Tromboseprofylaxe op de verpleegafdeling bij COVID-19
Uitgangsvraag
Wat is de plaats van tromboseprofylaxe bij COVID-19 patiënten op de verpleegafdeling?
Aanbeveling
Geef antistolling aan patiënten met COVID-19 op de verpleegafdeling en overweeg hierbij een standaard profylactische of intermediaire dosis LMWH, omdat de huidige bewijslast geen overtuigende meerwaarde aantoont van therapeutische antistolling.
Overwegingen
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
Er is literatuuronderzoek verricht naar de verschillen in klinische uitkomsten tussen 1) behandeling met therapeutische dosis antistolling versus standaard of intermediaire dosis tromboseprofylaxe, en 2) behandeling met intermediaire dosis tromboseprofylaxe versus standaard dosis tromboseprofylaxe, bij patiënten met COVID-19 op de verpleegafdeling. Tot op heden zijn 5 RCTs gevonden die de vergelijking in de eerste PICO – therapeutische dosis versus standaard of intermediaire dosis - hebben onderzocht. Er zijn geen studies gevonden die de vergelijking in de tweede PICO – intermediaire dosis versus standaard dosis - hebben onderzocht. Voor de uitwerking van de studies is naast een analyse van alle studies samen een onderverdeling gemaakt tussen de interventie met low-molecular-weight heparines (LMWHs) en/of ongefractioneerde heparines (UFHs) en de interventie met directe orale anticoagulantia (DOACs).
Cruciale uitkomstmaten
Op basis van de gevonden resultaten zou het gebruik van een therapeutische dosis antistolling met een LMWH/UFH kunnen resulteren in geen tot een klein verschil in mortaliteit vergeleken met een standaard of intermediaire dosis tromboseprofylaxe met heparine. Daarentegen lijkt het effect van het gebruik van een therapeutische dosis antistolling met een DOAC vergeleken met een standaard of intermediaire dosis tromboseprofylaxe met heparine de andere kant op te gaan: dit zou kunnen resulteren in een verhoogde mortaliteit. De bewijskracht voor beide subgroepen is laag.
Voor de cruciale uitkomstmaat veneuze trombo-embolie is het onduidelijk of een behandeling met therapeutische dosis antistolling met een LMWH/UFH of DOAC zou kunnen leiden tot een reductie in het optreden van veneuze trombo-embolie. De bewijskracht hiervoor is zeer laag. Als naar alle trombo-embolische complicaties tezamen wordt gekeken, kan er worden geconcludeerd dat gebruik van een therapeutische dosis antistolling met een LMWH/UFH zou kunnen resulteren in geen tot een klein verschil in het optreden van trombo-embolische complicaties vergeleken met een standaard profylactische of intermediaire dosis heparine. De bewijskracht is laag. Het is onduidelijk of een behandeling met therapeutische dosis met een DOAC zou kunnen leiden tot een reductie in het optreden van trombo-embolische complicaties vergeleken met een standaard of intermediaire dosis profylaxe met LMWH/UFH. De bewijskracht hiervoor is zeer laag.
Hetzelfde patroon gaat op voor de cruciale uitkomstmaat ernstige bloeding. Voor deze uitkomstmaat kan worden geconcludeerd dat het gebruik van een therapeutische dosis antistolling met een LMWH/UFH zou kunnen resulteren in geen tot een klein verschil in het optreden van een ernstige bloeding vergeleken met een standaard of intermediaire dosis tromboseprofylaxe met heparine. De bewijskracht is laag. Het is onduidelijk of een behandeling met therapeutische dosis met een DOAC zou kunnen leiden tot een toename in het optreden van een ernstige bloeding, vergeleken met een standaard of intermediaire dosis profylaxe met heparine. De bewijskracht hiervoor is zeer laag.
Belangrijke uitkomstmaten
Voor de belangrijke uitkomstmaat duur van ziekenhuis opname is geen bewijs gevonden voor de vergelijking tussen een therapeutische dosis met een LMWH/UFH en het gebruik van een standaard of intermediaire dosis tromboseprofylaxe met heparine. Voor de vergelijking tussen therapeutische dosis met een DOAC vergeleken met een standaard profylactische of intermediaire dosis heparine stelt de literatuur dat het gebruik van therapeutische dosis met een DOAC mogelijk zou kunnen resulteren in geen tot een klein verschil in de duur van ziekenhuis opname vergeleken met het gebruik van een standaard profylactische of intermediaire dosis heparine. De bewijskracht hiervoor was redelijk.
Voor opname op de intensive care unit (ICU) stelt de literatuur dat het gebruik van therapeutische dosis met een LMWH/UFH zou kunnen resulteren in geen tot een klein verschil in opname op de ICU vergeleken met het gebruik van een standaard of intermediaire dosis profylaxe met heparine. De bewijskracht hiervoor is laag. Er is geen bewijs gevonden voor de vergelijking tussen therapeutische dosis met een DOAC vergeleken met een standaard profylactische of intermediaire dosis met heparine.
Voor het aantal dagen overleving zonder orgaan ondersteuning stelt de literatuur dat het gebruik van therapeutische dosis met een LMWH/UFH zou kunnen resulteren in geen tot een klein verschil in het aantal dagen overleving zonder orgaan ondersteuning, vergeleken met het gebruik van een standaard of intermediaire dosis tromboseprofylaxe met heparine. De bewijskracht hiervoor is redelijk. Er is geen bewijs gevonden voor de vergelijking tussen therapeutische dosis met een DOAC vergeleken met een standaard of intermediaire dosis tromboseprofylaxe met heparine. Als wordt gekeken naar intubatie en mechanische ventilatie kan geconcludeerd worden dat het gebruik van zowel een therapeutische dosis met een LMWH/UFH als met een DOAC zou kunnen resulteren in geen tot een klein verschil in intubatie/mechanische ventilatie, vergeleken met het gebruik van een standaard of intermediaire dosis tromboseprofylaxe met heparine. De bewijskracht voor beide is redelijk.
Interpretatie
De interpretatie van de studies is om meerdere redenen complex. Er zijn geen studies gevonden die apart een therapeutische dosis met een intermediaire dosis tromboseprofylaxe met heparine vergeleken. Daarnaast zijn er ook geen studies die apart een intermediaire dosis met een standaard dosis tromboseprofylaxe met heparine hebben vergeleken. Daarom kan in de aanbeveling geen onderscheid worden gemaakt tussen de standaard dosis en intermediaire dosis tromboseprofylaxe met heparine. Voor bepaalde subgroepen, zoals leeftijd of D-dimeer niveau bij presentatie, kon geen onderscheid worden gevonden in de analyse van de studies. Daarom konden er geen specifieke aanbevelingen worden gedaan voor deze subgroepen. Alle drie de cruciale uitkomsten waren secundaire uitkomsten in de studies. De studies hadden samengestelde primaire eindpunten die onderling onvergelijkbaar bleken. Zo werden bijvoorbeeld non-invasieve en invasieve beademing samen genomen, of sterfte en trombotische complicaties. Door deze verschillende samengestelde uitkomstmaten kon deze data niet gepoold worden. Verder zijn de studies in andere landen dan Nederland onder verschillende omstandigheden uitgevoerd. Het is bijvoorbeeld de vraag of de patiënten betrokken in studies die in andere landen zijn uitgevoerd (zoals Brazilië), vergeleken kunnen worden met Nederlandse patiënten. Een ander belangrijk punt van overweging is het feit dat de behandeling van patiënten met COVID-19 in 2021 in Nederland veranderd is ten opzichte van 2020, het jaar waarin de studies zijn verricht. Zo is er nu standaardbehandeling met hoge doses steroïden en IL-6 remmers. In verschillende van de gevonden studies kreeg een substantieel deel van de patiënten bijvoorbeeld geen behandeling met steroïden en werd slechts een kleine minderheid behandeld met IL-6 remmers. Alle studies hadden een zogenaamd ‘open label design’, waardoor bias kan zijn opgetreden bij zachtere uitkomstmaten als veneuze tromboembolie en bloeding. De artsen wisten welke behandeling een patiënt kreeg, hetgeen de klinische verdenking en diagnostische strategie heeft kunnen beïnvloeden. Eindpunten werden slechts deels centraal of lokaal geadjudiceerd, waardoor de validiteit van de diagnose longembolie (overgrote meerderheid van de trombotische events) niet in alle studies is na te gaan. De inclusiecriteria tussen de studies waren ook erg wisselend, met soms -maar niet altijd - selectie van patiënten met hoge tot zeer hoge D-dimeerwaarden. Patiënten met een van tevoren ingeschat hoog bloedingsrisico werden uitgesloten. De incidentie van bloedingscomplicaties zou daarom bij toepassing van therapeutische antistolling in de dagelijkse praktijk hoger kunnen uitvallen. Dit alles maakt dat er nog kennislacunes zijn. Het zal moeten blijken of resultaten van nieuwe studies de conclusies van de samenvatting van de literatuur zullen veranderen.
Waarden en voorkeuren van patiënten (en evt. hun verzorgers)
Het doel van toedienen van antistolling is het voorkómen van veneuze en, in mindere mate, arteriële trombose. Er is geen verschil in toediening of impact van de standaard profylactische of intermediaire versus de therapeutische dosis LMWH; beide worden op dezelfde manier subcutaan geïnjecteerd. Er zijn geen subgroepen met andere uitkomsten gevonden.
Patiënten die een behandeling met medicijnen voor antistolling krijgen vinden complete en eenduidige informatievoorziening belangrijk, o.a. over de indicatie en de veiligheid en risico’s van de behandeling (onderwerpen die van belang zijn in de communicatie met patiënten zijn terug te vinden in de Landelijke Transmurale Afspraak antistollingszorg (https://lta-antistollingszorg.nl/communicatie-met-patienten).
Kosten (middelenbeslag)
Er is geen doorslaggevend verschil in de kosten voor de standaard profylactisch of therapeutisch gedoseerde LMWH of DOAC.
Aanvaardbaarheid, haalbaarheid en implementatie
Door de opzet en uitkomsten van de onderzochte studies is geen definitief bewijs gevonden voor de cruciale uitkomsten. In de door de werkgroep gevoerde discussies kwamen verschillende visies naar voren. Uiteindelijk was er volgens de werkgroep geen overtuigend bewijs om de aanbevelingen aan te passen, zoals geformuleerd in de leidraad COVID-19 coagulopathie van april 2020, en om het gebruik van therapeutische antistolling als standaardbehandeling aan te bevelen.
Rationale van de aanbeveling
De individuele studies hadden vaak gecombineerde uitkomstmaten, waarbij niet-invasieve beademing, IC opname, intubatie, trombotische complicaties, ECMO, CVVH en sterfte in verschillende combinaties waren samengenomen. Bij de analyses van de individuele eindpunten bleek het potentiële voordeel van therapeutische antistolling onder de van tevoren vastgestelde grens van klinische relevantie te liggen. Het effect van een therapeutische dosis antistolling werd derhalve niet klinisch relevant bevonden ten opzichte van een standaard profylactische- of intermediaire dosis antistolling. Eerder waren voor de Nederlandse situatie geen grenzen van klinische relevantie vastgesteld voor het instellen van tromboseprofylaxe. De werkgroep heeft zich bij het vaststellen van die grenzen geconformeerd aan de grenzen voor sterfte en IC opname, zoals vastgesteld door de SWAB werkgroep voor medicamenteuze behandeling van COVID-19. De grens voor een klinisch relevant verschil in trombotische complicaties werd indirect afgeleid uit de ACCP richtlijn tromboseprofylaxe uit 2012. Uit de onderzochte studies bleek ook dat therapeutische antistolling niet klinisch relevant meer schade berokkende dan een standaard profylactische- of intermediaire dosis: het optreden van bloedingen bleek niet klinisch relevant verschillend tussen de groepen. De werkgroep kwam na uitvoerige discussie in meerderheid, maar niet unaniem, tot de conclusie dat er geen grond is om af te wijken van de huidige standaard behandeling, zoals omschreven in de leidraad COVID-19 coagulopathie van april 2020 en de richtlijnmodules cardiovasculaire complicaties bij COVID-19 van maart 2021.
Onderbouwing
Achtergrond
Ondanks tromboseprofylaxe en een verbeterde behandeling van COVID-19 komen trombotische complicaties nog frequent voor, met een geschatte incidentie van 23-28% in ICU patiënten en 7-9 % in afdelingspatiënten (Jiménez, 2021; Tan, 2021; Nopp, 2021). Het is niet bekend wat de beste dosis van tromboseprofylaxe is (laag, intermediair of therapeutisch). Mede hierdoor verschillen ziekenhuisprotocollen.
Conclusies
1. Mortality
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
Low GRADE |
Treatment with therapeutic anticoagulation with LMWHs or UFHs may result in little to no difference in mortality when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Sholzberg, 2021; Marcos, 2021. |
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
Low GRADE |
Treatment with therapeutic anticoagulation with DOACs may result in an increase in mortality when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Lopes, 2021 |
Total group
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with therapeutic anticoagulation on mortality when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Sholzberg, 2021; Marcos, 2021; Lopes, 2021. |
2. Length of hospital stay
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
- GRADE |
No studies were found that could answer the question what the effect is of therapeutic anticoagulation with LMWHs or UFHs when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs on hospital length of stay in adult COVID-19 patients admitted to the hospital ward (not ICU).
|
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
Moderate GRADE |
Treatment with therapeutic anticoagulation with DOACs likely results in little to no difference in length of hospital stay when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Lopes, 2021. |
3. ICU-admission (yes/no)
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
Low GRADE |
Treatment with therapeutic anticoagulation with LMWHs or UFHs may result in little to no difference in ICU-admission when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Sholzberg, 2021; Marcos, 2021. |
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
- GRADE |
No studies were found that could answer the question what the effect is of therapeutic anticoagulation with DOACs when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs on ICU-admission in adult COVID-19 patients admitted to the hospital ward (not ICU).
|
4. Organ support
Organ support free days
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
Moderate GRADE |
Treatment with therapeutic anticoagulation with LMWHs or UFHs likely results in little to no difference in organ support free days when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Sholzberg, 2021. |
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
- GRADE |
No studies were found that could answer the question what the effect is of therapeutic anticoagulation with DOACs when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs on organ support free days in adult COVID-19 patients admitted to the hospital ward (not ICU). |
Organ support (intubation or mechanical ventilation)
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
Moderate GRADE |
Treatment with therapeutic anticoagulation with LMWHs or UFHs likely results in little to no difference in organ support (intubation or mechanical ventilation) when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Sholzberg, 2021. |
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
Moderate |
Treatment with therapeutic anticoagulation with DOACs likely results in little to no difference in organ support (intubation or mechanical ventilation) when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Lopes, 2021. |
Total group
|
Moderate GRADE |
Treatment with therapeutic anticoagulation likely results in little to no difference in organ support (intubation or mechanical ventilation) when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Sholzberg, 2021; Lopes, 2021. |
5. Venous thromboembolism
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with a therapeutic anticoagulation with LMWHs or UFHs on venous thromboembolism when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Sholzberg, 2021. |
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with a therapeutic anticoagulation with DOACs on venous thromboembolism when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Lopes, 2021. |
Thromboembolic complications (VTE/ATE)
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
Low GRADE |
Treatment with therapeutic anticoagulation with LMWHs or UFHs may result in little to no difference on thromboembolic complications (VTE/ATE) when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Sholzberg, 2021; Marcos, 2021. |
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with a therapeutic anticoagulation with DOACs on thromboembolic complications (VTE/ATE) when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Lopes, 2021. |
Total group
|
Low GRADE |
Treatment with therapeutic anticoagulation may result in little to no difference on thromboembolic complications (VTE/ATE) when compared to standard prophylactic/intermediate anticoagulation LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Sholzberg, 2021; Marcos, 2021; Lopes, 2021. |
6. Major bleeding
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
|
Low GRADE |
Treatment with therapeutic anticoagulation with LMWHs or UFHs may result in little to no difference in major bleeding when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Spyropoulos, 2021; Sholzberg, 2021; Marcos, 2021. |
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
|
Very low GRADE |
The evidence is very uncertain about the effect of treatment with a therapeutic anticoagulation with DOACs on major bleeding when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Source: Lopes, 2021. |
Total group
|
Low GRADE |
Treatment with therapeutic anticoagulation may result in little to no difference in major bleeding when compared to standard prophylactic/intermediate anticoagulation with LMWHs or UFHs in adult COVID-19 patients admitted to the hospital ward (not ICU).
Sources: Lawler, 2021; Spyropoulos, 2021; Sholzberg, 2021; Marcos, 2021; Lopes, 2021. |
Samenvatting literatuur
Description of studies: treatment with unfractionated or low-molecular-weight heparin in hospitalized COVID-19 patients
Lawler (2021) describes an open-label, international, adaptive, multiplatform RCT (mpRCT). In this mpRCT, three platforms (REMAPCAP, ATTACC and ACTIV-4a) evaluating therapeutic-dose anticoagulation with heparin were integrated. Patients were enrolled at 121 sites in 9 countries (the United States, Canada, the United Kingdom, Brazil, Mexico, Nepal, Australia, the Netherlands, and Spain) over a period of 8 months (April 2020-Jan 2021). A total of 1181 patients in the intervention group received a continuous intravenous therapeutic-dose anticoagulation with unfractionated or low-molecular-weight heparin up to 14 days or to hospital discharge and 1050 patients in the control group received usual-care thromboprophylaxis up to 14 days or hospital discharge (see evidence table for details about anticoagulation and thromboprophylaxis regimens). The study included hospitalized, non-critically ill COVID-19 patients, defined by the absence of critical care–level organ support at enrollment. The study population in the intervention group had a mean age of 59 years (SD 14.1) versus 58.8 years (SD 13.9) in the control group and the majority was male (60% in the intervention versus 57% in the control group). The study groups were balanced with respect to baseline characteristics. The length of follow-up was 28 to 90 days.
