PCEA met of zonder achtergrond infusie
Uitgangsvraag
Wat is de effectiviteit van patiënt gecontroleerde epidurale analgesie (PCEA) met continue achtergrond infusie ten opzichte van PCEA zonder continue achtergrondinfusie bij zwangere vrouwen met het verzoek tot behandeling van de baringspijn?
Aanbeveling
Kies bij PCEA bij voorkeur voor de bolus-only techniek indien de mogelijkheid tot extra bolustoedieningen gewaarborgd is.
Geef bij PCEA een achtergrondinfusie wanneer bij doorbraakpijn de toediening van een extra bolus enige tijd kan duren.
Overwegingen
De onderstaande overwegingen gelden in principe voor de gehele patiëntenpopulatie zoals geformuleerd in de uitgangsvraag.
Voor- en nadelen van de interventie en de kwaliteit van het bewijs
De literatuur toont geen significant of klinisch relevant verschil in effectiviteit en veiligheid tussen PCEA zonder achtergrondinfusie (oftewel bolus-only) in vergelijking tot PCEA met achtergrondinfusie. Tevredenheid van de zwangere vrouw is bij deze vergelijking een belangrijk aspect. Echter, satisfactie werd slechts beperkt in de literatuur gerapporteerd (1 RCT) met een zeer lage bewijskracht om hier richting aan te kunnen geven.
De kans op doorbraakpijn bij bolus-only techniek (in ieder geval bij de tot nu toe onderzochte instellingen) lijkt groter, hetgeen extra bolustoediening door zorgverleners noodzakelijk maakt. Met name op een drukke verlosafdeling of wanneer een anesthesioloog hiervoor naar het verloskamercomplex moet komen is de tijdsduur tussen extra pijnstillingswens en de uitvoering hiervan mogelijk erg lang. Daarentegen lijkt er een trend naar, weliswaar niet significante of klinisch relevante, obstetrische redenen om geen achtergrondinfusie toe te passen. Enige onderdeel van de uitkomstmaat bevallingsduur lijkt significant, namelijk de duur van de uitdrijvingsfase. Tevens wordt door Heesen (2015) beschreven dat er mogelijk minder vaginale kunstverlossingen noodzakelijk zijn wanneer geen achtergrondinfusie toegepast wordt en meer sectio’s wanneer wel achtergrondinfusie toegepast wordt. De forest plots in de literatuuranalyse geven ogenschijnlijk een trend hiervoor weer.
Wel wordt beschreven dat bij bolus-only de pijn tijdens de uitdrijving kan toenemen. Dit kan mogelijk geoptimaliseerd worden door de lockout periode aan te passen oftewel de toediening te individualiseren.
Bij het gebruik van een optimale concentratie van de infusievloeistof en pompinstellingen is er sprake van adequate pijnstilling, weinig noodzaak tot interventies, geen nadelige obstetrische uitkomsten en een hoge tevredenheid. De optimale concentratie van lokaal anesthetica met opioïden en de pompinstellingen zijn nog steeds onderwerp van discussie. Een hoog volume, lage concentratie met een lange lock-out lijkt geschikt gezien laag gebruik lokaal anesthetica en beste verspreiding in epidurale ruimte. Bij lage concentratie van lokaal anesthetica en opioïden met bolusvolume 5 tot 10 ml (zie de module ‘Epidurale analgesie: lokaal anestheticum’), lockout periodes tussen 10 en 20 minuten en een hoge perfusiedruk lijkt een achtergrondinfusie niet noodzakelijk (Halpern, 2009). Hierbij is het wel van belang adequate en tijdige extra bolussen door zorgverleners te waarborgen. (Voor dosering strategieën zie ook de module ‘Epidurale analgesie: doseringsstrategieën’)
Waarden en voorkeuren van patiënten
Indien een vrouw met baringspijn voor epidurale pijnbehandeling kiest is de mogelijkheid van zelf gecontroleerde pijnstilling te prefereren vanwege het op maat kunnen toedienen van de hoeveelheid medicatie en een mogelijk hoge tevredenheid. Dit laatste omdat de vrouw autonomie heeft over de toediening en er over het algemeen weinig interventies nodig zijn. Een placebo-effect van de bolusknop wordt in de literatuur beschreven (Haydon, 2011). Een mogelijk te ervaren nadeel van bolus-only voor de zwangere vrouw is de kans op doorbraakpijn, waarbij voor de zwangere vrouw tijdige aanvullende toediening van pijnstilling door de zorgverlener gewaarborgd moet zijn. Een mogelijk voordeel van bolus-only techniek is dat er overall minder medicatie nodig zal zijn. Zwangere vrouwen willen al tijdens de zwangerschap informatie over de verschillende vormen van pijnbehandeling, de voor- en de nadelen en wat dit betekent in hun situatie en willen hierover samen beslissen. Uit de meldactie blijkt dat dit nog onvoldoende in de praktijk gebeurt en vastgelegd wordt. Zie voor wijze van counselen en de vergelijking van de verschillende opties de module ‘Counseling: Samen beslissen‘.
Kosten (middelenbeslag)
Wanneer voor PCEA gekozen wordt zullen perfusiepompen met deze instellingsmogelijkheid aangeschaft moeten worden. PCEA met achtergrondinfusie leidt tot meer verbruik van lokaal anestheticum en opioïden. Wel zijn door de zelftoediening mogelijk minder interventies door zorgverleners nodig wat kostenbesparend zou kunnen werken. Wanneer bolus-only toegepast wordt bestaat de kans dat zorgverleners zich vrij moeten maken van andere werkzaamheden om een extra top-up te geven.
Aanvaardbaarheid voor de overige relevante stakeholders
Wanneer achtergrondinfusie weggelaten wordt of wanneer deze juist toegevoegd gaat worden zullen de zorgverleners rondom de barende vrouw hierover adequate scholing moeten krijgen. Als bezwaar zouden zij kunnen inbrengen dat zonder achtergrondinfusie meer interventies (dat wil zeggen toedieningen van extra bolus door zorgverlener) nodig zijn die niet altijd snel uitvoerbaar zijn op een drukke verlosafdeling.
Haalbaarheid en implementatie
Zie beschreven aspecten bij paragraaf kosten en aanvaardbaarheid overige stakeholders.
Elk ziekenhuis maakt een keuze voor het toepassen van één van de methoden per instelling om de veiligheid van de methode te waarborgen. In het maken van deze keuze kan de instelling bovenstaande factoren meewegen.
Aanbeveling-1
Rationale / balans tussen voor- en nadelen van de interventie
De werkgroep beveelt gebruik van PCEA bolus only aan, aangezien bij achtergrondinfusie onnodig hoge hoeveelheden lokaal anesthetica toegediend worden met kans op motorisch blok bij langdurig gebruik. Gezien de literatuur lijkt achtergrondinfusie voor pijnstilling niet noodzakelijk. Echter, doorbraakpijn komt vaker voor bij geen achtergrondinfusie, waardoor adequate en tijdige toediening van extra bolussen door zorgverlener gewaarborgd moet zijn.
Aanbeveling-2
Rationale / balans tussen voor- en nadelen van de interventie
In situaties waarbij tijdige toediening door zorgverlener niet gewaarborgd kan worden, ziet de werkgroep een indicatie om PCEA met achtergrond infusie te geven.
Onderbouwing
Achtergrond
Patiënt gecontroleerde epidurale analgesie als pijnbehandeling tijdens de bevalling wordt in Nederland nog niet overal toegepast. Continue epidurale analgesie met zo nodig op afroep extra bolus toediening door een verpleegkundige/verloskundige of een anesthesioloog is minder effectief en leidt tot meer gebruik van lokaal anesthetica en opioïden (zie de module ‘PCEA met achtergrondinfusie/ bolus only vs CEI’). De mogelijkheid voor patiënten om zelf een extra bolus toe te dienen met behulp van een pomp verbetert het effect van de pijnbehandeling, vergroot de tevredenheid en er is minder van de gebruikte infusievloeistof nodig. De vraag is of bolustoediening alleen voldoende is of dat een continue achtergrondinfusie noodzakelijk is voor een betere uitkomst.
Conclusies
|
GRADE |
Er lijkt geen verschil mate van pijnstilling tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Bremerich, 2005; Heesen, 2015; Paech, 1992; Petry, 2000; Srivastava, 2009) |
|
Laag GRADE |
Zwangeren behandeld met PCEA met achtergrondinfusie lijken minder vaak aanvullende pijnmedicatie door middel van bolustoediening door zorgverleners nodig te hebben dan zwangeren behandeld met PCEA zonder achtergrond infusie.
Bronnen: (Heesen, 2015; Paech, 1992; Petry, 2000; Srivastava, 2009) |
|
Zeer laag GRADE |
Het is onzeker of er een verschil is in tevredenheid ten aanzien pijnstilling tussen PCEA zonder achtergrondinfusie en PCEA met achtergrond infusie.
Bronnen: (Paech, 1992) |
|
Laag GRADE |
Er lijkt geen verschil in het risico op een vaginale kunstverlossing tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Heesen, 2015; Okutomi, 2009; Paech, 1992; Petry, 2000) |
|
Laag GRADE |
Er lijkt geen verschil in het risico op een sectio caesarea tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Heesen, 2015; Paech, 1992; Petry, 2000; Srivastava, 2009) |
|
Laag GRADE |
Er lijkt geen verschil in het risico op hypotensie tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Bremerich, 2005; Lim, 2008; Paech, 1992; Petry, 2000) |
|
Laag GRADE |
Er lijkt geen verschil in de mate van motorisch blok tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Heesen, 2015; Paech, 1992; Petry, 2000; Srivastava, 2009) |
|
Zeer laag GRADE |
Er lijkt geen verschil in het risico op misselijkheid tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Bremerich, 2005; Heesen, 2015; Okutomi, 2009) |
|
Laag GRADE |
Er lijkt geen verschil in de duur van de ontsluiting tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Heesen, 2015; Okutomi, 2009) |
|
Laag GRADE |
Zwangeren behandeld met PCEA zonder achtergrondinfusie lijken een kortere duur van de uitdrijving te hebben dan zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Heesen, 2015; Okutomi, 2009) |
|
Laag GRADE |
Er lijkt geen verschil in risico op jeuk tussen zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Bremerich, 2005; Heesen, 2015; Okutomi, 2009; Paech, 1992; Petry, 2000) |
|
Redelijk GRADE |
Er is waarschijnlijk geen verschil in pH a. umbilicalis van neonaten van zwangeren behandeld met PCEA zonder achtergrondinfusie versus zwangeren behandeld met PCEA met achtergrond infusie.
