
|
combinatie: P:smal AND P:volwassenen AND (O:predictive value OR O:persistence) AND (O:symptoms OR O:age of onset) AND O:diagnose AND O:diagnosis |
||
|
Medline |
3-5-2010 |
med100504 P smal age of onset |
|
Embase |
week 17 2010 |
emb100503 P smal age of onset |
|
PsycINFO |
12-4-2010 |
psy100503 P smal age of onset |
|
Cochrane |
geraadpleegd 4-5-2010 |
coc100504 P smal age of onset clinical trials |
|
combinatie: P:breed AND P:volwassenen AND O:validiteit dsm4 zonder de bij e gedownloade referenties |
||
|
Medline |
30-4-2010 |
med100503 P breed extra validiteit dsm4 |
|
Embase |
week 17 2010 |
emb100503 P breed extra validiteit dsm4 |
|
PsycINFO |
12-4-2010 |
psy100503 P breed extra validiteit dsm4 |
|
combinatie: P:smal AND P:volwassenen AND O:symptoms AND O:diagnosis AND O:diagnostiek dsm4 |
||
|
Medline |
30-4-2010 |
med100503 P smal dsm4 diagnostiek |
|
Embase |
week 17 2010 |
emb100503 P smal dsm4 diagnostiek |
|
PsycINFO |
12-4-2010 |
psy100503 P smal dsm4 diagnostiek |
|
Cochrane |
geraadpleegd 4-5-2010 |
coc100504 P smal dsm4 diagnostiek clinical trials |
|
Algemene opmerkingen: |
|