Spyropoulos (2021) describes a multicenter open label RCT evaluating the effects of therapeutic-dose low-molecular-weight heparin versus institutional standard prophylactic dose or intermediate-dose heparins for thromboprophylaxis in high-risk hospitalized patients with COVID-19. Patients were enrolled from March 8, 2020, through May 14, 2021, at 12 centers in the US. A total of 11649 patients were assessed for eligibility. Eligible patients consisted of hospitalized nonpregnant adults 18 years or older with COVID-19 diagnosed by nasal swab or serologic testing. Moreover, there was a requirement for supplemental oxygen per investigator judgment and a plasma D-dimer level greater than 4 times the upper limit of normal based on local laboratory criteria or a sepsis-induced coagulopathy score of 4 or greater. Two hundred and fifty-seven patients were randomized into the therapeutic dose group (n= 130) or standard prophylactic/intermediate dose group (n = 127). However, a part of these patients were admitted to the intensive care unit (ICU): 45 out of 129 patients in the therapeutic dose group and 38 out of 124 in the standard prophylactic dose group. The results of the analyses of major bleeding were reported separately based on ICU status. Treatment began after randomization and was stopped at hospital discharge or upon occurrence of a primary efficacy outcome, key secondary outcome, or principal safety outcome requiring study drug discontinuation. All patients without a primary or key secondary outcome event underwent lower extremity Doppler compression ultrasonography at hospital day 10 + 4 or at discharge if sooner. The length of follow-up was 30 +2 days after randomization. Patients in the therapeutic dose group had a mean age of 65.8 years (SD 13.9) versus 67.7 years (SD 14.1) in the standard prophylactic dose group and the small majority was male (52.7% in the intervention versus 54.8% in the control group). The study groups were comparable with respect to baseline characteristics. Because of the relatively great number of patients that were admitted to the ICU, only the outcome measure major bleeding was included in the current analysis.
Sholzberg (2021) described a randomized controlled, adaptive, open label clinical trial evaluating the effects of therapeutic unfractionated or low-molecular-weight heparin compared with standard prophylactic unfractionated or low-molecular-weight heparin among moderately ill patients with covid-19 and increased D-dimer levels admitted to hospital wards. Elevated D-dimer levels were defined as one D-dimer value above the upper limit of normal (within 5 days (i.e. 120 hours) of hospital admission), and either D-Dimer ≥2 times the upper limit of normal, or D-Dimer above the upper limit of normal and oxygen saturation ≤ 93% on room air. 465 adults were recruited between May 29, 2020, and April 12, 2021, at 28 hospitals in Brazil, Canada, Ireland, Saudi Arabia, United Arab Emirates, and the US. Eligible patients were randomized into the therapeutic dose heparin group (n = 228) or the standard prophylactic dose heparin group (n=237). Treatments were continued until hospital discharge, day 28 of treatment, or death. Patients in the therapeutic dose group had a mean age of 60.4 years (SD 14.1) versus 59.6 years (SD 15.5) in the standard prophylactic dose group and the small majority was male (53.9% in the intervention versus 59.5% in the control group). The study groups were comparable with respect to baseline characteristics.
Marcos (2021) described an open-label, multicenter RCT in adult patients with non-severe COVID-19 pneumonia and elevated D-dimer >500 ng/mL, who were hospitalized in a conventional ward. Patients were recruited and randomized at five Spanish hospitals. Patients were allocated to either the experimental arm (n = 33), which consisted of bemiparin treatment 115 IU/kg one a day, or the control arm (n = 33), which was standard prophylaxis with subcutaneous bemiparin 3,500 IU one a day. Treatments were continued for a period of 10 days, independently of hospital discharge. Patients in the intervention group had a mean age of 62.3 years (SD 12.2), versus 63.0 years (SD 13.7) in the control group. In the intervention group, the fast majority was male (n = 24, 72.7%), versus a small majority in the control group (n = 17, 53.1%). Overall, there was a good balance between both study arms.
Description of study: treatment with DOAC in hospitalized COVID-19 patients
Lopes (2021) described a pragmatic, open-label, multicenter RCT in patients hospitalized with COVID-19 and elevated D-dimer concentration (defined as D-dimer above the upper limit of normal) to assess whether in-hospital anticoagulation with rivaroxaban (20 mg once daily) for patients with a stable condition or enoxaparin (1 mg/kg twice daily) for patients with an unstable condition, followed by rivaroxaban for 30 days decreased the time to death, duration of hospitalization, or duration of supplemental oxygen support when compared with mainly in-hospital standard prophylactic dose anticoagulation with enoxaparin or unfractionated heparin. In total, 615 patients were allocated to receive the therapeutic anticoagulation or in-hospital standard prophylactic dose anticoagulation. Patients in the therapeutic group had a mean age of 56.7 years (SD 14.1), versus 56.5 years (SD 14.5) in the standard prophylactic group. The majority in both groups was male: 192 (62%) in the therapeutic group, versus 176 (58%) in the standard prophylactic dose group. At baseline, 23 out of 311 (7%) patients in the therapeutic dose group were defined as having a clinically unstable condition, versus 16 out of 304 (5%) patients in the standard prophylactic dose group. The study population contains a small number of clinically unstable patients (7%). This was taken into account when determining the level of evidence (indirectness).
Table 1. Overview of included RCTs that compared therapeutic dose anticoagulation with intermediate and/or standard dose anticoagulation in hospitalized COVID-19 patients, separated into subgroups
|
Author, year and trial name |
Intervention (I) and control (C) |
Sample size for analysis |
Doses of anticoagulants |
Follow-up |
||||
|
Therapeutic dose vs intermediate/standard prophylactic dose – LMWH/UFH |
||||||||
|
Lawler, 2021 |
I: Therapeutic dose with LMWH or UFH |
I: N=1181 |
REMAP-CAP |
ACTIV-4a |
ATTACC |
REMAP-CAP |
ACTIV-4a |
ATTACC |
|
C: Usual-care thromboprophylaxis |
REMAP-CAP |
ACTIV-4a |
ATTACC |
REMAP-CAP |
ACTIV-4a |
ATTACC |
||
|
Spyropoulos, 2021 |
I: therapeutic-dose LMWH (enoxaparin) |
I: N= 129 |
I: 1 mg/kg subcutaneously twice daily if CrCl was 30 mL/min/1.73 m2 or greater or 0.5 mg/kg twice daily if CrCl was 15-29 mL/min/ 1.73 m2 |
Study drug was administered for the duration of hospitalization, including patient transfers to ICU settings |
||||
|
C: institutional standard prophylactic dose or intermediate-dose heparins for thromboprophylaxis |
C: could include UFH, up to 22 500 IU subcutaneously (divided twice or thrice daily); enoxaparin, 30 mg or 40 mg subcutaneously once or twice daily (weight based enoxaparin 0.5 mg/kg subcutaneously twice daily was permitted but strongly discouraged); or dalteparin, 2500 IU or 5000 IU subcutaneously daily |
|||||||
|
Sholzberg, 2021 |
I: therapeutic heparin (LMWH or UFH) – Enoxaparin, dalteparin, fondaparinux, tinzaparin, UFH |
I: N= 228 |
Specific dosages specified in trial protocol for each type of heparin, depending on creatinine clearance and BMI. (see Table 1 and 2 supplementary file) |
Therapeutic heparin: |
||||
|
C: standard prophylactic dose heparins (LMWH or UFH) – Enoxaparin, dalteparin, fondaparinux, tinzaparin, UFH |
||||||||
|
Marcos, 2021 |
I: bemiparin |
I: N= 33 |
I: 115 IU/Kg once daily, adjusted to body weight (7,500 IU for patients between 50-70 Kg; 10,000 IU for patients weighing >70-100 Kg; 12,500 IU for patients who weighed >100 Kg). |
The assigned treatments were planned for a 10-day period, independently of early hospital discharge. After that period, thromboprophylaxis use was left at investigators’ choice. In case of ICU requirement during the study treatment period, it was at the discretion of the treating physician to continue the study drug or not, according to local practices. |
||||
|
C: standard prophylaxis with subcutaneous bemiparin |
C: 3,500 IU once daily |
|||||||
|
Therapeutic dose vs intermediate/standard prophylactic dose – DOAC |
||||||||
|
Lopes, 2021 |
I: therapeutic anticoagulation |
I: N= 311 |
I: Clinically stable patients = 20 mg once daily. A reduced dose of 15 mg once daily was used in patients with a creatinine clearance of 30–49 mL/min or those taking azithromycin. |
All patients in the therapeutic anticoagulation group continued treatment to day 30 with the same dose of rivaroxaban. |
||||
|
C: standard prophylactic dose anticoagulation – standard venous thromboembolism prophylaxis with enoxaparin or UFH during hospitalisation. Patients in this group could receive therapeutic anticoagulation if they developed a definitive clinical indication or at the discretion of the investigator if a high clinical suspicion of a thromboembolic event was raised and a confirmatory test was not available. |
C:? |
|||||||
Table 2. Overview of composite outcomes and results per study
|
Study |
Primary composite outcome |
Results |
|
Lawler (2021) Multiplatform trial |
Organ support free days as evaluated on an ordinal scale that combined in-hospital death and the number of days free of cardiovascular or respiratory organ support up to day 21 among patients who survived to hospital discharge. |
|
|
Sholzberg (2021) RAPID trial |
|
|
|
Spyropoulos (2021) HEP-COVID trial |
A composite of VTE, ATE, or death for the non-ICU stratum separately |
In the therapeutic dose heparin group, 14 out of 84 (16.7%) patients reported the composite outcome, versus 31 out of 86 (36.1%) patients in the standard prophylactic dose heparin group. The RD was 19.4% in favor of the therapeutic dose heparin group (95%CI -32.3% to 6.5%). |
|
Marcos (2021) BEMICOP trial |
A composite of death, admission at ICU, need of mechanical ventilation support, development of moderate/severe acute respiratory distress syndrome and venous or arterial thrombosis within 10 days |
In the therapeutic dose bemiparin group, 7 out of 32 (21.9%) patients reported the composite outcome, versus 6 out of 33 (18.2%) patients in the standard prophylactic dose bemiparin group. The RD was 3.7% in favor of the standard prophylactic dose bemiparin group (95%CI -15.8% to 23.1%). |
|
Lopes (2021) ACTION trial |
A composite outcome of time to death, duration of hospitalisation, or duration of supplemental oxygen use |
The win ratio for the stable patients stratum was 0.84 (95%CI 0.57 to 1.21), indicating a worse outcome in the therapeutic dose group. |
Results
1. Mortality
Figure 1: Mortality in hospitalized patients with COVID-19, divided in subgroups
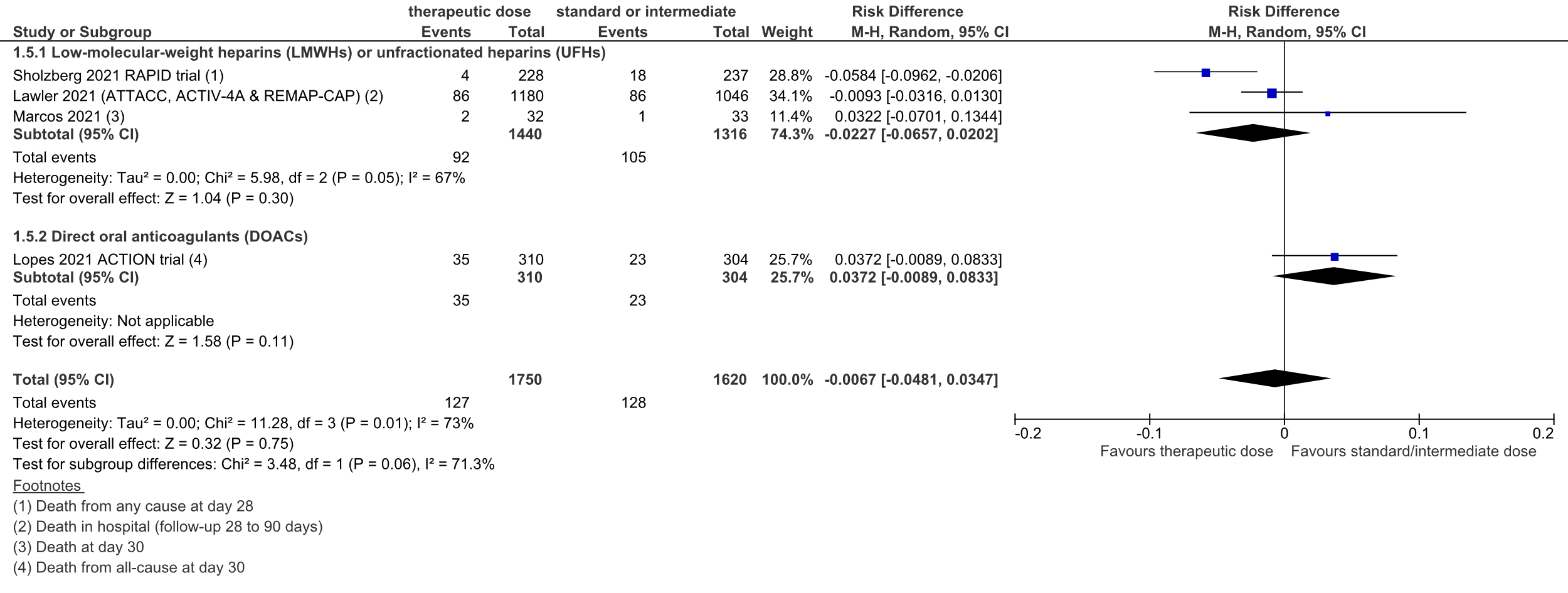
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
A total of 92 out of 1440 (6.4%) patients died in the therapeutic dose group, versus 105 out of 1316 (8.0%) in the standard prophylactic/intermediate dose group. The pooled risk difference (RD) was 2.3% in favor of the therapeutic dose group (95%CI -6.6% to 2.0%; figure 1). The corresponding NNT was 44. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure mortality was downgraded from high to low because of heterogeneity in the effect size (inconsistency, -1), and the confidence interval of the pooled RD crossing the lower threshold for clinical relevance (imprecision, -1).
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
A total of 35 out of 310 (11%) patients died in the therapeutic dose group, versus 23 out of 304 (8%) in the standard prophylactic/intermediate dose group. The pooled RD was 3.7% in favor of the standard prophylactic/intermediate dose group (95%CI -0.9% to 8.3%; figure 1). The corresponding NNT was 27. This was considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure mortality was downgraded from high to low because of the inclusion of a small number of patients admitted to the ICU (indirectness, -1), the inclusion of a single study and the confidence interval of the pooled RD crossing the upper threshold for clinical relevance (imprecision, -1).
Total group
Overall, 127 out of 1750 (7.3%) patients died in the therapeutic dose group, versus 128 out of 1620 (7.9%) in the standard prophylactic/intermediate dose group. The pooled RD was 0.7% in favor of the therapeutic dose group (95%CI -4.8% to 3.5%; figure 1). The corresponding NNT was 149. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure mortality was downgraded from high to very low because of heterogeneity in effect size (inconsistency, -1), and the confidence interval of the pooled RD crossing both thresholds (upper and lower) for clinical relevance (imprecision, -2).
2. Length of hospital stay
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
None of the studies reported length of hospital stay in both study groups.
However, Lawler (2021) did report the length of hospital stay for the total group of patients. The median hospital length of stay for all patients was 5 days (interquartile range 3-10). Lawler (2021) also reported an adjusted (age, sex, site, D-dimer group, and time epoch) hazard ratio of the time-to-event endpoint length of hospital stay truncated at 28 days of 1.03 (95%CI 0.95 to 1.13) in favor of the group treated with a therapeutic dose anticoagulation.
Sholzberg (2021) reported the number of hospital-free days alive. The mean number of hospital-free days alive was 19.8 days (SD 7.3) in the therapeutic dose group versus 18.4 (SD 9.2) in the standard prophylactic dose group. The mean difference was 1.40 days (95%CI -0.11 to 2.91).
Marcos (2021) reported hospital discharge in the first 10 days. In the therapeutic dose group, 21 out of 32 patients (65.6%) were discharged, versus 26 out of 33 patients (78.8%) in the standard prophylactic dose group.
Level of evidence
The level of evidence could not be determined, as none of the studies reported on length of hospital stay.
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
Lopes (2021) reported length of hospital stay at the end of 30 days. The mean length of stay was 8.1 days (SD 7.2) for the therapeutic dose group, versus 7.8 days (SD 7.5) for the standard prophylactic dose group. The mean difference was 0.3 days (95%CI -0.86 to 1.46). This difference was not considered to be clinically relevant.
Level of evidence
The level of evidence regarding the outcome measure length of hospital stay was downgraded from high to moderate because of the low number of patients (imprecision, -1).
3. ICU-admission (yes/no)
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
Figure 2: ICU-admission in hospitalized COVID-19 patients, divided in subgroups.
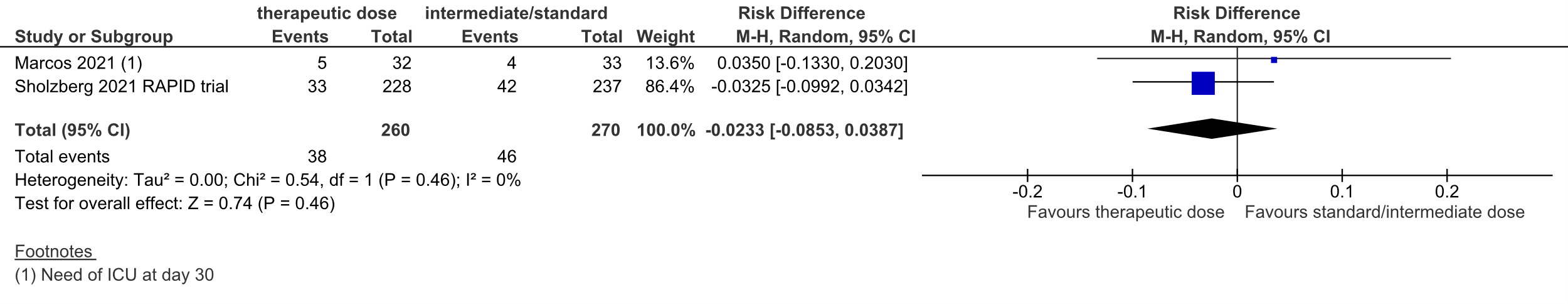
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Sholzberg (2021) reported ICU-admission. 33 out of 228 (14.5%) patients in the therapeutic dose group were admitted to the ICU, versus 42 out of 237 (17.7%) in the standard prophylactic dose group.