Bronnen: (Bremerich, 2005; Okutomi, 2009) |
|
Laag GRADE |
Er lijkt geen verschil in Apgarscore van neonaten van zwangeren behandeld met PCEA zonder achtergrondinfusie versus vrouwen behandeld met PCEA met achtergrond infusie.
Bronnen: (Heesen, 2015; Petry, 2000) |
Samenvatting literatuur
Beschrijving studies
De meta-analyse van Heesen (2015) is geïncludeerd en als uitgangspunt genomen. Heesen (2015) beoordeelde de methodologie van 13 RCT’s die PCEA met achtergrond infusie vergeleken met PCEA zonder achtergrond infusie, hiervan excludeerde Heesen (2015) 6 studies omdat deze niet geblindeerd waren. Alle 13 studies zijn beoordeeld of deze aan de PICO van de uitgangsvraag voldeden. Eén studie (Gambling, 1988) voldeed niet aan de PICO, omdat er geen controlegroep met PCEA zonder achtergrond infusie geïncludeerd werd. De overige 12 studies werden geïncludeerd in deze literatuuranalyse (Bremerich, 2005; Okutomi, 2009; Paech, 1992; Petry, 2000; Silvastava, 2009; Boselli, 2004; Brogly, 2011; Ferrante, 1994; Haydon, 2011; Lim, 2008; Missant, 2005; Vallejo, 2007).
Deze studies vergeleken PCEA met achtergrondinfusie versus PCEA zonder achtergrond infusie bij 1361 patiënten. De studies gebruikten verschillende doseringen en lock-out tijden (zie Evidencetabellen). Ook was er een gemixte populatie wat betreft pariteit, met als gevolg een mix van kortere en langere bevallingsduur.
Resultaten
Pijnintensiteit
Alle geïncludeerde studies onderzochten pijn, maar Okutomi (2009) rapporteerde geen resultaten. Doordat de uitkomstmaat pijnintensiteit op verschillende manieren (mediaan, gemiddelde, maximale pijn) en tijdstippen gerapporteerd werd, zijn de data niet gepoold. In zes studies uit de meta-analyse van Heesen (2015) werd bij 458 zwangeren geen significant verschil waargenomen tussen de twee groepen wanneer er gekeken werd naar de mediane of gemiddelde pijnscore. Eén studie (Lim, 2008) rapporteerde een significant (p< 0,01) lagere maximale VAS-score in de PCEA-groep met 5 mL/uur achtergrondinfusie (mediaan 2,0; range 0 tot 10) en de PCEA-groep met 10 mL/uur achtergrondinfusie (mediaan 0; range 0 tot 10) vergeleken met de PCEA-groep zonder achtergrondinfusie (mediaan 3,5; range 0 tot 10). Bremerich, 2005 rapporteerde hoe vaak de VAS score op alle meetmomenten hoger dan 4,0 was en vond dat patiënten zonder achtergrond infusie significant (p<0,001) vaker een VAS hoger dan 4,0 hadden (22,4%) dan patiënten met achtergrond infusie (7,5%). Paech (1992) rapporteerde een mediane pijn score (0 tot 100 schaal) van 7 (range 0 tot 86) punten in de PCEA-groep met achtergrond infusie versus 11 (range 0 tot 77) punten in de PCEA-groep zonder achtergrondinfusie.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat pijnintensiteit is met twee niveaus verlaagd gezien risico op bias door afwezigheid van blindering en het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Cross-over naar andere pijnmedicatie/ aanvullende pijnmedicatie (bolustoediening door zorgverleners)
Vier studies uit de meta-analyse van Heesen (2015) en 3 aanvullende RCT’s van Paech (1992), Petry (2000) en Srivatava (2009) (n=765 zwangeren) rapporteerden het aantal zwangeren die ten minste één extra bolustoediening door zorgverleners nodig hadden. Zwangeren behandeld met PCEA met achtergrond infusie hadden een 62% lagere kans op het nodig hebben van aanvullende bolusdoseringen in vergelijking met zwangeren behandeld met PCEA zonder achtergrondinfusie (RR 0,38, 95% BI: 0,29 tot 0,51). Er was geen sprake van heterogeniteit (I2 0%). Daarnaast rapporteerde één studie het mediane aantal dat een zorgverlener een bolus moest toedienen en vond geen verschil (mediaan 0 (range 0 tot 8) (PCEA plus achtergrond infusie) versus 0 (0 tot 6) (PCEA bolus-only)), p-waarde=0,57). Een andere studie rapporteerde het aantal aanvullende bolussen toegediend door zorgverleners, opgesplitst naar ontsluiting (16% (PCEA plus achtergrond infusie)) versus (15% (PCEA zonder achtergrond infusie)) en uitdrijving (respectievelijk 4% en 3%) van de bevalling en vond ook geen verschil. Een derde studie rapporteerde het gemiddelde aantal bolussen toegediend door zorgverleners en vond dat zwangeren in de PCEA zonder achtergrond infusie groep meer bolussen nodig hadden dan zwangeren in de PCEA met achtergrond infusie groep (gemiddelde (SEM): 43,3 (1,5) (PCEA plus 3 mL/ uur achtergrond infusie), 44,3 (1,7) (PCEA plus 6 mL/ uur achtergrond infusie) versus 54,3 (1,7) (PCEA bolus-only), p<0,004).
Figuur 1 Aanvullende pijnbehandeling (aantal patiënten met tenminste ten minste één extra bolustoediening door zorgverlener)
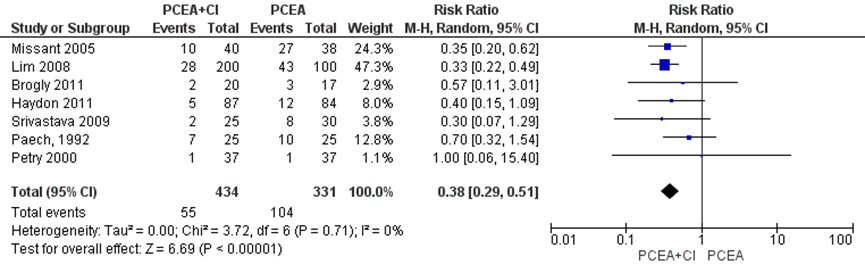
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen, 2015 aangevuld met RCT’s Paech (1992), Petry (2000) en Srivatava (2009)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat bolustoediening door zorgverleners is met twee niveaus verlaagd gezien het risico op bias door afwezigheid van blindering in 3 van de 7 studies, het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Tevredenheid ten aanzien van pijnstilling
Eén studie (Paech, 1992) rapporteerde de tevredenheid ten aanzien van pijnstilling bij 50 zwangeren, onderverdeeld in de categorieën zeer tevreden, goed, redelijk en ontevreden. Er werden geen significante verschillen gevonden tussen de groepen. Twintig van de zwangeren in de PCEA met achtergrondinfusie groep waren zeer tevreden versus 18 zwangeren in de PCEA-groep zonder achtergrondinfusie.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat tevredenheid ten aanzien van pijnstilling is met drie niveaus verlaagd gezien het risico op bias door afwezigheid van blindering (1 niveau) en het zeer geringe aantal patiënten (imprecisie, 2 niveaus). Het niveau van bewijskracht wordt gegradeerd als ‘zeer laag’.
Wijze van bevallen/ modus partus - vaginale kunstverlossing
Alle studies (n=7) geïncludeerd in de meta-analyse van Heesen (2015) namen vaginale kunstverlossing mee als uitkomstmaat. Deze data werden aangevuld met de resultaten van de RCT’s van Okutomi (2009), Paech (1992) en Petry (2000) (totaal n=1081 zwangeren). Het risico op een vaginale kunstverlossing bleek niet significant te verschillen tussen zwangeren met PCEA zonder achtergrond infusie en zwangeren met PCEA met achtergrondinfusie (RR 1,20, 95% BI: 0,95 tot 1,51). Er was geen sprake van heterogeniteit (I2 0%).
Figuur 2 vaginale kunstverlossing
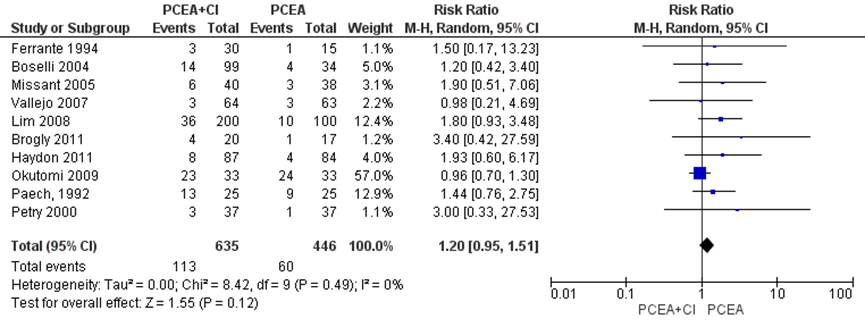
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen (2015) aangevuld met RCT’s Okutomi (2009), Paech (1992) en Petry (2000)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat vaginale kunstverlossing is met twee niveaus verlaagd gezien het brede betrouwbaarheidsinterval en het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Wijze van bevallen/ modus partus - sectio caesarea
Alle studies (n=7) geïncludeerd in de meta-analyse van Heesen (2015) namen de sectio caesarea mee als uitkomstmaat. Deze data werden aangevuld met de resultaten van de RCT’s van Srivastava (2009), Paech (1992) en Petry (2000) (totaal n=1070 zwangeren). Het risico op een sectio caesarea bleek niet significant te verschillen tussen zwangeren met of zonder achtergrond infusie (RR 0,83, 95% BI: 0,63 tot 1,10). Er was geen sprake van heterogeniteit (I2 0%).
Figuur 3 Sectio caesarea
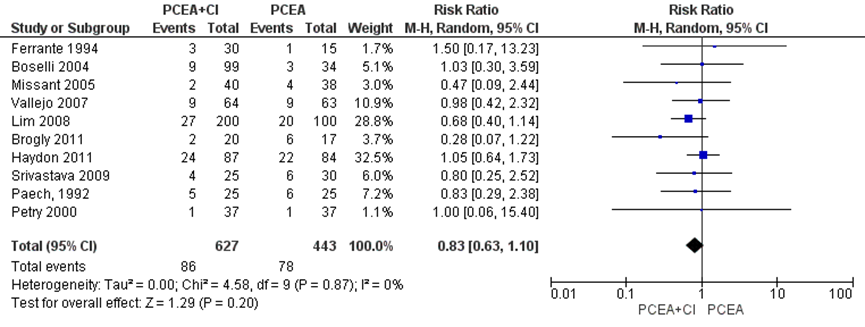
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen (2015) aangevuld met RCT’s Srivastava (2009), Paech (1992) en Petry (2000)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat sectio caesarea is met twee niveaus verlaagd gezien het brede betrouwbaarheidsinterval en het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Maternale complicatie sHypotensie
Eén studie uit het de meta-analyse van Heesen (2015) en 3 aanvullende RCT’s van Bremerich (2005), Paech (1992) en Petry (2000) rapporteerden de uitkomstmaat maternale hypotensie (n=490). Bremerich (2005) en Petry (2000) rapporteerden geen events. Lim (2008) en Paech (1992) vonden geen significante verschillen tussen zwangeren met PCEA met of zonder achtergrond infusie (RR 0,84, 95% BI 0,42 tot 1,66).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat hypotensie is met twee niveaus verlaagd gezien het risico op bias vanwege de afwezigheid van blindering in 3 van de 4 studies en het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Motorisch blok
Heesen (2015) rapporteerde de gepoolde uitkomst van zwangeren zonder motorisch blok versus enige mate van motorisch blok (niet nader gedefinieerd) in drie studies (n= 256 zwangeren). Er was geen significant verschil (RR 0,98, 95% BI 0,92 tot 1,06) tussen de zwangeren met PCEA met achtergrondinfusie versus zwangeren met PCEA zonder achtergrond infusie. Ook Bremerich (2005) rapporteerde geen significante verschillen tussen de groepen. Het maximale motorisch blok waargenomen, werd beschreven als het onvermogen om het gestrekte been te tillen, maar wel in staat de knie te bewegen, wat in 2/40 zwangeren met PCEA met achtergrondinfusie versus 1/40 zwangeren met PCEA zonder achtergrondinfusie voorkwam (P> 0,05). Ook de studie van Paech (1992) vond geen verschil in motorisch blok tussen beide groepen, waarbij in 8/25 van de PCEA met achtergrondinfusie en 9/25 in de PCEA zonder achtergrondinfusie-groep alleen de beweging van knie of voet mogelijk was. Petry (2000) rapporteerde dat 2 zwangeren in de PCEA-groep met achtergrondinfusie en 1 vrouw in de PCEA-groep zonder achtergrondinfusie moeite hadden om hun knie te strekken.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat motorisch blok is met twee niveaus verlaagd gezien het risico op bias vanwege de afwezigheid van blindering in 3 van de 6 studies en het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Misselijkheid
Twee studies uit de meta-analyse van Heesen (2015) namen misselijkheid mee als uitkomstmaat. Deze data werden aangevuld met de resultaten van de RCT van Bremerich (2005) en Okutomi (2009) (totaal n=491 zwangeren). Het risico op misselijkheid bleek niet significant te verschillen tussen zwangeren behandeld met PCEA zonder achtergrond infusie in vergelijking tot PCEA met achtergrondinfusie (RR 1,59, 95% BI: 0,85 tot 2,97). Er was geen sprake van heterogeniteit (I2 0%).
Figuur 4 Misselijkheid
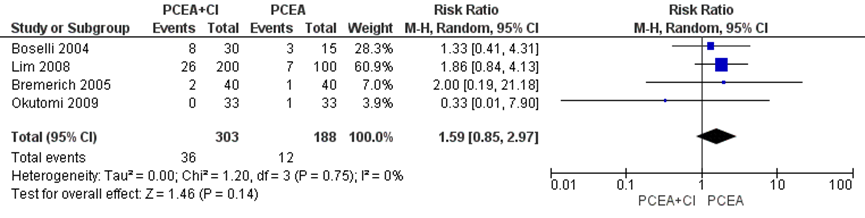
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen (2015) aangevuld met RCT Bremerich (2005) en Okutomi (2009)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat misselijkheid is met drie niveaus verlaagd gezien het risico op bias vanwege de afwezigheid van blindering in 3 van de 6 studies, het geringe aantal patiënten en het brede betrouwbaarheidsinterval (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘zeer laag’.
Bevallingsduur
Alle studies (n=7) geïncludeerd in de meta-analyse van Heesen (2015) namen de totale bevallingsduur mee als uitkomstmaat. Deze data werden aangevuld met de resultaten van de RCT’s van Srivastava (2009) en Paech (1992) (totaal n=996 zwangeren). Er werd geen significant verschil waargenomen in de totale bevallingsduur tussen zwangeren behandeld met PCEA zonder achtergrond infusie in vergelijking tot PCEA met achtergrondinfusie (gemiddeld verschil 0,61 minuten, 95% BI: -17,35 tot 18,58). Er was geen sprake van heterogeniteit (I2 0%).
Figuur 5 Totale bevallingsduur (minuten)
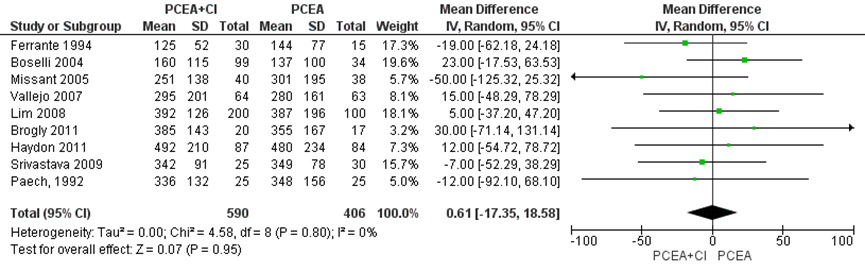
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen (2015) aangevuld met RCT’s Srivastava (2009) en Paech (1992)
De duur van de ontsluiting werd gerapporteerd in 3 studies van het systematische review van Heesen (2015) en aangevuld met de resultaten van de RCT van Okutomi (2009) (totaal n=371 zwangeren). Er werd geen significant verschil gevonden in de bevallingsduur in het 1e stadium tussen zwangeren behandeld met PCEA zonder achtergrond infusie in vergelijking tot PCEA met achtergrondinfusie (gemiddeld verschil -21,05 minuten, 95% BI: -68,89 tot 26,79). Er was sprake van een grote mate van heterogeniteit (I2 84%).
Figuur 6 Duur ontsluitingsfase (minuten)
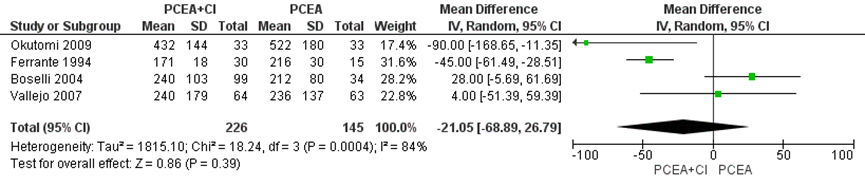
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen (2015) aangevuld met RCT Okutomi (2009)
De duur van de uitdrijving was significant langer in de PCEA-groep met achtergrondinfusie (beschreven in 4 studies van de systematische review van Heesen (2015) en aangevuld met de resultaten van de RCT van Okutomi (2009) totaal n=671 zwangeren) met een gemiddeld verschil van 12,29 minuten (95% BI: 5,18 tot 19,39). Er was geen sprake van heterogeniteit (I2 0%).
Figuur 7 Duur uitdrijving (minuten)
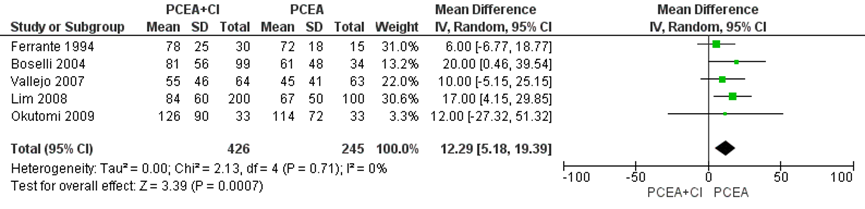
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen (2015) aangevuld met RCT Okutomi (2009)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat bevallingsduur is met twee niveaus verlaagd gezien de brede betrouwbaarheidsintervallen en het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Jeuk
Twee studies geïncludeerd in de meta-analyse van Heesen (2015) namen jeuk mee als uitkomstmaat. Deze data werden aangevuld met de resultaten van de RCT’s van Bremerich (2005), Okutomi (2009), Paech en Petry (2000) (totaal n=615 zwangeren). Er werd geen significant verschil waargenomen tussen zwangeren behandeld met PCEA zonder achtergrond infusie in vergelijking tot PCEA met achtergrondinfusie (RR 0,99, 95% BI: 0,82 tot 1,20). Er was geen sprake van heterogeniteit (I2 0%).