Marcos (2021) reported the need of ICU at day 10 and day 30. At day 10, 4 out of 32 patients (12.5%) needed ICU in the therapeutic dose group, versus 4 out of 33 patients (12.1%) in the standard prophylactic dose group. At day 30, 5 out of 32 patients (15.6%) needed ICU in the therapeutic dose group, versus 4 out of 33 patients (12.1%) in the standard prophylactic dose group.
Taken together, the pooled RD was 2.3% in favor of the therapeutic dose group (95%CI
-8.5% to 3.9%, figure 2). The corresponding NNT was 43. This was not considered to be a clinically relevant difference.
Lawler (2021) did not report ICU-admission.
Level of evidence
The level of evidence regarding the outcome measure ICU-admission was downgraded from high to low because of the low number of events and the confidence interval around the pooled RD crossing the lower threshold for clinical relevance (imprecision, -2).
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
Lopes (2021) did not report information on ICU-admission.
4. Organ support
Organ support free days
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
Sholzberg (2021) reported organ support free days alive. In the therapeutic dose group, patients had a mean of 25.8 (SD 6.2) organ support free days alive versus 24.1 (SD 8.8) days in the standard prophylactic dose group. The mean difference was 1.70 (95%CI 0.32 to 3.08) in favor of the therapeutic heparin group. This difference was not considered to be clinically relevant.
Lawler (2021) reported the organ support free days as evaluated on an ordinal scale that combined in-hospital death and the number of days free of cardiovascular or respiratory organ support up to day 21 among patients who survived to hospital discharge. Because the majority of patients in the two treatment groups survived until hospital discharge without receipt of critical care–level organ support, the median value for organ support–free days was 22 in both groups. Out of 1048 patients in the standard prophylactic dose group, 801 (76.4%) survived until hospital discharge without organ support during the first 21 days, as compared to 939 of 1171 patients (80.2%) in the therapeutic dose group. The RD was 3.8% in favor of the therapeutic dose group (95%CI 0.3% to 7.2%). However, no conclusions can be drawn based on this data.
Marcos (2021) did not report organ support free days as a separate outcome that matched how the working group defined it a priori, but solely as part of the primary composite outcome.
Level of evidence
The level of evidence regarding the outcome measure organ support free days (based on data from Sholzberg (2021)) was downgraded from high to moderate because of the confidence interval around the mean difference crossing the upper threshold for clinical relevance (imprecision, -1).
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
Lopes (2021) did not report organ support free days.
Organ support (intubation or mechanical ventilation)
Figure 3: intubation or mechanical ventilation in hospitalized COVID-19 patients, divided in subgroups.
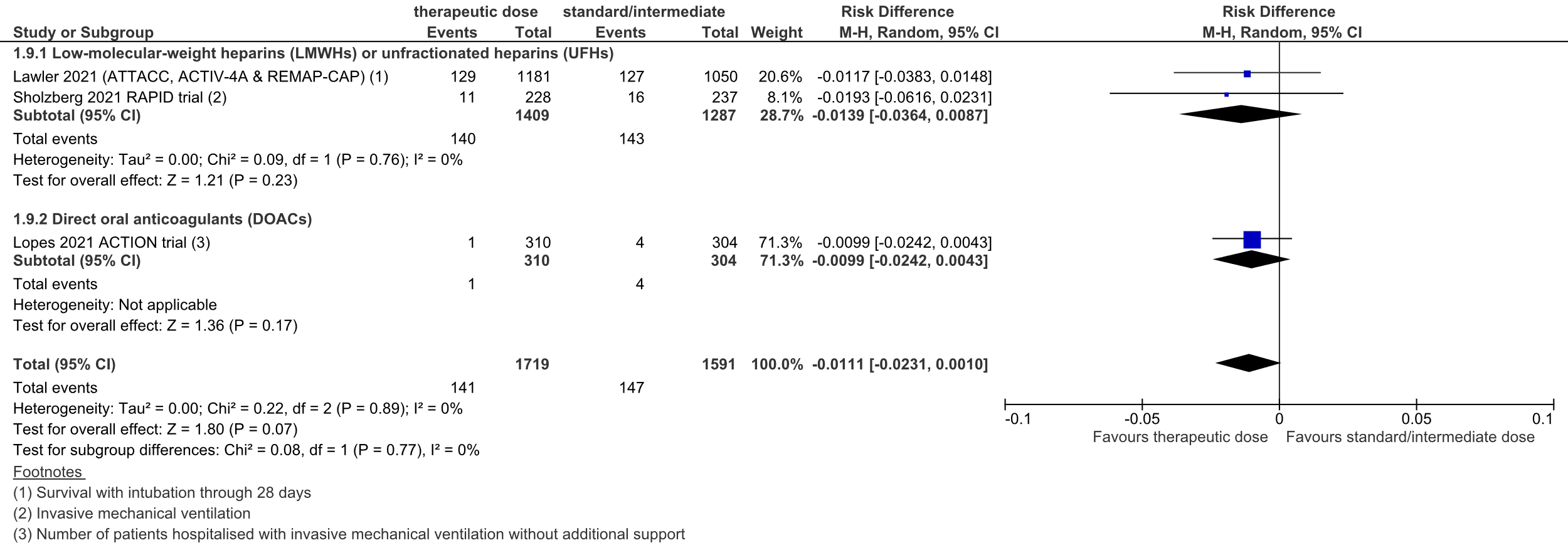
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
Lawler (2021) originally reported survival without intubation through 28 days. This was recalculated to survival with intubation through 28 days.
A total of 140 out of 1409 (9.9%) patients received intubation or invasive mechanical ventilation in the therapeutic dose group, versus 143 out of 1287 (11.1%) in the standard prophylactic/intermediate dose group. The pooled risk difference (RD) was 1.4% in favor of the therapeutic dose group (95%CI -3.6% to 0.9%; figure 3). The corresponding NNT was 72. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure organ support (intubation or mechanical ventilation) was downgraded from high to moderate, because of the small number of events (imprecision, -1).
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
A total of 1 out of 310 (0.3%) patients received invasive mechanical ventilation without additional support in the therapeutic dose group, versus 4 out of 304 (1.3%) in the standard prophylactic/intermediate dose group. The pooled risk difference (RD) was 1.0% in favor of the therapeutic dose group (95%CI -2.4% to 0.4%; figure 3). The corresponding NNT was 101. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure organ support (intubation or mechanical ventilation) was downgraded from high to moderate, because of the small number of events (imprecision, -1).
Total group
A total of 141 out of 1719 (8.2%) patients received intubation or invasive mechanical ventilation in the therapeutic dose group, versus 147 out of 1591 (9.2%) in the standard prophylactic/intermediate dose group. The pooled risk difference (RD) was 1.1% in favor of the therapeutic dose group (95%CI -2.3% to 0.1%; figure 3). The corresponding NNT was 90. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure organ support (intubation or mechanical ventilation) was downgraded from high to moderate, because of the small number of events (imprecision, -1).
5. Venous thromboembolism
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
Sholzberg (2021) reported the number of patients with venous thromboembolism (consisting of deep vein thrombosis and pulmonary embolism). In the therapeutic dose group, 2 out of 228 patients (0.9%) developed venous thromboembolism, versus 6 out of 237 (2.5%) in the standard prophylactic dose group. The RD was 1.7% in favor of the therapeutic dose group (95%CI -4.0% to 0.7%). The corresponding NNT was 61. This difference was not considered to be clinically relevant.
Lawler (2021) and Marcos (2021) did not report the number of patients with venous thromboembolism.
However, Lawler (2021) did report the number of events for deep venous thrombosis and pulmonary embolism separately. In the therapeutic dose group 6 events for deep venous thrombosis were reported, versus 7 events in the standard prophylactic dose group. For pulmonary embolism, 10 events were reported in the therapeutic dose group, versus 19 events in the standard prophylactic dose group.
Level of evidence
The level of evidence (based on data from Sholzberg (2021)) regarding the outcome measure venous thromboembolism was downgraded from high to very low because of the open-label study design (risk of bias, -1), the inclusion of one study with a small number of events (imprecision, -2).
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
Lopes (2021) reported the number of patients with venous thromboembolism (consisting of deep vein thrombosis and pulmonary embolism). 11 out of 310 (3.6%) patients in the therapeutic dose group versus 18 out of 304 (5.9%) patients in the standard prophylactic dose group developed venous thromboembolism. The RD was 2.4% in favor of the therapeutic dose group (95%CI -5.7% to 1.0%). The corresponding NNT was 42. This difference was not considered to be clinically relevant.
Level of evidence
The level of evidence regarding the outcome measure venous thromboembolism was downgraded from high to very low because of the open-label study design (risk of bias, -1), the inclusion of patients admitted to the ICU (indirectness, -1), the inclusion of only one study with a limited number of events and the confidence interval around the RD crossing the lower threshold for clinical relevance (imprecision, -1).
Thromboembolic complications (VTE/ATE)
Figure 4: thromboembolic complications in hospitalized COVID-19 patients, divided in subgroups.
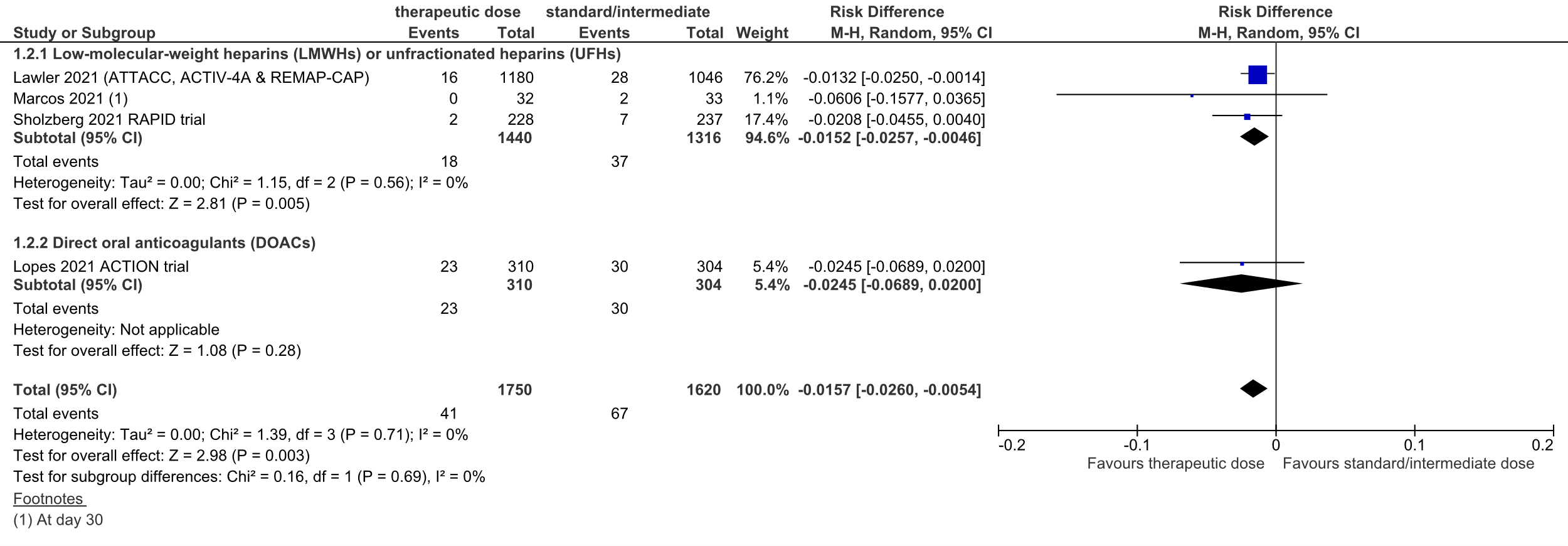
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
Lawler (2021) reported the number of patients with an in-hospital thrombotic event (defined as pulmonary embolism, myocardial infarction, ischemic cerebrovascular event, systemic arterial thromboembolism, and deep venous thrombosis): 16 out of 1180 (1.5%) patients experienced an in-hospital thrombotic event in the therapeutic dose group, versus 28 out of 1046 (2.7%) in the standard prophylactic dose group.
Sholzberg (2021) reported venous and arterial thromboembolism separately. When combined, 2 out of 228 (0.9%) patients developed VTE/ATE in the therapeutic dose heparin group, versus 7 out of 237 (3.0%) patients in the standard prophylactic dose heparin group.
Marcos (2021) reported arterial and venous thromboembolism (ATE/VTE) combined in one outcome measure at day 10 and day 30. At day 10, none of the patients in the intervention group developed ATE/VTE, versus 1 out of 33 patients (3.0%) in the control group. At day 30, none of the patients in the intervention group had developed ATE/VTE, versus 2 out of 33 patients (6.1%) in the control group.
Overall, the pooled RD was 1.5% in favor of the therapeutic dose heparin group (95%CI -2.6% to 0.5%, figure 4). The NNT was 66. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure thromboembolic complications was downgraded from high to low because of the open-label study designs (risk of bias, -1), and the small number of events (imprecision, -1).
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
Lopes (2021) also reported the number of patients with the composite thrombotic outcome (consisting of venous thromboembolism – deep vein thrombosis and pulmonary embolism, myocardial infarction, ischemic stroke, and major adverse limb event). 23 out of 310 (7.4%) patients in the therapeutic dose group versus 30 out of 304 (9.9%) patients in the standard prophylactic dose group reported the composite thrombotic outcome. The RD was 2.5% in favor of the therapeutic dose DOAC group (95%CI -6.9% to 2.0%, figure 4). The NNT was 41. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure thromboembolic complications was downgraded from high to very low because of the open-label study design (risk of bias, -1), the inclusion of patients admitted to the ICU (indirectness, -1), and the confidence interval around the RD crossing the lower threshold for clinical relevance (imprecision, -1).
Total group
In total, 41 out of 1750 (2.3%%) patients in the therapeutic dose group developed thromboembolic complications, versus 67 out of 1620 (4.1%) patients in the standard prophylactic/intermediate dose group. The pooled RD was 1.6% in favor of the therapeutic dose group (95%CI -2.6% to -0.5%; figure 4). The corresponding NNT was 64. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure thromboembolic complications was downgraded from high to low because of the open-label study designs (risk of bias, -1), and the low number of events (imprecision, -1).
6. Major bleeding
Figure 5: Major bleeding in hospitalized COVID-19 patients, divided in subgroups.
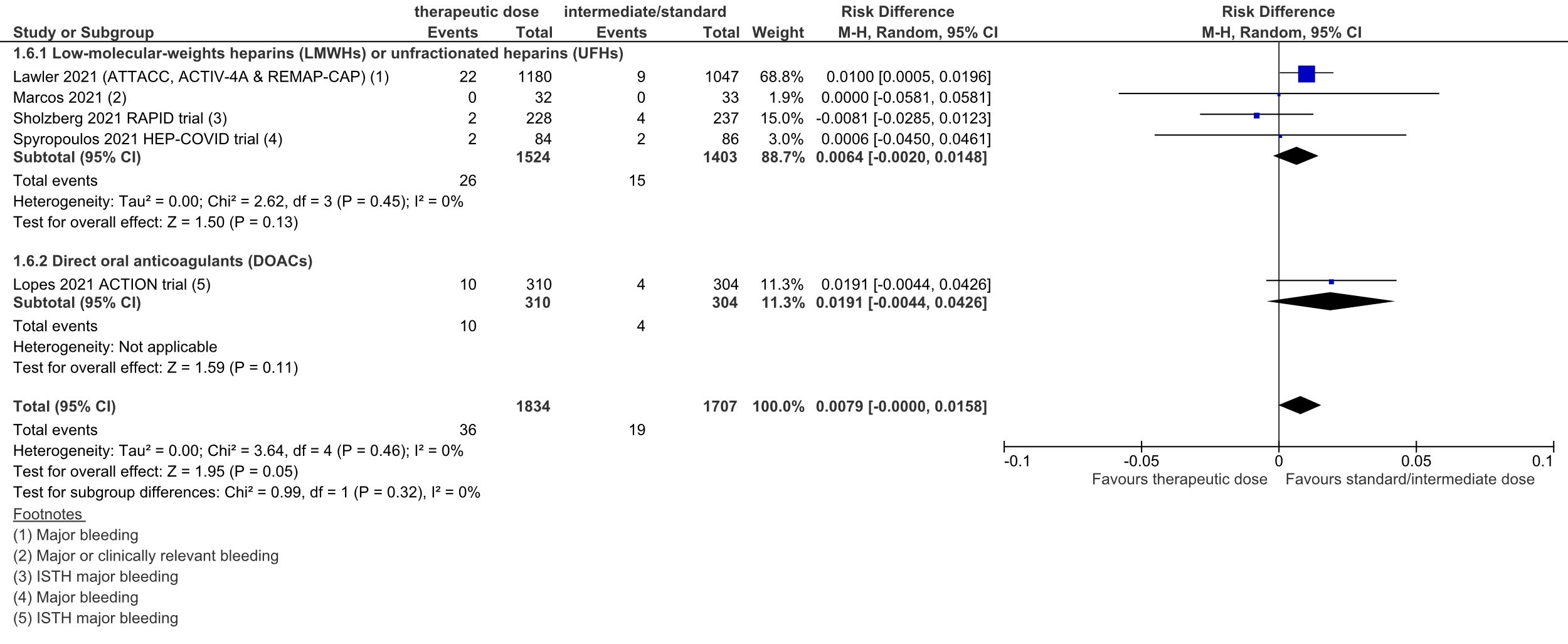
Z: p-value of overall effect; df: degrees of freedom; I2: statistical heterogeneity; CI: confidence interval
Therapeutic dose LMWHs/UFHs vs intermediate/standard prophylactic dose anticoagulation LMWHs/UFHs
In total, 26 out of 1524 (1.7%) patients in the therapeutic dose group developed major bleeding, versus 15 out of 1403 (1.1%) patients in the standard prophylactic/intermediate dose group. The pooled RD was 0.6% in favor of the standard prophylactic/intermediate dose group (95%CI -0.2% to 1.5%; figure 5). The corresponding NNH was 156. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure major bleeding was downgraded from high to low because of the open-label study designs (risk of bias, -1), and the low number of cases (imprecision, -1).
Therapeutic dose DOACs vs intermediate/standard prophylactic dose LMWHs/UFHs
In total, 10 out of 310 (3%) patients in the therapeutic dose group developed major bleeding, versus 4 out of 304 (1%) patients in the standard prophylactic/intermediate dose group. The RD was 1.9% in favor of the standard prophylactic/intermediate dose group (95%CI -0.4% to 4.3%, figure 5). The corresponding NNH was 52. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure major bleeding was downgraded from high to very low because of the open-label study design (risk of bias, -1), heterogeneity in the study population (indirectness, -1), inclusion of one study with a low number of cases (imprecision, -1).
Total group
In total, 36 out of 1834 (2.0%) patients in the therapeutic dose group developed major bleeding, versus 19 out of 1707 (1.1%) patients in the standard prophylactic/intermediate dose group. The pooled RD was 0.8% in favor of the standard prophylactic/intermediate dose group (95%CI 0.0% to 1.6%; figure 5). The corresponding NNH was 127. This was not considered to be a clinically relevant difference.
Level of evidence
The level of evidence regarding the outcome measure major bleeding was downgraded from high to low because of the open-label study designs (risk of bias, -1), and the low number of events (imprecision, -1).
Zoeken en selecteren
A systematic review of the literature was performed to answer the following question:
What is the efficacy and safety of anticoagulation therapy in COVID-19 patients admitted to the hospital (not ICU)?
PICO 1
P: all adult COVID-19 patients admitted to the hospital (not ICU) who are not already on chronic therapeutic anticoagulants
I: therapeutic dose (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin)
C: standard prophylactic dose or intermediate dose (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin) or no use of standard prophylactic dose or intermediate dose (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin)
O: mortality, major bleeding, venous thromboembolism, thromboembolic complications (venous and arterial thrombotic complications combined), length of hospital stay, ICU-admission (yes/no) and organ support free days
PICO 2
P: all adult COVID-19 patients admitted to the hospital (not ICU) who are not already on chronic therapeutic anticoagulants
I: intermediate dose (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin)
C: standard prophylactic dose (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin) or no use of standard prophylactic dose (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin)
O: mortality, major bleeding, venous thromboembolism, thromboembolic complications (venous and arterial thrombotic complications combined), length of hospital stay, ICU-admission (yes/no) and organ support free days
When possible subgroup analyses were performed to evaluate the efficacy and safety of the different types of anticoagulant.
We searched for standard dose of prophylaxis and intermediate dose of prophylaxis; the latter is typically a doubling of the standard dose of prophylaxis.
Relevant outcome measures
The guideline development group considered mortality, venous thromboembolism, thromboembolic complications (venous and arterial thrombotic complications combined) and major bleeding as critical outcome measures for decision making; and length of hospital stay, ICU-admission (yes/no), and organ support as important outcome measures for decision making.
A priori, the working group did not define organ support but used the definitions used in the studies.
The working group defined a risk difference of 3% as a minimal clinically (patient) important difference for mortality, venous thromboembolism, thromboembolic complications (venous and arterial thrombotic complications combined) and major bleeding; 3 days for length of hospital stay and organ support free days; and a risk difference of 5% for ICU-admission (yes/no) and a risk difference of 5% for organ support (yes/no).
Search and select (Methods)
The databases Medline (via OVID) and Embase (via Embase.com) were searched with relevant search terms until October 18th 2021. The detailed search strategy is depicted under the tab Methods. The systematic literature search resulted in 686 hits. Studies were selected based on the following criteria:
- randomized controlled trial (RCT)
- peer reviewed and published in indexed journal or pre-published
- comparing treatment with
- a therapeutic dose of anticoagulant (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin) with a standard prophylactic dose, intermediate dose, or no dose of anticoagulant (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin)
- an intermediate dose of anticoagulant (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin) with a standard prophylactic dose or no dose of anticoagulant (low molecular weight heparin, unfractionated heparin, direct oral anticoagulants, vitamin K-antagonists, aspirin)
- in non-critically ill patients with COVID-19
- <10% of patients admitted to the ICU.
Fourteen studies were initially selected based on title and abstract screening. After reading the full text, nine studies were excluded (see the table with reasons for exclusion under the tab Methods), and five studies were included.
Results
Five studies were included in the analysis of the literature. All studies investigated a therapeutic dose anticoagulant versus standard prophylactic or intermediate dose anticoagulant in COVID-19 patients admitted to the hospital (PICO 1). No studies were found that investigated intermediate dose anticoagulant versus standard prophylactic dose anticoagulant in COVID-19 patients admitted to the hospital (PICO 2). One of the studies included a small number of patients admitted to the ICU. Subgroups were made based on the type of anticoagulant used: low-molecular-weight heparins (LMWHs) and unfractionated heparins (UFHs), or direct oral anticoagulants (DOACs). Important study characteristics and results are summarized in the evidence tables. The assessment of the risk of bias is summarized in the risk of bias tables.
Referenties
- Jiménez D, García-Sanchez A, Rali P, Muriel A, Bikdeli B, Ruiz-Artacho P, Le Mao R, Rodríguez C, Hunt BJ, Monreal M. Incidence of VTE and Bleeding Among Hospitalized Patients With Coronavirus Disease 2019: A Systematic Review and Meta-analysis. Chest. 2021 Mar;159(3):1182-1196. doi: 10.1016/j.chest.2020.11.005. Epub 2020 Nov 17. PMID: 33217420; PMCID: PMC7670889.
- Lawler, P.R., et al., The REMAP-CAP, ACTIV-4a, and ATTACC Investigators (2021) Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with Covid-19. N Engl J Med DOI: 10.1056/NEJMoa2105911.
- Lopes RD, de Barros E Silva PGM, Furtado RHM, Macedo AVS, Bronhara B, Damiani LP, Barbosa LM, de Aveiro Morata J, Ramacciotti E, de Aquino Martins P, de Oliveira AL, Nunes VS, Ritt LEF, Rocha AT, Tramujas L, Santos SV, Diaz DRA, Viana LS, Melro LMG, de Alcântara Chaud MS, Figueiredo EL, Neuenschwander FC, Dracoulakis MDA, Lima RGSD, de Souza Dantas VC, Fernandes ACS, Gebara OCE, Hernandes ME, Queiroz DAR, Veiga VC, Canesin MF, de Faria LM, Feitosa-Filho GS, Gazzana MB, Liporace IL, de Oliveira Twardowsky A, Maia LN, Machado FR, de Matos Soeiro A, Conceição-Souza GE, Armaganijan L, Guimarães PO, Rosa RG, Azevedo LCP, Alexander JH, Avezum A, Cavalcanti AB, Berwanger O; ACTION Coalition COVID-19 Brazil IV Investigators. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): an open-label, multicentre, randomised, controlled trial. Lancet. 2021 Jun 12;397(10291):2253-2263. doi: 10.1016/S0140-6736(21)01203-4. Epub 2021 Jun 4. PMID: 34097856; PMCID: PMC8177770.
- Marcos M, Carmona-Torre F, Vidal Laso R, Ruiz-Artacho P, Filella D, Carbonell C, Jimenez-Yuste V, Schwartz J, Llamas P, Alegre F, Sádaba B, Núñez-Córdoba J, Yuste JR, Fernández-García J, Lecumberri R. Therapeutic vs. prophylactic bemiparin in hospitalized patients with non-severe COVID-19 (BEMICOP): an open-label, multicenter, randomized trial. Thromb Haemost. 2021 Oct 12. doi: 10.1055/a-1667-7534. Epub ahead of print. PMID: 34638151.
- Nopp S, Moik F, Jilma B, Pabinger I, Ay C. Risk of venous thromboembolism in patients with COVID-19: A systematic review and meta-analysis. Res Pract Thromb Haemost. 2020 Sep 25;4(7):1178–91. doi: 10.1002/rth2.12439. Epub ahead of print. PMID: 33043231; PMCID: PMC7537137.
- Sholzberg M, Tang GH, Rahhal H, AlHamzah M, Kreuziger LB, Ní Áinle F, Alomran F, Alayed K, Alsheef M, AlSumait F, Pompilio CE, Sperlich C, Tangri S, Tang T, Jaksa P, Suryanarayan D, Almarshoodi M, Castellucci L, James PD, Lillicrap D, Carrier M, Beckett A, Colovos C, Jayakar J, Arsenault MP, Wu C, Doyon K, Andreou ER, Dounaevskaia V, Tseng EK, Lim G, Fralick M, Middeldorp S, Lee AYY, Zuo F, da Costa BR, Thorpe KE, Negri EM, Cushman M, Jüni P; RAPID Trial investigators. Heparin for Moderately Ill Patients with Covid-19. medRxiv [Preprint]. 2021 Jul 12:2021.07.08.21259351. doi: 10.1101/2021.07.08.21259351. Update in: BMJ. 2021 Oct 14;375:n2400. PMID: 34268513; PMCID: PMC8282099.
- Spyropoulos AC, Goldin M, Giannis D, Diab W, Wang J, Khanijo S, Mignatti A, Gianos E, Cohen M, Sharifova G, Lund JM, Tafur A, Lewis PA, Cohoon KP, Rahman H, Sison CP, Lesser ML, Ochani K, Agrawal N, Hsia J, Anderson VE, Bonaca M, Halperin JL, Weitz JI; HEP-COVID Investigators. Efficacy and Safety of Therapeutic-Dose Heparin vs Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients With COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern Med. 2021 Oct 7:e216203. doi: 10.1001/jamainternmed.2021.6203. Epub ahead of print. PMID: 34617959; PMCID: PMC8498934.
- Tan BK, Mainbourg S, Friggeri A, Bertoletti L, Douplat M, Dargaud Y, Grange C, Lobbes H, Provencher S, Lega JC. Arterial and venous thromboembolism in COVID-19: a study-level meta-analysis. Thorax. 2021 Oct;76(10):970-979. doi: 10.1136/thoraxjnl-2020-215383. Epub 2021 Feb 23. PMID: 33622981; PMCID: PMC7907632.
Evidence tabellen
|
Study reference |
Study characteristics |
Patient characteristics |
Intervention (I) |
Comparison / control (C)
|
Follow-up |
Outcome measures and effect size
|
Comments |
|
Lawler, 2021
Integrated REMAPCAP, ATTACC and ACTIV-4a trial.
|
Type of study: open-label, international, adaptive, multiplatform RCT (mpRCR)
Setting and country: 121 sites in 9 countries (the United States, Canada, the United Kingdom, Brazil, Mexico, Nepal, Australia, the Netherlands, and Spain).
Funding and conflicts of interest: The trial was supported by multiple international funding organizations who had no role in the design, analysis or reporting of the trial result, apart from the ACTIV-4a protocol, which received input on design from professional staff members at the National Institutes of Health and from peer reviewers.
See publication for funding details. |
Inclusion criteria: Patients hospitalized with COVID-19 and who were not critically ill* (absence of critical care–level organ support at enrollment).
Exclusion criteria: Patients were ineligible for enrollment if 72 hours had elapsed since hospital admission for Covid-19 or since in-hospital confirmation of the presence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (ATTACC and ACTIV-4a) or if 14 days had elapsed since admission (REMAP-CAP). Patients were also excluded if hospital discharge was expected within 72 hours or if they had a clinical indication for therapeutic anticoagulation, a high risk of bleeding, receipt of dual antiplatelet therapy, or a known heparin allergy, including heparin-induced thrombocytopenia (HIT)
For inclusion criteria per sub trial, see bottom of column with outcomes
N total at baseline: N = 2244 randomized; primary analysis involved 2219 patients. After exclusion: Intervention: 1181 Control: 1050
Important characteristics: Age, mean (SD) I: 59.0 y (14.1) C: 58.8 y (13.9)
Sex, n/N (%) male I: 713/1181 (60.4%) C: 597/1050 (56.9%)
Country, n (%) (i/C) Canada 102 (8.6) / 83 (7.9) Brazil 234 (19.8) / 209 (19.9) Other§§ 177 (15.0) / 148 (14.1)
BMI, median (IQR) I: 29.8 (26.3–34.7) C: 30.3 (26.7–34.9)
Respiratory support, n/N (%) None I: 156 (13.2) C: 123 (11.7) Low-flow nasal cannula or face mask I: 789 (66.8) C: 696 (66.3) High-flow nasal cannula I: 25 (2.1) C: 28 (2.7) Noninvasive mechanical ventilation I: 21 (1.8) C: 24 (2.3) Unspecified**; In REMAP-CAP, levels of oxygen support (including no support) below the level of high-flow nasal cannula were not reported. I: 190 (16.1) C: 179 (17.0)
Preexisting condition — no./total no. (%) Hypertension I: 546/1023 (53.4) C: 447/892 (50.1) Diabetes mellitus I: 352/1181 (29.8) C: 311/1049 (29.6) Severe cardiovascular disease I: 123/1165 (10.6) C: 121/1038 (11.7) Chronic kidney disease I: 83/1173 (7.1) C: 69/1037 (6.7) Chronic respiratory disease I: 249/1132 (22.0) C: 212/988 (21.5) Immunosuppressive disease I: 105/1143 (9.2) C: 103/1005 (10.2)
Treatment — no./total no. (%) Antiplatelet agent I: 148/1140 (13.0) C: 111/1013 (11.0) Remdesivir I: 428/1178 (36.3) C: 383/1048 (36.5) Glucocorticoid I: 479/791 (60.6) C: 415/656 (63.3) Tocilizumab I: 6/1178 (0.5) C: 7/1048 (0.7)
Median laboratory value (IQR) Median d-dimer level relative to ULN at trial site I: 1.6 (0.9–2.6) C: 1.5 (1.0–2.7) Platelets — per mm3 I: 221,000 (171,000–290,000) C: 218,000 (172,500–289,000) Lymphocytes — per mm3 I: 900 (700–1300) C: 1000 (700–1400) Creatinine — mg/dl I: 0.9 (0.7–1.1) C: 0.9 (0.7–1.1)
Groups comparable at baseline. |
Therapeutic-dose anticoagulation with unfractionated or low-molecular-weight heparin up to 14 days or to hospital discharge. Unfractionated heparine: |
Usual-care pharmacologic thromboprophylaxis up to 14 days or hospital discharge. After this period, decisions regarding thromboprophylaxis are at discretion of treating clinician.
For ACTIV 4a any one of enoxaparin, dalteparin, tinzaparin, fondaparinux, or heparin according to local preference. Dose of agent specified to be consistent with guidelines for low dose thromboprophylaxis. |
Length of follow-up: 28 to 90 days
Loss-to-follow-up: Intervention: 19 (1.6%) Reasons 9 did not have confirmed COVID-19 9 withdrew consent 1 did not have outcome data available
Control: 6 (0.6%) Reasons 1 withdrew consent 2 did not have outcome data available 3 did not have confirmed COVID-19 |
Mortality: Death in hospital I: 86/1180 (7.3%) C: 86/1046 (8.2%)
Length of hospital stay: Hospital length of stay (time-to-event endpoint truncated at 28 days) in the total group of patients Hazard ratio (95% CI) 1.03 (0.94-1.13); the overall median (interquartile range) hospital length of stay following randomization was 5 (3, 10) days.
ICU-admission Nothing reported
Organ support free days Evaluated on an ordinal scale that combined in-hospital death and the number of days free of cardiovascular or respiratory organ support up to day 21 among patients who survived to hospital discharge I: 939/1171 patients (80.2%) C: 801/1048 patients (76.4%) Of the in the usual-care Adjusted odds ratio 1.27 (95% Credible Interval 1.03 to 1.58). Adjusted for age, sex, trial site, d-dimer cohort, and enrollment period
Survival without intubation through 28 days I: 1052/1181 (89.1%) C: 923/1050 (87.9%)
Venous thromboembolism (VTE) Number of patients with VTE not specifically reported.
Pulmonary embolism events I: 10 C: 19
Deep venous thrombosis events I: 6 C: 7
Number of patients with an in-hospital thrombotic event (defined as pulmonary embolism, myocardial infarction, ischemic cerebrovascular event, systemic arterial thromboembolism, and deep venous thrombosis). I: 16/1180 (1.5%) patients C: 28/1046 (2.7%) patients
Major bleeding I: 22 out of 1180 (1.9%) patients C: 9 out of 1047 (0.86%) patients
------------------------------------
Inclusion criteria per sub trial: REMAP-CAP:
ACTIV-4a:
ATTACC:
|
Definitions: * Moderate disease severity was defined as hospitalization for Covid-19 without the need for ICU-level care. ICU-level care was defined as the use of respiratory or cardiovascular organ support (oxygen delivered by high-flow nasal cannula, non-invasive or invasive mechanical ventilation, or the use of vasopressors or inotropes) in an ICU. [In ACTIV-4a, in which investigators found that ICU-level care was challenging to define during the pandemic, receipt of organ support, regardless of hospital setting, was used to define ICU-level care. Patients who were admitted to an ICU but without receiving qualifying organ support were considered to be moderately ill.]
Definitions of major thrombotic and any thrombotic events are described in the study protocol.
Remarks: The trial was stopped when prespecified criteria for the superiority of therapeutic dose anticoagulation were met.