Figuur 8 Jeuk
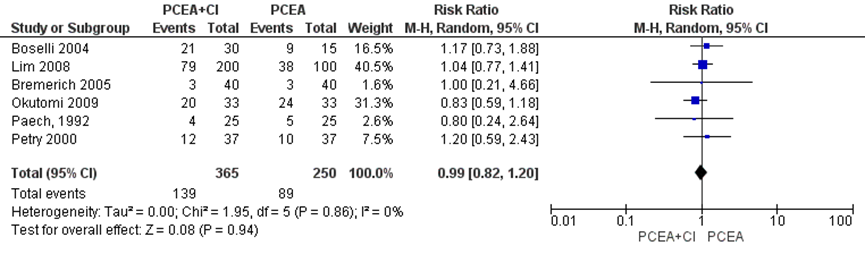
Z: p-waarde van gepoolde effect; df: degrees of freedom (vrijheidsgraden); I2: statistische heterogeniteit; CI: betrouwbaarheidsinterval. Bron: Heesen (2015) aangevuld met RCT’s Bremerich (2005), Okutomi (2009) en Paech (1992)
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat jeuk is met twee niveaus verlaagd gezien het risico op bias vanwege de afwezigheid van blindering in 4 van de 6 studies en het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Complicaties - neonatale complicaties
pH a. umbilicalis
Eén studie geïncludeerd in de meta-analyse van Heesen (2015) en twee RCT’s (Bremerich, 2005; Okutomi, 2009) rapporteerden de gemiddelde pH a. umbilicalis van neonaten (n=224 zwangeren). Er waren geen significante verschillen tussen de groepen (gemiddeld verschil 0,01, 95% BI: -0,01 tot 0,02).
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat pH a. umbilicalis is met één niveau verlaagd gezien het geringe aantal patiënten (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘redelijk’.
Apgarscore
Eén studie in de meta-analyse van Heesen (2005) rapporteerde de Apgarscore < 7 op 5 minuten bij 133 patiënten. Er was geen significant verschil tussen de twee groepen (RR: 0,69, 95% BI: 0,06 tot 7,34). Petry (2000) vond bij geen enkele neonaat een Apgarscore < 6 op 5 minuten.
Bewijskracht van de literatuur
De bewijskracht voor de uitkomstmaat Apgarscore < 7 bij 5 minuten is met twee niveaus verlaagd gezien het zeer geringe aantal patiënten en brede betrouwbaarheidsinterval (imprecisie). Het niveau van bewijskracht wordt gegradeerd als ‘laag’.
Geen van de studies naar het effect van PCEA zonder achtergrondinfusie versus PCEA met achtergrondinfusie rapporteerden de uitkomstmaten: cross-over naar andere pijnmedicatie, angst ten aanzien van pijn, borstvoeding, maternale complicaties: convulsie/ eclamptisch insult, fluxus post-partum, ademhalingsdepressie, temperatuur, urineretentie, postspinale punctie hoofdpijn, epiduraal abces/ hematoom, neonatale complicaties: opname NICU, foetale hartslag afwijkingen, temperatuur, mortaliteit, negatieve uitkomsten voor de baby op lange termijn en kosten.
Zoeken en selecteren
Om de uitgangsvraag te kunnen beantwoorden is er een systematische literatuuranalyse verricht naar de volgende zoekvraag:
Wat zijn de (on)gunstige effecten van PCEA met continue achtergrond infusie vergeleken met PCEA zonder continue achtergrond infusie bij zwangere vrouwen met het verzoek tot behandeling van de baringspijn?
P: zwangere vrouwen met het verzoek tot behandeling van de baringspijn die PCEA krijgen als pijnbehandeling;
I: PCEA (patient controlled epidural analgesia) met achtergrond infusie;
C: PCEA zonder achtergrond infusie;
O: zie onderstaand:
- Pijnintensiteit (VAS/ NRS-schaal of een ander gevalideerd instrument).
- Cross-over naar andere pijnmedicatie/aanvullende pijnmedicatie (bolustoediening door zorgverleners).
- Tevredenheid ten aanzien van pijnstilling (rapportcijfer/ VAS/ NRS-schaal of een ander gevalideerd instrument).
- Borstvoeding.
- Modus partus (vaginale kunstverlossing, sectio caesarea).
- Maternale complicaties: convulsie/ eclamptisch insult, fluxus postpartum, ademhalingsdepressie, bevallingsduur, hypotensie, temperatuur, urineretentie, misselijkheid/braken, jeuk, mate van motorisch blok, postspinale punctie hoofdpijn, epiduraal abces/ hematoom.
- Neonatale complicaties: opname NICU, Apgarscore < 7 bij 5 min, foetale hartslag afwijkingen, temperatuur, pH a. umbilicalis.
- Maternale/ neonatale sterfte.
- Negatieve uitkomsten voor de baby op lange termijn.
- Kosten.
Relevante uitkomstmaten
De werkgroep achtte pijnintensiteit, tevredenheid ten aanzien van pijnstilling, modus partus en maternale/ neonatale sterfte kritieke uitkomstmaten voor de besluitvorming. Consumptie van lokaal anestheticum/opioïd en mate van motorisch blok zijn voor de besluitvorming belangrijke uitkomstmaten.
De werkgroep definieerde de uitkomstmaten als volgt: pijnintensiteit (VAS/ NRS-schaal of een ander gevalideerd instrument) en tevredenheid (rapportcijfer/ VAS/ NRS-schaal of een ander gevalideerd instrument), waarbij scores door de patiënt zelf gedurende of direct na de bevalling gerapporteerd werden. Voor neonatale Apgarscore wordt de definitiescore van < 7 bij 5 minuten aangehouden. Voor de overige uitkomstmaten definieerde de werkgroep niet a priori de genoemde uitkomstmaten, maar hanteerde de in de studies gebruikte definities.
Naast significantie wordt de klinische besluitvorming vooral bepaald door de klinische relevantie van de waargenomen verschillen tussen behandelopties. Voor dichotome uitkomstmaten definieerde de werkgroep een minimaal klinisch (patiënt) relevant verschil volgens de grenzen van de GRADE-working group, namelijk een verschil in relatief risico van 25%. Voor de continue uitkomstmaten definieerde de werkgroep een verschil van 10% op pijnintensiteit of tevredenheid als een klinisch (patiënt) relevant verschil.
Zoeken en selecteren (Methode)
Het betreft een literatuurzoekactie gebaseerd op de systematische review van Heesen, 2015. In de databases Medline (OVID), Embase (via Embase.com) en de Cochrane Library (via Wiley) is op 18 december 2017 met relevante zoektermen gezocht naar studies die PCEA met achtergrond infusie vergeleken met PCEA zonder achtergrond infusie bij zwangere vrouwen met baringspijn. De zoekverantwoording is weergegeven onder het tabblad Verantwoording. De literatuurzoekactie leverde 15 treffers op. Studies werden geselecteerd op grond van de volgende selectiecriteria:
- Studiedesign RCT.
- Gepubliceerd tussen 1 januari 2012 en 18 december 2017.
- Gelijke concentratie lokaal anestheticum en opioïd in de interventie en controle groep.
- Beschrijven van minimaal één van de bovengenoemde uitkomstmaten.
Op basis van titel en abstract werden in eerste instantie 3 studies voorgeselecteerd. Na raadpleging van de volledige tekst, werden vervolgens 2 studies geëxcludeerd (zie exclusietabel onder het tabblad Verantwoording) en 1 systematisch review definitief geselecteerd (Heesen, 2015). De belangrijkste studiekarakteristieken en resultaten zijn opgenomen in de evidencetabellen. De beoordeling van de individuele studieopzet (risk of bias) is opgenomen in de risk of bias tabellen.
Referenties
- Heesen M, Böhmer J, Klöhr S, Hofmann T, Rossaint R, Straube S. The effect of adding a background infusion to patient-controlled epidural labor analgesia on labor, maternal, and neonatal outcomes: a systematic review and meta-analysis. Anesth Analg. 2015 Jul;121(1):149-58. doi: 10.1213/ANE.0000000000000743. Review. PubMed PMID: 25902319.
- Paech MJ. Patient-controlled epidural analgesia in labour - is a continuous infusion of benefit? Anaesth Intensive Care. 1992 Feb;20(1):15-20. PubMed PMID: 1609935.
- Gambling DR, Yu P, Cole C, McMorland GH, Palmer L. A comparative study of patient controlled epidural analgesia (PCEA) and continuous infusion epidural analgesia (CIEA) during labour. Can J Anaesth. 1988 May;35(3 ( Pt 1)):249-54. PubMed PMID: 3289769.
- Petry J, Vercauteren M, Van Mol I, Van Houwe P, Adriaensen HA. Epidural PCA with bupivacaine 0.125%, sufentanil 0.75 microgram and epinephrine 1/800.000 for labor analgesia: is a background infusion beneficial? Acta Anaesthesiol Belg. 2000;51(3):163-6. PubMed PMID: 11129615.
- Bremerich DH, Waibel HJ, Mierdl S, Meininger D, Byhahn C, Zwissler BC, Ackermann HH. Comparison of continuous background infusion plus demand dose and demand-only parturient-controlled epidural analgesia (PCEA) using ropivacaine combined with sufentanil for labor and delivery. Int J Obstet Anesth. 2005 Apr;14(2):114-20. PubMed PMID: 15795146.
- Boselli E, Debon R, Cimino Y, Rimmelé T, Allaouchiche B, Chassard D. Background infusion is not beneficial during labor patient-controlled analgesia with 0.1% ropivacaine plus 0.5 microg/ml sufentanil. Anesthesiology. 2004 Apr;100(4):968-72. PubMed PMID: 15087635.
- Okutomi T, Saito M, Mochizuki J, Amano K, Hoka S. A double-blind randomized controlled trial of patient-controlled epidural analgesia with or without a background infusion following initial spinal analgesia for labor pain. Int J Obstet Anesth. 2009 Jan;18(1):28-32. doi: 10.1016/j.ijoa.2008.06.006. Epub 2008 Nov 20. PubMed PMID: 19022653.
- Srivastava U, Gupta A, Saxena S, Kumar A, Singh S, Saraswat N, Mishra AR, Kannaujia A, Mishra S. Patient Controlled Epidural Analgesia during Labour: Effect of Addition of Background Infusion on Quality of Analgesia & Maternal Satisfaction. Indian J Anaesth. 2009 Dec;53(6):649-53. PubMed PMID: 20640091; PubMed Central PMCID: PMC2900073.
- Ferrante FM, Rosinia FA, Gordon C, Datta S. The role of continuous background infusions in patient-controlled epidural analgesia for labor and delivery. Anesth Analg. 1994 Jul;79(1):80-4. PubMed PMID: 8010458.
- Missant C, Teunkenst A, Vandermeersch E, Van de Velde M. Patient-controlled epidural analgesia following combined spinal-epidural analgesia in labour: the effects of adding a continuous epidural infusion. Anaesth Intensive Care. 2005 Aug;33(4):452-6. PubMed PMID: 16119485.
- Vallejo MC, Ramesh V, Phelps AL, Sah N. Epidural labor analgesia: continuous infusion versus patient-controlled epidural analgesia with background infusion versus without a background infusion. J Pain. 2007 Dec;8(12):970-5. Epub 2007 Aug 7. PubMed PMID: 17686658.
- Lim Y, Ocampo CE, Supandji M, Teoh WH, Sia AT. A randomized controlled trial of three patient-controlled epidural analgesia regimens for labor. Anesth Analg. 2008 Dec;107(6):1968-72. doi: 10.1213/ane.0b013e3181887ffb. PubMed PMID: 19020146.
- Brogly N, Schiraldi R, Vazquez B, Perez J, Guasch E, Gilsanz F. A randomized control trial of patient-controlled epidural analgesia (PCEA) with and without a background infusion using levobupivacaine and fentanyl. Minerva Anestesiol. 2011 Dec;77(12):1149-54. Epub 2011 May 30. PubMed PMID: 21623342.
- Haydon ML, Larson D, Reed E, Shrivastava VK, Preslicka CW, Nageotte MP. Obstetric outcomes and maternal satisfaction in nulliparous women using patient-controlled epidural analgesia. Am J Obstet Gynecol. 2011 Sep;205(3):271.e1-6. doi: 10.1016/j.ajog.2011.06.041. Epub 2011 Jun 17. PubMed PMID: 22071061.
- Halpern SH, Carvalho B. Patient-controlled epidural analgesia for labor. Anesth Analg. 2009 Mar;108(3):921-8. doi: 10.1213/ane.0b013e3181951a7f. Review. PubMed PMID: 19224805.
Evidence tabellen
Evidence table for intervention studies (randomized controlled trials and non-randomized observational studies (cohort studies, case-control studies, case series))1
This table is also suitable for diagnostic studies (screening studies) that compare the effectiveness of two or more tests. This only applies if the test is included as part of a test-and-treat strategy – otherwise the evidence table for studies of diagnostic test accuracy should be used.
|
Study reference |
Study characteristics |
Patient characteristics 2 |
Intervention (I) |
Comparison / control (C) 3
|
Follow-up |
Outcome measures and effect size 4 |
Comments |
|
Bremerich, 2005 |
Type of study: RCT
Setting: Johann Wolfgang Goethe- University Hospital
Country: Germany
Source of funding: not reported |
Inclusion criteria: 1. older than 18 years; 2. requesting epidural analgesia; 3. ASA physical status I or II; 4. >37 weeks of gestation; 5. singleton pregnancy; 6. cephalad presentation; 7. conversational fluency in German; 8. minimum cervical dilation before initiation of epidural analgesia was 3–4 cm with uterine contractions at regular intervals of 2–3 min.
Exclusion criteria: Common contraindications to epidural analgesia and the need for cesarean section during the study
N total at baseline: Intervention: 40 Control: 40
Important prognostic factors2: age (SD): I: 30 (6) C:30 (5)
Primiparae (%) I: 70 C: 52
Groups are comparable at baseline
|
Describe intervention (treatment/procedure/test):
Initial bolus containing ropivacaine 16 mg and sufentanil 10 µg
4 mL/h plus an hourly maximum of three 4-mL boluses on demand (lock-out time 20 min) of ropivacaine 0.16% plus sufentanil 0.5 µg /mL
|
Describe control (treatment/procedure/test):
Initial bolus containing ropivacaine 16 mg and sufentanil 10 µg
An hourly maximum of four 4-mL boluses (lock-out time 15 min) of ropivacaine 0.16% plus sufentanil 0.5 µg /mL |
Length of follow-up: 48 hours after delivery
Loss-to-follow-up: Intervention: 7 (17.5%) Reasons Cesarean delivery during study
Control: 7 (17.5%) Reasons Cesarean delivery during study
Incomplete outcome data: Intervention: none
Control: none
|
Outcome measures and effect size (include 95%CI and p-value if available):
Pain (VAS) >40 Measured every 5 min for the first 30 min after catheter insertion, after 60 min and subsequently every hour until delivery
I: 12 (7.5%) of the 161 time points C: 33 (22.4%) of the 147 time points P=0.0003
Maximum motor block Bromage 1, n I: 2 C: 1 No statistically significant difference between groups (p-value not reported)
Adverse events Nausea, n (n time points) I: 2 (3) C: 1 (2)
Pruritus I: 3 (6) C: 3(6)
No statistically significant difference between groups (p-value not reported)
Umbilical artery pH, mean (SD) I: 7.27 (0.07) C: 7.28 (0.06) |
|
|
Okutomi, 2009 |
Type of study: RCT
Setting: Kitasato University
Country: Japan
Source of funding: not reported |
Inclusion criteria: Nulliparous patients undergoing full-term elective induction of labor with a singleton fetus in the vertex presentation who desired neuraxial anesthesia for childbirth.
Exclusion criteria: Cesarean section
N total at baseline: Intervention: 33 Control: 33
Important prognostic factors2: age ± SD: I: 31 (5) C: 32 (4)
Groups are comparable at baseline
|
Describe intervention (treatment/procedure/test):
Bupivacaine 2.5 mg with fentanyl 25 µg
Continuous infusion of 6 mL/h with an hourly demand maximum of five 5-mL boluses of 0.1% ropivacaine with fentanyl 2 µg /mL |
Describe control (treatment/procedure/test):
Bupivacaine 2.5 mg with fentanyl 25 µg
5-mL demand dose of 0.1% ropivacaine with fentanyl 2 µg /mL with a 10-min lockout interval and a dose limit of 30 mL/h
|
Length of follow-up: until delivery
Loss-to-follow-up: none
Incomplete outcome data: none
|
Outcome measures and effect size (include 95%CI and p-value if available):
Labour duration (h) First stage, mean SD I: 7.2 ± 2.4 C: 8.7 ± 3.0 P<0.05
Second stage, mean SD I: 2.1 ± 1.5 C: 1.9 ± 1.2 33 No statistically significant difference between groups (p-value not reported)
Mode of delivery (n) Spontaneous I: 10 C: 9
Vacuum extraction I: 17 C: 17
Forceps extraction I: 6 C:7
Maternal adverse events, n (%) Hypotension I: 0 (0%) C: 0 (0%)
Nausea I: 0 (0%) C: 1 (3%)
Pruritus I: 20 (61%) C: 24 (73%)
No statistically significant difference between groups (p-value not reported)
Umbilical artery pH, mean (SD) I: 7.29 ± 0.04 C: 7.28 ± 0.03 |
Pain was assessed according to the method section, but results were not reported. |
|
Srivastava, 2009 |
Type of study: RCT
Setting: Research scholar Agra University
Country: India
Source of funding: not reported |
Inclusion criteria: ASA status I and II of mixed parity who were willing for epidural analgesia during labor, a singleton pregnancy with vertex presentation, no contraindication to epidural analgesia and who were able to use PCA pump.
Exclusion criteria:
N total at baseline: Intervention: 25 Control: 30
Important prognostic factors2: age ± SD: I: 24±5 C: 25±6
Primi/multi I: 11/14 C: 14/16
Groups are comparable at baseline |
Describe intervention (treatment/procedure/test):
Initial bolus dose of a 10 ml solution containing 0.125% bupivacaine + 2 mcg.ml-1 of fentanyl
Continuous epidural infusion of above solution at the rate of 10 ml/hr with demand bolus of 3 ml of above solution with lockout interval of 10 minutes with a maximum upper limit of epidural solution of 25ml/ hour |
Describe control (treatment/procedure/test):
Initial bolus dose of a 10 ml solution containing 0.125% bupivacaine + 2 mcg.ml-1 of fentanyl
8ml bolus of the above solution on demand with lockout interval of 20 minutes with a maximum upper limit of epidural solution of 25ml/ hour |
Length of follow-up: until delivery
Loss-to-follow-up: None
Incomplete outcome data: Not described
|
Outcome measures and effect size (include 95%CI and p-value if available):
Duration of labour (minutes), mean SD I: 342±91 C: 349±78
Vaginal/caesarean delivery, mean SD I: 21/4 C: 24/6
Pain Mean VAS during labour, mean SD I: 1.89±1.03 C: 1.96±1.08 p=0.32
Highest VAS, median (range) I: 5 (0 – 7) C: 6 (0 – 6) p<0.05
Rescue boluses, n patients I: 2 (8%) C: 8 (27%) P<0.05 |
|
|
Paech, 1992 |
Type of study: RCT
Setting: King Edward Memorial Hospital for Woman, Perth
Country: Australia
Source of funding: non-commercial |
Inclusion criteria: Women at term with a singleton cephalic fetus, no major medical or obstetric complications and in established labour were enrolled.
Exclusion criteria: Women delivering within two hours of attaching the PCA pump
N total at baseline: Intervention: 26 Control: 26
Important prognostic factors2: age ± SD: I: 26±6 C: 28±4
Primiparity,n (%): I: 18 (72%) C: 17 (68%)
Groups comparable at baseline? Yes
|
Describe intervention (treatment/procedure/test):
Epidural solution of 10 ml bolus of 0.125% bupivacaine (12.5 mg) containing fentanyl 5 mcg per ml. Allowance was made for an additional 4 ml bolus of 0.5% bupivacaine (20 mg).
PCEA 4 ml incremental bolus dose on demand of 0.125% plain bupivacaine plus fentanyl 3 mcg per ml (62.5 mg of bupivacaine and 150 mcg of fentanyl diluted to 50 ml with normal saline) with a lockout interval of 15 minutes.
CI 4 ml continuous infusion
|
Describe control (treatment/procedure/test):
Epidural solution of 10 ml bolus of 0.125% bupivacaine (12.5 mg) containing fentanyl 5 mcg per ml. Allowance was made for an additional 4 ml bolus of 0.5% bupivacaine (20 mg).