Authors conclusion: In noncritically ill patients with Covid-19, an initial strategy of therapeutic-dose anticoagulation with heparin increased the probability of survival to hospital discharge with reduced use of cardiovascular or respiratory organ support as compared with usual-care thromboprophylaxis.
|
|
Spyropoulos, 2021
HEP-COVID Randomized Clinical Trial
AND
corresponding Trial protocol design paper (Goldin, 2021) |
Type of study: Randomized controlled trial
Setting and country: Multicenter study in the US (12 centers)
Funding and conflicts of interest: Support from Feinstein Institutes for Medical Research, the Broxmeyer Fellowship in Clinical Thrombosis, and grant R24AG064191 from the National Institute on Aging. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. |
Inclusion criteria:
Exclusion criteria:
normalized ratio greater than 1.5
(CrCl) less than 15mL/min/1.73m2
25 000/μL
(HIT) within 100 days
Patients were stratified into two subgroups: non-intensive care unit patients and intensive care unit patients. ICU status was defined by mechanical ventilation, noninvasive positive pressure ventilation or high-flow nasal cannula, vasopressors, or vital sign monitoring more often than every 4 hours. ICU: 83 patients (32.8%) Non-ICU: 170 patients (67.2%)
N total at baseline: N = 257 participants randomized (130 to the intervention group and 127 to the control group) at baseline.
Important prognostic factors2:
Age, mean (SD): I: 65.8 (13.9) C: 67.7 (14.1)
Sex, No./total No. (%) male I: 68/129 (52.7%) C: 68/124 (54.8%)
BMI, mean (SD) I: 31.2 (9.3) C: 29.8 (13.6)
Race and ethnicity, No. (%) (I/C) Asian 11 (8.5) / 14 (11.3) Black 33 (25.6) / 37 (29.8) White 56 (43.4) / 46 (37.1) Multiracial/unknown 29 (22.5) / 27 (21.8)
ICU, No./total No. (%) I: 45/129 (34.9) C: 38/124 (30.6)
Comorbidities, No./total No. (%) (I/C) Hypertension 81/129 (62.8) / 70/123 (56.9) Heart failure 0 / 2/124 (1.6) Diabetes mellitus 51/128 (39.8) / 43/124 (34.7) Dyslipidemia 48/129 (37.2) / 39/124 (31.5) Coronary artery disease 7/129 (5.4) / 11/124 (8.9) Valvular heart disease 1/129 (0.8) / 3/124 (2.4) History of ischemic stroke 5/129 (3.9) / 3/124 (2.4) History of carotid occlusive disease 0 / 0 Peripheral artery disease 4/129 (3.1) / 1/124 (0.8) Chronic kidney disease 5/129 (3.9) / 4/124 (3.2) Chronic lung disease 9/129 (7.0) / 8/124 (6.5) Chronic liver disease/cirrhosis 2/129 (1.6) / 1/124 (0.8) Pulmonary hypertension 1/127 (0.8) / 2/124 (1.6)
VTE risk factors,, No./total No. (%) (I/C) Personal history of VTE 6/129 (4.7) / 2/124 (1.6) History of cancer 16/129 (12.4) / 10/124 (8.1) Active cancer 1/129 (0.8) / 4/124 (3.2) Autoimmune disease 1/128 (0.8) / 2/124 (1.6) Hormonal therapy/oral contraceptives 1/129 (0.8) / 1/124 (0.8) Known thrombophilia 0 / 0 Recent stroke with paresis 1/129 (0.8) / 1/124 (0.8)
Clinical scores, mean (SD) (I/C) IMPROVEDD VTE risk score 4.33 (1.48) / 4.22 (1.36) Sepsis-induced coagulopathy score 2.35 (0.73) / 2.31 (0.85)
Laboratory parameters, mean (SD) (I/C) White blood cell count, /μL 9600 (5800) / 9800 (8200) Platelets, ×103/μL 287.7 (119.8) / 269.7 (108.2) Serum creatinine, mg/dL 0.94 (0.45) / 1.00 (0.50) Prothrombin time, s 13.5 (1.6) /13.6 (2.6) D-dimer, ng/mL 3837 (6166) / 3183 (5409)
Medications prior to randomization, No./total No. (%)(I/C) Low-molecular-weight heparin 106/128 (82.8) / 97/124 (78.2) Unfractionated heparin 18/127 (14.2) / 23/121 (19.0) Remdesivir 93/129 (72.1) / 85/124 (68.6) Glucocorticoids 111/127 (87.4) / 93/123 (75.6) Antiplatelets 40/129 (31.0) / 24/124 (19.4)
Oxygen therapy, No./total No. (%) (I/C) Nasal cannula 80/129 (62.0) / 83/124 (66.9) Nonrebreather mask 12/129 (9.3) / 11/124 (8.9) Ventilation mask 4/129 (3.1) / 2/124 (1.6) High-flow or noninvasive positive-pressure ventilation 20/129 (15.5) / 19/124 (15.3) Invasive mechanical ventilation 8/129 (6.2) / 5/124 (4.0)
Length of hospital stay, mean (SD), d I: 12.2 (9.3)
Groups comparable at baseline? Yes
|
Describe intervention (treatment/procedure/test):
Patients in the therapeutic dose group received enoxaparin at a dose of 1 mg/kg subcutaneously twice daily if CrCl was 30 L/min/1.73m2 or greater or 0.5 mg/kg twice daily if CrCl was 15-29 mL/min/1.73m2. Study drug was administered for the duration of hospitalization, including patient transfers to ICU settings.
Study protocol (Goldin, 2021): Individual dose modification is not permitted unless the CrCl falls below 15 mL/min in the treatment arm (arm 0). In that case, conversion to dose-adjusted intravenous (IV) UFH is acceptable. The investigator is encouraged to convert back to treatment-dose enoxaparin as per protocol once the CrCl returns to values higher than or equal to 15 mL/min.
|
Describe control (treatment/procedure/test):
Patients in the standard-dose group received prophylactic or intermediate-dose heparin regimens per local institutional standard and could include UFH, up to 22500 IU subcutaneously (divided twice or thrice daily); enoxaparin, 30 mg or 40 mg subcutaneously once or twice daily (weight based enoxaparin 0.5mg/kg subcutaneously twice daily was permitted but strongly discouraged); or dalteparin, 2500 IU or 5000 IU subcutaneously daily. If CrCl fell below15 mL/min/ 1.73 m2, enoxaparin was converted to treatment-dose intravenous UFH until kidney function improved to CrCl greater than 15 mL/min/1.73 m2, when blinded-dose subcutaneous enoxaparin was resumed. Study drug was administered for the duration of hospitalization, including patient transfers to ICU settings. In the standard dose group, 76 patients (61.3%) received prophylactic doses of heparin (enoxaparin, ≤40mg daily), while 48 patients (38.7%) received intermediate doses of heparin (enoxaparin, 30 mg twice daily, 3 patients [2.4%]; enoxaparin, 40mg twice daily, 43 patients [34.7%]; enoxaparin, 0.5mg/kg twice daily, 2 patients [1.6%]).
Goldin, 2021: Dose modification is allowed in the prophylactic/intermediate group (arm 1) if the CrCl falls below15 mL/min so that UFH up to 22,500 U daily (i.e., UFH 5,000 U SQ BID or TID or 7,500 IU SQ BID or TID) can be used. The investigator is encouraged to convert back to prophylactic-/intermediate dose LMWH/UFH as per protocol once the CrCl returns to values higher than or equal to 15 mL/min. |
Length of follow-up: Until 30 + 2 days after randomization.
Loss-to-follow-up: 4 patients did not receive study drug (2 withdrew consent and 2 reached end points prior to the first dose). That resulted in 253 patients in the modified intention-to-treat population for analysis: Intervention: 129 Control: 124
The primary analysis was based on the modified intention to-treat population, followed by the per-protocol population.
|
Clinical Outcomes During the 30-Day, stratified for ICU and non-ICU:
Mortality Not reported
Length of hospital stay Not reported
ICU-admission Not reported
Organ support free days Not reported
Venous thromboembolism Not reported
Major bleeding I: 2/84 (2.4) C: 2/86 (2.3) RR (95% CI): 1.02 (0.15-7.10)
ICU patients: I: 4/45 (8.9) C: 0 RR (95% CI): 7.62 (0.42-137.03)
|
Author’s conclusions: Therapeutic dose LMWH reduced the composite of thromboembolism and death compared with standard heparin thromboprophylaxis without increased major bleeding among hospitalized patients with COVID-19 with very elevated D-dimer levels. The treatment effect was not seen in ICU patients. |
|
Sholzberg, 2021
RAPID trial |
Type of study: Randomized controlled, adaptive, open label clinical trial.
Setting and country: 28 hospitals in Brazil, Canada, Ireland, Saudi Arabia, United Arab Emirates, and US.
Funding and conflicts of interest: Funding limitations and covid-19 restrictions interfered with our ability to involve patient partners in setting the research question and in developing plans for recruitment, design, and implementation of the results of this study. |
Inclusion criteria: Moderately ill* patients admitted to hospital wards for covid-19. 1) Laboratory confirmed COVID-19 (diagnosis of SARS-CoV-2 via reverse transcriptase polymerase chain reaction as per the World Health Organization protocol or by nucleic acid based isothermal amplification) prior to hospital admission OR within first 5 days (i.e. 120 hours) after hospital admission; 2) Admitted to hospital for COVID-19; 3) One D-dimer value above the upper limit of normal (ULN) (within 5 days (i.e. 120 hours) of hospital admission) AND EITHER: a. D-Dimer ≥2 times ULN OR b. D-Dimer above ULN and Oxygen saturation ≤ 93% on room air; 4) > 18 years of age; 5) Informed consent from the patient (or legally authorized substitute decision maker).
Exclusion criteria: 1) pregnancy; 2) hemoglobin <80 g/L in the last 72 hours 3) platelet count <50 * 10^9/L in the last 72 hours 4) known fibrinogen <1.5 g/L (if testing deemed clinically indicated by the treating physician prior to the initiation of anticoagulation); 5) known INR >1.8 (if testing deemed clinically indicated by the treating physician prior to the initiation of anticoagulation); 6) patient already prescribed intermediate dosing of LMWH that cannot be changed (determination of what constitutes an intermediate dose is to be at the discretion of the treating clinician taking the local institutional thromboprophylaxis protocol for high risk patients into consideration); 7) patient already prescribed therapeutic anticoagulation at the time of screening [low or high dose nomogram UFH, LMWH, warfarin, direct oral anticoagulant (any dose of dabigatran, apixaban, rivaroxaban, edoxaban)]; 8) patient prescribed dual antiplatelet therapy, when one of the agents cannot be stopped safely; 9) known bleeding within the last 30 days requiring emergency room presentation or hospitalization; 10) known history of a bleeding disorder of an inherited or active acquired bleeding disorder; 11) known history of heparin-induced thrombocytopenia; 12) known allergy to UFH or LMWH; 1 3) admitted to the intensive care unit at the time of screening; 14) treated with non-invasive positive pressure ventilation or invasive mechanical ventilation at the time of screening; 15) Imminent death according to the judgement of the most responsible physician; 16) enrollment in another clinical trial of antithrombotic therapy involving hospitalized patients.
N total at baseline: 465 were randomized. Intervention: 228 Control: 237
Important prognostic factors2: Age, mean (SD): I: 60.4 (14.1) C: 59.6 (15.5)
Sex, n (%), Male: I: 123 (53.9%) C: 141 (59.5%)
Body mass index, mean (SD) I: 30.3 (6.4) C: 30.2 (7.0)
Duration of symptoms before hospital admission (days), mean (SD) I: 7.1 (5.1) C: 7.1 (5.2)
Duration of hospital admission before randomization (days), mean (SD) I: 1.5 (1.1) C: 1.4 (1.0)
Hypoxaemia at baseline, n (%) I: 190 (90.9) C: 203 (93.1)
High flow nasal cannula oxygen use, n (%) I: 14 (6.2)
Pre-existing conditions, n (%): Hypertension I: 108 (47.4) C: 117 (49.4)
Diabetes mellitus I: 83 (36.4) C: 77 (32.5)
Coronary artery disease I: 16 (7.0) C: 18 (7.6)
Heart failure I: 9 (3.9) C: 6 (2.5)
Atrial fibrillation I: 0 (0.0) C: 2 (0.8)
Cerebrovascular disease I: 10 (4.4) C: 9 (3.8)
Peripheral vascular disease I: 0 (0.0) C: 1 (0.4)
History of venous thromboembolism I: 3 (1.3) C: 2 (0.8)
Chronic pulmonary disease I: 36 (15.8) C: 27 (11.4)
Chronic kidney disease I: 20 (8.8) C: 13 (5.5)
Chronic liver disease I: 5 (2.2) C: 9 (3.8)
Cancer I: 13 (5.7) C: 19 (8.0)
Immunodeficiency I: 1 (0.4) C: 2 (0.8)
Autoimmune disease I: 6 (2.6) C: 11 (4.6)
Cognitive impairment I: 12 (5.3) C: 11 (4.6)
Mental illness I: 18 (7.9) C: 13 (5.5)
Active smoking I: 5 (2.2) C: 7 (3.0)
Drug history, n (%): Systemic corticosteroids I: 161 (70.6) C: 162 (68.4)
Antiplatelet agent I: 24 (10.5) C: 29 (12.2)
Previous covid-19 vaccine, n (%) I: 1 (0.4) C: 2 (0.8)
D-dimer distribution, n (%): <2.ULN I: 115 (50.4) C: 112 (47.3)
≥2-3.ULN I: 61 (26.8) C: 55 (23.2)
≥3-4.ULN I: 25 (11.0) C: 27 (11.4)
≥4.ULN I: 27 (11.8) C: 43 (18.1)
Groups comparable at baseline? Yes
|
Describe intervention (treatment/procedure/test):
Unfractionated heparin as used for the treatment of venous thromboembolism. Unfractionated heparin was administered using a weight based nomogram (bolus plus continuous infusion) with activated partial thromboplastin time or unfractionated heparin anti- Xa titration according to the centre specific protocols (that is, high dose nomogram).
Study treatment was started within 24 hours after randomization and continued until the first of hospital discharge, day 28, study withdrawal, or death. If a participant was admitted to ICU, continuation of the allocated treatment was recommended.
|
Describe control (treatment/procedure/test):
Dose capped prophylactic subcutaneous heparin (low molecular weight heparin or unfractionated heparin) adjusted for body mass index and creatinine clearance (see supplementary file for dosing).
Study treatment was started within 24 hours after randomization and continued until the first of hospital discharge, day 28, study withdrawal, or death. If a participant was admitted to ICU, continuation of the allocated treatment was recommended. |
Length of follow-up: 28 days
Loss-to-follow-up: Intervention: Out of 228 who were assigned to the intervention group, 6 did not receive the allocated intervention (clinician discretion and patient refusal). 4 were lost to follow-up after hospital discharge. There were 216 patients left for per protocol analysis (6 did not receive allocated intervention and 6 had a negative D-dimer result).
Control: Out of 237 who were assigned to the control group, 5 did not receive the allocated intervention (clinician discretion and patient refusal). 7 were lost to follow-up after hospital discharge. There were 227 patients left for per protocol analysis (5 did not receive allocated intervention and 5 had a negative D-dimer result).
|
Outcome measures and effect size (include 95%CI and p-value if available):
Mortality Death from any cause, follow-up 28 days I: 4 of 228 (1.8%) patients C: 18 of 237 (7.6%) patients
Length of hospital stay The mean number of hospital-free days. I: 19.8 days (SD 7.3) C: 18.4 days (SD 9.2) Mean difference: 1.40 days (95%CI -0.11 to 2.91)
ICU admission I: 33 out of 228 (14.5%) C: 42 out of 237 (17.7%) RR (95%CI): 0.82 (0.54 to 1.24)
Mean (SD) ICU-free days: I: 26.0 (6.1) C: 24.2 (8.8) Mean difference: 1.80 days (95%CI 0.43 to 3.17)
Organ support free days Mean (SD) organ support-free days I: 25.8 (6.2) I: 24.1 (8.8) Mean difference: 1.70 (95%CI 0.32 to 3.08)
Mean (SD) ventilator-free days I: 26.5 (5.6) C: 24.7 (8.5)
Mechanical ventilation I: 11/228 (4.8) C: 16/237 (6.8)
Venous thromboembolism Consisting of deep vein thrombosis and pulmonary embolism I: 2 (0.9) C: 6 (2.5) RR 0.35; 95%CI 0.07 to 1.70)
ATE (myocardial infarction) I: 0 (0%) C: 1 (0.4%)
Major bleeding ISTH major bleeding I: 2 (0.9) C: 4 (1.7)
|
Notes: * Moderate illness was defined as admission to hospital ward level of care (ie, not to ICU), not already mechanically ventilated, and not imminently requiring mechanical ventilation or critical care.