PCEA 4 ml incremental bolus dose on demand of 0.125% plain bupivacaine plus fentanyl 3 mcg per ml (62.5 mg of bupivacaine and 150 mcg of fentanyl diluted to 50 ml with normal saline) with a lockout interval of 15 minutes.
|
Length of follow-up: within 24 hours after delivery
Loss-to-follow-up: Intervention: None
Control: None
Incomplete outcome data: Intervention: 2 (4%) Reasons: 1 woman delivered within two hours of attaching the PCA pump, 1 woman was included in analysis until the time of her withdrawal due to cephalad sensory block exceeding T6.
Control: 1 (2%) Reasons: 1 woman delivered within two hours of attaching the PCA pump
|
Outcome measures and effect size (include 95%CI and p-value if available):
Pain score (0-100), median (range) I: 7 (0-86) C: 11 (0-77)
Satisfaction with pain relief During the first stage (Excellent/ good/ fair/ unsatisfactory) I: 20/ 4/ 1/ 0 C: 18/ 7/ 0/ 0
During the expulsion phase (Excellent/ good/ fair/ unsatisfactory) I: 6/ 6/ 2/ 2 C: 5/ 10/ 3/ 1 (no significant differences between groups, p-value not reported)
Mode of delivery (n) Spontaneous I: 7/ 25 C: 10/ 25
Instrumental I: 13/ 25 C: 9/ 25
Caesarean I: 5/ 25 C:6/ 25
Duration of labour (hour), mean SD I: 5.6 (2.2) C: 5.8 (2.6)
Maternal adverse events, n (%) Hypotension: I: 4/ 25 C: 2/ 25
Pruritus I: 4/ 25 C: 5/ 25
Grade 2/3 motor block I: 8/ 25 C: 9/ 25 (no significant differences between groups, p-value not reported)
Staff administered boluses, n patients I: 7 (28%) C: 10 (40%) P<0.55 |
|
|
Petry, 2000 |
Type of study: RCT
Setting: University hospital Antwerp
Country: Belgium
Source of funding: not reported |
Inclusion criteria: parturients requesting epidural analgesia during labor.
Exclusion criteria: Those presenting with cervical dilatation exceeding 5cm (for which intrathecal analgesia was selected) or those scheduled for trial of labor were excluded.
N total at baseline: Intervention: 40 Control: 40
Important prognostic factors2:
Groups comparable at baseline? Yes
|
Describe intervention (treatment/procedure/test):
A 10 ml loading dose consisting of bupicacaine 0.125% with sufentanil 0.75 µg/ml and epinephrine 1/800.000 was administered without a test does. If after 10 minutes initial VAS scores (0-10) were not below 2 or were not reduced by 50%, additional increments of 2 ml of the same mixture were administerd.
PCEA 3 ml (of the same mixture as described above) with a lock-out time of 12 minutes and a 1 hour limit of 10 ml.
CI: Basal rate of 3 ml/h.
|
Describe control (treatment/procedure/test):
A 10 ml loading dose consisting of bupicacaine 0.125% with sufentanil 0.75 µg/ml and epinephrine 1/800.000 was administered without a test does. If after 10 minutes initial VAS scores (0-10) were not below 2 or were not reduced by 50%, additional increments of 2 ml of the same mixture were administerd.
PCEA 3 ml (of the same mixture as described above) with a lock-out time of 12 minutes and a 1 hour limit of 10 ml.
|
Length of follow-up: Until delivery
Loss-to-follow-up: Intervention: None
Control: None
Incomplete outcome data: Intervention: 3 (7.5%) Reasons 2 patients delivered within the first hour after epidural catheter placement, 1 patient was not considered for statistical analysis because of technicalpump problems.
Control: 3 (7.5%) Reasons 2 patients delivered within the first hour after epidural catheter placement, 1 patient was not considered for statistical analysis because of technicalpump problems.
|
Outcome measures and effect size (include 95%CI and p-value if available):
Pain score (VAS) 2nd stage I: 2.4 (SD 0.4) C: 2.9 (SD 0.5)
Mode of delivery (n) Instrumental I: 3/ 37 C: 1/ 37
Caesarean I: 1/ 37 C:1/ 37
Maternal adverse events, n (%) Hypotension: I: 0 C: 0
Pruritus I: 12/ 37 C: 10/ 37
Motor block, defined as difficulty to flex knee I: 2/ 37 C: 1/ 37
Apgarscores All neonates had Apgarscores equal to or exceeding ≥9 at five minutes.
Rescue boluses, n patients I: 1 C: 1 |
|
95% CI: 95% confidence interval; CI: continuous infusion; PCEA: patient controlled epidural analgesia; RCT: randomized controlled trial; SD: standard deviation; VAS: visual analogue scale
Notes:
- Prognostic balance between treatment groups is usually guaranteed in randomized studies, but non-randomized (observational) studies require matching of patients between treatment groups (case-control studies) or multivariate adjustment for prognostic factors (confounders) (cohort studies); the evidence table should contain sufficient details on these procedures.
- Provide data per treatment group on the most important prognostic factors ((potential) confounders).
- For case-control studies, provide sufficient detail on the procedure used to match cases and controls.
- For cohort studies, provide sufficient detail on the (multivariate) analyses used to adjust for (potential) confounders.
Evidence table for systematic review of RCTs and observational studies (intervention studies)
Research question: wat zijn de (on)gunstige effecten van PCEA met continue achtergrond infusie vergeleken met PCEA zonder continue achtergrond infusie bij zwangere vrouwen met het verzoek tot behandeling van de baringspijn?
95% CI: 95% confidence interval; CI: continuous infusion; PCEA: patient controlled epidural analgesia; RCT: randomized controlled trial; RR: risk ratio; SD: standard deviation; SR: systematic review; VAS: visual analogue scale
Risk of bias table for intervention studies (randomized controlled trials)
|
Study reference
(first author, publication year) |
Describe method of randomisation1 |
Bias due to inadequate concealment of allocation?2
(unlikely/likely/unclear) |
Bias due to inadequate blinding of participants to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of care providers to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to inadequate blinding of outcome assessors to treatment allocation?3
(unlikely/likely/unclear) |
Bias due to selective outcome reporting on basis of the results?4
(unlikely/likely/unclear) |
Bias due to loss to follow-up?5
(unlikely/likely/unclear) |
Bias due to violation of intention to treat analysis?6
(unlikely/likely/unclear) |
|
Bremerich 2005 |
Closed opaque envelope allocation |
Unlikely |
Unlikely |
Likely |
Likely |
Unlikely |
Unlikely |
Unlikely |
|
Okutomi 2009 |
Not descibed |
Unclear |
Unlikely |
Unclear |
Unlikely |
Unlikely |
Unlikely |
Unlikely |
|
Srivastava 2009 |
Computer generated randomization |
Unclear |
Unclear |
Likely |
Likely |
Unlikely |
Unlikely |
Unlikely |
|
Paech 1992 |
Computer-derived random number sequence |
Unclear |
Unlikely |
Likely |
Likely |
Unlikely |
Unlikely |
Unlikely |
|
Petry 2000 |
Not described |
Unclear |
Likely |
Likely |
Likely |
Unlikely |
Unlikely |
Likely |
- Randomisation: generation of allocation sequences have to be unpredictable, for example computer generated random-numbers or drawing lots or envelopes. Examples of inadequate procedures are generation of allocation sequences by alternation, according to case record number, date of birth or date of admission.
- Allocation concealment: refers to the protection (blinding) of the randomisation process. Concealment of allocation sequences is adequate if patients and enrolling investigators cannot foresee assignment, for example central randomisation (performed at a site remote from trial location) or sequentially numbered, sealed, opaque envelopes. Inadequate procedures are all procedures based on inadequate randomisation procedures or open allocation schedules.
- Blinding: neither the patient nor the care provider (attending physician) knows which patient is getting the special treatment. Blinding is sometimes impossible, for example when comparing surgical with non-surgical treatments. The outcome assessor records the study results. Blinding of those assessing outcomes prevents that the knowledge of patient assignement influences the proces of outcome assessment (detection or information bias). If a study has hard (objective) outcome measures, like death, blinding of outcome assessment is not necessary. If a study has “soft” (subjective) outcome measures, like the assessment of an X-ray, blinding of outcome assessment is necessary.
- Results of all predefined outcome measures should be reported; if the protocol is available, then outcomes in the protocol and published report can be compared; if not, then outcomes listed in the methods section of an article can be compared with those whose results are reported.
- If the percentage of patients lost to follow-up is large, or differs between treatment groups, or the reasons for loss to follow-up differ between treatment groups, bias is likely. If the number of patients lost to follow-up, or the reasons why, are not reported, the risk of bias is unclear’.
- Participants included in the analysis are exactly those who were randomized into the trial. If the numbers randomized into each intervention group are not clearly reported, the risk of bias is unclear; an ITT analysis implies that (a) participants are kept in the intervention groups to which they were randomized, regardless of the intervention they actually received, (b) outcome data are measured on all participants, and (c) all randomized participants are included in the analysis.
Table of quality assessment for systematic reviews of RCTs and observational studies
Based on AMSTAR checklist (Shea, 2007; BMC Methodol 7: 10; doi:10.1186/1471-2288-7-10) and PRISMA checklist (Moher, 2009; PLoS Med 6: e1000097; doi:10.1371/journal.pmed1000097)
|
Study
First author, year |
Appropriate and clearly focused question?1
Yes/no/unclear |
Comprehensive and systematic literature search?2
Yes/no/unclear |
Description of included and excluded studies?3
Yes/no/unclear |
Description of relevant characteristics of included studies?4
Yes/no/unclear |
Appropriate adjustment for potential confounders in observational studies?5
Yes/no/unclear/not applicable |
Assessment of scientific quality of included studies?6
Yes/no/unclear |
Enough similarities between studies to make combining them reasonable?7
Yes/no/unclear |
Potential risk of publication bias taken into account?8
Yes/no/unclear |
Potential conflicts of interest reported?9
Yes/no/unclear |
|
Heesen 2015 |
Yes |
Yes |
Yes |
No, patient characteristics were not described |
Not applicable |
Yes |
Yes |
Unclear, source of funding for the included studies was not reported |
|
- Research question (PICO) and inclusion criteria should be appropriate and predefined.
- Search period and strategy should be described; at least Medline searched; for pharmacological questions at least Medline + EMBASE searched.
- Potentially relevant studies that are excluded at final selection (after reading the full text) should be referenced with reasons.
- Characteristics of individual studies relevant to research question (PICO), including potential confounders, should be reported.
- Results should be adequately controlled for potential confounders by multivariate analysis (not applicable for RCTs).
- Quality of individual studies should be assessed using a quality scoring tool or checklist (Jadad score, Newcastle-Ottawa scale, risk of bias table et cetera).
- Clinical and statistical heterogeneity should be assessed; clinical: enough similarities in patient characteristics, intervention and definition of outcome measure to allow pooling? For pooled data: assessment of statistical heterogeneity using appropriate statistical tests (for example Chi-square, I2)?
- An assessment of publication bias should include a combination of graphical aids (for example funnel plot, other available tests) and/or statistical tests (for example Egger regression test, Hedges-Olken). Note: If no test values or funnel plot included, score “no”. Score “yes” if mentions that publication bias could not be assessed because there were fewer than 10 included studies.
- Sources of support (including commercial co-authorship) should be reported in both the systematic review and the included studies. Note: To get a “yes,” source of funding or support must be indicated for the systematic review AND for each of the included studies.
Tabel Exclusie na het lezen van het volledige artikel
|
Auteur en jaartal |
Redenen van exclusie |
|
Drachtidi 2014 |
Abstract voor congres, geen full tekst beschikbaar |
|
Sng 2014 |
Voldoen niet aan PICO |
Verantwoording
Autorisatiedatum en geldigheid
Laatst beoordeeld : 03-07-2020
Laatst geautoriseerd : 03-07-2020
Geplande herbeoordeling : 01-01-2025
Bij het opstellen van de modules heeft de werkgroep een inschatting gemaakt over de maximale termijn waarop herbeoordeling moet plaatsvinden en eventuele aandachtspunten geformuleerd die van belang zijn bij een toekomstige herziening (update). De geldigheid van de richtlijnmodules komt eerder te vervallen indien nieuwe ontwikkelingen aanleiding zijn een herzieningstraject te starten.
De andere aan deze richtlijnmodule deelnemende wetenschappelijke verenigingen of gebruikers van de richtlijnmodule delen de verantwoordelijkheid en informeren de regiehouder over relevante ontwikkelingen binnen hun vakgebied.
|
Module[1] |
Regiehouder(s)[2] |
Jaar van autorisatie |
Eerstvolgende beoordeling actualiteit richtlijn[3] |
Frequentie van beoordeling op actualiteit[4] |
Wie houdt er toezicht op actualiteit[5] |
Relevante factoren voor wijzigingen in aanbeveling[6] |
|
PCEA met versus zonder achtergrond |
NVA/ NVOG |
2019 |
2024 |
Eens in de 5 jaar |
Regiehouder en relevante wetenschappelijke verenigingen |
Zodra PIEB landelijk wordt toegepast zullen de aanbevelingen in deze module opnieuw geëvalueerd moeten worden. |
[1] Naam van de module
[2] Regiehouder van de module (deze kan verschillen per module en kan ook verdeeld zijn over meerdere regiehouders)
[3] Maximaal na vijf jaar
[4] (half)Jaarlijks, eens in twee jaar, eens in vijf jaar
[5] regievoerende vereniging, gedeelde regievoerende verenigingen, of (multidisciplinaire) werkgroep die in stand blijft
[6] Lopend onderzoek, wijzigingen in vergoeding/organisatie, beschikbaarheid nieuwe middelen
Algemene gegevens
Deze richtlijn is ontwikkeld in samenwerking met:
- Patiëntenfederatie Nederland
De ontwikkeling van de richtlijnmodules werd ondersteund door het Kennisinstituut van de Federatie Medisch Specialisten en werd gefinancierd uit de Stichting Kwaliteitsgelden Medisch Specialisten (SKMS). De financier heeft geen enkele invloed gehad op de inhoud van de richtlijnmodules.
Samenstelling werkgroep
- Dr. W.L.M.C.M. Schellekens, extern voorzitter
- Drs. I.C.M. Beenakkers, anesthesioloog, werkzaam in het UMC Utrecht, NVA
- Drs. F.A. Klerk, anesthesioloog, werkzaam in het Diakonessenhuis Utrecht, NVA
- Drs. C.E. Kam-Endtz, anesthesioloog, werkzaam in het Haaglanden Medisch Centrum, NVA
- Dr. F.T.H. Lim, gynaecoloog, werkzaam in het IJssellandziekenhuis, NVOG
- Dr. L.M. Freeman, gynaecoloog, werkzaam in het Ikazia Ziekenhuis Rotterdam, NVOG
- Dr. J.M. Middeldorp, gynaecoloog, werkzaam in het Leids Universitair Medisch Centrum, NVOG
- Drs. A.G. Kaspers, kinderarts, werkzaam in het Medisch Spectrum Twente, NVK
- Drs. L.A.M. Moll, klinisch verloskundige, werkzaam in het St. Antonius Ziekenhuis Nieuwegein, KNOV
- Dr. J. de Boer, beleidsmedewerker bij KNOV
- Drs. S. Ratsma-Wesselius, Obstetrisch verpleegkundige, werkzaam bij het Amsterdam UMC, Locatie AMC, V&VN
- Dr. J.E. Nagtegaal, Ziekenhuisapotheker, werkzaam in het Meander Medisch Centrum, NVZA
- Dr. A.M.D.E. Timmerman, Klinisch Fysicus, werkzaam in het UMC Utrecht, NVKF
- Drs. J.C. Mooij, adviseur patiëntenbelang, Patiëntenfederatie Nederland.
Met ondersteuning van
- Dr. E.M.E. den Breejen, senior adviseur Kennisinstituut van de Federatie Medisch Specialisten
- Dr. W.J. Harmsen, adviseur Kennisinstituut van de Federatie Medisch Specialisten
Belangenverklaringen
De KNMG-code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling is gevolgd. Alle werkgroepleden hebben schriftelijk verklaard of zij in de laatste drie jaar directe financiële belangen (betrekking bij een commercieel bedrijf, persoonlijke financiële belangen, onderzoeksfinanciering) of indirecte belangen (persoonlijke relaties, reputatiemanagement, kennisvalorisatie) hebben gehad. Een overzicht van de belangen van werkgroepleden en het oordeel over het omgaan met eventuele belangen vindt u in onderstaande tabel. De ondertekende belangenverklaringen zijn op te vragen bij het secretariaat van het Kennisinstituut van de Federatie Medisch Specialisten.
|
Achternaam werkgroeplid |
Functie |
Nevenfuncties |
Gemelde belangen |
Ondernomen actie |
|
Schellekens |
gepensioneerd, ZZP: strategisch adviseur |
Lid RvT Diakonessenhuis, Utrecht |
Geen |
Geen |
|
Klerk |
Staf Anesthesiologie Diakonessenhuis Utrecht. |
bestuurslid Obst. Anesth. onbetaald |
Geen |
Geen |
|
Ratsma Wesselius |
Senior verpleegkundige Verloscentrum AMC |
Gastdocent VU Amstel Academie verpleegkundige vervolgopleidingen Obstetrie, betaald |
Geen |
Geen |
|
Beenakkers |
Anesthesioloog WKZ/UMCU |
Voorzitter sectie obstetrische anesthesie van de NVA. Onbetaald |
Echtgenoot werkzaam bij GSK |
Geen |
|
Nagtegaal |
Ziekenhuisapotheker Meander Medisch Centrum |
Beroepenveldcommissie Farmakunde Hogeschool Utrecht, onbetaald |
Geen |
Geen |
|
Timmerman |
Staffunctionaris Klinische Fysica & Patiëntveiligheid |
Lid NIVEL expertgroep infuustechnologie - advies maken kennistoets voor verpleegkundigen – onbetaald Lid ondernemingsraad UMC Utrecht - onbetaald |
EMRP Researcher Grant Metrology for Drug Delivery HLT07- REG1 €120,422.88 USPTO Applicaton #: #20160106909 Apparatus for simultaneous multiple medicament administration |
Geen |
|
Kam-Endtz |
Anesthesioloog Haaglanden MC |
Geen |
Geen |
Geen |
|
Middeldorp |
Gynaecoloog-perinatoloog |
Geen |
Geen |
Geen |
|
Moll |
Klinisch verloskundige/research verloskundige in het St. Antoniusziekenhuis in Nieuwegein |
Geen |
Geen |
Geen |
|
Freeman |
Gynaecoloog |
voorzitter multidiciplinaire werkgroep obstetrische anesthesie
|
Mijn promotieonderzoek naar epidurale analgesie en remifentanil is gesubsidieerd door ZonMw. Dit onderzoek is afgerond maar de resultaten zullen gebruikt worden in deze richtlijn |
Geen |
|
Mooij |
Beleidsmedewerker Patiëntenvereniging Nederland |
Vrijwilligerswerk (onbetaald) patiëntenorganisatie CCUVN |
Geen |
Geen |
|
Kaspers |
Kinderarts-neonatoloog, MST Enschede |
Geen |
Geen |
Geen |
|
De Boer |
Beleidsmedewerker richtlijnontwikkeling |
Geen nevenwerkzaamheden |
Geen |
Geen |
|
Lim |
gynaecoloog |
Geen |
Geen |
Geen |
Inbreng patiëntenperspectief
Er werd aandacht besteed aan het patiëntenperspectief door een afgevaardigde van de Patientenfederatie Nederland in de werkgroep te laten deelnemen. De conceptmodule is tevens voor commentaar voorgelegd aan de Patiëntenfederatie Nederland. Daarnaast is door de Patiëntenfederatie Nederland een achterbanraadpleging verricht, waarvan de uitkomsten zo veel mogelijk meegenomen zijn in de overwegingen van de modules.
Methode ontwikkeling
Evidence based
Implementatie
In de verschillende fasen van het ontwikkelproces is rekening gehouden met de implementatie van de richtlijnmodule en de praktische uitvoerbaarheid van de aanbevelingen. Daarbij is uitdrukkelijk gelet op factoren die de invoering van de module in de praktijk kunnen bevorderen of belemmeren. De implementatietabel is te vinden bij de aanverwante producten.