Author’s conclusions: The primary composite outcome of death, mechanical ventilation, or ICU admission with therapeutic heparin. However, therapeutic heparin was associated with a substantially decreased odds of all cause death and low risk of major bleeding. In conjunction with the recently published multiplatform trial,19 the RAPID trial therefore suggests that therapeutic heparin is beneficial in moderately ill patients with covid-19 admitted to hospital wards. |
|
Marcos, 2021 |
Type of study: open-label, multicenter, randomized, controlled trial
Setting and country: The study was conducted at 5 Spanish hospitals
Funding and conflicts of interest: RL reports investigation grants from Rovi, consultant fees from Aspen and Leo-Pharma and lecture fees from BMS, Boehringer Ingelheim, Daiichi-Sankyo, Leo Pharma, Rovi and Sanofi, outside of the submitted work. PRA reports investigation grants from Rovi, consultant fees from Viatris, BMS, Pfizer and Leo Pharma and lecture fees from BMS, Daiichi-Sankyo, Leo Pharma, Viatris and Rovi, outside of the submitted work. All other authors declare no competing interests.
|
Inclusion criteria: - Age > 18 years-old - Hospitalization at the conventional ward due to mild or moderate (CURB65< 2 points and Sat. O2>90%) COVID-19 pneumonia. Maximum allowed time between hospitalization and randomization is 48 hours - 3-4 points according to the WHO ordinal scale - Confirmed COVID-19 diagnosis by PCR or other validated test. - Baseline D-Dimer >500 ng/mL - Signed informed consent - The patient, according to investigator’s opinion, is able to deal with all the requirements of the clinical trial
Exclusion criteria: Patients who meet any of the following criteria cannot be included in the clinical trial: - Need of intensive care unit admission - Moderate or severe adult respiratory distress syndrome - Body weight <50Kg - Creatinine clearance (Cockroft-Gault) <30ml/min - Severe liver disease (elevation of hepatic enzymes 3 times above the upper limit of normal) - Thrombocytopenia <75,000/mm3 - History of coagulopathy or thrombocytopathy - Active bleeding of increased bleeding risk due to impairment of haemostasis - Recent (1 month) central nervous system o gastrointestinal bleeding - Lesions of surgery involving central nervous system, eyes or inner ear in the last 2 months - Presence of organic lesions with high bleeding risk (e.g. active peptic ulcer, hemorragic stroke, brain aneurism o tumor) - Planned surgery or interventional procedure requiring regional anesthesia - Uncontrolled arterial hypertension - Acute or subacute bacterial endocarditis - Need of therapeutic anticoagulation for other reasons (e.g. atrial fibrillation, valvular prosthesis, venous thromboembolism) - Need of antiplatelet therapy - Simultaneous participation in another clinical trial (use of drugs against COVID-19 in the setting of local clinical management protocols is allowed) - Previous history of heparin-induced thrombocytopenia - Hypersensitivity or allergy to sodic bemiparin, heparin, compounds of porcine origin or any of the excipients
N total at baseline: 72 patients were enrolled. 6 were excluded due to consent withdrawal or not meeting eligibility criteria. Intervention: 33 Control: 33
Important prognostic factors2:
Age, mean (SD): I: 62.3 (12.2) C: 63.0 (13.7)
Sex, n (%), Male: I: 24 (72.2%) C: 17 (53.1%)
BMI (kg/m2), median (IQR) I: 26.1 (24.1-28.8) C: 25.8 (24.0-29.4)
BMI >30; n (%) I: 4 (12.1) C: 5 (15.6)
Comorbidities, n (%) Hypertension I: 12 (36.3%) C: 10 (31.2%)
Diabetes mellitus I: 3 (9.1) C: 2 (6.3)
Chronic pulmonary disease I: 6 (18.2) C: 5 (15.6)
Cardiopathy I: 1 (3.0) C: 3 (9.3)
Previous arterial or venous thrombosis I: 0 C: 0
Current or former smoking habit I: 10 (30.3) C: 16 (50.0)
Cancer I: 1 (3.0) C: 1 (3.1)
Days since COVID-19 diagnosis, median (IQR) I: 6 (3-8) C: 5 (2-8)
Days since symptoms onset, median (IQR) I: 8 (6-10) I: 8 (7-10)
Status at inclusion Oxygen requirement; n (%) I: 18 (54.5) C: 20 (62.5)
D-dimer; median (IQR) I: 770 (590-1030) C: 780 (600-1125)
Ferritin; median (IQR) I: 1093 (514-1751) C: 518 (287-1248)
IL-6; median (IQR) I: 24.8 (5.1-57.9) C: 34.1 (15.7-77.7)
Brescia COVID-19 Score ≥2; n (%) I: 3 (9.1) C: 1 (3.1)
Sepsis-induced coagulopathy Score ≥4; n (%) I: 1 (3.0) C: 0
COVID-19 therapy, n (%) Steroids I: 30 (90.9) C: 32 (100)
Statins I: 20 (60.6) C: 23 (71.9)
Remdesivir I: 5 (15.2) C: 4 (12.5)
Tocilizumab I: 8 (24.2) C: 7 (21.9)
Extended prophylaxis after end of study treatment; n (%) I: 21 (63.6) C: 23 (71.9)
Duration (days); median (IQR) I: 10 (7-14) C: 10 (8-14)
Groups comparable at baseline? Yes
|
Describe intervention (treatment/procedure/test):
Therapeutic-dose bemiparin (115 IU/Kg daily) for 10 days independently of early hospital discharge, adjusted to body weight (7,500 IU for patients between 50-70 Kg; 10,000 IU for patients weighing >70-100 Kg; 12,500 IU for patients who weighed >100 Kg). |
Describe control (treatment/procedure/test):
Standard prophylaxis with subcutaneous bemiparin 3,500 IU once daily for 10 days independently of early hospital discharge |
Length of follow-up: On site study follow-up visits, that included laboratory assessments, were scheduled in days 5 (+ 1 day) and 10 (+ 1 day). A final follow-up visit was performed on day 30 (+ 2 days) either on site or by telephone interview.
Loss-to-follow-up: 33 were allocated to standard thromboprophylaxis and 33 to therapeutic-dose bemiparin. A patient in the therapeutic-dose arm did not start the assigned treatment due to ICU transfer before its first administration, and was excluded from primary analysis.
|
Outcome measures and effect size (include 95%CI and p-value if available):
Mortality Death (day 10), n (%) I: 0 C: 0
Death (day 30), n (%) I: 2 (6.3) C: 1 (3.0)
Length of hospital stay Hospital discharge in first 10 days, n (%) I: 21 (65.6) C: 26 (78.8)
ICU-admission Need of ICU (day 10), n (%) I: 4 (12.5) C: 4 (12.1)
Need of ICU (day 30), n (%) I: 5 (15.6) C: 4 (12.1) 5 (15.6)
Organ support free days Marcos reported the need of mechanical ventilation support and the development of moderate/severe acute respiratory distress syndrome as a part of the primary composite outcome at day 10 I: 7 out of 32 patients (21.9%) C: 6 out of 33 patients (18.2%) RR 1.20 (95%CI 0.45 to 3.19).
Venous and arterial thromboembolism ATE/VTE (day 10), n (%) I: 0 C: 1 (3.0)
ATE/VTE (day 30), n (%) I: 0 C: 2 (6.1)
Major bleeding Major or clinically relevant bleeding (day 10), n (%) I: 0 C: 0
|
Notes:
Author’s conclusions: In COVID-19 patients hospitalized with non-severe pneumonia but elevated D-dimer, the use of a 10-day course of therapeutic-dose bemiparin does not seem to improve clinical outcomes compared to prophylactic doses. |
|
Lopes, 2021
ACTION-trial |
Type of study: A pragmatic, open-label (with blinded adjudication), multicentre, randomized, controlled trial.
Setting and country: Brazil, 31 hospitals
Funding and conflicts of interest: The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. |
Inclusion criteria: 1. Patients with confirmed diagnosis of COVID-19 admitted to hospital 2. Duration of symptoms related to hospitalisation ≤14 days 3. Patients ≥ 18 years old 4. D-dimer above the ULN 5. Agreement to participate by providing the informed consent form.
Exclusion criteria: 1. Patients with indication for therapeutic anticoagulation during inclusion (e.g, diagnosis of VTE, AF, mechanical valve prosthesis) 2. Platelets <50,000/mm3 3. Use of ASA >100 mg 4. Use of P2Y12 inhibitor (clopidogrel, prasugrel, ticagrelor) 5. Chronic use of NSAIDs 6. Sustained uncontrolled systolic BP ≥180 mm Hg or diastolic BP ≥100 mm Hg 7. INR >1·5 8. Patients contraindicated to therapeutic anticoagulation (active bleeding, liver failure, blood dyscrasia or prohibitive haemorrhage risk as evaluated by the investigator) 9. Patients with DIC 10. History of haemorrhagic stroke or any intracranial bleeding at any time in the past or current intracranial neoplasm (benign or malignant), cerebral metastases, arteriovenous malformation, or aneurysm 11. Active cancer (excluding non-melanoma skin cancer) defined as cancer not in remission or requiring active chemotherapy or adjunctive therapies such as immunotherapy or radiotherapy 12. Hypersensitivity to rivaroxaban 13. Use of strong inhibitors of cytochrome P450 (CYP) 3A4 and/or P-gp (e.g., protease inhibitors, ketoconazole, itraconazole) and/or use of P-gp and strong CYP3A4 inducers (including, but not limited to, rifampin/rifampicin, rifabutin, rifapentine, phenytoin, phenobarbital, carbamazepine, or St. John's Wort) 14. Known HIV infection 15. Creatinine clearance.
N total at baseline: Intervention: 331 Control: 304
Important prognostic factors2: For example Age, mean (SD): I: 56.7 (14.1) C: 56.6 (14.5)
Sex, n (%), Male: I: 192 (62%) C: 176 (58%)
Body mass index, mean (SD), kg/m2: I: 30.3 (6.0) C: 30.3 (6.1)
Comorbidities, n (%) Asthma
Chronic lung disease I: 7 (2%) C: 12 (4%)
Malignant neoplasm I: 12 (4%) C: 4 (1%)
Diabetes 8 I: 3 (27%) C: 67 (22%)
Hypertension I: 151 (49%) C: 151 (50%)
Heart failure I: 8 (3%) C: 5 (2%)
Coronary disease I: 12 (4%) C: 16 (5%)
History of thromboembolism I: 2 (1%) C: 4 (1%)
Smoking habits, n (%) Never smoked I: 255 (82%) C: 241 (79%)
Current or former smoker I: 56 (18%) C: 63 (21%)
Clinical condition, n (%) Unstable I: 23 (7%) C: 16 (5%)
Stable I: 288 (93%) C: 288 (95%)
Time from symptom onset to randomization, days, median (IQR) I: 10.0 (9·0–12.0) C: 10.0 (8·0–12.0)
Time from symptom onset to hospital admission, days, median (IQR) I: 8.0 (6.0–10.0) C: 7.0 (6.0–9.0)
Time from hospital admission to randomization, days, median (IQR) I: 2.0 (1.0–3.0) C: 2.0 (1.0–3.0)
Oxygen support required, n (%) I: 236 (76%) C: 224 (74%)
Catheter or oxygen mask, n (%) I: 185 (59%) C: 184 (61%)
High-flow nasal cannula, n (%) I: 26 (8%) C: 22 (7%)
Tracheal intubation, n (%) I: 23 (7%) C: 15 (5%)
Non-invasive ventilation, n (%) I: 2 (1%) C: 3 (1%)
Disease state at baseline, n (%) Mild I: 30 (10%) C: 39 (13%)
Moderate I: 257 (83%)
Severe I: 24 (8%) C:16 (5%)
Anticoagulation before randomization, n (%) I: 285 (92%) C: 275 (90%)
Standard prophylactic dose, n (%) I: 175 (56%) C: 187 (62%)
Greater than standard prophylactic dose, n (%) I: 110 (35%) C: 88 (29%)
Baseline medication, n (%) Antiplatelet I: 22 (7%) C: 26 (9%)
Vasopressor I: 16 (5%) C: 8 (3%)
Systemic corticosteroids I: 257 (83%) C: 253 (83%)
D-dimer concentration, n (%) ≥1 ×upper limit of normal I: 311 (100%) C: 304 (100%)
≥3 ×upper limit of normal I: 84 (27%) C: 83 (27%)
Creatinine clearance, mL/min, median (IQR) I: 106.6 (82.9–143.4) C: 105.7 (76.9–145.1)
Groups comparable at baseline? Yes
|
Describe intervention (treatment/procedure/test): Therapeutic anticoagulation for 30 days (rivaroxaban if clinically stable or enoxaparin if clinically unstable).
Clinically stable patients assigned to receive therapeutic anticoagulation were given oral rivaroxaban at a dose of 20 mg once daily. A reduced dose of 15 mg once daily was used in patients with a creatinine clearance of 30–49 mL/min or those taking azithromycin. Patients were considered to be in a clinically unstable condition if they had COVID-19-related critical illness, a lifethreatening condition, a requirement for mechanical ventilation or vasopressors, or were unable (based on investigator assessment) to take oral medication. Those in an unstable condition received subcutaneous enoxaparin at a dose of 1 mg/kg twice per day, or intravenous unfractionated heparin at a dose to achieve a target anti-Xa concentration (0·3–0·7 IU/mL) or a corresponding target activated partial thromboplastin time (1·5–2·5 times the mean normal value). Unfractionated heparin was the preferred option for patients with renal dysfunction or disseminated intravascular coagulation. When these patients became stable, they were transitioned to oral rivaroxaban (20 mg or 15 mg, as described above). All patients in the therapeutic anticoagulation group continued treatment to day 30 with the same dose of rivaroxaban.
|
Describe control (treatment/procedure/test): In-hospital prophylactic anticoagulation (enoxaparin or unfractionated heparin).
Patients assigned to receive prophylactic anticoagulation were given standard venous thromboembolism prophylaxis with enoxaparin or unfractionated heparin during hospitalisation and could receive extended prophylaxis at the discretion of the treating physician (appendix p 7). Patients in this group could receive therapeutic anticoagulation if they developed a definitive clinical indication (eg, objectively confirmed deep vein thrombosis) or at the discretion of the investigator if a high clinical suspicion of a thromboembolic event was raised and a confirmatory test was not available. |
Length of follow-up: 30 days for study outcomes, and 60 days for additional safety information.
Loss-to-follow-up: Of 331 in intervention group, 1 was lost to follow-up before 30 days (withdrew consent, so 310 were included in primary analysis.
Of 304 in the control group, 1 received therapeutic coagulation, but 304 were still included in primary analysis.
|
Outcome measures and effect size (include 95%CI and p-value if available): During hospitalisation, data were collected daily.
Mortality Death, n/total (%) I: 35/310 (11%) C: 23/304 (8%) RR 1·49 (0·90–2·46)
Length of hospital stay at the end of 30 days, mean (SD) I: 8.1 days (SD 7.2) C: 7.8 days (SD 7.5)
ICU-admission Not reported
Organ support free days Not reported
Organ support The number of patients hospitalised with invasive mechanical ventilation without additional support. I: 1/310 (0.3%) C: 4/304 (1.3%)
Venous thromboembolism I: 11 (4%) C: 18 (6%) RR (95%CI): RR 0.60 (0.29–1.25)
Number of patients that reported the composite thrombotic outcome (consisting of venous thromboembolism – deep vein thrombosis and pulmonary embolism, myocardial infarction, stroke, and major adverse limb event). I: 23 out of 310 (7%) patients C: 30 out of 304 (10%) patients RR 0.75, 95%CI 0.45 to 1.26
Major bleeding, n (%) I: 10 (3%) C: 4 (1%) RR (95%CI): 2.45 (0.78-7.73)
|
Notes:
** Clinically unstable was defined as the presence of a COVID-19-related critical illness with an immediately life-threatening condition that would typically lead to intensive care unit admission
Author’s conclusion: in patients hospitalised with COVID-19 with elevated D-dimer concentration, initial in-hospital therapeutic anticoagulation with rivaroxaban for stable patients or enoxaparin for unstable patients followed by rivaroxaban through 30 days did not improve clinical outcomes and increased bleeding compared with in-hospital prophylactic anticoagulation. Thus, the use of therapeutic-dose rivaroxaban, and other direct oral anticoagulants, should be avoided in hospitalised patients with COVID-19 who do not have an evidence-based indication for oral anticoagulation.
|
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures
- Provide data per treatment group on the most important prognostic factors [(potential) confounders]
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders
Risk of Bias tabel
|
Study reference
(first author, publication year) |
Was the allocation sequence adequately generated? a
Definitely yes Probably yes Probably no Definitely no |
Was the allocation adequately concealed?b
Definitely yes Probably yes Probably no Definitely no |
Blinding: Was knowledge of the allocated interventions adequately prevented?c
Were patients blinded?
Were healthcare providers blinded?
Were data collectors blinded?
Were outcome assessors blinded?
Were data analysts blinded?
Definitely yes Probably yes Probably no Definitely no |
Was loss to follow-up (missing outcome data) infrequent?d
Definitely yes Probably yes Probably no Definitely no |
Are reports of the study free of selective outcome reporting?e
Definitely yes Probably yes Probably no Definitely no |
Was the study apparently free of other problems that could put it at a risk of bias?f
Definitely yes Probably yes Probably no Definitely no |
Overall risk of bias If applicable/necessary, per outcome measureg
LOW Some concerns HIGH
|
|
Lawler, 2021
|
Probably yes;
Reason: Central Internet-based systems to randomly assign patients were used. Treatments were initially randomly assigned in a 1:1 ratio. The ATTACC and REMAP-CAP designs specified the possibility of response-adap tive randomization, in which group-assignment ratios could be modified in a blinded fashion during the trial on the basis of response-adaptive interim analyses to favor the assignment of patients to the treatment group showing greater benefit. However, we did not think this influenced the prognostic balance between the two groups at baseline. |
Definitely yes;
Reason: Central Internet-based systems to randomly assign patients were used |
Definitely no;
Reason: open label trial |
Definitely yes;
Reason: Exclusion rate 1.6% and 0.6% in intervention group and control group respectively. Only 3 participants had no available outcome data. Data was not imputed, but patients were excluded from the analysis. |
Definitely yes;
Reason: Protocol adhered, relevant outcomes reported |
Definitely no;
Reason: enrollment was discontinued on the advice of the data and safety monitoring boards after a planned adaptive analysis of data from 1398 patients showed that the prespecified stopping criteria for superiority of therapeutic-dose anticoagulation had been reached
Intention to treat analysis is not clearly mentioned. |
HIGH (all outcome measures)
Reasons: - prespecified stopping criteria - no mentioning of ITT analysis - open label trial |
|
Spyropoulos, 2021 |
Definitely yes
Reason: Randomization well-described and well-performed
Study protocol The Feinstein Institutes Biostatistics Unit (Northwell Health) developed and implemented the randomization procedure using the Biostatistics Randomization Management System (BRMS). BRMS is a secure, HIPAA-compliant, web-based application that allows investigators to randomize subjects into randomized clinical trials (RCTs) using their personal computer. BRMS allows for multicenter, stratified, and single/double-blinded RCTs, using permuted blocks. Eligible patients will be stratified according to whether their level of care corresponds to intensive care unit (ICU) care or not. Subjects will be randomly assigned to the treatment arm (arm 0: treatment dose of LMWH) or the prophylactic-/ intermediate-dose arm (arm 1: prophylactic/intermediate dose of LMWH or UFH) in a 1:1 ratio.