Werkwijze
AGREE
Deze modules zijn opgesteld conform de eisen vermeld in het rapport Medisch Specialistische Richtlijnen 2.0 van de adviescommissie Richtlijnen van de Raad Kwaliteit. Dit rapport is gebaseerd op het AGREE II instrument (Appraisal of Guidelines for Research & Evaluation II; Brouwers, (2010)), dat een internationaal breed geaccepteerd instrument is. Voor een stap-voor-stap beschrijving hoe een evidence-based module tot stand komt wordt verwezen naar het stappenplan Ontwikkeling van Medisch Specialistische Richtlijnen van het Kennisinstituut van de Federatie Medisch Specialisten.
Knelpuntenanalyse
Uit inventarisatie van de knelpunten door de commissie van de NVA bleek dat er een noodzaak was voor revisie van deze richtlijnmodules. Tevens zijn tijdens een fysieke knelpunteninventarisatie knelpunten aangedragen door aanpalende stakeholders inclusief patiëntenorganisaties. Een verslag hiervan is opgenomen onder aanverwante producten.
Uitgangsvraag en uitkomstmaten
Op basis van de uitkomsten van de knelpuntenanalyse zijn door de werkgroepleden en de adviseur uitgangsvragen opgesteld. Vervolgens inventariseerde de werkgroep welke uitkomstmaten voor de patiënt relevant zijn. De werkgroep waardeerde deze uitkomstmaten volgens hun relatieve belang bij de besluitvorming rondom aanbevelingen, als cruciaal (kritiek voor de besluitvorming), belangrijk (maar niet kritiek) en onbelangrijk. Tevens definieerde de werkgroep tenminste voor de cruciale uitkomstmaten welke verschillen zij klinisch (patiënt) relevant vonden.
Strategie voor zoeken en selecteren van literatuur
Aan de hand van specifieke zoektermen werd gezocht naar gepubliceerde wetenschappelijke studies in (verschillende) elektronische databases. Tevens werd aanvullend gezocht naar studies aan de hand van de literatuurlijsten van de geselecteerde artikelen. In eerste instantie werd gezocht naar studies met de hoogste mate van bewijs. De werkgroepleden selecteerden de via de zoekactie gevonden artikelen op basis van vooraf opgestelde selectiecriteria. De geselecteerde artikelen werden gebruikt om de uitgangsvraag te beantwoorden. De geselecteerde databases waarin is gezocht en de gehanteerde selectiecriteria zijn te vinden in de module met desbetreffende uitgangsvraag. De zoekstrategie is opvraagbaar bij de Richtlijnendatabase, zie het tabblad ‘Zoekverantwoording’ voor verdere details.
Kwaliteitsbeoordeling individuele studies
Individuele studies werden systematisch beoordeeld, op basis van op voorhand opgestelde methodologische kwaliteitscriteria, om zo het risico op vertekende studieresultaten (risk of bias) te kunnen inschatten. Deze beoordelingen kunt u vinden in de Risk of Bias (RoB) tabellen. De gebruikte RoB instrumenten zijn gevalideerde instrumenten die worden aanbevolen door de Cochrane Collaboration: QUADAS II - voor diagnostisch onderzoek.
Samenvatten van de literatuur
De relevante onderzoeksgegevens van alle geselecteerde artikelen werden overzichtelijk weergegeven in evidencetabellen. De belangrijkste bevindingen uit de literatuur werden beschreven in de samenvatting van de literatuur.
Beoordelen van de kracht van het wetenschappelijke bewijs
De kracht van het wetenschappelijke bewijs werd bepaald volgens de GRADE-methode. GRADE staat voor ‘Grading Recommendations Assessment, Development and Evaluation’ (zie http://www.gradeworkinggroup.org/).
GRADE onderscheidt vier gradaties voor de kwaliteit van het wetenschappelijk bewijs: hoog, redelijk, laag en zeer laag. Deze gradaties verwijzen naar de mate van zekerheid die er bestaat over de literatuurconclusie (Schünemann, 2013).
|
GRADE |
Definitie |
|
Hoog |
|
|
Redelijk |
|
|
Laag |
|
|
Zeer laag |
|
In de gehanteerde generieke GRADE-methode werden de basisprincipes van de GRADE-methodiek toegepast: het benoemen en prioriteren van de klinisch (patiënt) relevante uitkomstmaten, een systematische review per uitkomstmaat, en een beoordeling van bewijskracht op basis van de vijf GRADE-criteria (startpunt hoog; downgraden voor risk of bias, inconsistentie, indirectheid, imprecisie, en publicatiebias).
Formuleren van de conclusies
Voor elke relevante uitkomstmaat werd het wetenschappelijk bewijs samengevat in een of meerdere literatuurconclusies waarbij het niveau van bewijs werd bepaald volgens de GRADE-methodiek. De werkgroepleden maakten de balans op van elke interventie (overall conclusie). Bij het opmaken van de balans werden de gunstige en ongunstige effecten voor de patiënt afgewogen. De overall bewijskracht wordt bepaald door de laagste bewijskracht gevonden bij een van de kritieke uitkomstmaten. Bij complexe besluitvorming waarin naast de conclusies uit de systematische literatuuranalyse vele aanvullende argumenten (overwegingen) een rol spelen, werd afgezien van een overall conclusie. In dat geval werden de gunstige en ongunstige effecten van de interventies samen met alle aanvullende argumenten gewogen onder het kopje 'Overwegingen'.
Overwegingen (van bewijs naar aanbeveling)
Om te komen tot een aanbeveling zijn naast (de kwaliteit van) het wetenschappelijke bewijs ook andere aspecten belangrijk en worden meegewogen, zoals de expertise van de werkgroepleden, de waarden en voorkeuren van de patiënt, kosten, beschikbaarheid van voorzieningen en organisatorische zaken. Deze aspecten worden, voor zover geen onderdeel van de literatuursamenvatting, vermeld en beoordeeld (gewogen) onder het kopje ‘Overwegingen’.
Formuleren van aanbevelingen
De aanbevelingen geven antwoord op de uitgangsvraag en zijn gebaseerd op het beschikbare wetenschappelijke bewijs en de belangrijkste overwegingen, en een weging van de gunstige en ongunstige effecten van de relevante interventies. De kracht van het wetenschappelijk bewijs en het gewicht dat door de werkgroep wordt toegekend aan de overwegingen, bepalen samen de sterkte van de aanbeveling. Conform de GRADE-methodiek sluit een lage bewijskracht van conclusies in de systematische literatuuranalyse een sterke aanbeveling niet a priori uit, en zijn bij een hoge bewijskracht ook zwakke aanbevelingen mogelijk. De sterkte van de aanbeveling wordt altijd bepaald door weging van alle relevante argumenten tezamen.
Randvoorwaarden (Organisatie van zorg)
Bij de ontwikkeling van de modules is expliciet rekening gehouden met de organisatie van zorg: alle aspecten die randvoorwaardelijk zijn voor het verlenen van zorg (zoals coördinatie, communicatie, (financiële) middelen, menskracht en infrastructuur). Randvoorwaarden die relevant zijn voor het beantwoorden van een specifieke uitgangsvraag maken onderdeel uit van de overwegingen bij de bewuste uitgangsvraag, randvoorwaarden die van invloed zijn op de implementatie van de aanbeveling zijn opgenomen in de implementatietabel.
Indicatorontwikkeling
Indicatoren over zwangerschap en geboorte zijn reeds onderdeel van de vervaardigde indicatoren bij de zorgstandaard integrale geboortezorg. Derhalve zijn er bij deze modules geen indicatoren ontwikkeld.
Kennislacunes
Tijdens de ontwikkeling van deze modules is systematisch gezocht naar onderzoek waarvan de resultaten bijdragen aan een antwoord op de uitgangsvraag. Er is nagegaan of (aanvullend) wetenschappelijk onderzoek gewenst is om de uitgangsvraag te kunnen beantwoorden. Mocht dit bij deze module het geval zijn, dan is er een aanbeveling voor het doen van onderzoek opgenomen in de Kennislacunes. Deze zijn te vinden onder de aanverwante producten.
Commentaar- en autorisatiefase
De conceptmodules werden aan de betrokken (wetenschappelijke) verenigingen, instanties en (patiënt) organisaties voorgelegd ter commentaar. De commentaren werden verzameld en besproken met de werkgroep. Naar aanleiding van de commentaren werden de conceptmodules aangepast en definitief vastgesteld door de werkgroep. De definitieve modules werden aan de deelnemende (wetenschappelijke) verenigingen en de Patiëntenfederatie Nederland voorgelegd voor autorisatie en door hen geautoriseerd dan wel geaccordeerd. De commentaartabel is op te vragen bij het Kennisinstituut via secretariaat@kennisinstituut.nl
Literatuur
Brouwers MC, Kho ME, Browman GP, et al. AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839-42. doi: 10.1503/cmaj.090449. Epub 2010 Jul 5. Review. PubMed PMID: 20603348.
Medisch Specialistische Richtlijnen 2.0 (2012). Adviescommissie Richtlijnen van de Raad Kwalitieit. https://richtlijnendatabase.nl/over_deze_site/richtlijnontwikkeling.html
Schünemann H, Brożek J, Guyatt G, et al. GRADE handbook for grading quality of evidence and strength of recommendations. Updated October 2013. The GRADE Working Group, 2013. Available from http://gdt.guidelinedevelopment.org/central_prod/_design/client/handbook/handbook.html.
Schünemann HJ, Oxman AD, Brozek J, et al. Grading quality of evidence and strength of recommendations for diagnostic tests and strategies. BMJ. 2008;336(7653):1106-10. doi: 10.1136/bmj.39500.677199.AE. Erratum in: BMJ. 2008;336(7654). doi: 10.1136/bmj.a139. PubMed PMID: 18483053.
Ontwikkeling van Medisch Specialistische Richtlijnen: stappenplan. Kennisinstituut van de Federatie Medisch Specialisten.
Wessels M, Hielkema L, van der Weijden T. How to identify existing literature on patients' knowledge, views, and values: the development of a validated search filter. J Med Libr Assoc. 2016 Oct;104(4):320-324. PubMed PMID: 27822157; PubMed Central PMCID: PMC5079497.
Zoekverantwoording
Zoekacties zijn opvraagbaar. Neem hiervoor contact op met de Richtlijnendatabase.