Spyropoulos, 2021: Randomization was performed using a secure web application and was stratified based on noncritical care (non– intensive care unit [ICU]) or critical care (ICU) status at the time of randomization. Participants were randomly assigned 1:1 to therapeutic-dose enoxaparin or institutional standard prophylactic or intermediate-dose heparins. |
Definitely yes
Reason:
Study protocol BRMS is a secure, HIPAA-compliant, web-based application that allows investigators to randomize subjects into randomized clinical trials (RCTs) using their personal computer.
Spyropoulos, 2021: The study pharmacists as well as data abstractors and designated randomization personnel (i.e., research coordinators and/or research nurses performing the randomization process) will be unblinded. |
Were patients blinded? Definitely yes
Were healthcare providers blinded? Definitely no
Were data collectors blinded? Definitely yes
Were outcome assessors blinded? Unknown
Were data analysts blinded? Unknown
Reason: Study protocol (Goldin, 2021): Due to the pragmatic nature of this study and pseudoblinded trial design, at the time of randomization the study subject and corresponding site PIs will be blinded (unaware of specific treatment arm to which the patient is assigned). The study pharmacists as well as data abstractors and designated randomization personnel (i.e., research coordinators and/or research nurses performing the randomization process) will be unblinded.
Spyropoulos, 2021: Patients and investigators were blinded to treatment assignment as much as possible.
Data were collected and adjudicated locally by blinded investigators via imaging, laboratory, and health record data.
Discussion part: Although both investigators and patients were blinded to study drug regimen, other unblinded personnel may have introduced bias affecting outcome ascertainment. |
Probably yes
Reason: There was no loss to follow-up. However, the primary analysis was based on the modified intention to-treat population, followed by the per-protocol population. We assumed that the modified ITT did not have an influence on the risk of bias, as the number of patients that was different between the ITT and modified ITT population was very small (1 in the intervention group and 3 in the control group). |
Definitely yes
Reason: Study protocol available, all reported outcomes in study protocol reported in trial. |
Probably yes
Reason: Funding/Support: This work was supported by the Feinstein Institutes for Medical Research, the Broxmeyer Fellowship in Clinical Thrombosis, and grant R24AG064191 from the National Institute on Aging. Role of the funder/Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Modified intention-to-treat analysis performed. However, only 1 and 3 patients in the intervention and control group, respectively, were not analysed. |
LOW (mortality, length of hospital stay, ICU-admission, organ support free days)
HIGH (venous thromboembolism, major bleeding)
Reasons: - patients and data collections blinded, but all other personnel including the healthcare providers were not blinded and could therefore have influenced the ‘soft’ outcome measures
|
|
Sholzberg, 2021 |
Definitely yes
Reason: We used web based central randomization with a computer generated random sequence of variable block sizes stratified by site and age (≤65 v >65 years) to assign patients in a 1:1 ratio to therapeutic heparin or prophylactic heparin. |
Definitely yes
Reason: Use of central randomization. |
Were patients blinded? Definitely no
Were healthcare providers blinded? Definitely no
Were data collectors blinded? Definitely no
Were outcome assessors blinded? Definitely no
Were data analysts blinded? Definitely yes
Reasons: Outcomes were blindly adjudicated.
An independent event adjudication committee, which was unaware of treatment assignments, adjudicated the components of the primary outcome, bleeding, thrombotic events, and cause of death. The event adjudication was not prespecified in the protocol. |
Definitely yes
Infrequent loss to follow-up (4 in intervention group and 7 in control group). Primary outcomes were analysed with ITT analysis.
|
Definitely yes
Study protocol available, most reported outcomes in study protocol reported in trial. Clear mentioning of components of the primary composite that were not included in the protocol but were prespecified as secondary outcomes in the statistical analysis plan; transparent.
IIT principle for primary outcomes. |
Definitely yes
The funders had no role in the trial design; conduct, collection, management, analysis, or interpretation of data; or in preparation or review of the manuscript or the approval of the manuscript for submission. |
LOW (mortality, length of hospital stay, ICU-admission, organ support free days)
HIGH (venous thromboembolism, major bleeding)
Reasons: - open-label trial, could possibly have influenced the ‘soft’ outcome measures
|
|
Marcos, 2021 |
Definitely yes
Reason: Randomization was performed in a 1:1 ratio using a central, electronic, automated system with permuted blocks of 4. |
Definitely yes
Reason: Randomization was performed in a 1:1 ratio using a central, electronic, automated system with permuted blocks of 4. |
Were patients blinded? Definitely no
Were healthcare providers blinded? Unknown
Were data collectors blinded? Definitely no (assuming these are the researchers)
Were outcome assessors blinded? Unknown
Were data analysts blinded? Unknown
Reason: There was no blinding of patients or investigators to group allocation.
However, no blinding likely did not affect the outcome measures we’re interested in, since these are ‘hard’ outcome measures. |
Definitely yes
Reason: There was 1 patient that did not start the assigned treatment due to transfer before its first administration and this patient was excluded from primary analysis. There was no other patient lost to follow-up. |
Probably yes
Reasons:
Study protocol available, although some figures missing. Study protocol describes the reported outcomes.
Efficacy and safety were evaluated in the modified intention to treat population (mITT), including all randomized patients that received at least one dose of the allocated treatment. è modified intention to treat analysis is doubtful. Not clearly described. |
Definitely yes
Reason: RL reports investigation grants from Rovi, consultant fees from Aspen and Leo- Pharma and lecture fees from BMS, Boehringer Ingelheim, Daiichi-Sankyo, Leo Pharma, Rovi and Sanofi, outside of the submitted work. PRA reports investigation grants from Rovi, consultant fees from Viatris, BMS, Pfizer and Leo Pharma and lecture fees from BMS, Daiichi-Sankyo, Leo Pharma, Viatris and Rovi, outside of the submitted work. All other authors declare no competing interests. |
LOW (mortality, length of hospital stay, ICU-admission, organ support free days)
HIGH (venous thromboembolism, major bleeding)
Reasons: - open-label trial, could possibly have influenced the ‘soft’ outcome measures
|
|
Lopes, 2021 |
Definitely yes
Reason: Randomization was done in a 1:1 ratio in permuted blocks of variable size, stratified according to clinical condition (stable or unstable), using a central, concealed, web-based, automated randomization system. |
Definitely yes
Reason: Randomization was done in a 1:1 ratio in permuted blocks of variable size, stratified according to clinical condition (stable or unstable), using a central, concealed, web-based, automated randomization system. |
Were patients blinded? Definitely no
Were healthcare providers blinded? Definitely no
Were data collectors blinded? Definitely no
Were outcome assessors blinded? Definitely no
Were data analysts blinded? Definitely yes
Reason: There was no masking of patients or investigators to group allocation.
However, there was a blinded adjudication process for the secondary outcomes using standard definitions, as well as regular site training and monitoring and sensitive triggers based on laboratory values, reports of adverse events, unknown causes of death, or changes in antithrombotic therapy, to ensure that no relevant events were missed. |
Definitely yes
Reason: Only 1 person in the intervention group withdrew consent and was therefore excluded from primary analysis. Out of 304 persons in the control group, 1 did not receive the allocation intervention but was nevertheless still analysed in this group. This gives no indication of bias. |
Definitely yes
Reason: Publication in line with protocol |
Definitely yes
Reason:
|
LOW (mortality, length of hospital stay, ICU-admission, organ support free days)
HIGH (venous thromboembolism, major bleeding)
Reasons: - open-label trial, could possibly have influenced the ‘soft’ outcome measures
|
Table of excluded studies
|
Author and year |
Reason for exclusion |
|
Connors, 2021 |
Outpatients, does not fit PICO |
|
Perepu, 2021 |
Exclusion, no difference made between ICU and non-ICU patients |
|
Diep, 2021 |
No publication available |
|
Ananworanich, 2021 |
Outpatients, does not fit PICO |
|
Cuker, 2021 |
Is not an RCT but a guideline based on observational studies |
|
Sadeghipour, 2021 |
Included in ICU submodule, but not in the non-ICU submodule |
|
Bikdeli, 2021 |
Included in ICU submodule, but not in the non-ICU submodule |
|
Goligher, 2021 |
Included in ICU submodule, but not in the non-ICU submodule |
|
Lemos, 2020 |
Included in ICU submodule, but not in the non-ICU submodule |
Verantwoording
Autorisatiedatum en geldigheid
Laatst beoordeeld : 09-02-2022
Laatst geautoriseerd : 09-02-2022
Geplande herbeoordeling :
Algemene gegevens
Van NVSHA is bestuurlijke goedkeuring ontvangen. Definitieve autorisatie volgt na de ALV in juni 2022.
De ontwikkeling/herziening van deze richtlijnmodule werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Kwaliteitsgelden Medisch Specialisten (SKMS).
De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodule.
Deze richtlijnmodule is ontwikkeld in samenwerking met:
- Nederlandse Vereniging voor Intensive Care
- Nederlandse Vereniging van Artsen voor Longziekten en Tuberculose
- Nederlandse Vereniging voor Cardiologie
- Nederlandse Vereniging voor Ziekenhuisapothekers
- Nederlandse Vereniging van Spoedeisende Hulp Artsen
- Nederlandse Vereniging voor Klinische Chemie en Laboratoriumgeneeskunde
- Harteraad
Samenstelling werkgroep
Voor het ontwikkelen van de richtlijnmodules is in 2020 een multidisciplinaire werkgroep ingesteld, bestaande uit vertegenwoordigers van alle relevante specialismen die betrokken zijn bij de behandeling van patiënten met COVID-19.
Werkgroep
- Prof. dr. M.V. Huisman (voorzitter), internist-vasculair geneeskundige, Leids Universitair Medisch Centrum (LUMC), NIV
- Dr. F.A. Klok, internist-vasculair geneeskundige, Leids Universitair Medisch Centrum (LUMC), NIV
- Prof. dr. H.C.J. Eikenboom, internist-vasculair geneeskundige/hematoloog, Leids Universitair Medisch Centrum (LUMC), NIV
- Prof. dr. S. Middeldorp, internist-vasculair geneeskundige, Radboud Universitair Medisch Centrum (Radboudumc), NIV
- Dr. M.J.H.A. Kruip, internist-hematoloog, Erasmus Medisch Centrum (Erasmus MC), NIV
- Prof. dr. K. Meijer, internist-hematoloog, Universitair Medisch Centrum Groningen (UMCG), NIV
- Dr. M. Coppens, internist-vasculair geneeskundige, Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Prof. dr. P.W. Kamphuisen, internist-vasculair geneeskundige, Tergooi Medisch Centrum (Tergooi MC), Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Dr. M.C.A. Müller, internist-intensivist, Amsterdam University Medical Centers (Amsterdam UMC), NVIC
- Dr. J.P.J. Wester, internist-intensivist, OLVG, NVIC
- Dr. R. Vink, internist-intensivist, Tergooi Medisch Centrum (Tergooi MC), NVIC
- Dr. L.M. van den Toorn, longarts, Erasmus Medisch Centrum (Erasmus MC), NVALT
- Dr. R.G. Tieleman, cardioloog, Martini Ziekenhuis, NVVC
- Dr. J. Diepstraten, ziekenhuisapotheker, Amphia ziekenhuis, NVZA
- Dr. N. van Rein, ziekenhuisapotheker, Leids Universitair Medisch Centrum (LUMC), NVZA
- Drs. N. Buenen, SEH-arts, Máxima Medisch Centrum (MMC), NVSHA
- Prof. dr. Ir. Y.C.M. Henskens, klinisch chemicus, Maastricht Universitair Medisch Centrum (Maastricht UMC), NVKC
Meelezer:
- Dr. I. Schalkers, Beleidsadviseur Patientenparticipatie, Harteraad
Onafhankelijke reviewers:
- Dr. E. van Dijk, neuroloog, Radboud Universitair Medisch Centrum, (Radboudumc), NVN
- Drs. P.R. van der Valk, internist-vasculair geneeskundige, Universitair Medisch Centrum Utrecht (UMC Utrecht), NIV
Met ondersteuning van:
- F.M. Janssen, MSc, junior adviseur, Kennisinstituut van Medisch Specialisten
- dr. S.N. Hofstede, senior adviseur, Kennisinstituut van Medisch Specialisten
- drs. I. van Dusseldorp, senior literatuurspecialist, Kennisinstituut van Medisch Specialisten
In 2020 is een multidisciplinair expertiseteam behandeling COVID-19 ingesteld, bestaande uit vertegenwoordigers van alle relevante die betrokken zijn bij de zorg voor patiënten met COVID-19. Dit expertiseteam fungeerde als stuurgroep, welke opdracht heeft gegeven tot het ontwikkelen van de module, alsmede fungeerde als klankbordgroep.
Stuurgroep (expertiseteam Behandeling COVID-19)
- Dr. L.M. van den Toorn (voorzitter), longarts, Erasmus Medisch Centrum (Erasmus MC), NVALT
- Dr. M.G.J. de Boer, internist-infectioloog, Leids Universitair Medisch Centrum (LUMC), SWAB/NIV)
- Drs. A.J. Meinders, internist-intensivist, St. Antonius Ziekenhuis, NVIC
- Prof. dr. D.W. de Lange, intensivist-toxicoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), NVIC
- Dr. C.H.S.B. van den Berg, infectioloog-intensivist Universitair Medisch Centrum Groningen (UMCG), NVIC
- Dr. S.U.C. Sankatsing, internist-infectioloog, Diakonessenhuis, NIV
- Dr. E.J.G. Peters, internist-infectioloog, Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. M.S. Boddaert, arts palliatieve geneeskunde, Leids Universitair Medisch Centrum (LUMC), IKNL
- Dr. P.L.A. Fraaij, kinderarts-infectioloog, Erasmus Medisch Centrum (Erasmus MC), Sophia Kinderziekenhuis, NVK
- Dr. E. van Leeuwen, gynaecoloog, Amsterdam University Medical Centers (Amsterdam UMC), NVOG
- Dr. J.J. van Kampen, arts-microbioloog, Erasmus Medisch Centrum (Erasmus MC), NVMM
- Dr. M. Bulatović-Ćalasan, internist allergoloog-immunoloog en klinisch farmacoloog, Universitair Medisch Centrum Utrecht (UMC Utrecht), Amsterdam University Medical Centers (Amsterdam UMC), NIV
- Drs. A.F.J. de Bruin, anesthesioloog-intensivist, St. Antonius Ziekenhuis, NVA
- Drs. A. Jacobs, klinisch geriater, Catharina Ziekenhuis, NVKG
- Prof. dr. P.H.M. van der Kuy, ziekenhuisapotheker, Erasmus Medisch Centrum (Erasmus MC), NVZA
Belangenverklaringen
De Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement) hebben gehad. Gedurende de ontwikkeling of herziening van een module worden wijzigingen in belangen aan de voorzitter doorgegeven. De belangenverklaring wordt opnieuw bevestigd tijdens de commentaarfase.
Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
Werkgroep
|
Achternaam werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Huisman (voorzitter) |
internist vasculaire geneeskunde Leids Universitair Medisch Centrum hoogleraar Interne Geneeskunde |
voorzitter commissie herziening Antitrombotisch Beleid - onbetaald voorzitter Dutch Thrombosis Network – onbetaald member European Society of Cardiology Guideline on Pulmonary Embolism – onbetaald member American College of chest Physicians VTE update – onbetaald |
- adviseur farmaceutische bedrijven die (nieuwe) antistollingsmiddelen maken - betaald; gelden gaan naar afdeling Interne Geneeskunde LUMC
- ZoNMw grant Dutch-AF - registry met onderzoek op het gebied van antistolling bij patienten met atriumfibrilleren; betaald, gelden gaan naar afdeling Interne Geneeskunde LUMC - ZonMw - Hartstichting grant Coronis - onderzoek naar herseninfarct bij COVID -19
- Research grants van farmaceutische bedrijven die (nieuwe) antistollingsmiddelen maken - betaald; gelden gaan naar afdeling Interne Geneeskunde LUMC |
Zie overkoepelende actie |
|
Klok |
Internist, LUMC, Leiden |
Algemeen commissiewerk voor ISTH, ESC, ERS. Richtlijncommissie voor ESC. Richtlijncommissie voor ASH die zich buigt over tromboseprofylaxe bij COVID-19 |
Unrestricted research grants from Bayer, Bristol-Myers Squibb, Boehringer-Ingelheim, MSD, Daiichi-Sankyo, Actelion, the Dutch thrombosis association, The Netherlands Organisation for Health Research and Development and the Dutch Heart foundation |
Zie overkoepelende actie |
|
Eikenboom |
Hoogleraar medisch specialist (fte 1,0) |
Sectie editor van tijdschrift HemaSphere (honorarium naar instituut)
|
- LSBR: Modelling Von Willebrand disease with patient-specific
- Sprekersgeld: Roche, Cellgene |
Zie overkoepelende actie |
|
Middeldorp |
- Internist-vasculaire geneeskunde |
Onbetaald: |
- Betaald adviseurschap NB - alle honoraria (personal fees) gaan naar mijn ziekenhuis, geen persoonlijk financieel belang of eigen stichting:
- Financiele bijdragen aan investigator-initiated onderzoek:
- Academisch belang:
Lid richtlijn COVID coagulopathie ISTH (gestart december 2021). |
Zie overkoepelende actie |
|
Kruip |
Internist-hematoloog Erasmus MC |
Medisch leider trombosedienst Star-shl, gedetacheerd vanuit het Erasmus MC (betaald) voorzitter Federatie Nederlandse Trombosediensten (FNT, onbetaald) |
- "Caging the dragon: translation approach to unravel and prevent COVID-19 associated thrombosis" gefinancieerd door ZonMW en Trombose Stichting Nederland
- Financiële bijdrage aan investigator-initiated onderzoek, gelden ten gunste van afdeling hematologie Erasmus MC: Sobi
- Sprekersvergoeding, gelden ten gunste van afdeling hematologie Erasmus MC: Bayer, Roche, Sobi, BMS
|
Zie overkoepelende actie |
|
Meijer |
Internist-hematoloog, UMCG |
Voorzitter Nederlandse Vereniging voor Hematologie, onbetaald |
- NWO: Ethische aspecten van gentherapie, binnen Symphony project - projectleider voor Symphony
- Rollen als voorzitter van de NVvH en gedeeld voorzitter Trombose Expertise Centrum Noord Nederland kunnen gezien worden als boegbeeldfunctie, ik heb daarmee belang DAT er een richtlijn komt, maar niet bij hoe de inhoud is.
- In de afgelopen drie jaar |
Zie overkoepelende actie |
|
Coppens |
Internist-vasculaire geneeskunde en hemofilie. Hoofd Antistollingscommissie. Amsterdam |
Voorzitter werkgroep trombose en hemostase, Nederlandse Vereniging van Internisten |
- Daiichi Sankyo. Lid Steering Committee van de ETNA-VTE Europe studie. Een - Bayer. Fase 1-2 onderzoek naar gentherapie voor hemofilie A.
- Het hoge risico op veneuze trombose bij COVID-19 en de zeldzame, maar ernstige |
Zie overkoepelende actie |
|
Kamphuisen |
Internist, Tergooi MC, 0,8 fte |
Geen |
Mijn wetenschappelijk onderzoek wordt gesubsidieerd door Tergooi MC, Trombose stichting Nederland, Nederlandse Hartstichting. Verder investigator grant van Daiichi Sankyo en Roche diagnostics |
Zie overkoepelende actie |
|
Müller |
internist-intensivist, Amsterdam UMC, lokatie AMC |
Geen |
Geen |
Geen |
|
Wester |
Internist-intensivist |
Geen |
Geen |
Geen |
|
Vink |
Internist-intensivist |
Geen |
Geen |
Geen |
|
Van den Toorn |
Voorzitter NVALT |
Geen |
Voorzitter NVALT, ik heb geen baat bij welke uitkomst dan ook |
Geen |
|
Tieleman |
Cardioloog, electrofysioloog Martiniziekenhuis Groningen 0,8 FTE en UMCG 0,2 FTE |
Voorzitter pijler Implementatie DCVA 0,1 FTE |
- Ik heb sponsoring, onderzoeksgeld en honoraria voor nascholing ontvangen van Boehringer Ingelheim, Bayer en BMS/Pfizer, Daichi Sankyo
- Boehringer Ingelheim: Covid en cardiovasculair lijden (CAPACITY studie) - geen projectleider |
Zie overkoepelende actie |
|
Diepstraten |
Ziekenhuisapotheker en Medisch manager Amphia Ziekenhuis |
SIG Hematologie NVZA onbetaald |
Stichting Phoenix |
Zie overkoepelende actie |
|
Van Rein |
Ziekenhuisapotheker, Klinische Farmacie en Toxicologie, LUMC (32 uur per week) |
Geen |
Trombose Stichting Nederland: Balans bloedingen trombose atrium fibrilleren patienten tijdens behandeling met antistolling optimaliseren - projectleider |
Geen |
|
Buenen |
SEH-arts KNMG - Máxima MC - vaste dienst |
Geen |
Geen |
Geen |
|
Henskens |
0,9 fte Klinisch chemicus, waarnemend hoofd Centraal Diagnostisch Laboratorium Maastricht UMC+ |
Voorzitter concilium NVKC sinds 2020, vacatiegelden |
Geen |
Geen |
Overkoepelende actie
Om potentiële belangenverstrengelingen te voorkomen is de module tegengelezen door twee onafhankelijke reviewers. Dit betreft één onafhankelijke reviewer van de regiehoudende vereniging (Nederlandse Internisten Vereniging) en één onafhankelijke reviewer van een wetenschappelijke vereniging die niet in de werkgroep zitting heeft genomen (Nederlandse Vereniging voor Neurologie). Zij controleren of de aanbeveling duidelijk volgt uit de overwegingen en niet mogelijk gekleurd wordt door potentiële belangen. Daarnaast is de module tegengelezen door de stuurgroep.
Stuurgroep
|
Achternaam stuurgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Van den Toorn (voorzitter) |
Voorzitter NVALT |
Geen |
Geen |
Geen actie nodig |
|
De Boer |
Internist-Infectioloog, senior medisch specialist, LUMC, afdeling infectieziekten |
- Voorzitter Stichting Werkgroep Antibioticabeleid (onkostenvergoeding) |
Geen |
Geen actie nodig |
|
Meinders |
Internist-intensivist, St.-Antonius ziekenhuis, Nieuwegein |
commissie werk |
Geen |
Geen actie nodig |
|
De Lange |
Afdelingshoofd Nationaal Vergiftigingen Informatie Centrum (NVIC) van het UMC Utrecht |
secretaris Stichting Nationale Intensive Care Evaluatie (Stichting NICE) (onbetaald) |
Geen |
Geen actie nodig |
|
Van den Berg |
Infectioloog-intensivist, UMCG |
Geen |
Geen |
Geen actie nodig |
|
Sankatsing |
Internist-infectioloog/internist-acute geneeskunde, Diakonessenhuis, Utrecht |
- Bestuurslid Nederlandse Vereniging van Internist-Infectiologen (NVII) (onbetaald). |
Geen |
Geen actie nodig |
|
Peters |
Internist - aandachtsgebieden infectieziekten en Acute Geneeskunde Amsterdam UMC, locatie Vumc. |
Wetenschappelijk Secretaris International Working Group on the Diabetic Foot (onbetaald) |
Geen |
Geen actie nodig |
|
Boddaert |
Medisch adviseur bij Integraal Kankercentrum Nederland (IKNL) en Palliatieve Zorg Nederland (PZNL) Arts palliatieve geneeskunde in LUMC |
Geen |
Geen |
Geen actie nodig |
|
Fraaij |
Kinderarts infectioloog- immunoloog, Erasmus MC-Sophia, Rotterdam |
Bestuur Stichting Infecties bij Kinderen (onbetaald) |
deelname aan RECOVER, European Union's Horizon 2020 research |
Geen actie nodig |
|
Van Leeuwen |
Gyaecoloog Amsterdam Universitair Medisch Centra |
Geen |
Geen |
Geen actie nodig |
|
Van Kampen |
Arts-microbioloog, afdeling Viroscience, Erasmus MC |
- associate editor antimicrobial resistance & infection control (onbetaald) - lid antibioticacommissie Erasmus MC (onbetaald) |
1. Mede uitvinder patent: 1519780601-1408/3023503 2. R01AI147330 (NIAID/NH) (HN onderzoek (1+2 niet gerelateerd aan COVID-19)
|
Geen actie nodig |
|
Bulatovic |
Internist allergoloog-immunoloog en klinische farmacoloog, UMC Utrecht en Diakonessenhuis Utrecht |
Functie 1: arts |
Geen |
Geen actie nodig |
|
De Bruin |
Anesthesioloog - Intensivist St. Antonius ziekenhuis Nieuwegein en Utrecht |
Geen |
Geen |
Geen actie nodig |
|
Jacobs |
Klinisch geriater en klinisch farmacoloog |
Geen |
Geen |
Geen actie nodig |
|
Van der Kuy |
Afdelingshoofd Ziekenhuisapotheek Erasmus MC |
Ziekenhuisapotheker/onder-zoeker Zuyderland MC (betaald) |
betrokkenheid bij 2 ZonMw gefinancieerde studies (CHECkUP, AMUSE) |
Geen actie nodig |
Meelezer
|
Achternaam meelezer |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Schalkers |
Beleidsadviseur bij Harteraad |
Geen |
Geen |
Geen actie nodig |
Onafhankelijke reviewers
|
Achternaam onafhankelijke reviewer |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Van der Valk |
Als internist werkzaam in het UMC Utrecht, bij van Creveldkliniek - centrum voor benigne hematologie. Expert centrum voor o.a. stollingsstoornissen. Het behelst patientenzorg en onderzoek (PI voor studies naar cardiovasculaire ziekten bij Hemofilie, gentherapie bij hemofilie) |
Lid van de richtlijn commissie NIV. Onbetaald. |
Geen |
Geen |
|
Van Dijk |
Neuroloog, Radboudumc |
Voorzitter bestuur Nederlandse Vereniging voor Neurologie (0,1fte, betaald aan Radboudumc) |
Geen |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door het meelezen van de patiëntenvereniging Harteraad. De verkregen input is meegenomen bij het opstellen van de module. De conceptrichtlijn is tevens voor commentaar voorgelegd aan Harteraad en de Patiëntenfederatie Nederland en de eventueel aangeleverde commentaren zijn bekeken en verwerkt.
Wkkgz & Kwalitatieve raming van mogelijke substantiële financiële gevolgen
Bij de richtlijn is conform de Wet kwaliteit, klachten en geschillen zorg (Wkkgz) een kwalitatieve raming uitgevoerd of de aanbevelingen mogelijk leiden tot substantiële financiële gevolgen. Bij het uitvoeren van deze beoordeling zijn richtlijnmodules op verschillende domeinen getoetst (zie het stroomschema).
Uit de kwalitatieve raming blijkt dat er waarschijnlijk geen substantiële financiële gevolgen zijn, zie onderstaande tabel.
Module |
Uitkomst raming |
Toelichting |
|
Module Tromboseprofylaxe op de afdeling |
Geen financiële gevolgen |
Hoewel uit de toetsing volgt dat de aanbevelingen breed toepasbaar zijn (>40.000 patiënten), volgt uit de toetsing dat het geen nieuwe manier van zorgverlening of andere organisatie van zorgverlening betreft, het geen toename in het aantal in te zetten voltijdsequivalenten aan zorgverleners betreft en het geen wijziging in het opleidingsniveau van zorgpersoneel betreft. Er worden daarom geen financiële gevolgen verwacht. |
Methode ontwikkeling
Evidence based
Werkwijze
AGREE
Deze richtlijnmodule is opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, 2010).
Knelpuntenanalyse en uitgangsvragen
Tijdens de COVID-19 pandemie zijn knelpunten op verschillende manieren geïnventariseerd:
- De expertiseteams benoemde de knelpunten in de zorg voor patiënten met COVID-19.
- Er is een mailadres geopend (covid19@demedischspecialist.nl) waar verschillende partijen knelpunten konden aandragen, die vervolgens door de expertiseteams geprioriteerd werden.
- Door de Federatie van Medisch Specialisten zijn webinars georganiseerd waarbij vragen konden worden ingestuurd. Deze vragen zijn na afloop van de webinars voorgelegd aan de expertiseteams en geprioriteerd.
Op basis van de uitkomsten van de bovenstaande knelpuntenanalyses zijn door de expertiseteams concept-uitgangsvragen opgesteld en definitief vastgesteld.
Uitkomstmaten
Na het opstellen van de zoekvraag behorende bij de uitgangsvraag inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn, waarbij zowel naar gewenste als ongewenste effecten werd gekeken. Hierbij werd een maximum van acht uitkomstmaten gehanteerd. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet cruciaal) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Methode literatuursamenvatting
Een uitgebreide beschrijving van de strategie voor zoeken en selecteren van literatuur en de beoordeling van de risk-of-bias van de individuele studies is te vinden onder ‘Zoeken en selecteren’ onder Onderbouwing. De beoordeling van de kracht van het wetenschappelijke bewijs wordt hieronder toegelicht.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/). De basisprincipes van de GRADE-methodiek zijn: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van de bewijskracht per uitkomstmaat op basis van de acht GRADE-domeinen (domeinen voor downgraden: risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias; domeinen voor upgraden: dosis-effect relatie, groot effect, en residuele plausibele confounding).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie, in het bijzonder de mate van zekerheid dat de literatuurconclusie de aanbeveling adequaat ondersteunt (Schünemann, 2013; Hultcrantz, 2017).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
Bij het beoordelen (graderen) van de kracht van het wetenschappelijk bewijs in richtlijnen volgens de GRADE-methodiek spelen grenzen voor klinische besluitvorming een belangrijke rol (Hultcrantz, 2017). Dit zijn de grenzen die bij overschrijding aanleiding zouden geven tot een aanpassing van de aanbeveling. Om de grenzen voor klinische besluitvorming te bepalen moeten alle relevante uitkomstmaten en overwegingen worden meegewogen. De grenzen voor klinische besluitvorming zijn daarmee niet één op één vergelijkbaar met het minimaal klinisch relevant verschil (Minimal Clinically Important Difference, MCID). Met name in situaties waarin een interventie geen belangrijke nadelen heeft en de kosten relatief laag zijn, kan de grens voor klinische besluitvorming met betrekking tot de effectiviteit van de interventie bij een lagere waarde (dichter bij het nuleffect) liggen dan de MCID (Hultcrantz, 2017).
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals aanvullende argumenten uit bijvoorbeeld de biomechanica of fysiologie, waarden en voorkeuren van patiënten, kosten (middelenbeslag), aanvaardbaarheid, haalbaarheid en implementatie. Deze aspecten zijn systematisch vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’ en kunnen (mede) gebaseerd zijn op expert opinion. Hierbij is gebruik gemaakt van een gestructureerd format gebaseerd op het evidence-to-decision framework van de internationale GRADE Working Group (Alonso-Coello, 2016a; Alonso-Coello 2016b). Dit evidence-to-decision framework is een integraal onderdeel van de GRADE methodiek.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk (Agoritsas, 2017; Neumann, 2016). De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen. De werkgroep heeft bij elke aanbeveling opgenomen hoe zij tot de richting en sterkte van de aanbeveling zijn gekomen.
In de GRADE-methodiek wordt onderscheid gemaakt tussen sterke en zwakke (of conditionele) aanbevelingen. De sterkte van een aanbeveling verwijst naar de mate van zekerheid dat de voordelen van de interventie opwegen tegen de nadelen (of vice versa), gezien over het hele spectrum van patiënten waarvoor de aanbeveling is bedoeld. De sterkte van een aanbeveling heeft duidelijke implicaties voor patiënten, behandelaars en beleidsmakers (zie onderstaande tabel). Een aanbeveling is geen dictaat, zelfs een sterke aanbeveling gebaseerd op bewijs van hoge kwaliteit (GRADE gradering HOOG) zal niet altijd van toepassing zijn, onder alle mogelijke omstandigheden en voor elke individuele patiënt.
|
Implicaties van sterke en zwakke aanbevelingen voor verschillende richtlijngebruikers |
||
|
|
Sterke aanbeveling |
Zwakke (conditionele) aanbeveling |
|
Voor patiënten |
De meeste patiënten zouden de aanbevolen interventie of aanpak kiezen en slechts een klein aantal niet. |
Een aanzienlijk deel van de patiënten zouden de aanbevolen interventie of aanpak kiezen, maar veel patiënten ook niet. |
|
Voor behandelaars |
De meeste patiënten zouden de aanbevolen interventie of aanpak moeten ontvangen. |
Er zijn meerdere geschikte interventies of aanpakken. De patiënt moet worden ondersteund bij de keuze voor de interventie of aanpak die het beste aansluit bij zijn of haar waarden en voorkeuren. |
|
Voor beleidsmakers |
De aanbevolen interventie of aanpak kan worden gezien als standaardbeleid. |
Beleidsbepaling vereist uitvoerige discussie met betrokkenheid van veel stakeholders. Er is een grotere kans op lokale beleidsverschillen. |
Organisatie van zorg
Bij de ontwikkeling van de richtlijnmodule is expliciet aandacht geweest voor de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, mankracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van deze specifieke uitgangsvraag zijn genoemd bij de overwegingen.
Commentaar- en autorisatiefase
De conceptrichtlijnmodule werd aan de betrokken (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werd de conceptrichtlijnmodule aangepast en definitief vastgesteld door de werkgroep. De definitieve richtlijnmodule werd aan de deelnemende (wetenschappelijke) verenigingen en (patiënt) organisaties voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd. Betrokken partijen (NVIC en NVVC) hebben geen bezwaar geuit tegen de inhoud van deze module.
Literatuur
Agoritsas T, Merglen A, Heen AF, Kristiansen A, Neumann I, Brito JP, Brignardello-Petersen R, Alexander PE, Rind DM, Vandvik PO, Guyatt GH. UpToDate adherence to GRADE criteria for strong recommendations: an analytical survey. BMJ Open. 2017 Nov 16;7(11):e018593. doi: 10.1136/bmjopen-2017-018593. PubMed PMID: 29150475; PubMed Central PMCID: PMC5701989.
Alonso-Coello P, Schünemann HJ, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Rada G, Rosenbaum S, Morelli A, Guyatt GH, Oxman AD; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 1: Introduction. BMJ. 2016 Jun 28;353:i2016. doi: 10.1136/bmj.i2016. PubMed PMID: 27353417.
Alonso-Coello P, Oxman AD, Moberg J, Brignardello-Petersen R, Akl EA, Davoli M, Treweek S, Mustafa RA, Vandvik PO, Meerpohl J, Guyatt GH, Schünemann HJ; GRADE Working Group. GRADE Evidence to Decision (EtD) frameworks: a systematic and transparent approach to making well informed healthcare choices. 2: Clinical practice guidelines. BMJ. 2016 Jun 30;353:i2089. doi: 10.1136/bmj.i2089. PubMed PMID: 27365494.
Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010 Dec 14;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348; PubMed Central PMCID: PMC3001530.
Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, Alper BS, Meerpohl JJ, Murad MH, Ansari MT, Katikireddi SV, Östlund P, Tranæus S, Christensen R, Gartlehner G, Brozek J, Izcovich A, Schünemann H, Guyatt G. The GRADE Working Group clarifies the construct of certainty of evidence. J Clin Epidemiol. 2017 Jul;87:4-13. doi: 10.1016/j.jclinepi.2017.05.006. Epub 2017 May 18. PubMed PMID: 28529184; PubMed Central PMCID: PMC6542664.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
Neumann I, Santesso N, Akl EA, Rind DM, Vandvik PO, Alonso-Coello P, Agoritsas T, Mustafa RA, Alexander PE, Schünemann H, Guyatt GH. A guide for health professionals to interpret and use recommendations in guidelines developed with the GRADE approach. J Clin Epidemiol. 2016 Apr;72:45-55. doi: 10.1016/j.jclinepi.2015.11.017. Epub 2016 Jan 6. Review. PubMed PMID: 26772609.
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Zoekverantwoording
Zoekacties zijn opvraagbaar. Neem hiervoor contact op met de Richtlijnendatabase.